

Abstract
Protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) is one of four known kinases that respond to cellular stress by deactivating the eukaryotic initiation factor 2 α (eIF2α) or other signal transduction cascades. Recently, both eIF2α and its kinases were found to play a role in normal and pathological brain function. Here, we show that reduction of either the amount or the activity of PERK, specifically in the CA1 region of the hippocampus in young adult male mice, enhances neuronal excitability and improves cognitive function. In addition, this manipulation rescues the age-dependent cellular phenotype of reduced excitability and memory decline. Specifically, the reduction of PERK expression in the CA1 region of the hippocampus of middle-aged male mice using a viral vector rejuvenates hippocampal function and improves hippocampal-dependent learning. These results delineate a mechanism for behavior and neuronal aging and position PERK as a promising therapeutic target for age-dependent brain malfunction.
SIGNIFICANCE STATEMENT We found that local reduced protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) expression or activity in the hippocampus enhances neuronal excitability and cognitive function in young normal mice, that old CA1 pyramidal cells have reduced excitability and increased PERK expression that can be rescued by reducing PERK expression in the hippocampus, and that reducing PERK expression in the hippocampus of middle-aged mice enhances hippocampal-dependent learning and memory and restores it to normal performance levels of young mice. These findings uncover an entirely new biological link among PERK, neuronal intrinsic properties, aging, and cognitive function. Moreover, our findings propose a new way to fight mild cognitive impairment and aging-related cognitive deterioration.
Keywords: aging, intrinsic properties, memory enhancement, PERK, translation regulation
Introduction
Translation of mRNA into protein can be divided into three phases: initiation, elongation, and termination, of which both the initiation and elongation phases are vital for memory consolidation (Gal-Ben-Ari et al., 2012; Gildish et al., 2012; Taha et al., 2013; Heise et al., 2017). Both the transition from short-term to long-term memory and synaptic plasticity are regulated by phosphorylation of eukaryotic initiation factor 2 (eIF2) at serine 51 of its α subunit (p-eIF2α). Specifically, reduction in levels of p-eIF2α results in facilitation of long-term memory formation, whereas an increase results in memory impairment (Costa-Mattioli et al., 2007). From a biochemical perspective, eIF2α phosphorylation reduces translation of most mRNAs, but paradoxically stimulates translation of mRNAs harboring upstream open reading frames (uORFs) in their 5′-UTR, such as activating transcription factor 4 (ATF4) and β-secretase 1 (BACE1) (Hooper et al., 2007; O'Connor et al., 2008).
In the brain, eIF2α can be phosphorylated by three well described kinases: protein kinase R (PKR), a double-stranded RNA-dependent protein kinase; PKR-like endoplasmic reticulum kinase (PERK); and general control nonderepressible 2 (GCN2) (Gal-Ben-Ari et al., 2012; Segev et al., 2015). Of these three kinases, PERK is the major one to determine basal p-eIF2α levels (Moreno et al., 2012; Trinh et al., 2012; Ounallah-Saad et al., 2014).
The role of PERK in synaptic plasticity and hippocampal-dependent memory has been studied using a PERK conditional knock-out (KO) mouse model in which PERK is deleted specifically in excitatory neurons. In these mice, overall memory strength is not affected but behavioral flexibility is impaired, a finding attributed to facilitated mGluR-mediated long-term depression (mGluR-LTD) (Trinh et al., 2012, 2014). Paradoxically, knocking down PERK in the cortex of adult rats resulted in enhanced memory and enhanced behavioral flexibility (Ounallah-Saad et al., 2014). Nevertheless, the effect of PERK reduction specifically in the CA1 of adult mice on hippocampal-dependent cognitive function remains unaddressed.
In addition to the function of PERK in learning and memory processes, it has also been implicated in age-dependent brain diseases. For example, decreasing p-eIF2α by reducing PERK activity or expression levels has been shown to rescue neurodegeneration in prion disease as well as memory deficits in the APP/PS1 Alzheimer's disease (AD) mouse models that overproduce amyloid beta (Ma et al., 2013; Moreno et al., 2013; Radford et al., 2015). Brain aging also alters calcium homeostasis, which can affect neuronal function negatively. For example, aging is correlated with reduced neuronal excitability that can be measured via modulation in afterhyperpolarization (AHP) and accommodation of hippocampal neurons (Disterhoft and Oh, 2007). Moreover, with age, medium AHP (mAHP) in CA1 neurons is significantly larger, thus causing lower neural excitability in aged rats (Disterhoft and Oh, 2007; Oh et al., 2010). Recent reports suggest that PERK regulates Gq-protein-coupled Ca2+ dynamics in pyramidal neurons independently of its function as an eIF2α kinase (Zhu et al., 2016). Another recent report suggests that PERK activation is correlated with memory deficits after traumatic brain injury and is mediated by novel targets of PERK independently of eIF2α (Sen et al., 2017).
Nevertheless, the role of PERK and its targets in neuronal intrinsic properties and normal aging remains unexplored. Consistent with the dual functions of PERK in learning processes and aging-dependent brain diseases, we tested two hypotheses. First, we tested the hypothesis that locally reduced PERK activity or expression in the CA1 region of the hippocampus results in better long-term memory. Second, we tested the hypothesis that reducing PERK in the CA1 region of middle-aged mice would have an ameliorating effect on both the declined cognitive performance and intrinsic neuronal excitability.
Materials and Methods
Animals
Male C57BL/6 mice (Harlan Laboratories) ∼12-week-old and weighing 20–25 g were used unless otherwise indicated. The mice were housed individually on a 12/12 h light/dark cycle and provided with water and standard rodent chow ad libitum. Animals were handled according to approved protocols and animal welfare regulations of the University of Haifa Institutional Ethics Committee. For the aging experiment, 8-month-old male mice were purchased from Harlan Laboratories and kept under the same conditions until the time of the experiment. For behavioral experiments, scrambled (SCR) animals were injected at either 3 or 10 months of age and assessed at 5 or 12 months. For electrophysiology SCR expansion, animals were injected at 4 months of age.
Stereotaxic administration of PERK shRNA adeno-associated virus (AAV)
To perform stereotaxic viral injections into the brain, mice were anesthetized with 5% isoflurane using an anesthesia system (HME109; Highland Medical Equipment). The mice were then placed in a rodent stereotaxic apparatus (Stereotaxic Alignment System Model 1900; Kopf Instruments) with continuous 1.5% isoflurane. A small incision was made in the head, the skin was reflected, and the skull was exposed to show the bregma and lambda, as well as the desired injection sites. The following injection coordinates targeting dorsal hippocampal CA1 were used (relative to bregma): −1.46 mm AP, ±1 mm ML, and −1.4 mm DV. A small drill hole was made in the skull over the injection site. A 10 μl Hamilton syringe with a 32-gauge stainless steel needle connected to a stereotaxic infusion pump was used to inject the virus into the brain. A total of 1 μl of PERK shRNA (Sigma-Aldrich clone TRCN28772) AAV (pAAV-EF1a-ZsGreen-shRNA, 1 × 1012 vg/ml, serotype 5) or SCR sequence AAV was bilaterally injected into the brain at a rate of 50 nl/min. The needle was left in place for 5 min before and after injection to allow for tissue relaxation and to minimize fluid retraction. Once the injection needle was withdrawn, the incision was closed with tissue adhesive (Vetbond; 3M) and the mouse was removed from the stereotaxic apparatus. The mice were given at least 4 weeks to recuperate and to allow infected neurons to express high contents of AAV before any behavior experiments were conducted.
