

Abstract
Strong human genetic evidence points to an essential contribution of the voltage-gated sodium channel Nav1.7 to pain sensation: loss of Nav1.7 function leads to congenital insensitivity to pain, whereas gain-of-function mutations in the SCN9A gene that encodes Nav1.7 cause painful neuropathies, such as inherited erythromelalgia, a syndrome characterized by episodic spontaneous pain. Selective Nav1.7 channel blockers thus hold promise as potential painkillers with improved safety and reduced unwanted side effects compared with existing therapeutics. To determine the maximum effect of a theoretically perfectly selective Nav1.7 inhibitor, we generated a tamoxifen-inducible KO mouse model enabling genetic deletion of Nav1.7 from adult mice. Electrophysiological recordings of sensory neurons from these mice following tamoxifen injection demonstrated the loss of Nav1.7 channel current and the resulting decrease in neuronal excitability of small-diameter neurons. We found that behavioral responses to most, but surprisingly not all, modalities of noxious stimulus are abolished following adult deletion of Nav1.7, pointing toward indications where Nav1.7 blockade should be efficacious. Furthermore, we demonstrate that isoform-selective acylsulfonamide Nav1.7 inhibitors show robust analgesic and antinociceptive activity acutely after a single dose in mouse pain models shown to be Nav1.7-dependent. All experiments were done with both male and female mice. Collectively, these data expand the depth of knowledge surrounding Nav1.7 biology as it relates to pain, and provide preclinical proof of efficacy that lays a clear path toward translation for the therapeutic use of Nav1.7-selective inhibitors in humans.
SIGNIFICANCE STATEMENT Loss-of-function mutations in the sodium channel Nav1.7 cause congenital insensitivity to pain in humans, making Nav1.7 a top target for novel pain drugs. Targeting Nav1.7 selectively has been challenging, however, in part due to uncertainties in which rodent pain models are dependent on Nav1.7. We have developed and characterized an adult-onset Nav1.7 KO mouse model that allows us to determine the expected effects of a theoretically perfect Nav1.7 blocker. Importantly, many commonly used pain models, such as mechanical allodynia after nerve injury, appear to not be dependent on Nav1.7 in the adult. By defining which models are Nav1.7 dependent, we demonstrate that selective Nav1.7 inhibitors can approximate the effects of genetic loss of function, which previously has not been directly established.
Keywords: acylsulfonamide, congenital insensitivity to pain, drug development, Nav1.7, pain, tamoxifen
Introduction
Voltage-gated sodium (Nav) channels are required for action potential (AP) generation and propagation in neurons and other electrically excitable cells (Hodgkin and Huxley, 1952; Hille, 2001). Nine voltage-gated sodium channel α subunits (Nav1.1-Nav1.9) are expressed in mammals, each with distinct biophysical properties and expression patterns. Strong human genetic evidence points to an essential contribution of the Nav1.7 subtype to pain sensation: loss of Nav1.7 function leads to congenital insensitivity to pain (CIP), whereas gain-of-function mutations in the SCN9A gene that encodes Nav1.7 cause painful neuropathies, such as inherited erythromelalgia, a syndrome characterized by episodic spontaneous pain and flushing (Cummins et al., 2004; Han et al., 2009; Eberhardt et al., 2014; Lampert et al., 2014), and paroxysmal extreme pain disorder, characterized by intense pain in the anal, orbital, and submandibular regions (Dib-Hajj et al., 2008; Estacion et al., 2008; Choi et al., 2011).
Local anesthetics, such as lidocaine, block pain and other sensations via nonselective inhibition of Nav channels in primary afferent nerve fibers of the somatosensory nervous system. Subtype-selective Nav channel blockers hold promise as potential novel pain drugs with improved safety and reduced unwanted side effects by offering a more targeted therapeutic approach. In particular, given the human genetic evidence that Nav1.7 is essential for pain and the fact that Nav1.7 expression is largely limited to the peripheral nervous system (Ahmad et al., 2007; Morinville et al., 2007), selective Nav1.7 inhibitors are expected to provide substantial efficacy while avoiding systemic or CNS toxicity.
To determine what would be the theoretical maximum effect of a perfectly selective Nav1.7 inhibitor, we undertook a genetic proof-of-concept study where we developed a tamoxifen-inducible Nav1.7 KO mouse allowing adult deletion of SCN9A. We demonstrate that this allows the rapid and efficient in vivo removal of SCN9A mRNA and the slower but still efficient removal of Nav1.7 protein from sensory neurons. Whole-cell voltage-clamp experiments demonstrate the expected removal of TTX-sensitive Nav channels from small-diameter/medium-diameter sensory neurons and the concomitant reduction in neuronal excitability of these neurons. We then show that behavioral responses to most, but surprisingly not all, modalities of noxious stimulus are abolished following Nav1.7 removal, pointing toward indications where Nav1.7 blockade should be efficacious. Furthermore, we demonstrate that two novel acylsulfonamide tool compounds that are selective inhibitors of Nav1.7 possess analgesic activity in preclinical mouse models that were determined to be Nav1.7-dependent. We believe our results have important implications, not only for understanding Nav1.7 function in the pain sensory nervous system, but also for driving new therapeutics to alleviate chronic pain.
Materials and Methods
Animals.
Experimental procedures involving animals were approved by Genentech's Institutional Animal Care and Use Committee and conducted in accordance with the recommendations of the International Association for the Study of Pain. Mice at least 8 weeks of age of both sexes were used. Nav1.7 cKO mice (pCAGG-CreERtg; SCN9AloxP/loxP) and control littermates (SCN9AloxP/loxP) were administered tamoxifen (three injections of 100 mg/kg separated by 24 h; 10 ml/kg i.p.; in corn oil) (Hayashi and McMahon, 2002; Nassar et al., 2004). WT mice for pharmacological studies were C57BL/6 strain (Charles River).
DRG acute culture and recording.
Six to 8 weeks after the tamoxifen dosing, mice were killed by CO2 inhalation and DRGs were isolated bilaterally. The ganglia were first incubated at 37°C and 5% CO2 with 1 mg/ml collagenase Type IV (Sigma-Aldrich C1889) for 40 min, followed by incubation with 0.05% trypsin (Sigma-Aldrich T9935) for 45 min. Ganglia were washed and then dissociated into single somata via trituration through a P200 pipette tip. The neurons were plated onto laminin-coated glass coverslips and incubated for 2 h at 37°C and 5% CO2 to allow adherence. Coverslips were then flooded with complete cell medium consisting of DMEM/Hams-F12, 10% heat-inactivated horse serum, 2 mm l-glutamine, 0.8% d-glucose, 100 units penicillin, and 100 mg/ml streptomycin. Voltage-clamp and current-clamp recordings were performed within 24 h after cells were plated.
RT-PCR.
To examine Nav1.7 tissue distribution, multiple tissues, including DRGs, were collected from Nav1.7 cKO mice (pCAGG-CreERtg; SCN9AloxP/loxP) and control littermates (SCN9AloxP/loxP) 6–8 weeks after tamoxifen dosing. DRGs were also harvested at other time points to determine the time course of Nav1.7 KO on RNA level. Dissected tissues were stored in RNAlater solution (Invitrogen, AM7021) at −20°C until required for analysis. Total RNA was isolated using an RNeasy Lipid Tissue Mini kit (QIAGEN, #74804) and further reverse transcribed into complementary DNA using a TaqMan gene expression kit (Ambion, Cells-to-CT kit, AM1728). qRT-PCR was performed in an ABI viiA7 system (Applied Biosystems). Gene expression was examined with a custom-designed TaqMan assay, which detects the expression of a SCN9A sequence crossing the boundary of exons 16 and 17 (primer sequence: TGATGGTCATGGTGATTGGG, forward; TGTTTGCGTCGGTGTCTTC, reverse). All other TaqMan assays were purchased from Thermo Fisher Scientific. The relative gene expression was quantified using the comparative Ct method. The ΔCt was calculated by subtraction of β-actin Ct value from that of a target gene, and the relative expression was determined using 2−ΔΔCt.
Western blotting.
Total DRG protein was extracted via homogenization in ice-cold lysis buffer containing 125 mm NaCl, 20 mm Tris, 1% DDM, and 1× protease inhibitor (Roche Diagnostics, #11836153011). After centrifugation at 4°C for 15 min at 12,000 rpm, the supernatant was collected and the protein concentration was adjusted using a BCA protein assay kit (Pierce #23225). Then 20 mg protein was loaded per lane on a 3%–8% Tris-acetate gel (Invitrogen, EA0378) and transferred to a PVDF membrane. Anti β-tubulin III antibody (Sigma-Aldrich, T8578, 1:1000) and anti-Nav1.7 monoclonal antibody (Antibodies, #75–103, 1:400) were diluted in blocking buffer and incubated overnight at 4°C. The signals were detected with an ECL plus kit (Pierce, #32132) and captured/analyzed by a Chemidoc MP imaging system (Bio-Rad).
Electrophysiological recordings.
Whole-cell patch-clamp recording on HEK293 cells stably expressing recombinant human Nav isoforms was performed as described previously (Ahuja et al., 2015). The membrane holding potentials for subtype selectivity experiments were as follows: −60 mV (Nav1.7 and Nav1.5), −40 mV (Nav1.1, Nav1.2, Nav1.3, and Nav1.6), and −50 mV (Nav1.4). For DRG cultures, total Na+ current was first recorded in the presence of Ca2+ and K+ channel blockers and then differentiated into TTX-sensitive and -resistant current by perfusion with 0.5 μm TTX. Mouse DRG Nav1.9 recordings were made from DRGs isolated form homozygous Nav1.8-cre knock-in mice (over 8 weeks of age) that lack endogenous Nav1.8 (thereby ensuring that remaining TTX-resistant current is from Nav1.9 channels). Peak current amplitudes were measured and normalized to cell membrane capacitance to obtain current density (pA/Cm). For current-clamp recordings, we injected a small hyperpolarizing current to set the starting potential close to −70 mV. The rheobase current necessary for AP generation was determined for each neuron by injecting a series of depolarizing current (10 ms) with progressively greater intensity (10 pA increment). All experiments were performed at room temperature. All internal and external solutions were at pH 7.3 and ∼300 mOsm. For voltage-clamp recordings, we used different external solutions for different preparations to adjust Na+ current amplitude to an acceptable level (1000–4000 pA). The bath solution for Nav stable cell lines contained the following (in mm): 140 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2, and 10 HEPES, whereas the bath solution for DRG neuron recording contained the following (in mm): 20 NaCl, 100 TEA-Cl, 40 choline-Cl, 3 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose and 30 μm CdCl2. The patch pipettes (2–4 mΩ) were back-filled with internal solution containing the following (in mm): 140 CsF, 10 NaCl, 5 EGTA, 10 HEPES, and 1.5 MgCl2. For current-clamp recording, the recording chamber was continuously perfused with bath solution containing the following (in mm): 140 KCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, and 10 glucose. The internal solution contained the following (in mm): 125 K-gluconate, 15 KCl, 10 HEPES, 3.0 Mg-ATP, 0.3 Na-GTP, 3.0 sodium phosphocreatine, and 0.2 EGTA. For Nav1.9 recordings in Nav1.8 KO mouse DRGs, the bath solution consisted of the following (in mm): 130 NaCl, 4.8 KCl, 1 MgCl2, 10 HEPES, 2 CaCl2, 1 CsCl, 10 glucose, 5 TEA-Cl, 0.0005 TTX, and 0.3 Cd2+; and the internal solution consisted of the following (in mm): 100 CsCl, 30 CsF, 10 HEPES, 10 EGTA, 0.5 CaCl2, 1 MgCl2, 4 MgATP, and 0.4 Na2GTP. In vitro pharmacology experiments were conducted using a Cellectricon Dynaflow (Cellectricon) all-glass perfusion system, which allows laminar flow of compounds and prevents stickiness issues that occur with plastic perfusion systems.
Behavioral testing.
The hot plate, tail immersion, hindpaw radiant heat (Hargreaves), acetic acid, capsaicin flinching, formalin, and von Frey tests were performed as described previously (Shields et al., 2006). The hot plate test was performed at 55°C with a cutoff of 30 s to avoid tissue damage. Tail immersion into 55°C water was performed with a cutoff of 5 s. In the acetic acid test, abdominal stretching events were counted during the first 30 min following intraperitoneal injection of 0.9% acetic acid in saline (10 ml/kg). Amount of time spent flinching, licking, or biting was measured for 5 min after the intraplantar injection of 1 μg capsaicin (10 μl; vehicle = 10% ethanol, 0.5% Tween 80). The formalin test was performed using an intraplantar injection of 2% formalin (10 μl in saline). In the Randall–Selitto test, mice were lightly restrained and a smooth pressure ramp was applied to the tail (Analgesymeter, Ugo Basile); a positive response was noted and the test stopped when the mouse struggled to remove its tail from the apparatus or vocalized. Three trials were run per mouse at least 5 min apart, and the cutoff was 500 g. Intradermal cheek injection of chloroquine (500 μg, 10 μl, in saline) or histamine (150 μg, 10 μl, in saline) was performed as described previously (Shimada and LaMotte, 2008). Behavioral observations were made for 20 min after injection. For intensity-response experiments, electrical shock was delivered through the floor grid of a fear conditioning box, and startle responses were measured by a near-infrared camera controlled by Video-Freeze software (Med Associates). The active avoidance test was performed as described previously (Weber et al., 2015). Behavioral responses to acetone evaporative cooling were measured by applying a drop of acetone to the plantar surface of the hindpaw using a syringe with no needle and counting the cumulative amount of time spent displaying nocifensive behavior during the first 20 s after application. General neurological function was performed as described previously (Crawley, 2000). Mice were considered to have passed the test if the standard reflexes were present or abnormal postures were absent. The rotarod test was administered using an accelerating paradigm from 4 to 40 rpm over 5 min. Cutoff for the wire hang test was 60 s. In the novel odor recognition test, mice were tested one at a time after 5 min acclimation to a fresh cage with a cotton swab soaked in water suspended from the cage top. The water-soaked cotton swab is replaced with one soaked in 1 of 3 odored solutions (lemon, vanilla, or rosemary extract) and number of approaches within 30 s are recorded. A new cotton swab soaked in the same odored solution is introduced for the second and third trials. Trials 4–6 are conducted using a second odor, and trial 7 uses a third odor. Fewer approaches upon subsequent presentation of the same odor are considered habituation to a decreasingly novel stimulus, whereas reinvigorated approach behavior is taken as evidence of olfactory discrimination. The open field test was performed by placing each mouse into a 40.5 (length) × 40.5 (width) × 38 cm (height) Plexiglas arena outfitted with photobeams to detect horizontal and vertical movement and recording behavior for 60 min; analysis used TopScan software (CleverSys). All studies were performed by experimenters blinded to genotype.
