

Abstract
Nosocomial infections have become alarming with the increase of multidrug-resistant bacterial strains of Acinetobacter baumannii. Being the causative agent in ~80% of the cases, these pathogenic gram-negative species could be deadly for hospitalized patients, especially in intensive care units utilizing ventilators, urinary catheters, and nasogastric tubes. Primarily infecting an immuno-compromised system, they are resistant to most antibiotics and are the root cause of various types of opportunistic infections including but not limited to septicemia, endocarditis, meningitis, pneumonia, skin, and wound sepsis and even urinary tract infections. Conventional experimental methods including typing, computational methods encompassing comparative genomics, and combined methods of reverse vaccinology and proteomics had been proposed to differentiate and develop vaccines and/or drugs for several outbreak strains. However, identifying proteins suitable enough to be posed as drug targets and/or molecular vaccines against the multidrug-resistant pathogenic bacterial strains has probably remained an open issue to address. In these cases of novel protein identification, the targets either are uncharacterized or have been unable to confer the most coveted protection either in the form of molecular vaccine candidates or as drug targets. Here, we report a strategic approach with the 3,766 proteins from the whole genome of A. baumannii ATCC19606 (AB) to rationally identify plausible candidates and propose them as future molecular vaccine candidates and/or drug targets. Essentially, we started with mapping the vaccine candidates (VaC) and virulence factors (ViF) of A. baumannii strain AYE onto strain ATCC19606 to identify them in the latter. We move on to build small networks of VaC and ViF to conceptualize their position in the network space of the whole genomic protein interactome (GPIN) and rationalize their candidature for drugs and/or molecular vaccines. To this end, we propose new sets of known proteins unearthed from interactome built using key factors, KeF, potent enough to compete with VaC and ViF. Our method is the first of its kind to propose, albeit theoretically, a rational approach to identify crucial proteins and pose them for candidates of vaccines and/or drugs effective enough to combat the deadly pathogenic threats of A. baumannii.
Keywords: Acinetobacter baumannii, nosocomial infection, vaccine candidates, drug targets, network analysis
Introduction
Nosocomial or hospital-acquired infections are among the multitude of diseases caused by the opportunistic pathogen Acinetobacter baumannii, one of the world's six most important multidrug-resistant (MDR) microorganisms identified in hospitals (Talbot et al., 2006; Lin and Lan, 2014). While critically ill patients in the intensive care unit (ICU) accounted for up to 20% of ventilator-associated pneumonia and bloodstream infections with A. baumannii (Fournier and Richet, 2006; Vincent et al., 2009), the mortality rates might reach up to 35% along with endocarditis, meningitis, skin and wound sepsis, and even urinary tract infections for immunosuppressive patients (Lin and Lan, 2014; Darvishi, 2016). The cases for treatment are complicated due to the fact that the severity of the infection depends on the site and the patient's susceptibility to such diseases (Antunes et al., 2014). Moreover, there exist numerous natural reservoirs for A. baumannii including natural and agricultural soil, vegetables, aquaculture, and other inanimate objects outside the hospital environment (Eveillard et al., 2013).
The complications, resulting in an array of diseases caused by A. baumannii, arise from a plethora of virulence factors used by the pathogen to access and colonize the host system. These include, but are obviously not limited to, porins, capsular polysaccharides, lipopolysaccharides, phospholipases, outer membrane vesicles (OMVs), metal acquisition systems, and protein secretion systems (Lee et al., 2017). Besides these, β-lactamases acquisition, efflux pumps up-regulation, aminoglycosides modification, target sites alteration, and permeability defects are the crucial factors guiding the mechanism of antibiotic resistance conferred by this pathogen (Lee et al., 2017). Further threats of infection arise from colonization outside the human host, mainly on medical devices, through the mechanism of biofilm production involving the associated pathways, proteins, secretion systems, and quorum sensing (Perez et al., 2007). With such a robust antibiotic resistance mechanism entailing a barrage of proteins comprising the host invading machinery, A. baumannii has been able to confer extensive drug resistance (XDR). In fact, such ability has gone to the extent of evading almost every new-generation antibiotic, including carbapenems, which used to be prescribed to treat MDR organismal infections (Viehman et al., 2014).
To cater to the need of addressing the urgent and pressing issues of antibiotic threats, vaccine development has been resorted to as one of the cost-effective and most promising strategies to prevent infections. This is due to the fact that inactivated whole cells and attenuated strains are able to elicit antibodies against multiple surface proteins, which can be utilized in the form of vaccines, to combat the antibiotic threats. In fact, efforts have been tested to utilize only parts of the pathogen without the administration of whole organisms. Thus, vaccines comprising multiple proteins of the bacterial outer membrane complex (OMC), OMVs, OmpA, auto-transporter (Ata), biofilm-associated protein (Bap), K1 capsular polysaccharide, and poly-N-acetyl-β-(1-6)-glucosamine (PNAG) have been shown to elicit antibodies and to induce protective immunity against infection, thereby giving promising results in early human clinical trials (Bertot et al., 2007; Fransen et al., 2007; Chiang et al., 2015). Moreover, recent studies to determine potential vaccine targets have delineated a combinatorial approach of in silico prediction tools with reverse vaccinology through comparative genome analysis and in vitro proteomics (Moriel et al., 2013; Singh et al., 2017). For instance, in silico analysis in A. baumannii helped the identification of a highly conserved outer membrane protein with β-barrel assembly, BamA, as the potential target for vaccine (Singh et al., 2017). However, the advent of new and emerging XDR strains of A. baumannii, which possibly arises from immune selection, might lead to antigen sequence variability and a down-regulation of the target antigens, thereby conferring poor “cross-protective efficacy” (Chiang et al., 2015). Therefore, the identification of potential antigens, expressed by the new and emerging A. baumannii strains during infections, still keeps the issue quite complex to be addressed.
Simplifying the complexity of diseases, caused by such XDR pathogens like A. baumannii, is thus challenging. Therefore, to unearth plausible antigenic proteins, potential enough to elicit an antibody response, a detailed analysis of the complex interaction of the proteins, involved in the disease phenomenon, might be helpful. Earlier attempts to computationally analyze such protein interaction networks or interactomes (PIN) of infectious diseases mostly focused on network centrality parametric values for the identification of candidate drug targets (Lahiri et al., 2014; Pan et al., 2015). Generally, in biological networks, centrality measures of degree (DC), betweenness (BC), closeness (CC), and eigenvector (EC) have been used extensively (Jeong et al., 2001; Lahiri et al., 2014; Pan et al., 2015). While DC gives a very basic understanding of the number of interacting partners of a particular protein, EC relates to the essential proteins interacting with other crucial partners in the disease phenomenon (Lahiri et al., 2014). Besides BC, EC has been shown to be a good target for drugs, albeit theoretically for infectious diseases and utilized in the identification of side effect-free drug targets of idiopathic diseases like cancer (Lahiri et al., 2014; Pan et al., 2015; Ashraf et al., 2018). However, in order to gain an insight into the global scenario of the disease complexity, the PIN needs to be inspected thoroughly for an effective analysis, potential enough to be translational in nature. Thus, the whole genome protein interactome (GPIN) has been utilized to prune and decompose to obtain a core of highly interacting proteins through the k-core analysis approach (Seidman, 1983). This coupled with the functional module-based cartographic analyses of the global network (Guimerà and Nunes Amaral, 2005a) has already been adopted to theoretically identify potential role players in bacterial infectious diseases (Pawar et al., 2017, 2018).
In this study, we have similarly delineated the relevance of the aforementioned centrality parametric measures for PIN of the claimed vaccine candidates and virulence factors (Moriel et al., 2013) of A. baumannii, namely, VaCAB and ViFAB. To this end, we have analyzed KeFAB, the PIN of key factors responsible for virulence and pathogenicity of A. baumannii (Chen et al., 2015). The top rankers of these three PINs were mapped onto the GPIN of A. baumannii to unravel their position in the network space and rationalize their candidature for drugs and/or molecular vaccines compared to our own sets of proposed candidates for vaccines. We consolidate our findings by the antigenic potential of these proteins along with their active sites for drug targets. In summary, we analyze the PIN of different relevant pathogenic proteins of A. baumannii to identify the plausible potential candidates for vaccines and/or drugs targets.
Materials and Methods
Acronyms and Terms Utilized
The present study on A. baumannii comprises a conglomerate of several terms pertaining to graph theory in general and network biology in particular. Table 1 lists them all in a comprehensive manner with an expansion of the acronym, where applicable, followed by a short description of the terms for the ease of reference of a broad interdisciplinary range of readers.
Table 1.
The acronyms and terms used in the present study.
| Acronyms/Terms | Description |
|---|---|
| VaC | Vaccine Candidates; proteins reported by Moriel et al. from Acinetobacter baumannii strain AYE and ATCC17978 |
| ViF | Virulent Factors; proteins reported by Moriel et al. from A. baumannii strain AYE and ATCC17978 |
| KeF | Key Factors; proteins retrieved from various databases and literature survey from strain AYE and ATCC17978 using common terms related to virulence |
| VFDB | Virulent Factors Database; database of several bacteria listing the known and/or predicted virulent factors as per literature |
| STRING | Search Tool for the Retrieval of Interacting Genes/Proteins; meta-database listing known and predicted protein–protein interactions from biological organisms |
| Cytoscape/Gephi | Interacting Software for visualizing node and edges of a graph and integrating them together |
| PIN | Protein Interaction Network; interaction pattern of proteins in a graphical form giving a network of nodes and edges |
| Interactome | A network of interacting entities; in the present study, this refers to Protein Interactome or PIN |
| Interactors | The molecules interacting in a network; in the present study, these are proteins interacting with VaC, ViF, or KeF |
| VaCAB | VaC PIN of A. baumannii; interactome of VaC with their interactors of strain ATCC19606 retrieved from STRING and visualized through Cytoscape/Gephi |
| ViFAB | ViF PIN of A. baumannii; interactome of ViF with their interactors of strain ATCC19606 retrieved from STRING and visualized through Cytoscape/Gephi |
| KeFAB | KeF PIN of A. baumannii; interactome of KeF with their interactors of strain ATCC19606 retrieved from STRING and visualized through Cytoscape/Gephi |
| SPIN | Small PIN of A. baumannii; this refers to VaCAB/ViFAB/KeFAB of A. baumannii strain ATCC19606 |
| GPIN | Genomic PIN of A. baumannii; interactome of the total number of proteins of whole genome of A. baumannii strain ATCC19606 retrieved from STRING and visualized through Cytoscape/Gephi |
| Network Analyser | Java Plug-in for Cytoscape for graph theoretical analysis of the network of nodes and edges |
| CytoNCA | Cytoscape Network Centrality Analyser; Cytoscape Java Plug-in for graph theoretical analysis of the network of nodes and edges |
| Centrality Measures | Graph theoretical measures for assessing the central character of nodes |
| BC/CC/DC/EC | Betweenness/Closeness/Degree/Eigenvector centrality measures; reflect measures for nodes of central passage/proximity/connectivity/weightage, respectively |
| k-Core | Maximally connected sub-graph of nodes having at least k-degree; achieved by gradually pruning all nodes of degree less than k |
| Modules | Sub-graphs of nodes having common aspects with respect to common biological functions |
| Functional Connectivity | Different classes of within-module and between-module connectivity |
| KFC | An approach combining k-core, functional connectivity, and centrality measures |
| Standalone BLAST | Offline Basic Local Alignment Search Tool; this comprises a downloadable version of the sequence alignment tool that can be installed locally on system and can be used with command lines to find the match/mismatch/gap between the given sequences |
| COG | Cluster of Orthologous Groups; similar groups of proteins having related functions |
Dataset Collection and Processing
Datasets for A. baumannii proteins were collected in different ways. The focus was given for either the strains AYE or ATCC17978, having either substantial reports or evidences for causing MDR. For the strain AYE, 168 proteins mentioned in the Supplementary Data 2 by Moriel et al. (2013) were collected and categorized as vaccine candidates (VaC, Supplementary Data 1) while 124 from Supplementary Datas 3–5 reported by the same group were collated as virulent factors (ViF, Supplementary Data 2). Other proteins were collected using keywords used by default for the available strains of ATCC17978 and AYE in VFDB (Chen et al., 2015). To this end, using the same keywords, further proteins of these strains were collected through direct literature search and the sequences of all these were retrieved from UniProt database (Apweiler et al., 2004). The last category proteins from VFDB, UniProt, and literature were parked under key factors (KeF, Supplementary Data 3). The counts of identifiers (IDs) for the various KeF proteins were 973 for ATCC17978, 92 for AYE, and 24 for no IDs found in the literature. Nine of these IDs, belonging to t-RNA, were eliminated, leaving a final total of 1,078 KeF (Figure 1).