Cannula implantation and microinjection of the PERK inhibitor GSK2606414
Animals were anesthetized under 5% isoflurane using an isoflurane anesthesia system (HME109; Highland Medical Equipment). Under aseptic conditions, a rodent stereotaxic apparatus (Stereotaxic Alignment System Model 1900; Kopf Instruments) with continuous 1.5% isoflurane anesthesia was used. For CA1 infusion, the cannula was implanted bilaterally into the CA1 region (from the bregma: −1.94 mm AP, ±1.0 mm ML, and −1.0 mm DV). The implanted cannula was cemented on the skull. Each cannula was fitted with a 28-gauge dummy cannula that extended 0.1 mm beyond the tip of the guide cannula to prevent blockage of the guide cannula. After cannulation, mice were allowed to recover for 7 d before infusion of PERK inhibitor or vehicle and behavior experiments. On conditioning day, randomly assigned mice were infused with a 100 nm concentration of the PERK inhibitor GSK2606414 (Rozpędek et al., 2017) dissolved in dimethyl sulfoxide (DMSO) and further diluted in saline to a final DMSO concentration of 0.5% via a 28-gauge infusion cannula connected by polyethylene (PE 20) tubing to a 10 μl Hamilton microsyringe. The infusion cannula protruded 0.25 mm beyond the guide cannula. For CA1 administration, infusion volume of 1 μl of 100 nm GSK2606414 or vehicle was delivered using a PHD2000 syringe pump (Harvard Apparatus) over the course of 1 min (at a rate of 1 μl/min). After infusion, the injection cannula was kept in the guide cannula for an additional minute to minimize dragging of infused solutes along the injection tract. Locations of cannula were verified in 30 μm coronal sections.
Trace fear conditioning
Model chambers measuring 25 × 25 × 25 cm internally (Panlab; Harvard Apparatus) were located inside a larger, insulated plastic cabinet that excluded external light and noise. For the trace-fear-conditioning protocol, mice were placed in a chamber (with a 20 W bulb and a 16 bar metal grid floor) for 120 s, after which the mice received a 2.9 kHz tone applied for 20 s at 80 dB (conditioned stimulus, CS) and a 0.5 mA foot shock applied for 2 s (unconditioned stimulus, US) 20 s at the end of the CS. This protocol was repeated 5 times and each tone–shock presentation was separated by a 2 min interval. After administration of the last shock, animals remained in the chamber for 2 min before they were taken back to the home cage. The chambers were cleaned with 10% ethanol between successive sets of mice. Animals from control and treatment groups were randomized between the four different chambers. For the context test, mice were placed in the conditioning chamber 24 h after conditioning and kept in the cage for 300 s without tone or foot shock. For the tone–trace test, animals were put in chambers 48 h after conditioning, but the grid floor was hidden with black plastic to create another context and the animals were presented with the tones as on the conditioning day. Animal behavior was recorded using an infrared camera and the data were analyzed by Freeze Frame 3.0 software (Coulbourn Instruments). The indication for fear memory was percentage of time spent freezing. The Shapiro–Wilk test was used as a numerical means of assessing normality. Independent-samples t test was used as a parametric test and the Mann–Whitney U test was used for the nonparametric equivalent. Exclusion/inclusion criteria were as follows: cannula and injection site were determined blindly, animals were numbered serially, and injection or cannulation sites were determined. Animals with misplaced injections were excluded. The experimenter who determined injection or cannula site was blinded to the treatment.
Preparation of total samples for biochemical analysis
At the end of the behavioral procedure, the brains were removed and snap-frozen in liquid nitrogen. CA1 punches were made using a cryostat (Leica) according to injection coordinates and were homogenized by 30 strokes in a glass–Teflon homogenizer in a lysis buffer containing 10 mm HEPES, pH 7.4, 2 mm EDTA, 2 mm EGTA, 0.5 mm DTT, 1% phosphatase inhibitor mixture (3-P0044; Sigma-Aldrich), and 1% protease inhibitor mixture (p2714; Sigma-Aldrich). Protein content was determined with the BCA Protein Assay Kit (GE Healthcare). Appropriate volumes of 2× SDS sample buffer (10% glycerol, 5% β-mercaptoethanol, 4% SDS, 120 mm Tris-HCl, pH 6.8) were added to the homogenates and samples were boiled for 5 min and stored at −80°C until further analysis.
Western blotting
Samples were prepared in SDS sample buffer and subjected to 7.5–10% SDS-PAGE and Western blot analysis. Each lane was loaded with an equal amount of protein (10 μg). After transfer to a nitrocellulose membrane, the blots were blocked for 1 h with 4% BSA in Tris-buffered saline plus 0.5% Tween 20 (TBST) at room temperature. They were then incubated overnight with the following primary antibodies: PKR (1:1000; Santa Cruz Biotechnology), eIF2α (1:1000; Cell Signaling Technology), p-(Ser51) eIF2α (1:1000; Cell Signaling Technology), PERK (1:1000; Cell Signaling Technology), cAMP response element-binding protein (CREB, 1:1000; Cell Signaling Technology), p-(Ser133) CREB (1:1000; Cell Signaling Technology), PSD95 (1:1000; Cell Signaling Technology), p-(Thr19) PSD95 (1:1000, Abcam), β-actin (1: 3000; Santa Cruz Biotechnology), and β-tubulin (1:30,000; Sigma-Aldrich). The blots were then subjected to 3 5 min washing steps in TBST, after which they were incubated with the corresponding HRP-conjugated secondary antibodies, goat anti-rabbit IgG, goat anti-mouse IgG, or rabbit anti-goat IgG (1:10,000; Millipore Bioscience Research Reagents), for 1 h at room temperature, followed by 3 10 min washing steps with TBST. Immunodetection was performed with the enhanced chemiluminescence EZ-ECL Kit (Biological Industries). The immunoblots were quantified with a CCD camera and Quantity One software (Bio-Rad). Each sample was measured relative to the background. Phosphorylation levels were calculated as the ratio between chemiluminescence values of the antibody directed against the phosphoproteins and those of the antibody directed against the phosphorylation-state-independent forms of the proteins.
RNA extraction and qRT-PCR
RNA was extracted from mouse hippocampus using TRI Reagent (Molecular Research Center) according to the manufacturer's protocol. RNA concentration and purity were evaluated using a Nanodrop 2000 (Thermo Fisher Scientific). The High Capacity Reverse Transcription Kit (ABI) was used according to the manufacturer's instructions for cDNA synthesis in a total volume of 20 μl using 1 μg of RNA as a template. qRT-PCR was performed in a total volume of 10 μl using the following TaqMan assays (Applied Biosystems): PERK (Mm00438700_m1) and HPRT (Mm00446968_m1). Reactions were performed in accordance with the manufacturer's instructions. qRT-PCR analysis was performed using the PCR System STEP-ONE plus (Applied Biosystems). Relative mRNA levels were calculated using the ΔΔCt method and the 2−ΔΔCt formula. Statistical analysis was performed on ΔCt values.
Electrophysiology
Tissue preparation.
Mice were killed by cervical dislocation or by anesthetizing with isoflurane and subsequent decapitation. Three-hundred-micrometer coronal brain slices were cut with a Campden-1000 Vibratome. Slices were cut in ice-cold sucrose-based cutting solution containing the following (in mm): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, 5 d-glucose, and 0.6 ascorbate. The slices were allowed to recover for 30 min at 37°C in artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 d-glucose, 2 CaCl2, and 1 MgCl2, followed by additional recovery for at least 30 min in ACSF at room temperature until electrophysiological recording. The solutions were constantly gassed with carbogen (95% O2 + 5% CO2).
Intracellular whole-cell recording.