Complete Freund's adjuvant (CFA) model.
An intraplantar injection of 10 μl of an emulsion of equal parts CFA and mineral oil was made to one hindpaw. Behavioral testing was performed at 24 h after injection for pharmacology studies or at the indicated time points for cKO mice. Paw thickness was measured after behavioral testing using a digital thickness gauge (Mitutoyo).
Surgical models.
Spared nerve injury (SNI) surgery entailed ligation and cutting of the sural and common peroneal branches of the sciatic nerve and was performed according to Shields et al. (2003). The incision model of postoperative pain was performed according to Pogatzki and Raja (2003).
Capsaicin-induced secondary mechanical allodynia.
A subcutaneous injection of 1 μg capsaicin was made at the ankle level. Mechanical withdrawal threshold was measured on the plantar surface of the hindpaw with von Frey filaments before and 30 min after capsaicin injection.
Drugs, dosing, and pharmacokinetic analysis.
GX-585 and GX-201 were formulated as a suspension in 0.5% methylcellulose/0.2% Tween 80 and administered by oral gavage in a volume of 10 ml/kg. Behavioral testing took place 1 h after dosing, as this was determined to be the Tmax in previous pharmacokinetic analysis. Immediately after the conclusion of behavioral testing, the mice were killed by CO2 inhalation. Blood samples were collected by cardiac puncture into K2EDTA-containing tubes and processed into plasma by centrifugation at 17,000 × g for 2 min and collection of supernatant. Plasma samples were frozen at −80°C until bioanalysis. Plasma concentrations of compounds were determined using liquid chromatography tandem mass spectrometry.
ISH.
DRGs from all segmental levels were harvested from 3 mice per genotype and pooled together for sectioning in a single block per genotype, with sections from both genotypes collected onto the same slide for uniformity of staining. ACD RNAScope 2.5 LS probe Mm-Scn9a(Nav1.7)-01, a 4zz pair probe is composed of 200 nt from target region 2812–3055 (Accession No: NM_018852.2) was used in ISH experiments. This probe binds to a region within exon-15, which is completely removed upon cre recombination. A fully automated ISH protocol from slide bake to counterstain was run using a BOND RX autostainer (Leica Microsystems). ACD RNAscope 2.5 LS Reagent Kit- RED catalog #322150 was used for signal amplification, and Bond Polymer Refine Red Detection catalog #DS9390 was used for red chromogen color development. Formalin-fixed paraffin-embedded tissue was sectioned at 4 μm and mounted onto slides, air dried overnight, and loaded onto BOND RX stainer. The slides were baked and dewaxed and the following script was executed: pretreatment, ER2 at 95°C for 15 minutes (Leica Microsystems) enzyme at 40°C for 15 minutes; probe hybridization, 120 minutes 42°C; ACD AMP 1 reagent 30 minutes 42°C; ACD AMP 2 reagent 15 minutes 42°C; ACD AMP 3 reagent 30 minutes 42°C; ACD AMP 4 reagent 15 minutes 42°C; ACD AMP 5 red reagent 15 minutes Ambient; ACD AMP 6 red reagent 15 minutes Ambient; mixed RED refine reagent 10 minutes Ambient; and hematoxylin reagent 5 minutes Ambient.
After hematoxylin counterstain, the slides were dried at 60°C for 1 h and coverslipped with Histomount. Slides were scanned using the SCN400 whole-slide imaging system (Leica Microsystems) at 200× magnification. Sections were analyzed using ImageJ. To quantify cross-sectional area of neuronal somata, two approximately perpendicular diameters (d1, d2) were measured, and the following formula was applied: area = π * (d1/2) × (d2/2).
Fos studies.
Mice were anesthetized with 2% isoflurane in O2 (0.5–1 L/min) throughout the duration of the experiment. Heat stimuli were applied by immersing one hindpaw into 55°C water 3 × 30 s with a 30 s interstimulus interval. Mechanical stimulus was a Randall–Selitto apparatus applied to one hindpaw ramped from 0 to 500 g three times with a 30 s interstimulus interval. Formalin (2%, 10 μl) was injected into the intraplantar space. Mice underwent cardiac perfusion with 4% PFA 1.5 h after stimulus. For electrical shock stimulation, awake mice were placed in fear conditioning boxes and received either 0.7 mA shock delivered through floor grids or no shock, then were immediately anesthetized with isoflurane and perfused 1.5 h later. For CFA mechanical allodynia experiments, mice were administered an intraperitoneal injection of CFA (10 μl, emulsion of equal parts CFA and saline) 24 h before stimulation. Stimulation consisted of walking on a rotarod at 5 rpm for 90 min. Mice were then perfused as above after a 30 min rest period in the home cage. Spinal cords were dissected, postfixed for 4 h in 4% PFA, cryopreserved in 30% sucrose overnight, and L4–6 segments were sectioned at 30 μm on a cryostat. Sections were processed for immunohistochemistry according to standard methods using rabbit anti-Fos primary antibody (Ab-5, Millipore; 1:1000 overnight) and biotinylated anti-rabbit secondary antibody (Vector; 1:200 for 2 h) before chromogenic development with nickel-enhanced diaminobenzidine (Sigma-Aldrich) using glucose oxidase as the hydrogen peroxide source. Sections were imaged with a 5500B microscope at 10× magnification (Leica Microsystems). At least 10 sections per animal were analyzed by manual counting of Fos-positive profiles by an experimenter blinded to genotype and treatment side.
Spinal microglia.
Seven days following SNI, mice were terminally anesthetized with 2.5% avertin and underwent cardiac perfusion with 4% PFA. Spinal cords were dissected, postfixed for 4 h in 4% PFA, cryopreserved in 30% sucrose overnight, and L4–6 segments were sectioned at 30 μm on a cryostat. Primary (anti-Iba1; 1:1000; Wako) and secondary (donkey anti-rabbit; 1:1000; Invitrogen) antibodies were applied according to standard methods. Sections were imaged with a 5500B microscope (Leica Microsystems) and analyzed for pixel density in the ipsilateral and contralateral dorsal horn using custom mirror-image ROIs per section in ImageJ. Ten sections were analyzed per animal with 4 animals per genotype.
Experimental design and statistical analyses.
Voltage-clamp and current-clamp data were analyzed using OriginPro8.5 (OriginLab). Behavioral data were analyzed using GraphPad Prism. For electrophysiological data, unless otherwise noted, statistical significance was determined using an independent two-tailed t test. For behavioral data, detailed statistical analysis is presented in Table 1. Error bars in the figures are presented as ± SEM. A p value of <0.05 was considered significant.
Table 1.
Statistical analysis of behavioral experiments
| Figure panel | Assay | Statistical test; findings | Post hoc analysis (adjusted p values) | No. of subjects |
|---|---|---|---|---|
| 5A, B | Neurological examination and tail suspension | χ2 test | NA | Control: n = 12 |
| Observed versus expected (10% failure rate) | cKO: n = 9 | |||
| Control: | ||||
| All tests: p > 0.9999 | ||||
| cKO: | ||||
| All tests: p > 0.9999 | ||||
| 5C | Negative geotaxis | Mann–Whitney U test | NA | Control: n = 12 |
| Control versus cKO | cKO: n = 9 | |||
| p = 0.3100 | ||||
| 5D | Rotarod | Mann–Whitney U test | NA | Control: n = 12 |
| Control versus cKO | cKO: n = 9 | |||
| p = 0.5071 | ||||
| 5E | Wire hang | Mann–Whitney U test | NA | Control: n = 12 |
| Control versus cKO | cKO: n = 9 | |||
| p = 0.1166 | ||||
| 5F | Novel odor recognition | Two-way repeated-measures ANOVA | Wilcoxson matched-pairs signed rank test | Control: n = 18 |
| Odor 1: | Control: | cKO: n = 16 | ||
| Genotype: p = 0.1205 | Trial 3 versus Trial 4: p = 0.0210 | |||
| Trial number: p < 0.0001 | Trial 6 versus Trial 7: | |||
| Interaction: p = 0.0450 | p = 0.0176 | |||
| Odor 2: | cKO: | |||
| Genotype: p = 0.2030 | Trial 3 versus Trial 4: p = 0.0459 | |||
| Trial number: p < 0.0001 | Trial 6 versus Trial 7: | |||
| Interaction: p = 0.9217 | p = 0.0459 | |||
| 6A, 7D | Hot plate | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 24 |
| Genotype: p < 0.0001 | Pretamoxifen versus 7 weeks post-tamoxifen | cKO: n = 18 | ||
| Time: p = 0.2376 | Control: p = 0.0014 | |||
| Interaction: p < 0.0001 | cKO: p < 0.0001 | |||
| 6B, 7E | Tail immersion | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 22 |
| Genotype: p < 0.0001 | Pretamoxifen versus 7 weeks post-tamoxifen | cKO: n = 16 | ||
| Time: p < 0.0001 | Control: p = 0.8191 | |||
| Interaction: p < 0.0001 | cKO: p < 0.0001 | |||
| 6C, 7F | Hargreaves test | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 24 |
| Genotype: p < 0.0001 | Pretamoxifen versus 7 weeks post-tamoxifen | cKO: n = 18 | ||
| Time: p < 0.0001 | Control: p = 0.2694 | |||
| Interaction: p = 0.0004 | cKO: p < 0.0001 | |||
| 6D | Randall-Selitto test | Mann–Whitney U test | NA | Control: n = 12 |
| Control versus cKO | cKO: n = 9 | |||
| p < 0.0001 | ||||
| 6E | Intraplantar capsaicin | Mann–Whitney U test | NA | Control: n = 8 |
| Control versus cKO | cKO: n = 11 | |||
| p < 0.0001 | ||||
| 6F | Acetic acid (i.p.) | Mann–Whitney U test | NA | Control: n = 12 |
| Control versus cKO | cKO: n = 8 | |||
| p = 0.0032 | ||||
| 6G, H | Formalin test, nocifensive behavior | Two-way ANOVA | Sidak's multiple-comparisons test | Control: n = 12 |
| Genotype: p = 0.0061 | Control versus cKO | cKO: n = 9 | ||
| Phase: p < 0.0001 | Phase I: p = 0.0069 | |||
| Interaction: p = 0.1405 | Phase II: p = 0.5503 | |||
| 6I | Formalin test, latency to onset | Mann–Whitney U test | NA | Control: n = 12 |
| Control versus cKO | cKO: n = 9 | |||
| p < 0.0001 | ||||
| 6J | Chloroquine scratching | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | |
| Genotype: p = 0.4825 | Pretamoxifen versus 8 weeks post-tamoxifen | |||
| Treatment: p = 0.0005 | Control: p = 0.5450 | |||
| Interaction: p = 0.0119 | cKO: p = 0.0003 | |||
| 6K | Histamine scratching | Mann–Whitney U test | NA | Control: n = 12 |
| Control versus cKO | cKO: n = 11 | |||
| p = 0.0381 | ||||
| 7A | von Frey threshold | Two-way repeated-measures ANOVA | NA | Control: n = 23 |
| Genotype: p = 0.0554 | cKO: n = 18 | |||
| Time: p = 0.6945 | ||||
| Interaction: p = 0.3343 | ||||
| 7B | Shock intensity-response relationship | Two-way ANOVA | Sidak's multiple-comparisons test | Control: n = 11 |
| Genotype: p = 0.1133 | Control versus cKO | cKO: n = 9 | ||
| Intensity: p < 0.0001 | 0.1 mA: p = 0.4528 | |||
| Interaction: p = 0.8191 | 0.3 mA: p = 0.9998 | |||
| 0.5 mA: p = 0.8528 | ||||
| 0.7 mA: p = 0.9356 | ||||
| 7C | Active avoidance learning | Two-way repeated-measures ANOVA | NA | Control: n = 12 |
| Genotype: p = 0.9687 | cKO: n = 8 | |||
| Block: p < 0.0001 | ||||
| Interaction: p = 0.2074 | ||||
| 8A | Fos induction, heat stimulus, laminae I-II | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 4 |
| Genotype: p = 0.0034 | Ipsilateral versus contralateral | cKO: n = 4 | ||
| Side: p = 0.0037 | Control: p = 0.0017 | |||
| Interaction: p = 0.0064 | cKO: p = 0.9275 | |||
| 8A | Fos induction, heat stimulus, lamina V | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 4 |
| Genotype: p = 0.1145 | Ipsilateral versus contralateral | cKO: n = 4 | ||
| Side: p = 0.0026 | Control: p = 0.0030 | |||
| Interaction: p = 0.0283 | cKO: p = 0.3537 | |||
| 8B | Fos induction, mechanical stimulus, laminae I-II | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 4 |
| Genotype: p = 0.0005 | Ipsilateral versus contralateral | cKO: n = 4 | ||
| Side: p = 0.0002 | Control: p < 0.0001 | |||
| Interaction: p = 0.0003 | cKO: p = 0.9443 | |||
