

 A new series of N-phenyl-4,5-dibromopyrrolamides was developed as inhibitors of bacterial DNA gyrase B with IC50 in the low nanomolar range.
A new series of N-phenyl-4,5-dibromopyrrolamides was developed as inhibitors of bacterial DNA gyrase B with IC50 in the low nanomolar range.
Abstract
Due to the rapid development of antimicrobial resistance, the discovery of new antibacterials is essential in the fight against potentially lethal infections. The DNA gyrase B (GyrB) subunit of bacterial DNA gyrase is an excellent target for the design of antibacterials, as it has been clinically validated by novobiocin. However, there are currently no drugs in clinical use that target GyrB. We prepared a new series of N-phenyl-4,5-dibromopyrrolamides and evaluated them against DNA gyrase and against the structurally and functionally similar enzyme, topoisomerase IV. The most active compound, 28, had an IC50 of 20 nM against Escherichia coli DNA gyrase. The IC50 values of 28 against Staphylococcus aureus DNA gyrase, and E. coli and S. aureus topoisomerase IV were in the low micromolar range. However, the compounds evaluated did not show significant antibacterial activities against selected Gram-positive and Gram-negative bacteria. Our results indicate that for potent inhibition of DNA gyrase, a combination of polar groups on the carboxylic end of the molecule and substituents that reach into the ‘lipophilic floor’ of the enzyme is required.
1. Introduction
Few antibacterials have reached the market over the last 50 years.1,2 At the same time, the rapid spread of antibacterial resistance has become a huge problem worldwide.3 Therefore, the search for new antibacterials, especially for those with new mechanisms of action or from new chemical classes, is extremely important. DNA gyrase is an enzyme that is not present in humans, but is essential for the survival of bacteria, as it is responsible for cleavage and reunion of the DNA molecule during DNA replication, transcription and recombination. In addition to DNA gyrase, bacteria have a second enzyme that is structurally and functionally similar to DNA gyrase, known as topoisomerase IV, which is also essential for bacterial cell division. Both of these enzymes are assembled as tetrameric complexes with the different subunits required for binding, cleavage and transport of DNA (DNA gyrase, 2× subunit A [GyrA]; topoisomerase IV, 2× subunit A [ParC]), and for hydrolysis of ATP (DNA gyrase, 2× subunit B [GyrB]; topoisomerase IV, 2× subunit B [ParE]).4–7 The most well-known inhibitors of DNA gyrase and topoisomerase IV are the fluoroquinolones, which act by binding to the GyrA subunit and stabilising the complex between the enzyme and the DNA molecule.8
There are currently no inhibitors of GyrB in clinical use that act through an ATP-competitive mechanism. Nevertheless, several classes of ATP-competitive DNA gyrase and/or topoisomerase IV inhibitors have been discovered in recent decades,4,9,10 such as pyridylureas,11 pyrimidinoindoles,12 benzimidazole ureas,13 benzothiazoles,14–17 pyrazolopyridones,18 and pyrrolamides19 (Fig. 1). Some of the new inhibitors have advanced to phase I clinical trials, although none of them have so far reached clinical use. Further research is required to produce drug candidates with stronger antibacterial activities and good absorption, distribution, metabolism and excretion (ADME) properties.
Fig. 1. Representative ATP-competitive DNA gyrase and topoisomerase IV inhibitors.

2. Results and discussion
2.1. Design
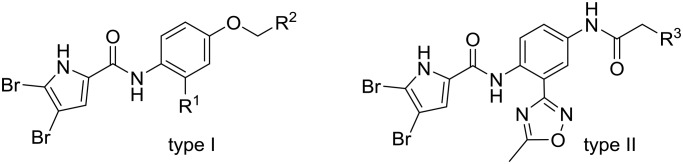
The present series of compounds was designed using the inhibitor A – E. coli GyrB structure as template (PDB code: ; 4ZVI),20 and the structure–activity relationship data derived from our previous N-phenylpyrrolamide GyrB inhibitors.21–24 We designed two types of compounds here, types I and II, where the most important functional groups that interact with amino acid residues in the binding pocket were retained, and additional substituents were introduced to attempt to increase the binding affinities (Fig. 2). To form hydrophobic contacts in the lipophilic pocket and to make important H-bonds with Asp73, a 4,5-dibromopyrrolamide moiety was attached (Fig. 2, inhibitor A, left side). Different groups were introduced to the central benzene ring to target either the ‘lipophilic floor’ of the enzyme (Fig. 2, type I, R1), or to establish polar interactions with the Glu50–Arg76 salt bridge or with Arg136 (Fig. 2, type 1, R2; type II, R3). To investigate the importance of hydrophobic interactions with the lipophilic floor, in the type I series, derivatives with benzyloxy substituents at position R1 were prepared (Fig. 2). Alternatively, in the type II series, a 5-methyl-1,2,4-oxadiazole moiety was attached to position R1 (Fig. 2). To the right-hand side of the type I and type II compounds illustrated in Fig. 2, different polar groups were introduced to form contacts with the Glu50–Arg76 salt bridge or with Arg136, such as terminal carboxylic or hydrazide groups. Additionally, as a slightly less acidic alternative for the carboxylic group, compounds with a 1,3,4-oxadiazole ring at R2 or R3 were prepared, with the aim of increasing the penetration of compounds into bacterial cells. In the type II series, a carbonyl group in the linker that connected the phenyl ring and the terminal acidic group was retained to form similar interactions with Glu50–Arg76 as the carbonyl group of inhibitor A (Fig. 2).
Fig. 2. General structures of type I and type II compounds as potential DNA gyrase B inhibitors, designed using the X-ray structure of inhibitor A bound to E. coli GyrB (PDB code: ; 4ZVI)20 as template.

2.2. Chemistry
Type I compounds 5–7 were synthesised according to Scheme 1. The synthetic procedure and analytical data for compounds 5 and 8 were reported previously.20 In the first step, 4-nitrophenol (1) was reacted with methyl 2-bromoacetate (2) to obtain compound 3. The nitro group of 3 was then reduced to an amino group by catalytic hydrogenation, and in the next step, the O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU)-promoted coupling of the amine 4 with 4,5-dibromopyrrole-2-carboxylic acid afforded compound 5. The final compound 7 was obtained using a two-step procedure; first the hydrazide 6 was formed using hydrazine monohydrate as the reagent, which was followed by a 1,1-carbonyldiimidazole-driven cyclisation and 1,3,4-oxadiazole ring formation.
Scheme 1. Reagents and conditions: (a) K2CO3, CH3CN, rt, 15 h; (b) H2, Pd/C, tetrahydrofuran, rt, 5 h; (c) 4,5-dibromo-pyrrole-2-carboxylic acid, TBTU, N-methylmorpholine, CH2Cl2, 50 °C, 15 h; (d) hydrazine monohydrate, ethanol, 80 °C, 15 h; (e) 1,1-carbonyldiimidazole, 1,4-dioxane, 101 °C, 15 h; (f) 2 M NaOH, tetrahydrofuran, rt, 15 h.

Type I compounds with benzyloxy substituents on the central benzene ring (14–17) were prepared according to Scheme 2 (see also Fig. 2, R1). First, the amino group of 9 was Boc protected, and the resulting compound 10 was then benzylated on the 2-OH group with benzyl bromide in the presence of potassium carbonate. The selective formation of the 2-benzyloxy derivative 11 was confirmed with 1H–13C HSQC (heteronuclear single-quantum correlation spectroscopy) and 1H–13C HMBC (heteronuclear multiple-bond correlation spectroscopy) nuclear magnetic resonance (ESI† Fig. S1). Next, the 4-OH group of compound 11 was alkylated using methyl bromoacetate, to obtain 12, after which the Boc protecting group was removed with gaseous hydrochloric acid, which resulted in compound 13. 4,5-Dibromopyrrol-2-carboxylic acid was attached to the amino group of 13 using TBTU as coupling reagent to obtain 14. The final compound 16 was synthesised first via the hydrazide formation, and then cyclisation to the 1,3,4-oxadiazole ring using 1,1-carbonyldiimidazole. Compound 17 was obtained by alkaline hydrolysis of the methyl ester group of 14.