Figure 1.

The flow chart for the whole process of the present study.
The datasets of the aforementioned VaC, ViF, and KeF proteins were converted into the counterparts of the corresponding A. baumannii strain ATCC19606, due to the reason of non-availability of ATCC17978 and AYE strains in the STRING 10.5 biological meta-database of protein interaction (Szklarczyk et al., 2016). A FASTA file containing a total of 3,376 protein sequences, downloaded from STRING for A. baumannii strain ATCC19606, was used as a database for standalone BLAST during mapping of the sequences of strain AYE onto strain 19606. Standalone BLAST is a non-graphic user interface version of BLAST that runs on command lines in Linux operating system (OS) and allows execution of BLAST locally on such OS for sequence alignment. The execution of such BLAST, returned 2,111 and 1,676 hits for VaC and ViF, respectively. A filtering of the topmost ones with the highest percentage of identities from these hits returned 168 and 123 proteins, respectively, of the strain 19606. A further threshold cutoff of 99–100% identity was set to select out the identical proteins of strain AYE in 19606 for the next set of analysis, leaving out other ambiguous ones (duplicates and/or percentage-wise less identical) to obtain 79 VaC and 78 ViF proteins of strain 19606. Similar approaches were adopted for selecting out the KeF proteins of the strain 19606 from those of the strains ATCC17978 and AYE. For this, 1,078 KeF protein sequences were executed in standalone BLAST to yield a total of 15,793 hits, from which the top-ranked 1,075 proteins, with highest percentage identity, were selected out. Final cutoff of 99–100% identity was used to remove ambiguous ones and duplicates to obtain 640 IDs for further processing (Figure 1).
Submission of these mapped proteins of ATCC19606 into STRING helped the retrieval of protein interaction datasets having the default medium (0.4) level confidence upon the interaction (period of access: August to September 2018). The detailed protein links file containing the interaction datasets for the whole genome of A. baumannii strain ATCC19606 was retrieved for the accession number 575584 in STRING. All interaction datasets for each category of VaC, ViF, and KeF proteins, hereafter used for PIN construction, have been listed in Supplementary Datas 1–3.
Interactome Construction
After the removal of duplicate interactions, all individual interaction data obtained as above were imported for construction and visualization of the small PINs (SPINs), which were named VaCAB, ViFAB, and KeFAB, and the whole genome PIN named GPIN, using Cytoscape version 3.6.0 (Shannon et al., 2003) and Gephi 0.9.2 (Bastian et al., 2009) (Figure 2; Supplementary Datas 1–3: Sheets 5–7). All interactomes were considered to be non-directional in nature to represent undirected graphs as G = (V, E), where V are finite set of vertices and E are edges in which e = (u,v) connecting two vertices (nodes), u and v, or proteins in the present context. Thus, the degree, d (v), indicates the number of interactions (physical and functional) a protein has with other proteins (Diestel, 2000).
Figure 2.
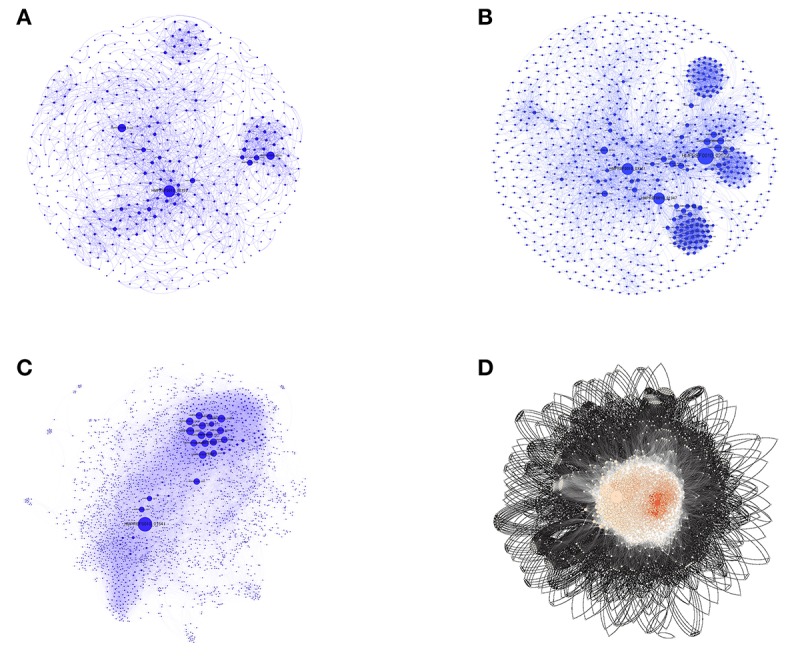
The three SPINs and GPINs of A. baumannii reflecting the degree of connectivity. SPINs are represented in blue spheres connected through blue-colored curved lines for (A) VaCAB, having vaccine candidates; (B) ViFAB, with virulent factors; and (C) KeFAB, with key factors each with their interactors. (D) GPIN with proteins represented in black spheres connected with black curved lines to form the interactome.
PIN Analyses
SPIN
Each of the constructed three SPINs were subsequently analyzed utilizing BC, CC, DC, and EC measures of centrality, commonly applied to biological networks (Pavlopoulos et al., 2011) (Supplementary Datas 1–3). This was done with the Cytoscape integrated java plugins, namely, Network Analyzer and CytoNCA (Assenov et al., 2007; Tang et al., 2015), utilizing the edge weights as the combined scores obtained from different parameters in STRING. These combined scores, which range from 0 to 1, generally convey the confidence of the protein's interaction with level of parametric evidences from gene neighborhood, gene fusion, gene co-occurrence, gene co-expression, experiments, annotated pathways, and text mining. Using the top 15 proteins appearing for each measures of analysis as mentioned above, a commonality of the proteins for all centrality measures was observed through Venny 2.1 (Oliveros, 2007–2015) (Figure 3).
Figure 3.

Venn diagram representation for the top-ranking network centrality measures of SPIN and GPIN of A. baumannii. (A,B) VaCAB, (C,D) ViFAB, (E,F) KeFAB, and (G) GPIN. Measures of four types of centrality are from CytoNCA and three types are from Network Analyzer. BC, CC, DC, and EC denote betweenness centrality, closeness centrality, degree centrality, and eigenvector centrality, respectively.
GPIN
The whole genome protein interaction network, GPIN, was analyzed by MATLAB version 7.11 (MATLAB Statistics Toolbox Release, 2010). An idea of the simplest of the technical aspects of the GPIN, i.e., the distributions of network degree (k), was obtained by plotting it against the Complementary Cumulative Distribution Function (CCDF) (Figure 4A). A k-core analysis of the whole genome was done by a network decomposition method that produces a gradually increasing cohesive sequence of sub-graphs and reveals the number of proteins having at least k-degree classified in K-shell as per their interacting patterns (Figures 4B,C) (Seidman, 1983). Further knowledge of the connectivity and participation of each protein, with respect to their functions, was obtained through analysis of the cartographic representation of the network topology that plots the within-module degree z score of the protein against its participation coefficient, P (Figure 5) (Guimerà and Amaral, 2005b). The participation of each protein in a modular network, sharing common biological function, gives rise to a concept of functional module having high intra-connectivity and sparse inter-connectivity (Vella et al., 2018). Calculations of such functional modules as per Rosvall method (Rosvall and Bergstrom, 2011) led to a major classification of the proteins of GPIN into non-hub and hub nodes, each having further sub-classes. These are ultra-peripheral (R1), peripheral (R2), non-hub connector (R3), and non-hub kinless (R4) for the former while provincial (R5), connector (R6), and kinless (R7) for the latter categories (Guimerà and Amaral, 2005b) (Figure 5; Supplementary Data 4).
Figure 4.

Network topological measures for the set of proteins from the GPIN of A. baumannii. (A) The degree distribution, (B) k-core distribution, and (C) K-shell sizes. CCDF denotes Complementary Cumulative Distribution Function.
Figure 5.

Cartographic representation for the set of classified proteins from the GPIN of A. baumannii. Designated quadrants from R1 to R7 (in random colors) comprise nodes in each representing different classes of proteins. Selected Vaccine Candidates, VaC (V), Virulent Factors, ViF (F), and Key Factors, KeF (K) proteins from network centrality analyzed SPIN (Small PIN) are mapped onto different quadrants as deemed fit in GPIN (Genome PIN). ∧ and * represent proteins shared between different centralities only and those also between different categories, respectively.
Vaccine and/or Drug Candidature Prediction
The network analyzed and shortlisted VaC, ViF, and KeF proteins along with their interactors from the three SPINs, namely, VaCAB, ViFAB, and KeFAB, were subjected to further analyses for predicting the plausible vaccine and/or drug candidates. To this end, the indispensable proteins of the GPIN were also taken into consideration as per the KFC method described by Ashraf et al. (2018). All such proteins were explored for their cellular localization, signal peptide prediction, COG classification, antigenic site prediction, followed by active site prediction. Cellular localization was analyzed by PSORTb v3.0.2 (Yu et al., 2010). Location of signal peptides was predicted using the server called SignalP 4.1 Server (Petersen et al., 2011). Lipoprotein signal peptides were predicted using the LipoP 1.0 Server (Juncker et al., 2003). For uncharacterized proteins, functional annotation with classification was done through the WebMGA server (Wu et al., 2011). Such COG classification from WebMGA was performed for all proteins, however, even if they are characterized, to maintain unanimity of comparison. To predict the antigenic potential of the candidate proteins, epitope prediction was done through the immune epitope database (IEDB) resource utilizing Bepipred Linear Epitope Prediction and maintaining a threshold cutoff of 0.75 for increased sensitivity (Vita et al., 2014). Furthermore, without any solved X-ray crystallographic or NMR 3D structures for the selected proteins, they were homology modeled and validated to pursue active site prediction studies. We have used Phyre2 (Kelley et al., 2015) and SWISS MODEL (Schwede et al., 2003) protein modeling servers to generate the structures and the integrated Procheck server in the latter, to evaluate them through Ramachandran plot, Q mean score, and z score. To identify the best structure of the different models generated by the abovementioned servers, we have performed consensus studies. These consensus models were finally utilized to determine the active sites or binding pockets of the selected proteins by the CASTp server (Computer Atlas of Surface Topology of protein) (Dundas et al., 2006). A comparative account of the results obtained from this section of the analysis is shown in Table 7.