| 8B | Fos induction, mechanical stimulus, lamina V | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 4 |
| Genotype: p = 0.9883 | Ipsilateral versus contralateral | cKO: n = 4 | ||
| Side: p = 0.2833 | Control: p = 0.0059 | |||
| Interaction: p = 0.0013 | cKO: p = 0.0393 | |||
| 8C | Fos induction, 2% formalin (i.pl.), laminae I-II | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 4 |
| Genotype: p = 0.0105 | Ipsilateral versus contralateral | cKO: n = 4 | ||
| Side: p = 0.0058 | Control: p = 0.0109 | |||
| Interaction: p = 0.1199 | cKO: p = 0.2700 | |||
| 8C | Fos induction, 2% formalin (i.pl.), lamina V | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 4 |
| Genotype: p = 0.4297 | Ipsilateral versus contralateral | cKO: n = 4 | ||
| Side: p = 0.0459 | Control: p = 0.2477 | |||
| Interaction: p = 0.9615 | cKO: p = 0.2260 | |||
| 8D | Fos induction, electrical stimulus, laminae I-II | Unpaired t test | NA | Control stimulated: n = 4 |
| Stimulated versus unstimulated subjects | Control unstimulated: n = 3 | |||
| Control: p = 0.0020 | cKO stimulated: n = 5 | |||
| cKO: p = 0.0038 | cKO unstimulated: n = 4 | |||
| 8D | Fos induction, electrical stimulus, lamina V | Unpaired t test | NA | Control stimulated: n = 4 |
| Stimulated versus unstimulated subjects | Control unstimulated: n = 3 | |||
| Control: p = 0.6420 | cKO stimulated: n = 5 | |||
| cKO: p = 0.1221 | cKO unstimulated: n = 4 | |||
| 9A | Acetone test, SNI pretamoxifen | Two-way repeated-measures ANOVA | Unpaired t test with Holm–Sidak multiple-comparisons correction | Control: n = 17 |
| Genotype: p = 0.0038 | Control versus cKO | cKO: n = 17 | ||
| Time: p = 0.0028 | BL: p = 0.9099 | |||
| Interaction: p = 0.0080 | D1: p = 0.9210 | |||
| D3: p = 0.9210 | ||||
| W1: p = 0.9210 | ||||
| W2: p = 0.9210 | ||||
| W4: p = 0.0095 | ||||
| W5: p = 0.0499 | ||||
| W7: p = 0.0499 | ||||
| W8: p = 0.0849 | ||||
| W9: p = 0.1101 | ||||
| W10: p = 0.1101 | ||||
| 9B | von Frey threshold, SNI pretamoxifen | Two-way repeated-measures ANOVA | NA | Control: n = 17 |
| Genotype: p = 0.1403 | cKO: n = 17 | |||
| Time: p < 0.0001 | ||||
| Interaction: p = 0.8480 | ||||
| 9C | Acetone test, SNI post-tamoxifen | Two-way repeated-measures ANOVA | Unpaired t test with Holm–Sidak multiple-comparisons correction | Control: n = 22 |
| Genotype: p = 0.0005 | Control versus cKO | cKO: n = 17 | ||
| Time: p = 0.0009 | BL: p = 0.3505 | |||
| Interaction: p = 0.2335 | W0.5: p = 0.0066 | |||
| W1: p = 0.0016 | ||||
| W2: p = 0.0028 | ||||
| 9D | von Frey threshold, SNI post-tamoxifen | Two-way repeated-measures ANOVA | NA | Control: n = 22 |
| Genotype: p = 0.5078 | cKO: n = 17 | |||
| Time: p < 0.0001 | ||||
| Interaction: p = 0.9924 | ||||
| 9F | Microgliosis, pixel density | Two-way ANOVA | NA | Control: n = 4 |
| Genotype: p = 0.0761 | cKO: n = 4 | |||
| Side: p = 0.0157 | ||||
| Interaction: p = 0.5383 | ||||
| 9G | Hargreaves test, CFA model | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 12 |
| Genotype: p < 0.0001 | Control: | cKO: n = 10 | ||
| Time: p < 0.0001 | BL versus D1: p < 0.0001 | |||
| Interaction: p < 0.0001 | BL versus D2: p < 0.0001 | |||
| BL versus D3: p < 0.0001 | ||||
| BL versus D4: p < 0.0001 | ||||
| BL versus D7: p = 0.0002 | ||||
| cKO: | ||||
| BL versus D1: p > 0.9999 | ||||
| BL versus D2: p = 0.8362 | ||||
| BL versus D3: p = 0.9510 | ||||
| BL versus D4: p = 0.6788 | ||||
| BL versus D7: p = 0.9837 | ||||
| 9H | von Frey threshold, CFA model | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 12 |
| Genotype: p = 0.0002 | Control: | cKO: n = 10 | ||
| Time: p < 0.0001 | BL versus D1: p < 0.0001 | |||
| Interaction: p = 0.1999 | BL versus D2: p < 0.0001 | |||
| BL versus D3: p < 0.0001 | ||||
| BL versus D4: p < 0.0001 | ||||
| BL versus D7: p < 0.0001 | ||||
| cKO: | ||||
| BL versus D1: p < 0.0001 | ||||
| BL versus D2: p < 0.0001 | ||||
| BL versus D3: p < 0.0001 | ||||
| BL versus D4: p < 0.0001 | ||||
| BL versus D7: p < 0.0001 | ||||
| 9I | Paw edema, CFA model | Two-way repeated-measures ANOVA | NA | Control: n = 12 |
| Genotype: p = 0.0530 | cKO: n = 10 | |||
| Time: p < 0.0001 | ||||
| Interaction: p = 0.1033 | ||||
| 9J | Fos induction, non-noxious mechanical stimulus (rotarod walking) in CFA model, laminae I-II | Two-way repeated-measures ANOVA Genotype: p = 0.5855 Side: p = 0.0039 Interaction: p = 0.2020 |
Sidak's multiple-comparisons test Ipsilateral versus contralateral Control: p = 0.0110 cKO: p = 0.1344 |
Control: n = 4 cKO: n = 4 |
| 9K | Hargreaves test, incision model | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 12 |
| Genotype: p < 0.0001 | Control: | cKO: n = 10 | ||
| Time: p = 0.0622 | BL versus 2 h: p = 0.0005 | |||
| Interaction: p = 0.0013 | BL versus D2: p = 0.0158 | |||
| BL versus D7: p > 0.9999 | ||||
| cKO: | ||||
| BL versus 2 h: p = 0.9799 | ||||
| BL versus D2: p > 0.9999 | ||||
| BL versus D7: p = 0.9973 | ||||
| 9L | von Frey threshold, incision model | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Control: n = 12 |
| Genotype: p = 0.5975 | Control: | cKO: n = 11 | ||
| Time: p < 0.0001 | BL versus 2 h: p = 0.0018 | |||
| Interaction: p = 0.9560 | BL versus D2: p = 0.0101 | |||
| BL versus D7: p > 0.9999 | ||||
| cKO: | ||||
| BL versus 2 h: p = 0.0131 | ||||
| BL versus D2: p = 0.1028 | ||||
| BL versus D7: p = 0.9743 | ||||
| 9M | Capsaicin-induced secondary mechanical allodynia | Wilcoxon matched-pairs signed rank test | NA | Control: n = 12 |
| BL versus 30 min | cKO: n = 10 | |||
| Control: p = 0.0210 | ||||
| cKO: p = 0.8457 | ||||
| 11A | Hot plate: GX-201 | Unpaired t test | NA | Vehicle: n = 12 |
| Vehicle versus GX-201 | GX-201: n = 9 | |||
| p = 0.0336 | ||||
| 11B | Hot plate: GX-585 | Unpaired t test | NA | Vehicle: n = 8 |
| Vehicle versus GX-585 | GX-585: n = 15 | |||
| p = 0.0064 | ||||
| 11C | Tail immersion: GX-201 | Unpaired t test | NA | Vehicle: n = 12 |
| Vehicle versus GX-201 | GX-201: n = 11 | |||
| p = 0.0113 | ||||
| 11D | Tail immersion: GX-585 | Unpaired t test | NA | Vehicle: n = 8 |
| Vehicle versus GX-585 | GX-585: n = 15 | |||
| p = 0.0114 | ||||
| 11E | Randall–Selitto test: GX-585 | Unpaired t test | NA | Vehicle: n = 8 |
| Vehicle versus GX-585 | GX-585: n = 15 | |||
| p = 0.0036 | ||||
| 11F | Randall–Selitto test: GX-201 | Unpaired t test | NA | Vehicle: n = 12 |
| Vehicle versus GX-201 | GX-201: n = 11 | |||
| p = 0.8214 | ||||
| 11G | Hargreaves test, CFA model: GX-201 | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Vehicle: n = 12 |
| Treatment: p = 0.0469 | Vehicle: | GX-201: n = 12 | ||
| Time: p < 0.0001 | Pre-CFA versus post-CFA, predose: p = 0.0002 | |||
| Interaction: p = 0.0012 | Pre-CFA versus post-CFA, postdose: p = 0.0002 | |||
| Post-CFA, predose versus post-CFA, postdose: | ||||
| p > 0.9999 | ||||
| GX-201: | ||||
| Pre-CFA versus post-CFA, predose: p < 0.0001 | ||||
| Pre-CFA versus post-CFA, postdose: p > 0.9999 | ||||
| Post-CFA, predose versus post-CFA, postdose: | ||||
| p < 0.0001 | ||||
| 11H | Hargreaves test, CFA model: GX-585 | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Vehicle: n = 6 |
| Treatment: p = 0.1107 | Vehicle: | GX-585: n = 6 | ||
| Time: p = 0.0203 | Pre-CFA versus post-CFA, predose: p = 0.1276 | |||
| Interaction: p = 0.8786 | Pre-CFA versus post-CFA, postdose: p = 0.6355 | |||
| Post-CFA, predose versus post-CFA, postdose: | ||||
| p = 0.5098 | ||||
| GX-585: | ||||
| Pre-CFA versus post-CFA, predose: p = 0.0918 | ||||
| Pre-CFA versus post-CFA, postdose: p = 0.9157 | ||||
| Post-CFA, predose versus post-CFA, postdose: | ||||
| p = 0.1885 | ||||
| 11I | Capsaicin-induced secondary mechanical allodynia:GX-585 | Two-way repeated-measures ANOVA | Sidak's multiple-comparisons test | Vehicle: n = 12 |
| Treatment: p = 0.0395 | Preinjection: | 10 mg/kg: n = 12 | ||
| Time: p < 0.0001 | Vehicle versus 10 mg/kg: p = 0.9993 | 30 mg/kg: n = 11 | ||
| Interaction: p = 0.8148 | Vehicle versus 30 mg/kg: p = 0.9785 | 60 mg/kg: n = 11 | ||
| Vehicle versus 60 mg/kg: p = 0.1026 | ||||
| Postinjection: | ||||
| Vehicle versus 10 mg/kg: p > 0.9999 | ||||
| Vehicle versus 30 mg/kg: p = 0.9284 | ||||
| Vehicle versus 60 mg/kg: p = 0.3197 | ||||
| 11J | Novel odor recognition | Two-way repeated-measures ANOVA | Wilcoxson matched-pairs signed rank test | Vehicle: n = 10 |
| Odor 1: | Vehicle: | GX-585: n = 10 | ||
| Treatment: p = 0.0173 | Trial 3 versus Trial 4: p = 0.1797 | |||
| Trial number: p = 0.0476 | Trial 6 versus Trial 7: p = 0.0625 | |||
| Interaction: p = 0.5307 | GX-585: | |||
| Odor 2: | Trial 3 versus Trial 4: p = 0.5000 | |||
| Treatment: p = 0.0012 | Trial 6 versus Trial 7: p = 0.8125 | |||
| Trial number: p = 0.3027 | ||||
| Interaction: p = 0.3586 |
Results
Characterization of tamoxifen-inducible Nav1.7 KO mice
We generated tamoxifen-inducible Nav1.7 KO mice (Nav1.7 cKO mice) by crossing mice bearing loxP sites flanking exons 15–16 of the SCN9A gene (Nassar et al., 2004) to a mouse containing a tamoxifen-inducible Cre-ERT transgene driven by the CMV early enhancer/chicken β actin promoter that expresses high levels of Cre-ERT in the brain and most peripheral tissues (Hayashi and McMahon, 2002). These mice had normal levels of Nav1.7 expression and normal nociceptive withdrawal responses before injection with tamoxifen. Following tamoxifen administration, levels of intact SCN9A mRNA isolated from DRGs of Nav1.7 cKO mice rapidly decreased >1000-fold at the earliest time point examined (1 d following the last of 3 consecutive days of tamoxifen dosing) (Fig. 1A). Levels of intact SCN9A mRNA isolated from DRGs of control littermates (SCN9AloxP/loxP but no Cre transgene) did not significantly change at any point within 6–8 weeks following tamoxifen dosing. We next examined a set of 20 tissues isolated from tamoxifen-treated control mice and found expression of SCN9A mRNA in sensory neurons (trigeminal ganglia [TGs], and DRGs), sympathetic ganglia (superior cervical ganglia [SCGs]), and the olfactory epithelium (OE) (Fig. 1B), as has been previously observed in WT mice (Ahn et al., 2011; Rupasinghe et al., 2012). On the other hand, in tamoxifen-treated Nav1.7 cKO mice, SCN9A mRNA was decreased to near undetectable levels in all tissues that normally express Nav1.7, with the exception of the OE, where no significant change in SCN9A mRNA was observed. We next used Western blotting to examine Nav1.7 protein levels present in DRG lysates following tamoxifen injection and found that the protein decays exponentially (in Nav1.7 cKO but not control mice) with a 0.98 ± 0.19 week exponential time-constant and reaches undetectable levels after 6 weeks (Fig. 1C,D). Expression levels of other Nav channels were not altered in the DRG (Fig. 1E), the TG (Fig. 1F), or the SCG (Fig. 1G), indicating lack of compensation due to upregulation of other voltage-gated sodium channels at the mRNA level. Using ISH, we examined the cellular profile of SCN9A mRNA expression in control DRGs and found it to be present in ∼90% of sensory neurons (838 of 933 DRG neurons) across the full range of soma diameters but mostly absent from sensory neurons from Nav1.7 cKO mice (46 positive for ISH signal of 770 DRG neurons counted; 6%) (Fig. 1H). In some (mostly medium- to large-diameter) neurons from Nav1.7 cKO (but not control) animals, the ISH probe appeared aggregated in the nucleus (113 of 770 neurons; 15%). It is not clear what this represents because the probe binds within exon 15, which has been deleted in Nav1.7 cKO mice.
