

Abstract
Amyloid-β (Aβ) drives the synaptic impairment and dendritic spine loss characteristic of Alzheimer's disease (AD), but how Aβ affects the actin cytoskeleton remains unknown and contentious. The actin-binding protein, cofilin-1 (cof1), is a major regulator of actin dynamics in dendritic spines, and is subject to phospho-regulation by multiple pathways, including the Rho-associated protein kinase (ROCK) pathway. While cof1 is implicated as a driver of the synaptotoxicity characteristic of the early phases of AD pathophysiology, questions remain about the molecular mechanisms involved. Cofilin-actin rods are observed in neurons exposed to Aβ oligomers (Aβo) and in tissue from AD patients, and others have described an increased cofilin phosphorylation (p-cof1) in AD patients. Here, we report elevated p-cof1 of the postsynaptic enriched fraction of synaptosomes from cortical samples of male APP/PS1 mice and human AD cases of either sex. In primary cortical neurons, Aβo induced rapid actin stabilization and increased p-cof1 in the postsynaptic compartment of excitatory synapses within 30 min. Fluorescence recovery after photobleaching of actin-GFP and calcium imaging in live neurons expressing active or inactive cof1 mutants suggest that cof1 phosphorylation is necessary and sufficient for Aβo-induced synaptic impairment via actin stabilization before the reported formation of cofilin-actin rods. Moreover, the clinically available and well-tolerated ROCK inhibitor, fasudil, prevented Aβo-induced actin stabilization, synaptic impairment, and synaptic loss by blocking cofilin phosphorylation. Aβo also blocked the LTP-induced insertion of the AMPAR subunit, GluA1, at the postsynaptic density, in a fasudil-sensitive manner. These data support an important role for ROCKs and cofilin in mediating Aβ-induced synaptic impairment.
SIGNIFICANCE STATEMENT We report that amyloid-β oligomers rapidly induce aberrant stabilization of F-actin within dendritic spines, which impairs synaptic strength and plasticity. Activation of the Rho-associated protein kinase (ROCK) pathway results in phosphorylation of cof1 and is sufficient to mediate Aβo-induced actin stabilization synaptic impairment and synaptic loss. Further, the ROCK inhibitor, fasudil, prevents cofilin phosphorylation, acute synaptic disruption, and synaptotoxicity in primary cortical neurons. Together, the herein presented data provide strong support for further study of the ROCK pathway as a therapeutic target for the cognitive decline and synaptotoxicity in Alzheimer's disease.
Keywords: actin, Alzheimer's disease, amyloid-β oligomers, cofilin, fasudil, Rho-associated protein
Introduction
Alzheimer's disease (AD) is the leading cause of dementia, yet current treatments provide only modest clinical benefit. Incidence of AD and its societal costs are expected to balloon in coming decades, as the world population ages (Alzheimer's Association, 2017). AD is characterized by accumulation of amyloid-β (Aβ) plaques and microtubule-associated protein tau inclusions. Aβ aggregates, and in particular low molecular weight oligomers (Aβo), are considered triggers of disease through a tau-dependent mechanism (Rapoport et al., 2002; Oddo et al., 2004; Tanzi and Bertram, 2005). Appearances of Aβo and tau-dependent dendritic spine loss are the features that correlate most directly with cognitive decline in AD (DeKosky and Scheff, 1990; Terry et al., 1991). Moreover, novel findings suggest that dendritic spine plasticity can provide cognitive resilience against dementia among the elderly with AD pathology (Boros et al., 2017). Last, acute exposure to Aβo impairs LTP in rodent tissue, predominantly by acting at the postsynaptic compartment (Kervern et al., 2012).
The actin cytoskeleton (CSK), enriched in dendritic spines, is central to the regulation of spine morphology, stability, and plasticity (Hotulainen and Hoogenraad, 2010). The neuronal actin-binding protein, cofilin-1 (cof1), is a major regulator of spine actin dynamics by severing filamentous actin (F-actin), leading to actin depolymerization (Rust, 2015). cof1 activity is regulated by phosphorylation of serine 3, rendering cof1 inactive. Synaptic activity-driven cof1 phosphorylation contributes to modifications of spine morphology (Rust et al., 2010; Noguchi et al., 2016). Further, functional loss of cof1 causes aberrant spine morphogenesis and impairs synaptic plasticity (Zhou et al., 2009; Gu et al., 2010). Constitutively active or inactive mutants of cof1 influence spine morphology and maturity in neurons (Yang et al., 1998; Niwa et al., 2002). Similarly, inactivation or ablation of the upstream regulators, Rho-associated protein kinase (ROCK) or LIM-kinase (LIMK1), also leads to increased cof1 activity, decreased spine F-actin, and immature spines or spine collapse (Niwa et al., 2002; Zhou et al., 2009; Swanger et al., 2015). cof1 is also necessary for the trafficking of the AMPARs at the postsynaptic density (PSD) during synaptic plasticity (Gu et al., 2010).
While several research groups have described the implication of cof1 in the pathophysiology of AD, the pathological modification of cof1 activity remains controversial. Indeed, cof1 was discovered to accumulate in senile plaques in AD tissue and AD mouse models (Bamburg and Bernstein, 2016). Additionally, recent publications suggest that active (i.e., dephosphorylated) cof1 forms aberrant cofilin-actin rods, which block axonal trafficking and may contribute to deficits in synaptic plasticity (Davis et al., 2011; Mendoza-Naranjo et al., 2012; Barone et al., 2014). Both Aβ fibrils and Aβo (i.e., dimers and trimers) promote cofilin-actin rod formation with prolonged exposure (Davis et al., 2011; Mendoza-Naranjo et al., 2012). However, brain tissue from AD patients and APP-expressing mouse models exhibit elevated ROCK levels and corresponding elevated levels of inactive p-cof1 (Herskowitz et al., 2013; Barone et al., 2014; Henderson et al., 2016; Han et al., 2017). Furthermore, in other neurodegenerative diseases characterized by an early synaptic loss, such as sporadic Creutzfeld-Jacob disease, upregulation of p-cof1 has been described (Zafar et al., 2018). These data highlight that cof1 exerts a pivotal role in the synaptotoxic process of neurodegenerative diseases (Heredia et al., 2006; Lacor et al., 2007; Herskowitz et al., 2013).
Here, we tested the hypothesis that phosphorylation-inactivation of cof1 induced by exposure to Aβo promotes synaptic deficits and impairs plasticity. We showed that there are elevated p-cof1 levels in PSD-enriched synaptosomes of APP/PS1 mice and human AD cases, consistent with overactivation of the ROCK pathway. In primary cortical cultures, while a 24 h exposure to Aβo decreases spine density (Lacor et al., 2007), we report that acute exposure to Aβo increases p-cof1 at the postsynaptic compartment and favors its distribution in the Triton-insoluble (TI) fractions, leading to a subsequent stabilization of spine F-actin and impairment of synaptic signaling and plasticity. Further, the clinically available and well-tolerated ROCK inhibitor, fasudil, can prevent these cof1-mediated synaptic deficits and synaptic loss.
Materials and Methods
Animals.
hAPP/PS1–21 (line 21) mice (APP/PS1) were used at 3 months of age for this study. APP/PS1 mice coexpress human APP carrying the K670N/M671L “Swedish” double mutation and HPS1 L166P with a threefold overexpression of human APP over endogenous mouse APP. Mice express the transgene under the control of a neuron-specific mThy-1 promoter element and were generated on a C57bl6 background. For immunohistochemical studies, Thy1-YFP-H (The Jackson Laboratory, B6 Cg-Tgn 2Jrs) mice were crossed with heterozygote APP/PS1 mice to generate C57BL/6Thy1-eYFP APP/PS1–21 mouse colony. All experiments involving animals were conducted in accordance with the policy of Institut des Neurosciences de Grenoble and French legislation, in compliance with the European Community Council Directive of November 24, 1986 (86/609/EEC). The research involving animals was authorized by the Direction Départementale de la protection des populations–Préfecture de l'Isère France and by the ethics committee of Institut des Neurosciences de Grenoble accredited by the French Ministry of Research.
Immunohistochemistry.
