

Abstract
Background
Muscle wasting and weakness in Duchenne muscular dystrophy (DMD) causes severe locomotor limitations and early death due in part to respiratory muscle failure. Given that current clinical practice focuses on treating secondary complications in this genetic disease, there is a clear need to identify additional contributions in the aetiology of this myopathy for knowledge‐guided therapy development. Here, we address the unresolved question of whether the complex impairments observed in DMD are linked to elevated mitochondrial H2O2 emission in conjunction with impaired oxidative phosphorylation. This study performed a systematic evaluation of the nature and degree of mitochondrial‐derived H2O2 emission and mitochondrial oxidative dysfunction in a mouse model of DMD by designing in vitro bioenergetic assessments that attempt to mimic in vivo conditions known to be critical for the regulation of mitochondrial bioenergetics.
Methods
Mitochondrial bioenergetics were compared with functional and histopathological indices of myopathy early in DMD (4 weeks) in D2.B10‐DMDmdx/2J mice (D2.mdx)—a model that demonstrates severe muscle weakness. Adenosine diphosphate's (ADP's) central effect of attenuating H2O2 emission while stimulating respiration was compared under two models of mitochondrial‐cytoplasmic phosphate exchange (creatine independent and dependent) in muscles that stained positive for membrane damage (diaphragm, quadriceps, and white gastrocnemius).
Results
Pathway‐specific analyses revealed that Complex I‐supported maximal H2O2 emission was elevated concurrent with a reduced ability of ADP to attenuate emission during respiration in all three muscles (mH2O2: +17 to +197% in D2.mdx vs. wild type). This was associated with an impaired ability of ADP to stimulate respiration at sub‐maximal and maximal kinetics (−17 to −72% in D2.mdx vs. wild type), as well as a loss of creatine‐dependent mitochondrial phosphate shuttling in diaphragm and quadriceps. These changes largely occurred independent of mitochondrial density or abundance of respiratory chain complexes, except for quadriceps. This muscle was also the only one exhibiting decreased calcium retention capacity, which indicates increased sensitivity to calcium‐induced permeability transition pore opening. Increased H2O2 emission was accompanied by a compensatory increase in total glutathione, while oxidative stress markers were unchanged. Mitochondrial bioenergetic dysfunctions were associated with induction of mitochondrial‐linked caspase 9, necrosis, and markers of atrophy in some muscles as well as reduced hindlimb torque and reduced respiratory muscle function.
Conclusions
These results provide evidence that Complex I dysfunction and loss of central respiratory control by ADP and creatine cause elevated oxidant generation during impaired oxidative phosphorylation. These dysfunctions may contribute to early stage disease pathophysiology and support the growing notion that mitochondria are a potential therapeutic target in this disease.
Keywords: Bioenergetics, Duchenne muscular dystrophy, Mitochondria, Reactive oxygen species, Respiration, Oxidative stress
Introduction
Duchenne muscular dystrophy (DMD) is a debilitating disease that causes progressive muscle atrophy and weakness. Immobility related to skeletal muscle weakness manifests in childhood while eventual respiratory muscle failure causes ~40% of deaths in early adulthood.1, 2 Inherited mutations in the gene dystrophin cause a loss in the normal function of this sarcolemmal‐cytoskeletal protein.3 Subsequent disruptions to cell membrane stability and cytoskeletal organization are linked to increased cytosolic calcium, oxidative stress, and cell death that are cellular hallmarks of this disease.2, 4 Moreover, a highly pro‐inflammatory environment worsens the myopathy.5 In this light, it has been proposed that mitochondrial dysfunctions arise from these multiple cellular stressors and may even reciprocate as a direct influence on muscle fibre degeneration.6, 7, 8
Given that there is no cure for DMD, identifying secondary contributions has been a major focus for guiding therapy development.9 Indeed, current clinical practice targets inflammation rather than the genetic mutation itself.9 To this end, compounds that target mitochondria have attenuated the myopathy in mouse models of this disease. For example, the quinone‐based idebenone—an electron shuttler10—improved cardiac function and exercise performance in mice11 as well as cardiac and respiratory function in DMD patients.12, 13, 14 These drugs were developed based in part on pre‐clinical evidence of impaired mitochondrial respiratory function in skeletal muscle of mouse models of DMD8, 15, 16 and of humans.15, 17 As such, identifying additional mitochondrial abnormalities could guide further therapy development.
As mitochondria naturally emit H2O2 during oxidative phosphorylation, impairments to efficient electron transport through the electron transport system could contribute to excessive H2O2 emission. To our knowledge, only one study to date has compared mitochondrial‐specific H2O2 emission in dystrophin‐deficient muscle to wild type (WT).8 This well‐designed investigation demonstrated the expected impairment in oxidative phosphorylation and calcium retention capacity (an index of permeability transition linked to apoptosis), but also reported a surprising decrease in mitochondrial H2O2 emission in C57bl/10‐mdx mice. However, a concomitant increase in oxidant buffering capacity was noted and could represent a compensatory response in this mouse model, suggesting that mitochondrial H2O2 may indeed be elevated in vivo, particularly in relation to impaired oxidative phosphorylation.8 Given these observations, it is uncertain if mitochondrial H2O2 is elevated in DMD.
The design of in vitro assessments of both mitochondrial H2O2 emission and respiration can be essential for capturing the degree of bioenergetic dysfunction in a disease. It is well established that adenosine diphosphate (ADP) lowers mitochondrial H2O2 emission while stimulating oxidative phosphorylation by virtue of reduced premature electron slip from the electron transport system.18 As such, it is possible that mitochondrial dysfunction could manifest as a loss of central control by ADP to govern both H2O2 emission and respiration during oxidative phosphorylation. Furthermore, it has also been proposed that creatine is essential for optimizing ADP stimulation of oxidative phosphorylation and attenuation of H2O2 emission. This is believed to occur through a process of ‘high energy phosphate shuttling’ facilitated by mitochondrial creatine kinase (mtCK) (Figure 2)19, 20, 21, 23 whereby the rapidly diffusing phosphocreatine export ultimately accelerates mitochondrial matrix ADP cycling, reduces H2O2 emission, and stimulates oxidative phosphorylation.22, 24 Overall, given the complexity of how bioenergetics are governed by ADP and creatine, it follows that a re‐design of in vitro protocols may reveal the extent to which dystrophin deficiency impairs the governance of mitochondrial H2O2 emission during oxidative phosphorylation.
Figure 2.
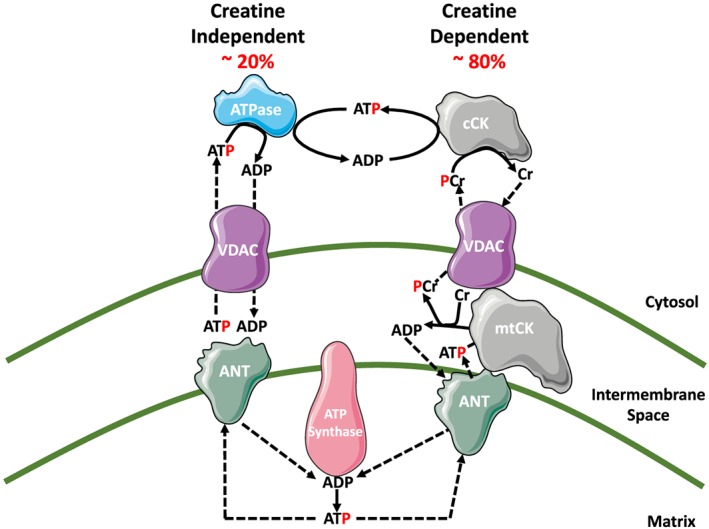
Schematic representation of energy transfer between mitochondria and cytosol. The leading model for energy exchange involves a creatine‐independent (−Cr) and creatine‐dependent (+Cr) pathway. The left side depicts the −Cr pathway whereby ADP/ATP transfer occurs solely through diffusion across voltage‐dependent anion carrier (VDAC) on the outer mitochondrial membrane and adenosine nucleotide translocase (ANT) on the inner mitochondrial membrane. The right side depicts the +Cr pathway where, in the presence of Cr, energy transfer is facilitated by mitochondrial creatine kinase (mtCK) through the transfer of the phosphate group from ATP to Cr, producing phosphocreatine (PCr) and ADP in the inner membrane space. The PCr is exported via VDAC into the cytosol while the ADP is directly recycled back via ANT into the mitochondrial matrix. Figure adapted from Aliev et al., 2011, Guzun et al., 2012, Myer et al., 1984, and Wallimann et al., 2011.19, 20, 21, 22
In this investigation, we determined the precise mechanism and degree of mitochondrial dysfunction in dystrophic locomotor muscles as well as respiratory muscle. The ability of ADP to attenuate H2O2 emission during oxidative phosphorylation was determined in the D2.B10‐DMDmdx/2J (D2.mdx) mouse. This model demonstrates severe muscle atrophy and weakness that more closely mimics human disease progression compared with the classic C57bl/10‐mdx mice due to attenuated muscle regeneration arising from a natural mutation in TGF‐β4 binding protein 4 in the background D2A strain.25, 26 By employing these methodological approaches, we provide new evidence that a central impairment in ADP and creatine's control of bioenergetics is linked specifically to increased Complex I‐supported H2O2 concurrent with impaired respiration. This observation is ubiquitous across locomotor and respiratory muscles and may be related to muscle weakness, atrophy, and cellular pathology in very early stages of myopathy in D2.mdx.
Materials and methods
Animal care
The 4‐week‐old D2.mdx animals originated from a colony maintained at York University (Toronto, Canada) and sourced from Jackson Laboratories (Bar Harbor, USA). Post‐weaning (28 days) animals were housed with male littermates until they underwent functional testing, at which time they were singly housed. Four‐week‐old DBA/2J WT mice were obtained directly from Jackson Laboratories because of breeding challenges with this strain. Upon arrival, DBA/2J mice were singly housed and given a minimum of 72 h to acclimatize before experiments were performed. All animals were provided access to standard chow and water ad libitum. All experiments and procedures were approved by the Animal Care Committee at York University (AUP Approval Number 2016‐18) in accordance with the Canadian Council on Animal Care.
Body composition and functional assays
Timeline of measurements
A variety of tests were performed on mice before surgical removal of muscle for in vitro analyses. Specifically, 2 days prior to surgery, whole body functional testing (cage hang time, forelimb grip strength, and voluntary wheel running) was performed. On the day of surgery, prior to muscle harvest, animals underwent a micro‐computed tomography scan for muscle volume (see below). Because of tissue limitation, a separate set of animals was used for histochemistry and transmission electron microscopy analysis. Finally, in order to avoid confounding influences of force/pressure measurements on mitochondrial bioenergetic analysis, a separate set of animals was used to assess oesophageal pressure transduction and in vivo force production.
In vivo micro‐computed tomography scans
In vivo body scans were performed using micro‐computed tomography (SkyScan 1278, Bruker‐microCT, Kontich, Belgium). Mice were anaesthetized and maintained with isoflurane anaesthetic during the scans. Mice were laid supine with hindlimbs extended and secured in place. Body scans were completed with a 100 μm resolution using a 0.5 mm aluminium filter at 50 kV and 996 μA, with a 30 ms exposure time and a 0.75° rotation step. Following scans, lower limb muscle volume was analysed by first reconstructing the image using NRecon (software version 1.7.0.4, Bruker‐microCT). Reconstructed images were then imported into CTAn (software version 1.15.4.0, Bruker‐microCT) where the region of interest was selected and analysed. The region of interest for hindlimb muscle volume was landmarked from the top of the patella to the ankle joint and was quantified by setting thresholds for fat‐free mass (76–102), expressed as volume (mm3) of lean mass/g body weight.