Interactor-Free Candidature Analyses
In order to determine the important vaccine candidates among a barrage of proteins proposed by Moriel et al. (2013), filtration was used to remove the influence of the interactors on the actual candidates upon their ranking. For the same, we have initially merged the VaCAB and ViFAB together, having their individual centrality-based analyzed data. Thereafter, the VaC and ViF interactors, of the respective PIN, were removed, leaving behind the actual candidates. As the PINs (VaCAB and ViFAB) were constructed from STRING data using the strain ATCC19606, we mapped back the centrality measures of the total VaC and ViF proteins onto the AYE strain of A. baumannii. This was followed by assigning the localization status of the total set of candidate proteins through the usage of PSORTb. Sorting was done based on localization followed by descending order of the centrality measures. Top five rankers, in each case, were used for creating a Venn diagram to find out the most promising candidates from the intersection of the centrality measures (Figure 1).
Results
The Candidate Proteins
In order to the identify the potential molecular vaccine candidates and drug targets of MDR A. baumannii, we have started with the available pathogenic strains, namely, AYE and ATCC17978, to accumulate the proclaimed proteins in categories of VaC, ViF, and KeF with counts of 168, 124, and 1,078, respectively (Supplementary Datas 1–3: Sheet 1). As these strains are not enlisted in the protein interaction database, STRING, we had to find the real counterparts of these proteins, from the only listed strain ATCC19606. An initial processing of the data through standalone blast from the total protein sets of ATCC19606 yielded 168, 123, and 1,075 VaC, ViF, and KeF candidates, respectively (Supplementary Datas 1–3: Sheet 3), which were further filtered for an exact match of 99–100% to result in 79, 78, and 640 proteins, respectively (Supplementary Datas 1–3: Sheet 4). Thus, all the candidate proteins were mapped onto ATCC19606 from other pathogenic strains. All data, hereafter, refer to strain ATCC19606 only, unless otherwise stated.
The Three Individual SPINs
To identify the crucial candidates among the three aforementioned sets of VaC, ViF, and KeF proteins of A. baumannii, we have taken an approach of building three different SPINs, each with its respective set of interaction data (Figure 2; Supplementary Datas 1–3: Sheets 5–7). Upon analyzing these SPINs through the Cytoscape plugins CytoNCA and Network Analyzer, the top rank holders as per each centrality measures are recorded (Supplementary Datas 1–3: Sheets 8–11), indicating their importance. A combined result from both the plugins are presented in Table 2, wherein protein entries are represented by their NCBI gene accession numbers with the suffix “HMPREF0010_” being replaced by “AB_” for the ease of further use. It is to be noted that there are overlaps of these protein entries across the different centrality measures and/or the different categories of VaCAB, ViFAB, and KeFAB. An overlap of the proteins across the measures within a particular category is represented by a “caret” (∧) notation while that from cross-categories is denoted with an “asterisk” (*). For instance, the protein AB_03493 is associated with the VaCAB category and is represented with V1*, due to its presence in the category of ViFAB (BC measure), despite its presence in two measures of BC and DC of VaCAB. On the contrary, AB_1947 is present in the BC, CC, and DC categories of ViFAB only and is represented by F5∧. The numbers of the proteins to denote such overlaps are put randomly, however, and do not indicate the actual ordering of the rank as depicted in Table 2. Each one of such overlapping proteins has been indicated in bold to stand out from the rest in the same measures and/or categories and has been utilized for further mapping in the whole genome context later (Figure 5). It is important to note that there are more overlaps between the VaCAB and ViFAB categories of proteins as indicated by the asterisk in the entries. On the contrary, proteins of KeFAB tend to have much fewer overlaps among other categories. The numbers of the top-ranking proteins from each of the categories and measures of centralities and their overlaps are reflected with Venn diagrams (Figure 3). The functions of these proteins, taken from UniProt, reflect those that have been mostly characterized, with some having proven and eminent roles as vaccine and drug targets (Supplementary Datas 1–3: Sheet 12).
Table 2.
The topmost proteins of A. baumannii SPIN and GPIN as per BC, CC, DC, and EC network centrality measures.
| Network | BC | CC | DC | EC |
|---|---|---|---|---|
| SPIN | ||||
| VaCAB |
AB_03493(V1*), AB_01517(V2∧), AB_01255(V3*), AB_03704(V4∧), AB_00353(V5*), AB_01334(V6∧), AB_00252, AB_01118, AB_01117, AB_01710, AB_00112, AB_00698, AB_00210, AB_03124, AB_00209 |
AB_03704(V4∧), AB_00353(V5*), AB_01334(V6∧), AB_00811(V7∧), AB_02146(V8∧), AB_00805(V9∧), AB_03351(V10*), AB_02143(F6*), AB_02888, AB_02124, AB_02677, AB_02145, AB_01785, AB_03499, AB_02178 |
AB_03493(V1*), AB_01517(V2∧), AB_01255(V3*), AB_03704(V4∧), AB_00353(V5*), AB_01334(V6∧), AB_00811(V7∧), AB_02146(V8∧), AB_00805(V9∧), AB_03351(V10*), AB_02233(V11∧), AB_03186(V12*), AB_03184(V13*), AB_03185(V14∧), AB_01388 |
AB_01255(V3*), AB_02233(V11∧), AB_03186(V12*), AB_03184(V13*), AB_03185(V14∧), AB_01226, AB_02909, AB_02910, AB_01257, AB_00660, AB_00760, AB_01258, AB_02683, AB_00675, AB_02914 |
| ViFAB |
AB_01641(F1*), AB_02522(F2*), AB_03351(F3*), AB_00353(F4*), AB_01947(F5∧), AB_02143(F6*), AB_03493(V1*) AB_01255(V3*), AB_00113, AB_01972, AB_00354, AB_01713, AB_01724, AB_02769, AB_02820 |
AB_02522(F2*), AB_03351(F3*), AB_00353(F4*), AB_01947(F5∧), AB_02143(F6*), AB_03184(F7*), AB_02069(F8∧), AB_00812, AB_02142, AB_01731, AB_02888, AB_01946, AB_01248, AB_02124, AB_01647 |
AB_01641(F1*), AB_02522(F2*), AB_03351(F3*), AB_00353(F4*), AB_01947(F5∧), AB_03184(F7*), AB_02069(F8∧), AB_02627(F9∧) AB_03186(V12*), AB_02909, AB_01226, AB_00660, AB_02910, AB_02683, AB_01361 |
AB_02627(F9∧), AB_02354, AB_02629, AB_01950, AB_03765, AB_00169, AB_01359, AB_01360, AB_01656, AB_01791, AB_01948, AB_01951, AB_02545, AB_02548, AB_02626 |
| KeFAB |
AB_01491(K1∧), AB_01641(K2*), AB_02522(K3*), AB_03512(K4∧), AB_03207(K5∧), AB_00427, AB_01800, AB_03553, AB_01281, AB_02769, AB_01114, AB_00382, AB_02314, AB_03088, AB_02152 |
AB_01491(K1∧), AB_01641(K2*), AB_02522(K3*), AB_03512(K4∧), AB_03207(K5∧), AB_00239(K6∧), AB_03022(K7∧), AB_02437(K8∧), AB_01461(K9∧), AB_01350(K10∧), AB_01047(K11∧), AB_03233(K12∧), AB_03046(K13∧), AB_02576(K14∧), AB_02252(K15∧) |
AB_01491(K1∧), AB_01641(K2*), AB_00239(K6∧), AB_03022(K7∧), AB_02437(K8∧), AB_01461(K9∧), AB_01350(K10∧), AB_01047(K11∧) AB_03233(K12∧), AB_03046(K13∧), AB_02576(K14∧), AB_02252(K15∧), AB_01051(K16∧), AB_01740(K17∧), AB_00091(K18∧) |
AB_01491(K1∧), AB_03022(K7∧), AB_02437(K8∧), AB_01461(K9∧), AB_01350(K10∧), AB_01047(K11∧) AB_03233(K12∧), AB_03046(K13∧), AB_02576(K14∧), AB_02252(K15∧), AB_01051(K16∧), AB_01740(K17∧), AB_00091(K18∧), AB_01789, AB_03031 |
| GPIN | ||||
|
AB_01641(F1*/K2*), AB_01590, AB_02383, AB_03076, AB_03532, AB_03595, AB_02409, AB_01611, AB_01613, AB_02391, AB_03107, AB_03129, AB_01437, AB_03092, AB_00006 |
AB_00110, AB_01918, AB_02296, AB_03731, AB_03732, AB_03317, AB_03318, AB_03726, AB_02992, AB_03666, AB_02700, AB_03651, AB_02374, AB_02371, AB_02372 |
AB_01491(K1∧), AB_01641(F1*/K2*), AB_03512(K4∧), AB_03207(K5∧), AB_01461(K9∧), AB_03233(K12∧), AB_03046(K13∧), AB_02576(K14∧), AB_01740(K17∧), AB_00091(K18∧), AB_03659, AB_03532, AB_01651, AB_00045, AB_01281 |
AB_01491(K1∧), AB_01641(F1*/K2*), AB_02522(F2*/K3*), AB_03512(K4∧), AB_03207(K5∧), AB_00239(K6∧), AB_03022(K7∧), AB_02437(K8∧), AB_01461(K9∧), AB_01350(K10∧), AB_01047(K11∧), AB_03233(K12∧), AB_01051(K16∧), AB_00091(K18∧), AB_02769 |
|
The bold cased proteins are overlapping either within same categories
or between others
in SPIN. Numbers are arbitrary.
Despite the overview obtained about the importance of the proteins from a comparative account within and between each of the aforementioned categories of VaCAB, ViFAB, and KeFAB, it is to be noted that these PINs are a mixture of the actual VaC, ViF, and KeF proteins along with their interactors that were extracted from STRING. Interestingly, of the 15 top rank holders of each category of PIN, the actual proteins were 9, 3, 7, and 1 as per BC, CC, DC, and EC measures of VaCAB, and the rest were their interactors. Similarly, for ViFAB, the actual VaC proteins were 6, 6, 3, and 1 for the same measures, respectively, as mentioned above. For KeFAB, however, the numbers of KeF proteins were almost the same throughout, being 4, 5, 5, and 5 for BC, CC, DC, and EC, respectively (Table 3). However, to decipher the effective drug or a vaccine candidate from the global perspective of the whole genome, the total sets of proteins interacting in the whole genome of A. baumannii (GPIN) were analyzed as described in the next section.
Table 3.