Male mice were deeply anesthetized and perfused intracardially with 0.9% NaCl followed by 35 ml 4% PFA in 0.1 m PBS, pH 7.3. Brains were rapidly removed, postfixed overnight at 4°C in 4% PFA, immersed in 20% sucrose in 0.1 m PBS at pH 7.5 overnight, frozen in isopentane, and stored at −30°C. Serial frontal sections (30 μm) were obtained with a cryostat microtome. Sections were blocked by incubation with 3% BSA in TBS-Tween-Triton (TBSTT) (0.1 m Tris base, 0.15 m NaCl, 0.1% Tween, 0.1% Triton X-100) for 30 min (dilution/blocking buffer) and incubated overnight at 4°C with anti-phospho-cofilin 1 antibody (Santa Cruz Biotechnology, rabbit polyclonal; 1:200). Tissue sections were washed in TBSTT and incubated for 2 h at room temperature with Cyanin 3-conjugated secondary antibodies. Sections were washed in TBSTT and mounted in fluorescent mounting medium. Sections were examined on an LSM 710 confocal laser-scanning microscope (Carl Zeiss), and images were acquired with an Airyscan module (Carl Zeiss) with an oil-immersion Plan Apochromat 63× objective (NA 1.46) to improve lateral resolution (∼140 nm) and signal-to-noise ratios. For illustration, images were merged with ImageJ software (RRID:SCR_003070). For quantitative analysis, ROIs were drawn to delimitate dendritic spine and the fluorescence intensity of p-cof1 staining measured using Analyze/Measure/Mean gray value functions of ImageJ.
Primary cultures of cortical neurons.
Mouse cortical neurons were cultured from 14- to 15-d-old OF1 embryos of either sex as previously described (Léveillé et al., 2008). Cerebral cortices were dissected, dissociated, and cultured in DMEM containing 5% FBS, 5% horse serum, and 2 mm glutamine (all from Sigma-Aldrich) on 24-well plates for biochemical experiments. Neurons were seeded on 35 mm glass-bottom dishes (MatTek) at a final concentration of two cortical hemispheres per dish for confocal experiments. All plates and dishes were coated with 0.1 mg/ml poly-d-lysine and 0.02 mg/ml laminin (Sigma-Aldrich). Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2/95% air. After 3–4 DIV, cytosine arabinoside (AraC, 10 μm; Sigma-Aldrich) was added to inhibit proliferation of non-neuronal cells in cultures used for biochemistry experiments; 98% of the cells were considered as neuronal. The day before the experiments, cells were washed in DMEM. Treatments were performed on neuronal cultures at 14–15 DIV.
Aβ oligomerization.
Amyloid β oligomers were prepared as previously described (Frandemiche et al., 2014). Briefly, human Aβ 1–42 peptides (Bachem) was resuspended in 1,1,1,3,3,3-hexafluoro-2-propanol (Sigma-Aldrich) to 1 mm until complete resuspension as previously described (Stine et al., 2003). Aβo was prepared by diluting Aβ to 1 mm in anhydrous DMSO (Sigma-Aldrich) and then to 100 μm in ice-cold, phenol red-free HEPES and bicarbonate-buffered saline solution (HBBSS) with immediate vortexing and ice bath sonication for 15 min, and then incubated at 4°C for 24 h with mild agitation.
Site-directed mutagenesis.
Constitutively inactive phosphomimetic (S3E) and active nonphosphorylatable (S3A, active) cof1-mCherry constructs were generated using quick change site-directed mutagenesis kit (Agilent Technologies), according to the instructions of the manufacturer, with the following primers: S3A (forward: 5′-atggccgccggtgtggctgtctcctg-3′; reverse: 5′-cagagacagccacaccctcggccat-3′), S3E (forward: 5′-atggccgagggtgtggctgtctcctg-3′; reverse: 5′-cagagacagccacaccctcggccat-3′).
Neuronal transfection.
Transfections were performed on cortical neuron cultures after 12–13 DIV with calcium phosphate precipitation technique. Growth medium (DMEM and sera) was removed and kept until the last transfection step. Cells were washed for 1–1.5 h in DMKY buffer containing the following (in mm): 1 kynurenic acid, 0.9 NaOH, 0.5 HEPES, 10 MgCl2, plus phenol red 0.05%, pH 7.4; 3 μg of the following plasmids: GCaMP6 fast, Lifeact-eGFP, actin-GFP, cof1-S3E-mCherry, and cof1-S3A-mCherry, mixed with CaCl2 (120 mm) in HBS containing the following (in mm): 25 HEPES, 140 NaCl, and 0.750 Na2HPO4, pH 7.06, and left for 15 min for DNA precipitation. Plasmids were applied for 30 min. Transfection medium was replaced with conditioned growth medium, and cultures were returned to the incubator until use at DIV 14–15.
Fluorescence recovery after photobleaching (FRAP) experiments.
FRAP was performed on cultured neurons (DIV 14–15) 48 h after transfection with actin-GFP. Images were acquired with an inverted Nikon Eclipse Ti C2 confocal microscope with a Nikon 60× water objective with a 1.33 numerical aperture. Actin-GFP was imaged for at least 20 s before being bleached at 405 nm. The subsequent fluorescence recovery was measured for 120 s (at 1 frame/s). Fluorescent signal analysis was performed with the Nikon software NIS Elements. Fluorescent signals from the dendritic spine heads were normalized to prebleach values. Average recovery plateau values were calculated using nonlinear regression (two-factor decay model) from individual spine recovery curves with GraphPad Prism 6.0 (RRID:SCR_002798).
Neuronal live calcium imaging.
Primary neurons were transfected at DIV 13 using the calcium phosphate technique. Imaging experiments were performed at DIV 14. Neurons were washed into HBBSS (containing the following in mm: 116 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgSO4 1.3 NaH2PO4, 12 HEPES, 5.5 glucose, 25 bicarbonate, and 10 μm glycine at pH 7.45) and moved to the stage of a Nikon Ti Eclipse C2 confocal microscope driven by NIS Elements software and equipped with a 60× 1.2 NA water-immersion lens. Cultures were continuously perfused with HBBSS in a closed-loop (for baseline and 100 nm Aβo application; total system volume of 12.5 ml) at a rate of 2 ml/min, and maintaining a constant culture volume of ∼2.5 ml. For synaptic activation with bicuculline (Bic) and 4-aminopyridine (4AP), the inlet tube was moved to a stock of 50 μm Bic + 2.5 mm 4AP in HBBSS for 15 s three times before and three times after incubation with the Aβo and/or fasudil treatment, and then to a tube of fresh HBBSS. The outlet tube of the perfusion pump was immediately directed to a waste container to avoid pharmacological contamination of the HBBSS stock. Event markers were applied to the imaging files as the Bic + 4AP solution reached the culture and when the application was finished. The Aβo and/or fasudil (30 μm) in HBBSS were applied by perfusion pump in a closed-loop for 15 min. For subsequent synaptic activation, the perfusion loop was opened to fresh HBBSS for 5 min before the subsequent series of three Bic + 4AP applications. Twelve-bit images of GCaMP6 fast fluorescence brought on by excitation with a 488 nm argon laser were acquired using a pinhole of 2.2 AU at 1 frames/s with a resolution of 512 × 512 to optimize scan speed and signal-to-noise ratio. Images were denoised and filtered before ROI extraction of the signal data using the Time Measurement function within NIS Elements software, and the data subsequently exported to MS Excel for calculation of the ΔF/F values, where F was the average baseline fluorescence value from within the ROI before experimentation. ΔF/F traces from each cell were entered into GraphPad Prism, after baseline subtraction, where area under curve values for each peak were normalized to the first Bic + 4AP (B4AP)-induced peak area. The three pretreatment and post-treatment responses were averaged by neuron and combined by group for statistical comparison.
Chemically induced LTP.
Chemically induced LTP (cLTP) was induced in primary cortical neurons as described previously (Frandemiche et al., 2014). Briefly, primary neurons were exposed to 50 μm Bic (a GABAa antagonist) with 2.5 mm 4AP (a weak potassium channel blocker) for 15–30 min, with or without 15 min pretreatment with Aβo (100 nm) and/or fasudil (30 μm). cLTP was verified by detecting increased GluA1 in the PSD-enriched synaptosomal fraction.
Subcellular fractionation and Western blotting. PSD fractions.
Cultured neurons, cortical samples from APP/PS1 male mice, or dorsolateral PFC samples from humans of either sex (Banco de Tejidos CIEN; for case details, see Table 1) were homogenized in cold buffer containing 0.32 m sucrose and 10 mm HEPES, pH 7.4. Samples were maintained at 4°C during all steps of the experiment. Homogenates were cleared at 1000 × g for 10 min to remove nuclei and large debris. The resulting supernatants were concentrated at 12,000 × g for 20 min to obtain a crude membrane fraction, which was then resuspended twice (4 mm HEPES, 1 mm EDTA, pH 7.4, 20 min at 12,000 × g). Then, the pellet was incubated (20 mm HEPES, 100 mm NaCl, 1% Triton X-100, pH 7.2) for 1 h at 4°C with mild agitation, and centrifuged at 12,000 × g for 20 min to pellet the synaptosomal membrane fraction. The supernatant was collected as the non-PSD fraction or Triton-soluble (TS) fraction. The pellet was then solubilized (20 mm HEPES, 0.15 m NaCl, 1% Triton X-100, 1% deoxycholic acid, 1% SDS, pH 7.5) for 1 h at 4°C and centrifuged at 10,000 × g for 15 min. The supernatant contained the PSD or TI fraction. The integrity of non-PSD fraction was verified by synaptophysin immunoblotting, and the integrity of the PSD-enriched fraction was verified by PSD-95 immunoblotting.
