

Abstract
Multiple sclerosis (MS) is a chronic inflammatory disorder of the CNS characterized by demyelination and axonal injury. Current therapies that mainly target lymphocytes do not fully meet clinical need due to the risk of severe side effects and lack of efficacy against progressive MS. Evidence suggests that MS is associated with CNS inflammation, although the underlying molecular mechanism is poorly understood. Transient receptor potential melastatin 2 (TRPM2), a Ca2+-permeable nonselective cation channel, is expressed at high levels in the brain and by immune cells, including monocyte lineage cells. Here, we show that TRPM2 plays a pathological role in experimental autoimmune encephalomyelitis (EAE), an animal model of MS. Knockout (KO) or pharmacological inhibition of TRPM2 inhibited progression of EAE and TRPM2-KO mice showed lower activation of Iba1-immunopositive monocyte lineage cells and neutrophil infiltration of the CNS than WT mice. Moreover, CXCL2 production in TRPM2-KO mice was significantly reduced at day 14, although the severity of EAE was the same as that in WT mice at that time point. In addition, we used BM chimeric mice to show that TRPM2 expressed by CNS-infiltrating macrophages contributes to progression of EAE. Because CXCL2 induces migration of neutrophils, these results indicate that reduced expression of CXCL2 in the CNS suppresses neutrophil infiltration and slows progression of EAE in TRPM2-KO mice. Together, the results suggest that TRPM2 plays an important role in progression of EAE pathology and shed light on its putative role as a therapeutic target for MS.
SIGNIFICANCE STATEMENT Current therapies for multiple sclerosis (MS), which mainly target lymphocytes, carry the risk of severe side effects and lack efficacy against the progressive form of the disease. Here, we found that the transient receptor potential melastatin 2 (TRPM2) channel, which is abundantly expressed in CNS-infiltrating macrophages, plays a crucial role in development of experimental autoimmune encephalomyelitis (EAE), an animal model of MS. EAE progression was suppressed by Knockout (KO) or pharmacological inhibition of TRPM2; this was attributed to a reduction in CXCL2 chemokine production by CNS-infiltrating macrophages in TRPM2-KO mice, resulting in suppression of neutrophil infiltration into the CNS. These results reveal an important role of TRPM2 in the pathogenesis of EAE and shed light on its potential as a therapeutic target.
Keywords: cxcl2, macrophage, multiple sclerosis, neutrophil, TRP channel, TRPM2
Introduction
Multiple sclerosis (MS) is a demyelinating disease of the CNS and is thought to have an autoimmune etiology. This is supported by the clinical efficacy of immunomodulatory agents that prevent lymphocyte infiltration into the CNS (e.g., fingolimod, a sphingosine-1-phosphate receptor modulator, and natalizumab, a humanized monoclonal antibody against α4 integrin; Steinman, 2014). However, severe adverse effects, such as severe infectious disease and progressive multifocal leukoencephalopathy (PML), can occur in patients receiving immunosuppressive therapies (Castro-Borrero et al., 2012). Although the etiology of PML in immunosuppressed patients is unclear, infection of the CNS by the JC virus is thought to be a risk factor; thus a reduction in T- and B-lymphocyte numbers in the CNS caused by immunosuppressive agents likely increases the risk (Beltrami and Gordon, 2014). Therefore, other types of therapy are required. Recent studies focused on not only adaptive immune cells such as T and B cells, but also on innate immune cells such as macrophages and neutrophils, cells that play a pathological role in MS and its animal equivalent, experimental autoimmune encephalomyelitis (EAE) (Mishra and Yong, 2016; Prinz and Priller, 2017). Recent studies report that activation of monocyte lineage cells such as macrophages and microglia in the CNS is characteristic of MS and EAE (Farez et al., 2009; Politis et al., 2012). However, emerging evidence indicates that neutrophils play an important role (Rumble et al., 2015; Pierson et al., 2018), as well as in other inflammatory white matter disorders such as neuromyelitis optica (NMO) (Lucchinetti et al., 2002; Juryńczyk et al., 2015). In this context, neutrophils in the blood of patients with MS as well as NMO show upregulated expression of adhesion molecules and increased migration, although their numbers are no different from those in healthy controls (Naegele et al., 2012; Hertwig et al., 2016). Consistent with these observations in humans, neutrophil depletion ameliorates EAE clinical scores significantly in mice (McColl et al., 1998). Given that neutrophils that infiltrate the CNS can cause extensive inflammation, demyelination, and axonal damage via production of cytotoxic mediators, inhibition of excessive neutrophil activation and migration into the CNS is important. However, the molecular relationships between activated monocyte lineage cells and infiltrating neutrophils and the precise mechanisms that regulate EAE progression remain unresolved. Moreover, there are no potential drug targets that suppress CNS inflammation during EAE progression.
Transient receptor potential melastatin 2 (TRPM2), a Ca2+-permeable and nonselective cation channel, is expressed in the brain at high levels (Nagamine et al., 1998) and broadly by immune cells (Sano et al., 2001). TRPM2 harbors a Nudix box motif in its intracellular C-terminal tail and is gated by intracellular ADP ribose (Perraud et al., 2001) and reactive oxygen species (ROS), including hydrogen peroxide (Hara et al., 2002; Kaneko et al., 2006). Previous studies focused extensively on the pathophysiological role of TRPM2 in immune cells. For example, TRPM2-mediated Ca2+ influx induces production of proinflammatory cytokines/chemokines by monocytes/macrophages and exacerbates inflammation in ulcerative colitis (Yamamoto et al., 2008) and ischemic stroke (Gelderblom et al., 2014). We previously reported that TRPM2 expressed by microglia and macrophages aggravates peripheral and spinal pronociceptive inflammatory responses in a mouse model of neuropathic pain (Haraguchi et al., 2012; Isami et al., 2013) and chronic cerebral hypoperfusion (Miyanohara et al., 2018), in which lipopolysaccharide/IFNγ-induced TRPM2 opening results in inflammatory responses in microglia via the p38/JNK pathway (Miyake et al., 2014). Given that ROS production and endogenous TLR2/4 agonists, including HMGB1, in the CNS are increased in EAE (Sun et al., 2015; Hasseldam et al., 2016), it is likely that TRPM2 expressed in monocytic lineage cells aggravates inflammatory processes in EAE. Here, we used WT and TRPM2-KO mice to demonstrate that TRPM2 has an important role in regulating chronic inflammation in the CNS by reducing neutrophil infiltration via suppression of a major neutrophil chemoattractant, CXCL2.
Materials and Methods
Mice.
All experiments were conducted in accordance with the ethical guidelines of the Kyoto University Animal Experimentation Committee and the guidelines of the Japanese Pharmacological Society. Female C57BL/6J mice (RRID:IMSR_JAX:000664) and TRPM2-KO mice (7–12 weeks old, 18–22 g) were maintained in-house. TRPM2-KO mice were generated as reported previously (Yamamoto et al., 2008) and backcrossed with C57BL/6J mice for 10 generations to eliminate any background effects on the phenotype. C57BL/6J mice and C57BL/6-Tg (CAG-EGFP)-transgenic mice (GFP-transgenic mice) were purchased from Japan SLC. All experiments in this study used WT C57BL/6 mice purchased from Japan SLC as controls; control mice were domesticated for at least 1 week under the same conditions as TRPM2-KO mice. Mice were kept at a constant ambient temperature of 22 ± 2°C under a 12 h light/dark cycle with ad libitum access to food and water.
EAE induction and symptom evaluation.
EAE was induced as described previously (So et al., 2015). Briefly, mice were immunized subcutaneously with 100 μg of myelin oligodendrocyte glycoprotein peptide 35–55 (MOG35-55; Eurofins Genomics) in complete Freund's adjuvant (Difco) containing 3 mg/ml Mycobacterium tuberculosis (H37Ra; Difco). Nonimmunized control mice received saline. Mice also received 10 μg/kg pertussis toxin (List Biological Laboratories) intraperitoneally on the day of immunization and again 48 h later. Mice were scored daily for clinical signs as follows: 0 = no clinical deficit; 1 = partial tail paralysis; 2 = full tail paralysis; 3 = partial hindlimb paralysis; 4 = full hindlimb paralysis; 5 = forelimb paresis; and 6 = dead. At day 16, mice were randomized into treatment/vehicle groups and intraperitoneally injected with 10 mg/kg miconazole (LKT Laboratories) or vehicle (saline containing 1% DMSO and 2% Tween 80) on a daily basis until day 28.