Scheme 2. Reagents and conditions: (a) Boc2O, Et3N, methanol, 0 °C → rt, 15 h; (b) benzyl bromide, K2CO3, CH3CN, 0 °C → rt, 15 h; (c) methyl bromoacetate, K2CO3, CH3CN, 0 °C → rt, 15 h; (d) HCl(g), diethyl ether, rt, 15 h; (e) 4,5-dibromo-pyrrole-2-carboxylic acid, TBTU, N-methylmorpholine, CH2Cl2, 50 °C, 16 h; (f) hydrazine monohydrate, ethanol, 80 °C, 15 h; (g) 1,1-carbonyldiimidazole, 1,4-dioxane, 101 °C, 15 h; (h) 2 M NaOH, tetrahydrofuran, rt, 15 h.

Synthesis of the type II compounds 25–28 is presented in Scheme 3. The amino group of 18 was initially di-Boc protected with di-tert-butyl dicarbonate (2.5 equivalents) in the presence of triethylamine. In the next step, nucleophilic addition of hydroxylamine to the cyano group of 19 formed compound 20. The N-hydroxycarbamimidoyl group of compound 20 was then cyclised to the 5-methyl-1,2,4-oxadiazole ring in the presence of acetic anhydride, to obtain compound 21. Tin(ii) chloride was used to reduce the nitro group of 21, and the resulting amino group of 22 was acylated with methyl malonyl chloride in the presence of potassium carbonate, to obtain compound 23. In the next step, the Boc protecting group was removed with 4 M hydrochloric acid in 1,4-dioxane, which resulted in compound 24. The carboxylic group of 4,5-dibromopyrrole-2-carboxylic acid was then activated with oxalyl chloride and attached to 24, to obtain the amide 25. The final compound 27 was prepared in a two-step reaction, initially using hydrazine monohydrate to form the hydrazide 26, and then 1,1-carbonyldiimidazole to form the 1,3,4-oxadiazole ring. To obtain compound 28, the methyl ester group of 25 was removed under alkaline conditions.
Scheme 3. Reagents and conditions: (a) Boc2O, DMAP, CH2Cl2/CH3CN, 70 °C, 24 h; (b) hydroxylamine hydrochloride, Et3N, ethanol, 65 °C, 15 h; (c) acetic anhydride, acetic acid, 85 °C, 15 h; (d) tin(ii) chloride, ethanol/ethyl acetate, 70 °C, 6 h; (e) methyl malonyl chloride, K2CO3, CH3CN, 0 °C, 12 h; (f) 4 M HCl in 1,4-dioxane, rt, 15 h; (g) 4,5-dibromo-pyrrole-2-carboxylic acid, oxalyl chloride, CH2Cl2, 15 h, then ii) 24, pyridine, CH2Cl2, rt, 6 h; (h) hydrazine monohydrate, ethanol/tetrahydrofuran, 80 °C, 15 h; (i) 1,1-carbonyldiimidazole, 1,4-dioxane/dimethylformamide, 100 °C, 15 h; (j) 2 M NaOH, THF, rt, 15 h.

2.3. Biological data
The enzyme inhibition data against DNA gyrase from E. coli for all of the final compounds are given in Table 1, either as residual activities (RA) at 10 μM, or as half-maximal inhibitory concentrations (IC50). The inhibitory data for compounds 5 and 8 were reported previously20 and are included in Table 1 for comparison.
Table 1. Inhibitory activities of the type I and type II compounds against DNA gyrase from E. coli.
| |||||
| Compound | Type | R1 | R2 | R3 | IC50 a (μM) or RA b (%) |
| 5 c | I | H | COOCH3 | — | IC50 = 88 ± 19 |
| 6 | I | H | CONHNH2 | — | RA = 100 |
| 7 | I | H |
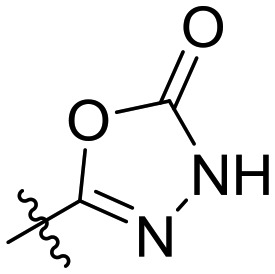
|
— | IC50 = 8.6 ± 2.4 |
| 8 c | I | H | COOH | — | IC50 = 1.4 ± 0.2 |
| 14 | I | OBzl | COOCH3 | — | RA = 100 |
| 15 | I | OBzl | CONHNH2 | — | RA = 100 |
| 16 | I | OBzl |
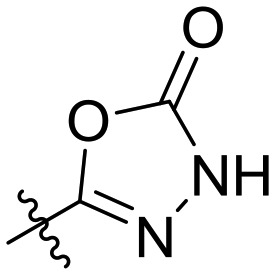
|
— | RA = 85 |
| 17 | I | OBzl | COOH | — | IC50 = 0.86 ± 0.02 |
| 25 | II | — | — | COOCH3 | IC50 = 0.086 ± 0.011 |
| 26 | II | — | — | CONHNH2 | IC50 = 0.14 ± 0.02 |
| 27 | II | — | — |
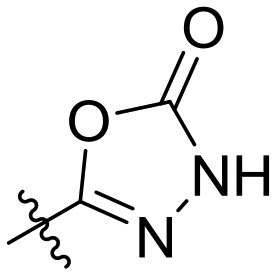
|
IC50 = 0.045 ± 0.022 |
| 28 | II | — | — | COOH | IC50 = 0.020 ± 0.009 |
| Novobiocin | IC50 = 0.17 ± 0.02 | ||||
aConcentration of compound that inhibits the enzyme activity by 50%.
bResidual activity of the enzyme at 10 μM concentration of the compound.
cPreviously published results.20
Compounds with carboxylic groups on R2 and R3 were more potent than their methyl ester precursors, probably because they can form stronger interactions with the Arg136 group. This can be seen by comparing compound 14, with a COOCH3 group at R2 (RA, 100%) with compound 17, with a carboxylic group at R2 (IC50, 0.86 μM), or by comparing the methyl ester 25 (IC50 = 0.086 μM) with its carboxylic analogue 28 (IC50, 0.020 μM). Similarly, compounds 15 (RA, 100%) and 26 (IC50, 0.14 μM) with a hydrazide group attached to R2 and R3, respectively, showed weaker activities against the E. coli DNA gyrase compared to their carboxylic analogues. This is probably because they cannot form ionic interactions with Arg136. The inhibitory activities of compounds 7 (IC50, 8.6 μM), 16 (RA, 85%) and 27 (IC50, 0.045 μM), which contained a 1,3,4-oxadiazole ring as a bioisostere for the carboxylic group, were higher than the activities of their methyl ester or hydrazide analogues, although also slightly lower than the activities of the corresponding carboxylic acids. The NH proton of the 1,3,4-oxadiazole ring has a low pKa, and it can be deprotonated under the test conditions, which will result in stronger polar interactions of these compounds with Arg136, compared to their methyl esters and hydrazides, although they will have weaker interactions compared to compounds with carboxylic groups. The most potent inhibitor from the type I series was compound 17 (IC50, 0.86 μM), which contains a carboxylic group on R2 and a benzyloxy substituent on the central phenyl ring (R1). The activity of compound 17 was approximately twice that of its analogue 8 without the benzyloxy substituent, which shows that relatively bulky lipophilic substituents can be tolerated at this position, and can form favourable interactions with the lipophilic floor of the enzyme. Interestingly, the inhibitory activity of compound 16 (RA, 85%) with a 1,3,4-oxadiazole ring on R2 and a benzyloxy substituent on the phenyl ring (R1) was lower than the activity of its analogue 7 without the benzyloxy group (IC50, 8.6 μM).