Voluntary wheel running
Animals were placed in individual cages equipped with a locked 14 cm diameter running wheel and rotation counter (VDO m3 bike computer, Mountain Equipment Co‐Op, Vancouver, Canada). After 24 h of acclimatization, wheels were unlocked, and distance run over 24 h was recorded.
Cage hang time
Animals were placed on top of a metal cage lid ~30 cm above soft bedding and positioned so that all four limbs grasped the cage. The cage lid was inverted so that the mouse was hanging, and a timer was started. Cage hang time was recorded for a maximum of 180 s. Animals that fell off prior to 180 s were given 3 min to recover before a second trial and third trial. The maximum hanging time for each animal was recorded.27
Forelimb grip strength
A metal grid was attached to a force transducer (Mark 10 Digital Force Gauge, Copiague, NY) set to peak tension mode. The force transducer was placed on the edge of a tabletop allowing the grid to extend past the table. Mice were removed from their cages by the tail and brought towards the grid until such time that the mice grasped the grid with their forepaws. Upon grasping, animals were pulled away from the grid until their grasp was broken. Peak tension was recorded, and the trial was repeated twice more. If the animal did not show resistance to the experimenter, the trial was not recorded. Maximum peak tension from the best of three trials was used for analysis.27
Oesophageal pressure transduction
Oesophageal pressure measurements were performed as previously described.28 Briefly, mice were anesthetized with an isoflurane/oxygen mix (2%:100%), and body temperature was maintained at 37°C. Mice were intubated using a 20‐gauge angiocatheter (Becton, Dickinson and Company) to maintain an open/unobstructed airway. Compressible tubing was attached to the angiocatheter to facilitate inducible airway occlusions. Oesophageal pressure (P eses) was measured with a 1.2F catheter (Transonic) recorded on LabScribe2 software (iWorx) and analysed with Spike2 software (Cambridge Electronic Design). Immediately after 2 min of baseline P es recording, the trachea was occluded, and P es was monitored for changes. Tracheal occlusions were initiated at end expiratory volume to ensure consistency of the diaphragm length–tension relationship. Maximum inspiratory pressure (PIocc) was evaluated after 30 s of occlusion when pressure generation reached a plateau.
In vivo hindlimb force production
In vivo force production of the hindlimb plantarflexor muscles was partially adapted from previous methods.29 In brief, mice were anesthetized with isoflurane and placed on a platform maintained at 37°C. The left foot was secured to a footplate attached to an Aurora Scientific 305C muscle lever (Aurora, Ontario, Canada) so that the ankle's axis of rotation coincided with that of the servomotor shaft. The knee was clamped in place such that the knee and ankle angles were both 90°. Contraction of the plantarflexors was controlled through percutaneous stimulation of the sciatic nerve. Peak isometric torque was determined by varying the voltage delivered to the sciatic nerve at a frequency of 60 Hz and 0.2 ms square wave pulse. Torque as a function of stimulation frequency was measured during seven isometric contractions at varying stimulation frequencies (40, 60, 80, 100, 120, 140, and 160 Hz). Maximum torque produced during the torque–frequency protocol was recorded and compared between groups.
Serum creatine kinase
Blood was collected through cardiac puncture and allowed to clot at room temperature for 30 min. Following clotting, blood was spun at 1000 g for 10 min, and serum was collected. Creatine kinase activity was measured spectrofluorometrically (QuantaMaster 80, HORIBA Scientific, Edison, NJ, USA) based on the autofluorescence of NADPH (excitation: 340 nm; emission: 450 nm) using the Pointe Scientific Serum Creatine Kinase kit (Pointe Scientific, Canton, MI, USA). Creatine kinase activity (U/L) was calculated from the rate of NADPH production (F/min) applied to a standard curve produced under the same conditions and normalized to volume dilution.
Selection of muscles for analyses
A subset of mice (n = 2) were injected i.p. with 1% Evans Blue Dye (Sigma, St Louis, MO, USA) (w/v) in phosphate‐buffered saline (pH 7.5) sterilized by passage through a Millex®‐GP 0.22 μm filter (Millipore, Bedford, MA, USA). Sixteen hour post injection, animals were sacrificed and muscles were harvested to allow for the visual inspection of dye uptake. We selected skeletal muscles that stained positive for Evans Blue Dye to ensure mitochondrial bioenergetics were assessed in muscle displaying a severe phenotype (Supporting Information, Figure S1). The vastus intermedius of the quadriceps (Quad), composed of IIA, IIX, and IIB fibres,30 and white gastrocnemius (WG), composed of predominantly of IIB fibres,30 were selected in addition to diaphragm, which also showed substantial membrane damage.
Mitochondrial bioenergetic assessments
Preparation of permeabilized muscle fibre bundles
This technique is partially adapted from previous methods and has been described elsewhere.31, 32, 33, 34 Briefly, muscles were removed from mice during anaesthesia with isoflurane. We performed all tissue harvests in approximately half an hour to minimize the chance of negative effects of isoflurane on mitochondrial function as has been reported with longer 6 h durations.35 The Quad and WG from the non‐stimulated leg as well as a strip of diaphragm were removed and immediately placed in ice‐cold BIOPS, containing (in mM) 50 MES Hydrate, 7.23 K2EGTA, 2.77 CaK2EGTA, 20 imidazole, 0.5 dithiothreitol, 20 taurine, 5.77 ATP, 15 PCr, and 6.56 MgCl2·6 H2O (pH 7.1). Each muscle was trimmed of connective tissue and fat and divided into several small muscle bundles (~2–7 mm, 1.0–2.5 mg wet weight). Each bundle was gently separated along the longitudinal axis to form bundles that were treated with 40 μg/mL saponin in BIOPS on a rotor for 30 min at 4°C. Bundles destined for Complex I and pyruvate dehydrogenase complex mitochondrial H2O2 emission (mH2O2) measurements (described below) were also treated with 35 μM 2,4‐dinitrochlorobenzene (CDNB) during the permeabilization step to deplete glutathione and allow for detectable rates of mH2O2.36 Following permeabilization, the permeabilized muscle fibre bundles (PmFB) were divided into three groups: (i) bundles intended for high‐resolution respirometry were placed in MiR05 containing (in mM) 0.5 EGTA, 10 KH2PO4, 3 MgCl2·6 H2O, 60 K‐lactobionate, 20 Hepes, 20 Taurine, 110 sucrose, and 1 mg/mL fatty acid free BSA (pH 7.1) while (ii) bundles intended for mH2O2 were placed in Buffer Z containing (in mM) 105 K‐MES, 30 KCl, 10 KH2PO4, 5 MgCl2·6 H2O, 1 EGTA, and 5 mg/mL BSA (pH 7.4). PmFB were washed on a rotor at 4°C in MiR05 or Buffer Z until the measurements were initiated. (iii) Bundles intended for calcium retention capacity were placed in Buffer Y + 1 mM EGTA containing (in mM) 250 sucrose, 10 tris–HCl, 20 tris‐base, 10 KH2PO4, and 0.5 mg/mL BSA and washed on a rotor at 4°C for 10 min. PmFB were then transferred to a second wash of Buffer Y + 10 μM blebbistatin (BLEB, see below) and were washed on a rotor at 4°C until measurements were initiated. Following permeabilization and washing, all mitochondrial bioenergetic measurements were performed in the same order from one animal to another and between groups to ensure consistent durations of the wash step.
Mitochondrial respiration
High‐resolution O2 consumption measurements were conducted in 2 mL of respiration medium (MiR05) using the Oroboros Oxygraph‐2k (Oroboros Instruments, Corp., Innsbruck, Austria) with stirring at 750 rpm at 37°C.33, 34, 37, 38, 39 Respiration medium contained 20 mM creatine to saturate mtCK or no creatine to prevent the activation of mtCK.37, 40 A third condition of 13.9 mM PCr and 9.1 mM Cr was used to provide an equilibrium across mtCK as occurs in skeletal muscle at rest in vivo.41 For ADP‐stimulated respiratory kinetics, standard procedures to determine complexes I and II‐supported respiration were employed.39 5 mM pyruvate, accompanied by 2 mM malate, was added as Complex I‐specific substrates (via generation of NADH to saturate electron entry into Complex I) followed by a titration of sub‐maximal ADP (25 and 500 μM) and maximal ADP (5 mM). Cytochrome c was added to test for mitochondrial membrane integrity, with all experiments demonstrating <10% increase in respiration. Finally, succinate (20 mM) was then added to saturate electron entry into Complex II. All experiments were conducted in the presence of 5 μM BLEB in the assay media to prevent spontaneous contraction of PmFB, which has been shown to occur in response to ADP at 37°C that alters respiration rates.37, 38, 42 Each protocol was initiated with a starting [O2] of ~350 μM and was completed before the oxygraph chamber [O2] reached 150 μM as done previously.33, 34, 37, 38, 39 Polarographic oxygen measurements were acquired in 2 s intervals with the rate of respiration derived from 40 data points and expressed as pmol/s/mg wet weight. PmFB were weighed in ~1.5 mL of tared BIOPS (ATP‐containing relaxing media) to ensure PmFB remained relaxed.
Mitochondrial H2O2 emission (mH2O2)
mH2O2 was determined fluorometrically (QuantaMaster 40, HORIBA Scientific) in a quartz cuvette with continuous stirring at 37°C, in 1 mL of Buffer Z supplemented with 10 μM Amplex Ultra Red, 0.5 U/mL horseradish peroxidase, 1 mM EGTA, 40 U/mL Cu/Zn‐SOD1, 5 μM BLEB, and 20 mM Cr. Site‐specific induction of mH2O2 was measured through the addition of either 10 mM pyruvate and 2 mM malate (NADH, Complex I), 10 mM succinate (FADH2, Complex I via reverse electron flux from Complex II) or 2.5 μM antimycin A (Complex III). Additionally, using 0.5 μM rotenone, a Complex I inhibitor, plus 10 mM pyruvate, electron slip specific to pyruvate dehydrogenase complex was also measured in CDNB‐treated fibres as noted earlier.36 Following the induction of State II mH2O2 by Complex I and Complex II substrates, a titration of ADP was added to progressively attenuate mH2O2. Complexe I and II‐supported mH2O2 were repeated with no creatine in the assay buffer to compare ADP's effects without mtCK‐mediated phosphate shuttling. All measurements were made in the presence of 1 μM BLEB to prevent ADP‐induced rigour as described earlier. The rate of mH2O2 emission was calculated from the slope (F/min), from a standard curve established with the same reaction conditions and normalized to fibre bundle dry weight, or citrate synthase content determined by western blot in muscle sampled from the same mouse as bioenergetics.