Electron microscopy.
Mice were deeply anesthetized with sodium pentobarbital (65 mg/kg body weight; Sumitomo Dainippon Pharm). Next, saline was perfused through the ascending aorta, followed by 4% paraformaldehyde (PFA). The spinal cords were then removed and dissected. After postfixing overnight at 4°C in 4% PFA/2% glutaraldehyde, the spinal cords were exposed to 1% osmium tetroxide in 0.1 m phosphate buffer, pH 7.4, for 2 h at room temperature, dehydrated by immersion in a series of graded ethanol solutions, and embedded in epoxy-resin (Luveak 812; Nacalai Tesque) according to the inverted beam capsule procedure. Samples were polymerized at 60°C for 3 d. Ultrathin sections were prepared on an ultramicrotome (EM UC6; Leica) and stained with uranyl acetate and lead citrate. Finally, sections were observed under a Hitachi H-7650 transmission electron microscope. Seven images obtained from each mouse were analyzed about axon number in a visual way and the mean result was regarded as an n = 1 data. Four mice were used for statistical analysis.
Flow cytometric analysis.
Mice were deeply anesthetized with sodium pentobarbital (65 mg/kg body weight; Sumitomo Dainippon Pharm) and killed. The spleen, draining lymph nodes (inguinal and axillary), and spinal cord were then removed. Spleens and lymph nodes were homogenized through a 40 μm cell strainer and centrifuged at 500 × g for 5 min at 4°C. The cells were the suspended in red blood lysis buffer (eBioscience) and kept stationary for 5 min at room temperature. After centrifugation (500 × g; 5 min; 4°C), the cells were resuspended in FACS buffer (eBioscience) and counted. To obtain enough cells for flow cytometry analysis, spinal cords from three mice were pooled and homogenized through a 100 μm cell strainer. After centrifugation (500 × g; 5 min; 4°C), the pellet was resuspended in complete RPMI medium containing 1 mg/ml collagenase D (Roche) and 10 μg/ml DNase I (Sigma-Aldrich) and incubated for 1 h at 37°C with gentle agitation. After centrifugation (500 × g; 5 min; room temperature), the pellet was resuspended in 5 ml of 70% Percoll (Sigma-Aldrich). Next, 5 ml of 40% Percoll was layered carefully on top of the cells (which were suspended in 70% Percoll) and then centrifuged at 600 × g for 20 min at room temperature with the brake off. The mononuclear cells were removed from the interface and centrifuged at 500 × g for 5 min at 4°C. After resuspension in FACS buffer, 1 × 106 cells were incubated for 10 min at 4°C with Fc block buffer (BD Biosciences) and then stained with both PE-conjugated hamster anti-mouse CD3 (BD Biosciences) and FITC-conjugated rat anti-mouse CD4 (BD Biosciences) antibodies. Cells were then incubated for 20 min at 4°C. For intracellular staining, cells were fixed and permeabilized with a FoxP3 staining buffer set (Miltenyi Biotec) and stained with T-bet monoclonal antibody (catalog #12-5825-82; eBioscience), ROR gamma (t) monoclonal antibody (catalog #12-6981-80; eBioscience) and Foxp3 monoclonal antibody (catalog #17-5773-80; eBioscience) for 20 min at 4°C. After centrifugation (500 × g; 5 min; 4°C), the cells were resuspended in FACS buffer and analyzed using FACS Aria II flow cytometer and FACSDiva software (BD Biosciences).
Histological examination.
Mice were deeply anesthetized with sodium pentobarbital (65 mg/kg body weight; Sumitomo Dainippon Pharm) and the ascending aorta perfused with saline followed by PFA. Spinal cords were then removed and dissected. After postfixing for 3 h in 4% PFA followed by cryoprotection overnight at 4°C in 15% sucrose, spinal cords were dehydrated with graded ethanol and xylene, embedded in paraffin, and cut into sections (5 μm thick) with a sliding microtome. Histological sections were stained with hematoxylin and eosin (H&E) or Luxol fast blue (LFB) and examined by light microscopy (BZ-9000; KEYENCE). To quantify demyelination, spinal cords were embedded in optimal cutting temperature (OCT) compound, cut into 16-μm-thick sections by a cryomicrotome (Leica), and stained with FluoroMyelin green fluorescent myelin stain (Invitrogen) and mounted with DAPI Fluoromount-G (Southern Biotech). The demyelinated and total areas of white matter were measured using ImageJ software. The final percentage value for demyelination was calculated using the following equation list: demyelinated area (%) = (demyelinated area in white matter)/(total white matter area) × 100 (%).
Immunohistochemistry.
Anesthetized mice were transcardially perfused with PBS and the lumbar spinal cord (L3–L5) was fixed in 4% PFA and embedded in the OCT compound. Coronal sections (16 μm thick) were cut using a cryomicrotome (Leica), blocked with PBS containing 3% bovine serum albumin, and permeabilized with 0.1% Triton X-100 in blocking solution. Appropriate primary antibodies were added overnight (at 4°C) at the following dilutions: anti-CD3 (catalog #555273, rat IgG, 1:200; BD Biosciences), anti-Iba1 (catalog #019-19741, rabbit IgG, 1:200; Wako), anti-GSTpi (catalog #311, rabbit IgG, 1:200; MBL), anti-GFAP (catalog #Z0334, rabbit IgG, 1:200; Dako), anti-Gr1 (catalog #RB6-8C5, rat IgG, 1:200; R&D Systems), and anti-CXCL2 (catalog #AF452, goat IgG, 1:200; R&D Systems). Secondary antibodies were as follows: Alexa Fluor 594 (catalog #A11058, anti-goat IgG, 1:200; Life Technologies) and Alexa Fluor 488 (catalog #A21206, anti-rabbit IgG, and catalog #A21208, anti-rat IgG, 1:200; Life Technologies). Secondary antibodies were added for 1.5 h at room temperature. Immunofluorescence was visualized under a laser scanning confocal imaging microscope (Fluoview FV10i; Olympus). The number of CD3 and Gr1-immunoreactive cells and the intensity of the Iba1-immunofluorescent signal in the white matter of the spinal cord were quantified in 600 × 600 μm2 fields using ImageJ software, as described previously (Munakata et al., 2013).
Quantitative real-time PCR.
Total RNA was extracted from the spinal cords (L3–L5) of EAE mice using the ISOGEN (Wako) method. Next, cDNA was synthesized using ReverTra Ace qPCR RT Master Mix (TOYOBO). Real-time quantitative PCR was performed using the StepOne real-time PCR system (Applied Biosystems) with Power SYBR Green PCR Master Mix (Applied Biosystems) in a final volume of 20 μl, as described previously (Shirakawa et al., 2017). The following oligonucleotide primers were used: 5′-GCA ATT ATT CCC CAT GAA CG-3′ and 5′-GGC CTC ACT AAA CCA TCC AA-3′ for 18s ribosomal RNA (18s rRNA); 5′-CGG AGC GGA CCA ACA GCA TCG TTT C-3′ and 5′-CAG GGT AGC CAT CCA CGG GCG GGT-3′ for Tbet; 5′-GGC AGA ACC GGC CCC TTA TC-3′ and 5′-TGG TCT GAC AGT TCG CGC AG-3′ for Gata3; 5′-AAG CTG AAG GCA GAG ACA GC-3′ and 5′-TGT TCT GGT TCC CCA AGT TC-3′ for Rorc; 5′-CCT GGT TGT GAG AAG GTC TTC G-3′ and 5′-TGC TCC AGA GAC TGC ACC ACT T-3′ for Foxp3; 5′-ATG CCT GAA GAC CCT GCC AAG-3′ and 5′-GGT CAG TTA GCC TTG CCT TTG-3′ for Cxcl2; and 5′-ACA GAC AAT GCC TGG ATC G-3′ and 5′-TGG ATC ATG AGT GTG CAG GT-3′ for Trpm2. The cycling conditions were as follows: 95°C for 10 min, followed by 35 cycles of 95°C for 15 s and 60°C for 60 s. The results were analyzed using Primer Express software (Applied Biosystems). The identity of the PCR product was confirmed by automated determination of the melting temperature. The results for each gene were normalized to 18s rRNA levels measured in parallel in each sample.