After the recovery period, slices were placed in the recording chamber and maintained at 34°C with continuous perfusion of carbogenated ACSF (2 ml/min). Brain slices containing the dorsal hippocampus were illuminated with infrared light and pyramidal cells were visualized under a differential interference contrast microscope with 60× or 40× water-immersion objectives mounted on a fixed-stage microscope (BX51-WI; Olympus). The image was displayed on a video monitor using a charge-coupled device (CCD) camera (Dage MTI). In AAV-injected animals, cells infected with AAV containing PERK—shRNA or SCR sequence shRNA (PERK-Scr)—were identified by GFP. Recordings were amplified by Multiclamp 700B or Axopatch 200B amplifiers and digitized with Digidata 1440 (Molecular Devices). The recording electrode was pulled from a borosilicate glass pipette (3–5 MΩ) using an electrode puller (P-1000; Sutter Instruments) and filled with a K-gluconate-based internal solution containing the following (in mm): 120 K-gluconate, 20 KCl, 10 HEPES, 2 MgCl2, 4 Na2ATP, 0.5 TrisGTP, and 14 phosphocreatine, osmolarity 290 mOsm, pH 7.3. The recordings were made from the soma of CA1 pyramidal cells. Liquid junction potential (+10 mV) was not corrected online. All current-clamp recordings were low-pass filtered at 10 kHz and sampled at 50 kHz. Pipette capacitance and series resistance were compensated and only cells with series resistance smaller than 20 MΩ were included in the dataset. The method for measuring active intrinsic properties was based on a modified version of previous protocols (Kaphzan et al., 2013; Chakraborty et al., 2017). For recordings with GSK2606414, the inhibitor was added to the pipette solution (100 nm).
Recording parameters.
Resting membrane potential (RMP) was measured 5 min after the beginning of whole-cell recording (rupture of the membrane under the recording pipette). The dependence of firing rate on the injected current was obtained by injection of current steps (of 1 s duration from −150 to +300 pA in 50 pA increments). Input resistance (Rin) was calculated from the voltage response to a hyperpolarizing current pulse (−150 pA). For measurements of a single action potential (AP), after initial assessment of the current required to induce an AP at 15 ms from the start of the current injection with large steps (50 pA), a series of brief depolarizing currents were injected for 10 ms in steps of 10 pA increments. The first AP that appeared on the 5 ms time point was analyzed. A curve of dV/dt was created for that trace and the 30 V/s point in the rising slope of the AP was considered as threshold (Chakraborty et al., 2017).
AP amplitude was measured from the equipotential point of the threshold to the spike peak, whereas AP duration was measured at the point of half-amplitude of the spike. The mAHP was measured using prolonged (3 s), high-amplitude (3 nA) somatic current injections to initiate time-locked AP trains of 50 Hz frequency and duration (10–50 Hz, 1 or 3 s) in pyramidal cells. These AP trains generated prolonged (∼20 s) AHPs, the amplitudes and integrals of which increased with the number of APs in the spike train. AHP was measured from the equipotential point of the threshold to the anti-peak of the same spike (Gulledge et al., 2013).
Series resistance, Rin, and membrane capacitance were monitored during the entire experiment. Changes >10% in these parameters were criteria for exclusion of data. All experimental analyses were performed in a blinded manner; the identity of the mouse from which neurons were recorded (PERK shRNA or SCR) was not known to the person conducting the experiments and measurements.
Statistical analysis
Experimenters were blinded to the group and treatment during testing and scoring. To decide the sample size for our behavioral, electrophysiological, imaging, and biochemical experiments, we followed the standard sample sizes used in similar experiments in each of the relevant fields in the literature. Calculated statistical values are presented as means ± SEM. Differences in mean values were assessed with the appropriate ANOVA (one-way or two-way repeated-measures) followed by multiple comparisons. For comparisons between two groups, data were analyzed using Student's t test. Differences between means were considered statistically significant if p < 0.05.
Results
Inhibition of PERK activity in CA1 region enhances hippocampal memory and neuronal excitability
In a previous study, we showed that reduced PERK activity or expression in specific cortical regions enhances memory and behavioral flexibility (Ounallah-Saad et al., 2014). Here, we first tested whether a similar phenomenon would be observed in the adult hippocampus, a brain structure necessary for different cognitive functions in human and rodents. Toward that aim, we reduced PERK activity in the adult hippocampus by bilaterally infusing a PERK-specific inhibitor, GSK2606414 (100 nm), to the CA1 region (Fig. 1a) 50 min before the acquisition phase (Fig. 1b) of a trace-fear-conditioning paradigm (Fig. 1c). Long-term memory was tested 24 h after conditioning by reexposing the animals to the training context. Tone–trace memory was tested 48 h after conditioning by putting the animals in a new context and presenting the tone from the training day. This behavioral paradigm was selected because working memory and declarative memory processes are engaged to facilitate formation of trace memory during the trace-fear-conditioning procedure, which is more difficult to acquire than standard delayed fear-conditioning procedures and critically engages the hippocampus (Bangasser et al., 2006; Connor and Gould, 2016). Several studies have shown that dorsal hippocampus lesions before and after the training impair the animal's ability to associate the CS and US stimuli when they are separated by the trace interval. However, animals with hippocampal lesions could associate CS and US in a delay situation in which no trace interval separates the CS and US (McEchron et al., 1998, 2000; Quinn et al., 2002). In addition, this paradigm is well characterized and enables testing hippocampal-dependent nonspatial learning and memory (Lugo et al., 2014), as well as detecting subtle learning and memory deficits and enhancements in mice that are not found using other fear-conditioning methods (Wiltgen et al., 2005; Lugo et al., 2014). In the tone–trace test conducted 48 h after conditioning, PERK inhibition resulted in enhanced freezing during tone presentation, as well as in the trace interval after the tone, compared with vehicle-treated animals (Fig. 1e). However, context memory was not altered, as demonstrated by a similar freezing percentage in both groups (Fig. 1d). In addition, reduction of PERK activity at the time of conditioning had no effect on extinction (data not shown).
Figure 1.
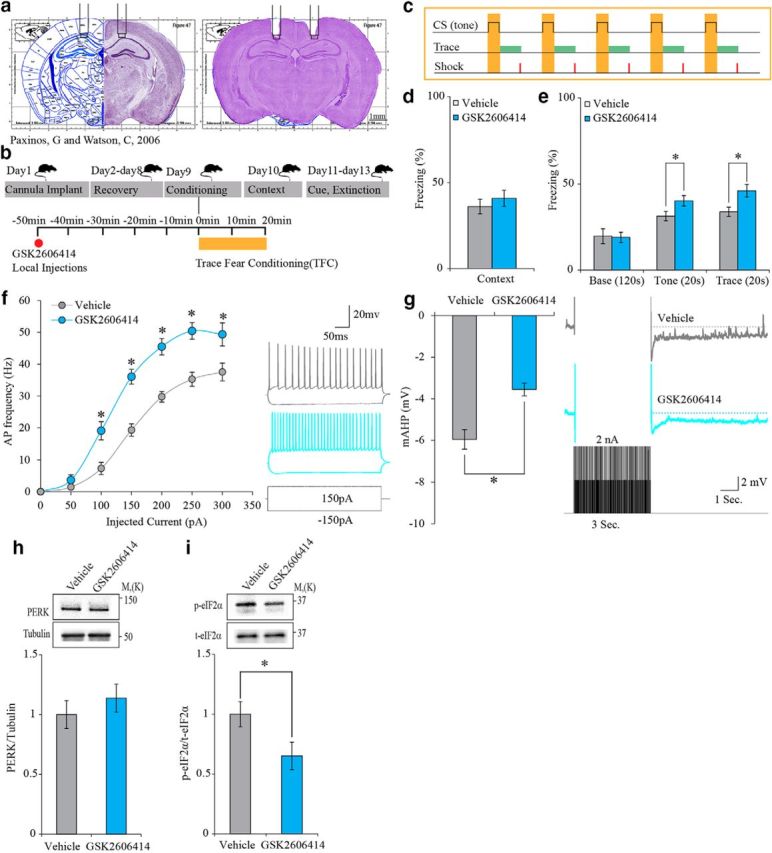
PERK inhibition (GSK2606414, 100 nm) in the CA1 enhances trace-fear-conditioning (TFC) memory and increases neuronal excitability. a, Cannulation site to CA1 region. b, Experimental design. c, TFC protocol. d, GSK2606414 has no effect on context memory (vehicle: n = 11; GSK2606414: n = 13; Mann–Whitney U test, U = 58.5, p = 0.451). e, GSK2606414 enhances tone memory. Inhibitor versus vehicle; GSK2606414 (*Mann–Whitney U test, U = 34, p = 0.03). GSK2606414 enhances trace memory in the 20 s trace interval after tone presentation. Inhibitor versus vehicle; GSK2606414 (independent-samples t test, t(21.086) = −2.672, *p = 0.014). f, GSK2606414 increases the firing index in treated neurons (n = 21) versus vehicle controls (n = 19) (two-way repeated measure ANOVA, F(1,18) = 41.24; *p = 4.8 × 10−6). Inset shows examples of traces obtained in response to ± 150 pA current steps with the vehicle (gray) or the inhibitor (magenta) in the recording pipette. g, GSK2606414 reduces mAHP in treated neurons (n = 21) versus vehicle controls (n = 19) (independent-samples t test, t(31.401) = −4.255, *p = 0.00017). Inset, Example traces showing AHP obtained in response to current protocol (see Materials and Methods) with the vehicle (gray) or the inhibitor (magenta) in the recording pipette. h, Total PERK protein levels are not changed in CA1 punches after GSK2606414 infusion (independent-samples t test, t(14) = −0.832, p = 0.419). i, GSK2606414 infusion into the CA1 region reduces p- eIF2α in adult mice (independent-samples t test, t(14) = 2.235, *p = 0.042).