Figure 1.
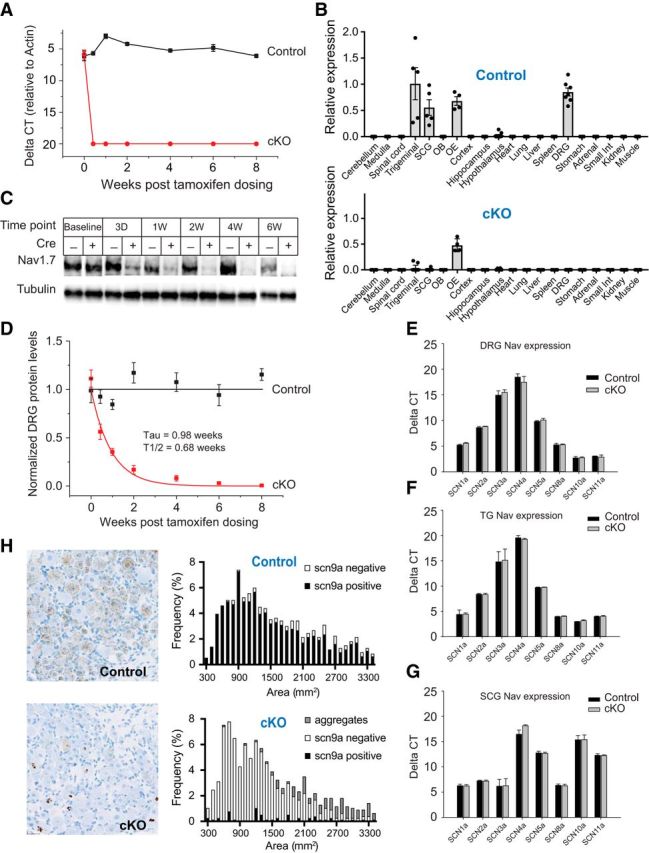
Expression distribution of scn9a mRNA and characterization of Nav1.7 deletion in cKO mice. A, Time course of loss of scn9a mRNA from DRG of Nav1.7 cKO mice (red circles). Signal is undetectable at all post-tamoxifen time points. Black squares represent control littermates. n = 6 mice/genotype. B, Tissue distribution of scn9a mRNA in Nav1.7 cKO mice and control mice. Relative expression of Nav1.7 cKO and control was normalized to the level observed in WT trigeminal ganglia. Tissues are as indicated. OB, Olfactory bulb; OE, olfactory epithelium. C, Example Western blot of Nav1.7 protein from DRG lysates 3 d and 1, 2, 4, or 6 weeks following tamoxifen injection. Tubulin immunostaining is shown as a loading control. D, Time course of loss of Nav1.7 protein from DRG of Nav1.7 cKO mice (red) as measured by Western blot. Decay constant (τ) = 0.98 ± 0.19 weeks. No detectable Nav1.7 protein loss was observed in DRGs from control mice (black). E–G, Expression of other Nav channel mRNAs in DRGs (E), TGs (F), and SCGs (G) following tamoxifen injection (after 8 weeks) from Nav1.7 cKO mice (gray) or control mice (black). No significant differences in Nav channel mRNA levels were observed (except for scn9a). H, Representative images of scn9a ISH in DRG from a Nav1.7 cKO mouse and a control mouse. scn9a mRNA was detected in 838 of 933 (90%) DRG neurons pooled from 3 control mice, representing all soma cross-sectional areas. Black bars represent scn9a-positive. White bars represent scn9a-negative. scn9a mRNA was detected in 46 of 770 (6%) DRG neurons pooled from 3 Nav1.7 cKO mice. Black bars represent scn9a-positive. White bars represent scn9a-negative. Gray bars represent nuclear aggregates. Error bars indicate SEM.
Electrophysiological properties of DRG neurons cultured from Nav1.7 cKO mice
To examine the effects of adult removal of Nav1.7 on DRG neuron electrophysiological properties, we patched small-, medium-, and large-diameter DRG neurons from either Nav1.7 cKO or control mice 6–8 weeks following tamoxifen injection. Small-, medium-, and large-diameter were defined as neurons with capacitance <30 pF, 30–60 pF, and >60 pF, respectively. We used 500 nm TTX to isolate TTX-sensitive and TTX-resistant components of the inward Nav currents recorded under whole-cell voltage-clamp (Fig. 2A). TTX-sensitive currents were significantly reduced in the absence of Nav1.7 for both small- and medium-diameter (but not large-diameter) DRG neurons (small: from 58 ± 12 pA/pF, n = 19 to 9.6 ± 3.8 pA/pF, n = 20, p = 0.0004; medium: from 88 ± 6.8 pA/pF, n = 17 to 46 ± 7.3 pA/pF, n = 16, p = 0.00024; large: from 108 ± 15 pA/pF, n = 7 to 97 ± 9 pA/pF, n = 12, p = 0.51) (Fig. 2B). The TTX-sensitive current density measured at an individual cell level was plotted versus membrane capacitance (as a measure of cell size), showing that many small-diameter DRG neurons isolated from Nav1.7 cKO (but not control) mice lack detectable TTX-sensitive currents (Fig. 2C). TTX-resistant currents were not significantly different between Nav1.7 cKO and control DRG neurons (small: from 71 ± 12 pA/pF, n = 19 to 96 ± 12 pA/pF, n = 20, p = 0.15; medium: from 38 ± 8.1 pA/pF, n = 19 to 25 ± 6.6 pA/pF, n = 17, p = 0.23; large: from 21 ± 15 pA/pF, n = 7 to 14 ± 8.4 pA/pF, n = 12, p = 0.68) (Fig. 2D,E).
Figure 2.

Voltage-gated sodium currents from DRG neurons derived from Nav1.7 cKO mice or control littermates. A, TTX-resistant and TTX-sensitive voltage-gated sodium currents were measured from small-, medium-, or large-diameter DRG neurons acutely isolated from either control (cre−) mice or Nav1.7 cKO (cre+) mice. Mice were dissected 7–9 weeks following tamoxifen dosing to ensure complete decay of Nav1.7 protein. Whole-cell voltage clamp was done using a holding voltage of −80 mV, then hyperpolarizing to −120 mV for 20 ms and depolarizing to 0 mV for 20 ms with a stimulus frequency of 1 Hz; 500 nm TTX was used to isolate the TTX-resistant component of the sodium current. B, TTX-sensitive current density measured from small-, medium-, and large-diameter DRG neurons obtained from control (cre−) or Nav1.7 cKO (cre+) mice. TTX-sensitive currents were found to be significantly decreased in small- and medium-diameter Nav1.7 cKO DRG neurons (but not large-diameter) relative to control DRG neurons. C, TTX-sensitive current density versus membrane capacitance as measured on a single-cell basis. D, TTX-resistant current density measured from small-, medium-, and large-diameter DRG neurons obtained from control (cre−) or Nav1.7 cKO (cre+) mice. No significant difference in TTX-resistant currents between DRG neurons from control versus Nav1.7 cKO mice was observed. Most large-diameter neurons did not have detectable TTX-resistant currents. E, TTX-resistant current density versus membrane capacitance as measured on a single-cell basis. ***p < 0.001. Error bars indicate SEM.
Current-clamp recordings were next conducted to determine the effects of removal of Nav1.7 on small-diameter DRG neuronal excitability. The resting membrane potential of small-diameter WT DRG neurons (−49.7 ± 1.3 mV, n = 13) was comparable with that of DRG neurons lacking Nav1.7 channels (−49.3 ± 2.3 mV, n = 14, p = 0.9) and was not altered with the application of 500 nm TTX (Fig. 3A,B). In our initial set of experiments, we allowed the neurons to maintain their natural resting membrane potentials. Short 10 ms injections of current were used to elicit single APs, and longer 500 ms current injections were used to examine spiking frequency. We found no significant difference between control neurons and Nav1.7 cKO neurons in several parameters examined, including AP voltage threshold (control: −21.5 ± 2.3 mV, n = 13; Nav1.7 cKO: −19.9 ± 1.9 mV, n = 14, p = 0.6) (Fig. 3C), AP current threshold (control: 85 ± 14.1 pA, n = 13; Nav1.7 cKO: 75.7 ± 10.7 pA, n = 15) (Fig. 3D), or the number of APs observed during a 500 ms current injection pulse delivered at the individual AP current threshold (control: 4.2 ± 0.8, n = 9; Nav1.7 cKO: 3.7 ± 0.5, n = 11, p = 0.6) (Fig. 3E). Furthermore, 500 nm TTX did not significantly alter neuronal excitability of small-diameter neurons, suggesting that neuronal excitability is determined entirely by TTX-resistant Nav channels under these conditions (Fig. 3A–E). Large-diameter DRG neurons, on the other hand, failed to fire APs in the presence of 500 nm TTX (Fig. 3A, right), demonstrating dependence on TTX-sensitive channels.
Figure 3.

Electrophysiological properties of DRG neurons derived from WT and Nav1.7 cKO mice without adjusting resting membrane potential. A, Current-clamp recordings of small-diameter DRG neurons acutely isolated from either control (cre−) mice or Nav1.7 cKO (cre+) mice. Mice were dissected 7–9 weeks following tamoxifen dosing to ensure complete decay of Nav1.7 protein. Cells were stimulated with short 10 ms depolarizing current pulses of increasing amplitude until the cell fired an AP (left) or with long 500 ms depolarizing current pulses to examine AP firing frequency (right) with and without 500 nm TTX. Large-diameter DRG neurons were also examined (far right). B, Resting membrane potential did not differ between genotypes and did not significantly change after adding 500 nm TTX. C, AP threshold voltage was measured before and after adding 500 nm TTX. AP threshold voltages did not differ between genotypes, and TTX had no significant effect. D, Current threshold for firing an AP was measured before and after adding 500 nm TTX. Current thresholds did not differ between genotypes, and TTX had no significant effect. E, Number of APs per 500 ms was measured before and after adding 500 nm TTX. No genotype difference was observed between control and Nav1.7 cKO neurons, and TTX had no significant effect. Error bars indicate SEM.
We next reexamined neural excitability of small-diameter DRG neurons while injecting current to maintain the resting membrane voltage at −70 mV where a much larger fraction of TTX-sensitive channels is available for activation and is likely closer to physiological resting membrane voltages, especially at the peripheral fibers where AP generation normally occurs. Additional current (in increasing steps) was then injected for 10 ms to determine how much current and voltage was required for AP generation (Fig. 4A). We found that the voltage threshold for firing APs was significantly increased (control: −31.0 ± 0.8 mV, n = 12; Nav1.7 cKO: −21.4 ± 1.5 mV, n = 14, p = 0.00001) (Fig. 4B) and the current necessary to generate an AP was significantly increased (control: 132.5 ± 5.4 pA, n = 12; Nav1.7 cKO: 189.3 ± 12.5 pA, n = 14, p = 0.0006) (Fig. 4C) in the absence of Nav1.7. Furthermore, the sensitivity to TTX of AP voltage and current thresholds was eliminated, suggesting that AP generation in small-diameter DRG neurons becomes dependent on TTX-resistant currents alone following Nav1.7 removal (Fig. 4A–C). The resting membrane potential, after being initially maintained at −70 mV, was unchanged following TTX application (Fig. 4D). Other parameters calculated from the AP waveform that were found to be sensitive to TTX, such as AP overshoot amplitude, 10%–90% peak rise rate, and AP half-width, lost their TTX sensitivity in the absence of Nav1.7 channels (Fig. 4E–G).