Cytokine analysis.
After anesthetized mice were transcardially perfused with PBS, lumbar spinal cords (L3–L5) were harvested and homogenized in ice-cold homogenizing buffer (PBS containing 0.1% Triton X-100 and 1% protease inhibitor mixture). The homogenates were centrifuged to remove debris and the protein concentration in the supernatant was measured using DC protein assay reagents (Bio-Rad). Cytokine levels in tissue homogenates were measured using a mouse cytokine ELISA kit (R&D Systems) according to the manufacturer's instructions.
Generation of BM (BM) chimeric mice.
Homozygous TRPM2−/− male mice were crossed with GFP-transgenic female mice to produce GFP+ TRPM2+/− mice. GFP+ TRPM2+/+ (WT) mice and GFP+ TRPM2−/− (TRPM2-KO) mice were obtained by hetero-mating of GFP+ TRPM2+/− female and male mice to obtain suitable BM donor mice. BM transplantation was performed as described previously (Isami et al., 2013; Miyanohara et al., 2018), with slight modifications. BM recipients were female 6-week-old C57BL/6J or TRPM2-KO mice. Recipient mice were lethally irradiated with 8 Gy of total body irradiation for 10 min. GFP+ WT or TRPM2-KO donor mice were then killed by decapitation, the femurs were isolated, and both ends were cut and placed into a microtube. The femurs were centrifuged at 400 × g for 10 min and the pellet of GFP+ BM cells was suspended in sterile PBS. Between 3 and 5 h after irradiation, WT or TRPM2-KO recipient mice received 1.5 × 107 BM cells by intravenous injection into the tail vein. WT recipient mice received WT donor mouse-derived GFP+ BM cells (WT-BM→WT) or TRPM2-KO donor mouse-derived GFP+ BM cells (TRPM2-KO-BM→WT), TRPM2-KO recipient mice received WT donor mouse-derived GFP+ BM cells (WT-BM→TRPM2-KO), and TRPM2-KO recipient mice received TRPM2-KO donor mouse-derived GFP+ BM cells (TRPM2-KO-BM→TRPM2-KO). All mice were housed in specific pathogen-free conditions with ad libitum access to autoclaved pellets and autoclaved water. After 6 weeks, female BM chimeric mice were used for EAE induction (age, 12 weeks).
Experimental design and statistical analysis.
Statistical analysis was performed using Prism5 software (GraphPad Software; RRID:SCR_002798). Details of the procedures used for statistical analyses (including which tests were performed, exact p-values, and sample sizes) and details about experimental design are provided in the Results section or in the legend to each figure. Briefly, comparisons between multiple experimental groups were made using one-way or two-way ANOVA with Tukey's or Dunnett's post hoc test as appropriate. For comparisons between a single experimental group and a control group, an unpaired Student's t test was used. p < 0.05 was considered to be significant. Data are expressed as the mean ± SEM. The assessor was blinded to all treatments throughout the study. In the quantitative analysis of Figures 1, 3, 4, and 5, each data point represents one sample (spinal cord section or extract) from one individual mouse. The number of animals used in each experiment is indicated in figure legends.
Figure 1.

EAE is less severe in TRPM2-KO mice. A, Mean clinical scores for WT and TRPM2-KO mice after induction of EAE using MOG35-55 peptide and pertussis toxin (number of mice = 8–10). **p < 0.01 (two-way ANOVA with Tukey's multiple-comparisons test). B, C, Representative images of lumbar spinal cord sections from immunized mice at day 16. Sections were stained with LFB (B) and H&E (C) to visualize demyelination and immune cell infiltration. Dashed boxes indicate the area magnified in the image below. D–G, Representative FluoroMyelin-stained images (D) and quantitative analyses (E) of lumbar spinal cord at day 16. Scale bar, 200 μm in D. Representative electronic microscope images (F) and quantitative analyses (G) of axons in lumbar spinal cord sections at day 21. Scale bar, 10 μm in F. Each data point was collected from a different animal. Four mice were used for statistical analysis. *p < 0.05 (unpaired Student's t test). Data are expressed as the mean ± SEM.
Figure 3.

Knocking out TRPM2 has no effect on T-cell infiltration into the spinal cord or on T-cell population in secondary lymphoid organs. A, Representative images of lumbar spinal cord sections from immunized mice at day 21 stained with an anti-CD3 antibody. Scale bar, 200 μm. B, Quantitative analysis of the CD3-immunopositive cells shown in A (number of sections = 3–6). C, Quantitative real-time PCR to examine expression of mRNA encoding helper T-cell lineage-specific transcription factors Tbet (Th1), Gata3 (Th2), Rorc (Th17), and Foxp3 (Treg). The relative amounts of mRNA extracted from lumbar spinal cords (at day 21) were normalized against 18s rRNA (number of spinal cords = 5–6). D, E, Representative FACS data for detection of CD3− (D) and CD4+ cells (E) from the spinal cord of naive, WT EAE, and TRPM2-KO EAE mice at day 21 and cumulative quantitative analysis from three to four mice for each group about CD3+ and CD4+ rate or cell number. Singlet and live cells based on forward and side scatter were analyzed. F–K, Representative FACS data for detection of helper T-cell lineage-specific transcription factors T-bet (F, G; Th1), ROR gamma (t) (H, I; Th17), and Foxp3 (J, K; Treg)+ cells from the lymph node (F, H, J) and the spleen (G, I, K) of WT and TRPM2-KO EAE mice at day 10 and cumulative quantitative analysis from 3 mice for each group about T-bet, ROR gamma (t), and Foxp3 positive rate. Singlet and CD4+ live cells based on forward and side scatter were analyzed. Data are expressed as the mean ± SEM.
Figure 4.

TRPM2-KO mice exhibit reduced macrophages/microglia activation and reduced neutrophil infiltration during the chronic phase of EAE. A–C, Representative sections of lumbar spinal cord from immunized mice obtained at days 7, 14, 21, and 28. Sections were stained with an anti-Iba1 antibody (A; macrophages/microglia), an anti-Gr1 antibody (B; neutrophils), and an anti-GSTpi antibody (C; oligodendrocytes). Scale bar, 100 μm. D–F, Quantitative analysis of Iba1+ cells (D), Gr1+ cells (E), and GSTpi+ cells (F) (number of mice = 3–10). *p < 0.05, ***p < 0.001 vs WT. Data are expressed as the mean ± SEM.
Figure 5.

Reduced cytokine production in the spinal cord of TRPM2-KO mice. A, B, Amounts of IL6, IFNγ, IL1β, and CXCL2 in the lumbar spinal cord of WT and TRPM2-KO mice at day 14 (A) and day 21 (B) (number of spinal cords = 4–9). C, D, Quantitative real-time PCR analysis of the relative amounts of Cxcl2 (C) and Trpm2 (D) in the lumbar spinal cord. The amount of mRNA extracted from the lumbar spinal cord at days 14 and 21 was normalized against 18s rRNA (number of spinal cords = 5–6). *p < 0.05, **p < 0.01 vs WT (A–C) or naive (D). Data are expressed as the mean ± SEM.
Results
KO or pharmacological blockade of TRPM2 attenuates neurological impairment and pathological outcome of EAE
To investigate the role of TRPM2 in MS, we first immunized WT and TRPM2-KO mice with MOG35-55 to induce EAE. Whereas WT mice gradually developed more severe EAE from the date of disease onset, TRPM2-KO mice were resistant. The day of onset was not different between WT and TRPM2-KO mice (Fig. 1A; p < 0.01, two-way ANOVA). To further differentiate pathological changes in EAE between both genotypes, we performed histologic analyses to examine inflammatory cell infiltration, demyelination, and axonal damage in the CNS. The spinal cords of TRPM2-KO mice showed preservation of myelination within the outmost white matter, as demonstrated by LFB staining (Fig. 1B) and FluoroMyelin (Fig. 1D,E; p < 0.05, unpaired Student's t test) at day 16, the peak time of the disease. Moreover, the number of inflammatory cells infiltrating the spinal cord of TRPM2-KO mice at day 16 was lower than that in WT mice (Fig. 1C). The spinal cords of WT mice contained fewer axons than those of naive mice; however, spinal cords from TRPM2-KO mice had more axons than those of WT mice (Fig. 1F,G; p < 0.05, unpaired Student's t test) at day 21. We observed less inflammatory cell infiltration, less myelin loss, and less axonal damage in spinal cords from TRPM2-KO mice.