Compounds of the type II series (25–28), which contain a 5-methyl-1,2,4-oxadiazole moiety attached to the central 4-aminoaniline ring (R1 of type I series), were approximately 100 fold more potent than the type I compounds. Their IC50 against E. coli DNA gyrase were in the low nanomolar range. The most potent compound from the type II series was compound 28, with an IC50 against E. coli DNA gyrase of 0.020 μM, and this was followed by its analogue 27 (IC50, 0.045 μM), with a 1,3,4-oxadiazole ring on R3. A possible reason for the higher activity of these type II compounds compared to the type I compounds is the 5-methyl-1,2,4-oxadiazole group, which can form hydrophobic or π-stacking interactions with the amino acid residues of the lipophilic floor of the enzyme. Another possible reason is additional polar interactions between the C O group in the linker that connects the phenyl ring and the terminal acidic group, and the Glu50–Arg76 salt bridge, similar to those seen for the C O group of inhibitor A in its crystal structure with E. coli GyrB (Fig. 2). We have shown the possibility of these interactions with docking of compound 27 into the GyrB active site (Fig. 3). Fig. 3 additionally shows the possibility of interactions between the NH and O groups 1,3,4-oxadiazole ring of 27 and Arg76 or Arg136. Thus, overall, it appears that for high activity against E. coli DNA gyrase, there is the need for a combination of a substituent that is directed towards the lipophilic floor of the enzyme, and a polar carboxylic group or its bioisostere on R2 or R3, with a possible additional C O or similar group in the linker.
Fig. 3. Binding mode of compound 27 (cyan) in the E. coli GyrB (grey) (PDB code: ; 4DUH),25 as predicted with GOLD.26 The crystal water molecule is shown as a red sphere. The predicted H-bonds are given as yellow dotted lines. The fig. was prepared using PyMOL.27.

Selected compounds were also tested against DNA gyrase from Staphylococcus aureus and against topoisomerase IV from E. coli and S. aureus. The inhibitory activities against those three enzymes were lower in comparison with E. coli DNA gyrase, probably because of the comparatively smaller sizes of the adenine binding pockets of these enzymes, which do not fit the large dibromopyrrole moiety perfectly.15 The most potent compound was again compound 28, with an IC50 of 1.4 μM against S. aureus DNA gyrase, 11 μM against E. coli topoisomerase IV, and 14 μM against S. aureus topoisomerase IV (Table 2).
Table 2. Inhibitory activities of selected compounds against DNA gyrase from S. aureus and topoisomerase IV from E. coli and S. aureus.
| Compound | IC50
a
(μM) or RA
b
(%) |
||
| S. aureus DNA gyrase | E. coli topoisomerase IV | S. aureus topoisomerase IV | |
| 8 c | RA = 88 | RA = 91 | RA = 100 |
| 17 | RA = 87 | RA = 84 | RA = 92 |
| 25 | IC50 = 63 ± 14 | RA = 72 | RA = 100 |
| 26 | RA = 80 | RA = 88 | RA = 98 |
| 27 | IC50 = 11 ± 2 | RA = 91 | RA = 60 |
| 28 | IC50 = 1.4 ± 0.4 | IC50 = 11 ± 3 | IC50 = 14 ± 4 |
| Novobiocin | IC50 = 0.041 ± 0.007 | IC50 = 11 ± 2 | IC50 = 27 ± 7 |
aConcentration of compound that inhibits the enzyme activity by 50%.
bResidual activity of the enzyme at 10 μM concentration of the compound.
cPreviously published results.20
Compounds 7, 16, 17 and 25–28 that were the most potent in the enzymatic assay, were evaluated for their antibacterial activities against two Gram-positive bacterial strains (Enterococcus faecalis American Type Culture Collection [ATCC] 29212 and S. aureus ATCC 25923) and two Gram-negative bacterial strains (E. coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853). These data are given in ESI† Table S1, and they are expressed as percentage inhibition of bacteria growth at 50 μM, for each tested compound. None of the tested compounds showed any particular inhibition of the bacterial strains used. The most active compound against E. faecalis was compound 17 with 25% inhibition, and the best compound against E. coli was 26 with 16% inhibition. The most probable reason for the lack of antibacterial activity is low intracellular concentration of compounds, which can be due to (i) insufficient penetration of compounds through the bacterial cell envelope, or (ii) active efflux of inhibitors because of efflux pumps. Additionally, for the higher antibacterial activity, probably also the on-target activities should be slightly improved.
3. Experimental section
3.1. Determination of inhibitory activities on DNA gyrase and topoisomerase IV
The assays for the IC50 determinations were performed according to previously reported procedures.21
3.2. Determination of antibacterial activity
Clinical control strains of E. faecalis (Gram positive; ATCC 29212), E. coli (Gram negative; ATCC 25922), P. aeruginosa (Gram negative; ATCC 27853), and S. aureus (Gram positive; ATCC 25923) were obtained from Microbiologics Inc. (St. Cloud, Minnesota, USA). Bacterial cultures were initiated on cation-adjusted Mueller Hinton agar (Becton Dickinson, Franklin Lakes, NJ, USA) slants, and prior to the assays, the suspensions were prepared in cation-adjusted Mueller Hinton broth (Becton Dickinson, Franklin Lakes, NJ, USA) and incubated at 37 °C for 16 h to 20 h at 100 rpm. Antimicrobial assays were performed by the broth microdilution method, in a 96-well plate format, according to the Clinical and Laboratory Standards Institute guidelines28 and to previously reported procedures.21
3.3. Molecular modelling
Protein and ligand preparation and ligand docking were performed using GOLD Suite v5.4,29 according to the previously reported procedures22 and are described in ESI.†
3.4. Chemistry
Chemicals were obtained from Acros Organics (Geel, Belgium), Sigma-Aldrich (St. Louis, MO, USA) and Apollo Scientific (Stockport, UK), and they were used without further purification. Analytical TLC was performed on silica gel Merck 60 F254 plates (0.25 mm), with visualisation with UV light and spray reagents. Column chromatography was carried out on silica gel 60 (particle size 240–400 mesh). HPLC analyses were performed on a 1100 system (Agilent Technologies, Santa Clara, CA, USA) with a UV-vis detector (G1365B), a thermostat (G1316A) and an autosampler (G1313A). A C18 column was used (Phenomenex Luna 5 μm, 4.6 × 150 mm), with a flow rate of 1.0 mL min–1. The eluent consisted of trifluoroacetic acid (0.1% in water) as solvent A and acetonitrile as solvent B. Melting points were determined on a Reichert hot stage microscope, and are uncorrected. 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz, respectively, on an AVANCE III 400 spectrometer (Bruker Corporation, Billerica, MA, USA) in DMSO-d6 or CDCl3, with TMS as the internal standard. The infrared spectra were recorded on a Fourier-transform infrared spectrometer (Thermo Nicolet Nexus 470 ESP; Thermo Fisher Scientific, Waltham, MA, USA). Mass spectra were obtained using a mass spectrometer (Q-TOF Premier; Micromass, Waters, Manchester, UK). The purity of the tested compounds was ≥95%, as established by HPLC.
3.5. Synthetic procedures
Methyl 2-(4-nitrophenoxy)acetate (3)
To a suspension of 4-nitrophenol (1, 1.50 g, 11.0 mmol) and potassium carbonate (1.52 g, 11.0 mmol) in acetonitrile (50 mL) methyl bromoacetate (2, 1.05 mL, 11.0 mmol) was added dropwise. The reaction mixture was stirred overnight at rt. Solvent was evaporated under reduced pressure and the solid residue was dissolved in ethyl acetate (100 mL). The organic phase was washed with water (2 × 20 mL) and brine (20 mL), dried over Na2SO4 and evaporated under reduced pressure. Yield: 80%; white crystals (2.12 g); mp: 98–100 °C (98–99 °C);30Rf (dichloromethane/methanol = 10/1): 0.84; 1H NMR (400 MHz, DMSO-d6): δ 3.72 (s, 3H, CH3), 5.02 (s, 2H, CH2), 7.12 (d, 2H, J = 9.2 Hz, Ar–H-2, 6), 8.21 (d, 2H, J = 9.2 Hz, Ar–H-3, 5); IR (ATR) ν = 3402, 3313, 3116, 2956, 1756, 1722, 1610, 1592, 1498, 1436, 1330, 1198, 1173, 1110, 1000, 856, 752 cm–1.