Table 1.
Human tissue case detailsa
| Diagnosis | Age (yr) | Gender | BRAAK | PMI (h) | Region |
|---|---|---|---|---|---|
| Control | 58 | M | 0 | 3 | FC |
| Control | 46 | F | NA | 3 | FC |
| Control | 43 | F | 0 | 5 | FC |
| AD | 81 | F | 5 | 2.5 | FC |
| AD | 86 | M | 5 | 5 | FC |
| AD | 79 | F | 5 | 6 | FC |
| AD | 84 | M | 6 | 6 | FC |
| AD | 89 | M | 5 | 6 | FC |
aNA, Not available; PMI, postmortem interval; FC, frontal cortex.
Soluble and cytoskeletal fractions.
The cellular fractions were prepared as described by Hinck et al. (1994). Primary cortical neurons (DIV 14) exposed to Aβo (100 nm for 15 min) were rinsed in HBBSS and homogenized in “cytoskeletal preserving” buffer (50 mm NaCl, 10 mm PIPES, pH 6.8, 3 mm MgCl2, 0.5% Triton X-100, 300 mm sucrose) supplemented with protease (Roche Diagnostics) and phosphatase (Sigma-Aldrich) inhibitor cocktails for 10 min at 4°C with gentle rocking. After centrifugation for 10 min at 14 000 rpm and 4°C, the supernatant constituted the TS fraction. The pellet was triturated in SDS buffer (20 mm Tris, pH 7.5, 5 mm EDTA, 2.5 mm EGTA, 1% SDS and boiled at 100°C for 10 min) and centrifuged for 10 min at 14,000 rpm at 4°C; the supernatant constituted the TI fraction. This fraction usually contained 5- to 8-fold less protein than the TS fraction, as determined by micro-BCA assay (Pierce Chemical). Equal amounts of proteins were routinely analyzed.
Immunoblotting.
Samples in loading buffer were boiled for 5 min (10 min for tissue samples), and equal amounts of proteins (10–25 μg, quantified by micro-BCA assay in duplicate for cell extracts, in triplicate for tissues) were resolved on 4%–20% gradient Bis-Tris polyacrylamide precast gels (Bio-Rad) + stain-free system for tissues (Bio-Rad) in denaturing conditions. Proteins were transferred to a PVDF membrane (EMD Millipore) for 35 min at 4°C. Membranes were blocked with Tris-buffered saline (10 mm Tris, 150 mm NaCl, pH 7.4) containing 0.02% Tween 20 and 3% BSA for 1 h at room temperature. Membranes were then incubated overnight at 4°C with the following primary antibodies.
Membranes were incubated with the HRP-conjugated secondary antibodies rabbit (1:40,000) and mouse (1:5000) (both from Sigma-Aldrich) for 45 min at room temperature. Specific proteins were visualized with an enhanced chemiluminescence ECL Detection System (Bio-Rad). Chemiluminescence detection was performed in a dark room with hyperfilms (GE Healthcare) and analyzed with ImageJ software (RRID:SCR_003070). For tissue analysis, chemiluminescence signals were normalized to protein loading acquired using Stain-free precast gels (Bio-Rad) illumination by the chemidoc system and analyzed with ImageLab (Table 2).
Table 2.
Antibody information
| Antibody | Species | Dilution | Supplier |
|---|---|---|---|
| Anti-Aβ17-24 4G8 | Mouse (monoclonal) | 1/500 | Covance (SIG-39220) |
| PSD-95 | Mouse (monoclonal) | 1/1000 | Abcam (ab2723) |
| Synaptophysin | Mouse (monoclonal) | 1/1000 | Abcam (ab8049) |
| p-Erk1/2 (Thr202/Tyr204) | Rabbit (polyclonal) | 1/500 | Santa Cruz Biotechnology (sc-16982) |
| Erk1/2 | Mouse (monoclonal) | 1/500 | Santa Cruz Biotechnology (sc-135900) |
| p-cof1 (Ser3) | Rabbit (polyclonal) | 1/500 | Santa Cruz Biotechnology (sc-21867) |
| Cof1 | Rabbit (polyclonal) | 1/500 | Santa Cruz Biotechnology (sc-33779) |
| GluA1 | Rabbit (polyclonal) | 1/1000 | EMD Millipore (AB1504) |
Experimental design and statistical analysis.
Results are expressed as the mean ± SEM from independent biological samples. Statistical analyses were performed with GraphPad Prism 6.0 (RRID:SCR_002798). Western blots were analyzed by Mann–Whitney U test, ANOVA with Tukey's post hoc test with correction for multiple comparisons, or two-way ANOVA with Tukey's post hoc test with correction for multiple comparisons; histological analysis by unpaired t test. Spine density quantification was analyzed by paired t test. FRAP data and calcium imaging were analyzed by two-way ANOVA using Aβ and drug treatment (e.g., fasudil or Bic + 4AP) or cof1 genotype as factors, followed by Tukey's multiple comparison tests with corrected p values, or as specified in the corresponding figure legend.
Results
Phospho-cofilin1 is elevated in AD model mice and human AD
In neurons, actin and actin binding proteins are enriched in the postsynaptic compartment of excitatory synapses. Thus, we performed brain tissue fractionation to isolate the postsynaptic compartment (PSD-enriched fraction). Then, we probed these fractions obtained from APP/PS1 model mice and human AD cases (Table 1) for PSD 95 and synaptophysin to verify the integrity of PSD-enriched and non-PSD fraction (Fig. 1A). Then, we compared the levels of cof1 and p-cof1 protein expression on the PSD-enriched fraction (Fig. 1B). Although total cof1 levels were similar in both APP/PS1 AD mice and human AD cases, we found a twofold increase of p-cof1 levels in transgenic APP/PS1 AD mice and threefold increase in human AD cases, compared with controls, consistent with previous reports (Barone et al., 2014). However, the increased in p-cof-1 observed in the human tissue PSD fraction might reflect the general increase in whole AD brain lysate compared with non-AD brains.
Figure 1.

Cortical samples from APP/PS1 mice and AD cases have elevated p-cof1. A, Western blot images of PSD-enriched (top) and non-PSD fraction (bottom) of cortical synaptosomes of APP/PS1 mice (left) and humans (right) showing the enrichment of presynaptic and postsynaptic marker in each fraction. B, Western blot images (top) along with associated quantification (bottom) showing elevated phospho-cofilin1 in APP/PS1 mice (left) compared with their nontransgenic littermates (Western blots n = 4; p-cof1 Mann–Whitney U = 6.0 (72,138) p = 0.0011; cof1 Mann–Whitney U = 43.0 (102,88) p = 0.9025) and in AD cases (right) compared with healthy controls (WB, n = 4; p-cof1 Mann–Whitney U = 0.0 (6,30) p = 0.0357; cof1 Mann–Whitney U = 7.0 (13,23) p = 1.000). *p < 0.05. ***p < 0.001. For case details, see Table 1.
To strengthen this result, we performed immunohistological staining against p-cof1 in 3-month-old Thy1-YFP × APP/PS1 mice, when senile plaques are sparse in the cortex (Radde et al., 2006). We observed a twofold increase in p-cof1 immunostaining in cortical dendritic spines of Thy-1-YFP × APP/PS1 animals compared with Thy-1-YFP mice (Fig. 2A–D). In addition, the Thy-1-YFP × APP/PS1 mice showed a significantly increased dendritic spine area (Fig. 2E). Together, these results highlight that AD patients and AD animal models have elevated p-cof1 located in the postsynaptic sites of excitatory synapses.
Figure 2.
