

 Fusidic acid is a natural product antibiotic used clinically, primarily against staphylococcal infections.
Fusidic acid is a natural product antibiotic used clinically, primarily against staphylococcal infections.
Abstract
Fusidic acid is a natural product antibiotic used clinically, primarily against staphylococcal infections. It has also exhibited antimycobacterial activity against Mycobacterium species, including Mycobacterium tuberculosis (Mtb). Novel C-21 fusidic acid amides were synthesized and evaluated for antimycobacterial activity in a drug repositioning approach for tuberculosis. The synthesized compounds exhibited good potency in MB7H9/CAS medium albeit showing low to no activity in MB7H9/ADC medium. The fusidic acid ethanamides were, generally, the most potent of the analogues evaluated for antimycobacterial activity (MIC90 < 10 μM) in the MB7H9/CAS medium. The lack of activity in the MB7H9/ADC medium was supported by strong binding interactions in the fusidic acid binding site of the human serum albumin (HSA) protein. The most potent antimycobacterial analogue was the N-(4-sulfamoylbenzyl)fusidic acid amide (1.26) with an MIC90 value of 2.71 μM.
Introduction
Tuberculosis (TB) is caused by the bacillus-type bacterium Mycobacterium tuberculosis (Mtb). According to the WHO Global tuberculosis report 2018, an estimated 1.6 million deaths were recorded in 2017. High disease prevalence was recorded in children and adults with compromised immune systems, especially HIV positive patients.1 TB can be cured and prevented. Its treatment is afflicted with a long chemotherapeutic regimen (at least 6 months) and a high drug dose (4 anti-TB first-line drugs: isoniazid, rifampicin, ethambutol and pyrazinamide). However, the lack of adherence to this exhaustive regimen, inaccurate prescriptions and poor quality drugs have led to resistance to single or multiple anti-TB drugs.2
Multidrug-resistant TB (MDR-TB), which is resistant to isoniazid and rifampicin, still thrives as a major contributor to the global reported TB cases in 2017.1 MDR-TB patients require a much longer treatment regimen (up to 2 years) with expensive and toxic second-line drugs (fluoroquinolones, kanamycin, capreomycin and amikacin). In extensively drug-resistant TB (XDR-TB) cases, the Mtb bacterium is resistant to most of the second-line anti-TB drugs. These patients may require the inclusion of the new anti-TB drugs, bedaquiline and delamanid, as part of their long MDR-TB chemotherapy regimen.1 Meanwhile, at least 25% of the world's population is reported to have latent TB, which awaits the development into transmissible forms of the disease due to a number of risk factors, including the immune system of the host being compromised. These drawbacks in TB treatment and prevention, namely the resistance to current anti-TB chemotherapy and latent TB infection, respectively, seem to suggest the prevalence of the disease in humankind for many generations to come if research to discover and develop new anti-TB drugs is not intensified.
Fusidic acid (Fig. 1) was first reported from the fungus Fusidium coccineum.3,4 It is the most potent member of the fusidane class of antibiotics and has been clinically used as an antibiotic since 1962. Fusidic acid is largely bacteriostatic but exhibits bactericidal activity at high concentrations. It interferes with the function of the protein elongation factor G (EF-G), thereby inhibiting protein synthesis in the bacteria. EF-G is necessary for the hydrolysis of guanosine triphosphate (GTP) to guanosine diphosphate (GDP) to provide energy for the translocation of the peptidyl-tRNA from the A site to the P site on the 50S unit of the ribosome. Fusidic acid binds to EF-G and the ribosome after hydrolysis of GTP, thus sterically blocking the next stage of protein synthesis.5
Fig. 1. Chemical structure of fusidic acid (1.0).