Mitochondrial calcium retention capacity
This assay is partially adapted from methods previously described.43 Mitochondrial calcium retention capacity measurements were completed spectrofluorometrically (QuantaMaster 80, HORIBA Scientific) in a cuvette with continuous stirring at 37°C, in 300 μL of Buffer Y containing 1 μM Calcium Green‐5N (Invitrogen), 2 μM thapsigargin, 5 mM 2‐deoxyglucose, 2 U/mL hexokinase, 20 mM creatine, 5 μM BLEB, and 40 μM EGTA. Prior to the initiation of each experiment, the cuvette was placed on a stir plate with 500 μL water and 10 mM EGTA. The water was then aspirated from the cuvette but not rinsed, leaving the EGTA coating on the cuvette walls to chelate any residual Ca2+ in the assay buffer. Minimum fluorescence was obtained following the addition of the PmFB and 5 mM glutamate +2 mM malate to the assay buffer. Calcium uptake was then initiated by a single 8 nmol pulse of CaCl2. Subsequent 4 nmol pulses of Ca2+ were added until mitochondrial permeability transition pore (PTP) opening was evident. Two 0.5 mM pulses of Ca2+ were then added to saturate the fluorophore and establish a fluorescent maximum. All experiments were conducted at 37°C. Changes in free Ca2+ in the cuvette during mitochondrial Ca2+ uptake were then calculated using the known Kd for Calcium Green‐5N and the equations established for calculating free ion concentrations using ion‐sensitive fluorophores.44 After the experiments, the fibres were rinsed in double deionized H2O, lyophilized in a freeze‐dryer (Labconco, Kansas City, MO, USA) for >4 h and weighed on a microbalance (Sartorius Cubis Microbalance, Gottingen, Germany).
In vitro muscle and blood analyses
Caspase activity
Enzymatic activities of caspase 3 and caspase 9 were measured fluorometrically using substrates Ac‐DVED‐AMC and Ac‐LEHD‐AMC (Enzo Life Sciences, Farmingdale, NY) as previously described.45, 46
Transmission electron microscopy
Fresh plantaris tissue was immediately fixed in 2% (v/v) glutaraldehyde in 0.1 mol/L sodium cacodylate buffer and processed as described previously.47 Eight representative micrographs from a unique fibre containing a portion of the subsarcolemmal region adjacent to the nucleus with most of the image containing the intermyofibrillar area were acquired at ×15 000 magnification. Quantification was achieved using the Nikon Elements software by manually outlining mitochondria and converting these to actual size using a calibration grid.48
Glutathione
Glutathione was assessed as previously described.49 GSH was assessed by UV‐HPLC monitoring of NEM‐GSH while GSSG was assessed by fluorescent‐HPLC by tracking O‐pthalymide (OPA, Sigma‐Aldrich, Oakville, Canada) tagged GSH through a flow‐through cuvette following GSSG conversion to GSH (FireflySci 8830, NY, USA) in a QuantaMaster 40 spectrofluorometer (HORIBA Scientific). All values were referenced to protein concentration and reported in μmol/g protein.
Western blotting
An aliquot of frozen Quad, WG, and diaphragm (10–30 mg) from each animal was homogenized in a plastic microcentrifuge tube with a tapered teflon pestle in ice‐cold buffer containing (mm) 40 Hepes, 120 NaCl, 1 EDTA, 10 NaHP2O7·10H2O pyrophosphate, 10 β‐glycerophosphate, 10 NaF, and 0.3% CHAPS detergent (pH 7.1 adjusted using KOH). Protein concentrations were determined using a bicinchoninic acid (BCA) assay (Life Technologies, Carlsbad, CA, USA). Fifty μg of denatured and reduced protein was subjected to 6–12% gradient SDS‐PAGE followed by transfer to low‐fluorescence polyvinylidene difluoride membrane. Membranes were blocked with LI‐COR Odyssey Blocking Buffer (LI‐COR, Lincoln NE, USA) and immunoblotted overnight (4°C) with antibodies specific for each protein. A commercially available monoclonal antibody was used to detect electron transport chain proteins (rodent OXPHOS Cocktail, ab110413; Abcam, Cambridge, UK, 1:250 dilution), including V‐ATP5A (55 kDa), III‐UQCRC2 (48 kDa), IV‐MTCO1 (40 kDa), II‐SDHB (30 kDa), and I‐NDUFB8 (20 kDa). Commercially available polyclonal antibodies were used to detect voltage‐dependent anion carrier 2 (VDAC 2) (32059, 33 kDa; Santa‐Cruz, 1:1000), adenine nucleotide translocase 1 (ANT 1) (ab180715, 32 kDa; Abcam, 1:1000), citrate synthase (ab96600; Abcam, 1:3000), MnSOD (ab13534; Abcam, 1:2000), 4‐hydroxynoneonal (4‐HNE) (ab46545; Abcam, 1:1000), and sarcomeric s‐mtCK (Dr Uwe Schlattner, Grenoble, France; 42 kDa, 1:1000). The mtCK antibody has been validated previously to confirm specificity.50 Protein carbonylation was detected using the OxyBlot Protein Oxidation Detection Kit (Millipore Sigma, Burlington, MA, USA).
After overnight incubation in primary antibodies, membranes were washed three times, for 5 min each time, in TBST and incubated for 1 h at room temperature with the corresponding infrared fluorescent secondary antibody (LI‐COR). Immunoreactive proteins were detected by infrared imaging (LI‐COR CLx; LI‐COR) and quantified by densitometry (ImageJ, http://imagej.nih.gov/ij/). All images were normalized to a whole membrane Amido Black total protein stain (A8181, Sigma). A double band was detected at 33 kDa for ANT 1 in skeletal muscle. Similar results were achieved when analysing either band. As such, the bottom band closer to 33 kDa has been reported.
Histochemistry
Immediately after removal from the animal, Quad and WG muscles were embedded in O.C.T compound (Tissue Tek), frozen in liquid nitrogen‐cooled isopentane, and stored at −80°C until analysis. Haematoxylin and eosin staining was performed on hindlimb muscle sections (8 μm thickness) to determine average cross‐sectional area (CSA) and the area of necrotic muscle fibres (i.e. fibres with fragmented sarcoplasm and/or areas of inflammatory cells). Three images spaced evenly throughout the muscle were used for analysis, and a total of 100 fibres per Quad and 100 fibres per WG were counted (n = 3 per group). Necrotic area was expressed as a percentage of the whole muscle section. To determine fibrotic area, skeletal muscle sections were stained with Masson's Trichrome (fibrotic regions stain blue for collagen) and expressed as a percentage of the whole muscle section. Quad and WG sections were imaged Nikon 90i‐eclipse microscope (Nikon Inc., Melville, NY) and analysed with the NIS Elements AR software (v4.6, Nikon).
For the diaphragm, a piece attached to the rib cage was immediately placed in 10% neutral buffered formalin for 72 h. After 72 h, the diaphragm was placed in 70% ethanol where it was preserved until processing using routine procedures. Processed diaphragm was embedded in paraffin wax (Thermo Fisher Scientific, Burlington, Canada) prior to sectioning (5 μm thickness). Diaphragm sections were stained with haematoxylin and eosin to measure CSA and necrotic area, and picrosirius red to measure fibrotic area (fibrotic regions stain red for collagen). Two to five images (four on average) were used for diaphragm analyses, and 200–250 fibres were used to determine CSA (n = 7 per group). Diaphragm sections were imaged using an Olympus FSX100 light microscope (Olympus, Richmond Hill, Canada) and analysed in cellSens software (Olympus) for necrosis and fibrosis analysis or ImageJ (http://imagej.nih.gov/ij/) for CSA analysis.
Statistics
Results are expressed as means ± SEM. The level of significance was established as P < 0.05 for all statistics. The D'Agostino–Pearson omnibus normality test was first performed to determine whether data resembled a Gaussian distribution. Given all data passed normality, parametric tests were then performed. For all data excluding State III respiration, State III mH2O2 emission, and torque–frequency data, statistical differences were analysed using unpaired t‐tests between WT and D2.mdx groups, within a given muscle where applicable. State III respiration, mH2O2 emission, and torque–frequency curves were analysed within a given muscle (where applicable) using a two‐way ANOVA with a main effect for group being reported (GraphPad Prism Software, La Jolla, CA, USA).
Results
Early onset of muscle wasting and weakness in D2.mdx mice
Body weight was 20% lower in D2.mdx mice (P < 0.05, Figure 1A). Diaphragm fibre CSA was 30% lower in D2.mdx, which demonstrates muscle atrophy at this young age. (P < 0.05, Figure 1C). In contrast, D2.mdx Quad CSA was increased by 42% (P < 0.05, Figure 1C) while WG CSA was not different between groups (Figure 1C) despite a 15% reduction in lower limb muscle volume (P = 0.07, Figure 1B) suggesting other lower limb muscles may have been atrophied. Serum creatine kinase activity was evaluated as a marker of net muscle damage with activity elevated by 392% in D2.mdx mice (P < 0.05, Figure 1D).
Figure 1.
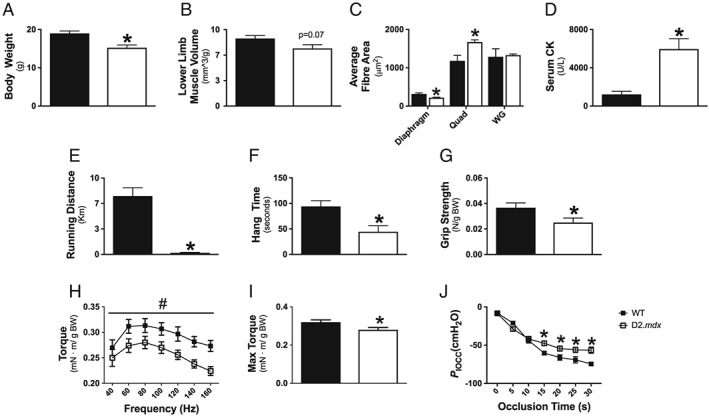
Evaluation of muscle wasting and weakness in D2.mdx mice. Body weight (A, n = 8–10), microCT analysis of lower limb muscle volume (B, n = 6–7), and muscle‐specific cross‐sectional area (CSA) using wheat germ stain for diaphragm (n = 6) and haematoxylin and eosin (H&E) for Quad (n = 3) and WG (n = 3) (C). Whole body muscle damage was quantified by serum creatine kinase (CK) activity (D, n = 5–7). Indices of muscle function were assessed by 24 h voluntary wheel running (E, n = 13–14), cage hang time (F, n = 10–11), and forelimb grip strength (G, n = 9–11). Lower hindlimb force production was assessed in vivo through torque–frequency relationship (H) followed by a max‐torque challenge (I, n = 5–6). Respiratory function in vivo was assessed through oesophageal pressure transduction to determine maximal inspiratory pressure (P Iocc) (J, n = 7). Results represent mean ± SEM; # P < 0.05 main effect compared with wild type (WT); * P < 0.05 compared with WT.
In conjunction with muscle wasting, muscle weakness was also evaluated through several approaches. Four‐week‐old D2.mdx mice demonstrated reduced 24 h voluntary wheel running (−98%, P < 0.05, Figure 1E), cage hang time (−53%, P < 0.05, Figure 1F), and forelimb grip strength (−32%, P < 0.05, Figure 1G). Given the voluntary nature of these in vivo assays and the potential for neurological contributions to performance, muscle‐specific measurements of force production were also performed. In vivo force production in the hindlimb plantarflexors revealed a main effect for decreases in D2.mdx torque production as a function of frequency (P < 0.05, Figure 1H) as well as a 13% decrease in maximum isometric torque (P < 0.05, Figure 1I) compared with WT. Diaphragm force production, measured through maximal inspiratory pressures produced during tracheal occlusion, revealed significant decreases in D2.mdx pressures from 15 to 30 s of tracheal occlusion (P < 0.05), but there were no differences at the earlier time points (0–10 s) of the occlusion protocol (Figure 1J).
