

Abstract
Accumulation of β-amyloid (Aβ) is one of the hallmarks of Alzheimer’s disease. The deposition of β-amyloid plaques is likely to start years in advance of manifestation of clinical symptoms, although the exact timing is unknown. Over the years, Aβ peptides undergo both post-translational modification and stereoisomerization. Analysis of the resulting stereoisomers is particularly challenging because of their identical elemental composition and similar physicochemical properties. Herein, we have utilized our recently developed structures for lossless ion manipulations ion mobility-mass spectrometry platform (SLIM IM-MS), in conjunction with serpentine ultralong path with extended routing (SUPER), to baseline resolve four distinct sets of Aβ17–28 tryptic peptide epimers on a rapid (~1 s) time scale. We discovered that sodium adduct ions, [M + H + Na]2+, allowed baseline SLIM SUPER IM resolution for all Aβ epimer sets assessed, while such baseline separations were unachievable for their [M + 2H]2+ doubly protonated ions.
Graphical Abstract
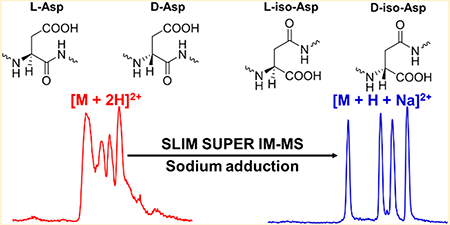
Alzheimer’s disease (AD) is manifested by a rapid decline in short-term memory and cognitive abilities.1–4 It is accepted that AD is associated with the aggregation and accumulation of amyloid in senile plaques, whose main component are amyloid β (Aβ) peptides.2–6 Typically, these peptides range from 38 to 42 amino acid residues in length, resulting from the proteolytic cleavage of amyloid precursor protein (APP), with Aβ40 and Aβ42 as the main components.2,3,5 Over extended periods the polypeptide chains in the aggregates are likely to accumulate a variety of chemical modifications.4,7–9 We focus here on a particular type of structural modification—stereoisomerization—because it has been suggested that these isomerizations may be used to determine the age of the protein.7,10–12 Though all of the amino acids, with exeption of glycine, racemize over time, we are specifically interested in Asp and Asn, collectively Asx residues, as they have the fastest rate, making them more useful for protein dating.13,14 These changes of Asx include the deamidation of L-asparagine (L-Asn) residues to L-aspartic acid (L-Asp), as well as the racemization/isomerization of L-Asp into D-Asp and its L/D-iso-Asp (β-Asp) forms through a succinimide intermediate (Figure 1),7,15–17 potentially providing information on the age of the Aβ plaques. Prior work using purified Aβ has demonstrated that it is indeed present in a substantially racemized form.2 The methodology described in that work is based on classical biochemical methods having significant challenges because of limited (arguably poor) resolution and throughput. Here, we have applied novel gas-phase ion manipulation techniques for the comprehensive characterization of mixtures of Aβ stereoisomers and envision this approach to be complementary to classical biochemical techniques. In this work, we were additionally interested in developing gas-phase separations methods that could in the future be coupled to solution-phase (e.g., liquid chromatography) separations for increased resolution, orthogonality, and detectability of the Aβ stereoisomers.
Figure 1.

Mechanism for deamidation of L-Asn to L-Asp, followed by racemization/isomerization of L-Asp into D-Asp and D/L-iso-Asp.
Although previously we analyzed the Aβ6–16 fragment,18 in which the Asp7 is subject to racemization/isomerization leading to four possible permutations (L-Asp, D-Asp, L-iso-Asp, and D-iso-Asp), here, we were interested in the Aβ17–28 fragment because of the more extensive information provided as well as the observed much better ionization Efficiency, potentially resulting in lower limits of quantification.19 However, racemization of Asx residues provides a rather complex mixture: this tryptic peptide contains Asp23, which is subject to racemization/isomerization, and Asn27, which can deamidate into Asp by the same racemization/isomerization process. This results in two Asp residues, each of which may be their D/L-Asp and/or D/L-iso-Asp forms, resulting in 20 total Aβ peptide epimers (16 with two Asp residues and 4 with one Asp and one Asn residue; the Asn-containing peptides were not assessed in this study and will be the subject of future work). Table 1 illustrates the 16 total peptides with two Asp residues as four, distinct, color-coded epimer sets. We hypothesized that this Aβ peptide fragment would potentially be more difficult to analyze than the one from our previous work18 because of having two Asx residues versus only one. Furthermore, attempting to resolve a set of 16 Aβ epimers is a much more formidable task. To better characterize which racemized/isomerized forms of Asp are present in the brain tissue of AD patients, better and faster methods are needed for their high-resolution separations. Our goal is to develop a high-throughput separation of the various Aβ17–28 epimers to be used in conjunction with mass spectrometry (MS) for future studies for monitoring Aβ accumulation levels in plasma, brain tissue, and serum.