Methyl 2-(4-aminophenoxy)acetate (4)
Compound 3 (2.07 g) was dissolved in tetrahydrofuran (50 mL) and Pd/C (0.414 g) was added. The reaction was stirred under H2 atmosphere for 5 h, the catalyst was filtered off and the solvent was removed under reduced pressure, to obtain 4 (1.73 g) as a yellow oil. Yield: 96%; yellow oil (1.73 g); mp: 215–217 °C; Rf (dichloromethane/methanol = 10/1): 0.29; 1H NMR (400 MHz, DMSO-d6): δ 3.68 (s, 3H, CH3), 4.60 (s, 2H, CH2), 4.68 (s, 2H, NH2), 6.49–6.51 (m, 2H, Ar–H-2, 6), 6.65 (d, 2H, J = 9.2 Hz, Ar–H-3, 5); IR (ATR) ν = 3456, 3434, 3359, 3046, 2953, 2922, 2856, 1748, 1509, 1440, 1214, 826 cm–1.
General procedure A. Synthesis of compounds 5 and 14 (compound 5 is given as an example)
A solution of 4,5-dibromo-pyrrole-2-carboxylic acid (245 mg, 0.911 mmol), TBTU (319 mg, 0.993 mmol) and N-methylmorpholine (273 μL, 2.48 mmol) in dichloromethane (20 mL) was stirred at rt for 30 min. Compound 3 (144 mg, 0.69 mmol) was added and the mixture was stirred at 50 °C overnight. The solvent was removed under reduced pressure, the residue was dissolved in ethyl acetate (20 mL) and the organic phase was washed with water (2 × 10 mL), brine (2 × 10 mL), dried over Na2SO4 and the solvent was removed under reduced pressure. To the crude product ether (10 mL) was added, the obtained suspension was sonicated and the undissolved solid was filtered off, washed with ether and dried.
Methyl 2-(4-(4,5-dibromo-1H-pyrrole-2-carboxamido)phenoxy)acetate (5)
Compound was prepared according to general procedure A. Yield: 46%; yellow crystals (165 mg); mp: 190–192 °C; Rf (ethyl acetate/petroleum ether = 1/2): 0.42; 1H NMR (400 MHz, DMSO-d6): δ 3.71 (s, 3H, CH3), 4.78 (s, 2H, CH2), 6.92–6.94 (m, 2H, Ar–H-2, 6), 7.19 (s, 1H, pyrrole-CH), 7.60 (d, 2H, J = 9.2 Hz, Ar–H-3, 5), 9.77 (s, 1H, NH), 12.88 (s, 1H, pyrrole-NH); 13C NMR (100 MHz, DMSO-d6): δ 51.77 (CH3), 64.71 (CH2), 98.03, 105.55, 113.44, 114.55, 121.49, 127.97, 132.38, 153.67, 157.07, 169.29; IR (ATR) ν = 3388, 3300, 3204, 2955, 2852, 1757, 1730, 1650, 1553, 1525, 1509, 1417, 1389, 1223, 1177, 1078, 973, 820, 750 cm–1; MS (ESI) m/z (%) = 428.9 ([M – H]–). HRMS for C14H11N2O4Br2: calculated 428.9086; found 428.9088; HPLC: tR = 19.554 min (98.6% at 280 nm).
General procedure B. Synthesis of compounds 6, 15 and 26 (compound 6 is given as an example)
Compound 5 (670 mg, 1.55 mmol) and hydrazine monohydrate (0.754 mL, 15.5 mmol) were dissolved in ethanol (10 mL) and the mixture was stirred at 80 °C overnight. The reaction mixture was cooled on an ice bath and the white precipitate was filtered off, washed with ethanol and dried to obtain 6 as white crystals (0.565 mg).
4,5-Dibromo-N-(4-(2-hydrazineyl-2-oxoethoxy)phenyl)-1H-pyrrole-2-carboxamide (6)
Compound was prepared according to general procedure B. Yield: 84%; white crystals (0.565 g); mp: 257–259 °C; Rf (dichloromethane/methanol = 20/1): 0.12; 1H NMR (400 MHz, DMSO-d6): δ 4.37 (br s, 2H, NH2), 4.47 (s, 2H, CH2), 6.95 (d, 2H, J = 9.2 Hz, Ar–H), 7.19 (s, 1H, pyrrole-CH), 7.59 (d, 2H, J = 9.2 Hz, Ar–H), 9.34 (s, 1H, NH), 9.77 (s, 1H, NH), 12.87 (s, 1H, pyrrole-NH); 13C NMR (100 MHz, DMSO-d6) δ 66.46 (CH2), 98.02, 105.54, 113.43, 114.70, 121.46, 12.99, 132.28, 153.93, 157.07, 166.67; IR (ATR) ν = 3436, 3303, 3119, 3072, 2950, 2861, 2687, 1649, 1605, 1512, 1419, 1391, 1224, 1075, 831 cm–1; MS (ESI) m/z (%) = 429.0 ([M – H]–). HRMS for C13H11N4O3Br2: calculated 428.9198; found 428.9201; HPLC: tR = 5.843 min (96.4% at 280 nm).
General procedure C. Synthesis of compounds 7, 16 and 27 (compound 7 is given as an example)
Compound 7 (150 mg, 0.35 mmol) and 1,1-carbonyldiimidazole (85 mg, 0.52 mmol) were suspended in 1,4-dioxane (15 mL), and the reaction mixture was heated at 101 °C overnight. The solvent was evaporated under reduced pressure and the solid residue was purified using column chromatography (dichloromethane/methanol = 20/1), to obtain 7 as white solid (71 mg).
4,5-Dibromo-N-(4-((5-oxo-4,5-dihydro-1,3,4-oxadiazol-2-yl)methoxy)phenyl)-1H-pyrrole-2-carboxamide (7)
Compound was prepared according to general procedure C. Yield: 45%; white solid (71 mg); mp: 260–262 °C; Rf (dichloromethane/methanol = 20/1): 0.17; 1H NMR (400 MHz, DMSO-d6): δ 5.04 (s, 2H, CH2), 7.04 (d, 2H, J = 8.8 Hz, Ar–H), 7.20 (s, 1H, pyrrole-CH), 7.63 (d, 2H, J = 8.8 Hz, Ar–H), 9.80 (s, 1H, NH), 12.53 (s, 1H, NH), 12.89 (s, 1H, pyrrole-NH); 13C NMR (100 MHz, DMSO-d6) δ 60.69 (CH2), 98.05, 105.63, 113.50, 115.12, 121.44, 127.94, 132.94, 152.92, 153.28, 154.60, 157.10; IR (ATR) ν = 3344, 3205, 1775, 1745, 1641, 1605, 1553, 1524, 1508, 1414, 1385, 1334, 1299, 1222, 1171, 1093, 939, 820, 742, 656; MS (ESI) m/z (%) = 455.0 ([M – H]–). HRMS for C14H9N4O4Br2: calculated 454.8991; found 454.8980; HPLC: tR: 9.517 min (98.1% at 280 nm).
General procedure D. Synthesis of compounds 8, 17, 28 (compound 28 is given as an example)
To a solution of compound 25 (51 mg, 0.096 mmol) in tetrahydrofuran (6 mL) 2 M NaOH (145 μL, 0.29 mmol) and water (2 mL) were added. The reaction mixture was stirred overnight at room temperature and neutralized with 1 M HCl. Solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate (10 mL). Organic phase was washed with 1 M HCl (2 × 7 mL), saturated solution of NaHCO3 (2 × 30 mL), brine (7 mL), dried over Na2SO4 and the solvent was removed under reduced pressure, to obtain 28 as grey solid (32 mg).