We next investigated whether pharmacological inhibition of TRPM2 after induction of EAE has a beneficial effect on the clinical score. Miconazole, a Food and Drug Administration-approved antifungal drug, effectively inhibits the TRPM2 channel (Togashi et al., 2008). To test the possibility that inhibition of TRPM2 modulates clinical disability, we started administration of miconazole after onset of disease (day 16 after immunization). We found that daily administration of miconazole (10 mg/kg, i.p.) throughout the chronic phase of the disease ameliorated disability in WT EAE mice compared with vehicle-treated WT EAE mice (Fig. 2A; p < 0.01, two-way ANOVA). In addition, miconazole did not result in additional clinical improvement in TRPM2-KO EAE mice compared with vehicle-treated TRPM2-KO EAE mice (Fig. 2B; p = 0.5403, two-way ANOVA), indicating that miconazole likely exerts its protective effects by targeting TRPM2. Miconazole administration preserved myelin as assessed by FluoroMyelin (Fig. 2C) and LFB staining (Fig. 2D) and inhibited inflammatory cell infiltration (Fig. 2E) of the spinal cord at day 21.
Figure 2.
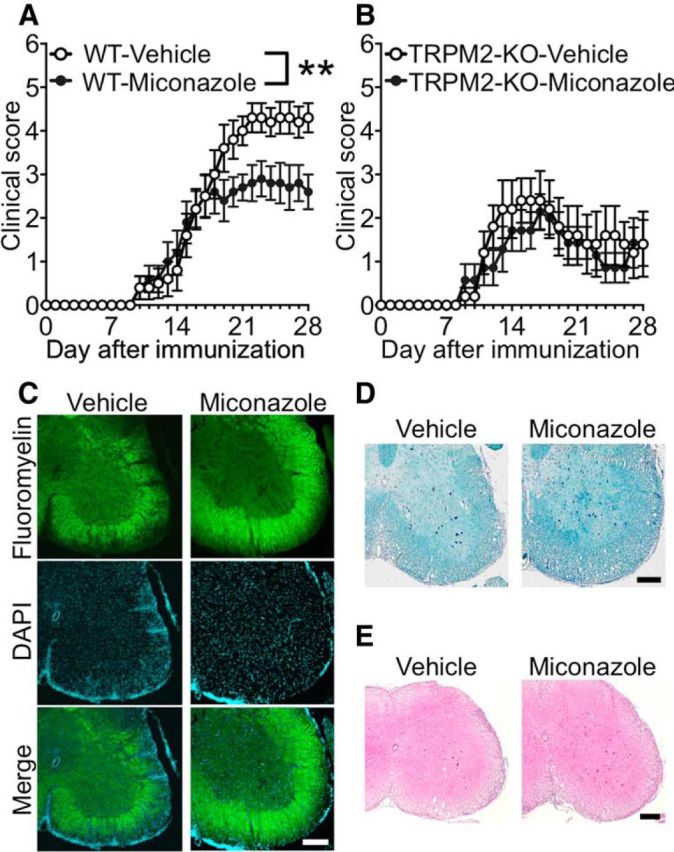
Pharmacological blockade of TRPM2 prevents EAE development. A, B, Mean clinical scores for WT (A) and TRPM2-KO (B) mice receiving a daily intraperitoneal injection of the TRPM2 inhibitor miconazole (10 mg/kg) or vehicle (DMSO in saline) from days 16 to 28 (number of mice = 5–10). Note that miconazole caused a significant reduction in the clinical score in WT mice, but had no significant effect in TRPM2-KO mice. C–E. Representative images of lumbar spinal cord sections from immunized WT mice receiving a daily intraperitoneal injection of miconazole or vehicle at day 21. Sections were stained with FluoroMyelin (C), LFB (D), or H&E (E) to visualize demyelination and immune cell infiltration. Scale bar, 200 μm. **p < 0.01 (two-way ANOVA, with Tukey's multiple-comparisons test). Data are expressed as the mean ± SEM.
Knocking out TRPM2 has no effect on T-cell function in the spinal cord, lymph nodes, and spleen of EAE mice
As TRPM2 deficiency suppresses T-cell proliferation induced by anti-CD3/CD28 in vitro (Melzer et al., 2012), we first investigated the effect of TRPM2 deficiency on T-cell function in vivo under pathological conditions. We found that genetic deletion of TRPM2 had no effect on the increased number of CD3+ cells infiltrating the spinal cord at the peak of disease, although the pathological phenotype was weaker in TRPM2-KO mice than in WT mice (Fig. 3A,B; WT vs TRPM2-KO: p = 0.6693, Dunnett's multiple-comparisons test). Next, to investigate whether TRPM2 deficiency affects the function of helper and regulatory T cells in the lumbar spinal cord, we examined changes in mRNA encoding transcription factors specific for helper and regulatory T cells (Tbet in Th1 cells; Gata3 in Th2 cells; Rorc in Th17 cells; and Foxp3 in Treg cells). However, there was no significant difference in mRNA expression between the genotypes (Fig. 3C; Tbet, p = 0.5296; Gata3, p = 0.8890; Rorc, p = 0.5847; Foxp3, p = 0.9851; unpaired Student's t test). To quantify T-cell infiltration into the spinal cord more precisely, we conducted flow cytometric analysis of spinal cords of both genotypes at day21. We observed no changes in the numbers of infiltrating CD3+ (Fig. 3D) and CD4+ (Fig. 3E) cells. Furthermore, when we assessed peripheral changes caused by genetic deletion of TRPM2, we did not observe any changes in the percentages of T-cell subpopulations, namely T-bet in Th1 cells (Fig. 3F,G), ROR gamma (t) in Th17 cells (Fig. 3H,I), and Foxp3 in Treg cells (Fig. 3J,K), draining lymph nodes (Fig. 3F,H,J), and spleen (Fig. 3G,I,K) of WT and TRPM2-KO mice.
Knocking out TRPM2 ameliorates the increased numbers of macrophages/microglia and neutrophils in the spinal cord of EAE mice
During the pathogenesis of EAE, abnormally activated microglia increase their production of proinflammatory cytokines and chemokines, which promotes infiltration of the CNS by peripheral leukocytes, including monocytes/macrophages and neutrophils (Mishra and Yong, 2016; Prinz and Priller, 2017). To investigate the effect of TRPM2 deficiency on infiltration by activation of these immune cells, we next analyzed the immunohistochemical expression of Iba1, a marker of macrophages/microglia, and Gr1, a marker of neutrophils, in the spinal cord at days 7, 14, 21, and 28. From day 14, both the immunoreactivity of Iba1 and the number of Gr1-immunopositive cells were higher in WT mice with EAE than in naive mice, which is the time point when the clinical symptoms are already manifest. Immunoreactivity of Iba1 and the number of Gr1-immunopositive cells peaked at day 21 and then settled down to a lower level, however, which is higher than the level observed in naive mice at day 28 (Fig. 4A–D). Iba1 immunoreactivity and the number of Gr1+ cells in TRPM2-KO EAE mice were lower than those in WT EAE mice at day 21 (Iba1, p < 0.05; Gr1, p < 0.05; Dunnett's multiple-comparisons test) and at day 28 (Iba1, p > 0.05; Gr1, p < 0.05; Dunnett's multiple-comparisons test), but not significantly different at days 7 and 14 (Fig. 4A–D). However, there was no difference in GFAP immunoreactivity, a marker of astrocytes, between the genotypes (data not shown). In addition, immunostaining for GSTpi, a marker of mature oligodendrocytes, revealed that TRPM2 deficiency had no effect on the number of oligodendrocytes present in the spinal cord during EAE disease (Fig. 4C,F), although the loss of myelin was less pronounced in TRPM2-KO mice than in WT mice. These results indicate that activation of TRPM2 plays a role in the progression, but not initiation, of CNS inflammation.