Table 1.
Four Sets of Aβ Peptide Epimers (Color Coded) Assessed in This Study, with the Asp Residue Identity in the Right Column
| 1) LVFFAEDVGSDK[+8] | L-Asp, L-Asp |
| 2) LVFFAEDVGS(dD)K[+8] | L-Asp, D-Asp |
| 3) LVFFAEDVGS(bD)K[+8] | L-Asp, L-isoAsp |
| 4) LVFFAEDVGS(dbD)K[+8] | L-Asp, D-isoAsp |
| 5) LVFFAE(dD)VGSDK[+8] | D-Asp, L-Asp |
| 6) LVFFAE(dD)VGS(dD)K[+8] | D-Asp, D-Asp |
| 7) LVFFAE(dD)VGS(bD)K[+8] | D-Asp, L-isoAsp |
| 8) LVFFAE(dD)VGS(dbD)K[+8] | D-Asp, D-isoAsp |
| 9) LVFFAE(bD)VGSDK[+8] | L-isoAsp, L-Asp |
| 10) LVFFAE(bD)VGS(dD)K[+8] | L-isoAsp, D-Asp |
| 11) LVFFAE(bD)VGS(bD)K[+8] | L-isoAsp, L-isoAsp |
| 12) LVFFAE(bD)VGS(dbD)K[+8] | L-isoAsp, D-isoAsp |
| 13) 1 VFFAF.(dbD)VGSDK[+8] | D-isoAsp, L-Asp |
| 14) LVFFAE(dbD)VGS(dD)K[+8] | D-isoAsp, D-Asp |
| 15) LVFFAE(dbD)VGS(bD)K[+8] | D-isoAsp, L-isoAsp |
| 16) LVFFAE(dbD)VGS(dbD)K[+8] | D-isoAsp, D-isoAsp |
Accurate characterization of D-, L-, and iso-Asp forms of epimers remains a major bottleneck in analytical chemistry because of their identical molecular weights and very similar structures.20–22 This structural similarity leads these epimers to have very similar molecular properties (e.g., hydrophobicity), and thus similar chromatographic retention. Reversed-phase liquid chromatography (LC) data is provided in the Supporting Information for each of these 16 Aβ epimers run individually; the observed retention times were the basis for grouping these epimers into their four groups. A single stage of MS provides no resolution of epimers since they all have identical mass-to-charge (m/z) ratios. The ever-growing suite of available MS fragmentation techniques (e.g., collision-induced dissociation, electron transfer/capture dissociation, radical-directed dissociation, and ultraviolet photodissociation) has given rise to the more confident characterization of such structurally similar biomolecules,5,16,23 but this comes at a cost of significantly reduced sensitivity and precision in quantification. Present techniques are ultimately limited by the separation prior to fragmentation because differences are typically small, making deconvolution of overlapping peaks challenging, and potentially leading to the inaccurate identification and reduced effectiveness of quantification for L- and D- and iso-aspartic acid residues in epimeric mixtures.
Ion mobility (IM) spectrometry has recently emerged as a powerful analytical tool not only to probe gas-phase structure but also to separate biomolecules in the gas phase on the basis of their mass to charge and shape (i.e., their mobilities).24,25 Previous IM-based studies of peptide epimers have elucidated a better characterization of D-amino acid-containing peptides and probed the oligomerization of such peptides, as well as their respective monomers.26–35 Unfortunately, although useful in developing a better understanding of the molecular structure/behavior of such biologically relevant species, the efforts fall short in baseline resolution of the various permutations of D/L-Asp and D/L-iso-Asp-containing epimers, largely because of the limited IM resolution of commercial platforms. This resolution bottleneck drives the need for new IM-MS-based platforms effective for currently intractable Aβ peptide epimer separations.