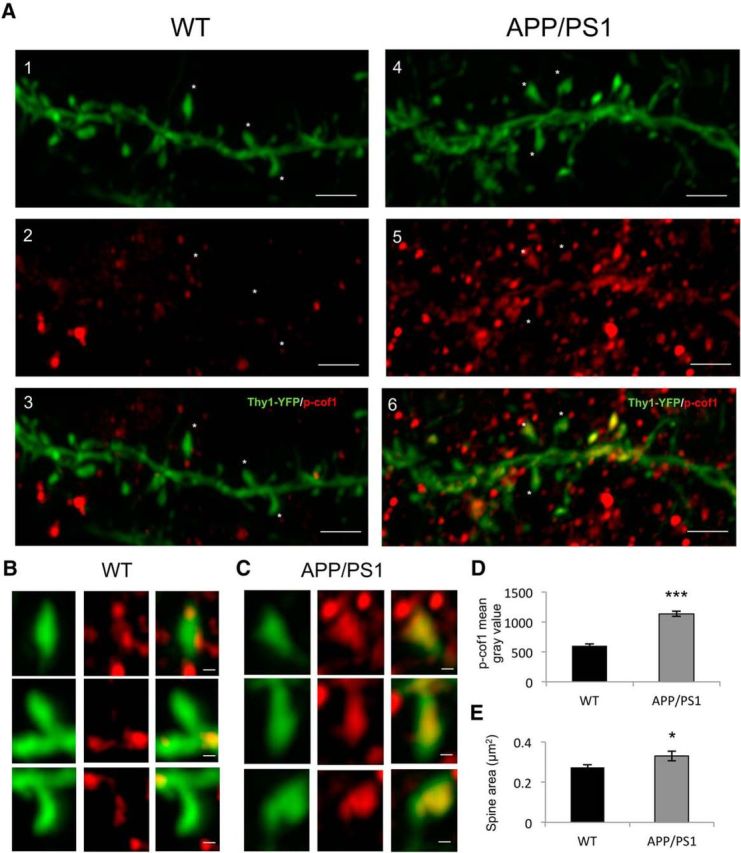
p-cof1 increases in dendritic spines of APP/PS1 mice. A, p-cof1 expression in cortical dendritic spines of Thy1-YFP WT (1–3) and Thy1-YFP APP/PS1 (4–6) mice. Airyscan images of WT and APP/PS1 cortical dendrites (green) showing general higher p-cof1 (red) immunostaining for APP/PS1 mouse. Scale bar, 2 μm. Stars indicate higher magnification of representative dendritic spines for either WT (B) and APP/PS1 (C) mice, showing clearly distinct distribution and higher expression level of p-cof1 in APP/PS1 dendritic spine. Scale bar, 0.2 μm. D, Measurement of p-cof1 staining intensity in WT versus APP/PS1 mice with their relative quantification (mean gray value ± SEM) showing a significative increase in transgenic mice (t values t(136) = 2.116, p = 0.0362, unpaired t test). E, Measurement of spine area in WT versus APP/PS1 mice with their relative quantification (mean ± SEM) showing a significant increase in transgenic mice (t values t(136) = 9.778, p < 0.0001, unpaired t test), n = 71 (WT) and 67 (APP/PS1) spines. *p < 0.05. ***p < 0.001.
Aβ oligomers induce phosphorylation of cof1 and block cLTP in primary culture neurons
To determine whether Aβo was involved in the increase of p-cof1 observed in AD animal models and in AD patients, we exposed mouse primary cortical neurons to 100 nm Aβo and/or a cLTP procedure. This method of cLTP induces synaptic potentiation similar to traditional methods of LTP (e.g., larger spines, increased PSD-95 in the PSD, recruitment of AMPAR subunit GluA1 to the PSD, enhanced EPSPs and activation of p-Erk1/2 signaling) (Blanpied et al., 2008; Behnisch et al., 2011; Frandemiche et al., 2014). We then isolated and probed the PSD-enriched fraction of synaptosomes for cof1 and p-cof1, Erk 1/2 and p-Erk1/2, AMPAR subunit GluA1, and PSD-95. Quantification of Western blots showed that Aβo alone caused an increase in total cof1 and p-cof1 in the PSD fraction, although Erk 1/2, p-Erk1/2, and GluA1 remain constant. In neurons exposed to B4AP (i.e., cLTP), we observed a large increase of p-Erk1/2 and GluA1 and moderate increase of p-cof1, cof1, and PSD-95. When neurons were pretreated with Aβo before cLTP, we found that Aβo precluded these GluA1 and PSD-95 and significantly impaired p-Erk1/2 increases (Fig. 3). Together, these data reveal that Aβo promote an increase of cof1 phosphorylation in the PSD fraction concomitant with the disruption of functional markers of synaptic plasticity.
Figure 3.

Aβo induces phosphorylation of cof1 and blocks cLTP in primary cortical neurons. A, Representative Western blots of PSD-enriched fraction of synaptosomes harvested from primary cortical neurons (DIV 14) exposed to cLTP in the presence or absence of Aβo (15 min pretreatment, 100 nm). B, Relative quantification of Western blots in A demonstrated robust increases in phospho-Erk1/2, GluA1, and PSD-95 induced by cLTP and blocked by pretreatment with Aβo. Aβo by itself caused an increase in cof1 and p-cof1 at the PSD. Two-way ANOVA and Tukey's post hoc test with correction for multiple comparisons for data from n = 5–10 independent cultures per group revealed significant interactions for cLTP and Aβo as factors for phospho-ERK1/2, cof1, p-cof1, GluA1, and PSD-95. p-Erk cLTP, F(1,20) = 94.79, p < 0.0001; Aβo treatment, F(1,20) = 5.173, p = 0.0341; interaction, F(1,20) = 12.50, p = 0.0021; Erk cLTP, F(1,16) = 0.8026, p = 0.3836; Aβo treatment, F(1,16) = 1.814, p = 0.1968; interaction, F(1,16) = 0.04813, p = 0.8291; PSD-95 cLTP, F(1,23) = 1.105, p = 0.3242; Aβo treatment, F(1,23) = 0.118, p = 0.7343; interaction, F(1,23) = 21.13, p = 0.0001; p-cof1 cLTP, F(1,24) = 0.0563, p = 0.8144; Aβo treatment, F(1,24) = 41.05, p < 0.0001; interaction, F(1,24) = 21.41, p = 0.0001; cof1 cLTP, F(1,24) = 7.975, p = 0.0094; Aβo treatment, F(1,24) = 42.52, p < 0.0001; interaction, F(1,24) = 6.33, p = 0.0190; GluA1 cLTP, F(1,26) = 7.014, p = 0.0136; Aβo treatment, F(1,26) = 4.971, p = 0.0346; interaction, F(1,26) = 4.454, p = 0.0446; followed by Tukey's post hoc test with correction for multiple comparisons. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
cof1 phosphorylation is necessary for Aβo-induced F-actin stabilization
The actin CSK, enriched in dendritic spines, is central to the regulation of spine morphology, stability, and thus, synaptic plasticity (Hotulainen and Hoogenraad, 2010). As cof1 is a major regulator of actin dynamics (Rust, 2015), we wondered whether the Aβo-induced increase of p-cof1 modifies actin dynamics and impairs synaptic plasticity. For this purpose, we first determined the distribution of p-cof1 and total cof1 in the neuronal CSK-enriched fractions in the presence of Aβo. This fraction, which has a weak solubility in Triton X-100 is named TI. Western blot analysis indicated that Aβo exposure (100 nm, 30 min) increased the proportion of actin in the TI fraction corresponding to F-actin (45% in CT vs 57% in Aβo-treated cells) (Fig. 4A). This process is correlated with recruitment of p-cof1 in the TI fractions (almost twofold increase), whereas no increase is observed for total cof1 (Fig. 4A); it suggests that Aβo exposure promotes an important change in the ratio p-cof1/cof1 leading to increased actin content in the CSK enriched fraction.
Figure 4.
