

Abstract
Microcirculatory damage is a common complication for those with vascular risk factors, such as diabetes. To resolve vascular insults, the brain's immune cells (microglia) must rapidly envelop the site of injury. Currently, it is unknown whether Type 1 diabetes, a condition associated with chronic immune system dysfunction, alters microglial responses to damage and what mechanisms are responsible. Using in vivo two-photon microscopy in adult male mice, we show that microglial envelopment of laser-induced cerebral microbleeds is diminished in a hyperglycemic mouse model of Type 1 diabetes, which could not be fully rescued with chronic insulin treatment. Microglia were important for vessel repair because reduced microglial accumulation in diabetic mice or near-complete depletion in healthy controls was associated with greater secondary leakage of the damaged vessel. Broadly suppressing inflammation with dexamethasone in diabetic mice but not healthy controls, significantly enhanced microglial responses to microbleeds and attenuated secondary vessel leakage. These enhancements were associated with changes in IFN-γ signaling because dexamethasone suppressed abnormally high levels of IFN-γ protein levels in brain and blood serum of diabetic mice. Further, blocking IFN-γ in diabetic mice with neutralizing antibodies restored normal microglial chemotaxic responses and purinoceptor P2ry12 gene expression, as well as mitigated secondary leakage. These results suggest that abnormal IFN-γ signaling disrupts microglial function in the diabetic brain, and that immunotherapies targeting IFN-γ can stimulate microglial repair of damaged vessels.
SIGNIFICANCE STATEMENT Although Type 1 diabetes is an established risk factor for vascular complications, such as microbleeds, and is known to hinder wound healing in the body, no study has examined how diabetes impacts the brain's innate immune reparative response (involving cells called microglia) to vascular injury. Here we show that microglial responses to brain microbleeds were diminished in diabetic animals, which also exacerbated secondary leakage from damaged vessels. These impairments were related to abnormally high levels of the proinflammatory cytokine IFN-γ because reducing IFN-γ with immunosuppressant drugs or blocking antibodies helped restore normal microglial responses and repair of damaged vessels. These data highlight the use of IFN-γ modulating therapeutics to enhance vascular repair in at-risk populations.
Keywords: diabetes, inflammation, interferon, microbleeds, microglia, repair
Introduction
Diabetes is quickly emerging as a global epidemic with ∼8.5% of the worldwide population living with some form of the disease (World Health Organization, 2016). The defining feature of both Type 1 and 2 diabetes is chronic dysregulation of blood glucose homeostasis. Elevated blood glucose is particularly damaging in that it leads to the generation of reactive oxygen species and glycation end products, which in turn disrupts the vascular endothelium and stimulates inflammatory processes (Vinik and Flemmer, 2002; Zherebitskaya et al., 2009; Volpe et al., 2018). Not surprisingly, diabetes, especially if not well controlled, leads to a host of vascular complications, such as cerebral microbleeds (CMBs). CMBs are prevalent in patients with vascular risk factors but have also been observed in the normal aging population (Cullen et al., 2005; Cordonnier et al., 2007; Kim and Lee, 2013; Shams et al., 2015). The presence of CMB in the brain has been linked with cognitive decline and increased risk of dementia (Seo et al., 2007; Hilal et al., 2014; Valenti et al., 2016). In contrast to patients with Type 2 diabetes where the association with CMBs has yielded mixed results (Moran et al., 2013; Brundel et al., 2014), Type 1 diabetic patients demonstrate greater prevalence of CMBs than healthy control subjects (Woerdeman et al., 2014).
Not only are diabetics at increased risk for vascular complications, it is also plausible that their brain's innate response to injury may also be compromised. For example, animal models of Type 1 and 2 diabetes show prolonged disruption of the blood–brain barrier (BBB) after ischemic and hemorrhagic stroke (Li et al., 2010; Ye et al., 2011; Reeson et al., 2015). Furthermore, these animals show increased rates of hemorrhagic transformation following ischemia (Won et al., 2011; Li et al., 2013). To properly repair damaged tissue, microglia are believed to play an important role (Glezer et al., 2007; Kierdorf and Prinz, 2013; Schwartz et al., 2013). Imaging studies have shown that ablating or blocking microglial responses is associated with greater tissue damage and BBB disruption following stroke or microvascular lesions (Hines et al., 2009; Lou et al., 2016; Szalay et al., 2016). Failure to properly repair and seal off damaged vessels exacerbates the disruptive impact that permeable blood vessels have on local synaptic structure and function (Zhang and Murphy, 2007; Cianchetti et al., 2013; Reeson et al., 2015; Taylor et al., 2015). Previous postmortem studies have suggested that microglial responses to major vascular insults are altered in diabetic animals and patients (Kondo et al., 2001; Kumari et al., 2007; Li et al., 2011), but the spatiotemporal dynamics of microglial responses is unknown. Whether the diabetic condition alters dynamic aspects of microglia responses to microvascular bleeds and what molecular mechanisms are responsible have not been explored.
Because it is well recognized that diabetes is associated with abnormal immune system function and impairs wound healing in the periphery (Kumari et al., 2007; Ebenezer et al., 2011; Cheng et al., 2013), we tested the hypothesis that microglial response dynamics to microvessel rupture would be compromised in an animal model of Type 1 diabetes. Our data reveal a significant impairment in microglial envelopment of damaged blood vessels which correlated with greater secondary vascular leakage. While these impairments could not be fully prevented with insulin treating diabetic mice with a broad-spectrum immunosuppressant, or more specifically with blocking antibodies to the proinflammatory cytokine IFN-γ, was effective in restoring normal microglial responses and repair of damaged vessels.
Materials and Methods
Animals.
Two- to 5-month-old male mice on a C57BL/6J background were used in this study. To image microglia in vivo, we examined two different mouse lines. First, we used mice that expressed enhanced green fluorescent protein (eGFP) in microglia due to placement of the eGFP reporter gene into the Cx3cr1 gene locus (Jung et al., 2000). As previously described, the eGFP reporter gene replaces the Cx3cr1 gene; therefore, mice heterozygous or homozygous for eGFP are considered Cx3cr1+/− or Cx3cr1−/−, respectively. To visualize microglia in mice that possess both Cx3cr1 alleles (Cx3cr1+/+), as well as validate the use of eGFP Cx3cr1+/− mice for our studies, we crossed a Cx3cr1 Cre-ERT2 driver line [Cx3cr1tm2.1(cre/ERT2)Litt/WGanJ)] with a Cre-dependent “Ai3-eYFP” reporter mouse [Gt(ROSA)26Sortm-CAG-eYFP] (Madisen et al., 2010). Offspring of this cross (Cx3cr1cre/+ × Ai3fl/+) possess both Cx3cr1 alleles (Cx3cr1+/+), express eYFP in microglia, and were used to compare microglial responses with Cx3cr1+/− or Cx3cr1−/− mice. Mice were group-housed in a 12 h light/dark cycle in ventilated racks in a humidity (RH 40%–55%) and temperature-controlled room (21°C–23°C) and given ad libitum access to food and water. All experiments were conducted in accordance with the guidelines set by the Canadian Council on Animal Care and approved by the local university Animal Care Committee. Reporting of this work complies with ARRIVE guidelines.
Induction and treatment of diabetic mice.
Mice were randomly assigned to 1 of the 3 experimental groups. Type 1 diabetes was induced in 2-month-old mice by intraperitoneal injections of streptozotocin (STZ) at a dose of 75 mg/kg dissolved in sodium citrate buffer, pH 4.5, once daily for 2 consecutive days following 5 h of food deprivation (Yamamoto et al., 1981; Lenzen, 2008). Nondiabetic mice consisted of those that received an injection of vehicle. For the first day after STZ injection, mice were supplied with 5% glucose in H2O to prevent acute hypoglycemia (Lenzen, 2008). To control blood glucose levels in a subgroup of diabetic mice (“diabetic + insulin” group), slow-release insulin pellets (0.1 U/24 h/implant, Lin-Bit, LinShin, Canada, Inc) were inserted subcutaneously 1 week following STZ injections after hyperglycemia was confirmed. If blood glucose levels exceeded 15 mmol/L, mice were lightly anesthetized with 1.3% isoflurane and another insulin pellet was inserted subcutaneously. Body weights and blood glucose levels were checked on a weekly basis using an Aviva Accu-Chek blood glucose meter by a drop of blood withdrawn from the tail vein. For our treatment intervention experiments, a subset of nondiabetic and hyperglycemic diabetic mice received 2 mg/kg dexamethasone (DEX; s.c.) or saline twice per day (12 h apart) for 5 d before imaging experiments. As previously described, in vivo neutralization of IFN-γ was achieved by two intravenous injections of IFN-γ antibody (clone XMG1.2, Bio X Cell; 300 μg dissolved in saline) 2–3 d apart during the 5 d period before inducing CMB. Diabetic controls received two intravenous injections of saline or mouse IgG1 isotype control (MOPC-21, Bio X Cell; 300 μg dissolved in saline).
Two-photon imaging and induction of microbleeds.
For imaging, two surgical preparations were used in this study: (1) an acute craniectomy-based preparation in which the skull was removed and animals were imaged immediately thereafter; or (2) a chronic cranial window preparation involving a craniectomy and the installation of a glass window 4–6 weeks before in vivo imaging. Our analysis indicated that there were no significant differences in the number of microglial cells or the area they occupied, turnover rate, and accumulation of microglial processes around the CMB between the two surgical preparations (statistics shown in Table 1). Because there were no differences in any of these parameters, data collected from both imaging preparations were pooled together. For both surgical preparations, mice were anesthetized with isoflurane (2% for induction and 1%–1.3% for maintenance) mixed with medical air (80% nitrogen, 20% oxygen) at a flow rate of 0.7 L/min. Body temperature was maintained at 37°C with a rectal thermo-probe and temperature feedback regulator. For craniectomy surgery, a circular (3–5 mm diameter) region of the skull over the somatosensory cortex was thinned with a high-speed dental drill and the skull was carefully removed with forceps. Gel foam soaked in HEPES-buffered ACSF was used to keep the brain moist throughout the surgical procedure. For acute imaging experiments, the brain was covered with 1.5% low-melt agarose dissolved in a HEPES-buffered ACSF and sealed with a glass coverslip (no. 1 thickness). For mice with a chronically implanted cranial window, the glass coverslip was placed over the craniectomy and fixed in place with cyanoacrylate glue and dental cement. The surrounding skin was glued to the edges of the cranial window, and the mice were allowed to recover under a heat lamp before being returned to their home cages.
Table 1.
Analysis of microglial process accumulation based on repeated-measures ANOVAs conducted at each distance from the CMB with surgical preparation (acute and chronic) as a factor over time (0–60 min)
| Distance | Surgical preparation | Time | Interaction |
|---|---|---|---|
| 5–10 μm | F(1,12) = 1.49, p = 0.25 | F(15,168) = 39.92, p < 0.001* | F(15,168) = 0.83, p = 0.64 |
| 10–15 μm | F(1,12) = 0.57, p = 0.47 | F(15,168) = 22.12, p < 0.001* | F(15,168) = 0.41, p = 0.97 |
| 15–20 μm | F(1,12) = 0.06, p = 0.82 | F(15,168) = 17.84, p < 0.001* | F(15,168) = 0.52, p = 0.92 |
| 20–25 μm | F(1,12) = 0.25, p = 0.63 | F(15,168) = 7.50, p < 0.001* | F(15,168) = 0.33, p = 0.99 |
| 25–30 μm | F(1,12) = 0.46, p = 0.51 | F(15,168) = 5.96, p < 0.001* | F(15,168) = 0.45, p = 0.96 |
| 30–35 μm | F(1,12) = 1.75, p = 0.21 | F(15,168) = 1.98, p < 0.05* | F(15,168) = 0.75, p = 0.72 |
*Significant main effects.