To test possible neuronal mechanisms underlying the cognitive enhancement observed by a reduction in PERK activity, we examined intrinsic neuronal properties using whole-cell patch-clamp recording from pyramidal CA1 neurons after PERK inhibition. PERK inhibition (GSK2606414, 100 nm, in the recording pipette) in acute mouse hippocampal slices resulted in increased neuronal excitability as reflected in increased AP firing frequency obtained in response to depolarizing current steps (Fig. 1f). Moreover, PERK inhibition caused a significantly reduced mAHP, another indication of increased neuronal excitability (Fig. 1g). The effect of the PERK inhibitor GSK2606414 on neuronal passive intrinsic properties was also examined. Neither RMP nor Rin was changed. However, PERK inhibition hyperpolarized the AP threshold and produced longer and narrower AP (data not shown). On the biochemical level, PERK inhibition had no effect on total PERK levels, but resulted in significant reduction in p-eIF2α levels (Fig. 1h,i).
Reduction in CA1 PERK expression level enhances hippocampal-dependent memory and neuronal excitability
To determine whether genetic reduction of PERK expression level yields similar results to those obtained with pharmacological inhibition of its activity, we bilaterally injected an AAV-expressing PERK-specific shRNA into the CA1 region of the hippocampus (Fig. 2a,b,e). The effect on hippocampal-dependent learning was examined using the trace-fear-conditioning paradigm as described in Figure 1. Indeed, the cognitive enhancement observed after genetically reducing PERK levels was similar to that observed after CA1-specific inhibition of PERK activity: whereas tone and trace memory were significantly enhanced in the animals injected with PERK shRNA AAV compared with control animals injected with SCR shRNA AAV (Fig. 2g), context memory was unaffected (Fig. 2f). The extinction of tone and trace memory in animals treated with PERK shRNA was normal (data not shown). As expected, animals injected with PERK shRNA AAV showed a reduction in both PERK expression level and p-eIF2α level in CA1 punches compared with SCR AAV-injected animals (Fig. 2c,d).
Figure 2.

Genetic reduction of PERK expression in the CA1 enhances trace-fear-conditioning memory. a, Representative illustration of target area for PERK knock-down. b, Structure of the plasmid used to express either SCR or PERK shRNA sequence. c, Total PERK levels are reduced in CA1 of animals injected with PERK shRNA (independent-samples t test, t(14) = 2.724, *p = 0.016). d, PERK knock-down reduces p-eIF2α level (independent-samples t test, t(14) = 2.28, *p = 0.039) in the CA1 region. e, Immunohistochemistry for EGFP reporter demonstrating that PERK shRNA AAV expression is restricted to the CA1 region of the hippocampus. f, Context memory is similar in animals injected with PERK shRNA or SCR-shRNA. g, Reduced PERK expression enhances tone (independent-samples t test, for tone: t(20) = −2.436, *p = 0.024) and trace (independent-samples t test, for trace: t(20) = −2.69, *p = 0.014) memory in animals injected with PERK shRNA compared with control animals injected with SCR-shRNA. Data are shown as mean freezing percentage ± SEM.
The changes in intrinsic properties were similar to those observed with the PERK inhibitor. To complement the behavioral data, 4-month-old mice were injected with PERK shRNA AAV or SCR sequence AAV into the CA1 region of hippocampus. The reduction of PERK expression levels in the CA1 region resulted in increased neuronal excitability (Fig. 3). AP firing frequency obtained in response to depolarizing current steps was significantly higher in neurons infected with PERK shRNA AAV compared with neurons infected with SCR AAV (Fig. 3a). In addition, neurons infected with PERK shRNA AAV showed a significant reduction in mAHP compared with neurons infected with SCR AAV (mean difference of 3.3 mV; Fig. 3b). In addition, the passive properties of neurons infected with PERK shRNA were examined. Similar to the inhibition of PERK activity, reducing perk expression levels had no effect on Rin, but hyperpolarized the AP threshold (data not shown). Interestingly, in contrast to acute inhibition of PERK activity with GSK2606414, chronic reduction in perk expression level hyperpolarized the RMP and had no effect on AP amplitude and AP midwidth compared with the neurons infected with SCR shRNA AAV (data not shown).
Figure 3.

Genetic reduction of PERK expression in the CA1 increases neuronal excitability. a, AP frequency was increased in PERK shRNA AAV-infected neurons (n = 14) versus SCR-shRNA AAV infected control neurons (n = 13) (two-way repeated-measures ANOVA, F(1,12) = 25.45; *p = 0.00029). Inset shows examples of voltage traces obtained in response to ±150 pA current steps in PERK shRNA AAV infected neuron (gray) or in SCR-shRNA AAV-infected neuron (blue). b, mAHP amplitude was smaller in PERK shRNA AAV-infected neurons (n = 14) versus SCR-shRNA AAV infected neurons (n = 13) (independent-samples t test, t(25) = 5.71, *p = 6 × 10−6). Examples for voltage traces are shown in the inset. Data are shown as mean freezing percentage ± SEM.
PERK reduction in the CA1 region of the hippocampus reverses memory deterioration and decreased neuronal excitability associated with age
Together, our results demonstrate that reduced PERK expression levels or activity improves hippocampal-dependent memory and increases neuronal excitability. Because PERK mRNA levels are increased in middle-aged mice (Fig. 4a), we next wished to determine whether PERK knock-down in the hippocampus of older (12-month-old) mice would have a beneficial effect on behavioral performance. To test this hypothesis, PERK shRNA AAV or SCR AAV was bilaterally injected into the CA1 region of the hippocampus of 10-month-old mice and behavioral performance was assessed at the age of 12 months. In addition, 3-month-old mice were bilaterally injected with SCR AAV into the CA1 region of the hippocampus and assessed for behavioral performance at the age of 5 months, thus enabling the assessment of the effect of age. In these experiments, we found that contextual memory, as measured in the trace-fear-conditioning paradigm, was impaired in 12-month-old animals injected with SCR AAV compared with 5-month-old animals injected with the same vector (Fig. 4b). However, administration of PERK shRNA AAV to 12-month-old animals prevented this age-induced memory decline, resulting in contextual memory performance similar to 5-month-old animals injected with SCR AAV (Fig. 4a). A similar rescue was observed in the tone–trace memory test, in which 12-month-old animals injected with PERK shRNA AAV performed at least as well as 5-month-old animals injected with SCR AAV, overcoming the age-induced memory impairment (Fig. 4c). Extinction was normal in both 12-month-old animals injected with PERK shRNA and with SCR control (data not shown).