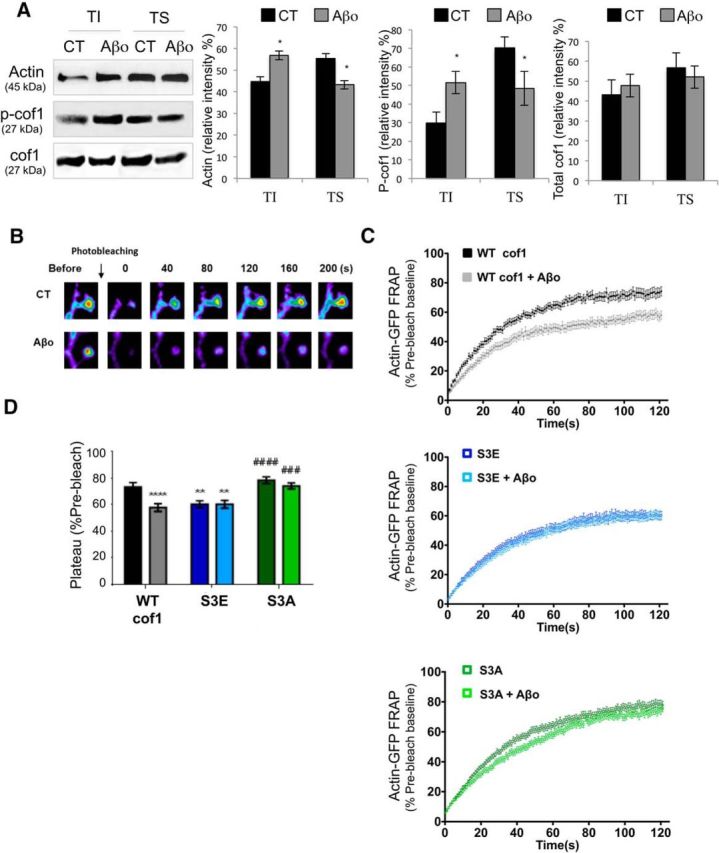
p-cof1 is distributed in the neuronal CSK-enriched fractions under Aβο treatment and is necessary and sufficient for Aβo-induced F-actin stabilization. A, Left, Representative Western blots of actin, p-cof1, and cof1 distribution in TI and TS fractions obtained from primary cortical neurons (DIV 14) in the presence or absence of Aβ oligomers (15 min pretreatment, 100 nm; Aβo). Right panels, Histograms showing that the percentage of molecules associated to either the TI or TS fractions was determined by densitometry analysis. Error bars indicate mean ± SEM. TI and TS actin, Mann–Whitney U = 0.0 (10,26), p = 0.0286; TI and TS p-cof, Mann–Whitney U = 5.000 (26,52), p = 0,0411; TI and TS cof, Mann–Whitney U = 7.000 (33,22), p = 0.3095. B, Representative time-lapse images of actin-GFP in dendritic spines before and after photobleaching showing actin stabilization after exposure to Aβo (images are 2.5 × 2.75 μm, width × height). C, Average FRAP curves for photobleached actin-GFP in dendritic spine heads of neurons expressing endogenous WT cof1 (top, black), inactive phosphomimetic cof1–S3E (S3E, top, blue), nonphosphorylatable cof1–S3A (S3A, bottom, green), treated 15 min with Aβo (100 nm, light), or vehicle (dark). D, Average recovery plateau values calculated using nonlinear regression (two-factor decay model) from individual spine recovery curves summarized in C, following Aβo (light bars) or vehicle treatment (dark bars). Two-way ANOVA revealed a significant interaction for Aβo and cof1 genotype as factors (cof1 genotype, F(2,330) = 20.94, p < 0.0001; Aβo treatment, F(1,330) = 14.21, p = 0.0002; interaction, F(2,330) = 7.675, p = 0.0006), followed by Tukey's post hoc test with correction for multiple comparisons. **p < 0.01, relative to vehicle-treated WT neurons. ***p < 0.001, relative to vehicle-treated WT neurons. ****p < 0.0001, relative to vehicle-treated WT neurons. ###p < 0.001, relative to Aβo-treated WT neurons. ####p < 0.0001, relative to Aβo-treated WT neurons. cof1–S3E also decreased recovery plateau values (i.e., increased the stable pool of F-actin in spines) while allowing no further effect of Aβo, suggesting that cof1 phosphorylation is sufficient for Aβo-induced actin stabilization. Preventing cof1 phosphorylation with the nonphosphorylatable mutant, cof1–S3A, had no effect on actin recovery and precluded Aβo-induced actin stabilization, consistent with cof1 inactivation being required for Aβo-induced F-actin polymerization. Error bars indicate mean ± SEM. n = 52 spines of 18–25 neurons from 6–9 independent cultures. *p < 0.05.
We next used FRAP imaging of neurons expressing actin-GFP to determine whether actin dynamics were altered in dendritic spines as a result of Aβo-induced cof1 phosphorylation. In neurons coexpressing mCherry-tagged cof1, Aβo decreased the FRAP plateau reached by 20% (Fig. 4B–D), consistent with a larger pool of stable F-actin in the dendritic spine. This was further confirmed performing FRAP of actin-GFP in neurons coexpressing cof1 phosphomimetic and nonphosphomimetic mutants, referred to as S3E and S3A, respectively. The phosphomimetic S3E cof1 mutant is constitutively inactive (i.e., this cof1 does not sever actin filaments) and mirrors the effects of Aβo on fluorescence recovery of actin in dendritic spines with no additive effect upon Aβo exposure (Fig. 4C,D). The nonphosphorylatable cof1-S3A mutant had no effect on actin stabilization, and precluded Aβo-induced actin stabilization, suggesting that Aβo-induced stabilization of spine actin required cof1 phosphorylation (Fig. 4C,D).
Inhibiting ROCK phosphorylation of cof1 with fasudil prevents Aβo-induced synaptotoxicity
Because p-cof1 was elevated concurrent with Aβo-impaired plasticity, we asked whether cof1 phosphorylation was necessary for this impairment. We first confirmed that the ROCK inhibitor, fasudil (30 μm), successfully blocked Aβo-induced p-cof1 increases in PSD-enriched synaptosomes (Fig. 5A). Fasudil prevented the Aβo-induced aberrant actin stabilization but did not affect actin stability relative to the vehicle-treated group (Fig. 5B,C). These FRAP results support a central role for p-cof1 mediating Aβo-induced actin stabilization. Thus, preventing cof1 phosphorylation induced by Aβo exposure alleviates the aberrant Aβ-induced actin stabilization. Jasplakinolide at 1 μm, which stabilizes preexisting actin filaments and nucleates actin polymerization, completely ablated spine actin recovery, serving as a positive control for F-actin stabilization (Fig. 5B).
Figure 5.

Aβo-induced phosphorylation of cof1 and F-actin stabilization is prevented by the RhoA kinase inhibitor, fasudil. A, Representative Western blots (left) and accompanying densitometric analysis (right) of cof1 and p-cof1 in the PSD-enriched fractions of synaptosomes from neurons treated with fasudil (Fas) and/or Aβo, confirmed that fasudil blocked ROCK-driven cof1 phosphorylation in this compartment. n = 4 independent neuronal cultures (ANOVA, p-cof1, F(3,16) = 34, p < 0.0001; cof1, F(3,12) = 4.125, p < 0.0317; both followed by Tukey's post hoc test with correction for multiple comparisons). B, Average FRAP curves for photobleached actin-GFP in dendritic spine heads of neurons (WT, black), treated for 15 min with Aβo (100 nm, WT + Aβo, gray), fasudil (30 μm, Fas, red), or fasudil and Aβo (Fas + Aβo, pink). Jasplakinolide (1 μm, brown) was used as a positive control for actin stabilization, evident by no actin-GFP recovery in spine heads. Symbols indicate mean. Error bars indicate the SEM. n = 16–52 spines from 5–9 independent cultures per group. C, Average recovery plateau values summarized in B, following Aβo (light bars) or vehicle treatment (dark bars) of neurons treated with vehicle (WT, black) and with fasudil (red) alone or concurrent with Aβo. Two-way ANOVA for recovery plateau values revealed a significant interaction for Aβo and fasudil as factors (Aβo treatment, F(1,186) = 19.06, p < 0.0001; fasudil treatment, F(1,186) = 11.28, p = 0.0010; interaction, F(1,186) = 8.302, p = 0.0044; followed by Tukey's post hoc test with correction for multiple comparisons). Error bars indicate mean ± SEM. n = 49–52 spines from 6–9 independent cultures. *p < 0.05. ****p < 0.0001.
We used the cLTP model to determine whether fasudil pretreatment (30 μm; 15 min before cLTP) prevents Aβo-induced impairment of synaptic plasticity. We observed that pretreatment with fasudil prevented Aβo blockade of cLTP-induced GluA1 insertion and Aβo induced increment of p-cof1 (Fig. 6A). Next, we evaluated whether ROCK-driven actin stabilization contributed to Aβo-induced synaptic impairments. To test this, we monitored calcium levels in neurons expressing the genetically encoded calcium indicator, GCaMP6fast, as they responded to 15 s pulses of B4AP before and after 15 min exposure to Aβo, or Aβo with fasudil. Aβo exposure impaired synaptically driven calcium responses of individual neurons relative to their own initial response to the first B4AP pulses (Fig. 6B–D), and the impairment was ameliorated by cotreatment with fasudil (Fig. 6B–D), whereas fasudil alone impaired the synaptic calcium response. To further validate the causal link between p-cof1 and Aβo-induced disruption of synaptic responses, we cotransfected neurons with the phosphomimetic cof1-S3E mutant or nonphosphorylatable cof1-S3A mutant and GcAMP6f in neurons subjected to B4AP stimulation. In these experimental conditions, we observed that expression of both cof1 mutants abolished the Aβo effect on B4AP-driven synaptic responses (Fig. 7A–C), validating the importance of cof1 phosphorylation to Aβo-induced disruption of synaptic responses.
Figure 6.