Although almost exclusively used as an anti-staphylococcal agent, fusidic acid has displayed antibacterial activity against a wide range of pathogens, except for Gram-negative bacilli.6,7 The antibacterial structure–activity relationship (SAR) of fusidic acid has revealed that the trans–syn–trans conformation of the tetracyclic triterpene backbone and the carboxylic acid and acetoxy groups are required for activity. The orientation of the lipophilic side chain and the carboxyl group around the Δ17,20 bond, rather than the double bond, has been demonstrated to be essential for antibacterial activity.8–11
Fusidic acid has also exhibited activity against some Mycobacterium species including Mtb with an MIC90 value of 31 μM.12–14 In an in vitro susceptibility study of 170 clinical isolates of Mtb to fusidic acid, only 3 of the 151 susceptible clinical isolates were resistant to fusidic acid with an MIC90 value >128 mg L–1 while 19 resistant isolates were susceptible to fusidic acid with an MIC90 value ≤16 mg L–1. With no cross resistance observed between fusidic acid and first-line anti-TB drugs, fusidic acid is a viable candidate for repositioning for TB.12 As part of our efforts to reposition fusidic acid for TB,15 we report the synthesis, antimycobacterial activity and cytotoxicity of novel C-21 amide analogues. Previous antibacterial SAR studies have revealed the importance of the carboxylic acid to activity.9 In order to establish the significance of the carboxylic acid to the antimycobacterial activity, we initially focused on amide derivatives as the amide functionality is one of the carboxylic acid bioisosteres.
Results and discussion
Chemistry
Fusidic acid C-21 ethanamides, anilides and benzyl amides were explored in the current work. The ethanamide analogues incorporated enantiomerically pure (R or S) and racemic N-aryl and (R)- and (S)-N-cyclohexyl ethanamines. The enantiomerically pure (R or S) ethanamines gave rise to pure enantiomeric (R or S) fusidic acid analogues. The aryl groups comprised phenyl, pyridyl, and pyrazinyl groups. Meanwhile, the choice of para-substituted groups of the benzyl amines was inspired by the Craig plot, which is a two-dimensional map of hydrophobicity (π) against the sigma constant (σ) of the Hammett equation or the steric term (ER) of the Taft equation.16–18 The Craig plot uses physicochemical descriptors to characterize molecular structure and facilitates the choice of substituents, which can span the appropriate chemical property space. This approach enabled exploration of the structure–antimycobacterial activity correlation based on the substituents on the phenyl ring.
The synthetic routes to arrive at 1-aryl/alkyl ethanamides (1.1–1.17) and anilides (1.18–1.23) and N-benzyl amides (1.24–1.28) of fusidic acid are outlined in Schemes 1 and 2, respectively. The C-21 amide derivatives were obtained by heating commercially sourced fusidic acid (1.0) and the respective amines in dichloromethane (DCM) in the presence of triethylamine (NEt3) as a base and propylphosphonic anhydride (T3P®) as a coupling reagent (Schemes 1 and 2a). The amide coupling reactions proceeded to completion in a time range of 5–24 hours depending on the amine used. Aqueous work-up to get rid of the reaction by-products afforded the crude organic residue, which upon further purification by normal phase silica preparative TLC gave the compounds in poor to good yields (10–70%). Pure (R)- and (S)-fusidic acid ethanamide derivatives were obtained from the corresponding amines. The racemic ethanamines afforded fusidic acid diastereomers (1.9–1.14) in a 1 : 1 to 3 : 2 ratio, except for 1.5 and 1.6 which were well separated and hence were isolated in their pure forms.
Scheme 1. Synthesis of fusidic acid ethanamide derivatives (1.1–1.17). Reagents and conditions: RNH2, T3P (50% w/v solution in EtOAc), NEt3, DCM, 35 °C, 5–24 h (depending on the amine).

Scheme 2. Synthesis of fusidic acid anilide (1.18–1.23) and N-benzyl amide derivatives (1.24–1.28). Reagents and conditions: (a) RNH2, T3P (50% w/v solution in EtOAc), NEt3, DCM, 35 °C, 5–24 h; (b) HATU, DCM, 30 °C, 2 h (for 1.23); (c) RNH2, KH2PO4, n-BuOH, 100 °C, 16 h (for 1.23).