In an effort to address these analytical challenges associated with limited resolution of conventional IM approaches, we have developed and implemented a higher resolution traveling wave (TW)-based IM-MS platform built upon structures for lossless ion manipulations (SLIM)36–39 and SLIM serpentine ultralong path with extended routing (SUPER) IM-MS.40 Herein, we describe the development of a SLIM SUPER IM-based approach, including the use of compression ratio ion mobility programming (CRIMP),36,38 for the high-resolution separation of four sets of Aβ17–28 peptide epimers, offering a potential rapid approach to probe Aβ accumulation in AD patients. We overcame the poor resolution of their doubly protonated, [M + 2H]2+, adducts by supplementing the electrospray buffer with sodium acetate, allowing all epimer sets to be baseline-resolved as their [M + H + Na]2+ adducts following SLIM SUPER IM separations with a CRIMP step to provide improved detection.41,42
EXPERIMENTAL SECTION
Experimental Conditions and Reagents.
All heavy-labeled (Lys residues) Aβ peptide epimers were acquired from New England Peptide (Gardner, MA, USA). All samples were prepared in 50/50 high-performance liquid chromatography grade water/methanol with 0.5% acetic acid (v/v). Final concentrations were 250 nM for the peptides and 25 nM sodium acetate (to ensure sodium cation attachment; higher sodium concentrations did not significantly increase the intensities of the sodiated peptides).
The TW SLIM SUPER IM-MS platform used in these experiments has been described in detail elsewhere.40 Briefly, samples were infused at flow rates of 500 nL/min for nanoelectrospray (3000 V; 110 °C inlet capillary) ionization. TW speed was 200 m/s and 30 V amplitude. Nitrogen gas pressure was maintained at 2.30 Torr in the ion funnel trap and 2.36 Torr in the SLIM chamber. All data were acquired with home-built software, and 25 individual separations were summed to produce the IM spectra shown unless otherwise indicated. No new, unexpected, or significant hazards were associated with this work.
In-SLIM Ion Accumulation.
As has been previously reported,36 ions can be accumulated in the SLIM module itself as opposed to an external ion funnel trap, with an extended trapping region providing significantly increased charge capacity. Briefly, the second traveling wave region (TW2) is halted (0 m/s velocity but the amplitude is kept at 30 V) during ion accumulation, while the first region (TW1) is kept under separation conditions (200 m/s at 30 V). This enabled ions to be accumulated in the first TW1 (9 m) region. Ions were accumulated for 1 s, in-SLIM, for all experiments.
Serpentine Ultralong Path with Extended Routing (SUPER) Separations.
SLIM SUPER IM separations are made possible through our switching region40 at the end of the TW2 region, just prior to ions exiting to the time-of-flight MS. At this point, two electrodes can switch between the voltage of the direct current guard or the normal traveling wave, to enable ions to be routed either to the TOF MS for detection or back to the start of the serpentine path for additional passes for SLIM SUPER IM separations. It is important to note that the path length for one complete pass is 13.5 m and ions are accumulated in-SLIM in the first 9 m of the SLIM module, leaving a 4.5 m separation path length.
Compression Ratio Ion Mobility Programming (CRIMP).
CRIMP36,38 was performed on the second pass of ions through the SLIM SUPER IM module and functioned as previously described.41,42 This enabled the otherwise lowered resolution due to the inherently broad peak width from the introduction of large ion populations (via in-SLIM ion accumulation) to be overcome. Briefly, ions undergo CRIMP by applying a stuttering/intermittent traveling wave in the second SLIM region, while maintaining separation conditions in the first traveling wave region. The second TW region stutters between 0 and 200 m/s at a rate determined by the selected compression ratio, with its amplitude maintained at 30 V. CRIMP was applied for 100 ms in each experiment so as to ensure all ion species (i.e., entire ion population) following in-SLIM accumulation were compressed to increase their signal intensity and signal-to-noise (S/N) ratio. Following CRIMP, normal separation conditions (200 m/s speed and 30 V amplitude are resumed for TW2) were in place, and no other CRIMP step is applied for the remainder of SLIM SUPER IM separation experiments. For an example of the benefit of applying a CRIMP step on narrowing the initial width of the ion population and increasing its signal intensity, please see the Supporting Information.
RESULTS AND DISCUSSION
Initial Survey of Metal Adduction Efficiency Following in-SLIM Ion Accumulation.