KO of TRPM2 attenuates CXCL2 production and release at the early stage of disease
Accumulating evidence suggests that proinflammatory cytokines and chemokines play key roles in the pathogenesis of EAE (Kothur et al., 2016). To investigate the effect of TRPM2 deficiency on the production and secretion of proinflammatory cytokines and chemokines, we analyzed levels of representative proinflammatory cytokines in the spinal cord by ELISA. Although at day 7 (predisease period), the level of IL6, IFNγ, and IL1β in the spinal cord of EAE mice of both genotypes was undetectable (data not shown), the amounts of IL6, IFNγ, and IL1β in the spinal cord of TRPM2-KO EAE mice at day 14 (the early stage of the disease) were markedly increased but similar to that in WT EAE mice (Fig. 5A; IL6, IFNγ, IL1β, p > 0.9999; Bonferroni's multiple-comparisons test). Surprisingly, the amounts of CXCL2 in TRPM2-KO EAE mice were markedly lower than those in WT EAE mice (Fig. 5A; CXCL2, p < 0.01; Bonferroni's multiple-comparisons test), although the EAE clinical scores for WT and TRPM2-KO mice at day 14 were comparable, as shown in Figure 1A. At day 21 (the peak stage of the disease), the amounts of IL6, IFNγ, and IL1β in TRPM2-KO EAE mice tended to be lower than those in WT EAE mice (Fig. 5B; IL6, p = 0.9610; IFNγ, p = 0.1356; IL1β, p = 0.1941; Bonferroni's multiple-comparisons test). The absolute amount of CXCL2 at day 21 was lower than that at day 14 (WT day 14: 95.81 ± 22.48 pg/mg protein, WT day 21: 22.23 ± 7.90 pg/mg protein, p = 0.0149; unpaired Student's t test); genetic deletion of TRPM2 tended to inhibit CXCL2 release (Fig. 5B). Similarly, marked inhibition of CXCL2 mRNA expression was observed at days 14 and 21 (Fig. 5C; day 14, p < 0.05; day 21, p = 0.1838, Dunnett's multiple-comparisons test). Moreover, expression of TRPM2 mRNA in the spinal cord of EAE mice was upregulated significantly at day 14 (Fig. 5D; naive vs day 14, p < 0.05; naive vs day 21, p = 0.0554; Dunnett's multiple-comparisons test). Together, these results suggest that upregulation of TRPM2 and the resulting production/release of CXCL2 precedes progression of EAE.
TRPM2 expressed by macrophages drives the pathogenesis of EAE
The production and secretion of CXCL2 by monocytes and macrophages are mediated mainly by the opening of TRPM2 channels; this mechanism underlies the pathophysiology of inflammatory diseases such as neuropathic pain (Haraguchi et al., 2012) and ulcerative colitis (Yamamoto et al., 2008). Therefore, we performed double-fluorescent labeling experiments to investigate CXCL2-expressing cell types. We found that CXCL2 protein was expressed in a population of Iba1+ cells (macrophages and microglia), but not in GFAP+ cells (astrocytes) or CD3+ cells (T-cell lineages; Fig. 6). Finally, we generated chimeric mice to determine whether endogenous CNS cells or CNS-infiltrating leukocytes are responsible for EAE induction. Irradiated WT and TRPM2-KO recipient mice were transplanted with WT or TRPM2-KO donor-mouse-derived GFP+ BM cells to generate four types of BM chimeric mouse (WT-BM→/WT recipient mice; WT-BM→/TRPM2-KO recipient mice; TRPM2-KO-BM→/WT recipient mice; and TRPM2-KO-BM→/TRPM2-KO recipient mice). Flow cytometry analysis revealed that >90% of the BM-derived cells in the blood of the chimeric mice had been replaced by GFP+ cells at 6 weeks after BM transplantation (Fig. 7A). TRPM2-KO-BM→/WT recipient mice and TRPM2-KO-BM→/TRPM2-KO recipient mice were resistant to EAE, whereas the EAE clinical score for WT-BM→/TRPM2-KO recipient mice was the same as that for WT-BM→/WT recipient mice (Fig. 7B; WT-BM→/WT vs WT-BM→/TRPM2-KO, p = 0.9444; WT-BM→/WT vs TRPM2-KO-BM→/WT, p < 0.01; WT-BM→/WT vs TRPM2-KO-BM→/TRPM2-KO, p < 0.01; two-way ANOVA). Furthermore, we performed immunohistochemical analysis of spinal cords from WT-BM→/WT recipient mice and found that the majority of CXCL2+ cells were Iba1+ and GFP+, indicating that central deficiency of TRPM2 is not involved in the pathogenesis of EAE, and that it is TRPM2 expressed in macrophages, but not microglia, that contributes to the pathogenesis of EAE by secreting chemokines that attract neutrophils.
Figure 6.

CXCL2 is primarily expressed by Iba1+ cells in the CNS. Representative lumbar spinal cord sections obtained from immunized mice at day 14 and stained with anti-Iba1 (top; macrophages/microglia), anti-GFAP (middle; astrocytes), anti-CD3 antibody (bottom; T cells), and anti-CXCL2 (center) antibodies are shown. Images of anti-CXCL2 antibody staining are shown in the center panels and merged images are shown in the right panels. Scale bar, 20 μm.
Figure 7.

TRPM2 in BM-derived cells contributes to disease progression of EAE. A, Flow cytometry analysis of BM-derived cells in WT/TRPM2-KO BM chimeric mice. Representative histograms showing GFP+ cells in WT (top, control) and BM chimeric mice (bottom). More than 90% of BM-derived cells in all examined chimeric mice were GFP+. B, Mean clinical scores of BM chimeras WT-BM→WT (open circles), WT-BM→TRPM2-KO (closed circles), TRPM2-KO-BM→WT (open squares), and TRPM2-KO-BM→TRPM2-KO (closed squares) after the induction of EAE (number of mice = 6–12). **p < 0.01 (two-way ANOVA with Tukey's multiple comparison test of WT-BM→/WT vs. TRPM2-KO-BM→/WT; WT-BM→/WT vs. TRPM2-KO-BM→/TRPM2-KO). C, Representative sections of lumbar spinal cord from immunized WT-BM→WT mice obtained at day 14 and stained with an anti-Iba1 antibody (macrophages/microglia) and an anti-CXCL2 antibody. Scale bar, 10 μm.
Discussion
Here, we provide the first evidence that TRPM2 expressed by macrophages mediates CXCL2 production and release in EAE lesions of the spinal cord, resulting in neutrophil infiltration into the CNS and aggravation of EAE pathology. This is supported by the following findings: (1) TRPM2 deletion and TRPM2 inhibition led to a significant reduction in the EAE clinical score; (2) TRPM2 deficiency had no effect on T-cell infiltration into both peripheral tissues and CNS tissue or on levels of mRNA encoding T-cell-specific transcription factors in the spinal cord; (3) TRPM2 deficiency led to a significant reduction in Iba1+ cells and Gr1+ cells in EAE lesions of the spinal cord; (4) TRPM2 deficiency led to a significant reduction in levels of CXCL2 at the early stage of disease; and (5) TRPM2-KO-BM→/WT recipient mice and TRPM2-KO-BM→/TRPM2-KO recipient mice were resistant to EAE, whereas the EAE clinical score in WT-BM→/TRPM2-KO recipient mice was similar to that in WT-BM→/WT recipient mice.