The 4-hydroxyanilide of fusidic acid (1.23) was obtained in two steps (Scheme 2b and c). The first step was the reaction between fusidic acid and the coupling agent HATU. After purification, the intermediate was then subjected to a nucleophilic substitution reaction by heating with p-hydroxyaniline in the presence of KH2PO4 as a base and n-BuOH as a solvent.
Antimycobacterial activity
In vitro antimycobacterial activity evaluation of all the synthesized analogues was conducted using the H37RvMa strain (ATCC 27294 virulent laboratory strain) of Mtb. The activity was evaluated at MIC90 (minimum concentration required to inhibit the growth of 90% of the bacterial population), using rifampicin as the reference drug. The synthesized compounds were tested in a concentration range of 0.244–125 μM. Two growth media were used, namely Middlebrook 7H9 (MB7H9) medium supplemented with albumin–dextrose–catalase (ADC; BD, BBL, Cat. No. 212352), 0.2% glucose (GLU) and 0.05% Tween 80 (TW), and Middlebrook 7H9 medium supplemented with 0.03% casitone (CAS), 0.4% glucose and 0.05% tyloxapol (TX). Albumin, the major serum protein, has been reported to bind strongly to drugs, thereby reducing their bioavailability in vivo. In fact, fusidic acid is highly protein bound (95–97%), sharing similar tetracyclic structures to adrenocorticoids and cholate and taurocholate bile salts.19–21 While the albumin medium enabled an analysis of the activities of the synthesized fusidic acid analogues, including the extent of protein binding compared to fusidic acid, the Casitone medium, as we envisaged, served as a better representation of the activity of the synthesized analogues. Similarly, based on reports of the effect of the detergent employed in the assay on the activity observed for the test compounds, two detergents, namely Tween 80 and tyloxapol, were used.22 To enable analysis of their SAR, all the compounds with MIC90 ≤ 10 μM were regarded as active (relative to fusidic acid (1.0), Table 1); those with MIC90 ranging from 10–20 μM had moderate activity; those with MIC90 = 20–125 μM were denoted as poorly active; compounds exhibiting MIC90 > 125 μM were considered inactive. Further, the structure–activity relationships (SARs) of the compounds are discussed below within their chemical classes as ethanamides, anilides and benzyl amides.
Table 1. In vitro antimycobacterial and cytotoxic activities of the fusidic acid analogues.

| |||||
| Compound | R | Yield (%) | Antimycobacterial activity, H37RvMa, MIC90 (μM) |
Cytotoxicity, IC50 (μM) | |
| MB7H9 GLU ADC TW a | MB7H9 GLU CAS TX a | CHO a | |||
| 1.0 | OH | — | 9.25 | 0.24 | — |
| 1.1 |