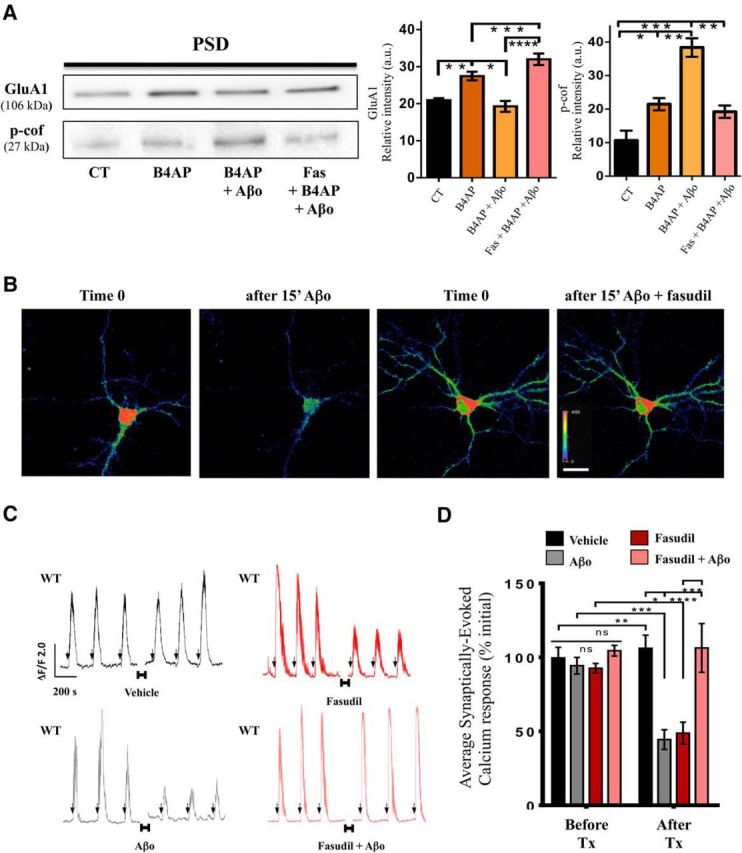
Fasudil inhibition of RhoA kinase prevents Aβo-induced phosphorylation of cof1 and Aβo-impaired cLTP. A, Representative Western blot images (left) and quantification (right) of GluA1 and p-cof in the PSD-enriched fraction of synaptosomes isolated from neurons exposed to vehicle (CT), cLTP (B4AP, 15 min, brown) alone, or cLTP after pretreatment (15 min) with Aβo (orange) or fasudil + Aβo (pink). Fasudil prevented the Aβo-induced GluA1 trafficking impairment and restored the p-cof values as determined by one-way ANOVA (GluA1, F(3,20) = 36.26, p < 0.0001; p-cof1, F(3,9) = 20.33, p = 0.0002) followed by Tukey's post hoc test with correction for multiple comparisons. n = 3–11 cultures per group. B, Representative images of primary cortical neurons (DIV 14) transfected with GCaMP6fast (an indicator of synaptically driven calcium responses) activated by 15 s pulses of B4AP before and after 15 min incubation with Aβo or fasudil + Aβo. C, Representative ΔF/F traces of GCaMP6fast fluorescence as an indicator of synaptically driven calcium responses induced by 15 s pulses of B4AP (arrows) in neurons expressing endogenous WT cof1 (WT, black), before and after 15 min incubation (indicated by the horizontal bars and break in the traces) with vehicle (dark) or Aβo (light), or coincubated with or without fasudil (red). D, Quantification of the B4AP-evoked synaptic calcium responses from C before (left bars, dark), and after (right bars, light) 15 min treatment (Tx) with vehicle (black), Aβo (gray), fasudil (red), or fasudil + Aβo (pink). Fasudil blocks the Aβo-induced synaptic impairment, as revealed by two-way ANOVA using time and treatment as factors (time, F(1,30) = 16.84, p = 0.0003; treatment, F(3,30) = 8.297, p = 0.0004; interaction, F(3,30) = 6.742, p = 0.0013; followed by Tukey's post hoc test with correction for multiple comparisons). n = 9–11 neurons, 5–7 neuronal cultures per group. *p < 0.05. **p < 0.01. ***p < 0.001. ****p < 0.0001.
Figure 7.

cof1 phospho-mutants prevent impairment of synaptic calcium responses induced by Aβo. A, Representative images of primary cortical neurons (DIV 14) transfected with GCaMP6fast (an indicator of synaptically driven calcium responses) and inactive cof1 phosphomimetic (S3E) or the nonphosphorylatable cof1 (S3A) during 15 s pulses of B4AP before and after 15 min incubation with Aβo. B, Representative ΔF/F traces of GCaMP6fast fluorescence as an indicator of synaptically driven calcium responses induced by 15 s pulses of B4AP (arrows) in neurons expressing the inactive cof1 phosphomimetic (top, S3E, blue), or the nonphosphorylatable cof1 (bottom, S3A, green), before and after 15 min incubation (indicated by the horizontal bars and break in the traces) with Aβo (100 nm). C, Quantification of the B4AP-evoked synaptic calcium responses from B, before (left bars, dark) and after (right bars, light) 15 min exposure to Aβo in neurons expressing the inactive cof1 phosphomimetic (S3E, blue) or the nonphosphorylatable cof1 (S3A, green). Both cof1 mutants block the Aβo-induced synaptic impairment (two-way ANOVA: time, F(1,17) = 0.0006, p = 0.983; treatment, F(1,17) = 3.257, p = 0.0889; interaction, F(1,17) = 0.8935, p = 0.142). Error bars indicate mean ± SEM. n = 9 or 10 neurons, 6 or 7 neuronal cultures per group.
Another cardinal feature of Aβo-induced synaptotoxicity is the decrease of excitatory synapse density. To address this issue, we further investigated whether a treatment with fasudil altered the effect of Aβo exposure on synaptic density. For this purpose, we monitored spine density in neurons transfected with LifeAct-GFP, a 16 amino acid peptide that binds selectively to F-actin. This approach allowed us to visualize dendritic spines and quantify spine density in neurons before and 24 h after Aβo exposure at 100 nm. We observed that Aβo reduced dendritic spine density by ∼20%. Treatment with fasudil (30 μm) successfully blocked Aβo-induced reduction in spine density (Fig. 8). These results confirm the pivotal role of p-cof1 in the synaptic loss induced by Aβo exposure.
Figure 8.

Fasudil treatment reverses Aβo-induced synaptotoxicity. A, Representative images of DIV 14–15 neurons expressing actin-GFP before (t 0) and after incubation 24 h with Aβo or fasudil + Aβo. Bottom panels, Magnified detail of the selected area in top panels, respectively. Scale bars: Top, 30 μm; Bottom, 10 μm. B, Relative quantification of spine density in neurons expressing actin-GFP before and after incubation 24 h with Aβo or fasudil + Aβo. A paired t test showed that 24 h exposition to Aβo caused decrease in the spine density that was blocked by fasudil (t values t(10) = 8.931, p < 0.0001). Error bars indicate mean ± SEM. n = 11 neurons, 3 cultures per group. *p < 0.05.
Discussion
In this study, we sought to decipher whether the actin CSK and its major regulatory protein at the synapse, cof1, are implicated in the synaptic impairment and synaptotoxicity induced by Aβo. We demonstrated that p-cof1 was elevated in the PSD-enriched fraction of cortical tissues from APP/PS1 mice, as well as frontal cortex from human AD cases. This increase in p-cof1 is mostly detected in the postsynaptic compartment of excitatory synapses. When we monitored mouse primary cultured neurons for cofilin-mediated actin stabilization and synaptic calcium impairment in response to Aβo exposure, we found that inactivation through phosphorylation of cof1 is a key component of Aβo-induced functional and structural synaptic impairment, leading to synaptic loss after 24 h. Furthermore, we also asked whether a well-tolerated inhibitor of the upstream ROCK could ameliorate the Aβo-induced deficits. ROCK inhibition prevents the Aβo-induced functional synaptic deficits and synaptic loss, supporting further development of the clinically well-tolerated compound, fasudil, and other more selective ROCK inhibitors.
Aβ-driven, tau-mediated, spine dysfunction and synapse loss are thought to underlie the cognitive impairment of AD dementia (Spires and Hyman, 2004; Cleary et al., 2005; Shankar et al., 2008). Spine loss is the best correlate of AD progression; however, acute disruption of synaptic plasticity is not accompanied by loss of spines (DeKosky and Scheff, 1990; Terry et al., 1991; Sze et al., 1997). Further, synaptic structural reorganization may provide cognitive resilience against dementia in elderly individuals with AD pathology (Boros et al., 2017). The actin CSK is central to spine dynamics, both structurally and functionally (Rust, 2015). Spine actin directly regulates receptor trafficking in/out of the postsynaptic density and perisynaptic membrane, thereby influencing the postsynaptic response to a given synapse (Malinow and Malenka, 2002; Gu et al., 2010). Changes in actin dynamics are also required for changes in synaptic strength (Ouyang et al., 2005). Spine actin directly regulates spine morphology and structural changes, facilitating the formation of new spines, growth and enlargement of strengthened spines, reduction of depressed spines, or collapse of those tagged for elimination (Hotulainen and Hoogenraad, 2010). Spine actin organization is tightly regulated, both spatially and temporally, by several signaling pathways that largely converge on cof1 as the key effector (Hotulainen and Hoogenraad, 2010; Rust et al., 2010; Rust, 2015). We observed that the PSD-enriched fraction of human AD cortex and that of cortex from the APP/PS1 mouse model of AD exhibit increased p-cof1 levels (Fig 1). This increase was also observed by immunohistochemistry in the structure that corresponds to PSD-positive fraction: the dendritic spine heads of the APP/PS1 mice (Fig 2). These features were also present in primary cortical cultures exposed to Aβo (Fig. 3). This finding fits well with previous reports of elevated p-cof1 in AD and APP/PS1 AD model mice (Heredia et al., 2006; Barone et al., 2014; Fowler et al., 2014), elevated LIMK in AD and following Aβo exposure (Heredia et al., 2006; Barone et al., 2014), and elevated ROCKs in AD model mice and AD (Petratos et al., 2008; Herskowitz et al., 2013; Henderson et al., 2016). Moreover, elevated ROCK activity has been tied directly to dendritic spine stability and memory impairment in mice (Bobo-Jiménez et al., 2017), and fasudil has protected against Aβ-induced cognitive impairment in rats (Sellers et al., 2018), strengthening the relevance of the present study to these features of AD and neurodegeneration.