Previously, we have reported on the use of SLIM SUPER IM for the separation of a single set of Aβ epimers (Aβ6–16),18 but it was unclear if a similar approach would hold up for other Aβ epimer sets, especially ones with more than one L-Asp residue subject to racemization/isomerization. Additionally, it remained uncertain how these four new sets of Aβ17–28 epimers would behave using in-SLIM ion accumulation (as well as if we could utilize lower concentrations in this study because of the increased charge capacity for ion introduction) followed by a subsequent CRIMP step because our previous work18 exclusively used ions introduced via the much lower ion charge capacity ion funnel trap.36
Because previous IM-MS studies have shown the potential utility of metal adducts43,44 for increasing resolution of challenging species, we also were interested in probing if common metal adducts (e.g., sodium and potassium cations) would improve separations of the typically doubly protonated Aβ peptide epimers. Additionally, on the basis of previous literature,45–47 we hypothesized that these specific Aβ fragment peptides may display some level of conformational flexibility (e.g., exist as distinct multiple conformers). Because metal ion adduction has shown promise for modifying the conformations of certain peptides,48,49 as well as affecting Aβ aggregation,50 we postulated that metal adduction could potentially prove useful for effectively annealing the multiple conformers into single energetically preferred species.
To probe if metal adduction would even be favorable for these species, we prepared a mixture containing final concentrations of 25 nM sodium acetate and 250 nM peptide mixture (peptides 9–12 from Table 1) and examined the mass spectrum (Figure 2) following 1 s of in-SLIM ion accumulation. We observe that several peptide–metal adducted species were indeed present in the mass spectrum (Figure 2), but also that [M + H + Na]2+ was relatively similar in intensity to [M + 2H]2+, implying a high Efficiency for metal adduction. The doubly sodiated, [M + 2Na]2+, adduct was less efficiently formed, as indicated by being lower in relative intensity as compared to the singly sodiated form, [M + H + Na]2+. A potassiated adduct, [M + H + K]2+, was also observed, indicating trace levels of potassium cation are potentially present in either our sodium acetate stock solution or in our emitter tip/infusion line. No singly charged ions were detected for any of the peptides assessed in this study.
Figure 2.

(A) Mass spectrum of peptide epimer mixture (9–12) after 1 s of in-SLIM ion accumulation. (B) Schematic of the SLIM SUPER IM module used in these experiments.
SLIM SUPER IM Separations of Aβ Peptide Epimer Sets.
Using these observed multiple adducts (both metalated and protonated), we set out to probe their IM separations with SLIM SUPER IM-MS. Our general workflow for all SLIM SUPER IM experiments presented here is as follows: (1) introduction of ions using 1 s of in-SLIM ion accumulation (see Experimental Section); (2) 4.5 m SLIM SUPER IM separation; (3) ions are then routed to the beginning of the serpentine path utilizing our ion switching region; (4) ions are subjected to CRIMP (see Experimental Section and Supporting Information) at the interface between the two TW regions; (5) ions exit through the serpentine path until the desired separation (total path length is [4.5 m + (13.5)(n)], where n is the number of passes) is achieved. It is important to note that CRIMP is performed only on the second pass during these SLIM SUPER IM separations.