|
65 | >125 | 8.90 | 45.95 |
| 1.2 |

|
60 | >125 | 11.00 | 47.61 |
| 1.3 |

|
25 | >125 | 7.59 | 47.40 |
| 1.4 |

|
20 | >125 | >125 | 8.24 |
| 1.5 |

|
33 | 41.52 | 9.80 | 45.15 |
| 1.6 |

|
8 | >125 | 10.10 | 18.16 |
| 1.7 |

|
9 | 38.83 | 7.10 | 42.88 |
| 1.8 |

|
17 | 15.73 | 9.10 | >50 |
| 1.9 |

|
58 | >125 | 2.91 | 47.90 |
| 1.10 |

|
40 | >125 | 19.82 | 45.30 |
| 1.11 |

|
25 | 32.26 | 7.20 | >50 |
| 1.12 |

|
48 | >125 | 17.87 | >50 |
| 1.13 |

|
48 | 125 | 13.03 | >50 |
| 1.14 |

|
40 | 62.50 | 17.49 | >50 |
| 1.15 |

|
28 | >125 | 13.90 | 36.84 |
| 1.16 |

|
35 | >125 | 4.50 | 44.69 |
| 1.17 |

|
43 | >125 | 7.45 | >50 |
| 1.18 |

|
20 | 43.21 | 6.20 | 46.13 |
| 1.19 |

|
19 | >125 | >125 | 42.68 |
| 1.20 |

|
21 | >125 | >125 | 47.12 |
| 1.21 |
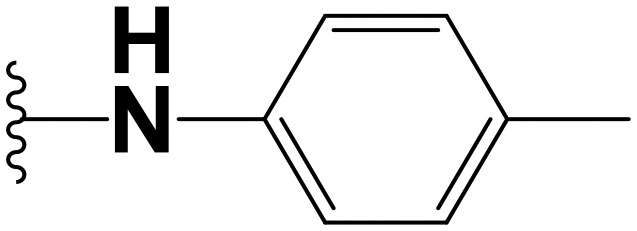
|
16 | 125 | >125 | >50 |
| 1.22 |

|
35 | 125 | 125 | 39.90 |
| 1.23 |

|
32 | >125 | 6.90 | 45.39 |
| 1.24 |
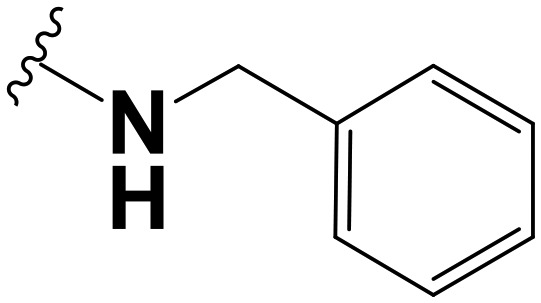
|
70 | 125 | >125 | 44.10 |
| 1.25 |