To visualize the cerebral vasculature; mice received an intravenous injection of 0.1 ml of either 0.5% Evans blue (Sigma-Aldrich), 4% Rhodamine-dextran (Sigma-Aldrich), or 1% Fluorescein-dextran dye (Sigma-Aldrich) in sterile saline solution. High-resolution in vivo two-photon images of fluorescently labeled microglia and blood vessels were collected in the somatosensory cortex of lightly anesthetized mice (∼1% isoflurane in air) with a 40× objective (NA = 0.8) using an Olympus FV1000MPE laser scanning microscope coupled to a mode-locked Ti:sapphire laser. For imaging eGFP- or eYFP-labeled microglia, the laser was tuned to 900 or 950 nm, respectively. Image stacks of fluorescently labeled blood vessels and microglia were collected at 1.5 μm z steps covering an area of 144 × 144 μm (800 × 800 pixels, 0.18 μm/pixel), averaging three images per plane. Image stacks were collected at 4 min intervals for 20 min before CMB and 60 min afterward. A flowing microvessel (∼4 μm in width), 50–100 μm from the pial surface was selected for laser-induced microablation. The CMB was induced with focal high-power illumination of microvessels (850 nm, ∼220 mW at back aperture, 3.4-μm-diameter region centered on vessel) for 5–6 s. Rupture of the microvessel was easily confirmed by the appearance of extravascular fluorescence emanating from the site of the bleed.
Analysis of targeted microvessel blood flow and diameter.
For determination of red blood cell (RBC) velocity, 3 sets of line scans (each taken 30–60 s apart) were conducted through the microvessel before baseline imaging. RBC velocity measurements were derived from the inverse slope (Δ time/Δ distance) of the linear paths of RBCs (Shih et al., 2012). Using ImageJ software, lumen diameter was measured as the width at half-maximal fluorescence intensity plotted orthogonal to the vessel (Shih et al., 2012). All analyses were conducted by an experimenter blind to treatment conditions.
Analysis of microglia structure and dynamics.
To measure microglia accumulation around the site of CMB, microglia image stacks were processed using FIJI-ImageJ 1.51 s software. Images from all time points (5 baseline and 15 post-CMB stacks) were median filtered (0.8 pixel radius). All images in a time series were aligned and corrected for any X-Y displacement using ImageJ plugin running rigid body stack registration. Three images above and below the center of the bleed (7 images total, z depth = 10.5 μm) were projected. Background pixel values were then subtracted from each image projection. To compare microglial process accumulation across mouse genotypes and conditions, background corrected images of microglia were thresholded using the automated “Li” plugin. To measure the accumulation of microglia processes around the CMB, 35 concentric circles spaced 1 μm apart were centered on the CMB in each binarized image in the time series by an observer blind to condition. Thresholded signal pixel values were binned into 5 μm radii and then expressed as percentage area. The innermost ring (0–5 μm) was excluded from analysis due to the appearance of autofluorescence from the laser-induced CMB. An estimation of microglial cell number was performed in a blinded manner by counting the number of microglial cell bodies in each image stack, excluding any soma that were clipped at the top or bottom of the stack.
To estimate total microglia process turnover every 4 min, binarized images of microglia were compared with the previous imaging time frame (e.g., 8 min frame − 4 min frame). This generated “turnover” images with three pixel values: positive values (black pixels or “gained” microglia signal), zero values (gray pixels or “no change”), or negative values (white pixels or “lost” microglia signal). Microglia turnover rates were calculated by summing the number of pixels gained and lost, and dividing by the total number of microglial signal pixels measured for each time point [(#Gained + #Lost)/Sum of total microglial pixels from 2 time points]. Baseline turnover rates were averaged over the 4 baseline imaging time points, whereas the turnover rates following CMB were calculated as the percentage change from baseline.
Microglial process velocity toward the CMB was estimated by tracing the linear distance of a process at baseline to where the process stopped advancing after ablation. If a primary process from a microglia had several branched tips, all were examined independently. Processes that moved out of the imaging plane were excluded. The velocity for all processes in a single imaging session (responding to one bleed) were averaged for each CMB.
The polarization of microglial processes was determined by drawing a reference line from the microglial soma to the center of the bleed (designated 0°), and then by determining the angle of each process relative to 0°. The angle for each microglial process was determined by drawing a line from where the primary process left the soma to the tip of each branch on the process. Values were binned in 30° increments (345°–15°, 15°–45°, etc.) and expressed as a percentage of total processes. For statistical analyses of polarity, we calculated the difference in percentage microglial processes in the 0° bin (i.e., those processes oriented directly toward the CMB) at 60 min versus baseline for each group. Only microglial processes with a main branch length of at least 5 μm were examined.
Assessment of secondary plasma dye leakage after microbleed.
To estimate secondary plasma leakage after CMB, mice were first injected with 1% FITC to visualize and ablate a microvessel, which was followed by Evans blue dye (0.5% in saline, i.v.) injection 30 min after CMB. We waited at least 30 min after inducing the original bleed because, by this time point, microglial processes had clearly aggregated around the CMB and group differences in aggregation had become apparent. Two-photon image stacks were collected within 5 min of Evans blue injection at 1.5 μm z steps covering an area of 317 × 317 μm (800 × 800 pixels, 0.397 μm/pixel). For analysis of extravascular fluorescence, we first subtracted a binarized image mask of fluorescently labeled blood vessels from the original average intensity z projection image. An automated custom macro calculated the average intensity of pixel values in 5 μm radii from the center of the CMB. The innermost measurements (<10 μm from CMB) were excluded because of extreme pixel values created by autofluorescent signal at site of damage as well as the edges of fluorescently labeled vessels (which could not be completely subtracted out from the mask). Because dye fluorescence within blood vessels was variable across animals and could influence the pixel intensity values of measured extravascular fluorescence, we normalized extravascular fluorescence as a percentage of intravascular fluorescence.
Depletion of microglia.
Standard mouse food chow (AIN-76A, Research Diets) was enriched with the colony stimulating factor 1 receptor antagonist (PLX5622) generously provided by Plexxikon. To extensively deplete microglia (Elmore et al., 2014), nondiabetic mice were fed a high dose of 1200 ppm PLX5622 mouse chow for 1–3 weeks and up to 12 h before imaging experiments. No obvious changes in mouse behavior or weight were detected in mice fed PLX5622 chow.
Multiplex and ELISA immunoassay.
Blood serum and brain tissue were extracted from nondiabetic and diabetic mice (4–5 weeks after induction of diabetes) and stored frozen at −80°C until used. Tissue was sonicated in ice-cold 0.1 m PBS-containing 1% Triton-X and 0.1% sodium deoxycholate, Protease Inhibitor Mixture (1:100; Sigma-Aldrich), and Phosphatase Inhibitor Mixture 2 (1:100; Sigma-Aldrich). Following centrifugation, the total protein concentration of each supernatant was measured using a Direct Detect Infrared Spectrometer (EMD Millipore). Cytokines and chemokines were then quantified using a mouse multiplex magnetic bead kit purchased from EMD Millipore used according to the manufacturer's recommendations. Each lysate sample, standard, and quality control was measured in duplicate. Plates were read using a MAGPIX instrument (Luminex), and results were analyzed using MILLIPLEX Analyst version 5.1 software (EMD Millipore).
Blood was collected following decapitation and allowed to clot by sitting undisturbed at room temperature for 15–20 min. The serum component was isolated by centrifugation at 1500 rpm for 10 min (4°C). The resulting supernatant was transferred into a clean Eppendorf and flash frozen in liquid nitrogen. Total protein content in serum samples was measured by BCA assay (Pierce, #23225). Serum was diluted 1:10, and IFN-γ concentrations were quantified using ELISA kits (IFN-γ: Abcam, #ab100689) according to the manufacturer's protocols. Absorbance was measured at 450 nm using a microplate reader (Infinite M200; TECAN), and IFN concentration was normalized to total protein concentration per sample (pg/mg total protein).
qRT-PCR.
Brain tissue was homogenized in Tri-Reagent (Ambion) via sonification, and RNA was isolated by phenol-chloroform extraction. cDNA was synthesized from 500 ng of DNase I (Invitrogen)-treated RNA and prepared using High Capacity cDNA Reverse Transcription Kits (Applied Biosystems). qRT-PCRs were assembled for genes of interest (e.g., P2ry12) using 4 μl of cDNA per 50 μl reaction via PowerUp SYBR Green Master Mix (Thermo Fisher Scientific), and samples were run on the StepOne Real-Time PCR system (Thermo Fisher Scientific). Expression levels were compared using the ΔΔCt method normalized to β-actin. Primers used for qRT-PCR were as follows: P2ry12: 5′-TACTGGGTAGAGTGTCATTATCGGT-3′ (forward) and 5′-TTGGTAGATGTGATGTGTGATTTGG-3′ (reverse); Infgr1: 3-GCTCTCCGTCCTCGTATTTCAC-5 (forward) and 5′-CCTGGCTCTCTCCATTCACAAC-3′ (reverse); Infgr2: 5′-GACTGTGAATGGGTGTGGGAAA-3′ (forward) and 5′-GACTGTGAATGGGTGTGGGAAA-3′ (reverse); β-actin: 5′-GCGGGCGACGATGCTC-3′ (forward) and 5′-GATACCTCTCTTGCTCTGGGC-3′ (reverse).
Immunohistochemistry.
Mice were overdosed with sodium pentobarbital and transcardially perfused with 10 ml of PBS (0.1 m PBS) followed by 10 ml of 4% PFA in PBS. Whole brains were submerged in 4% PFA and postfixed overnight, then transferred to a 0.1 m PBS solution. Coronal brain sections were cut at 50 μm on a Leica vibratome and stored in six well trays in PBS with 0.02% sodium azide. Sections were incubated in PBS with polyclonal rabbit anti IBA-1 (Wako, 1:500) overnight at room temperature, washed, and then immersed in secondary antibody (Cy5 anti-rabbit in PBS, 1:500) for 4–5 h at room temperature. Sections were washed, mounted on glass slides, coverslipped, and imaged with a confocal microscope. High-resolution image stacks of eGFP- and Cy5-labeled microglia in the somatosensory cortex were taken with a 20× objective (NA = 0.75, 1024 × 1024, 0.31 μm/pixel) with 1.5 μm z steps and Kalman filtering (mean = 2).
Design and statistical analysis.
Statistical analysis of the data was conducted in SPSS (IBM), Office Excel (Microsoft), or Prism 7 (GraphPad). Two-way repeated measures ANOVA was used for group comparisons on microglial process accumulation and dynamics. One-way ANOVA was used to assess group differences in microglia cell density, process turnover at baseline, velocity of process growth, RBC velocity, and microvessel width. Significant main effects from the ANOVA were followed up with independent samples t tests. All p values ≤ 0.05 were considered statistically significant. Data are presented as mean ± SEM.