SLIM SUPER IM separations (72 m) were performed for the Aβ epimeric mixture of peptides 5–8 (having D-Asp at residue 23 and either D-Asp, L-Asp, D-iso-Asp, or L-iso-Asp at residue 27) from Table 1, as their various doubly charged adducts (Figure 2). While it was observed that this Aβ epimeric mixture could not be baseline resolved as their doubly protonated adducts (Figure 3A), the [M + H + Na]2+ adducts were well resolved after a 72 m SLIM SUPER IM separation (Figure 2B). Additionally, the [M + H + K]2+ adducts displayed multiple mobility peaks, potentially indicating the presence of multiple distinct ion conformations for each peptide epimer (Figure 3C). Interestingly, the doubly sodiated species showed significantly higher resolution than their doubly protonated counterparts (Figure 3D). Because all adducts were subjected to the same separation conditions (and underwent the same total path length of separation) as well as being similar in their respective mobilities (i.e., have very similar arrival times), it was quite striking to note the difference in their respective peak widths (Figure 3A versus Figure 3B and 3D). The [M + 2H]2+ mobility peaks for each individual Aβ peptide were ~2–3 times broader in width than either [M + H + Na]2+ or [M + 2Na]2+. For example, peptide 6 had a peak width (essentially base to base) of ~8 ms as its [M + H + Na]2+ adduct, compared to the ~16 ms peak width of peptide 6 for the [M + 2H]2+ species (see Supporting Information for data of peptides run individually). Thus, the sodium cation may effectively lock in a certain conformation of these Aβ peptides, thus narrowing their peak widths and leading to their higher resolution SLIM SUPER IM separations. We are actively pursuing this metal ion adduction strategy to probe if it may be broadly applicable for other peptides. It was also interesting to note that the arrival time order of these four Aβ epimers (with a fixed D-Asp at residue 23) remained unchanged between the [M + 2H]2+ and [M + H + Na]2+ species (Asp27 order: D-Asp < D-iso-Asp < L-Asp < L-iso-Asp). For the [M + 2Na]2+ species, the Asp27 arrival time order (most compact to most elongated) observed was D-isoAsp < D-Asp < L-Asp < L-isoAsp, suggesting that attachment of two sodium cations may affect the conformational landscape and/or gas-phase structure of these Aβ peptides.
Figure 3.

SLIM SUPER IM separations of 72 m for the Aβ epimer 5–8 epimer set as their (A) [M + 2H]2+ adducts, (B) [M + H + Na]2+ adducts, (C) [M + H + K]2+ adducts, and (D) [M + 2Na]2+ adducts. For individual IM peak assignments, see the Supporting Information.
On the basis of the encouraging high-resolution separations of the four peptide epimers with a fixed D-Asp at residue 23, and varying Asp27, we set out to determine if a similar approach would hold up for the other epimer sets (1–4, 9–12, and 13–16) with permutations of isomerization/racemization at Asp23 (L-Asp, L-iso-Asp, and D-iso-Asp). We were also interested to see if the trend of the [M + H + Na]2+ adducts providing the highest resolution separation would apply for the other Aβ peptide sets. Because the potassium-based complexes showed no utility in the separation of Aβ peptides 5–8, they were not further pursued. Figure 4 illustrates the high-resolution SLIM SUPER IM separations of the other Aβ peptide epimer sets (1–4, 9–12, and 13–16) as their [M + 2H]2+ (A), [M + H + Na]2+ (B), and [M + 2Na]2+ (C) species. It is important to mention that the Aβ peptide epimer sets of 1–4 and 13–16 required only a 72 m SLIM SUPER IM separation for baseline resolution, while 99 m was needed to resolve the 9–12 epimer set. In examining the 1–4 and 9–12 epimer sets (who have a fixed L-Asp and L-iso-Asp at residue 23, respectively), it can be seen that only the [M + H + Na]2+ species were fully resolved for each Aβ peptide set. For the 13–16 set, which has a fixed D-iso-Asp at residue 23, each adduct (doubly protonated and both sodiated adducts) showed promise for complete epimeric separation, although higher resolution was observed for the [M + H + Na]2+ and [M + 2Na]2+ over [M + 2H]2+.
Figure 4.

SLIM SUPER IM separations of Aβ peptide epimer sets as (A) [M + 2H]2+ adducts, (B) [M + H + Na]2+ adducts, and (C) [M + 2Na]2+ adducts. Peptide epimer sets 1–4 and 13–16 were subjected to a 72 m SLIM SUPER IM separation, while 99 m of separation was needed for 9–12. For individual IM peak assignments, see the Supporting Information.
Again, from an analytical perspective, we globally observed the best resolution for all four unique peptide epimer sets as their [M + H + Na]2+ adducts, with all SLIM SUPER IM separations possible in ~1 s. Closer examination of the arrival time distributions revealed a highly significant effect for Aβ peptide 16: it was observed to be the most elongated structure (slowest ion/lowest mobility) for both the [M + 2H]2+ and [M + H + Na]2+ adducts, but the most compact (fastest ion/highest mobility) as its [M + 2Na]2+ adduct. Clearly, the modeling of the various protonated and sodiated structures for these Aβ peptide epimers would be informative.