The innermost core proteins of VaCAB, ViFAB, KeFAB, and GPIN as per the four centrality measures.
| Centrality measures | Protein ID | Protein type | Core | Protein ID | Protein type | Core | Protein ID | Protein type | Core | Protein ID | Protein type | Core |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BC | AB_03493 | VaC | 84 | AB_01641 | Interactor | 165 | AB_01491 | Interactor | 165 | AB_01590 | Genome | 66 |
| AB_01517 | VaC | 84 | AB_02522 | Interactor | 165 | AB_01641 | Interactor | 165 | AB_02383 | Genome | 85 | |
| AB_01255 | Interactor | 165 | AB_03351 | ViF | 165 | AB_02522 | Interactor | 165 | AB_03076 | Genome | 68 | |
| AB_03704 | VaC | 108 | AB_00353 | ViF | 163 | AB_03512 | KeF | 165 | AB_03532 | Genome | 165 | |
| AB_00353 | VaC | 163 | AB_01947 | Interactor | 165 | AB_03207 | Interactor | 165 | AB_01641 | Genome | 165 | |
| AB_01334 | VaC | 165 | AB_02143 | ViF | 123 | AB_00427 | Interactor | 165 | AB_03595 | Genome | 162 | |
| AB_00252 | VaC | 68 | AB_03493 | ViF | 86 | AB_01800 | Interactor | 165 | AB_02409 | Genome | 66 | |
| AB_01118 | Interactor | 70 | AB_01255 | Interactor | 165 | AB_03553 | KeF | 165 | AB_01611 | Genome | 22 | |
| AB_01117 | Interactor | 90 | AB_00113 | Interactor | 68 | AB_01281 | Interactor | 165 | AB_01613 | Genome | 66 | |
| AB_01710 | VaC | 91 | AB_01972 | Interactor | 112 | AB_02769 | Interactor | 165 | AB_02391 | Genome | 161 | |
| AB_00112 | Interactor | 68 | AB_00354 | ViF | 92 | AB_01114 | KeF | 165 | AB_03107 | Genome | 91 | |
| AB_00698 | VaC | 91 | AB_01713 | Interactor | 151 | AB_00382 | KeF | 165 | AB_03129 | Genome | 74 | |
| AB_00210 | Interactor | 98 | AB_01724 | ViF | 110 | AB_02314 | Interactor | 165 | AB_01437 | Genome | 30 | |
| AB_03124 | Interactor | 92 | AB_02769 | Interactor | 165 | AB_03088 | Interactor | 151 | AB_03092 | Genome | 85 | |
| AB_00209 | VaC | 102 | AB_02820 | Interactor | 147 | AB_02152 | Interactor | 165 | AB_00006 | Genome | 65 | |
| CC | AB_03704 | VaC | 108 | AB_02522 | Interactor | 165 | AB_01491 | Interactor | 165 | AB_00110 | Genome | 62 |
| AB_00353 | VaC | 163 | AB_03351 | ViF | 165 | AB_01641 | Interactor | 165 | AB_01918 | Genome | 5 | |
| AB_01334 | VaC | 165 | AB_00353 | ViF | 163 | AB_02522 | Interactor | 165 | AB_02296 | Genome | 5 | |
| AB_00811 | Interactor | 124 | AB_01947 | Interactor | 165 | AB_03512 | KeF | 165 | AB_03731 | Genome | 2 | |
| AB_02146 | Interactor | 112 | AB_02143 | ViF | 123 | AB_03207 | Interactor | 165 | AB_03732 | Genome | 2 | |
| AB_00805 | Interactor | 104 | AB_03184 | Interactor | 165 | AB_00239 | KeF | 165 | AB_03317 | Genome | 82 | |
| AB_03351 | Interactor | 165 | AB_02069 | Interactor | 165 | AB_03022 | Interactor | 165 | AB_03318 | Genome | 76 | |
| AB_02143 | Interactor | 123 | AB_00812 | ViF | 163 | AB_02437 | KeF | 165 | AB_03726 | Genome | 82 | |
| AB_02888 | Interactor | 123 | AB_02142 | ViF | 134 | AB_01461 | KeF | 165 | AB_02992 | Genome | 6 | |
| AB_02124 | Interactor | 117 | AB_01731 | ViF | 165 | AB_01350 | Interactor | 165 | AB_03666 | Genome | 4 | |
| AB_02677 | Interactor | 96 | AB_02888 | Interactor | 123 | AB_01047 | Interactor | 165 | AB_02700 | Genome | 6 | |
| AB_02145 | Interactor | 104 | AB_01946 | Interactor | 165 | AB_03233 | Interactor | 165 | AB_03651 | Genome | 2 | |
| AB_01785 | Interactor | 103 | AB_01248 | Interactor | 165 | AB_03046 | Interactor | 165 | AB_02374 | Genome | 7 | |
| AB_03499 | Interactor | 131 | AB_02124 | Interactor | 165 | AB_02576 | KeF | 165 | AB_02371 | Genome | 7 | |
| AB_02178 | Interactor | 165 | AB_01647 | Interactor | 165 | AB_02252 | Interactor | 165 | AB_02372 | Genome | 7 | |
| DC | AB_03493 | VaC | 84 | AB_01641 | Interactor | 165 | AB_01491 | Interactor | 165 | AB_01641 | Genome | 165 |
| AB_01517 | VaC | 84 | AB_02522 | Interactor | 165 | AB_01641 | Interactor | 165 | AB_03207 | Genome | 165 | |
| AB_01255 | Interactor | 165 | AB_03351 | ViF | 165 | AB_00239 | KeF | 165 | AB_03659 | Genome | 165 | |
| AB_03704 | VaC | 108 | AB_00353 | ViF | 163 | AB_03022 | Interactor | 165 | AB_03512 | Genome | 165 | |
| AB_00353 | VaC | 163 | AB_01947 | Interactor | 165 | AB_02437 | KeF | 165 | AB_03532 | Genome | 165 | |
| AB_01334 | VaC | 165 | AB_03184 | Interactor | 165 | AB_01461 | KeF | 165 | AB_01740 | Genome | 165 | |
| AB_00811 | Interactor | 124 | AB_02069 | Interactor | 165 | AB_01350 | Interactor | 165 | AB_01651 | Genome | 165 | |
| AB_02146 | Interactor | 112 | AB_02627 | Interactor | 165 | AB_01047 | Interactor | 165 | AB_02576 | Genome | 165 | |
| AB_00805 | Interactor | 104 | AB_03186 | Interactor | 165 | AB_03233 | Interactor | 165 | AB_03046 | Genome | 165 | |
| AB_03351 | Interactor | 165 | AB_02909 | Interactor | 165 | AB_03046 | Interactor | 165 | AB_01461 | Genome | 165 | |
| AB_02233 | VaC | 165 | AB_01226 | Interactor | 165 | AB_02576 | KeF | 165 | AB_03233 | Genome | 165 | |
| AB_03186 | Interactor | 165 | AB_00660 | Interactor | 165 | AB_02252 | Interactor | 165 | AB_01491 | Genome | 165 | |
| AB_03184 | Interactor | 165 | AB_02910 | Interactor | 165 | AB_01051 | Interactor | 165 | AB_00091 | Genome | 165 | |
| AB_03185 | Interactor | 165 | AB_02683 | ViF | 165 | AB_01740 | Interactor | 165 | AB_00045 | Genome | 74 | |
| AB_01388 | VaC | 102 | AB_01361 | Interactor | 165 | AB_00091 | KeF | 165 | AB_01281 | Genome | 165 | |
| EC | AB_01255 | Interactor | 165 | AB_02627 | Interactor | 165 | AB_01491 | Interactor | 165 | AB_02522 | Genome | 165 |
| AB_02233 | VaC | 165 | AB_02354 | Interactor | 165 | AB_03022 | Interactor | 165 | AB_02769 | Genome | 165 | |
| AB_03186 | Interactor | 165 | AB_02629 | Interactor | 165 | AB_02437 | KeF | 165 | AB_03233 | Genome | 165 | |
| AB_03184 | Interactor | 165 | AB_01950 | Interactor | 165 | AB_01461 | KeF | 165 | AB_00091 | Genome | 165 | |
| AB_03185 | Interactor | 165 | AB_03765 | Interactor | 165 | AB_01350 | Interactor | 165 | AB_03207 | Genome | 165 | |
| AB_01226 | Interactor | 165 | AB_00169 | Interactor | 165 | AB_01047 | Interactor | 165 | AB_01641 | Genome | 165 | |
| AB_02909 | Interactor | 165 | AB_01359 | Interactor | 165 | AB_03233 | Interactor | 165 | AB_03022 | Genome | 165 | |
| AB_02910 | Interactor | 165 | AB_01360 | Interactor | 165 | AB_03046 | Interactor | 165 | AB_02437 | Genome | 165 | |
| AB_01257 | Interactor | 163 | AB_01656 | Interactor | 165 | AB_02576 | KeF | 165 | AB_01350 | Genome | 165 | |
| AB_00660 | Interactor | 165 | AB_01791 | Interactor | 156 | AB_02252 | Interactor | 165 | AB_01047 | Genome | 165 | |
| AB_00760 | Interactor | 153 | AB_01948 | ViF | 165 | AB_01051 | Interactor | 165 | AB_01461 | Genome | 165 | |
| AB_01258 | Interactor | 153 | AB_01951 | Interactor | 165 | AB_01740 | Interactor | 165 | AB_00239 | Genome | 165 | |
| AB_02683 | Interactor | 165 | AB_02545 | Interactor | 165 | AB_00091 | KeF | 165 | AB_03512 | Genome | 165 | |
| AB_00675 | Interactor | 165 | AB_02548 | Interactor | 165 | AB_01789 | KeF | 165 | AB_01491 | Genome | 165 | |
| AB_02914 | Interactor | 165 | AB_02626 | Interactor | 165 | AB_03031 | Interactor | 165 | AB_01051 | Genome | 165 |
Shades of green in light are for interactors while those in dark are the candidates from each category.
The Complete GPIN
In an attempt to project the most probable candidates for vaccines and drug targets, we have further explored the GPIN. GPIN, built from the theoretically predicted and empirically found physical and functional interactions, was analyzed for its degree distribution in general and k-core distribution in particular. With an exponential curve for the cumulative degree distribution frequency (CCDF), the non-linear preferential attachment nature was evident (Figure 4A) (Vázquez, 2003). To this end, an idea of the important proteins forming the core of the genome was obtained through the k-core network decomposition. The shell size of the 165th innermost core had a conglomerate of 494 proteins (Figures 4B,C).
We have started analyzing the GPIN with the four common centrality measures as mentioned for SPIN (Table 2). A closer look at the top 15 from each measure reflects proteins mostly from the categories of KeF, two of which have overlaps with the ViFAB category (F1 and F2, Table 2). Around two-thirds of the top-ranking positions in DC are occupied by KeFAB proteins, while 99% of the top 15 EC measures comprise the KeFAB categories.
A detailed k-core analysis of the different proteins as per the centrality measures revealed other important facts. The numbers of VaCAB proteins belonging to the innermost core are 2, 3, 7, and 12 as per the top rank holders of BC, CC, DC, and EC measures (Table 3). Of these, the numbers of VaC proteins are 1, 1, 2, and 1 as per the same measures while the rest are their interactors (Table 3). This analysis filters out HMPREF0010_01334 as per the BC, CC, and DC measure overlaps and HMPREF0010_02233 as per EC, to be in the innermost core of the whole genome from the proclaimed VaC proteins. For the ViFAB proteins, 6, 10, 14, and 14 proteins, respectively, for BC, CC, DC, and EC measures belong to the 165th core, of which only 1, 2, 2, and 1, respectively, are the VaC proteins while the rest are their interactors (Table 3). This filters out HMPREF0010_03351 from BC, CC, and DC overlaps, while the new candidates HMPREF0010_01731, HMPREF0010_02683, and HMPREF0010_01948 stand out from CC, DC, and EC, respectively. The picture becomes different when it comes to the KeFAB where CC, DC, and EC all had only 165th core proteins from the top rankers while BC reflected 14 of them. Moreover, the actual KeF candidates are more in this PIN compared to the VaC and ViF proteins. The analysis filters out 4, 5, 5, and 5 KeF proteins from the BC, CC, DC, and EC measured top rankers (Table 3). This brings forth the candidates HMPREF0010_03512 from BC and CC, HMPREF0010_00239 from CC and DC, and HMPREF0010_00091 from DC and EC overlaps. Moreover, three candidates, namely, HMPREF0010_02437, HMPREF0010_01461, and HMPREF0010_02576, were unique in having overlaps across CC, DC, and EC measures, while HMPREF0010_01789 from EC and three candidates from BC, namely, HMPREF0010_03553, HMPREF0010_01114, and HMPREF0010_00382, were unique. It is important to note that sorting of the whole genome proteins as per the four centrality measures yielded 2, 0, 14, and 15, respectively, of which 0, 0, 4, and 2 proteins, respectively, belong to either the VaC, ViF, or KeF category while the rest are their interactors (Table 3).