Figure 4.

Genetic reduction of PERK expression in the CA1 region alleviates memory deficits in aging animals and increases neuronal excitability of middle-aged neurons beyond young neurons expressing SCR controls. a, PERK mRNA relative quantity is increased in 13-month-old animals compared with 4-month-old animals. b, Context memory is impaired in 12-month-old animals injected with a SCR lentivirus compared with 5-month-old animals injected with the same vector. PERK knock-down in 12-month-old animals restores context text memory, comparable to 5-month-old injected with the SCR vector [one-way ANOVA F(2,24) = 3.728, *p = 0.039, Tukey's post hoc test *p = 0.043 SCR (5 months); SCR (12 months)]. c, Freezing during tone test is similar between the three groups. PERK knock-down increases trace freezing in 12-month-old animals versus 12-month-old SCR controls, comparable to 5-month-old animals treated with a SCR vector [SCR (5 months), one-way ANOVA, F(2,24) = 4.24, *p = 0.026, Tukey post hoc test, *p = 0.021]. d, Aging decreases AP frequency (12-month SCR, n = 18 vs 5-month SCR, n = 13; Bonferroni post hoc, *p = 0.04). PERK knock-down (n = 21) reverses this reduction to levels comparable to 5-month-old animals treated with a SCR vector Bonferroni post hoc (*p = 0.001; two-way repeated-measures ANOVA, F(2,11) = 26.526; *p = 6.1 × 10−5). e, Aging increases mAHP (12-month SCR, n = 18 vs 5-month SCR, n = 13 Tukey post hoc, *p = 0.0048). PERK knock-down reduces mAHP in 12-month-old animals (n = 21) versus 5-month-old and 12-month-old SCR controls (*Tukey's post hoc, p = 3.65 × 10−8; *one-way ANOVA, F(2,49) = 66.59; p = 1.06 × 10−14). The 5-month SCR group used in d and e is the same as in Figure 3a and b.
On the physiological level, AP frequency in infected neurons derived from 12-month-old animals injected with SCR AAV was lower than in infected neurons from 5-month-old animals treated with the same vector shown in Figure 3a (the 5-month SCR group used in Fig. 4d,e is the same as in Fig. 3a,b), reflecting normal physiological changes that accompany aging (Fig. 4d), as described previously (Kumar and Foster, 2007; Gant and Thibault, 2009). However, this aging-induced decrease in AP frequency was prevented in infected neurons derived from 12-month-old animals treated with PERK shRNA AAV and AP frequency was even higher in these animals compared with 5-month-old animals treated with SCR AAV (Fig. 4d). Furthermore, the effect of normal aging on neuronal excitability was reflected in the mAHP that was significantly increased in neurons derived from 12-month-old animals treated with SCR AAV compared with neurons derived from 5-month-old SCR injected animals shown in Figure 3b (Fig. 4e). However, treatment with PERK shRNA AAV not only reversed this increase in 12-month-old animals but also resulted in significantly smaller mAHP than in the 5-month-old animals treated with SCR AAV (Fig. 4e). When assessing passive properties, RMP in neurons from 12-month-old animals infected with either PERK shRNA AAV or SCR AAV was of similar levels and more hyperpolarized compared with neurons derived from 5-month-old animals treated with SCR AAV (data not shown). A molecular pathway known to regulate memory and neuronal intrinsic properties is CREB. A recent study has shown that PERK stimulation inactivates CREB and destabilizes PSD95 in dendritic spines (Sen et al., 2017). To determine whether CREB is the signaling pathway downstream of the effects of PERK that leads to modifications in neuronal intrinsic properties in middle-aged mice, we injected another group of 11-month-old animals with PERK shRNA or SCR control to the CA1 region of the hippocampus. Our results show that, whereas administration of PERK shRNA reduced expression levels of both PERK and eIF2α phosphorylation in the CA1 region of the hippocampus (Fig. 5a,b, in agreement with Fig. 2), no change was observed in the expression levels of p-CREB (Ser 133; Fig. 5c), total PSD95, or its phosphorylated form at threonine-19 (Fig. 5d,e).
Figure 5.

CA1 expression levels of p-CREB, total PSD95, and p-PSD95 do not change after PERK knock-down in middle-aged mice. Total levels of a, PERK and b, p-eIF2α (S51) are reduced in the CA1 region of PERK shRNA-injected middle-aged mice (12-month-old) [PERK shRNA: n = 5, SCR: n = 4; PERK: independent-samples t test, t(6.928) = 5.695, *p = 0.0008; p-eIF2α (S51): independent-samples t test, t(4.851) = 4.348, *p = 0.0079]. Levels of p-CREB (S133) (c), total PSD95 (d), or p-PSD95 (e) in the CA1 region do not change after administration of PERK shRNA to the CA1 of middle-aged mice [p-CREB: independent-samples t test, t(4.595) = 0.9977, p = 0.368; total PSD95: independent-samples t test, t(4.494) = 0.0188, p = 0.9986); p-PSD95 (T19): independent-samples t test, t (5.633) = 2.292, p = 0.0646].
Discussion
In the current work, we show that local inhibition of PERK activity by infusing a PERK-specific inhibitor (GSK2606414) into the CA1 region of the hippocampus of wild-type mice enhances trace and tone memories using the trace-fear-conditioning paradigm (Fig. 1), whereas the extinction of the trace and tone memories remained normal. Similar results of enhanced trace and tone memories with normal memory extinction were also obtained following prolonged reduction in perk expression level, achieved by genetically knocking down PERK in the CA1 region using PERK-specific shRNA (Fig. 2). This phenotype was distinct from that of the PERK KO mouse model under the CaMKII promoter, characterized by impaired hippocampal-dependent flexibility, with no effect on memory strength (Trinh et al., 2012, 2014). The different phenotype observed in the present study suggests that the role of PERK in cognitive function depends on the brain area, cell type manipulated, and behavioral paradigm used. For example, injecting viral vector expressing shRNA against PERK into the CA1 region of the hippocampus, as described in the present study, reduces the expression level of PERK in both excitatory and inhibitory neurons, not only in excitatory neurons (Trinh et al., 2012, 2014). In addition, different behavioral paradigms have been used in the studies performed using the PERK KO mice than the one used in the present study. Interestingly, pkr KO mice, another eIF2α kinase, have a phenotype that is specific to interneurons (Zhu et al., 2011). It is thus possible that the effect is dependent on specific neuronal subtype populations.
In accordance with the enhanced behavioral performance observed after either PERK inhibition (using GSK2606414) or decreased PERK expression (using a viral vector), both manipulations resulted in increased neuronal excitability, manifested as significant reduction in mAHP amplitude and increase in AP firing frequency (Figs. 1, 3). This result indicates that PERK plays a role in the homeostasis of neuronal intrinsic properties. Assuming that aging can be biochemically defined as prolonged metabolic stress, the link between aging and changes in neuronal intrinsic properties can be mediated by PERK, which serves as a main cellular mechanism to respond to endoplasmic reticulum (ER) stress. Further research is required to better define additional PERK targets during normal aging.