|
50 | 125 | >125 | 40.20 |
| 1.26 |

|
10 | 62.27 | 2.71 | 30.20 |
| 1.27 |

|
17 | 125 | 125 | >50 |
| 1.28 |

|
56 | 125 | >125 | >50 |
| Rifampicin | 9.00 nM | 0.02 | |||
| Emetine | 0.02 | ||||
aMB7H9 GLU ADC TW (Middlebrook 7H9 medium supplemented with glucose, albumin–dextrose–catalase, and Tween); MB7H9 GLU ADC TW (Middlebrook 7H9 medium supplemented with glucose, casitone, and tyloxapol); CHO (Chinese hamster ovarian cell line).
The fusidic acid ethanamide analogues were either poorly active or inactive in the protein-based MB7H9/ADC medium, except for compound 1.8 which showed moderate activity (MIC90 = 15.73 μM). We hypothesized that the poor activity could be attributed to strong binding to albumin since all the analogues (except for 1.4) showed improved potency in the non-protein-based medium. In general, analogues with polar groups and/or substituents in close proximity to the amide bond showed inhibitory activity in the MB7H9/ADC medium, suggesting that they were less strongly bound to albumin. This is particularly exemplified in the activities of 1.7, 1.8, 1.11 and 1.14, which displayed MIC90 values of 38.83, 15.73, 32.26, and 62.5 μM, respectively (Table 1).
The enantiomerically pure fusidic acid ethanamide analogues exhibited comparable activity (MIC90 = 7–12 μM) in the MB7H9/CAS medium, except for compound 1.4 which was inactive. Generally, the S-enantiomer configuration at the benzylic position exhibited better potency than the corresponding R-enantiomer. The cyclohexyl congener 1.16 was the most potent analogue amongst the diastereomeric fusidic acid ethanamides in the MB7H9/CAS medium, exhibiting about 2-fold better activity (MIC90 = 4.50 μM). Meanwhile, all the diastereomeric ethanamide analogues exhibited moderate activity (MIC90 = 13–20 μM), except for compounds 1.9 (MIC90 = 2.91 μM) and 1.11 (MIC90 = 7.20 μM). The 4-pyridinyl analogue 1.13 was the most active (MIC90 = 13.03 μM) analogue amongst the nitrogen heterocyclic ethanamide fusidic acid analogues. The 3-pyridinyl and the 2,5-pyrazinyl congeners exhibited comparable activity (MIC90 = ∼17 μM).
Amongst the fusidic acid anilide analogues, it was observed that substitution at the ortho position favored activity in both the MB7H9/ADC and MB7H9/CAS media. This is exemplified by comparison of the activities of the three methoxy-substituted anilide regioisomers 1.18, 1.19 and 1.20 (Table 1). Compound 1.18 with the methoxy group ortho to the amide bond displayed good activity (MIC90 = 6.20 μM) in the non-protein-based medium but poor activity (MIC90 = 43.21 μM) in the protein-based medium. However, the meta- and para-substituted congeners were not potent in both media. Similarly, compounds 1.21 and 1.22, which bear lipophilic electron-releasing and electron-withdrawing groups, respectively, were equally inactive in both media. Meanwhile, it was observed that activity in the MB7H9/CAS medium was restored if the para position bears a hydrophilic electron-donating group capable of forming hydrogen bonds, as exemplified by 1.23 (MIC90 = 6.90 μM).
Amongst the fusidic acid benzyl amide analogues, it was observed that all the analogues, including the unsubstituted benzyl amide 1.24, were inactive in both the MB7H9/ADC and MB7H9/CAS media, except for compound 1.26 (MB7H9/ADC: MIC90 = 62.27 μM; MB7H9/CAS: MIC90 = 2.71 μM), which bears a sulfonamide group at the para position. This seems to suggest the importance, to antimycobacterial activity, of hydrophilic-electron withdrawing substituent groups on the phenyl/benzyl ring.
Cytotoxicity
Cytotoxicity profiling of the synthesized compounds on mammalian cells was carried out using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay at a maximum concentration of 50 μM of the test compounds. All the synthesized compounds displayed low to no cytotoxicity (IC50 > 30 μM) at the maximum concentration, except for the ethanamide analogue 1.4 (IC50 = 8.24 μM). Meanwhile, compound 1.6 exhibited moderate cytotoxicity (IC50 = 18.16 μM). The most potent fusidic acid antimycobacterial analogue, 1.26, exhibited an acceptable cytotoxic activity (IC50 = 30.20 μM) against the Chinese hamster ovarian cell line (CHO-K1; American Type Culture Collection, Cat No. CCL-61). Measurement of antimycobacterial activity as MIC90 and cytotoxicity as IC50 could not allow for an assessment of the selectivity indices of the compounds.
HSA docking studies
To validate the hypothesis that the lack of in vitro antimycobacterial activity of some synthesized fusidic acid analogues in the MB7H9/ADC medium (albeit with good potency in the 7H9/CAS medium) is attributable to strong serum albumin binding, molecular docking studies were conducted on the fusidic acid analogues with human serum albumin (HSA) (Fig. 2). Bovine serum albumin (present in the MB7H9/ADC medium) is similar to HSA.23 The crystal structure of HSA complexed with fusidic acid (PDB ID: ; 2VUF) was acquired from the Protein Data Bank (PDB).24 The reported orientation of fusidic acid in the predefined binding site of HSA was reproduced and the docking protocols validated and performed using Glide25 of Schrödinger 2018-3.26 The co-crystallised fusidic acid revealed the favorable interactions with amino acid residues. Tyr161 forms crucial hydrogen bond (H-bond) interactions with both hydroxyls (at C-3 and C-11) of fusidic acid, which are necessary for HSA binding. Other characteristic interactions observed are between the acetate ester carbonyl and Arg117, while the carbonyl of the carboxylic acid interacted by H-bonding with Arg186.24
Fig. 2. Molecular docking of compound 1.3 in human serum albumin (HSA). The contact residues are shown and labelled. The yellow dotted lines illustrate the hydrogen bond interactions.