2-(4-(4,5-Dibromo-1H-pyrrole-2-carboxamido)phenoxy)acetic acid (8)
Compound was prepared according to general procedure D. Reported previously.20
tert-Butyl (2,4-dihydroxyphenyl)carbamate (10)
To a solution of 2,4-dihydroxybenzenaminium chloride (9, 2.75 g, 17.1 mmol) and triethylamine (2.61 mL, 18.7 mmol) in glacial acetic acid (100 mL) cooled on an ice bath, a solution of di-tert-butyl dicarbonate (5.57 g, 25.5 mmol) in methanol (5 mL) was added dropwise. The reaction mixture was stirred overnight at rt, the solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate (100 mL). Organic phase was washed with water (2 × 30 mL), 10% citric acid (2 × 30 mL) and brine (2 × 30 mL), dried over Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified with flash column chromatography (dichloromethane/methanol = 20/1) to obtain 10 (2.51 g) as a dark oil. Yield, 68%; dark oil (2.51 g); Rf (dichloromethane/methanol = 20/1): 0.16; 1H NMR (400 MHz, DMSO-d6): δ 1.43 (s, 9H, 3 × CH3), 6.14–6.17 (m, 1H, Ar–H-5), 6.30 (d, 1H, J = 2.8 Hz, Ar–H-3), 7.14 (d, 1H, J = 8.4 Hz, Ar–H-6), 7.66 (s, 1H, NH), 9.04 (s, 1H, OH), 9.40 (s, 1H, OH); 13C NMR (100 MHz, DMSO-d6): δ 28.11 (3 × CH3), 78.46, 102.71, 105.57, 117.58, 123.88, 149.74, 153.59, 154.45; IR (ATR) ν = 3307, 2979, 2934, 1680, 1609, 1523, 1452, 1368, 1284, 1246, 1152, 1112, 1054, 971, 841, 802, 732 cm–1; MS (ESI) m/z (%) = 224.1 ([M – H]–). HRMS for C11H14NO4: calculated 224.0923; found 224.0922.
tert-Butyl (2-(benzyloxy)-4-hydroxyphenyl)carbamate (11)
To a suspension of compound 10 (2.50 g, 11.9 mmol) and potassium carbonate (3.29 g, 23.7 mmol) in acetonitrile (100 mL) benzyl bromide (1.41 mL, 11.9 mmol) was added dropwise at 0 °C, and the reaction mixture was stirred overnight at rt. Solvent was removed under reduced pressure and the solid residue was dissolved in ethyl acetate (100 mL). The organic phase was washed with water (3 × 20 mL), saturated solution of NaHCO3 (2 × 20 mL) and brine (20 mL), dried with Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified with column chromatography (dichloromethane/ethyl acetate = 20/1) to obtain 11 (1.30 g) as a white solid. Yield: 36%; white solid (1.30 g); mp: 135–136 °C; Rf (dichloromethane/ethyl acetate = 20/1): 0.20; 1H NMR (400 MHz, DMSO-d6): δ 1.42 (s, 9H, 3 × CH3), 5.06 (s, 2H, CH2), 6.28–6.31 (m, 1H, Ar–H-5), 6.30 (d, 1H, J = 2.8 Hz, Ar–H-3), 7.16 (d, 1H, J = 8.8 Hz, Ar–H-6), 7.33–7.40 (m, 3H, 3 × Ar–H), 7.47–7.49 (m, 2H, 2 × Ar–H), 7.83 (s, 1H, NH), 9.29 (s, 1H, OH); 13C NMR (100 MHz, DMSO-d6): δ 28.12 (3 × CH3), 69.30 (CH2), 78.29, 100.68, 106.43, 118.69, 125.44, 127.37, 127.69, 128.28, 137.10, 151.99, 153.68, 155.14; IR (ATR) ν = 3358, 3289, 3032, 2979, 1680, 1597, 1524, 1507, 1457, 1363, 1281, 1242, 1177, 1153, 1107, 1013, 974, 832, 702, 628 cm–1; MS (ESI) m/z (%) = 314.1 ([M – H]–). HRMS for C18H20NO4: calculated 314.1392; found 314.1401.
Methyl 2-(3-(benzyloxy)-4-((tert-butoxycarbonyl)amino)phenoxy)acetate (12)
To a suspension of compound 11 (500 mg, 1.63 mmol) and potassium carbonate (450 mg, 3.25 mmol) in acetonitrile (20 mL) methyl bromoacetate (155 μL, 1.63 mmol) was added dropwise at 0 °C, and the reaction mixture was stirred overnight at rt. Solvent was removed under reduced pressure and the solid residue was dissolved in ethyl acetate (20 mL). The organic phase was washed with water (3 × 10 mL), saturated solution of NaHCO3 (2 × 10 mL) and brine (10 mL), dried with Na2SO4, and the solvent was removed under reduced pressure to obtain 12 (0.610 g) as a yellow solid. Yield: 99%; yellow solid (610 mg); mp: 101–103 °C; Rf (dichloromethane/methanol = 20/1): 0.79; 1H NMR (400 MHz, DMSO-d6): δ 1.43 (s, 9H, 3 × CH3), 3.69 (s, 3H, CH3), 4.75 (s, 2H, CH2), 5.13 (s, 2H, CH2), 6.30–6.46 (m, 1H, Ar–H-5), 6.68 (d, 1H, J = 2.4 Hz, Ar–H-3), 7.31–7.41 (m, 4H, 4 × Ar–H–), 7.50–7.51 (m, 2H, 2 × Ar–H), 7.96 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6): δ 28.09 (3 × CH3), 51.75 (CH3), 64.60 (CH2), 69.59 (CH2), 78.60, 100.82, 104.93, 121.10, 124.67, 127.50, 127.78, 128.32, 136.91, 151.54, 153.46, 155.06, 169.18; IR (ATR) ν = 3431, 3061, 3033, 2981, 2957, 1753, 1718, 1617, 1521, 1485, 1429, 1364, 1243, 1165, 1127, 1009, 826, 773, 701 cm–1; MS (ESI) m/z (%) = 388.2 ([M – H]+). HRMS for C21H26NO6: calculated 388.1760; found 388.1767.
2-(Benzyloxy)-4-(2-methoxy-2-oxoethoxy)benzenaminium chloride (13)
Compound 12 (0.550 g, 1.45 mmol) was dissolved in diethyl ether (20 mL), HCl(g) was bubbled through the solution for 40 min and the reaction mixture was stirred overnight at rt. Precipitated crystals were filtered off, washed with cold diethyl ether (2 × 10 mL) and dried. Yield: 42%; white crystals (185 mg); mp: 199–200 °C; Rf (ethyl acetate/petroleum ether = 1/1): 0.55; 1H NMR (400 MHz, DMSO-d6): δ 3.69 (s, 3H, CH3), 4.84 (s, 2H, CH2), 5.25 (s, 2H, CH2), 6.58–6.60 (m, 1H, Ar–H-5), 6.87 (d, 1H, J = 2.4 Hz, Ar–H-3), 7.40–7.44 (m, 4H, 4 × Ar–H), 7.57–7.59 (m, 2H, 2 × Ar–H), 7.91 (s, 3H, NH3+); 13C NMR (100 MHz, DMSO-d6): δ 51.84 (CH3), 64.77 (CH2), 69.94 (CH2), 101.38, 105.70, 114.11, 124.49, 127.67, 127.99, 128.38, 136.16, 152.16, 158.08, 168.88; IR (ATR) ν = 2846, 2623, 2588, 1744, 1633, 1506, 1448, 1304, 1234, 1200, 1165, 1067, 997, 834, 751 cm–1; MS (ESI) m/z (%) = 288.1 ([M – H]+). HRMS for C16H18NO4: calculated 288.1236; found 288.1241.