Results
Heterozygous Cx3cr1-eGFP mice show normal microglial responses to microbleed
To study microglial dynamics in vivo, we first determined whether microglia in the heterozygous Cx3cr1-eGFP reporter strain responded in a normal manner to CMB since “knock-in” of the eGFP gene replaced a Cx3cr1 gene. This is an important confound to address because previous reports have shown that microglial activity is altered in Cx3cr1 knock-out mice (Dénes et al., 2008; Liu et al., 2010; Tang et al., 2014). Therefore, we compared microglial response dynamics in heterozygous or homozygous Cx3cr1-eGFP mice (referred to as Cx3cr1+/− and Cx3cr1−/−, respectively, in Fig. 1) to a Cx3cr1-Cre driver line crossed with a Cre-dependent YFP reporter line (referred to as Cx3cr1+/+). As expected, expression of eGFP was restricted to microglia in the brain of Cx3cr1-eGFP mice (Fig. 1A). There were no significant genotype differences in the density (Fig. 1B; F(2,24) = 0.09, p = 0.92) or percentage area occupied by microglia (Fig. 1C; F(2,24) = 2.54, p = 0.10). Microglial response dynamics to CMB were assessed for 20 min before and 60 min after CMB (Fig. 1D). Consistent with previous studies (Davalos et al., 2005; Nimmerjahn et al., 2005), laser ablation of a microvessel led to the rapid accumulation of microglial processes around the lesion (Fig. 1D). Although Cx3cr1−/− mice tended to have slightly elevated microglial accumulation for regions most proximal to the CMB (see 10–15 μm in Fig. 1E), process accumulation was very similar between Cx3cr1+/+ and Cx3cr1+/− mice (Fig. 1E, compare black and yellow lines). Because Cx3cr1+/− mice showed normal microglial responses and the GFP signal was superior to the Cre-dependent YFP reporter mouse line, we used heterozygous Cx3cr1-eGFP mice to study the impact of diabetes on microglial responses to CMB.
Figure 1.

Cx3cr1+/− eGFP reporter mice show normal microglial responses to CMB. A, Confocal images demonstrating that eGFP labeled microglia colocalize with Iba-1, a classical marker of microglia. Bar graphs show the density of microglia (B) and average percentage area (C) of microglia in each genotype. D, In vivo time lapse images showing microglial responses to CMB in a Cx3cr1+/− mouse given intravenous Evans blue dye to label blood vessels. E, Graphs show the progressive accumulation of microglial processes (expressed as percentage area) in 5 μm radii around the CMB in Cx3cr1+/+ (7 CMBs from 6 mice), Cx3cr1+/− (14 CMBs from 13 mice), and Cx3cr1−/− (6 CMBs from 4 mice) mice. The center of the bleed (0–5 μm) was omitted from the analysis due to the presence of auto-fluorescent signal. *p < 0.05 compares Cx3cr1+/+ to Cx3cr1−/− at 10–15 μm. Scale bars: A,D,E = 20 μm. Data were analyzed with one-way ANOVA (B–C) or two-way repeated-measures ANOVA with post hoc t tests (E).
Diabetes impairs microglial envelopment of the microbleed
Microglial responses to CMB were examined in three experimental groups: nondiabetic controls and STZ-induced diabetic mice treated with or without insulin (Fig. 2A). As expected, blood glucose levels in the untreated diabetic group were chronically elevated relative to nondiabetic and insulin-treated diabetic mice (Fig. 2B). To determine whether any basal differences in microglial labeling existed between experimental groups, we estimated the percentage area occupied by microglia in binary images (Fig. 2C) and the number of cells per mm3 before the induction of microbleeds. There were no significant differences between experimental groups in microglial area (Fig. 2D; F(2,48) = 1.37, p = 0.26) or density (Fig. 2E; F(2,48) = 1.25, p = 0.30). Following CMB, in vivo time lapse imaging revealed a rapid and progressive time-dependent accumulation of microglial processes around the site of the CMB (Fig. 2F,G). While there were no group differences in microglial envelopment of the CMB at the innermost regions (5–15 μm in Fig. 2F,G), hyperglycemic diabetic mice showed significantly fewer microglial processes enveloping the CMB at more distal sites (Fig. 2G, compare blue with red lines from 15 to 35 μm; statistics in Table 2). Treating diabetic mice with insulin partially prevented this deficit in accumulation (Fig. 2G; statistics in Table 3). These results show that microglia envelopment of microbleeds is reduced in untreated diabetics, which can be partially reversed by maintaining normal blood glucose levels with insulin.
Figure 2.
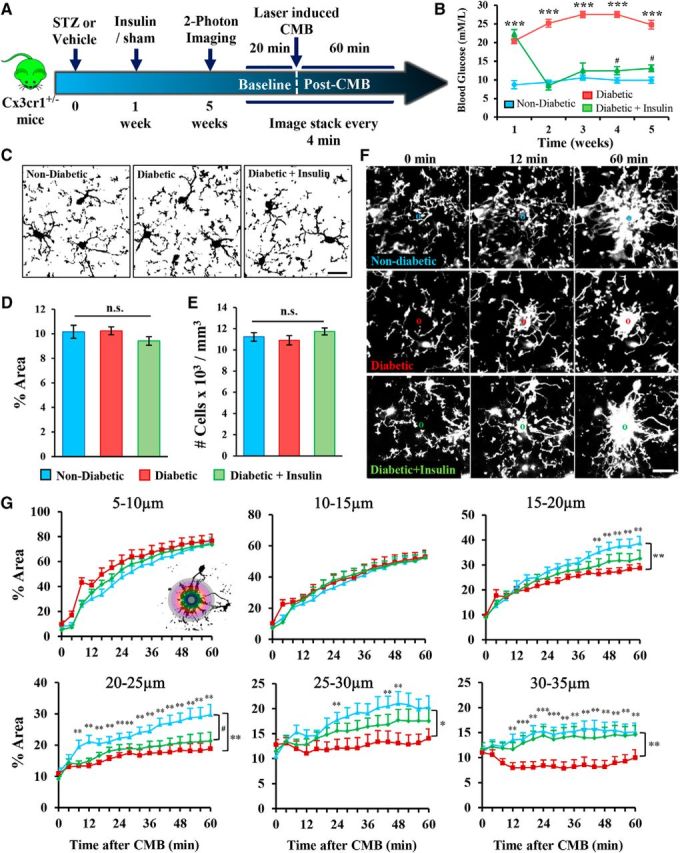
Diabetes attenuates microglia process envelopment of the microbleed. A, Experimental timeline for imaging the effect of diabetes on microglial responses to CMB. B, Average blood glucose level for each experimental group was assessed at weekly intervals. ***p < 0.001, nondiabetic versus diabetic. #p < 0.05, nondiabetic versus diabetic + insulin. ##p < 0.01, nondiabetic versus diabetic + insulin. ###p < 0.001, nondiabetic versus diabetic + insulin. C, Representative binarized images of microglia in each group at baseline. D, There were no significant group differences in average percentage area of microglia at baseline. E, Bar graph represents there were no differences in the density of microglial cells. F, Representative maximum intensity z projection images of microglia in each group immediately before CMB (0 min) or 12 and 60 min afterward. G, Graphs represent time- and distance-dependent changes in microglia process accumulation around the CMB (binned in 5 μm rings) in nondiabetic (14 CMBs from 13 mice), diabetic (11 CMBs from 10 mice), and diabetic mice treated with insulin (16 CMBs from 10 mice). *p < 0.05, nondiabetic versus diabetic. **p < 0.01, nondiabetic versus diabetic. ***p < 0.001, nondiabetic versus diabetic. #p < 0.05, nondiabetic versus diabetic + insulin. Scale bars: C, F, 20 μm. Data were analyzed with one-way ANOVA (D, E) or two-way repeated-measures ANOVA with post hoc t tests (B, G).
Table 2.
Analysis of microglial process accumulation based on repeated-measures ANOVAs conducted at each distance from the CMB with condition (nondiabetic and diabetic) as a factor over time (0–60 min)
| Distance | Condition | Time | Interaction |
|---|---|---|---|
| 5–10 μm | F(1,23) = 2.76, p = 0.11 | F(15,345) = 98.11, p < 0.001* | F(15,345) = 1.45, p = 0.11 |
| 10–15 μm | F(1,23) = 0.47, p = 0.50 | F(15,345) = 62.07, p < 0.001* | F(15,345) = 0.43, p = 0.97 |
| 15–20 μm | F(1,23) = 8.17, p < 0.01* | F(15,345) = 39.49, p < 0.001* | F(15,345) = 3.48, p < 0.001* |
| 20–25 μm | F(1,23) = 10.16, p < 0.01* | F(15,345) = 14.54, p < 0.001* | F(15,345) = 2.43, p < 0.01* |
| 25–30 μm | F(1,23) = 5.21, p < 0.05* | F(15,345) = 6.87, p < 0.001* | F(15,345) = 4.49, p < 0.001* |
| 30–35 μm | F(1,23) = 11.16, p < 0.01* | F(15,345) = 1.09, p = 0.36 | F(15,345) = 3.94, p < 0.001* |
*Significant main effects.
Table 3.
Analysis of microglial process accumulation based on repeated-measures ANOVAs conducted at each distance from the CMB with condition (nondiabetic and diabetic + insulin) as a factor over time (0–60 min)
| Distance | Condition | Time | Interaction |
|---|---|---|---|
| 5–10 μm | F(1,28) = 0.21, p = 0.65 | F(15,392) = 118.31, p < 0.001* | F(15,392) = 0.63, p = 0.84 |
| 10–15 μm | F(1,28) = 0.13, p = 0.71 | F(15,392) = 78.10, p < 0.001* | F(15,392) = 0.58, p = 0.88 |
| 15–20 μm | F(1,28) = 1.56, p = 0.22 | F(15,392) = 42.92, p < 0.001* | F(15,392) = 1.34, p = 0.18 |
| 20–25 μm | F(1,28) = 6.08, p < 0.05* | F(15,392) = 13.28, p < 0.001* | F(15,392) = 1.43, p = 0.14 |
| 25–30 μm | F(1,28) = 1.12, p = 0.30 | F(15,392) = 9.51, p < 0.001* | F(15,392) = 0.56, p = 0.90 |
| 30–35 μm | F(1,28) = 0.21, p = 0.64 | F(15,392) = 3.51, p < 0.001* | F(15,392) = 0.19, p = 0.99 |
*Significant main effects.
Because uncontrolled diabetes was associated with reduced accumulation of microglia processes around the CMB, we questioned whether these microglia were simply less dynamic as opposed to less responsive. To answer this, we quantified microglia process turnover every 4 min (Fig. 3A). Consistent with previous work (Fuhrmann et al., 2010), approximately one-third of microglial processes changed every 4 min, and there were no significant differences between groups before inducing CMB (Fig. 3B). Following induction of CMB, turnover rates increased equivalently in all groups for the first 12 min and then fell below baseline rates thereafter, presumably because many processes had reached the site of CMB and started to stabilize (Fig. 3A,C). Microglia turnover rates remained steady throughout the 60 min imaging period in the absence of ablation (Fig. 3C, gray line). The velocity of microglial process growth toward the CMB was also not significantly different between experimental groups (Fig. 3D; F(2,31) = 0.43, p = 0.64). However, the polarity of microglial processes toward the CMB (Fig. 3E) was significantly reduced in hyperglycemic diabetic mice (nondiabetic vs diabetic: t(18) = 5.78, p < 0.001), which could be ameliorated with insulin treatment (diabetic vs diabetic + insulin: t(22) = 3.39, p < 0.01). These results indicate that uncontrolled diabetes does not alter baseline motility or the capacity for microglia to remodel their processes after CMB, but it does affect chemotaxic process growth toward the CMB.