Elevated Complex I‐induced mH2O2 during oxidative phosphorylation in D2.mdx mice
We first determined whether the central role of ADP in stimulating respiration and attenuating mH2O2 emission was altered. Complex I‐supported respiration and mH2O2 emission was stimulated with NADH generated by pyruvate (5 mM) and malate (2 mM) in the presence of low/physiological (25 μM), high sub‐maximal (500 μM), and saturating concentrations (5 mM) of ADP. This titration was repeated with (+Cr) and without (−Cr) 20 mM creatine to drive energy transfer dependent or independent of mtCK, respectively (as depicted in Figure 2).
In both −Cr and +Cr, ADP‐stimulated respiration was decreased at various ADP concentrations in diaphragm (−12 to −46%), Quad (−47 to −72%), and WG (−27 to −53%) in D2.mdx vs. WT (P < 0.05, Figure 3). Similar impairments in respiration were seen when mtCK flux was maintained by both PCr and Cr (Supporting Information, Figure S2). mH2O2 was elevated during oxidative phosphorylation in both −Cr and +Cr across low and high [ADP] in D2.mdx diaphragm (+17 to 88%, P < 0.05), Quad (+19 to 197%, P < 0.05), and WG (+18 to +74%, P < 0.05) vs. WT (Figure 4).
Figure 3.

Complex I‐supported respiration in D2.mdx mice. State III respiration, supported by complex I substrates pyruvate (5 mM) and malate (2 mM), was assessed in the absence (creatine independent) and presence (creatine dependent) of 20 mM creatine at physiological (25 μM), sub‐maximal (500 μM), and maximal (5000 μM) [ADP]. Assessments of bioenergetic function were completed in diaphragm (A,B), Quad (C,D), and WG (E,F) muscles. Results represent means ± SEM; n = 8–14; * P < 0.05 compared with wild type (WT).
Figure 4.
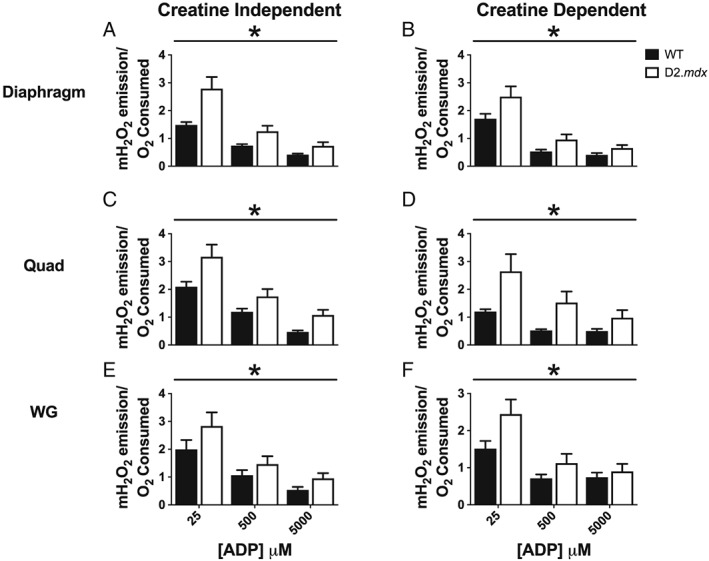
mH2O2 emission during oxidative phosphorylation in D2.mdx mice. State III mH2O2, supported by complex I substrates pyruvate (5 mM) and malate (2 mM), was assessed in the absence (creatine independent) and presence (creatine dependent) of creatine at physiological (25 μM), sub‐maximal (500 μM), and maximal (5000 μM) [ADP]. Assessments of bioenergetic function were completed in diaphragm (A,B), Quad (C,D), and WG (E,F) muscles. Results represent means ± SEM; n = 8–14; * P < 0.05 compared with wild type (WT).
We also determined the capacity for Complex I‐supported mH2O2 emission in the absence of ADP. This test promotes maximal electron slip and oxidant generation at Complex I given ADP is not present to attenuate membrane potential and promote forward electron flux. As such, the test reveals the capacity of oxidant generation by Complex I independent of the influence of ADP (State II). Respiration is driven by proton leak through uncoupling pathways independent of ATP synthase. Proton leak‐dependent respiration trended lower in D2.mdx diaphragm (−31%, P = 0.06) and was significantly lower in D2.mdx Quad (−58%, P < 0.05) and WG (−39%, P < 0.05) vs. WT (Supporting Information, Figure S3A). Concurrent pyruvate/malate‐supported mH2O2 in the absence of ADP was unchanged in diaphragm, elevated by 110% in Quad (P < 0.05), and tended to be higher in WG (+47%, P = 0.09) vs. WT (Supporting Information, Figure S3B).
We then determined the degree to which other pathways support mH2O2. Complex III (2.5 μM Antimycin A) and pyruvate dehydrogenase‐supported mH2O2 (10 mM pyruvate, 0.5 μM Rotenone to inhibit Complex I) were decreased in the Quad (−53%, P = 0.05) and WG (−60%, P < 0.05), while the diaphragm showed a trend for increased Complex III mH2O2 (+35%, P = 0.11) and no differences in pyruvate dehydrogenase‐supported mH2O2 between D2.mdx and WT (Supporting Information, Figure S4A and S4B). Normalizing mH2O2 to Complex III subunit content (Supporting Information, Figure S4C) or pyruvate dehydrogenase E1α content (Supporting Information, Figure S4D) altered rates such that no differences existed in mH2O2 emission in any muscle vs. WT.
Impaired creatine sensitivity in oxidative D2.mdx muscle
Given impaired bioenergetics were seen in both creatine‐independent and creatine‐dependent models of phosphate shuttling, we also compared whether creatine insensitivity existed in D2.mdx to gain insight on the role of mtCK in this disease. As seen in Table 1, at 500 μM ADP, creatine tended to increase respiration in both diaphragm (+43%, P = 0.06) and Quad (+57%, P = 0.08) and decreased mH2O2 (diaphragm: −37%, P < 0.05; Quad: −56%, P < 0.01) in WT consistent with its proposed role in facilitating phosphate shuttling (Figure 2). In contrast, +Cr had no effect on respiration at 500 μM in any D2.mdx muscle vs. −Cr or on mH2O2 in Quad or WG, although it lowered mH2O2 in D2.mdx diaphragm (−42%, P < 0.05; Table 1) suggesting mtCK and phosphate shuttling are impaired in D2.mdx in addition to loss of control by ADP (Figure 2).
Table 1.
Impairment in creatine sensitivity with 500 μM ADP in DMD
| Wild type | D2.mdx | |||||
|---|---|---|---|---|---|---|
| −Cr | +Cr |
P‐value (−Cr vs. +Cr) |
−Cr | +Cr |
P‐value (−Cr vs. +Cr) |
|
| JO2 (pmol·s−1·mg−1 wet wt) | ||||||
| Diaphragm | 67.2 ± 5.2 | 95.8 ± 10.2 | <0.05 | 57.6 ± 7.2 | 63.7 ± 6.4 | 0.94 |
| Quad | 21.2 ± 2.2 | 33.2 ± 5.5 | 0.07 | 12.7 ± 2.1 | 12.9 ± 2.2 | 0.53 |
| WG | 28.7 ± 3.5 | 28.1 ± 4.4 | 0.94 | 18.9 ± 3.0 | 16.7 ± 1.1 | 0.53 |
| mH2O2/O2 consumed | ||||||
| Diaphragm | 0.73 ± 0.06 | 0.53 ± 0.07 | <0.05 | 1.25 ± 0.21 | 0.72 ± 0.10 | <0.05 |
| Quad | 1.19 ± 0.11 | 0.52 ± 0.05 | <0.01 | 1.76 ± 0.26 | 1.54 ± 0.40 | 0.64 |
| WG | 1.09 ± 0.19 | 0.72 ± 0.10 | 0.16 | 1.46 ± 0.28 | 1.11 ± 0.25 | 0.37 |
Creatine dependent (+Cr), creatine independent (−Cr), quadriceps (Quad), white gastrocnemius (WG).
Values represent mean ± SEM (n = 8–14).
Increased susceptibility to permeability transition pore opening and elevations in mitochondria‐derived apoptosis in D2.mdx Quad
Previous work has shown that calcium overload leads to PTP opening more rapidly in dystrophin‐deficient diaphragm and tibialis anterior muscle from C57bl/10‐mdx mice.8, 51 We next determined whether reduced calcium retention capacity was related to the elevated mH2O2 during oxidative phosphorylation seen in the D2.mdx mice (Figure 5A,B). D2.mdx diaphragm and WG muscle showed no differences in the [Ca2+] required to induce PTP opening vs. WT (Figure 5B) whereas Quad required 62% less [Ca2+] vs. WT (P < 0.05, Figure 5B). Caspase 9 activity was also elevated in Quad (+25%, P < 0.05) suggesting the reduced calcium retention capacity is related to mitochondrial‐induced apoptosis in this muscle unlike WG (Figure 5C). Interestingly, despite no differences in calcium retention capacity, caspase 9 activity tended to be elevated in D2.mdx diaphragm (+43%, P = 0.08; Figure 5C). These elevations in caspase 9 did not result in activation of its downstream target, caspase 3, in any muscle (Figure 5D).
Figure 5.
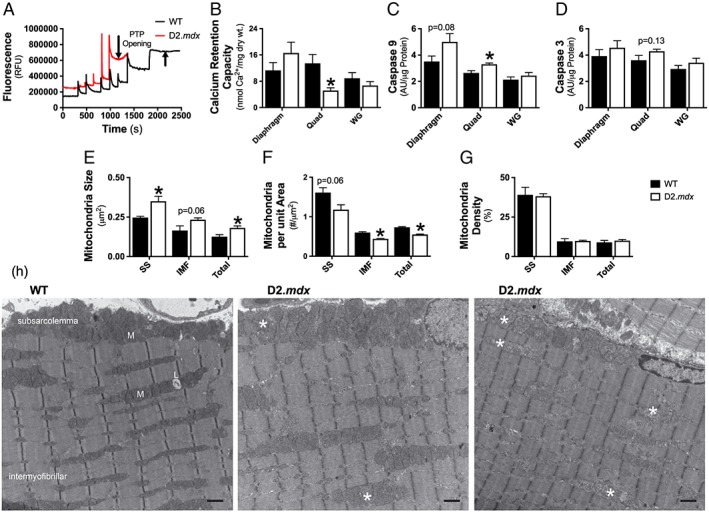
Evaluation of mitochondrial calcium retention capacity, content, and disbursement. Calcium retention capacity, an index of susceptibility to permeability transition pore opening (A), was assessed in permeabilized fibres from diaphragm, Quad, and WG muscle (B); n = 6–8. Mitochondrial induction of apoptosis was evaluated through caspase 9 activity (C) followed by its downstream target caspase 3 (D); n = 7. Transmission electron micrographs were acquired in the plantaris muscle and analysed for mitochondrial size (E, mean area, μm2), distribution (F, # per μm2), and density (G, μm2 × # per μm2 × 100) within the subsarcolemmal (SS) area and intermyofibrillar (IMF) area of the muscle. Representative images (H) demonstrate the large accumulation of swollen mitochondria (*) in D2.mdx mice relative to the normal conformation of wild type (WT) mitochondria (M); scale bar = 1 μm, n = 2–3. Results represent mean ± SEM; * P < 0.05 compared with WT.