tert-Butyl-(1-(tert-butyloxy)carbonyl)(2-cyano-4-nitrophenyl)carbamate (19)
To a solution of 2-amino-5-nitrobenzonitrile (18, 3.00 g, 18.4 mmol) in a mixture of dichloromethane (30 mL) and acetonitrile (30 mL) di-tert-butyl dicarbonate (10.0 g, 45.8 mmol) and DMAP (50.0 g, 0.41 mol) were added and the mixture was heated at 70 °C for 24 h. The solvent was evaporated and the solid residue was dissolved in ethyl acetate (50 mL). Organic phase was washed with water (2 × 20 mL), saturated solution of NaHCO3 (2 × 20 mL), 0.5 M HCl (2 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by crystallization from diethyl ether (30 mL). Yield: 40%; pale yellow crystals (5.2 g); mp: 87–88 °C; Rf (dichloromethane/petroleum ether = 8/2): 0.19; 1H NMR (400 MHz, DMSO-d6) δ 1.39 (s, 18H, 2 × C(CH3)3), 7.93–7.95 (d, 1H, J = 8.8 Hz, Ar–H-6), 8.57–8.60 (dd, 1H, J1 = 8.8 Hz, J2 = 2.4 Hz, Ar–H-5), 8.90 (d, J = 2.44 Hz, 1H, Ar–H-3); 13C NMR (100 MHz, DMSO-d6): δ 27.30 (C(CH3)3), 84.01 (C(CH3)3), 112.93, 114.34, 128.48, 129.01, 131.29, 146.58, 146.71, 149.08; IR (ATR) ν = 3080, 2978, 2936, 2237, 1798, 1529, 1368, 1347, 1276, 1254, 1150, 1095, 845, 832, 786, 776 cm–1; MS (ESI) m/z (%) = 362.1 ([M – H]–).
tert-Butyl (E)-(2-(N′-hydroxycarbamimidoyl)-4-nitrophenyl)carbamate (20)
To a solution of compound 19 (4.00 g, 11.0 mmol) in absolute ethanol (50 mL), hydroxylamine hydrochloride (2.29 g, 33.0 mmol) and triethylamine (6.10 mL, 44.0 mmol) were added. The reaction mixture was heated overnight at 65 °C. The solvent was evaporated and the solid residue was dissolved in ethyl acetate (50 mL). Organic phase was washed with water (2 × 20 mL), saturated solution of NaHCO3 (2 × 20 mL), 0.5 M HCl (2 × 20 mL) and brine (2 × 20 mL), dried with Na2SO4 and the solvent was removed under reduced pressure. Yield: 53%; pale yellow solid (3.33 g); mp: 155–156 °C; Rf (dichloromethane/methanol = 20/1): 0.60; 1H NMR (400 MHz, DMSO-d6): δ 1.50 (s, 9H, C(CH3)3), 6.44 (s, 2H, NH2), 8.23–8.26 (m, 1H, Ar–H), 8.45–8.51 (m, 2H, 2 × Ar–H), 10.36 (s, 1H, OH/NH), 11.16 (s, 1H, OH/NH); 13C NMR (100 MHz, DMSO-d6): δ 27.82 (C(C[combining low line]H3)3), 80.96 (C[combining low line](CH3)3), 118.00, 118.79, 123.36, 124.88, 140.93, 143.18, 151.02, 151.80; IR (ATR) ν = 3367, 2982, 1735, 1597, 1497, 1368, 1339, 1309, 1233, 1131, 900, 827, 771, 747, 706 cm–1; MS (ESI) m/z (%) = 297.1 ([M – H]+).
tert-Butyl (2-(5-methyl-1,2,4-oxadiazol-3-yl)-4-nitrophenyl)carbamate (21)
Compound 20 (2.86 g, 9.65 mmol) was dissolved in glacial acetic acid (30 mL), acetic anhydride was added (1.37 mL, 14.5 mmol) and the mixture was heated overnight at 85 °C. The solvent was evaporated and the solid residue was dissolved in ethyl acetate (40 mL). Organic phase was washed with water (2 × 20 mL), saturated solution of NaHCO3 (2 × 20 mL), 0.5 M HCl (2 × 20 mL) and brine (2 × 20 mL), dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude product was purified with flash column chromatography (ethyl acetate/petroleum ether = 1/8 to 1/4). Yield: 37%; yellow solid (1.6 g); mp: 139–142 °C; Rf (ethyl acetate/petroleum ether = 1/4): 0.50; 1H NMR (400 MHz, DMSO-d6): δ 1.52 (s, 9H, C(CH3)3), 2.76 (s, 3H, CH3), 8.42 (dd, 1H, J1 = 9.2 Hz, J2 = 2.8 Hz, Ar–H), 8.50 (d, 1H, J = 9.2 Hz, Ar–H), 8.81 (d, 1H, J = 2.8 Hz, Ar–H-3), 9.83 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6): δ 11.98 (CH3), 27.73 (C(CH3)3), 81.69 (C(CH3)3), 113.75, 119.23, 124.80, 127.26, 141.45, 143.30, 151.51, 165.68, 177.35; IR (ATR) ν = 3293, 3076, 2977, 1725, 1579, 1547, 1512, 1341, 1284, 1237, 1131, 1048, 1025, 871, 852, 765, 683, 640 cm–1; MS (ESI) m/z (%) = 221.1 ([M – H]+).
tert-Butyl (4-amino-2-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)carbamate (22)
To a solution of compound 21 (1.32 g, 4.11 mmol) in a mixture of ethanol (25 mL) and ethyl acetate (25 mL) tin(ii) chloride (3.89 g, 20.5 mmol) was added and the reaction mixture was heated at 70 °C for 6 h. Saturated solution of sodium hydrogen phosphate (20 mL) was added and the mixture was heated at 70 °C for an additional hour. Solvent was evaporated and the solid residue was dissolved in ethyl acetate (30 mL). Organic phase was washed with water (2 × 20 mL) and brine (20 mL), dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude product was purified with flash column chromatography (ethyl acetate/petroleum ether = 1/2). Yield: 82%; white solid (1.08 g); mp: 154–155 °C; Rf (ethyl acetate/petroleum ether = 1/1): 0.42; 1H NMR (400 MHz, DMSO-d6): δ 1.43 (s, 9H, C(CH3)3), 2.76 (s, 3H, CH3), 5.20 (s, 2H, NH2), 6.72 (dd, 1H, J1 = 8.8 Hz, J2 = 2.8 Hz, Ar–H-5), 7.24 (d, 1H, J = 2.8 Hz, Ar–H-3), 7.57–7.59 (m, 1H, Ar–H-6), 8.76 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6): δ 11.30 (CH3), 28.05 (C(CH3)3), 78.93 (C(CH3)3), 113.60 (2 × C, overlapping signals), 117.25, 123.45, 126.33, 144.96, 152.88, 167.26, 176.18; IR (ATR) ν = 3432, 3357, 2978, 2932, 2360, 2340, 1713, 1528, 1525, 1513, 1296, 1281, 1246, 1231, 1156, 1056, 879, 764, 662 cm–1; MS (ESI) m/z (%) = 291.1 ([M – H]+), HRMS for C14H19N4O3: calculated 291.1457, found 291.1452.