Figure 3.
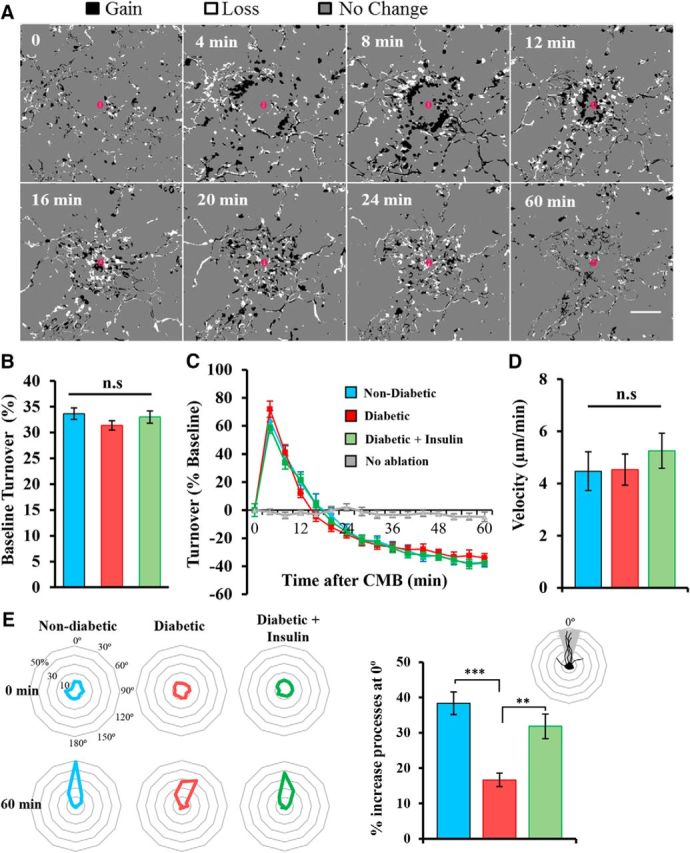
Diabetes alters microglial polarity but not velocity or process dynamics. A, Montage showing microglial process turnover in response to CMB in a nondiabetic mouse. Pixels are expressed as processes gained (black), lost (white), or no change (gray). There is a wave of gain (black) that is followed by a wave of loss (white) as the processes extend toward the CMB. B, Percentage change in microglia process turnover every 4 min at baseline in nondiabetic (14 CMBs from 13 mice), diabetic (11 CMBs from 10 mice), and diabetic mice treated with insulin (16 CMBs from 10 mice). C, Percentage change in microglia process turnover after CMB normalized to baseline turnover. D, Bar graph represents the velocity of process growth toward the lesion. E, Radial plots indicate the polarity of microglial processes before and 60 min after CMB (oriented at 0° relative to each microglia). Microglia in diabetic mice were significantly less polarized after CMB than nondiabetic and insulin-treated diabetic mice. **p < 0.01, ***p < 0.001. Scale bar: A, 20 μm. Data were analyzed with one-way ANOVA with post hoc t tests (B, D, E) or two-way repeated-measures ANOVA (C).
To ensure that any group differences in microglial responses to CMB were not explained by any differences in the properties of microvessels targeted, we assessed RBC flow velocity and lumen width before ablation (Fig. 4A). There were no significant differences between experimental groups in RBC flow velocity or lumen width (Fig. 4B,C). These results suggest that experimental differences in microglial process accumulation cannot be explained by differences in the microvessels targeted for ablation.
Figure 4.
Microvessels targeted for CMB do not differ between groups. A, Representative average intensity z projection images show plasma fluorescence before and after induction of CMB. Line scans through each vessel were used to calculate RBC velocity. B, C, Bar graphs represent RBC velocity (mm/s) and width of vessels targeted for CMB. n.s., Not significant based on one-way ANOVA. Scale bar: A, 25 μm.
Diabetes exacerbates secondary vessel leakage after microbleed
Next, we wanted to assess what impact a reduced microglial response in diabetic mice would have on microvessel integrity and repair using secondary plasma leakage as an indicator of this process, as previously described (Lou et al., 2016). Mice from each experimental group were injected with Evans blue dye 30 min after the induction of CMB and the extravasation of dye was assessed immediately thereafter (Fig. 5A). In addition, we examined the extreme condition of depleting almost all microglia in the cortex of nondiabetic and diabetic mice to provide more direct evidence that microglia were indeed playing a role in repairing damaged microvessels. Microglia were depleted by feeding nondiabetic and diabetic mice chow containing colony-stimulating factor 1 receptor antagonist (PLX5622) for 1–3 weeks before imaging (Fig. 5B). In general, microvessels leaked Evans blue dye from the vessel segment adjacent to the CMB (Fig. 5C) with an average displacement of 16.8 ± 2 μm from the site of the original bleed. Analysis of secondary leakage sites revealed significantly elevated extravascular fluorescence in diabetic and microglia depleted groups relative to the nondiabetic controls (Fig. 5D,E). Differences in secondary vessel leakage could not be explained by systematic differences in blood flow (F(4,29) = 0.44, p = 0.77) or width (F(4,29) = 1.19, p = 0.33) of targeted microvessels (Fig. 5F,G). These results suggest that the blunted or absent microglial response to CMB in diabetic or microglial depleted mice, respectively, leads to increased secondary leakage from damaged microvessels.
Figure 5.
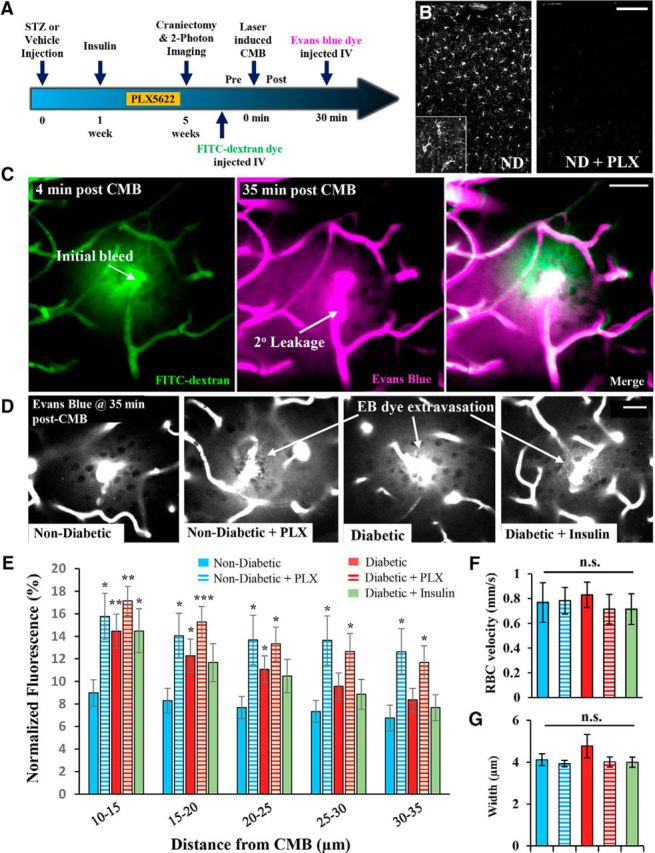
Diabetic and microglia-depleted mice show greater secondary microvessel leakage. A, Timeline of experiments conducted. Only a subset of nondiabetic and diabetic mice were fed PLX5622 chow to deplete microglia. B, IBA-1 immunostaining for microglia in the cortex of nondiabetic (ND) mice fed regular (left) or PLX5622 (right) chow. C, Representative images show FITC-dextran fluorescence (green) associated with the initial induction of CMB and Evans blue dye fluorescence (magenta) when injected 30 min after CMB. Evans Blue dye egresses from sites adjacent to the original ablation site (see arrow for “2° leakage”). D, Representative images showing extravagated Evans blue dye fluorescence 35 min after induction of CMB. E, Normalized extravascular Evans blue dye fluorescence in nondiabetic (6 CMB in 5 mice), nondiabetic + PLX5622 (9 CMB in 5 mice), diabetic (6 CMB in 4 mice), diabetic + PLX5622 (7 CMB in 4 mice), and diabetic + insulin mice (6 CMB in 3 mice). F, G, Graphs show baseline RBC velocity (mm/s) and width for vessels targeted in each group. *p < 0.05 compared with nondiabetic mice fed standard diet. **p < 0.01 compared with nondiabetic mice fed standard diet. ***p < 0.005 compared with nondiabetic mice fed standard diet. Scale bars: B, 100 μm; C, D, 20 μm. E, F, Data were analyzed with ANOVA with post hoc t tests.
DEX enhances microglial responses and reduces vessel leakage
Given that diabetes is associated with abnormal immune system function (Pradhan et al., 2011; Sickmann et al., 2012), and insulin therapy could only partially ameliorate microglial impairments and secondary vessel leakage, we explored the possibility that treating diabetic mice with an immunosuppressant might reinvigorate microglial responses to injury. To test this hypothesis, we treated hyperglycemic diabetic mice with the broad-spectrum immunosuppressant DEX or vehicle for 5 d before CMB. DEX treatment generally suppressed inflammation-related cytokines and chemokines, especially IFN-γ in the brain of diabetic mice relative to those given vehicle (Fig. 6A; F(1,21) = 26.18, p < 0.001; proteins measured 4–5 weeks after induction of diabetes). DEX treatment did not significantly alter the level of hyperglycemia in diabetic mice (diabetic + vehicle: 24.64 ± 1.07 mmol/L vs diabetic + DEX: 24.56 ± 1.37 mmol/L; t(15) = 0.4, p = 0.96). As shown in Figure 6B, microglial process accumulation around the CMB in DEX-treated diabetic mice was significantly enhanced relative to vehicle-treated diabetic mice (Fig. 6C, compare magenta with red lines; statistics shown in Table 4) and was indistinguishable from healthy nondiabetic controls. The DEX-mediated enhancement of microglia process accumulation was limited to diabetic mice, as there was no further enhancement in nondiabetic mice given DEX (Fig. 6C, compare blue and black lines). There were no significant group differences in microglial process turnover rates (Fig. 6D; F(3,35) = 0.27, p = 0.85) and velocity of growth toward the lesion (F(3,31) = 0.24, p = 0.86). However, DEX treatment in diabetic mice appeared to restore the normal polarization of microglial processes toward the CMB (Fig. 6E). Corresponding with the improvement in microglial responses, secondary dye leakage near the CMB was significantly reduced in DEX-treated diabetic mice relative to vehicle-treated mice (Fig. 6F,G). Microvessels targeted for ablation were not significantly different between groups in terms of blood velocity (F(3,23) = 0.41, p = 0.75) and width (F(3,23) = 1.34, p = 0.28). In summary, our data indicate that DEX treatment in diabetic animals can mitigate abnormal microglial responses and secondary plasma leakage from damaged microvessels.
Figure 6.