The docked complexes showed that some analogues bind quite strongly to HSA. In addition to the characteristic interactions, the carbonyl and NH of the amide linker form H-bond interactions with Arg186 and Leu115, respectively, which lead to an increase in the binding energy of the analogues. The absence of the methyl linker puts the lipophilic terminal in close proximity to Arg186. This creates a pi–cation interaction increasing the binding energy as observed in the change of potency between 1.3 and 1.22. The lipophilic compounds fit well into the binding pocket, as previously hypothesized.24 The meta and para substitutions are preferred for HSA binding as observed in 1.19 and 1.20, respectively. The ortho substitution may distort HSA binding as in 1.18. The presence of bulky and polar substituents as observed in compounds 1.8 and 1.26 reduced HSA binding. These significant binding interactions of the fusidic acid analogues with HSA may lead to reduced bioavailability of the compounds for the site of infection. The predictions from these docking studies are important to make informed decisions into future SAR explorations aimed at reducing the HSA binding of fusidic acid analogues while increasing the potency in the quest to reposition fusidic acid for TB.
Conclusion
C-21 fusidic acid ethanamides, anilides and benzyl amides were synthesized as novel antimycobacterial agents against the H37RvMa strain of Mtb. Antimycobacterial activity was favored by the presence of bulky and/or polar substituents, notably, in close proximity to the amide bond. The antimycobacterial activity data obtained for the synthesized analogues reported herein, compared to fusidic acid, seem to suggest the importance of the C-21 carboxylic acid functionality to the activity. However, more analogues need to be explored to validate this hypothesis. Furthermore, based on the comparatively poor or no activity of the synthesized compounds to that of fusidic acid in the MB7H9/ADC medium, it is concluded that the analogues exhibited strong binding interactions with HSA compared to fusidic acid. We observed that the strong binding interaction between HSA and the fusidic acid amide analogues was favored by lipophilic substituents on the phenyl moiety, which exhibited the lack of, or poor, antimycobacterial activity in the MB7H9/ADC medium albeit having good potency in the MB7H9/CAS medium. Meanwhile, the compounds exhibited acceptable cytotoxic activity inhibitory concentrations against the CHO cell line.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The authors are grateful for the support of the University of Cape Town, South African Medical Research Council (SAMRC) and the South African Research Chairs Initiative (SARChI) of the Department of Science and Technology administered through the South African National Research Foundation (K. C.). We thank Dr Dale Taylor of the Drug Discovery and Development Centre (H3D) for the cytotoxicity data generated on the compounds.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00161a
References
- WHO, Global tuberculosis report, 2018, http://www.who.int/news-room/fact-sheets/detail/tuberculosis.
- Silva D. R., Dalcolmo M., Tiberi S., Arbex M. A., Munoz-Torrico M., Duarte R., D'Ambrosio L., Visca D., Rendon A., Gaga M. J. Bras. Pneumol. 2018;44(2):153–160. doi: 10.1590/S1806-37562017000000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godtfredsen W. O., Jahnsen S., Lorck H., Roholt K., Tybring L. Nature. 1962;193:987. doi: 10.1038/193987a0. [DOI] [PubMed] [Google Scholar]