Methyl 3-((4-((tert-butoxycarbonyl)amino)-3-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)amino)-3-oxopropanoate (23)
To a suspension of compound 22 (0.844 g, 2.91 mmol) and potassium carbonate (804 mg, 5.81 mmol) in acetonitrile (20 mL) methyl malonyl chloride (343 μL, 3.19 mmol) in acetonitrile (5 mL) was added dropwise and the reaction mixture was stirred at rt overnight. The solvent was evaporated and the solid residue was dissolved in ethyl acetate (20 mL). Organic phase was washed with water (2 × 15 mL), saturated solution of NaHCO3 (2 × 15 mL) and brine (2 × 15 mL), dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude product was purified with flash column chromatography (ethyl acetate/petroleum ether = 1/2). Yield: 43%; white solid (550 mg); mp: 143 °C; Rf (ethyl acetate/petroleum ether = 1/1): 0.29; 1H NMR (400 MHz, DMSO-d6): δ 1.47 (s, 9H, C(CH3)3), 2.71 (s, 3H, CH3), 3.49 (s, 2H, CH2), 3.67 (s, 3H, OCH3), 7.67 (dd, 1H, J1 = 9.2 Hz, J2 = 2.4 Hz, Ar–H-5), 8.06 (d, 1H, J = 8.8 Hz, Ar–H-6), 8.38 (d, 1H, J = 2.0 Hz, Ar–H-3), 9.26 (s, 1H, NH), 10.37 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6): δ 11.94 (CH3), 27.92 (C(CH3)3), 43.36 (CH2), 51.98 (OCH3), 78.96 (C(CH3)3), 115.02, 119.66, 120.95, 122.61, 133.21, 134.01, 152.29, 163.98, 166.80, 168.04, 176.66; IR (ATR) ν = 3275, 3109, 2976, 2360, 2340, 1746, 1729, 1662, 1561, 1512, 1391, 1362, 1295, 1267, 1230, 1155, 1048, 1018, 766, 751 cm–1; MS (ESI) m/z (%) = 389.1 ([M – H]–), HRMS for C18H21N4O6: calculated 389.1461, found 389.1454.
Methyl 3-((4-amino-3-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)amino)-3-oxopropanoate (24)
To a solution of compound 23 (50 mg, 0.13 mmol) in 1,4-dioxane (4 mL) 4 M HCl (320 μL, 1.28 mmol) in 1,4-dioxane was added and the mixture was stirred overnight at rt. The solvent was evaporated under reduced pressure, to the solid residue ether (15 mL) was added, the obtained suspension was sonicated and the undissolved solid was filtered off, washed with ether and dried. Yield: 84%; grey solid (42 mg); mp: 122–123 °C; Rf (dichloromethane/methanol/ammonia = 20/1/0.01): 0.61; 1H NMR (400 MHz, DMSO-d6): δ 2.69 (s, 3H, CH3), 3.48 (s, 2H, CH2), 3.57 (s, 3H, OCH3), 5.21 (s, 2H, NH2), 7.08 (d, 1H, J = 8.0 Hz, Ar–H-5), 7.53 (dd 1H, J1 = 8.8 Hz, J2 = 2.4 Hz, Ar–H-6), 8.28 (d, 1H, J = 2.4 Hz, Ar–H-2), 10.34 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6): δ 11.85 (CH3), 43.24 (CH2), 51.93 (OCH3), 110.66, 118.59, 120.04, 123.79, 130.70, 139.14, 163.60, 167.05, 168.20, 176.04; IR (ATR) ν = 3531, 3469, 3226, 3051, 2847, 2580, 1727, 1717, 1582, 1553, 1508, 1362, 1346, 1264, 1131, 833, 717 cm–1; MS (ESI) m/z (%) = 291.1 ([M – H]+), HRMS for C13H15N4O4: calculated 291.1093, found 291.1092.
Methyl 3-((4-(4,5-dibromo-1H-pyrrole-2-carboxamido)-3-(5-methyl-1,2,4-oxadiazol-3-yl)phenyl)amino)-3-oxopropanoate (25)
To a solution of 4,5-dibromopyrrol-2-carboxylic acid (1.30 mg, 1.01 mmol) in dichloromethane (7 mL) 2 M oxalyl chloride in dichloromethane (1.66 mL, 1.66 mmol) was added and the mixture was stirred overnight under an argon atmosphere. The solvent was evaporated, fresh dichloromethane (5 mL), pyridine (2.5 mL) and compound 23 (161 mg, 0.55 mmol) were added and the mixture was stirred at rt for 6 h. The solvent was evaporated and the solid residue was dissolved in ethyl acetate (15 mL). Organic phase was washed with water (2 × 10 mL), saturated solution of NaHCO3 (2 × 10 mL) and brine (10 mL), dried over Na2SO4 and the solvent was removed under reduced pressure. Yield: 38%; white solid (176 mg); mp: 225–227 °C; Rf (ethyl acetate/petroleum ether = 2/1): 0.21; 1H NMR (400 MHz, DMSO-d6): δ 2.72 (s, 3H, CH3), 3.51 (s, 2H, CH2), 3.68 (s, 3H, OCH3), 7.03 (s, 1H, pirol-CH), 7.72 (dd 1H, J1 = 9.0 Hz, J2 = 2.6 Hz, Ar–H-6), 8.16–8.19 (d, 1H, J = 8.8 Hz, Ar–H-5), 8.43 (d, 1H, J = 2.4 Hz, Ar–H-2), 10.28 (s, 1H, NH), 10.46 (s, 1H, NH), 13.07 (s, 1H, pyrrole-NH); 13C NMR (100 MHz, DMSO-d6): δ 11.96 (CH3), 43.41 (CH2), 52.01 (OCH3), 98.44, 106.52, 113.29, 117.17, 119.60, 122.33, 123.60, 127.95, 132.25, 135.23, 157.02, 164.13, 166.73, 168.03, 176.83; IR (ATR) ν = 3231, 3116, 2955, 2360, 1725, 1671, 1608, 1584, 1524, 1390, 1350, 1324, 1303, 1252, 1217, 1168, 808, 751, 636 cm–1; MS (ESI) m/z (%) = 537.9 ([M – H]–), HRMS for C18H14N5O5Br2: calculated 537.9362, found 537.9370; HPLC: tR: 12.046 min (95.4% at 280 nm).
4. Conclusions
In the present study, we prepared two new series of N-phenyl-4,5-dibromopyrrolamides as potential inhibitors of GyrB. The most potent compound, 28, had an IC50 of 20 nM against DNA gyrase from E. coli. Additionally, compound 27, an analogue of 28 that contains a 1,3,4-oxadiazole ring as the carboxylic group bioisostere had an IC50 value of 40 nM. The structure–activity relationship data obtained suggest that for potent DNA gyrase inhibition, acidic or polar groups on R2 or R3 are crucial. Furthermore, this activity can be largely improved with the introduction of lipophilic groups to R1. The tested compounds did not show significant antibacterial activities, which is most likely due to their too low intracellular concentrations, either because of the problems with cell entry or because of the activity of efflux pumps. Since a positive correlation between the antibacterial activity of GyrB inhibitors and their pKa has been observed previously,11 the penetration of compounds can possibly be improved by increasing the pKa of functionalities that interact with Arg76 or Arg136, e.g. by introducing less acidic groups to positions R2 or R3. Overall, in the design of new GyrB inhibitors, in addition to improving their on-target activities, the focus will have to be on optimising the size, shape and electronic properties of compounds to obtain their higher intracellular concentration and to avoid efflux.
Abbreviations
- ATCC
American Type Culture Collection
- GyrA
DNA gyrase A
- GyrB
DNA gyrase B
- RA
Residual activity
- TBTU
O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the Slovenian Research Agency (Grant No. P1-0208), and Academy of Finland (Grant No. 277001, 304697 and 312503). We thank Anja Sirc for the help with the synthesis, Michaela Barančoková for the help with biochemical evaluation, and Heidi Mäkkylä for her technical assistance in the antibacterial assays. We thank Dr. Christopher Berrie for proofreading the manuscript.