It is currently unknown how inhibition or decreased expression of PERK leads to increased neuronal excitability. One possibility is that PERK exerts its effect via CREB signaling (Sen et al., 2017). As we and others have shown, genetic or pharmacologic reduction of PERK decreases phosphorylation of eIF2α in neurons in vitro and in vivo (Ounallah-Saad et al., 2014; Bruch et al., 2017). Phosphorylation of eIF2α, in turn, activates CREB by inhibiting the CREB repressor ATF4 and enhances memory performances in different learning tasks by decreasing the threshold for neuronal excitability (Costa-Mattioli et al., 2005, 2007; Stern et al., 2013; Devi and Ohno, 2014). Different CREB mutant mice with loss of CREB function have impaired memory formation and long-term potentiation (LTP), whereas enhanced CREB function in CREB-DIEDML mice and CREB-Y134F mice show improved memory formation. In addition, CREB-VP16 mice in which CREB is expressed constitutively displayed a lowered threshold of long-lasting LTP induction. These findings have been shown to be mediated by regulating the expression of c-fos, activity-regulated cytoskeleton-associated protein (Arc), and brain-derived neurotrophic factor (BDNF) (Sheng et al., 1991; Finkbeiner et al., 1997; Kawashima et al., 2009; Kida, 2012). Conversely, we found that PERK reduction in the CA1 region of the hippocampus does not affect CREB phosphorylation, suggesting that CREB activity is not downstream of PERK under normal conditions. However, we cannot rule out that specific neuronal subtypes are differentially regulated because our tissue samples contain different cells.
Interestingly, PERK has been shown to regulate insulin secretion and calcium dynamics in β cells of the pancreas (Wang et al., 2013a). In addition, acute inhibition of PERK reduced the rise in intracellular Ca2+ concentration in response to activation of acetylcholine, metabotropic glutamate, and bradykinin-2 receptor in primary cortical neurons. This was shown to occur by increased Ca2+ release from the ER mediated by inositol triphosphate receptor in parallel with decreased extracellular Ca2+ influx mediated by Gq-protein-coupled receptors (Zhu et al., 2016).
These findings raise the possibility that PERK may affect neuronal excitability at least in part by regulating intracellular calcium dynamics. Further research is required to better characterize this possible link between calcium and PERK in the brain and its effect on cognitive performance in health and disease. Therefore, manipulation of PERK levels or activity may modulate neuronal plasticity by regulating and controlling neuronal excitability in parallel and possibly independently of its role as the major eIF2α-regulating kinase. It was found recently that PERK deletion alleviates mGluR-LTD impairment in aging AD animals (Yang et al., 2016). However, our findings present an additional cellular mechanism that is distinct from LTP- and LTD-related mechanisms. Furthermore, we show here that reducing perk expression levels rescues age-dependent deficits, as measured both on the behavioral level and on the physiological level (Fig. 4). This is consistent with the reported increase in hippocampal phosphorylation levels of eIF2α with age in both rats and mice and in young AD model mice (Segev et al., 2013, 2016).
The relationships among aging, neuronal intrinsic properties, and cognitive function are complex. Changes in PERK levels correlated with age may underlie some of the processes that lead to cognitive deterioration by altering mAHP in CA1 neurons (Disterhoft and Oh, 2006, 2007). Consistent with the literature, we observed that mAHP is increased in the aging hippocampus (Disterhoft and Oh, 2007; Kumar and Foster, 2007; Gant and Thibault, 2009). The functional consequence of this age-related increase in mAHP is that CA1 neurons fire fewer APs in response to a prolonged depolarizing stimulus; that is, the intrinsic neuronal excitability of these neurons is reduced by the larger mAHP.
mAHP is activated by Ca2+ ions released from the ER via Ca2+-induced Ca2+ release (CICR) mechanisms and Ca2+ ions that enter the cell via voltage-gated calcium channels (VGCCs). VGCCs are modulated by Gq proteins, which govern calcium-dependent neurotransmission and synaptic communication (Bernheim et al., 1991; Mintz and Bean, 1993; Catterall and Few, 2008).
In normal aging, mAHP is significantly increased (Disterhoft and Oh, 2006, 2007). There are numerous potential reasons for this increase, which may lead to cognitive deficits (Oh et al., 2010). Examples include increased L-type VGCCs (Thibault and Landfield, 1996; Norris et al., 1998; Power et al., 2002), increased CICR (Kumar and Foster, 2004; Gant et al., 2006), increased calcineurin activity and expression (Foster et al., 2001) that leads to a reduction in CREB activation (Bito et al., 1996; Mons et al., 2004), and reduced Na+/K+-ATPase activity (Koçak et al., 2002).
Our data show that, in young (5-month-old) animals, pharmacological as well as genetic reduction of PERK activity or levels resulted in significantly reduced mAHP, increased excitability, and improved cognitive performance. Moreover, genetic reduction of PERK expression in the CA1 region of the hippocampus in aging (12-month-old) mice rescued memory deterioration, reduced the mAHP, and increased AP frequency. This is the first time, to our knowledge, that reducing PERK levels has been reported to affect neuronal intrinsic excitability properties and to rescue cognitive deficits related to normal aging. Further research is required to determine the specific molecular changes that result from PERK reduction and lead to the observed cellular changes and correlated observed cognitive changes.
Aging is the main risk factor for developing neurodegenerative disorders involving cognitive disabilities (Segev et al., 2013, 2015, 2016; Wang et al., 2013b). Recent studies have highlighted PERK as a potential therapeutic target for several neurodegenerative diseases such as prion disease, AD, and frontotemporal dementia (Moreno et al., 2012; Ma et al., 2013; Devi and Ohno, 2014; Radford et al., 2015; Penke et al., 2016). One possible mechanism through which PERK is thought to mediate brain diseases in aging is via its role in the regulation of the unfolded protein response, an ER stress response that affects cell fate (Ma et al., 2013; Moreno et al., 2013). The ER, along with maintaining protein folding/unfolding, controls cellular Ca2+ homeostasis and can therefore affect neuronal excitability (De Felice and Lourenco, 2015).
Interestingly, both Ca2+/calmodulin-dependent protein phosphatase calcineurin (CaN) and ATPase functionality are modulated by binding PERK. Therefore, CaN may serve as a link among PERK activation, Ca2+ regulation, synaptic plasticity, cell survival, and cognition (Bollo et al., 2010). For example, overexpression of CaN in young adult animals leads to altered synaptic function and memory retention deficits (Mansuy et al., 1998; Winder et al., 1998; Zhuo et al., 1999) remarkably similar to those observed in normal aged rodents (Sharp et al., 1985; Foster, 1999). In addition, age-related impairments in neural function and cognition may involve increased CaN activity as a result of Ca2+ dyshomeostasis (Kumar and Foster, 2005). Furthermore, expression levels of CaN and its coactivator, Na+/K+-ATPase, are inversely correlated with the severity of AD (Wu et al., 2010; Karch et al., 2013).
In recent reports, oral treatment of prion disease and of frontotemporal dementia rodent models with PERK inhibitor GSK206414 resulted in the rescue of the pathology and associated neurodegeneration (Moreno et al., 2013; Radford et al., 2015), with diabetes as a side effect (Wang et al., 2013a, 2014). Therefore, targeting PERK systemically to treat age-related cognitive deficits and neurodegeneration will require a better understanding of the drug dynamics to allow improved drug design that would make it more specific to brain cells. Gene therapy in the brain using AAV, as described in the current study, may be a useful tool for achieving local specificity. Nevertheless, substantial studies are required to assess the safety and potential adverse events that may arise when using gene-modifying therapies. The notion that neuronal cells can be rejuvenated and that age-dependent cognitive decline can be restored by manipulating PERK position PERK as a major target with which to develop therapies for mild cognitive impairment and neurodegeneration.
Footnotes
This work was supported by the Israeli Ministry of Science, Technology, and Space (MOST 3-12080 to K.R.); the Israel Science Foundation (ISF 1003/12, ISF-IDRC 2395/2015 to K.R.); the Wolfson Charitable Trust (K.R.); the Ministry of Science and Technology (Eshkol Postdoctoral Fellowship to H.O.-S.); the Tauber Foundation (Fellowship to H.O.-S.); and the Israeli Planning and Budgeting Committee Program Fellowships for Outstanding Post-Doctoral Fellows from China and India (V.S.). We thank the members of the K.R. laboratory, specifically Dr. Shunit Gal Ben-Ari, for critical reading of this manuscript; Noah Cohen for valuable contributions to this manuscript; and Dr. Axten J. and G.S.K. for providing the PERK small-molecule inhibitor GSK2606414.