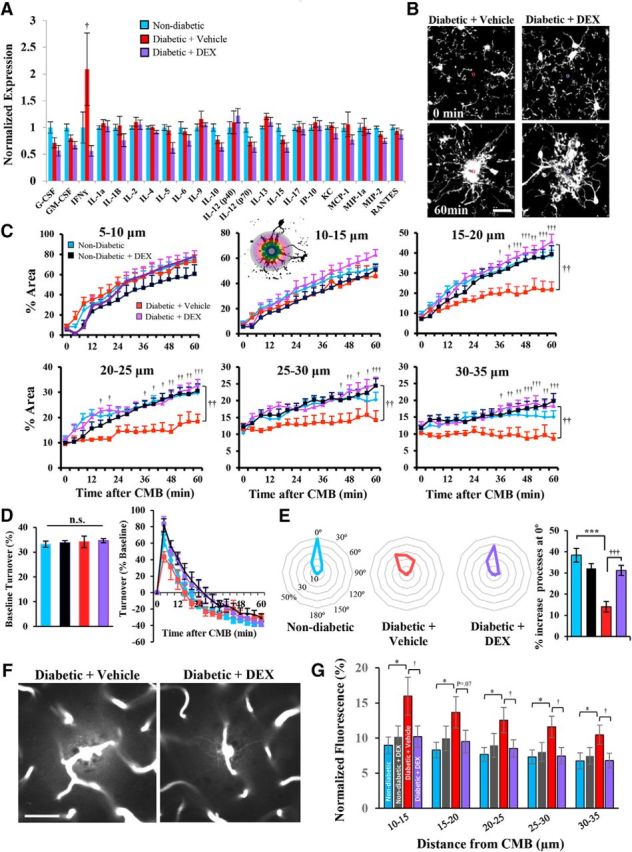
DEX enhances microglia responses and attenuates vessel leakage in diabetic mice. A, Multiplex immunoassay for brain cytokine and chemokine expression in nondiabetic and diabetic mice treated with vehicle or DEX. B, Representative in vivo images showing microglial responses to CMB in diabetic mice given saline or DEX for 5 d before imaging. C, Analysis of microglial process accumulation over time in 5 μm radii from the CMB in naive nondiabetic (14 CMBs from 13 mice), nondiabetic + DEX (10 CMBs from 5 mice), diabetic + vehicle (5 CMBs from 3 mice), and diabetic + DEX mice (9 CMBs from 5 mice). D, Graphs represent percentage microglial process turnover every 4 min at baseline (left) and after CMB (right). E, Plots represent enhanced polarization of microglial processes 60 min after CMB in diabetic mice treated with DEX. F, Representative images showing secondary leakage of Evans blue-labeled plasma 35 min after CMB. G, Quantification of normalized extravascular fluorescence in nondiabetic (6 CMB in 5 mice), nondiabetic + DEX (8 CMBs from 4 mice), diabetic + vehicle (10 CMBs from 5 mice), and diabetic + DEX-treated mice (8 CMBs from 4 mice). †p < 0.05, diabetic + vehicle versus diabetic + DEX. ††p < 0.01, diabetic + vehicle versus diabetic + DEX. †††p < 0.001, diabetic + vehicle versus diabetic + DEX. *p < 0.05, nondiabetic versus diabetic + vehicle. **p < 0.01, nondiabetic versus diabetic + vehicle. ***p < 0.001, nondiabetic versus diabetic + vehicle. Scale bars: B, 20 μm; F, 40 μm. Data were analyzed by one-way ANOVA (D, E, G) or two-way repeated-measures ANOVA (C) with post hoc t tests.
Table 4.
Analysis of microglial process accumulation based on repeated-measures ANOVAs conducted at each distance from the CMB with condition (diabetic + vehicle and diabetic + DEX) as a factor over time (0–60 min)
| Distance | Condition | Time | Interaction |
|---|---|---|---|
| 5–10 μm | F(1,12) = 0.04, p = 0.83 | F(15,180) = 62.27, p < 0.001* | F(15,180) = 2.28, p < 0.01* |
| 10–15 μm | F(1,12) = 1.53, p = 0.24 | F(15,180) = 32.82, p < 0.001* | F(15,180) = 1.52, p = 0.10 |
| 15–20 μm | F(1,12) = 19.45, p < 0.001* | F(15,180) = 21.44, p < 0.001* | F(15,180) = 4.53, p < 0.001* |
| 20–25 μm | F(1,12) = 17.67, p < 0.001* | F(15,180) = 11.03, p < 0.001* | F(15,180) = 1.93, p < 0.05* |
| 25–30 μm | F(1,12) = 15.60, p < 0.001* | F(15,180) = 5.051, p < 0.001* | F(15,180) = 1.54, p = 0.09 |
| 30–35 μm | F(1,12) = 16.81, p < 0.001* | F(15,180) = 1.31, p = 0.20 | F(15,180) = 2.31, p < 0.01* |
*Significant main effects.
Role for IFN-γ in abnormal microglial responses to CMB
Because broadly suppressing the immune system with DEX was sufficient to prevent abnormal microglial responses in diabetic mice, our next goal was to identify a specific signaling pathway mediating this phenomenon. Motivated by the results of our brain cytokine screen (Fig. 6A) and recent work showing that microglial dysfunction in certain autoimmune diseases is related to abnormal expression of blood-borne IFNs that permeate into the brain (Pan et al., 1997; Filiano et al., 2016; Bialas et al., 2017), we focused our attention on the proinflammatory cytokine IFN-γ. Given that circulating leukocytes are a major source of IFN-γ, we first measured IFN-γ expression in blood serum 4–5 weeks after the induction of diabetes. Vehicle-treated diabetic mice had significantly elevated IFN-γ levels in blood serum relative to nondiabetic mice (Fig. 7A; t(11) = 1.90, p < 0.05), which was reduced with DEX treatment. Because it is possible that STZ injection alone could induce inflammation and alter IFN-γ levels independent of hyperglycemia, we also examined IFN-γ levels in blood serum of STZ-injected mice that did not develop hyperglycemia. Serum IFN-γ levels were not significantly altered in these mice relative to nondiabetic controls (t(9) = 1.34, p = 0.21). Based on these findings, we injected hyperglycemic diabetic mice with neutralizing antibodies to IFN-γ (over 5 d before CMB). To validate this approach (Heink et al., 2017), we showed that diabetic mice treated with neutralizing antibody exhibited significantly lower IFN-γ in blood serum (Fig. 7A). Similar to the effects of DEX treatment, blocking IFN-γ in hyperglycemic diabetic mice enhanced microglial process accumulation around the CMB (Fig. 7B; statistics summarized in Table 5). Microglial process dynamics before CMB were slightly, but significantly, elevated in IFN-γ IgG-treated diabetic mice (Fig. 7C), but were no different from controls after the induction of CMB (Fig. 7D; main effect of treatment: F(1,210) = 0.93, p = 0.34). Treating diabetic mice with IFN-γ neutralizing antibody significantly increased the polarity of microglial processes toward the CMB (t(11) = 4.92, p < 0.001) but did not alter microglia process velocity (t(11) = 0.67, p = 0.26). Importantly, treating diabetic mice with IFN-γ neutralizing antibody significantly reduced secondary vessel leakage after CMB (Fig. 7E; main effect of treatment: F(1,110) = 21.8, p < 0.001). These treatment-related effects could not be explained by systematic differences in blood glucose levels (t(17) = 0.28, p = 0.38), or RBC velocity (t(19) = 0.06, p = 0.48) and width (t(19) = 0.30, p = 0.52) of targeted microvessels. Thus, treatment of diabetic mice with neutralizing antibodies to IFN-γ can restore normal microglial responses to vascular injury.
Figure 7.
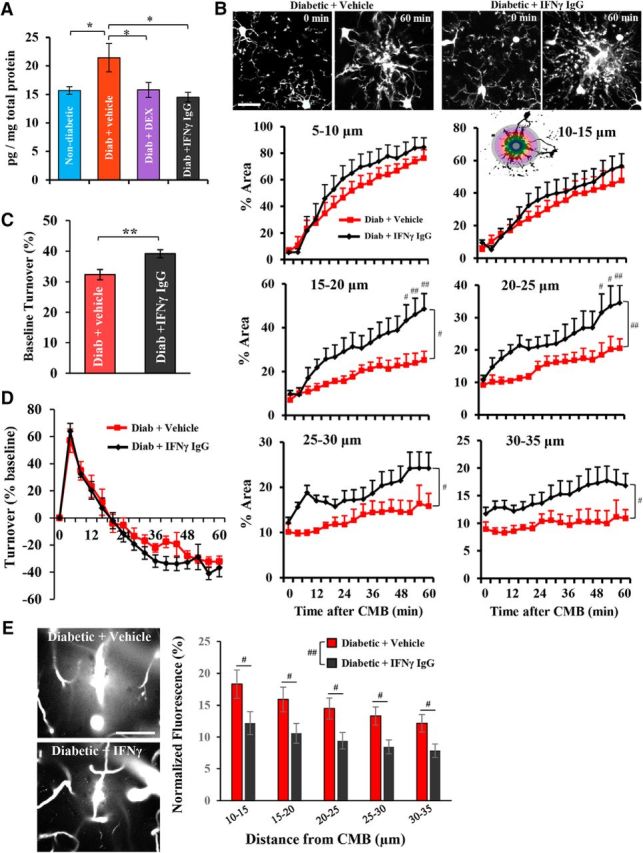
Role for IFN-γ in abnormal microglial responses to CMB. A, IFN-γ protein expression in blood serum. B, Top, Two-photon images of microglial responses before (0 min) and 60 min after CMB. Bottom, Analysis of microglial process accumulation in diabetic + vehicle or isotype control IgG (9 CMBs from 5 mice), and diabetic + IFN-γ blocking IgG (11 CMBs from 6 mice)-treated mice as a function of distance from the center of the CMB. C, Microglia process turnover every 4 min before the induction of CMB. D, Microglia process turnover (normalized to baseline) over 60 min period after CMB. E, Left, Representative in vivo image projections of secondary vessel leakage collected 35 min after CMB. Right, Quantification of normalized extravascular dye fluorescence in diabetic mice treated with vehicle or isotype control IgG (13 CMBs from 7 mice) or IFN-γ blocking IgG (10 CMBs from 5 mice). *p ≤ 0.05, **p < 0.01, ** #p < 0.05, diabetic + vehicle versus diabetic + IFN-γ blocking IgG. ##p < 0.01, diabetic + vehicle versus diabetic + IFN-γ blocking IgG. Scale bars: B, 20 μm; E, 50 μm. B, D, E, Data were analyzed by two-way ANOVA with post hoc t tests.
Table 5.
Analysis of microglial process accumulation based on repeated-measures ANOVAs conducted at each distance from the CMB with condition (diabetic + vehicle and diabetic + IFNγ-blocking IgG) as a factor over time (0–60 min)
| Distance | Condition | Time | Interaction |
|---|---|---|---|
| 5–10 μm | F(1,15) = 0.38, p = 0.54 | F(15,270) = 67.65, p < 0.001* | F(15,270) = 0.73, p = 0.74 |
| 10–15 μm | F(1,15) = 0.39, p = 0.53 | F(15,270) = 31.47, p < 0.001* | F(15,270) = 0.57, p = 089 |
| 15–20 μm | F(1,15) = 6.85, p < 0.05* | F(15,270) = 23.63, p < 0.001* | F(15,270) = 3.44, p < 0.001* |
| 20–25 μm | F(1,15) = 9.08, p < 0.01* | F(15,270) = 13.00, p < 0.001* | F(15,270) = 1.50, p = 0.11 |
| 25–30 μm | F(1,15) = 5.92, p < 0.05* | F(15,270) = 7.37, p < 0.001* | F(15,270) = 1.04, p = 0.41 |
| 30–35 μm | F(1,15) = 6.23, p < 0.05* | F(15,270) = 2.97, p < 0.001* | F(15,270) = 0.77, p = 0.71 |
*Significant main effects.