CONCLUSIONS
Herein, we have demonstrated SLIM SUPER IM-MS for resolving four challenging sets of Aβ peptide epimers incorporating two Asp residues labile to racemization/isomerization. While these peptides were not separated as their doubly protonated species, all epimer sets could be resolved as their [M + H + Na]2+ species in ~1 s. Compared to the previously reported separation of Aβ6–16 peptide epimers,18 here, we were able to achieve ~4-fold greater sensitivity because of the combination of in-SLIM ion accumulation followed by a subsequent CRIMP step as compared to ion introduction via the ion funnel trap, increasing the signal-to-noise ratios of measurements. Additionally, in this study we also developed a metal ion adduction strategy helping to resolve these Aβ17–28 epimers, which were unable to be baseline separated as their doubly protonated adducts.18 The four unique sets of four Aβ epimers containing two Asx residues provided a much greater separation challenge as compared to those in our previous work,18 demonstrated by the need for both sodium cation adduction and longer separation path lengths (up to 99 m for one set of four epimers). It is important to mention that such baseline separations of epimeric mixtures would not be achievable with a 1 m drift tube IM platform because of its ~8–10 times lower IM resolution compared to that of our SLIM SUPER IM platform with the 72 to 99 m of total separation path length used (as achievable resolution scales with the square root increase in path length40).
In this work we also noted significant differences in protonated versus sodiated peptide mobility peak widths (sodiated ones being far narrower than their protonated counterparts) that would not be evident with lower resolution platforms, and suggesting potentially much more effective approaches for analysis, as well as additional information on peptide conformers and potential insights regarding their sequence. We are in the process of further probing sodium adduction, along with computational efforts, on other peptide analytes to determine its effect on observed IM peak widths, especially with the higher resolution separations afforded by our SLIM SUPER IM platform.
We emphasize that there still remains considerable room for improvement, both in sensitivity through further optimization of the in-SLIM ion accumulation as well as other aspects of this work. In this regard, we are currently developing front-end liquid chromatography separations that are anticipated to additionally aid in deconvoluting more complex mixtures of peptide epimers, either through direct online coupling to our SLIM SUPER IM separations or through offline fractionation (see the Supporting Information for preliminary SLIM SUPER IM separations of this mixture without LC). By further improving our in-SLIM ion accumulation strategy, we anticipate overcoming the limitation because of the spreading of the peptide epimer signal across various adduct species (protonated and sodiated adducts). Additionally, online-LC-SLIM SUPER IM-MS separations are highly feasible and would provide sufficient chromatographic peak sampling. Specifically, a chromatographic peak of ~20–30 s in width would permit us to sample ~10–15 points across an LC peak with our presented SLIM SUPER IM separation methodology. This is based on the fact that each of these presented SLIM SUPER IM separations are a total of 2 s in duration (1 s of in-SLIM ion accumulation + 1 s of IM separation). These 10–15 points across a chromatographic peak are similar in magnitude to the number of summed separations in our presented direct infusion data (25 summed separations), but with the added benefit of analyte focusing from LC prior to ionization and introduction into the SLIM IM module. In addition, we note that the speed could be further increased in the future by conducting the ion accumulation step in tandem with a separation, and essentially reducing the time needed from 2 to 1 s. As an example (see Supporting Information), a two-dimensional (2-D) plot of the 12 peptide epimers subjected to 72 m of SLIM SUPER IM separation (i.e., 1–4, 5–8, and 13–16) is shown with retention time from LC on the x-axis and arrival time on the y-axis. This 2-D plot demonstrates the orthogonality of LC and SLIM SUPER IM separations; some peptides (e.g., 3 and 7) are unresolved by IM but are resolved by LC, and other peptides (e.g., 3 and 4) are unresolved by LC but well resolved by IM. Additionally, these infusion experiments used only ~200 pg of sample, highlighting the sensitivity of such measurements; coupling online-LC to these separations should significantly increase the limits of detection. We are actively working to couple online-LC separation to our SLIM SUPER IM separations to fully resolve all of these Aβ17–28 epimers. We envision this SLIM SUPER IM approach to have broad utility for the separation of other challenging peptide epimers and, more specifically, for clinical applications in probing Aβ accumulation in AD patients as a complementary technique to existing, classical, biochemical techniques.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institute of General Medical Sciences (P41 GM103493) and the National Cancer Institute of the NIH (R33 CA217699). Experiments were performed at the W.R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a DOE national scientific user facility at the Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle under Contract DE-AC05–76RL0 1830 for the DOE.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.8b04696.