Furthermore, to identify the candidate VaC, ViF, and KeF proteins in the network topological space of A. baumannii, we have classified the protein sets of GPIN and represented them cartographically (Figure 5; Supplementary Data 4). Essentially, one part of such classification, namely, z score, is based on their regional connectivity with other similar proteins having a similar biological function, which is referred to as the functional module. The other part, namely, the participation coefficient, P, deals with the participation of these proteins with other functional modules, either related or non-related. Thus, there are seven such quadrants that are formed and termed R1 to R7. Noticeably, the proteins of VaCAB and ViFAB are spread throughout R1–R3 in the network space, while those of KeFAB are mostly concentrated in R5 (Figure 5; Supplementary Data 4). Interestingly, R4 and R6 did not show any mapping of the VaC, ViF, or KeF proteins or their interactors, represented by V, F, and K comprising the full VaCAB, ViFAB, and KeFAB SPIN (Figure 5; Supplementary Data 4). Furthermore, there were no proteins from the GPIN that occupied the R7 quadrant, having the highest P or z scores. With a more focused analysis to find out the most indispensable proteins of GPIN, we have performed the KFC method mentioned by Ashraf et al. (2018). These KFC proteins all belong to the innermost 165th core and had R5 as their classifying P vs. z quadrant and high EC scores (Table 3; Supplementary Data 4).
The Candidature Prediction
To ascertain the plausible candidature of the proteins of VaCAB, ViFAB, KeFAB, and GPIN as either vaccine or drug targets, we have reclassified them as VaC, ViF, and KeF, along with their interactors and KFC proteins, as mentioned in the previous section. This projects 2 VaC with 13 interactors, 4 ViF with 29 interactors, 19 KeF with 41 interactors from the 165th core and 10 KFC proteins for further analyses (Tables 3, 4). Among these, the cellular localization could not be determined for one VaC, two ViF, five KeF, and two R6 proteins, respectively. Notably, one candidate from VaC, namely, AB_02233, belongs to the outer membrane, while the periplasmic and extracellular comprise the ViF category. The remaining categories of KeF, KFC, and R6 mostly comprise cytoplasmic proteins having few cytoplasmic membrane proteins for KeF and R6 as well (Table 7). To this end, only one KeF (AB_01691) did not reflect any COG classification for the aforementioned categories. On the contrary, 50% of R6 proteins did not reflect any COG, being uncharacterized. Interestingly, only one protein from each of the VaC, ViF, and KeF categories, namely, AB_02233, AB_03351, and AB_01758, respectively, predicted a signal peptide while KFC and R6 did not show any. Moreover, only one lipoprotein cleavage site was predicted for ViF, KeF, and R6, namely, AB_03351, B_01758, and AB_00641, respectively, while VaC has two proteins (AB_01334 and AB_02233) showing the same compared to KFC, having none. Furthermore, about 50% of the proteins of the category KeF and KFC indicated an overlap of the antigenic site with the active pocket site while proteins of VaC, ViF, and R6 had no such overlap for candidature prediction (Table 7).
Table 4.
The candidate proteins of VaC, ViF, KeF, and KFC along with their interactors in A. baumannii ATCC19606 strain.
| Category | VaCAB | ViFAB | KeFAB | KFC | |||
|---|---|---|---|---|---|---|---|
| Protein type | VaC | Interactors | ViF | Interactors | KeF | Interactors | |
| AB_01334 | AB_00660 | AB_03351 | AB_00169 | AB_03689 | AB_01249 | AB_02522 | |
| AB_02233 | AB_00675 | AB_01731 | AB_00660 | AB_03360 | AB_01784 | AB_02769 | |
| AB_01226 | AB_02683 | AB_01226 | AB_03276 | AB_03688 | AB_03233 | ||
| AB_01255 | AB_01948 | AB_01248 | AB_03274 | AB_03695 | AB_00091 | ||
| AB_02178 | AB_01255 | AB_03221 | AB_03697 | AB_03207 | |||
| AB_02683 | AB_01359 | AB_03191 | AB_03371 | AB_01641 | |||
| AB_02909 | AB_01360 | AB_02008 | AB_03354 | AB_03022 | |||
| AB_02910 | AB_01361 | AB_01991 | AB_03349 | AB_02437 | |||
| AB_02914 | AB_01641 | AB_01903 | AB_03269 | AB_01350 | |||
| AB_03184 | AB_01647 | AB_01875 | AB_03268 | AB_01047 | |||
| AB_03185 | AB_01656 | AB_01854 | AB_03266 | ||||
| AB_03186 | AB_01946 | AB_01851 | AB_03257 | ||||
| AB_03351 | AB_01947 | AB_01810 | AB_03245 | ||||
| AB_01950 | AB_01791 | AB_03244 | |||||
| AB_01951 | AB_01758 | AB_03209 | |||||
| AB_02069 | AB_01757 | AB_02016 | |||||
| AB_02124 | AB_01723 | AB_01961 | |||||
| AB_02354 | AB_01691 | AB_01953 | |||||
| AB_02522 | AB_01682 | AB_01952 | |||||
| AB_02545 | AB_01931 | ||||||
| AB_02548 | AB_01899 | ||||||
| AB_02626 | AB_01885 | ||||||
| AB_02627 | AB_01876 | ||||||
| AB_02629 | AB_01868 | ||||||
| AB_02909 | AB_01834 | ||||||
| AB_02910 | AB_01829 | ||||||
| AB_03184 | AB_01817 | ||||||
| AB_03186 | AB_01816 | ||||||
| AB_03765 | AB_01796 | ||||||
| AB_01794 | |||||||
| AB_01793 | |||||||
| AB_01787 | |||||||
| AB_01784 | |||||||
| AB_01755 | |||||||
| AB_01734 | |||||||
| AB_01733 | |||||||
| AB_01725 | |||||||
| AB_01721 | |||||||
| AB_01720 | |||||||
| AB_01711 | |||||||
| AB_01665 | |||||||
VaC, ViF, KeF, and KFC denote Vaccine candidates, Virulent Factors, Key Factors, and K-core-Functional modularity-Centrality, respectively. The prefix of the accession numbers for the proteins has been replaced by AB.
The Final Selection
Finally, to assess for the relevance of the final set of proteins (Table 4), in virulence and pathogenicity of Acinetobacter, we have cross-examined through UniProt, PDB, and PubMed, for their role, either predictive or empirical, in Acinetobacter and/or other gram-negative bacterial pathogens. Two unique VaC proteins, namely, AB_01334 and AB_02233, are predicted to encode tetratricopeptide repeat protein and peptidase M16 inactive domain protein, respectively (Table 7). Annotations for other ViF proteins like thiol:disulfide interchange protein (AB_03351), malate dehydrogenase (AB_02683), and 50S ribosomal protein L7/L12 (AB_01948) were inferred from homology while AB_01731 was experimentally verified to have the function of a nucleoside diphosphate kinase (Table 7). The KeF proteins are shown to be a conglomerate of different types engaged in different biological process starting from carbohydrate, amino acid, and DNA metabolism to even those involved in signal transduction, cell wall synthesis, ribosomal and translational machineries, as well as some uncharacterized proteins (AB_02008 and AB_01691; Table 7). On the contrary, proteins of the KFC class are only concentrated on carbohydrate, amino acid, and fatty acid metabolism with the inclusion of one (AB_01641) having DNA polymerase with 5′-3′ exonuclease activity. Again, proteins of the R6 category are mostly either uncharacterized or belong to the transcriptional regulator family with the inclusion of one CRISPR-associated protein (AB_01430).
All the corresponding candidates from ATCC19606 have been mapped onto ATCC17978 and AYE strains of A. baumannii as reflected in Table 5 for the ease of use by future researchers. Moreover, to determine the orthologous presence of these proteins in the human host, their pairwise identities (PI) and query coverages (QC) are reflected in Table 6. Notably, the VaC proteins have considerably low PI and QC, which is exactly opposite in nature to all but one of the proteins of KFC, having high QC and PI. Intriguingly, ViF, KeF, and R6 comprise a mixture of unique proteins having no human counterpart as well as those having moderate to high PI. Of these, however, only proteins of ViF have very high QC as well (Table 6).
Table 5.
Corresponding candidates of A. baumannii ATCC19606 in ATCC17978 and AYE strains.
| STRAIN_19606 | STRAIN_AYE | STRAIN17978 | |
|---|---|---|---|
| VaC | HMPREF0010_01334 | ABAYE2977 | – |
| HMPREF0010_02233 | ABAYE0990 | – | |
| ViF | HMPREF0010_03351 | ABAYE3833 | – |
| HMPREF0010_01731 | ABAYE3267 | – | |
| HMPREF0010_02683 | ABAYE0465 | – | |
| HMPREF0010_01948 | ABAYE3490 | – | |
| KeF | HMPREF0010_03689 | – | A1S_0003 |
| HMPREF0010_03360 | – | A1S_0028 | |
| HMPREF0010_03276 | – | A1S_0061 | |
| HMPREF0010_03274 | – | A1S_0063 | |
| HMPREF0010_03221 | – | A1S_0114 | |
| HMPREF0010_03191 | – | A1S_0147 | |
| HMPREF0010_02008 | – | A1S_0217 | |
| HMPREF0010_01991 | – | A1S_0236 | |
| HMPREF0010_01903 | – | A1S_0334 | |
| HMPREF0010_01875 | – | A1S_0364 | |
| HMPREF0010_01854 | – | A1S_0388 | |
| HMPREF0010_01851 | – | A1S_0391 | |
| HMPREF0010_01810 | – | A1S_0428 | |
| HMPREF0010_01791 | – | A1S_0447 | |
| HMPREF0010_01758 | – | A1S_0469 | |
| HMPREF0010_01757 | – | A1S_0470 | |
| HMPREF0010_01723 | – | A1S_0506 | |
| HMPREF0010_01691 | – | A1S_0561 | |
| HMPREF0010_01682 | – | A1S_0571 | |
| KFC | HMPREF0010_02522 | – | – |
| HMPREF0010_02769 | – | – | |
| HMPREF0010_03233 | – | – | |
| HMPREF0010_00091 | – | A1S_2232 | |
| HMPREF0010_03207 | – | – | |
| HMPREF0010_01641 | – | – | |
| HMPREF0010_03022 | – | – | |
| HMPREF0010_02437 | – | A1S_3280 | |
| HMPREF0010_01350 | – | – | |
| HMPREF0010_01047 | – | – |
Table 6.
The human counterparts of the corresponding VaC, ViF, KeF, KFC, and R6 candidates of A. baumannii ATCC19606 strain.
VaC, ViF, KeF, and KFC denote Vaccine candidates, Virulent Factors, Key Factors, K-core-Functional modularity-Centrality and Function-R6, respectively. The prefix of the accession numbers for the proteins has been replaced by AB.
Discussion
The sole aim of this study is to look out for plausible vaccine and/or drug candidates among a plethora of proteins from the whole genome of A. baumannii. In this context, the work proposed by Moriel et al. (2013) has mentioned an array of proteins as candidates for vaccines to test out in real-life scenario. Besides these, several researchers have proposed different virulence factors crucial for the XDR A. baumannii, probably potential enough to be targeted as drugs. Moreover, the VFDB presents lists of several such factors as well. Considering a filtration to shortlist just few of these would probably be a good idea to save the time and money of future researchers in this field of study. Thus, network analysis is being considered in order to sort and identify, albeit theoretically, the most probable candidates among them.