To understand how abnormal IFN-γ signaling in diabetic mice could disrupt microglial process chemotaxis toward the CMB, we used qRT-PCR to quantify purinergic receptor P2ry12 and IFN-γ receptor 1 and 2 (Ifngr1 and Ifngr2) gene expression in brain tissue. We focused on P2RY12 because it is primarily expressed in microglia and has a well-established role in mediating microglial chemotaxis to tissue damage and repair of the BBB (Haynes et al., 2006; Lou et al., 2016). Our analysis revealed that untreated diabetes is associated with a significant downregulation of P2ry12 gene expression (Fig. 8). Furthermore, we show that DEX- or IFN-γ-blocking antibody treatment in diabetic mice not only restored microglial process chemotaxis but also normalized P2RY12 gene expression. Gene expression for Ifngr1 and Ifngr2 was not significantly changed across experimental treatment groups (Fig. 8). Last, analyses of P2ry12, Ifngr1, or Ifngr2 gene expression in STZ injected mice that did not develop hyperglycemia revealed no significant changes in transcript expression (P2RY12: t(16) = 0.36, p = 0.72; Ifngr1: t(9) = 1.13, p = 0.28; Ifngr2: t(8) = 1.35, p = 0.21).
Figure 8.

P2ry12 mRNA expression correlates with diabetes-related impairments and rescue of microglial process chemotaxis. Graphs represent normalized (to β-actin) Ifngr1, Ifngr2, or P2ry12 receptor expression mRNA expression in somatosensory cortex. P2ry12, but not Ifngr1, Ifngr2 mRNA expression, was significantly reduced in untreated diabetic mice, which could be reversed with DEX treatment or IFN-γ-blocking antibodies. *p < 0.05 nondiabetic versus untreated diabetic mice.
Discussion
Microglia rapidly respond to stroke or injury-related signals (Masuda et al., 2011; Rosidi et al., 2011; del Zoppo et al., 2012) that can protect or enhance repair processes after injury (Glezer et al., 2007; Szalay et al., 2016; Yuan et al., 2016). In support of this idea, depletion of microglia or pharmacological inhibition of microglial chemotaxic responses exacerbates neuronal injury after laser ablation (Hines et al., 2009), stroke (Lalancette-Hébert et al., 2012; Fernández-López et al., 2016; Szalay et al., 2016), and impedes the repair of the BBB (Lou et al., 2016). Indeed, we found that depleting almost all microglia in healthy nondiabetic animals significantly increased secondary vessel leakage after CMB. However, the relationship between microglial accumulation around the CMB and the extent to which damaged vessels continued to leak blood plasma was more nuanced in diabetic animals. For instance, microglial accumulation in hyperglycemic (untreated) diabetic mice was relatively normal at the center of the CMB (5–15 μm radius) but significantly reduced at more distal sites relative to nondiabetic mice. These mice also showed greatest vessel permeability ∼35 min after CMB, which may be related to the fact that secondary plasma leakage typically emanated 15–20 μm from the center of the CMB, where fewer microglial processes would be present in diabetic mice. It is interesting to note that enhancing microglial accumulation specifically within these more distal regions (15–35 μm from center of CMB) with DEX- or IFN-γ-blocking antibodies normalized the degree of vessel leakage after CMB. The functional consequences of this failure to seal off damaged microvessels in diabetic animals is not well understood at this time. Because extravasation of blood plasma through leaky or ruptured vessels is associated with local loss of dendritic spines (Zhang and Murphy, 2007; Reeson et al., 2015; Taylor et al., 2015) and perturbation of sensory-evoked neuronal activity (Cianchetti et al., 2013), it is possible that the disruptive impact of microbleeds on brain circuitry would be exacerbated in diabetic animals.
The idea of using immune-suppressing drugs to enhance the brains' innate immune response to injury may seem counterintuitive. However, in light of our cytokine analysis data and others showing that both Type 1 and 2 diabetes is linked with abnormal expression of certain inflammatory cytokines and chemotactic factors (Pradhan et al., 2011; Dey et al., 2014; Oubaha et al., 2016), it is conceivable that a blunted microglial response may be related to a reduced sensitivity to injury/inflammation-related signals (Davalos et al., 2005, 2012; Kim and Cho, 2016). In this scenario, chronic IFN-γ inflammation in diabetic mice may downregulate the expression of receptors on microglia critical for mediating chemotactic responses (Kettenmann et al., 2011; Tay et al., 2017). Thus, when vascular injury occurs, microglia only grow toward the center of the lesion where chemotactic factors are highest, bypassing or ignoring weaker signals further away from the CMB. This could explain why microglia accumulation near the center of the CMB was relatively normal in diabetic mice but reduced at more distal sites. Indeed, using qRT-PCR, we found that untreated diabetes was associated with a significant reduction in brain P2ry12 mRNA expression compared with nondiabetic controls, which could be reversed by treating diabetic mice with DEX- or IFN-γ-neutralizing antibodies. This impairment in microglial chemotactic responses and associated reduction in P2ry12 mRNA expression mirrored what has been described with systemic inflammation (Gyoneva et al., 2014; Holtman et al., 2015) and aging (Damani et al., 2011; Hickman et al., 2013; Rawji et al., 2016). It is also interesting to note that diabetes-related changes to chemotaxic behavior (i.e., reduced process orientation and envelopment of bleeds) were dissociable from basal motility of processes, often referred to as “surveillance” activity. Indeed, several studies have shown that these microglial behaviors are separate and controlled by different mechanisms (Hines et al., 2009; Madry et al., 2018). Future studies will be needed to address how diabetes and perhaps specifically IFN-γ differentially affect microglial behaviors.
Consistent with the hypothesis that chronic elevation of inflammatory cytokines in diabetic mice underlies changes in microglial responses to injury, administering a broad-spectrum immunosuppressant (DEX) to diabetic mice normalized microglia responses. Although we recognize that glucocorticoids have pleiotropic actions and microglia express glucocorticoid receptors (Carrillo-de Sauvage et al., 2013; Holtman et al., 2014), the fact that DEX treatment had no effect in healthy nondiabetic controls suggests that it likely acts via modulation of the immune system, rather than a direct effect on microglia. Moreover, DEX treatment normalized levels of IFN-γ in the blood serum of diabetic mice, implying that maintaining healthy levels of IFN-γ signaling could be a critical factor. In support of this mechanism, repeated intravenous injections of an IFN-γ blocking antibody in diabetic mice were sufficient to mimic the beneficial effects of DEX and normalize microglial responses and vessel leakage following CMB. The idea that elevated IFN-γ in blood could alter microglial behavior is supported by data showing that IFN-γ permeates the BBB (Pan et al., 1997) and microglia highly express IFN-γ receptors (Lavin et al., 2014; Zhang et al., 2014). Further, these findings build on accumulating evidence that chronic elevation of blood-borne IFNs can influence microglial behavior, often with detrimental changes in brain function associated with aging, autoimmune diseases such as lupus, or neuropathic pain (Tsuda et al., 2009; Baruch et al., 2014; Goldmann et al., 2016; Bialas et al., 2017).
Identifying the exact source of elevated IFN-γ and the forces driving its increase in diabetic animals will require further study. First, it is well established that proinflammatory CD4+ and CD8+ T helper and natural killer cells in the blood are the primary sources of IFN-γ (de Weerd and Nguyen, 2012). However, there is some evidence that microglia, at least in cell culture, can produce small amounts of IFN-γ (Kawanokuchi et al., 2006). Therefore, our finding of elevated IFN-γ in blood serum of diabetic mice is presumably derived from T helper and natural killer cells. In the brain, however, it is possible that IFN-γ is derived from both leukocytes and resident microglia. One predominant hypothesis regarding why IFN-γ levels would be elevated in diabetic mice is that metabolic processes in vascular endothelial cells are chronically perturbed due to exposure to hyperglycemic conditions. This is plausible because glucose uptake in endothelial cells occurs via facilitated diffusion through glucose transporters rather than insulin-dependent transporters. As a result, intracellular glucose concentrations are abnormally high in endothelial cells, which leads to the generation of reactive oxygen species, advanced glycation end products, and ultimately damages the integrity of the vascular endothelium (Vinik and Flemmer, 2002). Thus, the vascular endothelium is in a chronic state of injury, thereby recruiting the adaptive immune response which enhances IFN-γ production. Indeed, there is an extensive body of literature implicating abnormal IFN-γ production and signaling with the pathogenesis of Type 1 diabetes (see Wang et al., 1997; Marwaha et al., 2014). It is important to note that the link between elevated IFN-γ and diabetes is not restricted to just the STZ model but has been described in other rodent models of diabetes (Rabinovitch et al., 1996; Hwang et al., 2014; Vuong et al., 2017), as well as in humans with Type 1 diabetes (Nicoletti et al., 2002; Alizadeh et al., 2006; Kaas et al., 2012; Allam et al., 2014).
Treating hyperglycemic mice with insulin (∼1 week after STZ injection) was only partially successful in restoring normal microglial responses to CMB and secondary microvessel leakage. While there is evidence that insulin can reverse some neurological complications associated with diabetes (Biessels et al., 1999), our findings are generally consistent with previous work showing that insulin therapy cannot fully rescue diabetes-related impairments in blood flow dynamics, sensory circuit remodeling, or BBB disruption after stroke (Sweetnam et al., 2012; Tennant and Brown, 2013; Reeson et al., 2015). One caveat with our study was that slow-release insulin pellets were not perfect in maintaining blood glucose levels within the normal range, as levels were slightly but significantly elevated relative to nondiabetic mice. Therefore, the partial improvement in diabetic mice may be related to the fact blood glucose levels were close to, but not quite, normal. In addition to regulating blood glucose levels, insulin signaling could directly influence microglia function. Microglia express insulin growth factor receptor 1 (IGFR1) and possibly insulin receptor/IGFR1 heteromers (Varewijck and Janssen, 2012). If circulating insulin is important for maintaining microglial chemotactic responses, then disrupting insulin signaling in our mouse model of diabetes could account for microglial dysfunction. It is worth stating that we do not suspect that the administration of STZ in our mouse model caused any direct injury to the brain or microglia because we have previously shown that equivalent or higher doses of STZ do not produce any signs of cell death or toxicity in the brain (Sweetnam et al., 2012). Moreover, we compared IFN-γ serum protein levels and brain P2RY12 gene expression in mice that were injected with STZ but did not become hyperglycemic relative to those that were injected with vehicle solution. In both of these nondiabetic controls, we found no significant differences in either dependent measure, suggesting that STZ injection alone (without hyperglycemia or at least abnormal glucose regulation) cannot mimic effects described in our hyperglycemic diabetic group.
In conclusion, we have applied real-time in vivo imaging to assess the impact of diabetes on microglial response dynamics to CMB. Using two-photon microscopy, we show that chronic hyperglycemia in the STZ model of Type 1 diabetes leads to decreased microglial process accumulation and polarization around the site of microvascular injury, concomitant with increased leakage of blood plasma from damaged microvessels. These impairments were partially prevented by lowering blood glucose levels with insulin. However, treating hyperglycemic diabetic mice with DEX or an IFN-γ blocking antibody was sufficient to restore normal microglial responses to CMB and mitigate secondary microvessel leakage. To our knowledge, this is the first study to implicate abnormal IFN signaling as a critical factor behind impaired wound healing in the diabetic brain.