The authors declare no competing financial interests.
References
- Bangasser DA, Waxler DE, Santollo J, Shors TJ (2006) Trace conditioning and the hippocampus: the importance of contiguity. J Neurosci 26:8702–8706. 10.1523/JNEUROSCI.1742-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheim L, Beech DJ, Hille B (1991) A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron 6:859–867. 10.1016/0896-6273(91)90226-P [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell 87:1203–1214. 10.1016/S0092-8674(00)81816-4 [DOI] [PubMed] [Google Scholar]
- Bollo M, Paredes RM, Holstein D, Zheleznova N, Camacho P, Lechleiter JD (2010) Calcineurin interacts with PERK and dephosphorylates calnexin to relieve ER stress in mammals and frogs. PLoS One 5:e11925. 10.1371/journal.pone.0011925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruch J, Xu H, Rösler TW, De Andrade A, Kuhn PH, Lichtenthaler SF, Arzberger T, Winklhofer KF, Müller U, Höglinger GU (2017) PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol Med 9:371–384. 10.15252/emmm.201606664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Few AP (2008) Calcium channel regulation and presynaptic plasticity. Neuron 59:882–901. 10.1016/j.neuron.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Chakraborty D, Truong DQ, Bikson M, Kaphzan H (2017) Neuromodulation of axon terminals. Cereb Cortex 24:1–9. 10.1093/cercor/bhx158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor DA, Gould TJ (2016) The role of working memory and declarative memory in trace conditioning. Neurobiol Learn Mem 134:193–209. 10.1016/j.nlm.2016.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, Imataka H, Cuello AC, Seidah N, Sossin W, Lacaille JC, Ron D, Nader K, Sonenberg N (2005) Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 436:1166–1173. 10.1038/nature03897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, Krnjević K, Lacaille JC, Nader K, Sonenberg N (2007) eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129:195–206. 10.1016/j.cell.2007.01.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Lourenco MV (2015) Brain metabolic stress and neuroinflammation at the basis of cognitive impairment in Alzheimer's disease. Front Aging Neurosci 7:94. 10.3389/fnagi.2015.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M (2014) PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer's disease. Neurobiol Aging 35:2272–2281. 10.1016/j.neurobiolaging.2014.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM (2006) Pharmacological and molecular enhancement of learning in aging and Alzheimer's disease. J Physiol Paris 99:180–192. 10.1016/j.jphysparis.2005.12.079 [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM (2007) Alterations in intrinsic neuronal excitability during normal aging. Aging Cell 6:327–336. 10.1111/j.1474-9726.2007.00297.x [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19:1031–1047. 10.1016/S0896-6273(00)80395-5 [DOI] [PubMed] [Google Scholar]
- Foster TC. (1999) Involvement of hippocampal synaptic plasticity in age-related memory decline. Brain Res Brain Res Rev 30:236–249. 10.1016/S0165-0173(99)00017-X [DOI] [PubMed] [Google Scholar]
- Foster TC, Sharrow KM, Masse JR, Norris CM, Kumar A (2001) Calcineurin links Ca2+ dysregulation with brain aging. J Neurosci 21:4066–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal-Ben-Ari S, Kenney JW, Ounalla-Saad H, Taha E, David O, Levitan D, Gildish I, Panja D, Pai B, Wibrand K, Simpson TI, Proud CG, Bramham CR, Armstrong JD, Rosenblum K (2012) Consolidation and translation regulation. Learn Mem 19:410–422. 10.1101/lm.026849.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Thibault O (2009) Action potential throughput in aged rat hippocampal neurons: regulation by selective forms of hyperpolarization. Neurobiol Aging 30:2053–2064. 10.1016/j.neurobiolaging.2008.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant JC, Sama MM, Landfield PW, Thibault O (2006) Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci 26:3482–3490. 10.1523/JNEUROSCI.4171-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildish I, Manor D, David O, Sharma V, Williams D, Agarwala U, Wang X, Kenney JW, Proud CG, Rosenblum K (2012) Impaired associative taste learning and abnormal brain activation in kinase-defective eEF2K mice. Learn Mem 19:116–125. 10.1101/lm.023937.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Dasari S, Onoue K, Stephens EK, Hasse JM, Avesar D (2013) A sodium-pump-mediated afterhyperpolarization in pyramidal neurons. J Neurosci 33:13025–13041. 10.1523/JNEUROSCI.0220-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise C, et al. (2017) eEF2K/eEF2 pathway controls the excitation/inhibition balance and susceptibility to epileptic seizures. Cereb Cortex 27:2226–2248. 10.1093/cercor/bhw075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper C, Markevich V, Plattner F, Killick R, Schofield E, Engel T, Hernandez F, Anderton B, Rosenblum K, Bliss T, Cooke SF, Avila J, Lucas JJ, Giese KP, Stephenson J, Lovestone S (2007) Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci 25:81–86. 10.1111/j.1460-9568.2006.05245.x [DOI] [PubMed] [Google Scholar]
- Kaphzan H, Buffington SA, Ramaraj AB, Lingrel JB, Rasband MN, Santini E, Klann E (2013) Genetic reduction of the α1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep 4:405–412. 10.1016/j.celrep.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Jeng AT, Goate AM (2013) Calcium phosphatase calcineurin influences tau metabolism. Neurobiol Aging 34:374–386. 10.1016/j.neurobiolaging.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Okuno H, Nonaka M, Adachi-Morishima A, Kyo N, Okamura M, Takemoto-Kimura S, Worley PF, Bito H (2009) Synaptic activity-responsive element in the Arc/Arg3.1 promoter essential for synapse-to-nucleus signaling in activated neurons. Proc Natl Acad Sci U S A 106:316–321. 10.1073/pnas.0806518106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida S. (2012) A functional role for CREB as a positive regulator of memory formation and LTP. Exp Neurobiol 21:136–140. 10.5607/en.2012.21.4.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koçak H, Oner P, Oztaş B (2002) Comparison of the activities of Na(+), K(+)-ATPase in brains of rats at different ages. Gerontology 48:279–281. 10.1159/000065249 [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC (2004) Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol 91:2437–2444. 10.1152/jn.01148.2003 [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC (2005) Intracellular calcium stores contribute to increased susceptibility to LTD induction during aging. Brain Res 1031:125–128. 10.1016/j.brainres.2004.10.023 [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC (2007) Neurophysiology of old neurons and synapses. In: Brain Aging: Models, Methods, and Mechanisms (Riddle DR, editor). Boca Raton, FL: Taylor and Francis. [PubMed] [Google Scholar]
- Lugo JN, Smith GD, Holley AJ (2014) Trace-fear-conditioning in mice. J Vis Exp 85. 10.3791/51180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E (2013) Suppression of eIF2α kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nat Neurosci 16:1299–1305. 10.1038/nn.3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansuy IM, Mayford M, Jacob B, Kandel ER, Bach ME (1998) Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memory. Cell 92:39–49. 10.1016/S0092-8674(00)80897-1 [DOI] [PubMed] [Google Scholar]
- McEchron MD, Bouwmeester H, Tseng W, Weiss C, Disterhoft JF (1998) Hippocampectomy disrupts auditory trace-fear-conditioning and contextual fear-conditioning in the rat. Hippocampus 8:638–646. [DOI] [PubMed] [Google Scholar]
- McEchron MD, Tseng W, Disterhoft JF (2000) Neurotoxic lesions of the dorsal hippocampus disrupt auditory-cued trace heart rate (fear) conditioning in rabbits. Hippocampus 10:739–751. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Bean BP (1993) GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron 10:889–898. 10.1016/0896-6273(93)90204-5 [DOI] [PubMed] [Google Scholar]