Given the decrease in D2.mdx Quad calcium retention capacity and subsequent increase in mitochondria‐derived apoptotic signalling, further analysis of mitochondria structure and morphology was performed. Because of tissue limitations, the plantaris muscle, which displays a similar fibre type composition to the vastus intermedius component of the Quad used for calcium retention capacity,30 was used for transmission electron microscopy analysis. D2.mdx plantaris muscle was found to contain swollen mitochondria (Figure 5E), regardless of the mitochondrial location (subsarcolemmal vs. intermyofibrillar). D2.mdx plantaris muscle also contained less mitochondria per unit area (Figure 5F), but due to their larger size, no differences in mitochondria density was evident (Figure 5G).
Increased redox buffering with no change in markers of cellular oxidative stress in D2.mdx mice
Content of the reduced form of glutathione (GSH) content was increased in D2.mdx Quad (+55%, P < 0.05) and WG (+38%, P < 0.05), and tended to be higher in diaphragm (+8%, P = 0.09), indicative of a compensatory increase in redox buffering (Figure 6A) consistent with increased mH2O2 noted earlier. WG muscle demonstrated elevated levels of oxidized glutathione (GSSG) content (+50%, P < 0.05) relative to WT, while no differences were found in Quad or diaphragm (Figure 6B). However, total glutathione (GSH + 2×GSSG) was elevated or tended to be elevated in all muscles (diaphragm: +8%, P = 0.09; Quad: +54%, P < 0.05; WG: +38%, P < 0.05; Figure 6C) predominately because of increases in reduced GSH content. Importantly, the ratio of GSH:GSSG was unchanged in all muscles, suggesting the elevated mH2O2 was buffered to prevent cellular oxidation at this early time point (Figure 6D). Indeed, western blotting for lipid peroxidation (Figure 6E) and protein carbonylation (Figure 6F), indicators of whole cell oxidative stress, revealed no differences between D2.mdx and WT muscle. Mitochondrial matrix MnSOD was lower in Quad (−29%, P < 0.05) and WG (−22%, P < 0.05) with no change in diaphragm being observed (Figure 6G).
Figure 6.

Evaluation of redox buffering capacity and whole cell oxidative status. Glutathione was measured in muscle homogenate using HPLC‐UV for the detection of GSH (A) and HPLC‐fluorescence for the detection of GSSG (B). The GSH:GSSG ratio (C) and total glutathione (D) were calculated from GSH and GSSG measurements; n = 6. Whole muscle lysate was used for the quantification of proteins modified through lipid peroxidation (E) and carbonylation (F) as well as the quantification of protein content of MnSOD (G); n = 6–8. Results represent mean ± SEM; * P < 0.05 compared with wild type (WT).
Muscle‐specific alterations in key regulators of energy shuttling
Given the alterations in State III respiration and mH2O2 in D2.mdx muscle, we next quantified key regulators of energy shuttling. ANT 1 and VDAC 2, situated in the inner and outer mitochondrial membranes, respectively (Figure 2), were unchanged in diaphragm muscle but were significantly decreased in D2.mdx Quad (ANT 1: −48%, P < 0.05; VDAC 2: −33%, P < 0.05) and WG (ANT 1: −53%, P < 0.05; VDAC 2: −36%, P < 0.05) relative to WT (Figure 7A,B). Similarly, mtCK content was unchanged in diaphragm but significantly lower in D2.mdx Quad (−45%, P < 0.05) and WG (−38%, P < 0.05; Figure 7C). Citrate synthase protein content was reduced in Quad only (−23%, P < 0.05; Figure 7D). Normalizing respiration to citrate synthase content (Supporting Information, Figure S5) as validated previously52 generally did not explain the changes in respiration observed when normalized per mg weight seen in Figure 3. Quantification of the complexes comprising the electron transport chain (ETC) was also completed to provide an index of ETC content. ETC subunit contents were unchanged relative to WT in both diaphragm (Figure 7E) and WG (Figure 7G) muscle apart from a decrease in Complex II subunit content in D2.mdx diaphragm (−15%, P = 0.06; Figure 7E). However, Quad demonstrated decreases or trends for decreases in subunit content of Complex I (−38%, P < 0.05), II (−36%, P = 0.08), III (−34%, P < 0.05), IV (−55%, P < 0.05), and V (−19%, P = 0.09) as well as the sum of all five complex subunits (−37%, P < 0.05; Figure 7F).
Figure 7.

Muscle‐specific changes in essential regulators of bioenergetics in D2.mdx mice. Protein content of adenine nucleotide translocase 1 (ANT 1) on the inner mitochondrial membrane (A), voltage‐dependent anion carrier 2 (VDAC 2) on the outer mitochondrial membrane (B), mitochondrial creatine kinase (mtCK) found in the inner membrane space (C), and citrate synthase that catalyses the first reaction in the citric acid cycle (D). Protein content of electron transport chain components was also quantified in diaphragm (E), Quad (F), and WG (G). Results represent mean ± SEM; n = 8–12; * P < 0.05 compared with wild type (WT).
Bioenergetic impairments are associated with elevations in muscle damage in D2.mdx muscle
Muscle‐specific membrane damage was evaluated though Evans Blue Dye staining in a separate subset of animals. Sixteen hour post injection of 1% Evans Blue Dye, animals were sacrificed, and muscle was visually inspected for dye uptake. Dye uptake, which occurs in muscles with damaged cell membranes,53 was evident in the Quad, WG, and tricep brachii muscles with minimal uptake occurring in the soleus, plantaris, extensor digitorum longus, and the tibialis anterior. A striated pattern of dye uptake was evident in the diaphragm muscle (Supporting Information, Figure S1).
Necrotic area was elevated in diaphragm (5.5% vs. 0% of total area vs. WT, P < 0.05; Figure 8A), Quad (31.8% vs. 0.6%, P < 0.05; Figure 8B), and WG (48.2% vs. 0.2%, P = 0.06; Figure 8C). Fibrotic area was also elevated in diaphragm (12.7% vs. 8.4% of total area vs. WT, P < 0.05; Figure 8D), Quad (17.0% vs. 6.3%, P < 0.05; Figure 8E), and WG (22.0% vs. 3.5%, P < 0.05; Figure 8F).
Figure 8.
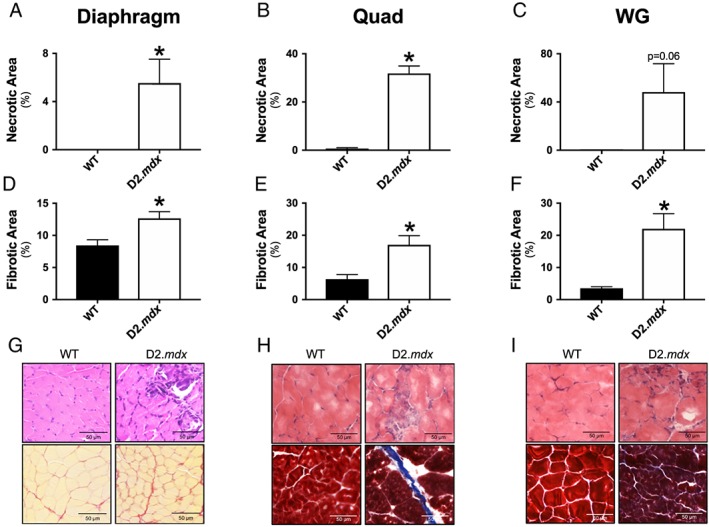
Quantification of muscle damage in D2.mdx mice. Necrosis was evaluated in diaphragm (A), Quad (B), and WG (C) using haematoxylin and eosin (H&E) staining in all muscles. Fibrosis was evaluated in diaphragm (D) using picrosirius red and in Quad (E) and WG (F) using Masson's trichrome. Results represent mean ± SEM; n = 3–6; * P < 0.05 compared with wild type (WT). Representative images of diaphragm (G, magnification, ×40), Quad (H, magnification, ×20), and WG (I, magnification, ×20).
Discussion
Oxidative stress has long been implicated as a contributor to the myopathy of DMD. However, identifying the precise source of elevated oxidants has remained controversial. In this study, we demonstrate that mH2O2 is elevated concurrent with impaired oxidative phosphorylation specific to Complex I‐supported respiration. An impairment in the ability of ADP to both stimulate respiration and attenuate mH2O2 was central to these impairments. These bioenergetic deficits occurred early in the disease process (4 weeks of age) and were associated with severe myopathy in skeletal muscles and diaphragm. By identifying a potential new relationship between elevated mH2O2, impaired oxidative phosphorylation and myopathy in diaphragm (respiratory) and limb muscle (locomotor; see summary Figure 9), these findings suggest that DMD may be a candidate for therapeutics that attenuate mH2O2 54 and improve mitochondrial respiratory control.
Figure 9.

Summary of the relationship between impaired mitochondrial bioenergetics and myopathy in Duchenne muscular dystrophy (DMD). Specific dysfunctions in Complex I and central respiratory control by ADP and creatine cause elevated oxidant generation during impaired oxidative phosphorylation in association with a loss of muscle force.
Several factors warranted a re‐evaluation of mH2O2 in DMD. First, an elegant study by Godin et al.8 demonstrated an unexpected decrease in mH2O2 in the tibialis anterior of the classic C57bl/10‐mdx mouse, but this occurred concurrently with increased redox buffering capacity suggesting compensation occurred in response to an underlying redox stress.8 Furthermore, the degree of mitochondrial dysfunction may differ across muscles in relation to the extent of myopathy that warrants comparisons of muscle types, particularly given the known regenerative capacities of the classic C57bl/10‐mdx model.55, 56 The design of in vitro protocols to measure mitochondrial respiration and mH2O2 must also be considered given (i) ADP governs both processes concurrently, (ii) disease‐related stressors may alter sensitivity to low ADP concentrations to a different extent than saturating conditions, (iii) mitochondrial‐cytosolic energy exchange is thought to occur through either direct ADP/ATP cycling or Cr/PCr shuttling via mtCK, and (iv) there are multiple sources of oxidant generation that contribute to net mH2O2. By controlling for these factors, a general pattern of impaired Complex I activity, creatine insensitivity, and ADP insensitivity in the regulation of mH2O2 and respiration was identified in both dystrophic respiratory and locomotor muscles.