Figure 4.
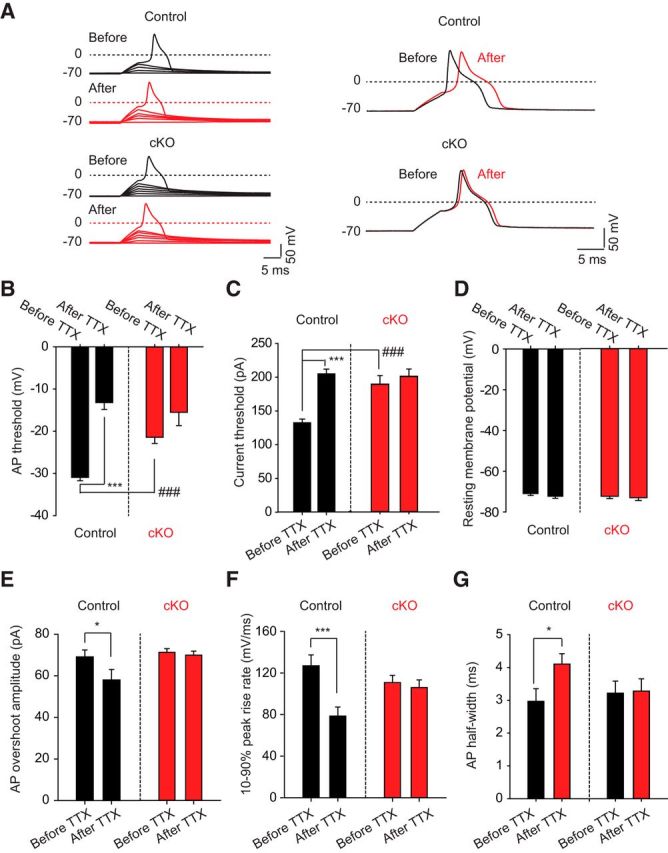
Electrophysiological properties of DRG neurons derived from WT and Nav1.7 cKO mice. A, Current-clamp recordings of small-diameter DRG neurons acutely isolated from either control (cre.neg) mice or Nav1.7 cKO (cre.pos) mice. Mice were dissected 7–9 weeks following tamoxifen dosing to ensure complete decay of Nav1.7 protein. Current was injected to maintain a resting membrane potential of −70 mV in these recordings. Cells were stimulated with depolarizing current pulses of increasing amplitude until the cell fired an AP. The overall shape of the AP did not differ between genotypes. B, AP threshold voltage was measured before and after adding 500 nm TTX. Neurons from Nav1.7 cKO mice have significantly more depolarized AP threshold voltages relative to neurons from control mice. Furthermore, TTX significantly shifted the AP threshold voltage to more depolarized voltages in control neurons but not Nav1.7 cKO neurons. C, Current threshold for firing an AP was measured before and after adding 500 nm TTX. Neurons from Nav1.7 cKO mice required significantly larger current pulses to fire an AP relative to neurons from control mice. Furthermore, TTX significantly increased the amount of current needed to fire an AP in control neurons but not Nav1.7 cKO neurons. D, Resting membrane potential, set to −70 mV before the addition of TTX, did not significantly change after adding 500 nm TTX. E, AP overshoot amplitude was measured before and after adding 500 nm TTX. No difference was observed between control and Nav1.7 cKO neurons; however, addition of TTX significantly decreased AP overshoot amplitude in control neurons but not Nav1.7 cKO neurons. F, The 10%–90% peak rise rate was measured before and after adding 500 nm TTX. No difference was observed between control and Nav1.7 cKO neurons; however, addition of TTX significantly decreased upstroke slope in control neurons but not Nav1.7 cKO neurons. G, AP half-width was measured before and after adding 500 nm TTX. No difference was observed between control and Nav1.7 cKO neurons; however, addition of TTX significantly increased AP half-width in control neurons but not Nav1.7 cKO neurons. Significance (paired two-tailed t test): *p < 0.05; ***p < 0.001; ###p < 0.001. Error bars indicate SEM.
Adult-onset deletion of Nav1.7 results in profound insensitivity to most modalities of noxious stimulus
Deletion of Nav1.7 in adult mice is expected to provide preclinical proof of concept that would allow us to predict what would be the action of a theoretical perfectly selective and maximally potent Nav1.7 inhibitor administered to adult human pain patients. We therefore tested our Nav1.7 inducible KO mice in a battery of tests of general neurological function and acute nociception. Except where noted, mice were tested 6–8 weeks after tamoxifen injection.
Nav1.7 cKO mice displayed normal reflexes in a rodent neurological examination (Fig. 5A,B); normal behavior in the negative geotaxis (Fig. 5C), rotarod (Fig. 5D), and wire hang tests (Fig. 5E); and normal olfaction in a novel odor recognition test (Fig. 5F). In sharp contrast, Nav1.7 cKO mice had greatly prolonged latency to respond to noxious heat in the hot plate, tail immersion, and Hargreaves tests, often not responding at all up to the cutoff time at which the test was stopped to prevent tissue damage (Fig. 6A–C). Additionally, cKO mice had greatly increased escape threshold in response to noxious pressure applied to the tail (Randall–Selitto test; Fig. 6D). Nav1.7 cKO mice did not display nocifensive flicking or licking behavior upon hindpaw injection of capsaicin, a chemical algogen, and displayed few abdominal constrictions after intraperitoneal injection of acetic acid in a model of visceral pain (Fig. 6E,F). In the formalin test, flicking and licking behavior was observed in both control and cKO mice. During the first phase (0–10 min after injection), Nav1.7 cKO mice had a longer latency to onset and spent approximately half as much time as controls engaged in this behavior. Although the second phase (10–60 min after injection) appeared delayed in cKO mice, they nonetheless displayed similar levels of formalin-induced behavior compared with controls (Fig. 6G–I). Scratching behavior in response to pruritogens was also diminished in Nav1.7 cKO mice. When chloroquine was injected intradermally to the cheek, it induced robust scratching behavior in mice of both genotypes before the administration of tamoxifen, and this response was strongly diminished at 6 weeks after tamoxifen only in cKO mice (Fig. 6J). Similarly, scratching behavior following intradermal cheek injection of histamine was mostly absent in Nav1.7 cKO mice (Fig. 6K). On the other hand, threshold to withdraw the paw from von Frey filaments was not significantly altered in cKO animals (Fig. 7A). We were also surprised to find that electrical stimuli delivered through a floor grid evoked intensity-dependent escape responses that were identical between genotypes, and that Nav1.7 cKO mice were equally proficient to learn an active avoidance paradigm, indicating that they can use noxious electrical stimuli as an aversive teaching signal in a manner indistinguishable from control littermates (Fig. 7B,C).
Figure 5.

General neurological function and olfaction are intact in Nav1.7 cKO mice. A, Nav1.7 cKO mice perform similarly to control littermates in several tests of general neurological function. B, Mice of both genotypes displayed a normal motor and postural phenotype in the tail suspension test. C, Negative geotaxis, a measure of vestibulomotor reflexes, was normal in Nav1.7 cKO mice. D, Latency to fall from a rotating beam was unaltered by deletion of Nav1.7. E, In the wire hang test of motor coordination, Nav1.7 cKO and control littermates performed similarly. F, In the novel odor recognition test, mice are presented with three olfactory cues. Habituation to repeated presentation of the same odor, as well as increased approaches to a novel odor, are prominent features of this test that indicate correct olfactory function. Performance on this test was identical between genotypes, reflecting intact olfaction in Nav1.7 cKO mice. Error bars indicate SEM.
Figure 6.
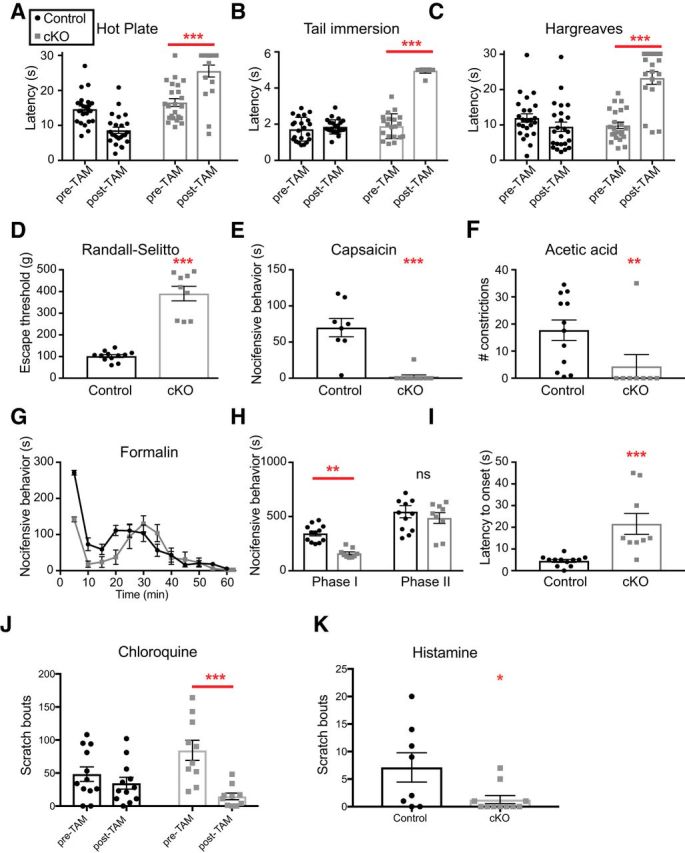
Nav1.7 cKO mice are insensitive to most modalities of acute noxious stimulus. A, Hot plate test, 55°C. Control and Nav1.7 cKO mice have similar withdrawal latency before tamoxifen administration; after tamoxifen-mediated deletion, cKO mice show prolonged latency to respond. Cutoff = 30 s. B, Tail immersion test, 55°C. Cutoff = 5 s. C, Hindpaw radiant heat (Hargreaves) test. Cutoff = 30 s. D, Randall–Selitto test (pressure applied to the tail). Cutoff = 750 g. E, Intraplantar capsaicin (1 μg in 10 μl) induced nocifensive behavior measured in the first 5 min after injection in control, but not Nav1.7 cKO, mice. F, Intraperitoneal injection of dilute acetic acid (0.9% in saline, 10 ml/kg) induced abdominal constrictions in control, but not Nav1.7 cKO, mice. G–I, Formalin test (2% formalin i.pl.). G, Time course of flinching, licking, and biting behaviors scored in 5 min bins for 1 h. H, Nav1.7 cKO mice display approximately half as much formalin-induced behavior as control mice in Phase 1 (0–10 min) and an equivalent amount in Phase 2 (10–60 min). I, Latency to begin displaying nocifensive behavior after formalin injection is increased in Nav1.7 cKO mice. J, Scratch bouts in response to intradermal cheek injection of the nonhistaminergic pruritogen chloroquine were markedly suppressed after adult-onset deletion of Nav1.7 compared with control conditions. K, Scratch bouts in response to intradermal cheek injection of histamine were significantly reduced in Nav1.7 cKO mice compared with control littermates. *p < 0.05, **p < 0.01, ***p < 0.001. Statistical analysis described in Table 1. Error bars indicate SEM.
Figure 7.

Nav1.7 cKO mice are sensitive to some noxious stimuli and time course of behavioral effects. A, von Frey test of mechanical threshold. Tamoxifen treatment did not change the response of Nav1.7 cKO mice to von Frey filaments. Cutoff = 7.5 g. B, Intensity-response relationship to electrical stimuli delivered through a floor grid did not differ between genotypes. C, Nav1.7 cKO mice learn an active avoidance paradigm where electrical stimuli are an effective aversive teaching signal in a manner indistinguishable from control mice. n = 24 control, 18 cKO mice. D–F, Time courses to develop insensitivity to the hot plate test (D), the tail immersion test (E), and the Hargreaves test (F) when measured at weekly intervals following tamoxifen dosing. Statistical analysis described in Table 1. Error bars indicate SEM.
One striking observation we made is that insensitivity to noxious stimuli develops gradually over the course of several weeks after tamoxifen administration in Nav1.7 cKO mice (Fig. 7D–F). Withdrawal latencies in the hot plate, tail immersion, and Hargreaves assays began increasing steadily between 2 and 4 weeks and then plateaued near the assay cutoff by 6–8 weeks after tamoxifen injection. Beyond 8 weeks after tamoxifen, cKO mice began to develop skin lesions that were consistent with self-inflicted scratch wounds, as they appeared even on single-housed animals and could be delayed by trimming the nails of the hindpaws. We speculate that this occurrence may mirror accidental self-injury in human Nav1.7 loss-of-function CIP individuals due to loss of the protective function of pain to limit bodily damage.
We tested whether there may be a potential toxic effect of tamoxifen in control mice that would cause a decrease in latency on the hot plate test over time (Figs. 6, 7). Indeed, we believe the decrease in latency is due to a learning effect: with repeated weekly exposure to the hot plate apparatus, the control mice learn to escape (jump) from the noxious surface with shorter and shorter latency. However, to directly test whether tamoxifen exposure affects hot plate latency in WT mice, we performed the following experiment. Eight-to-10-week-old C57BL/6 mice were either injected with tamoxifen or left uninjected (n = 10/group). Four weeks later, they were tested in a single session on the hot plate test. Naive and tamoxifen-exposed mice had indistinguishable latencies to respond (naive: 6.25 ± 0.84 s, n = 10; tamoxifen-exposed: 8.28 ± 0.95 s, n = 9; p = 0.1265).