- Mons N, Segu L, Nogues X, Buhot MC (2004) Effects of age and spatial learning on adenylyl cyclase mRNA expression in the mouse hippocampus. Neurobiol Aging 25:1095–1106. 10.1016/j.neurobiolaging.2003.10.014 [DOI] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR (2012) Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485:507–511. 10.1038/nature11058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 5:206ra138. 10.1126/scitranslmed.3006767 [DOI] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC (1998) Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci 18:3171–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R (2008) Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60:988–1009. 10.1016/j.neuron.2008.10.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Oliveira FA, Disterhoft JF (2010) Learning and aging related changes in intrinsic neuronal excitability. Front Aging Neurosci 2:2. 10.3389/neuro.24.002.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounallah-Saad H, Sharma V, Edry E, Rosenblum K (2014) Genetic or pharmacological reduction of PERK enhances cortical-dependent taste learning. J Neurosci 34:14624–14632. 10.1523/JNEUROSCI.2117-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penke B, Bogár F, Fülöp L (2016) Protein folding and misfolding, endoplasmic reticulum stress in neurodegenerative diseases: in trace of novel drug targets. Curr Protein Pept Sci 17:169–182. 10.2174/1389203716666151102104653 [DOI] [PubMed] [Google Scholar]
- Power JM, Wu WW, Sametsky E, Oh MM, Disterhoft JF (2002) Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro. J Neurosci 22:7234–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JJ, Oommen SS, Morrison GE, Fanselow MS (2002) Post-training excitotoxic lesions of the dorsal hippocampus attenuate forward trace, backward trace, and delay fear-conditioning in a temporally specific manner. Hippocampus 12:495–504. 10.1002/hipo.10029 [DOI] [PubMed] [Google Scholar]
- Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR (2015) PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol 130:633–642. 10.1007/s00401-015-1487-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozpędek W, Pytel D, Dziki Ł, Nowak A, Dziki A, Diehl JA, Majsterek I (2017) Inhibition of PERK-dependent pro-adaptive signaling pathway as a promising approach for cancer treatment. Pol Przegl Chir 89:7–10. [PubMed] [Google Scholar]
- Segev Y, Michaelson DM, Rosenblum K (2013) ApoE ε4 is associated with eIF2α phosphorylation and impaired learning in young mice. Neurobiol Aging 34:863–872. 10.1016/j.neurobiolaging.2012.06.020 [DOI] [PubMed] [Google Scholar]
- Segev Y, Barrera I, Ounallah-Saad H, Wibrand K, Sporild I, Livne A, Rosenberg T, David O, Mints M, Bramham CR, Rosenblum K (2015) PKR inhibition rescues memory deficit and ATF4 overexpression in apoe ε4 human replacement mice. J Neurosci 35:12986–12993. 10.1523/JNEUROSCI.5241-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segev Y, Livne A, Mints M, Rosenblum K (2016) Concurrence of high fat diet and APOE gene induces allele specific metabolic and mental stress changes in a mouse model of alzheimer's disease. Front Behav Neurosci 10:170. 10.3389/fnbeh.2016.00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, Gupta R, Kaiser H, Sen N (2017) Activation of PERK elicits memory impairment through inactivation of CREB and downregulation of PSD95 after traumatic brain injury. J Neurosci 37:5900–5911. 10.1523/JNEUROSCI.2343-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PE, McNaughton BL, Barnes CA (1985) Enhancement of hippocampal field potentials in rats exposed to a novel, complex environment. Brain Res 339:361–365. 10.1016/0006-8993(85)90105-2 [DOI] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME (1991) CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 252:1427–1430. 10.1126/science.1646483 [DOI] [PubMed] [Google Scholar]
- Stern E, Chinnakkaruppan A, David O, Sonenberg N, Rosenblum K (2013) Blocking the eIF2α kinase (PKR) enhances positive and negative forms of cortex-dependent taste memory. J Neurosci 33:2517–2525. 10.1523/JNEUROSCI.2322-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha E, Gildish I, Gal-Ben-Ari S, Rosenblum K (2013) The role of eEF2 pathway in learning and synaptic plasticity. Neurobiol Learn Mem 105:100–106. 10.1016/j.nlm.2013.04.015 [DOI] [PubMed] [Google Scholar]
- Thibault O, Landfield PW (1996) Increase in single L-type calcium channels in hippocampal neurons during aging. Science 272:1017–1020. 10.1126/science.272.5264.1017 [DOI] [PubMed] [Google Scholar]
- Trinh MA, Kaphzan H, Wek RC, Pierre P, Cavener DR, Klann E (2012) Brain-specific disruption of the eIF2α kinase PERK decreases ATF4 expression and impairs behavioral flexibility. Cell Rep 1:676–688. 10.1016/j.celrep.2012.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh MA, Ma T, Kaphzan H, Bhattacharya A, Antion MD, Cavener DR, Hoeffer CA, Klann E (2014) The eIF2α kinase PERK limits the expression of hippocampal metabotropic glutamate receptor-dependent long-term depression. Learn Mem 21:298–304. 10.1101/lm.032219.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, McGrath BC, Kopp RF, Roe MW, Tang X, Chen G, Cavener DR (2013a) Insulin secretion and Ca2+ dynamics in β-cells are regulated by PERK (EIF2AK3) in concert with calcineurin. J Biol Chem 288:33824–33836. 10.1074/jbc.M113.503664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Munoz EE, Zhu S, McGrath BC, Cavener DR (2014) Perk gene dosage regulates glucose homeostasis by modulating pancreatic β-cell functions. PLoS One 9:e99684. 10.1371/journal.pone.0099684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZZ, Zhang Y, Liu YQ, Zhao N, Zhang YZ, Yuan L, An L, Li J, Wang XY, Qin JJ, Wilson SP, O'Donnell JM, Zhang HT, Li YF (2013b) RNA interference-mediated phosphodiesterase 4D splice variants knock-down in the prefrontal cortex produces antidepressant-like and cognition-enhancing effects. Br J Pharmacol 168:1001–1014. 10.1111/j.1476-5381.2012.02225.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltgen BJ, Sanders MJ, Ferguson C, Homanics GE, Fanselow MS (2005) Trace-fear-conditioning is enhanced in mice lacking the delta subunit of the GABAA receptor. Learn Mem 12:327–333. 10.1101/lm.89705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER (1998) Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell 92:25–37. 10.1016/S0092-8674(00)80896-X [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT (2010) Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci 30:2636–2649. 10.1523/JNEUROSCI.4456-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Zhou X, Zimmermann HR, Cavener DR, Klann E, Ma T (2016) Repression of the eIF2α kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer's disease. Neurobiol Aging 41:19–24. 10.1016/j.neurobiolaging.2016.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Huang W, Kalikulov D, Yoo JW, Placzek AN, Stoica L, Zhou H, Bell JC, Friedlander MJ, Krnjević K, Noebels JL, Costa-Mattioli M (2011) Suppression of PKR promotes network excitability and enhanced cognition by interferon-γ-mediated disinhibition. Cell 147:1384–1396. 10.1016/j.cell.2011.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, McGrath BC, Bai Y, Tang X, Cavener DR (2016) PERK regulates Gq protein-coupled intracellular Ca(2+) dynamics in primary cortical neurons. Mol Brain 9:87. 10.1186/s13041-016-0268-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Zhang W, Son H, Mansuy I, Sobel RA, Seidman J, Kandel ER (1999) A selective role of calcineurin aalpha in synaptic depotentiation in hippocampus. Proc Natl Acad Sci U S A 96:4650–4655. 10.1073/pnas.96.8.4650 [DOI] [PMC free article] [PubMed] [Google Scholar]