Concurrent impairments in Complex I and governance by adenosine diphosphate
At 4 weeks of age, D2.mdx diaphragm, Quad, and WG muscle exhibit elevated Complex I‐supported mH2O2 during oxidative phosphorylation. This pattern appeared to be due to several factors. First, the maximal rate of mH2O2 supported by Complex I was increased as seen in the absence of ADP's influence on membrane potential (State II conditions). The impairments at Complex I were not explained by altered protein content of the NDUFB8 subunit. This suggests post‐translational modifications may have occurred. Indeed, Complex I activity can be decreased by oxidation of specific cysteine residues57 but the degree to which this occurs in DMD remains to be determined. Also, while superoxide production prior to dismutation to H2O2 was not assessed, the increases in H2O2 emission during respiration are suggestive of increased superoxide production particularly given MnSOD was lower in Quad and WG. It is also possible that the lower leak respiration in the absence of ADP reflects less uncoupling, which may contribute to elevated membrane potential‐dependent Complex I superoxide production.18
Furthermore, low/physiological to high concentrations of ADP were unable to attenuate mH2O2 to the same extent as WT. The impaired control by ADP in skeletal muscle may not be related to changes in mitochondrial content given ETC markers were unchanged in diaphragm and WG similar to what has been reported recently in C57bl/10‐mdx mouse soleus and extensor digitorum longus.58 Rather, this may be explained partially by limited ADP/ATP exchange due to decreased protein expression of VDAC 2 and ANT 1 in Quad and WG, or octameric mtCK, the regulator of high energy phosphate shuttling. There was no change in the content of these proteins in diaphragm, which suggests that post‐translational regulation of these proteins may have been altered. Indeed, all three proteins are redox sensitive.59, 60, 61 ANT and VDAC also have known phosphorylation sites62, 63 although the physiological relevance of this regulatory paradigm remains unknown. Furthermore, mtCK is highly susceptible to oxidative modifications, leading to enzymatic inactivation and octamer dissociation, as well as formation of crystalline mitochondrial inclusion bodies, all resulting in loss of function.64, 65 The present findings suggest future examination of post‐translational regulation of Complex I, isoforms of ANT 1 and VDAC 2, as well as mtCK may reveal how multiple cellular stressors in DMD lead to the observed mitochondrial dysfunctions. Lastly, while no other site of mitochondrial oxidant generation tested in this investigation demonstrated altered mH2O2 (reverse electron flux from Complex II, Complex III, and pyruvate dehydrogenase), additional sites could be considered in future investigations at this early age.66, 67
To our knowledge, this is the first study to demonstrate impairments in ADP‐stimulated respiration at both low and maximal kinetics in D2.mdx diaphragm muscle. The respiratory impairments in Quad and WG are consistent with previous reports in humans15, 17 and C57bl/10‐mdx skeletal muscle.8, 15, 16, 68 However, the role of creatine‐dependent phosphate shuttling through mtCK was not assessed, which means these earlier studies only report impairments in creatine‐independent ADP/ATP cycling similar to the present findings, albeit with one exception that also reported impaired creatine sensitivity in oxidative muscles.69 Given the creatine‐dependent model is predicted to be the dominant system of energy exchange,22 the lack of effect of creatine on respiration in D2.mdx may be particularly detrimental to muscle function, in particular, at high workloads with repeated contractions (Table 1). As noted earlier, changes in ANT 1, VDAC 2, and mtCK might contribute to this loss of control by ADP in Quad and WG, but alternative mechanisms are likely for diaphragm, further highlighting the importance of considering post‐translational modifications.
Possible relationships between mitochondrial bioenergetics and myopathy in Duchenne muscular dystrophy
Mitochondrial induction of apoptosis and necrosis has been linked to several diseases that also exhibit elevated mitochondrial ROS and impaired oxidative phosphorylation.70, 71, 72, 73, 74, 75 It is thought that impairments in both processes can contribute to the induction of PTP formation as can occur under conditions of calcium overload.76 Such PTP formation has already been implicated in DMD myopathy in response to calcium stress.8, 51 However, the synergistic link with mH2O2 had not been demonstrated. Indeed, an increase in mitochondrial [Ca2+] alone is relatively ineffective at inducing PTP formation, but mitochondrial sensitivity to [Ca2+] can be drastically increased through elevated ROS.77 Furthermore, the increases in caspase 9 seen in Quad are consistent with previous reports of greater cytochrome c release in dystrophic muscle78—a process that is known to be triggered in part by mitochondrial ROS during induction of the PTP.79 Overall, the present findings suggest that elevated mH2O2 contributes to this calcium‐overload induction of PTP in Quad but not necessarily in diaphragm or WG at this early stage of the disease (Figure 4). Surprisingly, Quad displayed hypertrophy based on fibre CSA, which may be compensation to caspase 9‐mediated cell death. Indeed, this early hypertrophy is consistent with observations in Quad of young boys with DMD that was later followed by atrophy in advanced stages of the disease.80
In contrast to the present study, previous work has shown heightened sensitivity to Ca2+‐induced PTP in diaphragm in C57bl/10‐mdx mice. Methodological differences may contribute to these divergent findings as previous studies have produced divergent results depending on whether PTP opening was assessed in terms of elapsed time in response to a single bolus of Ca2+ 51 or identifying the minimal concentration required to trigger PTP as in the present study.8, 51 These discrepant findings suggest the approach used to determine PTP sensitivity to Ca2+ can change the conclusion and that comparison to other proxies of PTP are warranted.
It is possible that calcium‐independent modulators of mitochondrial dysfunction and induction of cell death are present in DMD. While not assessed in this study, mH2O2 also activates necrosis in a PTP‐independent manner by activating nuclear factor‐kappa B (NF‐κB)81 and up‐regulating the pro‐inflammatory cytokine tumour necrosis factor‐alpha (TNF‐α).82, 83 Indeed, antioxidants that lower mH2O2 have been shown to prevent muscle wasting in a TNF‐α induced model of cachexia,84 and an anti‐TNF‐α antibody (Remicade) reduces muscle necrosis and inflammation in C57bl/10‐mdx mice.85 Further examination of alternative mechanisms by which impaired mitochondrial bioenergetics contributes to cell death in DMD may be fruitful considering the necrosis observed in the present study (diaphragm, WG, and Quad) was not consistently linked to a calcium‐induction of PTP, particularly in diaphragm that displayed atrophy without caspase 9 activation.
Perspectives on redox homeostasis in Duchenne muscular dystrophy: reconciling with previous literature
While cytoskeletal‐based NADPH oxidase activity is increased in dystrophin‐deficient mouse skeletal muscle,86 the contribution of mitochondrial‐specific ROS across all muscle types has received surprisingly little attention. To our knowledge, only one previous study has directly measured mH2O2 in skeletal muscle of dystrophin‐deficient mice. Using tibialis anterior muscle from 6‐week‐old C57bl/10‐mdx mice, Godin et al. demonstrated a surprising decrease in mH2O2.8 This decrease appeared to be due to increases in mH2O2 scavenging capacity that may be related to extensive muscle regeneration at this age in C57bl/10‐mdx mice that occurs after an earlier period of peak necrosis at 3–4 weeks.55, 56 This robust increase in scavenging capacity may reflect a compensation in response to an underlying elevation in mH2O2. As such, we re‐evaluated the relationship between mH2O2 and myopathy in a model with less regenerative capacity25, 26 and in muscles displaying membrane damage (Supporting Information, Figure S1). While a direct comparison of mitochondrial bioenergetics in muscles with and without damaged cell membranes was not performed, heterogeneous rates and degrees of myopathy between muscles should be taken into consideration when evaluating the contribution of the mitochondria to the myopathy. It is also possible that the differences between these previous observations and the present investigation may be due to the age (6 vs. 4 weeks currently) or the muscle analysed (tibialis anterior previously), but this remains to be determined.
Contrary to previous work in young C57bl/10‐mdx mice that showed a more oxidized GSH:GSSG ratio,87, 88 the present investigation revealed no changes in the glutathione ratio in all muscles analysed. Despite the differences in mH2O2 emission rates, both the present study and the work by Godin et al. demonstrate increases in redox buffering capacity. Similarly, in accordance with previous findings,8, 88 no differences in lipid peroxidation were detected through 4‐HNE or protein carbonylation levels between WT and D2.mdx muscles. These results suggest that the increases in buffering capacity are sufficient to tolerate the stress created by elevated mH2O2 and prevent detectable oxidative damage from occurring at this early time point. However, it is plausible that the buffering capacity of the cell will become overwhelmed and result in oxidative damage as the disease progresses. Furthermore, 4‐HNE and carbonylation are measures of irreversible oxidative damage, and therefore, the present investigation does not rule out H2O2 acting as a reversible redox signal within the cell. Future investigation into the time‐course of redox stress in the context of both reversible and irreversible modifications in this D2.mdx model of DMD are warranted.
Conclusions
In summary, this investigation identified elevations in Complex I‐supported mH2O2 during oxidative phosphorylation concurrent with impaired ADP and creatine control of respiration in D2.mdx diaphragm, Quad, and WG muscle. These impairments were associated with a greater propensity for calcium‐induced PTP and caspase 9 activation in Quad, but the degree to which they are related to apoptosis in other muscles requires further investigation. The potential association between these mitochondrial impairments and myopathy at an early stage of DMD may guide the pre‐clinical testing of mitochondrial‐targeted therapeutics to restore normal mH2O2 and Complex I‐supported oxidative phosphorylation, particularly by rescuing ADP and creatine control of bioenergetics. While such an approach would not target the primary cause of the disease, the addition of mitochondrial therapies warrants consideration given existing standards of care already focus on treating other secondary complications of DMD in the absence of genetic therapies.
Funding
Funding was provided to C.G.R.P. and T.J.H. by the National Science and Engineering Research Council (no. 436138‐2013 and no. 2018‐06324, respectively) and an Ontario Early Researcher Award (C.G.R.P., no. 2017‐0351) with infrastructure supported by Canada Foundation for Innovation, the James. H. Cummings Foundation, and the Ontario Research Fund. M.C.H. was supported by an NSERC CGS‐PhD scholarship. S.V.R. was supported by an Ontario Graduate Scholarship. P.C.T. was supported by an NSERC CGS‐PhD scholarship. I.A.R. was supported by an Ontario Graduate Scholarship. C.M.F.M. was supported by an NSERC PGS‐PhD and the Michael DeGroote Doctoral Scholarship of Excellence.
Conflicts of interest
None declared.
Author Contributions
C. G. R. P., M. C. H., S. V. R., J. A. S., and T. J. H. contributed to the rationale and study design. All authors conducted experiments and/or analysed all data. C. G. R. P. and M. C. H. wrote the manuscript. All authors contributed to the interpretation of the data and manuscript preparation. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Supporting information
Figure S1. Evaluation of sarcolemmal damage by Evans Blue Dye staining in D2.mdx. Representative images of Evans Blue Dye (EBD) staining in WT and D2.mdx skeletal muscles 16 hours following an injection of 1% EBD solution at 5 μl/g body weight.
Figure S2. ADP‐stimulated respiration in the presence of phosphocreatine and creatine. 13.9 mM phosphocreatine (PCr) and 9.1 mM creatine (Cr) were added to the assay media to assess state III respiration supported by Complex‐I substrates pyruvate (5 mM) and malate (2 mM) in Diaphragm (A), Quad (B) and WG (C). Results represent mean ± SEM; n = 10–12; * p < 0.05 compared to WT.
Figure S3. Evaluation of state II respiration and mH2O2. State II (no ADP; proton leak) respiration (A) and mH2O2 emission (B) were initiated using Complex‐I substrates pyruvate (5 mM) and malate (2 mM) in Diaphragm, Quad and WG muscles. Results represent mean ± SEM; n = 10–12; * p < 0.05 compared to WT.
Figure S4. Additional sites of state II mH2O2 emission. Complex‐III derived mH2O2 was assessed using Complex‐III inhibitor antimycin A (2.5 μM). Data was expressed per mg muscle weight (A) as well as normalized to Complex‐III content (B). Pyruvate dehydrogenase complex (PDC) derived mH2O2 was assessed using pyruvate (10 mM) and Complex‐I inhibitor rotenone (0.5 μM). Data was expressed per mg muscle weight (C) as well as normalized to PDH‐E1α content (D). Results represent mean ± SEM; n = 8–12; * p < 0.05 compared to WT.