In parallel experiments, we used Fos-like immunoreactivity in the superficial dorsal horn as an alternative measure of activation of ascending nociceptive pathways. Applying noxious heat or pinch or injecting formalin to the hindpaw evoked a large increase in Fos-positive profiles in ipsilateral laminae I-II in control animals but had no significant effect in cKO mice (Fig. 8A–C). Likewise, Fos immunoreactivity in the spinal cords of mice that had received noxious electrical stimulation of the hindpaws was significantly reduced in cKO mice, although we still observed significant increases in Fos-positive profiles after electrical stimulation in mice of both genotypes relative to unstimulated mice (Fig. 8D). These data corroborate and extend our behavioral results, and indicate that insensitivity to noxious stimuli arises from a failure of transmission of neural signaling across the first synapse in the ascending nociceptive pathway.
Figure 8.

Evidence for reduced activation of spinal nociceptive pathways in Nav1.7 cKO mice. Noxious heat (A), strong mechanical pressure (B), or intraplantar formalin injection (C) applied to the hindpaw induces an increase in Fos-immunoreactive profiles in the ipsilateral laminae I-II in control, but not Nav1.7 cKO, mice. D, Compared with unstimulated mice, electrical shock delivered through a floor grid induced a significant increase in Fos-positive profiles in laminae I-II of both control and Nav1.7 cKO mice. n = 4 mice/group. Black bars represent control littermates. White bars represent Nav1.7 cKO mice. Statistical analysis described in Table 1. Error bars indicate SEM.
We were especially interested to determine how Nav1.7 cKO mice would behave in models of chronic pain conditions. In one set of experiments, mice that had not yet been administered tamoxifen underwent the SNI model of neuropathic pain (Fig. 9A,B). We confirmed that mice of both genotypes began to display hypersensitivity to cooling and mechanical stimuli using acetone and von Frey filaments, respectively. Two weeks after SNI surgery, all mice were injected with tamoxifen (arrows) and behavioral testing continued at weekly intervals. Indeed, neuropathic allodynia to cooling stimuli was reversed in Nav1.7 cKO mice, but not control littermates, within several weeks following tamoxifen administration (Fig. 9A). Surprisingly, neuropathic mechanical hypersensitivity remained (Fig. 9B). In a separate experiment, we took baseline measurements of response to acetone and von Frey filaments, administered tamoxifen, waited 6 weeks, and then performed SNI surgeries (Fig. 9C–F). Nav1.7 cKO mice were protected from developing cool allodynia in this preventative paradigm (Fig. 9C). Again, however, both genotypes became strongly hypersensitive to von Frey filaments, although Nav1.7 cKO mice had slightly elevated withdrawal thresholds in this assay (Fig. 9D).
Figure 9.
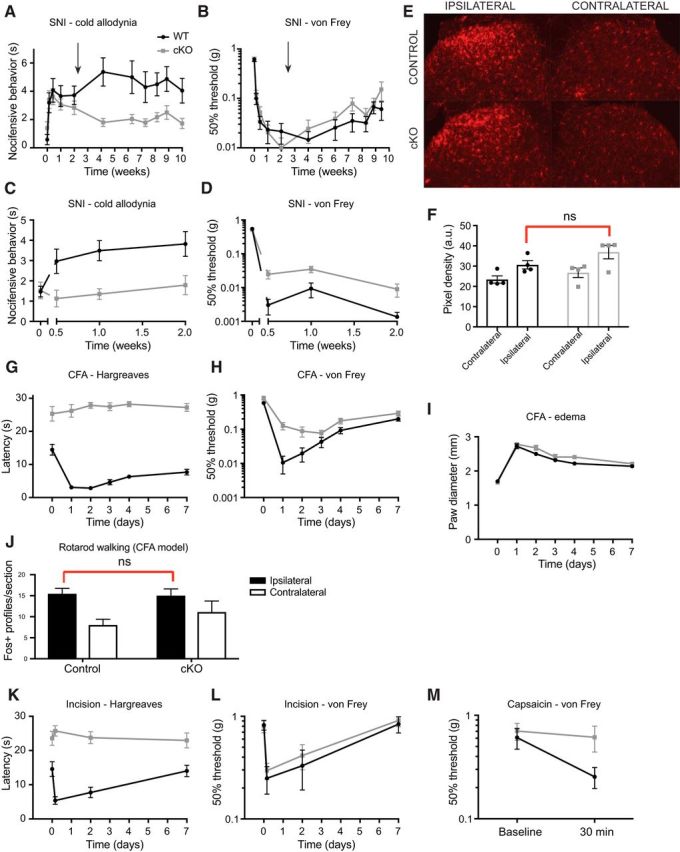
Mechanical allodynia is surprisingly preserved in several models of chronic pain. A, Mice developed cooling-evoked allodynia in the SNI model of neuropathic pain as measured by responses to acetone evaporation. After 2 weeks, tamoxifen was administered (arrow) to induce deletion of Nav1.7. Nav1.7 cKO mice (open symbols) showed a gradual reversal of cooling-evoked allodynia. B, Established neuropathic mechanical allodynia was not reversed by Nav1.7 deletion. C, When Nav1.7 was deleted before SNI (arrow), cKO mice were protected from developing cooling-evoked allodynia. D, Mechanical allodynia developed mostly normally (though to a slightly reduced extent) when SNI took place after Nav1.7 deletion. E, Representative sections of spinal cord dorsal horn from control and Nav1.7 cKO mice at 3 d after SNI. Microglia, stained for Iba-1, are activated ipsilateral to the nerve injury in both genotypes. F, Quantification of Iba-1 staining intensity in mice that have undergone SNI. Increase in pixel density ipsilateral to the injury is similar in control and Nav1.7 cKO mice. G, Heat hypersensitivity in the CFA model is absent in Nav1.7 cKO mice. H, Mechanical hypersensitivity in the CFA model develops to a mostly similar degree in both genotypes. I, Hindpaw edema after CFA injection is similar in both genotypes. J, Fos-like immunoreactivity in the spinal cord dorsal horn following CFA inflammation and exposure to a normally non-noxious mechanical stimulus (forced walking). Ipsilateral to an inflammatory insult, the number of Fos-positive profiles is increased specifically in laminae I-II upon mechanical stimulation in both control and Nav1.7 cKO mice, serving as an additional indication of intact mechanical allodynia in the absence of Nav1.7. K, In the incision model of postoperative pain, heat hypersensitivity is absent in Nav1.7 cKO mice. L, Incision-induced mechanical hypersensitivity in Nav1.7 cKO mice is comparable with control mice. M, Unlike control littermates, Nav1.7 cKO mice do not develop mechanical allodynia secondary to an extraterritorial capsaicin injection. Statistical analysis described in Table 1. Error bars indicate SEM.
We examined spinal microglial activation, an anatomical hallmark of nerve-injury induced neuropathic pain models, and found it to be similar in mice of both genotypes when they underwent SNI surgery 6 weeks after tamoxifen administration (Fig. 9E,F). Thus, lack of Nav1.7 before nerve injury does not prevent spinal microglia from becoming activated; and indeed, the preserved microglial activation may provide an explanation for the intact mechanical hypersensitivity we observed in the SNI model in Nav1.7 cKO mice.
In the CFA model of inflammatory pain, hindpaw CFA injection leads to localized edema and hypersensitivity to both heat and mechanical stimuli that lasts for ∼1 week in control mice. Mice in which Nav1.7 has been deleted have prolonged latencies to respond to radiant heat applied to the hindpaw and do not become sensitized to heat after CFA injection (Fig. 9G). In contrast, Nav1.7 cKO mice still display CFA-induced mechanical hypersensitivity as measured using von Frey filaments as well as normal paw edema formation (Fig. 9H,I). Furthermore, using Fos induction in laminae I-II of the spinal cord dorsal horn as an additional endpoint, we found evidence for a functional anatomical correlate of the retained mechanical allodynia in the CFA model. Control or Nav1.7 cKO mice were administered CFA to one hindpaw, then 24 h later were made to walk on a rotarod apparatus, a normally non-noxious mechanical stimulus that does not induce Fos-like immunoreactivity in laminae I-II in intact animals, although it is induced in deeper laminae. We found no statistically significant difference between genotypes in the number of cell profiles in which Fos was induced ipsilateral to the CFA inflammation (Fig. 9J), consistent with our behavioral observation that mechanical allodynia is mostly preserved in Nav1.7 cKO mice in the CFA model.
The incision model of postoperative pain also features both heat and mechanical hypersensitivity and may be translationally relevant. When Nav1.7 cKO mice were tested in this model, we again observed an absence of response to noxious heat and a failure to sensitize in this modality, yet they developed mechanical hypersensitivity that was indistinguishable from control littermates (Fig. 9K,L).
Finally, we examined the contribution of Nav1.7 to the development of secondary mechanical allodynia at the hindpaw plantar surface after a subcutaneous injection of capsaicin extraterritorially at the ankle. We found that this form of mechanical allodynia depends on the presence of Nav1.7, as mice in which the channel has been deleted are protected from developing mechanical allodynia in this model (Fig. 9M).
Structure and selectivity of acylsulfonamide inhibitors of Nav1.7
We next decided to determine whether selective inhibitors of Nav1.7 could alter pain behavior in WT mice in models shown to be Nav1.7-dependent based on our KO studies. We chose to examine the effects of two acylsulfonamide inhibitors, GX-201 and GX-585 (Fig. 10A). These compounds are potent inhibitors of Nav1.7, as demonstrated in whole-cell patch-clamp of HEK293 cells expressing human Nav1.7. Given the slow off-rates of these potent but slow inhibitors, we used kinetics measurements to more accurately determine potencies (Fig. 7B). In cases where the inhibitors had fast off-rates, such as on the Nav1.5 channel (Fig. 7C), equilibrium IC50 measurements were performed in addition to kinetics potency measurements. No significant differences were observed between kinetics measurements of potency and IC50 measurements when both were determined, as expected for inhibitors with a single binding site. Such potency determinations demonstrate that both GX-201 and GX-585 are selective for Nav1.7 over all other human Nav isoforms (Fig. 10D,E,H), with the more selective GX-201 being >30-fold selective for mouse Nav1.7 over all other isoforms (with the exception of Nav1.4, where it was found to be 13-fold selective). The potencies of GX-201 and GX-585 were also examined on the native mouse Nav1.7 channel in recordings from small-diameter mouse DRG neurons (TTX_S; Fig. 10H). No significant differences in potency were observed between native and overexpressed (HEK293 cell line) mouse Nav1.7. We also verified that selectivity is retained in the mouse by examining compound potencies on medium-diameter/large-diameter DRG neurons obtained from the Nav1.7 cKO mouse where only non-Nav1.7 TTX-sensitive channels are present (TTX_S(KO); Fig. 10H). GX-201 and GX-585 bind to a similar site in the domain IV voltage-sensing domain (VSD4) as arylsulfonamides, such as GX-674 (Ahuja et al., 2015) as demonstrated by mutagenesis of the S4 arginines in VSD4 (Fig. 10F). The selectivity of GX-201 is mostly dependent on three residues in the DIV S1-S2 loop region (Y1537, W1538, and V1541), which differ between Nav isoforms (Fig. 10G).
Figure 10.
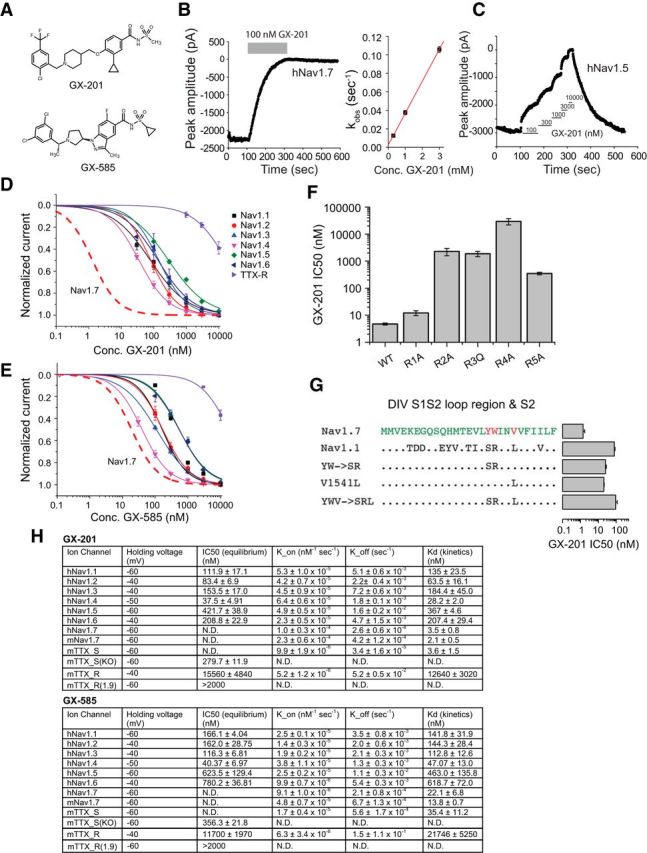
Selectivity profiles of GX-585 and GX-201. A, Chemical structures of GX-201 and GX-585. B, Example trace showing a voltage-clamp recording of human Nav1.7 where each point represents the peak current amplitude during a 20 ms pulse to 0 mV. The blocking effect of 100 nm GX-201 as well as the very slow off-rate are shown. Right, Linear relationship between K_obs and GX-201 concentration, which was used to calculate the on-rate (K_on). C, Example trace showing a measurement of the IC50 of GX-201 on human Nav1.5. D, IC50 curves showing the selectivity pattern of GX-201 against human Nav1.1–1.8 channels. Red dashed trace represents the expected IC50 curve for GX-201 on hNav1.7 based on the Kd measured using kinetics. E, Selectivity pattern for GX-585 against human Nav1.1–1.8 channels. F, IC50 values of GX-201 on WT hNav1.7 and S4 voltage-sensor arginine mutants. G, IC50 values of GX-201 on hNav1.7, hNav1.1, and hNav1.7 mutants showing the residues driving selectivity. H, Table showing holding voltages used for each channel subtype, equilibrium IC50 values, kinetics parameters, and calculated Kd values. mTTX_S and mTTX_S(KO) refer to TTX-sensitive channels recorded in WT and Nav1.7 cKO mouse DRG neurons, respectively. mTTX_R and mTTX_R(1.9) refer to mouse TTX-resistant channels recorded in WT and Nav1.8 KO DRG neurons, respectively. In the absence of Nav1.8, the TTX-resistant current is carried entirely by Nav1.9 channels. Error bars indicate SEM.