In contrast, other researchers have reported that p-cof is decreased in APP/PS1 mice at 7 months of age (Davis et al., 2011; Woo et al., 2015). These apparently contradictory results came from whole-tissue lysates, whereas we have analyzed cof1 and p-cof1 found within the PSD-enriched fraction of synaptosomes and in the CSK-enriched fraction (TI fraction). It is not surprising that the postsynaptic compartment (known to be targeted very rapidly by Aβo) would have earlier and differential results. Further, it seems that they have reciprocal influence. Elevated and overactive ROCKs, in response to APP and/or Aβo, lead to further production of Aβ (Herskowitz et al., 2013; Henderson et al., 2016). ROCK1 and ROCK2 knockdown with siRNA or genetic ablation decreased Aβ production, whereas overexpression of either ROCK increased Aβ production (Herskowitz et al., 2013; Henderson et al., 2016). The role and influence of cof1 are complex, and it will be important to resolve differential timing of cof1 in/activation and rod formation with actin at the spine and whole-cell level in in vitro models, mouse models of AD, and human AD pathological progression.
Synaptic plasticity is associated with changes to AMPAR numbers at the postsynaptic density directly underlying changes to synaptic responses (Malinow and Malenka, 2002). LTP is associated with increases in AMPARs at the PSD, whereas LTD is associated with decreases in AMPARs at the PSD (Kopec et al., 2006; Anggono and Huganir, 2012). Our model of cLTP induced a robust increase in the AMPAR subunit, GluA1, detected in the PSD-enriched fraction of synaptosomes, consistent with previous reports using this and other models of cLTP (Fig. 4) (Kopec et al., 2006; Blanpied et al., 2008; Frandemiche et al., 2014). Also consistent with our previous report, Aβo blocked the cLTP-induced insertion of GluA1 at the PSD (Frandemiche et al., 2014) and reduced cLTP activation of Erk, suggesting impairment of the synaptic response.
Congruent with increased p-cof1, we also found that Aβo caused an increase in stabilized F-actin, detected by the increased amount of actin in the TI fraction (i.e., the CSK fraction) (Fig. 4A) and the decrease in globular actin in the TS fraction. Subsequently, actin-GFP FRAP recovery plateaus following Aβo exposure are reduced (Fig. 5). Together, these results reveal that Aβo exposure promotes a shift of globular actin toward fibrilization leading to abnormal dynamics of the synaptic actin CSK. The Aβo-altered actin dynamic was mimicked in neurons expressing the phosphomimetic cof1 mutant, cof1-S3E (Fig. 4). The cof1-S3E mutant also blocked any further increase in stable F-actin with Aβo exposure, suggesting p-cof1 as the common and sufficient effector. Importantly, the nonphosphorylatable cof1 mutant, cof1-S3A, had no effect on synaptic actin dynamics, suggesting that maintaining stable spine structure does not require cof1 inactivation. Changes to spine structure and function, however, require a temporal sequence of cof1 dephosphorylation/phosphorylation (Gu et al., 2010; Calabrese et al., 2014; Noguchi et al., 2016). Expression of cofilin-S3A also precluded Aβo-increased stability of F-actin, confirming that inactivation of cof1 is required. Finally, fasudil reversed the Aβo-induced stabilization of actin and subsequent synaptic loss observed after a 24 h exposure. This finding strengthens the conclusion that phosphorylation of cof1 mediates Aβo-induced actin stabilization and the subsequent synaptic loss.
Monitoring somatic calcium responses driven by brief bouts of synaptic stimulations brought on by Bic4AP exposure revealed that Aβo acutely disrupts the postsynaptic response. cLTP following Aβo exposure did not show an activity-driven increase in p-cof1 relative to cLTP alone (Fig. 3), underscoring the importance of tight temporal regulation of cof1 and the actin CSK. Indeed, Aβo blocked both GluA1 insertion at the PSD and synaptic calcium responses. Fasudil treatment reversed both Aβo-induced synaptic dysfunctions. These results suggest that, by restoring a pool of active cofilin-1, fasudil treatment promoted a recovery of the synaptic plasticity potential in the presence of Aβo and abolished the synaptic loss observed in neurons exposed to Aβo for 24 h. The fact that fasudil inhibition of the ROCK pathway alone reduces the postsynaptic calcium responses is in accordance with a previous report (González-Forero et al., 2012). It may reflect the consequences of the alteration of the physiological balance between of active cof1/p-cof1 on excitatory neurotransmission and synaptic plasticity. Together, these results underscore the importance of balanced cytoskeletal regulation in the postsynaptic compartment and unveil the therapeutic potential of targeting rho kinase in AD.
Collectively, our results support further study of actin CSK dynamics, and drugs targeting ROCK regulation thereof, as a therapeutic target for neuronal dysfunction in AD. Fasudil, a clinically approved drug, is a timely candidate for this purpose, although studies of prolonged fasudil treatment have not been performed in AD. According to our results, the development and study of more selective ROCK inhibitors are warranted.
Footnotes
This work was supported by the Fondation Neurodis, Fondation Bettencourt, Institut National de la Santé et de la Recherche Médicale, and Agence Nationale de la Recherche (ANR MALAAD program). Human postmortem tissue was obtained from the Banco de Tejidos, suppress fundation CIEN, Madrid, Spain. All experiments were performed in accordance with local animal welfare committee (Comité Local Grenoble Institute Neurosciences, C2EA-04) and European Union guidelines (Directive 2010/63/EU). Every precaution was taken to minimize stress and the number of animals used in each series of experiments.
The authors declare no competing financial interests.
References
- Anggono V, Huganir RL (2012) Regulation of AMPA receptor trafficking and synaptic plasticity. Curr Opin Neurobiol 22:461–469. 10.1016/j.conb.2011.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's Association (2017) Alzheimer's disease facts and figures. Alzheimers Dement 13:325–373. 10.1016/j.jalz.2017.02.001 [DOI] [Google Scholar]
- Bamburg JR, Bernstein BW (2016) Actin dynamics and cofilin-actin rods in Alzheimer disease. Cytoskeleton 73:477–497. 10.1002/cm.21282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone E, Mosser S, Fraering PC (2014) Inactivation of brain cofilin-1 by age, Alzheimer's disease and gamma-secretase. Biochim Biophys Acta 1842:2500–2509. 10.1016/j.bbadis.2014.10.004 [DOI] [PubMed] [Google Scholar]
- Behnisch T, Yuanxiang P, Bethge P, Parvez S, Chen Y, Yu J, Karpova A, Frey JU, Mikhaylova M, Kreutz MR (2011) Nuclear translocation of jacob in hippocampal neurons after stimuli inducing long-term potentiation but not long-term depression. PLoS One 6:e17276. 10.1371/journal.pone.0017276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied TA, Kerr JM, Ehlers MD (2008) Structural plasticity with preserved topology in the postsynaptic protein network. Proc Natl Acad Sci U S A 105:12587–12592. 10.1073/pnas.0711669105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobo-Jiménez V, Delgado-Esteban M, Angibaud J, Sánchez-Morán I, de la Fuente A, Yajeya J, Nägerl UV, Castillo J, Bolaños JP, Almeida A (2017) APC/CCdh1-Rock2 pathway controls dendritic integrity and memory. Proc Natl Acad Sci U S A 114:4513–4518. 10.1073/pnas.1616024114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boros BD, Greathouse KM, Gentry EG, Curtis KA, Birchall EL, Gearing M, Herskowitz JH (2017) Dendritic spines provide cognitive resilience against Alzheimer's disease. Ann Neurol 82: 602–614. 10.1002/ana.25049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese B, Saffin JM, Halpain S (2014) Activity-dependent dendritic spine shrinkage and growth involve downregulation of cofilin via distinct mechanisms. PLoS One 9:e94787. 10.1371/journal.pone.0094787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH (2005) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 8:79–84. 10.1038/nn1372 [DOI] [PubMed] [Google Scholar]
- Davis RC, Marsden IT, Maloney MT, Minamide LS, Podlisny M, Selkoe DJ, Bamburg JR (2011) Amyloid beta dimers/trimers potently induce cofilin-actin rods that are inhibited by maintaining cofilin-phosphorylation. Mol Neurodegener 6:10. 10.1186/1750-1326-6-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol 27:457–464. 10.1002/ana.410270502 [DOI] [PubMed] [Google Scholar]