- Godtfredsen W. O., Rastrup-Andersen N., Vangedal S., Ollis W. D. Tetrahedron. 1979;35(20):2419–2431. [Google Scholar]
- Dobie D., Gray J. Arch. Dis. Child. 2004;89(1):74–77. doi: 10.1136/adc.2003.019695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R. N., Mendes R. E., Sader H. S., Castanheira M. Clin. Infect. Dis. 2011;52(suppl_7):S477–S486. doi: 10.1093/cid/cir163. [DOI] [PubMed] [Google Scholar]
- FUCITHALMIC® (fusidic acid) Product Monograph, version 1.0 (2014.02.03), http://methapharm.com/wp-content/uploads/2015/11/Fucithalmic-PM00025061.pdf, (accessed Aug 13, 2018).
- Godtfredsen W. O., von Daehne W., Tybring L., Vangedal S. J. Med. Chem. 1966;9(1):15–22. doi: 10.1021/jm00319a004. [DOI] [PubMed] [Google Scholar]
- von Daehne W., Godtfredsen W. O., Rasmussen P. R. Adv. Appl. Microbiol. 1979;25:95–146. doi: 10.1016/s0065-2164(08)70148-5. [DOI] [PubMed] [Google Scholar]
- Duvold T., Sørensen M. D., Björkling F., Henriksen A. S., Rastrup-Andersen N. J. Med. Chem. 2001;44(19):3125–3131. doi: 10.1021/jm010899a. [DOI] [PubMed] [Google Scholar]
- Duvold T., Jørgensen A., Andersen N. R., Henriksen A. S., Dahl Sørensen M., Björkling F. Bioorg. Med. Chem. Lett. 2002;12(24):3569–3572. doi: 10.1016/s0960-894x(02)00797-7. [DOI] [PubMed] [Google Scholar]
- Cicek-Saydam C., Cavusoglu C., Burhanoglu D., Hilmioglu S., Ozkalay N., Bilgic A. Clin. Microbiol. Infect. 2001;7(12):700–702. doi: 10.1046/j.1469-0691.2001.00341.x. [DOI] [PubMed] [Google Scholar]
- Van Caekenberghe D. J. Antimicrob. Chemother. 1990;26(3):381–386. doi: 10.1093/jac/26.3.381. [DOI] [PubMed] [Google Scholar]
- Witzig R. S., Franzblau S. G. Antimicrob. Agents Chemother. 1993;37(9):1997–1999. doi: 10.1128/aac.37.9.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigondu E. M., Wasuna A., Warner D. F., Chibale K. Bioorg. Med. Chem. 2014;22(16):4453–4461. doi: 10.1016/j.bmc.2014.06.012. [DOI] [PubMed] [Google Scholar]
- Hansch C. Acc. Chem. Res. 1969;2(8):232–239. [Google Scholar]
- Taft Jr. R. W., Steric Effects in Organic Chemistry, ed. M. S. Newman, Wiley, New York, NY, 1956. [Google Scholar]
- Craig P. N. J. Med. Chem. 1971;14(8):680–684. doi: 10.1021/jm00290a004. [DOI] [PubMed] [Google Scholar]
- Godtfredsen W., Roholt K., Tybring L. Lancet. 1962;279(7236):928–931. doi: 10.1016/s0140-6736(62)91968-2. [DOI] [PubMed] [Google Scholar]
- Turnidge J. Int. J. Antimicrob. Agents. 1999;12:S23–S34. doi: 10.1016/s0924-8579(98)00071-5. [DOI] [PubMed] [Google Scholar]
- Collignon P., Turnidge J. Int. J. Antimicrob. Agents. 1999;12:S45–S58. doi: 10.1016/s0924-8579(98)00073-9. [DOI] [PubMed] [Google Scholar]
- Singh V., Dhar N., Pató J., Kolly G. S., Korduláková J., Forbak M., Evans J. C., Székely R., Rybniker J., Palčeková Z. Mol. Microbiol. 2017;103(1):13–25. doi: 10.1111/mmi.13535. [DOI] [PubMed] [Google Scholar]
- Steinhardt J., Krijn J., Leidy J. G. Biochemistry. 1971;10(22):4005–4015. doi: 10.1021/bi00798a001. [DOI] [PubMed] [Google Scholar]
- Zunszain P. A., Ghuman J., McDonagh A. F., Curry S. J. Mol. Biol. 2008;381(2):394–406. doi: 10.1016/j.jmb.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., Mainz D. T. J. Med. Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 2018-3, Glide S. R., Schrondinger, LLC: New York, NY, 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.