Footnotes
This work was supported by Canadian Institutes of Health Research, Heart and Stroke Foundation, Natural Sciences and Engineering Research Council of Canada, and Canadian Foundation for Innovation to C.E.B. S.T. was supported by Canadian Institutes of Health Research graduate fellowship. We thank Dr. Kerry Delaney for technical advice; and Taimei Yang for managing our mouse colony.
The authors declare no competing financial interests.
References
- Alizadeh BZ, Hanifi-Moghaddam P, Eerligh P, van der Slik AR, Kolb H, Kharagjitsingh AV, Pereira Arias AM, Ronkainen M, Knip M, Bonfanti R, Bonifacio E, Devendra D, Wilkin T, Giphart MJ, Koeleman BP, Nolsøe R, Mandrup Poulsen T, Schloot NC, Roep BO, Roep BO (2006) Association of interferon-γ and interleukin 10 genotypes and serum levels with partial clinical remission in type 1 diabetes. Clin Exp Immunol 145:480–484. 10.1111/j.1365-2249.2006.03172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allam G, Alsulaimani AA, Alghamdi H, Alswat H, Edrees BM, Ahmad I, Nasr A (2014) Changes in the levels of cytokines in both diabetic/non-diabetic type I children living in a moderate altitude area in Saudi Arabia. High Alt Med Biol 15:380–387. 10.1089/ham.2014.1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch K, Deczkowska A, David E, Castellano JM, Miller O, Kertser A, Berkutzki T, Barnett-Itzhaki Z, Bezalel D, Wyss-Coray T, Amit I, Schwartz M (2014) Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346:89–93. 10.1126/science.1252945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas AR, Presumey J, Das A, van der Poel CE, Lapchak PH, Mesin L, Victora G, Tsokos GC, Mawrin C, Herbst R, Carroll MC (2017) Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature 546:539–543. 10.1038/nature22821 [DOI] [PubMed] [Google Scholar]
- Biessels GJ, Cristino NA, Rutten GJ, Hamers FP, Erkelens DW, Gispen WH (1999) Neurophysiological changes in the central and peripheral nervous system of streptozotocin-diabetic rats: course of development and effects of insulin treatment. Brain 122:757–768. 10.1093/brain/122.4.757 [DOI] [PubMed] [Google Scholar]
- Brundel M, Reijmer YD, van Veluw SJ, Kuijf HJ, Luijten PR, Kappelle LJ, Biessels GJ (2014) Cerebral microvascular lesions on high-resolution 7-tesla MRI in patients with type 2 diabetes. Diabetes 63:3523–3529. 10.2337/db14-0122 [DOI] [PubMed] [Google Scholar]
- Carrillo-de Sauvage MÁ, Maatouk L, Arnoux I, Pasco M, Sanz Diez A, Delahaye M, Herrero MT, Newman TA, Calvo CF, Audinat E, Tronche F, Vyas S (2013) Potent and multiple regulatory actions of microglial glucocorticoid receptors during CNS inflammation. Cell Death Differ 20:1546–1557. 10.1038/cdd.2013.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Singh V, Krishnan A, Kan M, Martinez JA, Zochodne DW (2013) Loss of innervation and axon plasticity accompanies impaired diabetic wound healing. PLoS One 8:e75877. 10.1371/journal.pone.0075877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianchetti FA, Kim DH, Dimiduk S, Nishimura N, Schaffer CB (2013) Stimulus-evoked calcium transients in somatosensory cortex are temporarily inhibited by a nearby microhemorrhage. PLoS One 8:e65663. 10.1371/journal.pone.0065663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordonnier C, Al-Shahi Salman R, Wardlaw J (2007) Spontaneous brain microbleeds: systematic review, subgroup analyses and standards for study design and reporting. Brain 130:1988–2003. 10.1093/brain/awl387 [DOI] [PubMed] [Google Scholar]
- Cullen KM, Kócsi Z, Stone J (2005) Pericapillary haem-rich deposits: evidence for microhaemorrhages in aging human cerebral cortex. J Cereb Blood Flow Metab 25:1656–1667. 10.1038/sj.jcbfm.9600155 [DOI] [PubMed] [Google Scholar]
- Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT (2011) Age-related alterations in the dynamic behavior of microglia. Aging Cell 10:263–276. 10.1111/j.1474-9726.2010.00660.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758. 10.1038/nn1472 [DOI] [PubMed] [Google Scholar]
- Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K (2012) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3:1227. 10.1038/ncomms2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Zoppo GJ, Frankowski H, Gu YH, Osada T, Kanazawa M, Milner R, Wang X, Hosomi N, Mabuchi T, Koziol JA (2012) Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J Cereb Blood Flow Metab 32:919–932. 10.1038/jcbfm.2012.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dénes A, Ferenczi S, Halász J, Környei Z, Kovács KJ (2008) Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab 28:1707–1721. 10.1038/jcbfm.2008.64 [DOI] [PubMed] [Google Scholar]
- de Weerd NA, Nguyen T (2012) The interferons and their receptors-distribution and regulation. Immunol Cell Biol 90:483–491. 10.1038/icb.2012.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A, Hao S, Erion JR, Wosiski-Kuhn M, Stranahan AM (2014) Glucocorticoid sensitization of microglia in a genetic mouse model of obesity and diabetes. J Neuroimmunol 269:20–27. 10.1016/j.jneuroim.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebenezer GJ, O'Donnell R, Hauer P, Cimino NP, McArthur JC, Polydefkis M (2011) Impaired neurovascular repair in subjects with diabetes following experimental intracutaneous axotomy. Brain 134:1853–1863. 10.1093/brain/awr086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–397. 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-López D, Faustino J, Klibanov AL, Derugin N, Blanchard E, Simon F, Leib SL, Vexler ZS (2016) Microglial cells prevent hemorrhage in neonatal focal arterial stroke. J Neurosci 36:2881–2893. 10.1523/JNEUROSCI.0140-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiano AJ, Xu Y, Tustison NJ, Marsh RL, Baker W, Smirnov I, Overall CC, Gadani SP, Turner SD, Weng Z, Peerzade SN, Chen H, Lee KS, Scott MM, Beenhakker MP, Litvak V, Kipnis J (2016) Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 535:425–429. 10.1038/nature18626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J (2010) Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci 13:411–413. 10.1038/nn.2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glezer I, Simard AR, Rivest S (2007) Neuroprotective role of the innate immune system by microglia. Neuroscience 147:867–883. 10.1016/j.neuroscience.2007.02.055 [DOI] [PubMed] [Google Scholar]
- Goldmann T, Blank T, Prinz M (2016) Fine-tuning of type I IFN-signaling in microglia: implications for homeostasis, CNS autoimmunity and interferonopathies. Curr Opin Neurobiol 36:38–42. 10.1016/j.conb.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoneva S, Davalos D, Biswas D, Swanger SA, Garnier-Amblard E, Loth F, Akassoglou K, Traynelis SF (2014) Systemic inflammation regulates microglial responses to tissue damage in vivo. Glia 62:1345–1360. 10.1002/glia.22686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D (2006) The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 9:1512–1519. 10.1038/nn1805 [DOI] [PubMed] [Google Scholar]
- Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C, Husterer V, Croxford AL, Möller-Hackbarth K, Bartsch HS, Sotlar K, Krebs S, Regen T, Blum H, Hemmer B, Misgeld T, Wunderlich TF, Hidalgo J, Oukka M, Rose-John S, et al. (2017) Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH 17 cells. Nat Immunol 18:74–85. 10.1038/ni.3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–1905. 10.1038/nn.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilal S, Saini M, Tan CS, Catindig JA, Koay WI, Niessen WJ, Vrooman HA, Wong TY, Chen C, Ikram MK, Venketasubramanian N (2014) Cerebral microbleeds and cognition: the epidemiology of dementia in Singapore study. Alzheimer Dis Assoc Disord 28:106–112. 10.1097/WAD.0000000000000015 [DOI] [PubMed] [Google Scholar]
- Hines DJ, Hines RM, Mulligan SJ, Macvicar BA (2009) Microglia processes block the spread of damage in the brain and require functional chloride channels. Glia 57:1610–1618. 10.1002/glia.20874 [DOI] [PubMed] [Google Scholar]
- Holtman IR, Noback MA, Bijlsma M, Duong KN, van der Geest MA, Ketelaars PT, Boersma S, Dubbelaar ML, Brouwer N, Vainchtein ID, Eggen BJL, Boddeke HW (2014) Glia open access database. http://bioinf.nl:8080/GOAD2/. Accessed May 18, 2017. [DOI] [PubMed] [Google Scholar]
- Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Möller T, Orre M, Kamphuis W, Hol EM, Boddeke EW, Eggen BJ (2015) Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta Neuropathol Commun 3:31. 10.1186/s40478-015-0203-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang IK, Choi JH, Nam SM, Park OK, Yoo DY, Kim W, Yi SS, Won MH, Seong JK, Yoon YS (2014) Activation of microglia and induction of pro-inflammatory cytokines in the hippocampus of type 2 diabetic rats. Neurol Res 36:824–832. 10.1179/1743132814Y.0000000330 [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20:4106–4114. 10.1128/MCB.20.11.4106-4114.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas A, Pfleger C, Kharagjitsingh AV, Schloot NC, Hansen L, Buschard K, Koeleman BP, Roep BO, Mortensen HB, Alizadeh BZ, Alizadeh BZ (2012) Association between age, IL-10, IFN-γ, stimulated C-peptide and disease progression in children with newly diagnosed Type1 diabetes. Diabet Med 29:734–741. 10.1111/j.1464-5491.2011.03544.x [DOI] [PubMed] [Google Scholar]
- Kawanokuchi J, Mizuno T, Takeuchi H, Kato H, Wang J, Mitsuma N, Suzumura A (2006) Production of interferon-gamma by microglia. Mult Scler 12:558–564. 10.1177/1352458506070763 [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91:461–553. 10.1152/physrev.00011.2010 [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Prinz M (2013) Factors regulating microglia activation. Front Cell Neurosci 7:44. 10.3389/fncel.2013.00044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Lee SH (2013) Cerebral microbleeds: their associated factors, radiologic findings, and clinical implications. J Stroke 15:153–163. 10.5853/jos.2013.15.3.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Cho S (2016) Microglia and monocyte-derived macrophages in stroke. Neurotherapeutics 13:702–718. 10.1007/s13311-016-0463-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo F, Asanuma M, Miyazaki I, Kondo Y, Tanaka K, Makino H, Ogawa N (2001) Progressive cortical atrophy after forebrain ischemia in diabetic rats. Neurosci Res 39:339–346. 10.1016/S0168-0102(00)00233-9 [DOI] [PubMed] [Google Scholar]
- Kumari R, Willing LB, Krady JK, Vannucci SJ, Simpson IA (2007) Impaired wound healing after cerebral hypoxia-ischemia in the diabetic mouse. J Cereb Blood Flow Metab 27:710–718. 10.1038/sj.jcbfm.9600382 [DOI] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S, Kriz J (2012) Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci 32:10383–10395. 10.1523/JNEUROSCI.1498-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I (2014) Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159:1312–1326. 10.1016/j.cell.2014.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzen S. (2008) The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 51:216–226. 10.1007/s00125-007-0886-7 [DOI] [PubMed] [Google Scholar]