Individually run Aβ peptides and IM spectra showing benefit of initial CRIMP step on second pass; preliminary LC data for these Aβ peptides (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Rajendran L; Honsho M; Zahn TR; Keller P; Geiger KD; Verkade P; Simons K Proc. Natl. Acad. Sci. U. S. A 2006, 103, 11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Roher AE; Lowenson JD; Clarke S; Wolkow C; Wang R; Cotter RJ; Reardon IM; Zurcher-Neely HA; Heinrikson RL; Ball MJ; et al. J. Biol. Chem 1993, 268, 3072–3083. [PubMed] [Google Scholar]
- (3).Gouras GK; Olsson TT; Hansson O Neurotherapeutics 2015, 12, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Murphy MP; LeVine H 3rd J. Alzheimer’s Dis 2010, 19, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sargaeva NP; Lin C; O’Connor PB Anal. Chem 2009, 81, 9778–9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yu L; Petyuk VA; Gaiteri C; Mostafavi S; Young-Pearse T; Shah RC; Buchman AS; Schneider JA; Piehowski PD; Sontag RL; Fillmore TL; Shi T; Smith RD; De Jager PL; Bennett DA Ann. Neurol 2018, 84, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lyons B; Friedrich M; Raftery M; Truscott R Anal. Chem 2016, 88, 2675–2684. [DOI] [PubMed] [Google Scholar]
- (8).Padayachee E; Ngqwala N; Whiteley CG J. Enzyme Inhib. Med. Chem 2012, 27, 356–364. [DOI] [PubMed] [Google Scholar]
- (9).Rufenacht P; Guntert A; Bohrmann B; Ducret A; Dobeli HJ Mass Spectrom 2005, 40, 193–201. [DOI] [PubMed] [Google Scholar]
- (10).Truscott RJW; Schey KL; Friedrich MG Trends Biochem. Sci 2016, 41, 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Moro ML; Collins MJ; Cappellini E Biochem. Soc. Trans 2010, 38, 539. [DOI] [PubMed] [Google Scholar]
- (12).Stabler TV; Byers SS; Zura RD; Kraus VB Arthritis. Res. Ther 2009, 11, R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).McCudden CR; Kraus VB Clin. Biochem 2006, 39, 1112–1130. [DOI] [PubMed] [Google Scholar]
- (14).Bada JL; Helfman PM World Archaeology 1975, 7, 160–173. [DOI] [PubMed] [Google Scholar]
- (15).Radkiewicz JL; Zipse H; Clarke S; Houk KN J. Am. Chem. Soc 1996, 118, 9148–9155. [DOI] [PubMed] [Google Scholar]
- (16).Cournoyer JJ; Pittman JL; Ivleva VB; Fallows E; Waskell L; Costello CE; O’Connor PB Protein Sci. 2005, 14, 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li B; Borchardt RT; Topp EM; VanderVelde D; Schowen RL J. Am. Chem. Soc 2003, 125, 11486–11487. [DOI] [PubMed] [Google Scholar]
- (18).Zheng X; Deng L; Baker ES; Ibrahim YM; Petyuk VA; Smith RD Chem. Commun 2017, 53, 7913–7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kim JS; Ahn H-S; Cho SM; Lee JE; Kim Y; Lee C Anal. Chim. Acta 2014, 840, 1–9. [DOI] [PubMed] [Google Scholar]
- (20).Bai L; Sheeley S; Sweedler JV Bioanal. Rev 2009, 1, 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ha S; Kim I; Takata T; Kinouchi T; Isoyama M; Suzuki M; Fujii N PLoS One 2017, 12, e0189972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jansson ET J. Sep. Sci 2018, 41, 385–397. [DOI] [PubMed] [Google Scholar]
- (23).Tao Y; Quebbemann NR; Julian RR Anal. Chem 2012, 84, 6814–6820. [DOI] [PubMed] [Google Scholar]
- (24).Gabelica V; Marklund E Curr. Opin. Chem. Biol 2018, 42, 51–59. [DOI] [PubMed] [Google Scholar]
- (25).Kemper PR; Dupuis NF; Bowers MT Int. J. Mass Spectrom 2009, 287, 46–57. [Google Scholar]