With the target being set out for the pathogenic strains like AYE and ATCC17978, the main hindrance was the lack of protein interaction datasets for these strains in the STRING database. This led to the mapping of the proteins from these strains onto ATCC19606. We started with an initial categorization of the protein sets proposed by Moriel et al. (2013) based on their results. One set, representing the A. baumannii antigenic proteins identified through reverse vaccinology approach, was denoted as Vaccine Candidates (VaC). The other set was named Virulence Factors (ViF) and comprised all other proteins listed by the same group including OMVs and secretome, potentially insoluble proteins and periplasmic proteins found in OMV and secretome of A. baumannii. Initial filtration through standalone BLAST led to almost the same number of proteins in ATCC19606. A stringent filtering approach with 99–100% identity cutoff threshold, however, was adopted to rule out any ambiguity of the protein functions upon such conversion from the former strains to the latter. Similar strategies were adopted for database- and literature-retrieved proteins searched through keywords and were named Key Factors (KeF).
As the proteins would always be interacting with others to manifest their functions, a PIN construction was the next move for the VaC, ViF, and KeF protein sets to yield the three SPINs namely, VaCAB, ViFAB, and KeFAB. These were analyzed for their importance through the four centrality parameters BC, CC, DC, and EC, often utilized for biological network analysis (Jeong et al., 2001; Lahiri et al., 2014; Pan et al., 2015; Pawar et al., 2017, 2018; Ashraf et al., 2018). Among them, DC reflects the simple network connectedness of any protein, while for a virulent phenotype, CC might bring out the functional proximity of a protein with others. Moreover, BC might help in reflecting the bridging of different functionally important groups of virulent proteins, thereby posing its importance to be targeted for therapeutic purposes. EC, however, might reflect the connectivity of the most important proteins with other important proteins in a virulent network, thereby posing them to be indispensable for therapeutic targets. Notably, there were overlaps between VaC and ViF categories of proteins across different centrality measures (Table 2), probably indicating a faint line of difference between them, which actually were set by this study and not by Moriel et al. (2013). Moreover, there were very few overlaps of VaC and ViF proteins with those of KeF sets, probably indicating the uniqueness of the former groups compared to the common KeF proteins already reported in the literature and database searched through keywords (Table 2).
The functional aspects of the candidate proteins would be best put forward through the whole genome global scenario for which the GPIN was constructed (Figure 2D) and analyzed by the network centrality and other topological parametric measures (Supplementary Data 4). The initial characterization of the GPIN was done through the exponential decay of the degree distribution, P(k), of a particular node upon connecting to k other nodes, for large values of k. Such construction at least confirms the non-random (Erds and Rényi, 1960) or non-small-world nature (Watts and Strogatz, 1998) of the GPIN, if not completely following the power law (Albert et al., 2000) and becoming scale free (Figure 4A). Hereafter, the constructed GPIN is analyzed with the four centrality measures. Proteins of KeFAB categories occupying most of the EC and DC measures probably indicate the importance of either of these measures in bringing out the top rankers of KeFAB proteins (Table 2). Moreover, with no appearance among the top 15 important categories, the VaCAB and ViFAB proteins might not be so essential from the whole genome perspective (Table 2).
The actual set of proteins important from the GPIN perspective are probably reflected by the innermost core of the proteins brought about by the k-core/K-shell topological parameters (Figures 4B,C). The concept of the importance of the innermost k-core lies in the fact that k-core is a subnetwork with a minimum number of k-links such that the 165th innermost core would have 165 connections of each of those proteins lying in that core (Figure 4B; Supplementary Data 4). Essentially, the number of proteins in this case is 498 (Figure 4C; Supplementary Data 4). With such a large inner core member proteins having high connectivity, the core tends to be highly interactive and, thus, robust in nature (Alvarez-Hamelin et al., 2005). This could be indicative of the tight control resistance mechanism of this MDR/XDR species of A. baumannii. It is important to note that the relation between a K-shell and k-core is that the former is the part of the latter but not of the (k + 1)-core, such that the former is a set of nodes having exactly k-links. This brings out the fact that there are a lower number of proteins (interacting among their partners) with lower k-core that belongs to the outer shell, and thus, the innermost core would have the maximum number of proteins needed to be decomposed to affect the global network of A. baumannii. Interestingly, only limited VaC and ViF proteins (2 and 4, respectively) belong to the 165th innermost core identified through all different centrality measures (Table 3). These numbers are 12–13% of the total proteins of VaCAB and ViFAB including the VaC and ViF proteins along with their interactors. In comparison to these, ~32% of the total proteins of KeFAB are important as KeF (Table 3). These probably tells us that only few proteins are important from VaC and ViF compared to KeF categories. Thus, considering just these few proteins as either vaccines or drug targets may not be sufficient to break the robust inner core compared to a large number of options available for KeF proteins.
With some preliminary idea about the centrality and k-core measures, we moved on to delve deep into the functional connectivity, R, of the modules formed in the GPIN. Such connectivity of the proteins within and between the functional modules is represented cartographically by P-values and z score, respectively, across the x- and y-axis. This results in the lowest values of P and z for R1 and the highest for R7. The classifications are thus named ultra-peripheral proteins (R1) and peripheral proteins (R2), which can be detached with convenience from the network. Moreover, the non-hub connectors (R3) might be involved in connecting fundamental sets of interactions while the non-hub kinless proteins (R4) connect other proteins evenly distributed across the modules without forming hubs themselves. Furthermore, the provincial hub proteins, R5, have many within-module connections, whereas the connector hubs, R6 proteins connect most of the other modules, and thus are probably most conserved in terms of decomposition as well as evolution. Finally, the kinless hubs, R7 proteins, show the highest connection within and between the members of the GPIN such that they could be the most essential ones to be maintained by the pathogen for its very survival. Taking into account the importance of such functional modules, we have sorted the GPIN as per the KFC method adopted by Ashraf et al. (2018). We found the 165th core getting aligned with the functional module R5 proteins having the highest EC values (Table 3; Supplementary Data 4: Sheet 2).
With a consolidated set of VaC, ViF, KeF, and KFC proteins, we have set our ultimate goal to determine their antigenic potential and predicted some probable active sites. As cellular location plays an important role in conferring the potential of proteins as vaccines and/or drugs, we have attempted for the same and identified the COG classes for them, keeping in view the uncharacterized proteins taken as reference for the R6 categories. Interestingly, most of the proteins of VAC, ViF, KeF, and KFC could be classified as per COG, suggesting their likelihood to share similar functions. To this end, the antibody epitope site prediction shared 50% similarities with the predicted active sites of the validated homology modeled structures of KeF and KFC proteins, suggesting their plausibility to be used either as vaccine candidates or as drug candidates. On the contrary, VaC, ViF, and R6 proteins were unique enough and would probably have to be experimentally verified for their actual candidature. All such VaC, ViF, KeF, and KFC proteins were mapped back from ATCC19606 strains to the strains of AYE or ATCC17978 for the ease of quick referral for future researchers (Table 5). Besides the aforementioned list in Table 4, a comparison of the proteins with their Homo sapiens counterpart has been made as reflected in Table 6. This was done to ensure that the proteins of A. baumannii are different from their host, either fully or largely, and thus could be used as targets.
A comparison of the data from Tables 6, 7 shows that the protein with the accession ID AB_02233 is a good candidate for vaccine, residing on the outer membrane, having a signal peptide, and bearing much less similarity with its human counterpart. Similarly, the protein AB_03351 is a good candidate for drugs, having periplasmic location, a signal peptide, and no match at all with the human host proteins. A very difficult comparison is posed for the KeF proteins, which largely present themselves as either cytoplasmic or cytoplasmic membrane. The protein with the accession number AB_01758 could have some potential though its similarity with the human counterpart rules out such possibility. Proteins of the KFC category, surprisingly, do not present themselves as good candidates at all, all being cytoplasmic, having no signal peptides, and bearing huge similarity with their human homologs. The protein AB_02522 in this category having much less query coverage (3%), however, does not stand a chance either, owing to its cellular localization in the cytoplasm. Similarly, most cytoplasmic proteins of the R6 category present themselves as poor candidates for vaccines and/or drugs. One uncharacterized protein, viz., AB_00210 of unknown cellular localization in this category could be of some potential due to the absence of any match with human counterpart.
Table 7.
Protein analyses of selected VaC, ViF, KeF, and KFC proteins of A. baumannii for cellular localization, COG classification, antigenicity, and active site predictions.