Figure S5. Intrinsic Respiratory Capacity. State III respiration, supported by Complex‐I substrates pyruvate (5 mM) and malate (2 mM), was assessed in the absence (Creatine Independent) and presence (Creatine Dependent) of 20 mM creatine at physiological (25 μM), sub‐maximal (500 μM) and maximal (5000 μM) [ADP] and normalized to citrate synthase content. Assessments of bioenergetic function were completed in Diaphragm (a‐b), Quad (c‐d) and WG (e‐f) muscles. Results represent means ± SEM; n = 5–8; * p < 0.05 compared to WT.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle.89
Hughes M. C., Ramos S. V., Turnbull P. C., Rebalka I. A., Cao A., Monaco C. M. F., Varah N. E., Edgett B. A., Huber J. S., Tadi P., Delfinis L. J., Schlattner U., Simpson J. A., Hawke T. J., and Perry C. G. R. (2019) Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2O2 emission during impaired oxidative phosphorylation, Journal of Cachexia, Sarcopenia and Muscle, 10: 643–661. 10.1002/jcsm.12405.
References
- 1. Emery AE. Duchenne muscular dystrophy—Meryon's disease. Neuromuscul Disord 1993;3:263–266. [DOI] [PubMed] [Google Scholar]
- 2. Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve 2000;23:1456–1471. [DOI] [PubMed] [Google Scholar]
- 3. Emery A. Genetic heterogeneity in Duchenne muscular dystrophy. Am J Med Genet 1987;26:235–236. [DOI] [PubMed] [Google Scholar]
- 4. Allen DG, Whitehead NP, Froehner SC. Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev 2016;96:253–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bridges LR. The association of cardiac muscle necrosis and inflammation with the degenerative and persistent myopathy of MDX mice. J Neurol Sci 1986;72:147–157. [DOI] [PubMed] [Google Scholar]
- 6. Rybalka E, Timpani CA, Cooke MB, Williams AD, Hayes A. Defects in mitochondrial ATP synthesis in dystrophin‐deficient Mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014;9:e115763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Timpani CA, Hayes A, Rybalka E. Revisiting the dystrophin‐ATP connection: how half a century of research still implicates mitochondrial dysfunction in Duchenne muscular dystrophy aetiology. Med Hypotheses 2015;85:1021–1033. [DOI] [PubMed] [Google Scholar]
- 8. Godin R, Daussin F, Matecki S, Li T, Petrof BJ, Burelle Y. Peroxisome proliferator‐activated receptor γ coactivator 1‐α gene transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post‐necrotic mdx mouse skeletal muscle. J Physiol 2012;590:5487–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guiraud S, Davies KE. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr Opin Pharmacol 2017;34:36–48. [DOI] [PubMed] [Google Scholar]
- 10. Gillis JC, Benfield P, McTavish D. Idebenone. Drugs Aging 1994;5:133–152. [DOI] [PubMed] [Google Scholar]
- 11. Buyse GM, Van der Mieren G, Erb M, D'Hooge J, Herijgers P, Verbeken E, et al. Long‐term blinded placebo‐controlled study of SNT‐MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance. Eur Heart J 2009;30:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buyse GM, Voit T, Schara U, Straathof CS, D'Angelo MG, Bernert G, et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): a double‐blind randomised placebo‐controlled phase 3 trial. Lancet 2015;385:1748–1757. [DOI] [PubMed] [Google Scholar]
- 13. Buyse GM, Voit T, Schara U, Straathof CS, D'Angelo MG, Bernert G, et al. Treatment effect of idebenone on inspiratory function in patients with Duchenne muscular dystrophy. Pediatr Pulmonol 2017;52:508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buyse GM, Goemans N, van den Hauwe M, Thijs D, de Groot IJ, Schara U, et al. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: results from a 12 month, double‐blind, randomized placebo‐controlled trial. Neuromuscul Disord 2011;21:396–405. [DOI] [PubMed] [Google Scholar]
- 15. Kuznetsov AV, Winkler K, Wiedemann F, von Bossanyi P, Dietzmann K, Kunz WS. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin‐deficient mdx mouse. Mol Cell Biochem 1998;183:87–96. [DOI] [PubMed] [Google Scholar]
- 16. Schuh RA, Jackson KC, Khairallah RJ, Ward CW, Spangenburg EE. Measuring mitochondrial respiration in intact single muscle fibers. Am J Physiol Regul Integr Comp Physiol 2012;302:R712–R719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sperl W, Skladal D, Gnaiger E, Wyss M, Mayr U, Hager J, et al. High resolution respirometry of permeabilized skeletal muscle fibers in the diagnosis of neuromuscular disorders. Mol Cell Biochem 1997;174:71–78. [PubMed] [Google Scholar]
- 18. Nicholls DG, Ferguson SJ. Bioenergetics 4. Elsevier; 2013. [Google Scholar]
- 19. Aliev M, Guzun R, Karu‐Varikmaa M, Kaambre T, Wallimann T, Saks V. Molecular system bioenergics of the heart: experimental studies of metabolic compartmentation and energy fluxes versus computer modeling. Int J Mol Sci 2011;12:9296–9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guzun R, Gonzalez‐Granillo M, Karu‐Varikmaa M, Grichine A, Usson Y, Kaambre T, et al. Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and mtCK within mitochondrial interactosome. Biochimica et Biophysica Acta (BBA) ‐ Biomembranes 2012;1818:1545–1554. [DOI] [PubMed] [Google Scholar]
- 21. Wallimann T, Tokarska‐Schlattner M, Schlattner U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011;40:1271–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meyer RA, Sweeney HL, Kushmerick MJ. A simple analysis of the “phosphocreatine shuttle”. Am J Physiol Cell Physiol 1984;246:C365–C377. [DOI] [PubMed] [Google Scholar]
- 23. Schlattner U, Kay L, Tokarska‐Schlattner M. Mitochondrial proteolipid complexes of creatine kinase In Harris JR, Boekema EJ, eds. Membrane Protein Complexes: Structure and Function. Singapore: Springer Singapore; 2018. p 365–408. [DOI] [PubMed] [Google Scholar]
- 24. Meyer LE, Machado LB, Santiago APSA, da‐Silva WS, De Felice FG, Holub O, et al. Mitochondrial creatine kinase activity prevents reactive oxygen species generation: antioxidant role of mitochondrial kinase‐dependent adp re‐cycling activity. J Biol Chem 2006;281:37361–37371. [DOI] [PubMed] [Google Scholar]
- 25. S‐i F, Morikawa D, Yamamoto Y, Yoshida T, Sumie N, Yamaguchi M, et al. Genetic background affects properties of satellite cells and mdx phenotypes. Am J Pathol 2010;176:2414–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coley WD, Bogdanik L, Vila MC, Yu Q, Van Der Meulen JH, Rayavarapu S, et al. Effect of genetic background on the dystrophic phenotype in mdx mice. Hum Mol Genet 2016;25:130–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aartsma‐Rus A, van Putten M. Assessing functional performance in the Mdx mouse model. J Vis Exp JoVE 2014;85:51303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Foster AJ, Platt MJ, Huber JS, Eadie AL, Arkell AM, Romanova N, et al. Central‐acting therapeutics alleviate respiratory weakness caused by heart failure‐induced ventilatory overdrive. Sci Transl Med 2017;9. [DOI] [PubMed] [Google Scholar]
- 29. Warren GL, Stallone JL, Allen MR, Bloomfield SA. Functional recovery of the plantarflexor muscle group after hindlimb unloading in the rat. Eur J Appl Physiol 2004;93:130–138. [DOI] [PubMed] [Google Scholar]
- 30. Bloemberg D, Quadrilatero J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS ONE 2012;7:e35273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tonkonogi M, Harris B, Sahlin K. Mitochondrial oxidative function in human saponin‐skinned muscle fibres: effects of prolonged exercise. J Physiol 1998;510:279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kuznetsov AV, Tiivel T, Sikk P, Kaambre T, Kay L, Daneshrad Z, et al. Striking differences between the kinetics of regulation of respiration by ADP in slow‐twitch and fast‐twitch muscles in vivo . Eur J Biochem 1996;241:909–915. [DOI] [PubMed] [Google Scholar]
- 33. Ydfors M, Hughes MC, Laham R, Schlattner U, Norrbom J, Perry CG. Modelling in vivo creatine/phosphocreatine in vitro reveals divergent adaptations in human muscle mitochondrial respiratory control by ADP after acute and chronic exercise. J Physiol 2016;594:3127–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perry CGR, Kane DA, Lanza IR, Neufer PD. Methods for assessing mitochondrial function in diabetes. Diabetes 2013;62:1041–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y, Dong Y, Wu X, Lu Y, Xu Z, Knapp A, et al. The mitochondrial pathway of anesthetic isoflurane‐induced apoptosis. J Biol Chem 2010;285:4025–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fisher‐Wellman KH, Gilliam LA, Lin CT, Cathey BL, Lark DS, Neufer PD. Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2‐emitting source under conditions of nutrient overload. Free Radic Biol Med 2013;65:1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perry CGR, Kane DA, Lin C‐T, Kozy R, Cathey BL, Lark DS, et al. Inhibiting myosin‐ATPase reveals dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem J 2011;437(): 10.1042/BJ20110366:215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hughes MC, Ramos SV, Turnbull PC, Nejatbakhsh A, Baechler BL, Tahmasebi H, et al. Mitochondrial bioenergetics and fibre type assessments in microbiopsy vs Bergstrom percutaneous sampling of human skeletal muscle. Front Physiol 2015;6: 10.3389/fphys.2015.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pesta D, Gnaiger E. High‐resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle In Palmeira CM, Moreno AJ, eds. Mitochondrial Bioenergetics: Methods and Protocols. Totowa, NJ: Humana Press; 2012. p 25–58. [DOI] [PubMed] [Google Scholar]
- 40. Walsh B, Tonkonogi M, Söderlund K, Hultman E, Saks V, Sahlin K. The role of phosphorylcreatine and creatine in the regulation of mitochondrial respiration in human skeletal muscle. J Physiol 2001;537:971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perry CGR, Heigenhauser GJF, Bonen A, Spriet LL. High‐intensity aerobic interval training increases fat and carbohydrate metabolic capacities in human skeletal muscle. Appl Physiol Nutr Metab 2008;33:1112–1123. [DOI] [PubMed] [Google Scholar]
- 42. Perry CGR, Kane DA, Herbst EAF, Mukai K, Lark DS, Wright DC, et al. Mitochondrial creatine kinase activity and phosphate shuttling are acutely regulated by exercise in human skeletal muscle. J Physiol 2012;590:5475–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fisher‐Wellman KH, Mattox TA, Thayne K, Katunga LA, La Favor JD, Neufer PD, et al. Novel role for thioredoxin reductase‐2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J Physiol 2013;591:3471–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsien RY. Fluorescent indicators of ion concentrations. Methods Cell Biol 1989;30:127–156. [DOI] [PubMed] [Google Scholar]