- Li G, Xu X, Wang D, Wang J, Wang Y, Yu J (2011) Microglial activation during acute cerebral infarction in the presence of diabetes mellitus. Neurol Sci 32:1075–1079. 10.1007/s10072-011-0632-2 [DOI] [PubMed] [Google Scholar]
- Li W, Prakash R, Kelly-Cobbs AI, Ogbi S, Kozak A, El-Remessy AB, Schreihofer DA, Fagan SC, Ergul A (2010) Adaptive cerebral neovascularization in a model of type 2 diabetes: relevance to focal cerebral ischemia. Diabetes 59:228–235. 10.2337/db09-0902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Qu Z, Prakash R, Chung C, Ma H, Hoda MN, Fagan SC, Ergul A (2013) Comparative analysis of the neurovascular injury and functional outcomes in experimental stroke models in diabetic Goto-Kakizaki rats. Brain Res 1541:106–114. 10.1016/j.brainres.2013.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J (2010) CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J Neurosci 30:17091–17101. 10.1523/JNEUROSCI.4403-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou N, Takano T, Pei Y, Xavier AL, Goldman SA, Nedergaard M (2016) Purinergic receptor P2RY12-dependent microglial closure of the injured blood–brain barrier. Proc Natl Acad Sci U S A 113:1074–1079. 10.1073/pnas.1520398113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Wook Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madry C, Kyrargyri V, Arancibia-Cárcamo IL, Jolivet R, Kohsaka S, Bryan RM, Attwell D (2018) Microglial ramification, surveillance, and interleukin-1β release are regulated by the two-pore domain K+ channel THIK-1. Neuron 97:299–312.e6. 10.1016/j.neuron.2017.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marwaha AK, Tan S, Dutz JP (2014) Targeting the IL-17/IFN-γ axis as a potential new clinical therapy for type 1 diabetes. Clin Immunol 154:84–89. 10.1016/j.clim.2014.06.006 [DOI] [PubMed] [Google Scholar]
- Masuda T, Croom D, Hida H, Kirov SA (2011) Capillary blood flow around microglial somata determines dynamics of microglial processes in ischemic conditions. Glia 59:1744–1753. 10.1002/glia.21220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran C, Phan TG, Chen J, Blizzard L, Beare R, Venn A, Münch G, Wood AG, Forbes J, Greenaway TM, Pearson S, Srikanth V (2013) Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care 36:4036–4042. 10.2337/dc13-0143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti F, Conget I, Di Mauro M, Di Marco R, Mazzarino MC, Bendtzen K, Messina A, Gomis R (2002) Serum concentrations of the interferon-γ-inducible chemokine IP-10/CXCL10 are augmented in both newly diagnosed type I diabetes mellitus patients and subjects at risk of developing the disease. Diabetologia 45:1107–1110. 10.1007/s00125-002-0879-5 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, Germain MA, Bourdel G, Popovic N, Rezende FA, Kaufman RJ, Mallette FA, Sapieha P (2016) Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med 8:362ra144. 10.1126/scitranslmed.aaf9440 [DOI] [PubMed] [Google Scholar]
- Pan W, Banks WA, Kastin AJ (1997) Permeability of the blood–brain and blood–spinal cord barriers to interferons. J Neuroimmunol 76:105–111. 10.1016/S0165-5728(97)00034-9 [DOI] [PubMed] [Google Scholar]
- Pradhan L, Cai X, Wu S, Andersen ND, Martin M, Malek J, Guthrie P, Veves A, Logerfo FW (2011) Gene expression of pro-inflammatory cytokines and neuropeptides in diabetic wound healing. J Surg Res 167:336–342. 10.1016/j.jss.2009.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch A, Suarez-Pinzon W, El-Sheikh A, Sorensen O, Power RF (1996) Cytokine gene expression in pancreatic islet-infiltrating leukocytes of BB rats: expression of Th1 cytokines correlates with β-cell destructive insulitis and IDDM. Diabetes 45:749–754. 10.2337/diab.45.6.749 [DOI] [PubMed] [Google Scholar]
- Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW (2016) Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 139:653–661. 10.1093/brain/awv395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeson P, Tennant KA, Gerrow K, Wang J, Weiser Novak S, Thompson K, Lockhart KL, Holmes A, Nahirney PC, Brown CE (2015) Delayed inhibition of VEGF signaling after stroke attenuates blood–brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J Neurosci 35:5128–5143. 10.1523/JNEUROSCI.2810-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosidi NL, Zhou J, Pattanaik S, Wang P, Jin W, Brophy M, Olbricht WL, Nishimura N, Schaffer CB (2011) Cortical microhemorrhages cause local inflammation but do not trigger widespread dendrite degeneration. PLoS One 6:e26612. 10.1371/journal.pone.0026612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Kipnis J, Rivest S, Prat A (2013) How do immune cells support and shape the brain in health, disease, and aging? J Neurosci 33:17587–17596. 10.1523/JNEUROSCI.3241-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SW, Hwa Lee B, Kim EJ, Chin J, Sun Cho Y, Yoon U, Na DL (2007) Clinical significance of microbleeds in subcortical vascular dementia. Stroke 38:1949–1951. 10.1161/STROKEAHA.106.477315 [DOI] [PubMed] [Google Scholar]
- Shams S, Martola J, Granberg T, Li X, Shams M, Fereshtehnejad SM, Cavallin L, Aspelin P, Kristoffersen-Wiberg M, Wahlund LO (2015) Cerebral microbleeds: different prevalence, topography, and risk factors depending on dementia diagnosis: the Karolinska Imaging Dementia Studay. AJNR Am J Neuroradiol 36:661–666. 10.3174/ajnr.A4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Driscoll JD, Drew PJ, Nishimura N, Schaffer CB, Kleinfeld D (2012) Two-photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. J Cereb Blood Flow Metab 32:1277–1309. 10.1038/jcbfm.2011.196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann HM, Waagepetersen HS, Schousboe A, Benie AJ, Bouman SD (2012) Brain glycogen and its role in supporting glutamate and GABA homeostasis in a type 2 diabetes rat model. Neurochem Int 60:267–275. 10.1016/j.neuint.2011.12.019 [DOI] [PubMed] [Google Scholar]
- Sweetnam D, Holmes A, Tennant KA, Zamani A, Walle M, Jones P, Wong C, Brown CE (2012) Diabetes impairs cortical plasticity and functional recovery following ischemic stroke. J Neurosci 32:5132–5143. 10.1523/JNEUROSCI.5075-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalay G, Martinecz B, Lénárt N, Környei Z, Orsolits B, Judák L, Császár E, Fekete R, West BL, Katona G, Rózsa B, Dénes Á (2016) Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 7:11499. 10.1038/ncomms11499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Gan Y, Liu Q, Yin JX, Liu Q, Shi J, Shi FD (2014) CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation 11:26. 10.1186/1742-2094-11-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay TL, Savage JC, Hui CW, Bisht K, Tremblay MÈ (2017) Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J Physiol 595:1929–1945. 10.1113/JP272134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SL, Trudeau D, Arnold B, Wang J, Gerrow K, Summerfeldt K, Holmes A, Zamani A, Brocardo PS, Brown CE (2015) VEGF can protect against blood brain barrier dysfunction, dendritic spine loss and spatial memory impairment in an experimental model of diabetes. Neurobiol Dis 78:1–11. 10.1016/j.nbd.2015.03.022 [DOI] [PubMed] [Google Scholar]
- Tennant KA, Brown CE (2013) Diabetes augments in vivo microvascular blood flow dynamics after stroke. J Neurosci 33:19194–19204. 10.1523/JNEUROSCI.3513-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Masuda T, Kitano J, Shimoyama H, Tozaki-Saitoh H, Inoue K (2009) IFN-gamma receptor signaling mediates spinal microglia activation driving neuropathic pain. Proc Natl Acad Sci U S A 106:8032–8037. 10.1073/pnas.0810420106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti R, Del Bene A, Poggesi A, Ginestroni A, Salvadori E, Pracucci G, Ciolli L, Marini S, Nannucci S, Pasi M, Pescini F, Diciotti S, Orlandi G, Cosottini M, Chiti A, Mascalchi M, Bonuccelli U, Inzitari D, Pantoni L, Pantoni L (2016) Cerebral microbleeds in patients with mild cognitive impairment and small vessel disease: The Vascular Mild Cognitive Impairment (VMCI)-Tuscany Study. J Neurol Sci 368:195–202. 10.1016/j.jns.2016.07.018 [DOI] [PubMed] [Google Scholar]
- Varewijck AJ, Janssen JA (2012) Insulin and its analogues and their affinities for the IGF1 receptor. Endocr Relat Cancer 19:F63–F75. 10.1530/ERC-12-0026 [DOI] [PubMed] [Google Scholar]
- Vinik A, Flemmer M (2002) Diabetes and macrovascular disease. J Diabetes Complications 16:235–245. 10.1016/S1056-8727(01)00212-4 [DOI] [PubMed] [Google Scholar]
- Volpe CM, Villar-Delfino PH, Dos Anjos PM, Nogueira-Machado JA (2018) Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis 9:119. 10.1038/s41419-017-0135-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong B, Odero G, Rozbacher S, Stevenson M, Kereliuk SM, Pereira TJ, Dolinsky VW, Kauppinen TM (2017) Exposure to gestational diabetes mellitus induces neuroinflammation, derangement of hippocampal neurons, and cognitive changes in rat offspring. J Neuroinflammation 14:1–13. 10.1186/s12974-017-0859-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, André I, Gonzalez A, Katz JD, Aguet M, Benoist C, Mathis D (1997) Interferon-gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci U S A 94:13844–13849. 10.1073/pnas.94.25.13844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2016) Global report on diabetes. Geneva: World Health Organization [Google Scholar]
- Woerdeman J, van Duinkerken E, Wattjes MP, Barkhof F, Snoek FJ, Moll AC, Klein M, de Boer MP, IJzerman RG, Serné EH, Diamant M (2014) Proliferative retinopathy in type 1 diabetes is associated with cerebral microbleeds, which is part of generalized microangiopathy. Diabetes Care 37:1165–1168. 10.2337/dc13-1586 [DOI] [PubMed] [Google Scholar]
- Won SJ, Tang XN, Suh SW, Yenari MA, Swanson RA (2011) Hyperglycemia promotes tissue plasminogen activator-induced hemorrhage by increasing superoxide production. Ann Neurol 70:583–590. 10.1002/ana.22538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Uchigata Y, Okamoto H (1981) Streptozotocin and alloxan induce DNA strand breaks and poly(ADP-ribose) synthetase in pancreatic islets. Nature 294:284–286. 10.1038/294284a0 [DOI] [PubMed] [Google Scholar]
- Ye X, Chopp M, Cui X, Zacharek A, Cui Y, Yan T, Shehadah A, Roberts C, Liu X, Lu M, Chen J (2011) Niaspan enhances vascular remodeling after stroke in type 1 diabetic rats. Exp Neurol 232:299–308. 10.1016/j.expneurol.2011.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J (2016) TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90:724–739. 10.1016/j.neuron.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Murphy TH (2007) Imaging the impact of cortical microcirculation on synaptic structure and sensory-evoked hemodynamic responses in vivo. PLoS Biol 5:1152–1167. 10.1371/journal.pbio.0050119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zherebitskaya E, Akude E, Smith DR, Fernyhough P (2009) Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes 58:1356–1364. 10.2337/db09-0034 [DOI] [PMC free article] [PubMed] [Google Scholar]