| String-id | Protein description: function | Localization | COG analysis | Cleavage position | Epitope analysis | Active site analysis | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PSORTb | Accession | Name | SignalP | LipoP | Peptide | Start | End | length | |||
| VaCAB | |||||||||||
| AB_01334 | Tetratricopeptide repeat protein: DNA binding | Unknown | COG5010 | TadD | 32,N | 31–32 | KHANDPQL | 547 | 554 | 8 | 511–523 |
| AB_02233 | Peptidase M16 inactive domain protein: metal ion binding; metalloendopeptidase activity | Outer Membrane | COG0612 | PqqL | 24,Y | 23–24 | KDKPKTLDQTDVKAEPLKDPKVY | 454 | 476 | 23 | 404–442 |
| ViFAB | |||||||||||
| AB_03351 | Thiol:disulfide interchange protein protein disulfide oxidoreductase activity | Periplasmic | COG1651 | DsbG | 23,Y | 22–23 | GKVEVP | 38 | 43 | 6 | 67–81 |
| QGEDGK | 184 | 189 | 6 | ||||||||
| AB_01731 | Nucleoside diphosphate kinase: ATP binding; metal ion binding; nucleoside diphosphate kinase activity | Extracellular | COG0105 | Ndk | 48,N | – | NAAHGSDSVAS | 114 | 124 | 11 | 104–128 |
| AB_02683 | Malate dehydrogenase: L-malate dehydrogenase activity | Unknown | COG0039 | Mdh | 28,N | – | GESLKDKINDPAW | 204 | 216 | 13 | 225–244 |
| AB_01948 | 50S ribosomal protein L7/L12: structural constituent of ribosome | Unknown | COG0222 | RplL | 51,N | – | APAGGAAAAAEEQSE | 42 | 56 | 15 | 38–50 |
| KeFAB | |||||||||||
| AB_03689 | DNA replication and repair protein RecF: ATP binding; single-stranded DNA binding | Cytoplasmic | COG1195 | RecF | 30,N | – | DPQSTDI | 114 | 120 | 7 | 112–165 |
| AB_03360 | Alkanesulfonate monooxygenase: alkanesulfonate monooxygenase activity | Cytoplasmic | COG2141 | COG2141 | 37,N | – | TWGEPPAAV | 198 | 206 | 9 | 140–172 |
| ALVGDPETV | 306 | 314 | 9 | 356–365 | |||||||
| AB_03276 | Bacterial sugar transferase: transferase activity, transferring glycosyl groups | Cytoplasmic Membrane | COG2148 | WcaJ | 30,N | – | FDAQGNPLPDEARI | 66 | 79 | 14 | 42–97 |
| AB_03274 | UDP-glucose 6-dehydrogenase: NAD binding; UDP-glucose 6-dehydrogenase activity | Cytoplasmic | COG1004 | Ugd | 17,N | – | KENTSSTHN | 307 | 315 | 9 | 306–399 |
| AB_03221 | Phosphopantetheine attachment domain protein: phosphopantetheine binding | Unknown | COG0236 | AcpP | 25,N | – | PETIDPDQKF | 24 | 33 | 10 | 32–67 |
| AB_03191 | ATP synthase F0, I subunit: hydrolase activity | Cytoplasmic Membrane | COG3312 | AtpI | 31,N | – | AR | 67 | 68 | 2 | 68–74 |
| AB_02008 | Uncharacterized protein | Unknown | COG2960 | COG2960 | 55,N | – | DEPKKD | 14 | 19 | 6 | 32–43 |
| AB_01991 | Response regulator receiver domain protein: DNA binding | Cytoplasmic | COG2197 | CitB | 26,N | – | SDTQQSPFDS | 139 | 148 | 10 | 131–174 |
| AB_01903 | Ribosome maturation factor RimP: Required for maturation of 30S ribosomal subunits | Cytoplasmic | COG0779 | COG0779 | 21,N | – | PVDENAEPVINEDGEVEQG | 46 | 64 | 19 | 60–61 |
| AB_01875 | DnaJ domain protein unfolded protein binding+C4 | Cytoplasmic | COG0484 | DnaJ | 62,N | – | GFGGGQQQYQRQ | 117 | 128 | 12 | 101–195 |
| AB_01854 | S-(Hydroxymethyl)glutathione synthase: carbon-sulfur lyase activity | Unknown | COG3791 | COG3791 | 46,N | – | TPLDQK | 104 | 109 | 6 | 72–115 |
| AB_01851 | 50S ribosomal protein L31 type B: structural constituent of ribosome | Cytoplasmic | COG0254 | RpmE | 20,N | – | QTKQTKEYQG | 30 | 39 | 10 | 40–41 |
| AB_01810 | Endonuclease/ exonuclease/ phosphatase family protein: endonuclease activity; exonuclease activity | Cytoplasmic | COG3021 | COG3021 | 23,N | – | PKPPSPTEAKDSTL | 208 | 221 | 14 | 245–291 |
| AB_01791 | 50S ribosomal protein L33: structural constituent of ribosome | Cytoplasmic | COG0267 | RpmG | 21,N | – | KNKRTM | 21 | 26 | 6 | 22–31 |
| AB_01758 | Sel1 repeat protein | Unknown | COG0790 | COG0790 | 21,Y | 20–21 | ASNGDNR | 120 | 126 | 7 | 97–129 |
| AB_01757 | Methionine biosynthesis protein MetW | Cytoplasmic | COG2226 | UbiE | 31,N | – | IK | 12 | 13 | 2 | 8–18 |
| NQ | 72 | 73 | 2 | 81–84 | |||||||
| AB_01723 | GTPase Der: GTP binding | Cytoplasmic Membrane | COG1160 | COG1160 | 31,N | – | SENPFEGRKSQVDERTA | 434 | 450 | 17 | 119–150 |
| AB_01691 | Uncharacterized protein | Unknown | NA | NA | 21,N | – | TMKPNNHSTETNTPPAI | 36 | 52 | 17 | 69–74 |
| AB_01682 | AP endonuclease, family 2: endonuclease activity; isomerase activity | Cytoplasmic | COG3622 | Hfi | 29,N | – | PGRHEPDTAQI | 211 | 221 | 11 | 206–242 |
| KFC | |||||||||||
| AB_02522 | Glutamate synthase [NADPH], large subunit: glutamate synthase (NADPH) activity | Cytoplasmic | COG0067 | GltB | 60,N | – | GRSNSGEGGEDPARY | 896 | 910 | 15 | 867–996 |
| COG0069 | GltB | ||||||||||
| COG0070 | GltB | ||||||||||
| AB_02769 | Putative fatty acid oxidation complex subunit alpha: 3-hydroxyacyl-CoA dehydrogenase activity; lyase activity | Cytoplasmic | COG1250 | FadB | 26,N | – | YKIPGGDPKTPA | 216 | 227 | 12 | 239–297 |
| AB_03233 | Methylmalonate-semialdehyde dehydrogenase (Acylating): methylmalonate-semialdehyde dehydrogenase (acylating) activity | Cytoplasmic | COG1012 | PutA | 70,N | – | GKTLADAEGD | 106 | 115 | 10 | 141–147 |
| AB_00091 | Methylmalonate-semialdehyde dehydrogenase (Acylating): methylmalonate-semialdehyde dehydrogenase (acylating) activity | Cytoplasmic | COG1012 | PutA | 44,N | – | ARKQPVYNPATGEIS | 41 | 55 | 15 | 140–145 |
| AB_03207 | GMP synthase [glutamine-hydrolyzing]: ATP binding; GMP synthase (glutamine-hydrolyzing) activity; pyrophosphatase activity | Cytoplasmic | COG0518 | GuaA | 23,N | – | GPESVHEEGSPRA | 61 | 73 | 13 | 60–92 |
| COG0519 | GuaA | ||||||||||
| AB_01641 | DNA polymerase I: 3′-5′ exonuclease activity; DNA binding; DNA-directed DNA polymerase activity | Cytoplasmic | COG0749 | PolA | 20,N | – | VKPAQTIETEDQASLTSQDDQLG | 303 | 325 | 23 | 599–687 |
| AB_03022 | Putative aminobutyraldehyde dehydrogenase: oxidoreductase activity, acting on the aldehyde or oxo group of donors, NAD or NADP as acceptor | Cytoplasmic | COG1012 | PutA | 36,N | – | EVYAQSPNATEAEV | 32 | 45 | 14 | 33–97 |
| AB_02437 | Succinate-semialdehyde dehydrogenase [NAD(P)+]: succinate-semialdehyde dehydrogenase [NAD(P)+] activity | Cytoplasmic | COG1012 | PutA | 61,N | – | DGRQEGSTQGPLI | 318 | 330 | 13 | 330–386 |
| AB_01350 | Succinate-semialdehyde dehydrogenase [NAD(P)+]: succinate-semialdehyde dehydrogenase [NAD(P)+] activity | Cytoplasmic | COG1012 | PutA | 38,N | – | NATVPVSNPATGEEIG | 26 | 41 | 16 | 59–73 |
| AB_01047 | Succinate-semialdehyde dehydrogenase [NAD(P)+]: succinate-semialdehyde dehydrogenase [NAD(P)+] activity | Cytoplasmic | COG1012 | PutA | 16,N | – | GLGREGSKY | 459 | 467 | 9 | 410–476 |
| R6 | |||||||||||
| AB_00210 | Uncharacterized protein | Unknown | COG2911 | COG2911 | 21,N | – | VEQQPTSAPSSPK | 4 | 16 | 13 | |
| AB_00797 | Transcriptional regulator, TetR family: DNA binding | Cytoplasmic | COG1309 | AcrR | 39,N | – | EKA | 78 | 80 | 3 | |
| AB_03124 | Putative phage uncharacterized protein domain protein | Cytoplasmic Membrane | COG5412 | COG5412 | 40,N | – | DPTKPIEPPKPPKLGL GTAPPNPKLGIGTGE KDDKGGSKSSAKS KAEQEAKERQRQAEQ | 494 | 552 | 59 | 587–599 |
| AB_02872 | Uncharacterized protein | Cytoplasmic | NA | NA | 45,N | – | QQHAGESVKKNRKAQSIKS GYDESAEQSVELEED FEQYAAEQQQAR EQAKQQRQQQKREQAEQM | 122 | 185 | 64 | 65–89 |
| AB_02571 | Uncharacterized protein | Cytoplasmic | NA | NA | 42,N | – | SIDPEQVED | 118 | 126 | 9 | |
| QQAENPKKG | 294 | 302 | 9 | ||||||||
| AB_00406 | Uncharacterized protein | Cytoplasmic Membrane | NA | NA | 11,N | – | SGSARPGFS | 139 | 147 | 9 | 57–136 |
| AB_00641 | Uncharacterized protein | Cytoplasmic | NA | NA | 23,N | 17–18 | AEEKPTEKTEKTS TIKATEQPPKEEN | 22 | 47 | 26 | 16–97 |
| AB_01430 | CRISPR-associated protein, Csy2 family | Unknown | NA | NA | 32,N | – | RQAQDQENTAHA | 227 | 238 | 12 | 110–178 |
| AB_01223 | Transcriptional regulator, TetR family: DNA binding | Cytoplasmic | COG1309 | AcrR | 33,N | – | ESNQDDQ | 131 | 137 | 7 | |
| AB_01974 | Transcriptional regulator, TetR family: DNA binding | Cytoplasmic | COG1309 | AcrR | 11,N | – | EFPANSSERSSVKQ | 112 | 125 | 14 | |
A completely different approach of interactor-free centrality-based ranking of the different classes of the candidates proposed by Moriel et al. (2013) unanimously pulls out AB_00353 from proteins of the outer membrane class, AB_01731, from those of the extracellular region and a set of five proteins from the periplasmic region category (Supplementary Data 5). Among these, the protein AB_00353 or BamA has already been found by some in silico approach earlier and reported to elicit high IgG antibody titer with the production of opsonizing antibodies against a virulent MDR clinical isolate using a murine pneumonia model (Singh et al., 2017). The other protein, AB_01731, coding for nucleoside diphosphate kinase, has also been reported by another group through reverse vaccinology approach (Chiang et al., 2015). Of the five periplasmic proteins, HMPREF0010_03351 (Supplementary Data 5) (reflected as AB_03351 in Table 7) has been found out by our detailed interactome-based approach as well. These proteins have the potential to be used as either vaccine candidates, for the outer membrane proteins, or drugs, for other periplasmic or cytoplasmic proteins. Unknown or uncharacterized proteins from different aforementioned categories (Table 7), however, can be worked upon by future researchers for more prospective candidates as well. Thus, a mixed-bag result has come out of the analyses done through our two approaches. Of the interactome-based approach, where the interactors have been considered as well, the actual candidates were sorted through network centrality and protein signature analyses and the results need to be further experimentally validated as most of the candidates are novel and not reported earlier. The other, interactor-free approach, considered the candidates from their cellular localization and network centrality analyses unanimously bring out two candidates already reported earlier. The success of future researchers in targeting a protein for vaccine candidates and/or drugs would, thus, depend on further experimental validation for any category of proteins.
Conclusion
The study is based on the concept of utilizing the already proclaimed vaccine candidates and virulent and other key pathogenic factors to sort and filter them to a useful list of most probable final candidates. Essentially, the work revolved around a network biological approach of analyzing the conceived networks of the aforementioned proteins and mapping them onto the whole genome perspective to shortlist the candidates. To this end, established methods of antigenicity and active site prediction have been added to produce the final list for further experimental validation.
Author Contributions
The analyses and the study were conceptualized, planned, and designed by CL. Data generated by RM and SM were analyzed by CL and supported by SM and RM with tabulation. Artwork was done by RM. CL wrote the manuscript aided by inputs from SP, SM, and DG in general and RM and SM for the Materials and Methods section.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the support of Sunway University, Selangor, Malaysia, and IMSc, Chennai, India, for providing the computational facilities. The authors immensely acknowledge the contribution made by Md. Izhar Ashraf in providing some initial network data for further analyses. They also wish to thank Indhuja Thirumudi, Ng Jia Wei, and Vaibhav Kandale for their preliminary initiatives to earlier forms of this work which metamorphosed to the current state.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00203/full#supplementary-material
Excel sheets showing the data collection, processing, interactome construction, analysis, and functions of vaccine candidates, VaC of A. baumannii.
Excel sheets showing the data collection, processing, interactome construction, analysis, and functions of virulent factors, ViF of A. baumannii.
Excel sheets showing the data collection, processing, interactome construction, analysis, and functions of key factors, KeF of A. baumannii.