- Fowler SW, Chiang AC, Savjani RR, Larson ME, Sherman MA, Schuler DR, Cirrito JR, Lesné SE, Jankowsky JL (2014) Genetic modulation of soluble abeta rescues cognitive and synaptic impairment in a mouse model of Alzheimer's disease. J Neurosci 34:7871–7885. 10.1523/JNEUROSCI.0572-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I, Lanté F, Buisson A (2014) Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci 34:6084–6097. 10.1523/JNEUROSCI.4261-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Forero D, Montero F, García-Morales V, Domínguez G, Gómez-Pérez L, García-Verdugo JM, Moreno-López B (2012) Endogenous rho-kinase signaling maintains synaptic strength by stabilizing the size of the readily releasable pool of synaptic vesicles. J Neurosci 32:68–84. 10.1523/JNEUROSCI.3215-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ (2010) ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci 13:1208–1215. 10.1038/nn.2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F, Zhuang TT, Chen JJ, Zhu XL, Cai YF, Lu YP (2017) Novel derivative of Paeonol, Paeononlsilatie sodium, alleviates behavioral damage and hippocampal dendritic injury in Alzheimer's disease concurrent with cofilin1/phosphorylated-cofilin1 and RAC1/CDC42 alterations in rats. PLoS One 12:e0185102. 10.1371/journal.pone.0185102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BW, Gentry EG, Rush T, Troncoso JC, Thambisetty M, Montine TJ, Herskowitz JH (2016) Rho-associated protein kinase 1 (ROCK1) is increased in Alzheimer's disease and ROCK1 depletion reduces amyloid-beta levels in brain. J Neurochem 138:525–531. 10.1111/jnc.13688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia L, Helguera P, de Olmos S, Kedikian G, Solá Vigo F, LaFerla F, Staufenbiel M, de Olmos J, Busciglio J, Cáceres A, Lorenzo A (2006) Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer's disease. J Neurosci 26:6533–6542. 10.1523/JNEUROSCI.5567-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz JH, Feng Y, Mattheyses AL, Hales CM, Higginbotham LA, Duong DM, Montine TJ, Troncoso JC, Thambisetty M, Seyfried NT, Levey AI, Lah JJ (2013) Pharmacologic inhibition of ROCK2 suppresses amyloid-beta production in an Alzheimer's disease mouse model. J Neurosci 33:19086–19098. 10.1523/JNEUROSCI.2508-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck L, Näthke IS, Papkoff J, Nelson WJ (1994) Dynamics of cadherin/catenin complex formation: novel protein interactions and pathways of complex assembly. J Cell Biol 125:1327–1340. 10.1083/jcb.125.6.1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Hoogenraad CC (2010) Actin in dendritic spines: connecting dynamics to function. J Cell Biol 189:619–629. 10.1083/jcb.201003008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervern M, Angeli A, Nicole O, Léveillé F, Parent B, Villette V, Buisson A, Dutar P (2012) Selective impairment of some forms of synaptic plasticity by oligomeric amyloid-beta peptide in the mouse hippocampus: implication of extrasynaptic NMDA receptors. J Alzheimers Dis 32:183–196. 10.3233/JAD-2012-120394 [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R (2006) Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci 26:2000–2009. 10.1523/JNEUROSCI.3918-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL (2007) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci 27:796–807. 10.1523/JNEUROSCI.3501-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillé F, El Gaamouch F, Gouix E, Lecocq M, Lobner D, Nicole O, Buisson A (2008) Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J 22:4258–4271. 10.1096/fj.08-107268 [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126. 10.1146/annurev.neuro.25.112701.142758 [DOI] [PubMed] [Google Scholar]
- Mendoza-Naranjo A, Contreras-Vallejos E, Henriquez DR, Otth C, Bamburg JR, Maccioni RB, Gonzalez-Billault C (2012) Fibrillar amyloid-beta1–42 modifies actin organization affecting the cofilin phosphorylation state: a role for Rac1/cdc42 effector proteins and the slingshot phosphatase. J Alzheimers Dis 29:63–77. 10.3233/JAD-2012-101575 [DOI] [PubMed] [Google Scholar]
- Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T (2002) Control of actin reorganization by slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108:233–246. 10.1016/S0092-8674(01)00638-9 [DOI] [PubMed] [Google Scholar]
- Noguchi J, Hayama T, Watanabe S, Ucar H, Yagishita S, Takahashi N, Kasai H (2016) State-dependent diffusion of actin-depolymerizing factor/cofilin underlies the enlargement and shrinkage of dendritic spines. Sci Rep 6:32897. 10.1038/srep32897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM (2004) Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43:321–332. 10.1016/j.neuron.2004.07.003 [DOI] [PubMed] [Google Scholar]
- Ouyang Y, Wong M, Capani F, Rensing N, Lee CS, Liu Q, Neusch C, Martone ME, Wu JY, Yamada K, Ellisman MH, Choi DW (2005) Transient decrease in F-actin may be necessary for translocation of proteins into dendritic spines. Eur J Neurosci 22:2995–3005. 10.1111/j.1460-9568.2005.04521.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petratos S, Li QX, George AJ, Hou X, Kerr ML, Unabia SE, Hatzinisiriou I, Maksel D, Aguilar MI, Small DH (2008) The beta-amyloid protein of Alzheimer's disease increases neuronal CRMP-2 phosphorylation by a rho-GTP mechanism. Brain 131:90–108. 10.1093/brain/awm260 [DOI] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jäggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Hölscher C, Mathews PM, Jucker M (2006) Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7:940–946. 10.1038/sj.embor.7400784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A 99:6364–6369. 10.1073/pnas.092136199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MB. (2015) ADF/cofilin: a crucial regulator of synapse physiology and behavior. Cell Mol Life Sci 72:3521–3529. 10.1007/s00018-015-1941-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MB, Gurniak CB, Renner M, Vara H, Morando L, Görlich A, Sassoè-Pognetto M, Banchaabouchi MA, Giustetto M, Triller A, Choquet D, Witke W (2010) Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J 29:1889–1902. 10.1038/emboj.2010.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers KJ, Elliott C, Jackson J, Ghosh A, Ribe E, Rojo AI, Jarosz-Griffiths HH, Watson IA, Xia W, Semenov M, Morin P, Hooper NM, Porter R, Preston J, Al-Shawi R, Baillie G, Lovestone S, Cuadrado A, Harte M, Simons P, et al. (2018) Amyloid β synaptotoxicity is wnt-PCP dependent and blocked by fasudil. Alzheimers Dement 14:306–317. 10.1016/j.jalz.2017.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14:837–842. 10.1038/nm1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Hyman BT (2004) Neuronal structure is altered by amyloid plaques. Rev Neurosci 15:267–278. 10.1515/REVNEURO.2004.15.4.267 [DOI] [PubMed] [Google Scholar]
- Stine WB, Dahlgren KN, Krafft GA, LaDu MJ (2003) In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem 278:11612–11622. 10.1074/jbc.m210207200 [DOI] [PubMed] [Google Scholar]
- Swanger SA, Mattheyses AL, Gentry EG, Herskowitz JH (2015) ROCK1 and ROCK2 inhibition alters dendritic spine morphology in hippocampal neurons. Cell Logist 5:e1133266. 10.1080/21592799.2015.1133266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ (1997) Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 56:933–944. 10.1097/00005072-199708000-00011 [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120:545–555. 10.1016/j.cell.2005.02.008 [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580. 10.1002/ana.410300410 [DOI] [PubMed] [Google Scholar]
- Woo JA, Zhao X, Khan H, Penn C, Wang X, Joly-Amado A, Weeber E, Morgan D, Kang DE (2015) Slingshot-cofilin activation mediates mitochondrial and synaptic dysfunction via abeta ligation to beta1-integrin conformers. Cell Death Differ 22:921–934. 10.1038/cdd.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K (1998) Cofilin phosphorylation by LIM-kinase 1 and its role in rac-mediated actin reorganization. Nature 393:809–812. 10.1038/31735 [DOI] [PubMed] [Google Scholar]
- Zafar S, Younas N, Sheikh N, Tahir W, Shafiq M, Schmitz M, Ferrer I, Andréoletti O, Zerr I (2018) Cytoskeleton-associated risk modifiers involved in early and rapid progression of sporadic Creutzfeldt-Jakob disease. Mol Neurobiol 55:4009–4029. 10.1007/s12035-017-0589-0 [DOI] [PubMed] [Google Scholar]
- Zhou Z, Meng Y, Asrar S, Todorovski Z, Jia Z (2009) A critical role of rho-kinase ROCK2 in the regulation of spine and synaptic function. Neuropharmacology 56:81–89. 10.1016/j.neuropharm.2008.07.031 [DOI] [PubMed] [Google Scholar]