- 45. Campbell TL, Mitchell AS, McMillan EM, Bloemberg D, Pavlov D, Messa I, et al. High‐fat feeding does not induce an autophagic or apoptotic phenotype in female rat skeletal muscle. Exp Biol Med (Maywood) 2015;240:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dam AD, Mitchell AS, Rush JW, Quadrilatero J. Elevated skeletal muscle apoptotic signaling following glutathione depletion. Apoptosis 2012;17:48–60. [DOI] [PubMed] [Google Scholar]
- 47. Nilsson MI, MacNeil LG, Kitaoka Y, Suri R, Young SP, Kaczor JJ, et al. Combined aerobic exercise and enzyme replacement therapy rejuvenates the mitochondrial–lysosomal axis and alleviates autophagic blockage in Pompe disease. Free Radic Biol Med 2015;87:98–112. [DOI] [PubMed] [Google Scholar]
- 48. Tarnopolsky MA, Rennie CD, Robertshaw HA, Fedak‐Tarnopolsky SN, Devries MC, Hamadeh MJ. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol Regul Integr Comp Physiol 2007;292:R1271–R1278. [DOI] [PubMed] [Google Scholar]
- 49. Mandel ER, Dunford EC, Abdifarkosh G, Turnbull PC, Perry CGR, Riddell MC, et al. The superoxide dismutase mimetic tempol does not alleviate glucocorticoid‐mediated rarefaction of rat skeletal muscle capillaries. Physiol Rep 2017;5 10.14814/phy2.13243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schlattner U, Mockli N, Speer O, Werner S, Wallimann T. Creatine kinase and creatine transporter in normal, wounded, and diseased skin. J Investig Dermatol 2002;118:416–423. [DOI] [PubMed] [Google Scholar]
- 51. Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol 2012;181:583–592. [DOI] [PubMed] [Google Scholar]
- 52. Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 2012;590:3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol 1997;139:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol 2016;14:238 10.1038/nrcardio.2016.203–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome‐linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A 1984;81:1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McGreevy JW, Hakim CH, McIntosh MA, Duan D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech 2015;8:195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hurd TR, Prime TA, Harbour ME, Lilley KS, Murphy MP. Detection of reactive oxygen species‐sensitive thiol proteins by redox difference gel electrophoresis: implications for mitochondrial redox signaling. J Biol Chem 2007;282:22040–22051. [DOI] [PubMed] [Google Scholar]
- 58. Barker RG, Wyckelsma VL, Xu H, Murphy RM. Mitochondrial content is preserved throughout disease progression in the mdx mouse model of Duchenne muscular dystrophy, regardless of taurine supplementation. Am J Physiol Cell Physiol 2017;314:C483–C491. [DOI] [PubMed] [Google Scholar]
- 59. Koufen P, Rück A, Brdiczka D, Wendt S, Wallimann T, Stark G. Free radical‐induced inactivation of creatine kinase: influence on the octameric and dimeric states of the mitochondrial enzyme (Mib‐CK). Biochem J 1999;344:413–417. [PMC free article] [PubMed] [Google Scholar]
- 60. Lemasters JJ, Holmuhamedov E. Voltage‐dependent anion channel (VDAC) as mitochondrial governator—thinking outside the box. Biochimica et Biophysica Acta (BBA) ‐ Molecular Basis of Disease 2006;1762:181–190. [DOI] [PubMed] [Google Scholar]
- 61. O‐Uchi J, Ryu S‐Y, Jhun BS, Hurst S, Sheu S‐S. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid Redox Signal 2014;21:987–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kerner J, Lee K, Tandler B, Hoppel CL. VDAC proteomics: post‐translation modifications. Biochim Biophys Acta 2012;1818:1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Feng J, Lucchinetti E, Enkavi G, Wang Y, Gehrig P, Roschitzki B, et al. Tyrosine phosphorylation by Src within the cavity of the adenine nucleotide translocase 1 regulates ADP/ATP exchange in mitochondria. Am J Physiol Cell Physiol 2010;298:C740–C748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schlattner U, Wallimann T. Octamers of mitochondrial creatine kinase isoenzymes differ in stability and membrane binding. J Biol Chem 2000;275:17314–17320. [DOI] [PubMed] [Google Scholar]
- 65. Schlattner U, Tokarska‐Schlattner M, Wallimann T. Mitochondrial creatine kinase in human health and disease. Biochimica et Biophysica Acta (BBA) ‐ Molecular Basis of Disease 2006;1762:164–180. [DOI] [PubMed] [Google Scholar]
- 66. Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol 2010;45:466–472. 10.1016/j.exger.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Menazza S, Blaauw B, Tiepolo T, Toniolo L, Braghetta P, Spolaore B, et al. Oxidative stress by monoamine oxidases is causally involved in myofiber damage in muscular dystrophy. Hum Mol Genet 2010;19:4207–4215. [DOI] [PubMed] [Google Scholar]
- 68. Passaquin A‐C, Renard M, Kay L, Challet C, Mokhtarian A, Wallimann T, et al. Creatine supplementation reduces skeletal muscle degeneration and enhances mitochondrial function in mdx mice. Neuromuscul Disord 2002;12:174–182. [DOI] [PubMed] [Google Scholar]
- 69. Braun U, Paju K, Eimre M, Seppet E, Orlova E, Kadaja L, et al. Lack of dystrophin is associated with altered integration of the mitochondria and ATPases in slow‐twitch muscle cells of MDX mice. Biochim Biophys Acta 2001;1505:258–270. [DOI] [PubMed] [Google Scholar]
- 70. Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial‐targeted antioxidants protect skeletal muscle against immobilization‐induced muscle atrophy. J Appl Physiol (1985) 2011;111:1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Powers SK, Hudson MB, Nelson WB, Talbert EE, Min K, Szeto HH, et al. Mitochondria‐targeted antioxidants protect against mechanical ventilation‐induced diaphragm weakness. Crit Care Med 2011;39:1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Szeto HH. Mitochondria‐targeted cytoprotective peptides for ischemia‐reperfusion injury. Antioxid Redox Signal 2008;10:601–619. [DOI] [PubMed] [Google Scholar]
- 73. Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia‐reperfusion injury. FASEB J 2005;19:1088–1095. [DOI] [PubMed] [Google Scholar]
- 74. Singh K, Hood DA. Effect of denervation‐induced muscle disuse on mitochondrial protein import. Am J Physiol Cell Physiol 2011;300:C138–C145. [DOI] [PubMed] [Google Scholar]
- 75. Pollock N, Staunton CA, Vasilaki A, McArdle A, Jackson MJ. Denervated muscle fibers induce mitochondrial peroxide generation in neighboring innervated fibers: Role in muscle aging. Free Radic Biol Med 2017;112:84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bernardi P, von Stockum S. The permeability transition pore as a Ca(2+) release channel: new answers to an old question. Cell Calcium 2012;52:22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res 2004;61:372–385. [DOI] [PubMed] [Google Scholar]
- 78. Giacomotto J, Brouilly N, Walter L, Mariol M‐C, Berger J, Ségalat L, et al. Chemical genetics unveils a key role of mitochondrial dynamics, cytochrome c release and IP3R activity in muscular dystrophy. Hum Mol Genet 2013;22:4562–4578. [DOI] [PubMed] [Google Scholar]
- 79. Cai J, Yang J, Jones D. Mitochondrial control of apoptosis: the role of cytochrome c. Biochimica et Biophysica Acta (BBA) ‐ Bioenergetics 1998;1366:139–149. [DOI] [PubMed] [Google Scholar]
- 80. Cros D, Harnden P, Pellissier JF, Serratrice G. Muscle hypertrophy in Duchenne muscular dystrophy. J Neurol 1989;236:43–47. [DOI] [PubMed] [Google Scholar]
- 81. Kumar A, Boriek AM. Mechanical stress activates the nuclear factor‐kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J 2003;17:386–396. [DOI] [PubMed] [Google Scholar]
- 82. Schulze‐Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem 1992;267:5317–5323. [PubMed] [Google Scholar]
- 83. Festjens N, Vanden Berghe T, Vandenabeele P. Necrosis, a well‐orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochimica et Biophysica Acta (BBA) ‐ Bioenergetics 2006;1757:1371–1387. [DOI] [PubMed] [Google Scholar]
- 84. Buck M, Chojkier M. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO J 1996;15:1753–1765. [PMC free article] [PubMed] [Google Scholar]
- 85. Grounds MD, Torrisi JO. Anti‐TNFα (Remicade®) therapy protects dystrophic skeletal muscle from necrosis. FASEB J 2004;18:676–682. [DOI] [PubMed] [Google Scholar]
- 86. Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, et al. Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci Signal 2012;5 10.1126/scisignal.2002829:ra56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dudley RW, Danialou G, Govindaraju K, Lands L, Eidelman DE, Petrof BJ. Sarcolemmal damage in dystrophin deficiency is modulated by synergistic interactions between mechanical and oxidative/nitrosative stresses. Am J Pathol 2006;168:1276–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dudley RW, Khairallah M, Mohammed S, Lands L, Des Rosiers C, Petrof BJ. Dynamic responses of the glutathione system to acute oxidative stress in dystrophic mouse (mdx) muscles. Am J Physiol Regul Integr Comp Physiol 2006;291:R704–R710. [DOI] [PubMed] [Google Scholar]
- 89. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Evaluation of sarcolemmal damage by Evans Blue Dye staining in D2.mdx. Representative images of Evans Blue Dye (EBD) staining in WT and D2.mdx skeletal muscles 16 hours following an injection of 1% EBD solution at 5 μl/g body weight.
Figure S2. ADP‐stimulated respiration in the presence of phosphocreatine and creatine. 13.9 mM phosphocreatine (PCr) and 9.1 mM creatine (Cr) were added to the assay media to assess state III respiration supported by Complex‐I substrates pyruvate (5 mM) and malate (2 mM) in Diaphragm (A), Quad (B) and WG (C). Results represent mean ± SEM; n = 10–12; * p < 0.05 compared to WT.
Figure S3. Evaluation of state II respiration and mH2O2. State II (no ADP; proton leak) respiration (A) and mH2O2 emission (B) were initiated using Complex‐I substrates pyruvate (5 mM) and malate (2 mM) in Diaphragm, Quad and WG muscles. Results represent mean ± SEM; n = 10–12; * p < 0.05 compared to WT.
Figure S4. Additional sites of state II mH2O2 emission. Complex‐III derived mH2O2 was assessed using Complex‐III inhibitor antimycin A (2.5 μM). Data was expressed per mg muscle weight (A) as well as normalized to Complex‐III content (B). Pyruvate dehydrogenase complex (PDC) derived mH2O2 was assessed using pyruvate (10 mM) and Complex‐I inhibitor rotenone (0.5 μM). Data was expressed per mg muscle weight (C) as well as normalized to PDH‐E1α content (D). Results represent mean ± SEM; n = 8–12; * p < 0.05 compared to WT.
Figure S5. Intrinsic Respiratory Capacity. State III respiration, supported by Complex‐I substrates pyruvate (5 mM) and malate (2 mM), was assessed in the absence (Creatine Independent) and presence (Creatine Dependent) of 20 mM creatine at physiological (25 μM), sub‐maximal (500 μM) and maximal (5000 μM) [ADP] and normalized to citrate synthase content. Assessments of bioenergetic function were completed in Diaphragm (a‐b), Quad (c‐d) and WG (e‐f) muscles. Results represent means ± SEM; n = 5–8; * p < 0.05 compared to WT.