Excel sheets showing the data collection, processing, interactome construction, analysis, and functions of whole genome proteins of A. baumannii.
Excel sheets showing the strain mapping, cellular localization, and centrality measures along with Venn diagram for the total set of candidate proteins of A. baumannii.
References
- Albert R., Jeong H., Barabasi A. L. (2000). Error and attack tolerance of complex networks. Nature 406:378. 10.1038/35019019 [DOI] [PubMed] [Google Scholar]
- Alvarez-Hamelin J. I., Dall'Asta L., Barrat A., Vespignani A. (2005). K-core decomposition of internet graphs: hierarchies, self-similarity and measurement biases. arXiv. 10.3934/nhm.2008.3.371 [DOI] [Google Scholar]
- Antunes L. C., Visca P., Towner K. J. (2014). Acinetobacter baumannii: evolution of a global pathogen. Pathog. Dis. 71, 292–301. 10.1111/2049-632X.12125 [DOI] [PubMed] [Google Scholar]
- Apweiler R., Bairoch A., Wu C. H., Barker W. C., Boeckmann B., Ferro S., et al. (2004). UniProt: The universal protein knowledgebase. Nucleic Acids Res. 32(Suppl.1), D115–D119. 10.1093/nar/gkh131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf M. I., Ong S. K., Mujawar S., Pawar S., More P., Paul S., et al. (2018). A side-effect free method for identifying cancer drug targets. Sci. Rep. 8:25042. 10.1038/s41598-018-25042-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assenov Y., Ramírez F., Schelhorn S. E., Lengauer T., Albrecht M. (2007). Computing topological parameters of biological networks. Bioinformatics 24, 282–284. 10.1093/bioinformatics/btm554 [DOI] [PubMed] [Google Scholar]
- Bastian M., Heymann S., Jacomy M. (2009). Gephi: an open source software for exploring and manipulating networks. Icwsm 8, 361–362. [Google Scholar]
- Bertot G. M., Restelli M. A., Galanternik L., Urey R. C. A., Valvano M. A., Grinstein S. (2007). Nasal immunization with Burkholderia multivorans outer membrane proteins and the mucosal adjuvant adamantylamide dipeptide confers efficient protection against experimental lung infections with B. multivorans and B. cenocepacia. Infect. Immun. 75, 2740–2752. 10.1128/IAI.01668-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Zheng D., Liu B., Yang J., Jin Q. (2015). VFDB 2016: hierarchical and refined dataset for big data analysis−10 years on. Nucleic Acids Res. 44, D694–D697. 10.1093/nar/gkv1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang M. H., Sung W. C., Lien S. P., Chen Y. Z., Lo A. F., Huang J. H., et al. (2015). Identification of novel vaccine candidates against Acinetobacter baumannii using reverse vaccinology. Hum. Vaccines Immunother. 11, 1065–1073. 10.1080/21645515.2015.1010910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvishi M. (2016). Virulence factors profile and antimicrobial resistance of Acinetobacter baumannii strains isolated from various infections recovered from immunosuppressive patients. Biomed. Pharmacol. J. 9, 1057–1062. 10.13005/bpj/1048 [DOI] [Google Scholar]
- Diestel R. (2000). Graph Theory. Heidelberg: Springer-Verlag. [Google Scholar]
- Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., Liang J. (2006). CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34(Suppl.2), W116–W118. 10.1093/nar/gkl282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erds P., Rényi A. (1960). On the evolution of random graphs. Publ. Math. Inst. Hung. Acad. Sci. 5, 17–61. [Google Scholar]
- Eveillard M., Kempf M., Belmonte O., Pailhoriès H., Joly-Guillou M. L. (2013). Reservoirs of Acinetobacter baumannii outside the hospital and potential involvement in emerging human community-acquired infections. Int. J. Infect. Dis. 17, e802–e805. 10.1016/j.ijid.2013.03.021 [DOI] [PubMed] [Google Scholar]
- Fournier P. E., Richet H. (2006). The epidemiology and control of Acinetobacter baumannii in health care facilities. Clin. Infect. Dis. 42, 692–699. 10.1086/500202 [DOI] [PubMed] [Google Scholar]
- Fransen F., Boog C. J., van Putten J. P., van der Ley P. (2007). Agonists of Toll-like receptors 3, 4, 7, and 9 are candidates for use as adjuvants in an outer membrane vaccine against Neisseria meningitidis serogroup B. Infect. Immun. 75, 5939–5946. 10.1128/IAI.00846-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimerà R., Amaral L. A. (2005b). Cartography of complex networks: modules and universal roles. J. Stat. Mech. Theory Exp. 2005:P02001. 10.1088/1742-5468/2005/02/P02001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimerà R., Nunes Amaral L. A. (2005a). Functional cartography of complex metabolic networks. Nature 433:895. 10.1038/nature03288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H., Mason S. P., Barabási A. L., Oltvai Z. N. (2001). Lethality and centrality in protein networks. Nature 411:41. 10.1038/35075138 [DOI] [PubMed] [Google Scholar]
- Juncker A. S., Willenbrock H., Von Heijne G., Brunak S., Nielsen H., Krogh A. (2003). Prediction of lipoprotein signal peptides in gram-negative bacteria. Protein Sci. 12, 1652–1662. 10.1110/ps.0303703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protocols 10:845. 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri C., Pawar S., Sabarinathan R., Ashraf M. I., Chand Y., Chakravortty D. (2014). Interactome analyses of Salmonella pathogenicity islands reveal SicA indispensable for virulence. J. Theor. Biol. 363, 188–197. 10.1016/j.jtbi.2014.08.013 [DOI] [PubMed] [Google Scholar]
- Lee C. R., Lee J. H., Park M., Park K. S., Bae I. K., Kim Y. B., et al. (2017). Biology of Acinetobacter baumannii: pathogenesis, antibiotic resistance mechanisms, and prospective treatment options. Front. Cell. Infect. Microbiol. 7:55. 10.3389/fcimb.2017.00055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M. F., Lan C. Y. (2014). Antimicrobial resistance in Acinetobacter baumannii: From bench to bedside. World J. Clin. Cases 2, 787–814. 10.12998/wjcc.v2.i,12.787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATLAB and Statistics Toolbox Release (2010). The MathWorks, Inc. Natick, MA. [Google Scholar]
- Moriel D. G., Beatson S. A., Wurpel D. J., Lipman J., Nimmo G. R., Paterson D. L., et al. (2013). Identification of novel vaccine candidates against multidrug-resistant Acinetobacter baumannii. PLoS ONE. 8:e77631. 10.1371/journal.pone.0077631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveros J. C. (2007–2015). Venny. An Interactive Tool for Comparing Lists with Venn's Diagrams. Available online at: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed January 31, 2019).
- Pan A., Lahiri C., Rajendiran A., Shanmugham B. (2015). Computational analysis of protein interaction networks for infectious diseases. Briefings Bioinf. 17, 517–526. 10.1093/bib/bbv059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlopoulos G. A., Secrier M., Moschopoulos C. N., Soldatos T. G., Kossida S., Aerts J., et al. (2011). Using graph theory to analyze biological networks. BioData Mining 4, 10. 10.1186/1756-0381-4-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar S., Ashraf M. I., Mehata K. M., Lahiri C. (2017). Computational identification of indispensable virulent proteins of Salmonella Typhi CT18, in Current Topics in Salmonella and Salmonellosis, ed Mares M. (InTech Publishers; ), 21–39. [Google Scholar]
- Pawar S., Ashraf M. I., Mujawar S., Mishra R., Lahiri C. (2018). In silico identification of the indispensable quorum sensing proteins of multidrug resistant Proteus mirabilis. Front. Cell. Infect. Microbiol. 8:269. 10.3389/fcimb.2018.00269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez F., Hujer A. M., Hujer K. M., Decker B. K., Rather P. N., Bonomo R. A. (2007). Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 51, 3471–3484. 10.1128/AAC.01464-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen T. N., Brunak S., von Heijne G., Nielsen H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- Rosvall M., Bergstrom C. T. (2011). Multilevel compression of random walks on networks reveals hierarchical organization in large integrated systems. PLoS ONE. 6:e18209. 10.1371/journal.pone.0018209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwede T., Kopp J., Guex N., Peitsch M. C. (2003). SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385. 10.1093/nar/gkg520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman S. B. (1983). Network structure and minimum degree. Soc. Networks 5, 269–287. 10.1016/0378-8733(83)90028-X [DOI] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Capalash N., Sharma P. (2017). Immunoprotective potential of BamA, the outer membrane protein assembly factor, against MDR Acinetobacter baumannii. Sci. Rep. 7:12411. 10.1038/s41598-017-12789-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., et al. (2016). The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016:gkw937 10.1093/nar/gkw937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot G. H., Bradley J., Edwards J. E., Jr., Gilbert D., Scheld M., Bartlett J. G. (2006). Bad bugs need drugs: an update on the development pipeline from the antimicrobial availability task force of the infectious diseases society of America. Clin. Infect. Dis. 42, 657–668. 10.1086/499819 [DOI] [PubMed] [Google Scholar]
- Tang Y., Li M., Wang J., Pan Y., Wu F. X. (2015). CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 127, 67–72. 10.1016/j.biosystems.2014.11.005 [DOI] [PubMed] [Google Scholar]
- Vázquez A. (2003). Growing network with local rules: preferential attachment, clustering hierarchy, and degree correlations. Phys. Rev. E 67:056104. 10.1103/PhysRevE.67.056104 [DOI] [PubMed] [Google Scholar]
- Vella D., Marini S., Vitali F., Di Silvestre D., Mauri G., Bellazzi R. (2018). MTGO: PPI network analysis via topological and functional module identification. Sci. Rep. 8:5499. 10.1038/s41598-018-23672-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viehman J. A., Nguyen M. H., Doi Y. (2014). Treatment options for carbapenem-resistant and extensively drug-resistant Acinetobacter baumannii infections. Drugs 74, 1315–1333. 10.1007/s40265-014-0267-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J. L., Rello J., Marshall J., Silva E., Anzueto A., Martin C. D., et al. (2009). International study of the prevalence and outcomes of infection in intensive care units. JAMA 302, 2323–2329. 10.1001/jama.2009.1754 [DOI] [PubMed] [Google Scholar]
- Vita R., Overton J. A., Greenbaum J. A., Ponomarenko J., Clark J. D., Cantrell J. R., et al. (2014). The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 43, D405–D412. 10.1093/nar/gku938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts D. J., Strogatz S. H. (1998). Collective dynamics of ‘small-world’ networks. Nature 393:440. 10.1038/30918 [DOI] [PubMed] [Google Scholar]
- Wu S., Zhu Z., Fu L., Niu B., Li W. (2011). WebMGA: A customizable web server for fast metagenomic sequence analysis. BMC Genomics 12:444. 10.1186/1471-2164-12-444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N. Y., Wagner J. R., Laird M. R., Melli G., Rey S., Lo R., et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615. 10.1093/bioinformatics/btq249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Excel sheets showing the data collection, processing, interactome construction, analysis, and functions of vaccine candidates, VaC of A. baumannii.
Excel sheets showing the data collection, processing, interactome construction, analysis, and functions of virulent factors, ViF of A. baumannii.
Excel sheets showing the data collection, processing, interactome construction, analysis, and functions of key factors, KeF of A. baumannii.
Excel sheets showing the data collection, processing, interactome construction, analysis, and functions of whole genome proteins of A. baumannii.
Excel sheets showing the strain mapping, cellular localization, and centrality measures along with Venn diagram for the total set of candidate proteins of A. baumannii.