Nav1.7-selective inhibitors are antinociceptive in WT mice
We sought to determine whether pharmacological blockade of Nav1.7 using GX-201 and GX-585 has an effect on nociceptive thresholds in normal C57BL/6 mice. Because GX-585 and GX-201 are structurally quite similar acylsulfonamide Nav1.7 inhibitors, sharing a similar binding site on the Nav1.7 protein and possessing similar selectivities and pharmacokinetic properties, etc., we have used them more or less interchangeably in pharmacological experiments. In subsequently described studies, wherever we show effects for both compounds, it is meant as a confirmation of results with a second compound; wherever we show effects for just one or the other compound, it is because we only tested one.
Mice were dosed by oral gavage 1 h before behavioral testing. We monitored mice for changes in locomotor behavior that could indicate a sedative effect of the compounds. GX-585 suppressed ambulation and rearing in the open field test at high doses, similarly to gabapentin. We found that this effect was driven by a small number of individual mice that had high total plasma concentrations of GX-585 (>95 μm); thus, in all subsequent experiments, mice with plasma exposures >95 μm were excluded on the basis of potentially confounding locomotor effects.
Response latency on the hot plate and tail immersion tests was significantly elevated in mice dosed with GX-585 and GX-201 compared with vehicle-treated control mice (Fig. 11A–D). Additionally, GX-585 elevated escape threshold in the Randall–Selitto test (Fig. 11E). No significant difference was seen in von Frey threshold between mice dosed with GX-201 or vehicle (Fig. 11F). These data are consistent with our findings in adult-onset Nav1.7 cKO mice and indicate that selective pharmacological inhibition of Nav1.7 leads to acute antinociceptive effects.
Figure 11.
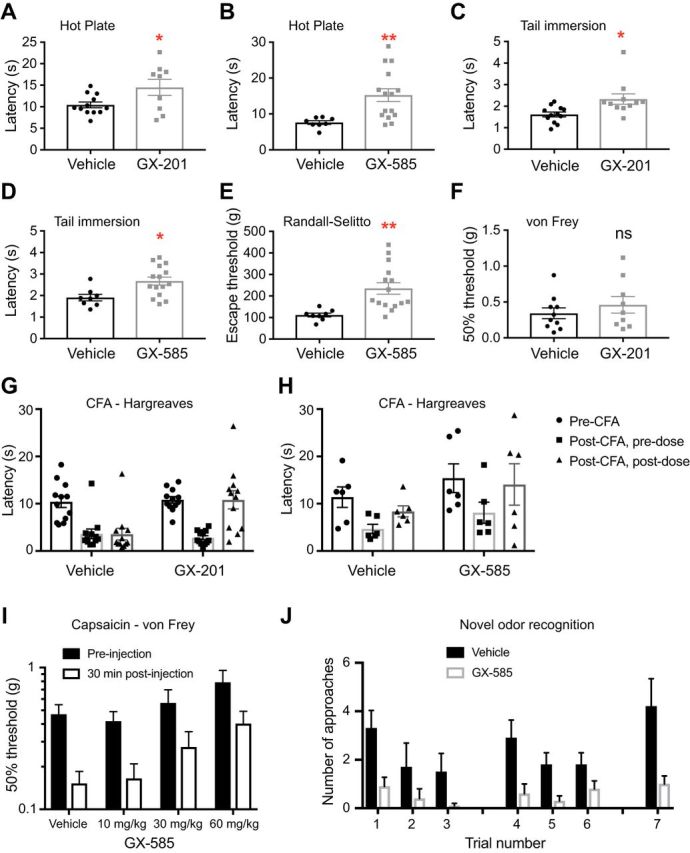
Nav1.7-selective inhibitors have antinociceptive and analgesic properties in WT mice. A, B, Response latency on the 55°C hot plate test was prolonged after oral administration of GX-201 (A) or GX-585 (B), but not vehicle. C, D, Latency to withdraw the tail from a 55°C water bath was significantly longer after dosing of GX-201 (C) or GX-585 (D) compared with vehicle. E, Escape threshold in the Randall–Selitto assay was elevated in mice administered GX-585 compared with vehicle-treated controls. F, von Frey threshold was not significantly different between mice treated with GX-201 or vehicle, as expected because Nav1.7 cKO mice also do not differ from WT littermates in this assay. A, C, F, GX-201 (30 mg/kg, p.o., average total plasma concentration 10.5 ± 0.7 μm). B, D, E, GX-585 (60 mg/kg, p.o., average total plasma concentration 49.3 ± 5.4 μm). G, H, Baseline withdrawal latency from a radiant heat stimulus was recorded; then an intraplantar injection of CFA was performed. Withdrawal latencies were recorded again 24 h later (post-CFA, predose). GX-201 (30 mg/kg, p.o., average total plasma concentration 16.1 ± 1.5 μm; G) or GX-585 (60 mg/kg, p.o., average total plasma concentration 67.9 ± 15.6 μm; H) were administered, and testing was performed again 1 h later (post-CFA, postdose). Both Nav1.7 inhibitors reversed established hypersensitivity back to near pre-CFA baseline levels. I, C57BL/6 mice (n = 12/group) were administered the indicated dose of GX-585, and precapsaicin baseline von Frey thresholds were measured at the plantar footpad 1 h later. Capsaicin was injected extraterritorially at the ankle, and plantar von Frey thresholds were measured again after 30 min. GX-585 provided a dose-dependent protection from developing mechanical allodynia in this model. Average total plasma concentrations were 15.7 ± 3.1, 31.1 ± 5.3, and 60.8 ± 8.0 μm for 10, 30, and 60 mg/kg doses, respectively. J, C57BL/6 mice were dosed with vehicle (0.5% methylcellulose/0.2% Tween 80) or 60 mg/kg GX-585 1 h before testing. Vehicle-treated mice (black bars) demonstrated the expected pattern of habituation/dishabituation to the presentation of novel/familiar odor stimuli. In contrast, GX-585-treated mice were relatively indifferent to odor presentation. This may indicate perturbed olfaction after systemic administration of the drug. *p < 0.05, **p < 0.01. Statistical analysis described in Table 1. Error bars indicate SEM.
Nav1.7-selective inhibitors are efficacious in mouse models of chronic pain
On the basis of our experiments with Nav1.7 cKO mice, we identified two chronic pain models to which Nav1.7 makes a large apparent contribution: heat hypersensitivity in the setting of CFA inflammation and mechanical allodynia secondary to capsaicin injection. We tested whether our selective Nav1.7 inhibitors would be efficacious to block exaggerated responsiveness in these models. In the CFA model, all mice tested 24 h after induction of inflammation had developed strong hypersensitivity to a radiant heat stimulus compared with their pre-CFA baselines. Oral administration of a single dose of GX-201 or GX-585 reversed this established hypersensitivity back to near baseline by 1 h after dose (Fig. 11G,H). In mice that were administered GX-585 before capsaicin injection, we observed a dose-dependent protection from developing secondary mechanical allodynia (Fig. 11I).
We also investigated the potential for systemic administration of a Nav1.7 inhibitor to perturb olfaction. Mice were dosed orally with either vehicle or GX-585 and underwent a novel odor recognition task 1 h later. We observed that, whereas vehicle-treated mice appeared generally able to perform this task, GX-585-treated mice were relatively indifferent to the presentation of odors and made fewer approaches on average during each of the trials (Fig. 11J). While on the one hand this might foreshadow a clinically relevant side effect to be expected in patients, it may also be interpreted as a monitorable sign that has potential application as a biomarker for proof-of-activity studies in humans.
These results demonstrate clear evidence of analgesic efficacy acutely after a single dose of either of two selective acylsulfonamide Nav1.7 inhibitors in multiple preclinical models of chronic pain.
Discussion
We characterized adult-onset Nav1.7 cKO mice, intended to mimic the expected effects of dosing an adult human pain patient with a potent Nav1.7-selective inhibitor while circumventing potential developmental effects of Nav1.7 deletion. We developed isoform-selective Nav1.7 small-molecule inhibitors with antinociceptive and analgesic properties in normal animals and in chronic pain models. Collectively, these data deepen the knowledge surrounding Nav1.7 biology related to pain and provide preclinical proof of efficacy, laying a clear path toward translation for the therapeutic use of Nav1.7-selective inhibitors in humans.
The Nav1.7 expression pattern we observed in mice is in good agreement with previous reports (Toledo-Aral et al., 1997; Morinville et al., 2007; Fukuoka et al., 2008). While tamoxifen robustly depletes SCN9A mRNA from sensory and sympathetic neurons, KO was inefficient in the OE for unknown reasons. This is presumably the basis for preserved olfaction in Nav1.7 cKO mice (Fig. 5F). By ISH, almost all DRG neurons from control animals express SCN9A, consistent with previous literature (Fukuoka et al., 2008). In contrast, in Nav1.7 cKO mice, qPCR indicates > 1000-fold decrease in SCN9A, and in DRG sections only ∼6% of neurons retain a normal-appearing ISH signal. Although behavioral phenotypes of other Nav1.7 cKOs have been described in the literature (Nassar et al., 2004; Minett et al., 2012), careful quantification of SCN9A depletion in DRG was overlooked. However, it is expected that SCN10ACre/+; SCN9AoxP/loxP mice may still express SCN9A in 25% of DRG neurons (Fukuoka et al., 2008; Shields et al., 2012), whereas AVILCre/+; SCN9AloxP/loxP mice may still express SCN9A in 11% of DRG neurons (Lau et al., 2011). To our knowledge, the mice described here represent the most complete Nav1.7 cKO so far, which is consistent with the more complete insensitivity-to-pain phenotype we observe.
In contrast to SCN9A mRNA, which is rapidly depleted from DRG tissue, Nav1.7 protein is strikingly long-lived. We found it still detectable by Western blot weeks after tamoxifen-induced gene recombination, and our behavioral time course studies indicate that the remaining Nav1.7 is able to maintain some electrogenic function for up to 6 weeks after its production is halted. Evolutionarily, a high turnover rate for a large 24 transmembrane-domain protein would be energetically costly. According to proteome dynamics studies, Nav proteins are quite stable in mouse brain (turnover rate of ∼0.05 d) largely consistent with our observations in the peripheral nervous system (Price et al., 2010). It may also be that very little Nav1.7 protein is required to support function (i.e., a high safety factor might exist before failure occurs).
Electrophysiologically, we show that Nav1.7 carries the majority of TTX-sensitive current in small- to medium-diameter DRG neurons. Indeed, many small-diameter DRG neurons appear to completely lack detectable TTX-sensitive Nav current in the absence of Nav1.7. This is in contrast to large-diameter neurons that still show normal TTX-sensitive current without Nav1.7. Current-clamp recordings of small-diameter DRG neurons indicate that Nav1.7 is required to maintain normal voltage and current thresholds for AP generation under conditions where Nav1.7 is active (resting membrane potential −70 mV). While TTX-resistant channels are able to support AP firing in the absence of Nav1.7 and even in the presence of TTX, it is not clear that this happens in vivo given the Nav1.7 dependence to nociception.
While the insensitivity-to-pain phenotype in our Nav1.7 cKO mice is quite robust for most modalities, we were surprised to find that behavioral responses were still intact upon presentation of some stimuli that are normally considered noxious. Mechanical withdrawal thresholds in the von Frey test were not affected by deletion of Nav1.7, for example, similar to results obtained with germline KO mice in this test (Gringas et al., 2014). Could this assay actually be a read-out of touch sensation rather than pain? Touch sensation is reportedly normal in human CIP patients (Cox et al., 2006). Similarly, paw-directed behavior was mostly preserved in the formalin test in the absence of Nav1.7. Subcutaneous formalin injection is a nonphysiological stimulus that nonspecifically activates sensory neurons of all functional types (Braz and Basbaum, 2010) and causes paw-directed behavior, even in mice in which the majority of nociceptors have been ablated (Shields et al., 2010). We suggest that paresthesias evoked by nonspecific activation of functionally diverse sensory neurons may elicit formalin-induced behaviors in Nav1.7 cKO mice and that the formalin test may not be a suitably specific assay of nociception. Our finding that Fos is not induced in laminae I-II in Nav1.7 cKO mice upon formalin injection, while the mice still display mostly normal paw-directed behaviors, is consistent with this interpretation.