Many CXC chemokines act mainly on neutrophils and many CC chemokines act mainly on monocytes. Of the known CXCL chemokines (systematically numbered from 1 to 15), CXCL1 and CXCL2 (and its homolog CXCL8 in humans) mainly attract neutrophils by binding to CXCR2 receptors (Zlotnik and Yoshie, 2000). CXC chemokines and their specific receptors play roles in acute inflammation and in chronic inflammatory and autoimmune diseases such as MS. For example, CXCL8 expression is upregulated in CSF and serum of MS patients (Lund et al., 2004; Ishizu et al., 2005;). IFNβ is used widely as a first-line treatment for MS; however, some patients are nonresponders. Indeed, IFNβ-resistant EAE is highly related to expression of CXCR2 and the lymphotoxin-β receptor (LTβR), leading to the production of CXCL8 (Mikami et al., 2014). In addition, peripheral blood mononuclear cells from patients with IFNβ-resistant MS show upregulated expression of CXCL8, CXCR2, and LTβR (Inoue et al., 2016). Moreover, genetic silencing of CXCR2 (Carlson et al., 2008), blockade of CXCR2 (Kerstetter et al., 2009), and neutrophil-specific gene deletion of CXCR2 (Liu et al., 2010) show that CXCR2 mediates the pathogenesis and pathology of EAE and related demyelination diseases. Antibodies that neutralize CXCR2 almost completely inhibit neutrophil infiltration into inflammatory sites, whereas neutralizing either CXCL1 or CXCL2 only partially inhibits neutrophil infiltration (Tanimoto et al., 2007). These findings are consistent with our own finding that Gr1-immunopositive neutrophil infiltration into the CNS of TRPM2-KO mice was largely suppressed, whereas CXCL2 production in the spinal cord was markedly attenuated, suggesting that CXCL2 is a major chemoattractant for neutrophils in the EAE model. However, most studies, including those mentioned above, suggesting that suppression of neutrophil migration into the CNS inhibits progression of EAE report delayed EAE onset. This is probably because neutrophils induce disruption of the blood–brain barrier (Carlson et al., 2008). Here, we show that there was no delay in the onset of EAE in TRPM2-KO mice. The discrepancy between our results and those of others is unclear; however, it is plausible that CXCL2 may be produced and released during the progressive phase of EAE, a theory supported by the fact that CXCL2, but not CXCL1, is upregulated robustly after EAE onset (Carlson et al., 2008). Together, these results suggest that TRPM2 on macrophages, which were activated by some kind of local inflammation after disease onset, exclusively mediates CXCL2 production, leading to further neutrophil infiltration and CNS demyelination.
As described above, inhibiting CXCR2 attenuates the severity of EAE; however, some studies warn that completely inhibiting CXCR2 signaling and completely removing neutrophils may lead to a disappointing result. For example, CXCR2 signaling protects oligodendrocytes and restricts demyelination in a mouse model of viral-induced demyelination (Hosking et al., 2010) and CXCR2 KO mice show abnormal phenotypes (such as a poor ability to respond to environmental stress, low reproduction, and low body weight; Semple et al., 2010) and exaggerated macrophage-dependent acute inflammatory responses (Dyer et al., 2017). In addition, systemic suppression of neutrophil activation and migration increases the risk of severe infections, including sepsis (Tavares-Murta et al., 2002). Therefore, TRPM2 may be a good therapeutic target for preventing the progression of MS because it can be activated locally within CNS lesions. In addition, inhibiting TRPM2 may reduce neutrophil infiltration into the CNS.
It has become apparent that neutrophils are key player not only in acute inflammation, but also in chronic inflammatory diseases including MS (Caielli et al., 2012; Moliné-Velázquez et al., 2016). However, the pathophysiological contribution of neutrophils in MS patients is still a matter of debate; neutrophils themselves are rarely found in the brain of MS patients (∼4% of MS patients), but are generally rich in the CNS tissue of NMO patients (∼56% of NMO patients; Lucchinetti et al., 2002). Conversely, the plasma levels of the neutrophil chemokines CXCL1, CXCL5, and neutrophil elastase correlates with clinical and radiological measures of CNS injury in MS (Rumble et al., 2015) and granulocyte colony-stimulating factor is upregulated in acute MS lesions (Lock et al., 2002). Neutrophils in the blood of patients with MS show upregulated expression of adhesion molecules and increased activation such as migration, production of reactive oxygen species (respiratory burst), and degranulation compared with healthy controls and these changes related to neutrophil functionality are more evident than those seen in NMO patients (Naegele et al., 2012; Hertwig et al., 2016). The reason that neutrophils themselves are rarely found in the autopsied brains of MS patients may be because the short-living and high turnover nature of neutrophils. Further investigation is warranted to elucidate neutrophil invasion into the CNS lesion during MS.
It remains unclear which molecules activate TRPM2 on macrophages. The most feasible are ROS, which activate TRPM2 directly (Hara et al., 2002). In addition, increased ROS production in the CNS plays a role in the pathogenesis and progression of EAE (Choi et al., 2015; Hasseldam et al., 2016). ROS are produced locally and abundantly by microglia in the brain of MS patients and infiltrating macrophages secrete NADPH oxidase and myeloperoxidase, substances that impose severe and chronic oxidative stress on oligodendrocytes and axons within the lesions (Mahad et al., 2015). In fact, edaravone, a well known ROS scavenger, ameliorates the pathogenesis of EAE (Moriya et al., 2008). Because ROS-mediated physiological signals and defensive responses are necessary for homeostasis and health, long-term administration of ROS scavengers must be performed with caution. Therefore, unlike therapy for ischemic disease, the pathology of which is characterized by acute and marked oxidative stress, ROS scavengers are not a suitable therapy for chronic inflammatory diseases like MS. Another possible activator of TRPM2 in MS and EAE is the PARP-1 signaling pathway; ADP ribose within this pathway directly activates TRPM2 (Perraud et al., 2001). Free ADP ribose is generated in the cytoplasm, where it activates TRPM2 when PARP-1 is expressed to repair DNA damage induced by excessive ROS production after ischemic injury (Shimizu et al., 2013). In addition, increased activation of PARP-1 in monocytes plays a pathological role in MS and EAE (Farez et al., 2009). According to that study, oxysterols (present at higher levels in the serum of MS patients than in that of healthy controls) activate microglia and macrophages, thereby inducing CNS inflammation via the TLR2-PARP-1 axis. These results are concordant with our own results showing that inhibiting TRPM2 under inflammatory conditions suppresses activation of macrophages. Therefore, it is feasible that higher levels of oxysterols activate TRPM2 in macrophages indirectly through the TLR2-PARP-1-ADP ribose axis in EAE. Therefore, we plan future studies to clarify whether excessive ROS production in the CNS activates TRPM2 directly or indirectly.
Although the EAE model (in which mice are immunized with the MOG35-55 peptide) is dependent on helper T cells, especially Th17 cells, we did not observe suppressed migration of helper T cells to the CNS of TRPM2-KO mice. This indicates that cells other than helper T cells drive the pathology of EAE in mice immunized with MOG35-55, which is in agreement with other reports examining macrophages/microglia (Zhou et al., 2015). Nevertheless, a recent report shows that TRPM2 modulates T-cell proliferation after T-cell receptor stimulation (Melzer et al., 2012), although we demonstrated here that there were no differences in the percentages of T cells in secondary lymphoid organs or in the level of T-cell infiltration into the CNS between WT and TRPM2-KO mice in vivo. The reason for the differences between our data and those presented in the above-mentioned study is unclear; it may be because the previous study demonstrated the interesting phenotype only in in vitro cultures.
We showed that miconazole, a TRPM2 inhibitor (Togashi et al., 2008), suppresses progression of EAE pathology in WT mice, but does not affect the pathology of TRPM2-KO mice. Likewise, a recent report shows that administration of miconazole from the point of peak EAE severity leads to an improvement in pathology (Najm et al., 2015). In that study, the investigators suggested that miconazole acts directly on oligodendrocyte progenitor cells (OPCs) to promote remyelination. Because therapies that target lymphocytes are successful to some degree, research interest is shifting to therapies aimed at remyelination therapies; however, no study has detected TRPM2 expression on OPCs and it is unclear whether TRPM2 is functional in these cells. Because we demonstrated that the severity of EAE in chimeric TRPM2-KO mice was similar to that in WT mice, we consider that TRPM2 expression by OPCs will have a minor impact on the pathology of EAE, at least under our experimental conditions. Here, we observed that miconazole administration suppressed disease progression rather than inducing pathological recovery from the peak of disease (Fig. 2), implying that miconazole-mediated inhibition of TRPM2 suppressed inflammation in the CNS, thereby suppressing disease progression. To clarify whether TRPM2 is involved in remyelination, it would be better to use a cuprizone- or lysophosphatidylcholine-induced demyelination mouse model because they manifest the three distinct phases: the demyelination phase, the remyelination phase, and the remyelination complete phase (Miron et al., 2013; Berghoff et al., 2017). Therefore, in the future, we intend to examine remyelination in such WT and TRPM2-KO mouse models.