- (26).Jeanne Dit Fouque K.; Garabedian A; Porter J; Baird M; Pang X; Williams TD; Li L; Shvartsburg A; Fernandez-Lima F Anal. Chem 2017, 89, 11787–11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pang X; Jia C; Chen Z; Li LJ Am. Soc. Mass Spectrom 2017, 28, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Srebalus CA; Li J; Marshall WS; Clemmer DE Anal. Chem 1999, 71, 3918–3927. [DOI] [PubMed] [Google Scholar]
- (29).Wu C; Siems WF; Asbury GR; Hill HH Anal. Chem 1998, 70, 4929–4938. [DOI] [PubMed] [Google Scholar]
- (30).Wu C; Siems WF; Klasmeier J; Hill HH Anal. Chem 2000, 72, 391–395. [DOI] [PubMed] [Google Scholar]
- (31).Daly S; Kulesza A; Poussigue F; Simon A-L; Choi CM; Knight G; Chirot F; MacAleese L; Antoine R; Dugourd P Chem. Sci 2015, 6, 5040–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Soper MT; DeToma AS; Hyung S-J; Lim MH; Ruotolo BT Phys. Chem. Chem. Phys 2013, 15, 8952–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Soper-Hopper MT; Eschweiler JD; Ruotolo BT ACS Chem. Biol 2017, 12, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bleiholder C; Do TD; Wu C; Economou NJ; Bernstein SS; Buratto SK; Shea J-E; Bowers MT J. Am. Chem. Soc 2013, 135, 16926–16937. [DOI] [PubMed] [Google Scholar]
- (35).Young LM; Cao P; Raleigh DP; Ashcroft AE; Radford SE J. Am. Chem. Soc 2014, 136, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Deng L; Garimella SVB; Hamid AM; Webb IK; Attah IK; Norheim RV; Prost SA; Zheng X; Sandoval JA; Baker ES; Ibrahim YM; Smith RD Anal. Chem 2017, 89, 6432–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Deng L; Ibrahim YM; Baker ES; Aly NA; Hamid AM; Zhang X; Zheng X; Garimella SVB; Webb IK; Prost SA; Sandoval JA; Norheim RV; Anderson GA; Tolmachev AV; Smith RD ChemistrySelect 2016, 1, 2396–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Garimella SV; Hamid AM; Deng L; Ibrahim YM; Webb IK; Baker ES; Prost SA; Norheim RV; Anderson GA; Smith RD Anal. Chem 2016, 88, 11877–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Deng L; Ibrahim YM; Hamid AM; Garimella SV; Webb IK; Zheng X; Prost SA; Sandoval JA; Norheim RV; Anderson GA; Tolmachev AV; Baker ES; Smith RD Anal. Chem 2016, 88, 8957–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Deng L; Webb IK; Garimella SVB; Hamid AM; Zheng X; Norheim RV; Prost SA; Anderson GA; Sandoval JA; Baker ES; Ibrahim YM; Smith RD Anal. Chem 2017, 89, 4628–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chouinard CD; Nagy G; Webb IK; Garimella SVB; Baker ES; Ibrahim YM; Smith RD Anal. Chem 2018, 90, 11086–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nagy G; Chouinard CD; Attah IK; Webb IK; Garimella SVB; Ibrahim YM; Baker ES; Smith RD Electrophoresis 2018, 39, 3148–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Huang Y; Dodds ED Anal. Chem 2013, 85, 9728–9735. [DOI] [PubMed] [Google Scholar]
- (44).Gaye MM; Nagy G; Clemmer DE; Pohl NL Anal. Chem 2016, 88, 2335–2344. [DOI] [PubMed] [Google Scholar]
- (45).Sgourakis NG; Yan Y; McCallum SA; Wang C; Garcia AE J. Mol. Biol 2007, 368, 1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Larini L; Shea JE Biophys. J 2012, 103, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Manea M; Kalaszi A; Mezo G; Horvati K; Bodor A; Horvath A; Farkas O; Perczel A; Przybylski M; Hudecz FJ Med. Chem 2008, 51, 1150–1161. [DOI] [PubMed] [Google Scholar]
- (48).Dilger JM; Valentine SJ; Glover MS; Ewing MA; Clemmer DE Int. J. Mass Spectrom 2012, 330–332, 35–45. [Google Scholar]
- (49).Kohtani M; Jarrold MF; Wee S; O’Hair RA J. J. Phys. Chem. B 2004, 108, 6093–6097. [Google Scholar]
- (50).Kim AC; Lim S; Kim YK Int. J. Mol. Sci 2018, 19, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.