Surprisingly, we observed that electrical stimuli evoke both intensity-dependent startle and active avoidance in mice lacking Nav1.7, indicating that shock can act as an aversive teaching signal, even in the absence of this sodium channel. Additionally, an electrical stimulus was the only modality of acute nociception we tested that was capable of significant Fos induction in laminae I-II of Nav1.7 cKO mice. Although the magnitude of Fos induction was much reduced in Nav1.7 cKO mice compared with WT mice, this amount of signal transmission through the nociceptive circuitry may be sufficient to drive active avoidance behavior. The remaining Fos induction may be due to electrical stimuli depolarizing nerve endings sufficiently to bypass the normal requirement for Nav1.7 as a receptor-potential amplifier, allowing some Nav1.7-independent AP generation.
We were also surprised to find that most forms of mechanical allodynia, including in neuropathic, inflammatory, and postoperative pain models, are resistant to loss of Nav1.7. This striking observation is at odds with reports of human Nav1.7 loss-of-function CIP individuals who feel no pain despite injuries and inflammation. Possibly, Nav1.7 plays a greater role in mechanical allodynia in humans than mice due to species differences in expression. Alternatively, lack of functional Nav1.7 throughout development may result in an altered organization of nociceptive circuitry that accounts for the difference. Interestingly, mechanical allodynia in neuropathic pain models was reported to be retained in the SCN10ACre/+; SCN9AloxP/loxP mouse and the AVILCre/+; SCN9AloxP/loxP mouse but not in the WNT1Cre/+; SCN9AloxP/loxP mouse (Nassar et al., 2004; Minett et al., 2012), where Nav1.7 is inactivated in both sensory as well as sympathetic neurons. In our adult-inducible Nav1.7 cKO mice, mechanical allodynia is not altered despite inactivation of Nav1.7 in both sensory and sympathetic neuron populations (other aspects of sympathetic function were not examined).
One form of mechanical allodynia is abolished when Nav1.7 is deleted in the adult (i.e., extraterritorial mechanical allodynia secondary to capsaicin injection). In this model, it is believed that capsaicin activates TRPV1-expressing nociceptors, which then unleash a flood of excitation in the dorsal horn causing transient central sensitization. In Nav1.7 cKO mice, the lack of Nav1.7 may prevent the initial barrage such that central sensitization and its associated mechanical allodynia do not develop.
Here we have confirmed and extended the phenotype of global Nav1.7 KO mice reported by (Gingras et al., 2014). In both cases, the mice were insensitive to noxious heat and strong pressure, but had normal von Frey thresholds. Heat hypersensitivity in the CFA model was also abolished in both studies (Gingras et al., 2014 did not report mechanical allodynia data). The main difference is that the global KO mice had a much-reduced response to formalin compared with what we observed. Potentially, this reflects a developmental consequence of lacking Nav1.7, or it could be due to background strain (C57BL/6 in our study and CD1 in Gingras et al., 2014). In the aggregate, comparison of the two studies leads to the interpretation that adult-onset loss of Nav1.7 function can cause robust insensitivity to pain.
Our pharmacological studies provide clear proof of concept that acute Nav1.7-selective blockade results in pain relief. We observed antinociception to noxious heat and mechanical stimuli at drug exposure levels that were not sedative. The animals did not reach assay cutoff values, likely due to incomplete block of Nav1.7 at the doses tested. Furthermore, the antinociceptive effects of the compounds were observed within 1 h of dosing. This acute action supports the idea that simple pharmacological blockade of Nav1.7 channels is sufficient to increase pain threshold in vivo, and thus there is no need to invoke alternative mechanisms of action (e.g., endogenous opioids) (Minett et al., 2015; Isensee et al., 2017). Even more compelling than the compounds' effect on acute nociception is the analgesic efficacy they provide in two mouse models of chronic pain. While it is unclear how translationally relevant heat hypersensitivity is to common chronic pain disorders in humans, we are nevertheless encouraged by our positive and robust preclinical efficacy data.
In conclusion, we compiled comprehensive data that demonstrate genetic and pharmacological evidence for a critical contribution of Nav1.7 to most modalities of acute and chronic pain. We developed novel isoform-selective acylsulfonamide Nav1.7 inhibitors that have suitable drug-like properties for use in vivo and possess antinociceptive and analgesic properties in preclinical pain models. We believe this work represents an important step forward in our understanding of the biology of Nav1.7 and the pain sensory nervous system, and significant progress toward new potential therapeutic options for chronic pain patients.
Footnotes
All data needed to evaluate the conclusions in this paper are present in the paper and/or supplemental materials. We thank Elizabeth Lu for assistance with breeding and genotyping mice; and Evera Wong for assistance with generating cDNA constructs to express mutant Nav1.7 channels.
All authors are employed at Genentech, Inc., a for-profit biopharmaceutical company.
References
- Ahmad S, Dahllund L, Eriksson AB, Hellgren D, Karlsson U, Lund PE, Meijer IA, Meury L, Mills T, Moody A, Morinville A, Morten J, O'Donnell D, Raynoschek C, Salter H, Rouleau GA, Krupp JJ (2007) A stop codon mutation in SCN9A causes lack of pain sensation. Hum Mol Genet 16:2114–2121. 10.1093/hmg/ddm160 [DOI] [PubMed] [Google Scholar]
- Ahn HS, Black JA, Zhao P, Tyrrell L, Waxman SG, Dib-Hajj SD (2011) Nav1.7 is the predominant sodium channel in rodent olfactory sensory neurons. Mol Pain 7:32. 10.1186/1744-8069-7-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja S, Mukund S, Deng L, Khakh K, Chang E, Ho H, Shriver S, Young C, Lin S, Johnson JP Jr, Wu P, Li J, Coons M, Tam C, Brillantes B, Sampang H, Mortara K, Bowman KK, Clark KR, Estevez A, et al. (2015) Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 350:aac5464. 10.1126/science.aac5464 [DOI] [PubMed] [Google Scholar]
- Braz JM, Basbaum AI (2010) Differential ATF3 expression in dorsal root ganglion neurons reveals the profile of primary afferents engaged by diverse noxious chemical stimuli. Pain 150:290–301. 10.1016/j.pain.2010.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JS, Boralevi F, Brissaud O, Sánchez-Martín J, Te Morsche RH, Dib-Hajj SD, Drenth JP, Waxman SG (2011) Paroxysmal extreme pain disorder: a molecular lesion of peripheral neurons. Nat Rev Neurol 7:51–55. 10.1038/nrneurol.2010.162 [DOI] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG (2006) An SCN9A channelopathy causes congenital inability to experience pain. Nature 444:894–898. 10.1038/nature05413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. (2000) What's wrong with my mouse? Behavioral phenotyping of transgenic and knockout mice. Ed 2 Hoboken, NJ: John Wiley and Sons. [Google Scholar]
- Cummins TR, Dib-Hajj SD, Waxman SG (2004) Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J Neurosci 24:8232–8236. 10.1523/JNEUROSCI.2695-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Estacion M, Jarecki BW, Tyrrell L, Fischer TZ, Lawden M, Cummins TR, Waxman SG (2008) Paroxysmal extreme pain disorder M1627K mutation in human Nav1.7 renders DRG neurons hyperexcitable. Mol Pain 4:37. 10.1186/1744-8069-4-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt M, Nakajima J, Klinger AB, Neacsu C, Hühne K, O'Reilly AO, Kist AM, Lampe AK, Fischer K, Gibson J, Nau C, Winterpacht A, Lampert A (2014) Inherited pain: sodium channel Nav1.7 A1632T mutation causes erythromelalgia due to a shift of fast inactivation. J Biol Chem 289:1971–1980. 10.1074/jbc.M113.502211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, Dib-Hajj SD, Benke PJ, Te Morsche RH, Eastman EM, Macala LJ, Drenth JP, Waxman SG (2008) NaV1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders. J Neurosci 28:11079–11088. 10.1523/JNEUROSCI.3443-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka T, Kobayashi K, Yamanaka H, Obata K, Dai Y, Noguchi K (2008) Comparative study of the distribution of the α-subunits of voltage-gated sodium channels in normal and axotomized rat dorsal root ganglion neurons. J Comp Neurol 510:188–206. 10.1002/cne.21786 [DOI] [PubMed] [Google Scholar]
- Gingras J, Smith S, Matson DJ, Johnson D, Nye K, Couture L, Feric E, Yin R, Moyer BD, Peterson ML, Rottman JB, Beiler RJ, Malmberg AB, McDonough SI (2014) Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS One 9:e105895. 10.1371/journal.pone.0105895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Dib-Hajj SD, Lin Z, Li Y, Eastman EM, Tyrrell L, Cao X, Yang Y, Waxman SG (2009) Early- and late-onset inherited erythromelalgia: genotype-phenotype correlation. Brain J Neurol 132:1711–1722. 10.1093/brain/awp078 [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244:305–318. 10.1006/dbio.2002.0597 [DOI] [PubMed] [Google Scholar]
- Hille B. (2001) Ion channels of excitable membranes. Sunderland, MA: Sinauer. [Google Scholar]
- Hodgkin AL, Huxley AF (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117:500–544. 10.1113/jphysiol.1952.sp004764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isensee J, Krahé L, Moeller K, Pereira V, Sexton JE, Sun X, Emery E, Wood JN, Hucho T (2017) Synergistic regulation of serotonin and opioid signaling contributes to pain insensitivity in Nav1.7 knockout mice. Sci Signal 10:eaah4874. 10.1126/scisignal.aah4874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert A, Eberhardt M, Waxman SG (2014) Altered sodium channel gating as molecular basis for pain: contribution of activation, inactivation, and resurgent currents. In: Voltage gated sodium channels (Ruben PC, ed), pp 91–110. Berlin: Springer. [DOI] [PubMed] [Google Scholar]
- Lau J, Minett MS, Zhao J, Dennehy U, Wang F, Wood JN, Bogdanov YD (2011) Temporal control of gene deletion in sensory ganglia using a tamoxifen-inducible advillin-CreERT2 recombinase mouse. Mol Pain 7:100. 10.1186/1744-8069-7-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett MS, Pereira V, Sikandar S, Matsuyama A, Lolignier S, Kanellopoulos AH, Mancini F, Iannetti GD, Bogdanov YD, Santana-Varela S, Millet Q, Baskozos G, MacAllister R, Cox JJ, Zhao J, Wood JN (2015) Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat Commun 6:8967. 10.1038/ncomms9967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett MS, Nassar MA, Clark AK, Passmore G, Dickenson AH, Wang F, Malcangio M, Wood JN (2012) Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun 3:791. 10.1038/ncomms1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morinville A, Fundin B, Meury L, Juréus A, Sandberg K, Krupp J, Ahmad S, O'Donnell D (2007) Distribution of the voltage-gated sodium channel Na(v)1.7 in the rat: expression in the autonomic and endocrine systems. J Comp Neurol 504:680–689. 10.1002/cne.21484 [DOI] [PubMed] [Google Scholar]
- Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH, Wood JN (2004) Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A 101:12706–12711. 10.1073/pnas.0404915101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogatzki EM, Raja SN (2003) A mouse model of incisional pain. Anesthesiology 99:1023–1027. 10.1097/00000542-200310000-00041 [DOI] [PubMed] [Google Scholar]
- Price JC, Guan S, Burlingame A, Prusiner SB, Ghaemmaghami S (2010) Analysis of proteome dynamics in the mouse brain. Proc Natl Acad Sci U S A 107:14508–14513. 10.1073/pnas.1006551107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupasinghe DB, Knapp O, Blomster LV, Schmid AB, Adams DJ, King GF, Ruitenberg MJ (2012) Localization of nav 1.7 in the normal and injured rodent olfactory system indicates a critical role in olfaction, pheromone sensing and immune function. Channels (Austin) 6:103–110. 10.4161/chan.19484 [DOI] [PubMed] [Google Scholar]
- Shields SD, Eckert WA, Basbaum AI (2003) Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J Pain 4:465–470. 10.1067/s1526-5900(03)00781-8 [DOI] [PubMed] [Google Scholar]
- Shields SD, Mazario J, Skinner K, Basbaum AI (2006) Anatomical and functional analysis of aquaporin 1, a water channel in primary afferent neurons. Pain 131:8–20. 10.1016/j.pain.2006.11.018 [DOI] [PubMed] [Google Scholar]
- Shields SD, Cavanaugh DJ, Lee H, Anderson DJ, Basbaum AI (2010) Pain behavior in the formalin test persists after ablation of the great majority of nociceptors. Pain 151:422–429. 10.1016/j.pain.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Ahn HS, Yang Y, Han C, Seal RP, Wood JN, Waxman SG, Dib-Hajj SD (2012) Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system: Pain 153:2017–2030. 10.1016/j.pain.2012.04.022 [DOI] [PubMed] [Google Scholar]
- Shimada SG, LaMotte RH (2008) Behavioral differentiation between itch and pain in mouse. Pain 139:681–687. 10.1016/j.pain.2008.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I, Halegoua S, Mandel G (1997) Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A 94:1527–1532. 10.1073/pnas.94.4.1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Wu T, Hanson JE, Alam NM, Solanoy H, Ngu H, Lauffer BE, Lin HH, Dominguez SL, Reeder J, Tom J, Steiner P, Foreman O, Prusky GT, Scearce-Levie K (2015) Cognitive deficits, changes in synaptic function, and brain pathology in a mouse model of normal aging. eNeuro 2:pii: ENEURO.0047-15.2015. 10.1523/eneuro.0047-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]