In conclusion, we show here that knocking out TRPM2 improves EAE severity without delaying disease onset by suppressing production of CXCL2 and inhibiting infiltration of neutrophils into the CNS. Moreover, pharmacological inhibition of TRPM2 with miconazole reduces EAE severity after onset. The cells responsible for producing CXCL2 in the CNS of chimeric mice are Iba1+ monocytic cells, most likely macrophages. Therefore, the TRPM2 channel is a potential therapeutic target for MS and TRPM2 antagonists may be a relatively safe and effective therapy for MS.
Note Added in Proof: The graphs in panels 5A and B were accidentally switched. They have now been corrected.
Footnotes
This work was supported by MEXT/JSPS KAKENHI (Grant 17K19486 to H.S. and Grant 24390016 to S.K.); the Naito Foundation, the Takeda Science Foundation, the Ono Medical Research Foundation, and FY 2018 Kyoto University Research Development Program ISHIZUE. The irradiation experiments were supported by the Radiation Biology Center Cooperative Research Program (Kyoto University). We thank Dr. Yoshio Bando (Akita University) for helpful advice and discussion about the EAE experiments and Keiko Okamoto-Furuta and Haruyasu Kohda (Division of Electron Microscopic Study, Center for Anatomical Studies, Graduate School of Medicine, Kyoto University) for technical assistance with electron microscopy.
The authors declare no competing financial interests.
References
- Beltrami S, Gordon J (2014) Immune surveillance and response to JC virus infection and PML. J Neurovirol 20:137–149. 10.1007/s13365-013-0222-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghoff SA, Gerndt N, Winchenbach J, Stumpf SK, Hosang L, Odoardi F, Ruhwedel T, Böhler C, Barrette B, Stassart R, Liebetanz D, Dibaj P, Möbius W, Edgar JM, Saher G (2017) Dietary cholesterol promotes repair of demyelinated lesions in the adult brain. Nat Commun 8:14241. 10.1038/ncomms14241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caielli S, Banchereau J, Pascual V (2012) Neutrophils come of age in chronic inflammation. Curr Opin Immunol 24:671–677. 10.1016/j.coi.2012.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson T, Kroenke M, Rao P, Lane TE, Segal B (2008) The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med 205:811–823. 10.1084/jem.20072404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Borrero W, Graves D, Frohman TC, Flores AB, Hardeman P, Logan D, Orchard M, Greenberg B, Frohman EM (2012) Current and emerging therapies in multiple sclerosis: a systematic review. Ther Adv Neurol Disord 5:205–220. 10.1177/1756285612450936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BY, Kim JH, Kho AR, Kim IY, Lee SH, Lee BE, Choi E, Sohn M, Stevenson M, Chung TN, Kauppinen TM, Suh SW (2015) Inhibition of NADPH oxidase activation reduces EAE-induced white matter damage in mice. J Neuroinflammation 12:104. 10.1186/s12974-015-0325-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer DP, Pallas K, Medina-Ruiz L, Schuette F, Wilson GJ, Graham GJ (2017) CXCR2 deficient mice display macrophage-dependent exaggerated acute inflammatory responses. Sci Rep 7:45423. 10.1038/srep45423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farez MF, Quintana FJ, Gandhi R, Izquierdo G, Lucas M, Weiner HL (2009) Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat Immunol 10:958–964. 10.1038/ni.1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Melzer N, Schattling B, Göb E, Hicking G, Arunachalam P, Bittner S, Ufer F, Herrmann AM, Bernreuther C, Glatzel M, Gerloff C, Kleinschnitz C, Meuth SG, Friese MA, Magnus T (2014) Transient receptor potential melastatin subfamily member 2 cation channel regulates detrimental immune cell invasion in ischemic stroke. Stroke 45:3395–3402. 10.1161/STROKEAHA.114.005836 [DOI] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y (2002) LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 9:163–173. 10.1016/S1097-2765(01)00438-5 [DOI] [PubMed] [Google Scholar]
- Haraguchi K, Kawamoto A, Isami K, Maeda S, Kusano A, Asakura K, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2012) TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J Neurosci 32:3931–3941. 10.1523/JNEUROSCI.4703-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasseldam H, Rasmussen RS, Johansen FF (2016) Oxidative damage and chemokine production dominate days before immune cell infiltration and EAE disease debut. J Neuroinflammation 13:246. 10.1186/s12974-016-0707-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertwig L, Pache F, Romero-Suarez S, Stürner KH, Borisow N, Behrens J, Bellmann-Strobl J, Seeger B, Asselborn N, Ruprecht K, Millward JM, Infante-Duarte C, Paul F (2016) Distinct functionality of neutrophils in multiple sclerosis and neuromyelitis optica. Mult Scler 22:160–173. 10.1177/1352458515586084 [DOI] [PubMed] [Google Scholar]
- Hosking MP, Tirotta E, Ransohoff RM, Lane TE (2010) CXCR2 signaling protects oligodendrocytes and restricts demyelination in a mouse model of viral-induced demyelination. PLoS One 5:e11340. 10.1371/journal.pone.0011340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Chen PH, Siecinski S, Li QJ, Liu C, Steinman L, Gregory SG, Benner E, Shinohara ML (2016) An interferon-β-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat Neurosci 19:1599–1609. 10.1038/nn.4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isami K, Haraguchi K, So K, Asakura K, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2013) Involvement of TRPM2 in peripheral nerve injury-induced infiltration of peripheral immune cells into the spinal cord in mouse neuropathic pain model. PLoS One 8:e66410. 10.1371/journal.pone.0066410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizu T, Osoegawa M, Mei FJ, Kikuchi H, Tanaka M, Takakura Y, Minohara M, Murai H, Mihara F, Taniwaki T, Kira J (2005) Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain 128:988–1002. 10.1093/brain/awh453 [DOI] [PubMed] [Google Scholar]
- Juryńczyk M, Craner M, Palace J (2015) Overlapping CNS inflammatory diseases: differentiating features of NMO and MS. J Neurol Neurosurg Psychiatry 86:20–25. 10.1136/jnnp-2014-308984 [DOI] [PubMed] [Google Scholar]
- Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, Takada Y, Kume T, Katsuki H, Mori Y, Akaike A (2006) A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 101:66–76. 10.1254/jphs.FP0060128 [DOI] [PubMed] [Google Scholar]
- Kerstetter AE, Padovani-Claudio DA, Bai L, Miller RH (2009) Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp Neurol 220:44–56. 10.1016/j.expneurol.2009.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothur K, Wienholt L, Brilot F, Dale RC (2016) CSF cytokines/chemokines as biomarkers in neuroinflammatory CNS disorders: a systematic review. Cytokine 77:227–237. 10.1016/j.cyto.2015.10.001 [DOI] [PubMed] [Google Scholar]
- Liu L, Belkadi A, Darnall L, Hu T, Drescher C, Cotleur AC, Padovani-Claudio D, He T, Choi K, Lane TE, Miller RH, Ransohoff RM (2010) CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci 13:319–326. 10.1038/nn.2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L (2002) Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 8:500–508. 10.1038/nm0502-500 [DOI] [PubMed] [Google Scholar]
- Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, Trebst C, Weinshenker B, Wingerchuk D, Parisi JE, Lassmann H (2002) A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 125:1450–1461. 10.1093/brain/awf151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund BT, Ashikian N, Ta HQ, Chakryan Y, Manoukian K, Groshen S, Gilmore W, Cheema GS, Stohl W, Burnett ME, Ko D, Kachuck NJ, Weiner LP (2004) Increased CXCL8 (IL-8) expression in multiple sclerosis. J Neuroimmunol 155:161–171. 10.1016/j.jneuroim.2004.06.008 [DOI] [PubMed] [Google Scholar]