Footnotes
†Electronic supplementary information (ESI) available: Antibacterial data; full experimental procedures; NMR spectra of the tested compounds. See DOI: 10.1039/c9md00224c
References
- Klahn P. and Brönstrup M., in How to Overcome the Antibiotic Crisis : Facts, Challenges, Technologies and Future Perspectives, ed. M. Stadler and P. Dersch, Springer International Publishing, Cham, 2016, pp. 365–417. [Google Scholar]
- Brown E. D., Wright G. D. Nature. 2016;529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- Tagliabue A., Rappuoli R. Front. Immunol. 2018;9:1068. doi: 10.3389/fimmu.2018.01068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisacchi G. S., Manchester J. I. ACS Infect. Dis. 2015;1:4–41. doi: 10.1021/id500013t. [DOI] [PubMed] [Google Scholar]
- Collin F., Karkare S., Maxwell A. Appl. Microbiol. Biotechnol. 2011;92:479–497. doi: 10.1007/s00253-011-3557-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiravan M. K., Khilare M. M., Nikoomanesh K., Chothe A. S., Jain K. S. J. Enzyme Inhib. Med. Chem. 2013;28:419–435. doi: 10.3109/14756366.2012.658785. [DOI] [PubMed] [Google Scholar]
- Tomašić T., Mašič L. P. Curr. Top. Med. Chem. 2014;14:130–151. doi: 10.2174/1568026613666131113153251. [DOI] [PubMed] [Google Scholar]
- Aldred K. J., Kerns R. J., Osheroff N. Biochemistry. 2014;53:1565–1574. doi: 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcik M., Tomašič T., Zidar N., Zega A., Kikelj D., Mašič L. P., Ilaš J. Expert Opin. Ther. Pat. 2019;29:171–180. doi: 10.1080/13543776.2019.1575362. [DOI] [PubMed] [Google Scholar]
- Barančoková M., Kikelj D., Ilaš J. Future Med. Chem. 2018;10:1207–1227. doi: 10.4155/fmc-2017-0257. [DOI] [PubMed] [Google Scholar]
- Basarab G. S., Manchester J. I., Bist S., Boriack-Sjodin P. A., Dangel B., Illingworth R., Sherer B. A., Sriram S., Uria-Nickelsen M., Eakin A. E. J. Med. Chem. 2013;56:8712–8735. doi: 10.1021/jm401208b. [DOI] [PubMed] [Google Scholar]
- Tari L. W., Li X., Trzoss M., Bensen D. C., Chen Z., Lam T., Zhang J., Lee S. J., Hough G., Phillipson D., Akers-Rodriguez S., Cunningham M. L., Kwan B. P., Nelson K. J., Castellano A., Locke J. B., Brown-Driver V., Murphy T. M., Ong V. S., Pillar C. M., Shinabarger D. L., Nix J., Lightstone F. C., Wong S. E., Nguyen T. B., Shaw K. J., Finn J. PLoS One. 2013;8:e84409. doi: 10.1371/journal.pone.0084409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillot A.-L., Tiran A. L., Shannon D., Krueger E., Liao Y., O'Dowd H., Tang Q., Ronkin S., Wang T., Waal N., Li P., Lauffer D., Sizensky E., Tanoury J., Perola E., Grossman T. H., Doyle T., Hanzelka B., Jones S., Dixit V., Ewing N., Liao S., Boucher B., Jacobs M., Bennani Y., Charifson P. S. J. Med. Chem. 2014;57:8792–8816. doi: 10.1021/jm500563g. [DOI] [PubMed] [Google Scholar]
- Gjorgjieva M., Tomašič T., Barančokova M., Katsamakas S., Ilaš J., Tammela P., Peterlin Mašič L., Kikelj D. J. Med. Chem. 2016;59:8941–8954. doi: 10.1021/acs.jmedchem.6b00864. [DOI] [PubMed] [Google Scholar]
- Tomašič T., Katsamakas S., Hodnik ž., Ilaš J., Brvar M., Solmajer T., Montalvão S., Tammela P., Banjanac M., Ergović G., Anderluh M., Mašič L.L. P.P., Kikelj D. J. Med. Chem. 2015;58:5501–5521. doi: 10.1021/acs.jmedchem.5b00489. [DOI] [PubMed] [Google Scholar]
- Tomašič T., Barančoková M., Zidar N., Ilaš J., Tammela P., Kikelj D. Arch. Pharm. 2018;351:1700333. doi: 10.1002/ardp.201700333. [DOI] [PubMed] [Google Scholar]
- Tomašič T., Mirt M., Barančoková M., Ilaš J., Zidar N., Tammela P., Kikelj D. Bioorg. Med. Chem. 2017;25:338–349. doi: 10.1016/j.bmc.2016.10.038. [DOI] [PubMed] [Google Scholar]
- Cross J. B., Zhang J., Yang Q., Mesleh M. F., Romero J. A. C., Wang B., Bevan D., Poutsiaka K. M., Epie F., Moy T., Daniel A., Shotwell J., Chamberlain B., Carter N., Andersen O., Barker J., Ryan M. D., Metcalf C. A., Silverman J., Nguyen K., Lippa B., Dolle R. E. ACS Med. Chem. Lett. 2016;7:374–378. doi: 10.1021/acsmedchemlett.5b00368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basarab G. S., Hill P. J., Garner C. E., Hull K., Green O., Sherer B. A., Dangel P. B., Manchester J. I., Bist S., Hauck S., Zhou F., Uria-Nickelsen M., Illingworth R., Alm R., Rooney M., Eakin A. E. J. Med. Chem. 2014;57:6060–6082. doi: 10.1021/jm500462x. [DOI] [PubMed] [Google Scholar]
- Zidar N., Macut H., Tomašič T., Brvar M., Montalvão S., Tammela P., Solmajer T., Peterlin Mašič L., Ilaš J., Kikelj D. J. Med. Chem. 2015;58:6179–6194. doi: 10.1021/acs.jmedchem.5b00775. [DOI] [PubMed] [Google Scholar]
- Zidar N., Tomašič T., Macut H., Sirc A., Brvar M., Montalvão S., Tammela P., Ilaš J., Kikelj D. Eur. J. Med. Chem. 2016;117:197–211. doi: 10.1016/j.ejmech.2016.03.079. [DOI] [PubMed] [Google Scholar]
- Durcik M., Tammela P., Barančoková M., Tomašič T., Ilaš J., Kikelj D., Zidar N. ChemMedChem. 2018;13:186–198. doi: 10.1002/cmdc.201700549. [DOI] [PubMed] [Google Scholar]
- Durcik M., Lovison D., Skok ž., Durante Cruz C., Tammela P., Tomašič T., Benedetto Tiz D., Draskovits G., Nyerges Á., Pál C., Ilaš J., Peterlin Mašič L., Kikelj D., Zidar N. Eur. J. Med. Chem. 2018;154:117–132. doi: 10.1016/j.ejmech.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiz D. B., Skok ž., Durcik M., Tomašič T., Mašič L. P., Ilaš J., Zega A., Draskovits G., Révész T., Nyerges Á., Pál C., Cruz C. D., Tammela P., žigon D., Kikelj D., Zidar N. Eur. J. Med. Chem. 2019;167:269–290. doi: 10.1016/j.ejmech.2019.02.004. [DOI] [PubMed] [Google Scholar]
- Brvar M., Perdih A., Renko M., Anderluh G., Turk D., Solmajer T. J. Med. Chem. 2012;55:6413–6426. doi: 10.1021/jm300395d. [DOI] [PubMed] [Google Scholar]
- GOLD Suite v5.4 is available from The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ (UK), http://www.ccdc.cam.ac.uk.
- PyMOL, Delano Scientific LLC, San Francisco, CA, http://pymol.sourceforge.net.
- CLSI, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that GrowMethods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow AerobicallyAerobically, Approved Standard - Ninth Edition. CLSI document M07-A9, Clinical Standards Institute, Wayne, PA, 2012. [Google Scholar]
- Jones G., Willett P., Glen R. C., Leach A. R., Taylor R. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Oguchi M., Wada K., Honma H., Tanaka A., Kaneko T., Sakakibara S., Ohsumi J., Serizawa N., Fujiwara T., Horikoshi H., Fujita T. J. Med. Chem. 2000;43:3052–3066. doi: 10.1021/jm990522t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.