- Mahad DH, Trapp BD, Lassmann H (2015) Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 14:183–193. 10.1016/S1474-4422(14)70256-X [DOI] [PubMed] [Google Scholar]
- McColl SR, Staykova MA, Wozniak A, Fordham S, Bruce J, Willenborg DO (1998) Treatment with anti-granulocyte antibodies inhibits the effector phase of experimental autoimmune encephalomyelitis. J Immunol 161:6421–6426. [PubMed] [Google Scholar]
- Melzer N, Hicking G, Göbel K, Wiendl H (2012) TRPM2 cation channels modulate T cell effector functions and contribute to autoimmune CNS inflammation. PLoS One 7:e47617. 10.1371/journal.pone.0047617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikami Y, Matsuzaki H, Horie M, Noguchi S, Jo T, Narumoto O, Kohyama T, Takizawa H, Nagase T, Yamauchi Y (2014) Lymphotoxin β receptor signaling induces IL-8 production in human bronchial epithelial cells. PLoS One 9:e114791. 10.1371/journal.pone.0114791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, van Wijngaarden P, Wagers AJ, Williams A, Franklin RJM, Ffrench-Constant C (2013) M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16:1211–1218. 10.1038/nn.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra MK, Yong VW (2016) Myeloid cells–targets of medication in multiple sclerosis. Nat Rev Neurol 12:539–551. 10.1038/nrneurol.2016.110 [DOI] [PubMed] [Google Scholar]
- Miyake T, Shirakawa H, Kusano A, Sakimoto S, Konno M, Nakagawa T, Mori Y, Kaneko S (2014) TRPM2 contributes to LPS/IFNγ-induced production of nitric oxide via the p38/JNK pathway in microglia. Biochem Biophys Res Commun 444:212–217. 10.1016/j.bbrc.2014.01.022 [DOI] [PubMed] [Google Scholar]
- Miyanohara J, Kakae M, Nagayasu K, Nakagawa T, Mori Y, Arai K, Shirakawa H, Kaneko S (2018) TRPM2 channel aggravates CNS inflammation and cognitive impairment via activation of microglia in chronic cerebral hypoperfusion. J Neurosci 38:3520–3533. 10.1523/JNEUROSCI.2451-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moliné-Velázquez V, Vila-Del Sol V, de Castro F, Clemente D (2016) Myeloid cell distribution and activity in multiple sclerosis. Histol Histopathol 31:357–370. 10.14670/HH-11-699 [DOI] [PubMed] [Google Scholar]
- Moriya M, Nakatsuji Y, Miyamoto K, Okuno T, Kinoshita M, Kumanogoh A, Kusunoki S, Sakoda S (2008) Edaravone, a free radical scavenger, ameliorates experimental autoimmune encephalomyelitis. Neurosci Lett 440:323–326. 10.1016/j.neulet.2008.05.110 [DOI] [PubMed] [Google Scholar]
- Munakata M, Shirakawa H, Nagayasu K, Miyanohara J, Miyake T, Nakagawa T, Katsuki H, Kaneko S (2013) Transient receptor potential canonical 3 inhibitor Pyr3 improves outcomes and attenuates astrogliosis after intracerebral hemorrhage in mice. Stroke 44:1981–1987. 10.1161/STROKEAHA.113.679332 [DOI] [PubMed] [Google Scholar]
- Naegele M, Tillack K, Reinhardt S, Schippling S, Martin R, Sospedra M (2012) Neutrophils in multiple sclerosis are characterized by a primed phenotype. J Neuroimmunol 242:60–71. 10.1016/j.jneuroim.2011.11.009 [DOI] [PubMed] [Google Scholar]
- Nagamine K, Kudoh J, Minoshima S, Kawasaki K, Asakawa S, Ito F, Shimizu N (1998) Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 54:124–131. 10.1006/geno.1998.5551 [DOI] [PubMed] [Google Scholar]
- Najm FJ, Madhavan M, Zaremba A, Shick E, Karl RT, Factor DC, Miller TE, Nevin ZS, Kantor C, Sargent A, Quick KL, Schlatzer DM, Tang H, Papoian R, Brimacombe KR, Shen M, Boxer MB, Jadhav A, Robinson AP, Podojil JR, Miller SD, Miller RH, Tesar PJ (2015) Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature 522:216–220. 10.1038/nature14335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM (2001) ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by nudix motif homology. Nature 411:595–599. 10.1038/35079100 [DOI] [PubMed] [Google Scholar]
- Pierson ER, Wagner CA, Goverman JM (2018) The contribution of neutrophils to CNS autoimmunity. Clin Immunol 189:23–28. 10.1016/j.clim.2016.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis M, Giannetti P, Su P, Turkheimer F, Keihaninejad S, Wu K, Waldman A, Malik O, Matthews PM, Reynolds R, Nicholas R, Piccini P (2012) Increased PK11195 PET binding in the cortex of patients with MS correlates with disability. Neurology 79:523–530. 10.1212/WNL.0b013e3182635645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Priller J (2017) The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci 20:136–144. 10.1038/nn.4475 [DOI] [PubMed] [Google Scholar]
- Rumble JM, Huber AK, Krishnamoorthy G, Srinivasan A, Giles DA, Zhang X, Wang L, Segal BM (2015) Neutrophil-related factors as biomarkers in EAE and MS. J Exp Med 212:23–35. 10.1084/jem.20141015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K (2001) Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293:1327–1330. 10.1126/science.1062473 [DOI] [PubMed] [Google Scholar]
- Semple BD, Kossmann T, Morganti-Kossmann MC (2010) Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab 30:459–473. 10.1038/jcbfm.2009.240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Macey TA, Quillinan N, Klawitter J, Perraud AL, Traystman RJ, Herson PS (2013) Androgen and PARP-1 regulation of TRPM2 channels after ischemic injury. J Cereb Blood Flow Metab 33:1549–1555. 10.1038/jcbfm.2013.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa H, Katsumoto R, Iida S, Miyake T, Higuchi T, Nagashima T, Nagayasu K, Nakagawa T, Kaneko S (2017) Sphingosine-1-phosphate induces Ca2+ signaling and CXCL1 release via TRPC6 channel in astrocytes. Glia 65:1005–1016. 10.1002/glia.23141 [DOI] [PubMed] [Google Scholar]
- So K, Haraguchi K, Asakura K, Isami K, Sakimoto S, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2015) Involvement of TRPM2 in a wide range of inflammatory and neuropathic pain models. J Pharmacol Sci 127:237–243. 10.1016/j.jphs.2014.10.003 [DOI] [PubMed] [Google Scholar]
- Steinman L. (2014) Immunology of relapse and remission in multiple sclerosis. Annu Rev Immunol 32:257–281. 10.1146/annurev-immunol-032713-120227 [DOI] [PubMed] [Google Scholar]
- Sun Y, Chen H, Dai J, Zou H, Gao M, Wu H, Ming B, Lai L, Xiao Y, Xiong P, Xu Y, Gong F, Zheng F (2015) HMGB1 expression patterns during the progression of experimental autoimmune encephalomyelitis. J Neuroimmunol 280:29–35. 10.1016/j.jneuroim.2015.02.005 [DOI] [PubMed] [Google Scholar]
- Tanimoto N, Terasawa M, Nakamura M, Kegai D, Aoshima N, Kobayashi Y, Nagata K (2007) Involvement of KC, MIP-2, and MCP-1 in leukocyte infiltration following injection of necrotic cells into the peritoneal cavity. Biochem Biophys Res Commun 361:533–536. 10.1016/j.bbrc.2007.07.060 [DOI] [PubMed] [Google Scholar]
- Tavares-Murta BM, Zaparoli M, Ferreira RB, Silva-Vergara ML, Oliveira CH, Murta EF, Ferreira SH, Cunha FQ (2002) Failure of neutrophil chemotactic function in septic patients. Crit Care Med 30:1056–1061. 10.1097/00003246-200205000-00017 [DOI] [PubMed] [Google Scholar]
- Togashi K, Inada H, Tominaga M (2008) Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br J Pharmacol 153:1324–1330. 10.1038/sj.bjp.0707675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y (2008) TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 14:738–747. 10.1038/nm1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Cai W, Jin M, Xu J, Wang Y, Xiao Y, Hao L, Wang B, Zhang Y, Han J, Huang R (2015) 18β-glycyrrhetinic acid suppresses experimental autoimmune encephalomyelitis through inhibition of microglia activation and promotion of remyelination. Sci Rep 5:13713. 10.1038/srep13713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O (2000) Chemokines: a new classification system and their role in immunity. Immunity 12:121–127. 10.1016/S1074-7613(00)80165-X [DOI] [PubMed] [Google Scholar]