

Abstract
Background
Muscle wasting occurs in response to various physiological and pathological conditions, including ageing and Duchenne muscular dystrophy (DMD). Transforming growth factor‐β1 (TGF‐β1) contributes to muscle pathogenesis in elderly people and DMD patients; inhibition of TGF‐β1 signalling is a promising therapeutic strategy for muscle‐wasting disorders. Hemojuvelin (HJV or Hjv as the murine homologue) is a membrane‐bound protein that is highly expressed in skeletal muscle, heart, and liver. In hepatic cells, Hjv acts as a coreceptor for bone morphogenetic protein, a TGF‐β subfamily member. The aim of this study was to investigate whether Hjv plays an essential role in muscle physiological and pathophysiological processes by acting as a coreceptor for TGF‐β1 signalling.
Methods
Conventional and conditional Hjv knockout mice as well as mdx and aged mice transfected with Hjv overexpression vector were used to study the role of Hjv in muscle physiology and pathophysiology. qRT‐PCR, western blotting, and immunohistochemistry examinations were conducted to evaluate gene, protein, and structural changes in vivo and in vitro. Exercise endurance was determined using treadmill running test, and muscle force was detected by an isometric transducer. RNA interference, immunoprecipitation, and dual‐luciferase reporter assays were utilized to explore the mechanism by which Hjv regulates TGF‐β1 signalling in skeletal muscle.
Results
Conventional and conditional Hjv knockout mice displayed muscle atrophy, fibrosis, reduced running endurance, and muscle force. HJV was significantly down‐regulated in the muscles of DMD patients (n = 3, mean age: 11.7 ± 5.7 years) and mdx mice as well as in those of aged humans (n = 10, 20% women, mean age: 75.1 ± 9.5 years) and mice. Overexpression of Hjv rescued dystrophic and age‐related muscle wasting. Unlike its function in hepatic cells, the bone morphogenetic protein downstream phosphorylated p‐Smad1/5/8 signalling pathway was unchanged, but TGF‐β1, TGF‐β receptor II (TβRII), and p‐Smad2/3 expression were increased in Hjv‐deficient muscles. Mechanistically, loss of Hjv promoted activation of Smad3 signalling induced by TGF‐β1, whereas Hjv overexpression inhibited TGF‐β1/Smad3 signalling by directly interacting with TβRII on the muscle membrane.
Conclusions
Our findings identify an unrecognized role of HJV in skeletal muscle by regulating TGF‐β1/Smad3 signalling as a coreceptor for TβRII. Unlike the TGF‐β1/Smad3 pathway, HJV could be a reliable drug target as its expression is not widespread. Novel therapeutic strategies could potentially be devised to interfere only with the muscle function of HJV to treat DMD and age‐related muscle wasting.
Keywords: Hemojuvelin, Duchenne muscular dystrophy, Age‐related muscle wasting, Coreceptor, TGF‐β1 signalling
Introduction
Skeletal muscle is the most abundant tissue in the human body, accounting for approximately 40% of total body weight.1 The primary functions of skeletal muscle are to generate force, maintain posture, and serve as a major site for glucose metabolism.1 Muscle wasting occurs in response to a variety of physiological and pathological conditions and is highly detrimental to the human body.2 For example, elderly people are known to undergo a progressive loss of muscle mass, which is associated with reduced muscle force, decreased physical activity, insulin resistance, and increased risk of fracture.3, 4 Progressive muscle wasting and weakness also occur in Duchenne muscular dystrophy (DMD), an inherited X‐linked recessive disorder caused by a mutation in the dystrophin gene. The lack of dystrophin results in increased sarcolemma fragility, leading to chronic inflammation, ongoing degeneration, and regeneration cycles as well as fibrosis. Most DMD patients die of respiratory failure because of loss of muscle force in the diaphragm.5, 6 Thus, maintenance of muscle mass and composition is crucial for healthy ageing and increasing life expectancy of DMD patients.
Transforming growth factor‐β (TGF‐β) superfamily members have profound effects on muscle development, physiology, and pathophysiology.7, 8, 9 Signalling transduction is initiated when TGF‐β superfamily ligands bind with their specific type II receptors. The activated type II receptor transphosphorylates type I receptor, which subsequently transduces the signal by phosphorylating the carboxyl terminus of receptor‐regulated (R)‐Smads. In most cell types, TGF‐β signals via phosphorylation of Smad2 and Smad3, whereas bone morphogenetic protein (BMP) signals via phosphorylation of Smad1, Smad5, and Smad8.8 In addition to playing a major role in regulating muscle mass, TGF‐β/Smad signalling is also involved in the pathogenesis of various muscle‐wasting disorders.9, 10 For example, TGF‐β1 is up‐regulated and promotes fibrosis formation in the muscles of DMD patients.9, 11 In aged muscles, excess TGF‐β1 is produced, which impairs the regenerative capacity of muscle satellite cells, thus accelerating degeneration and leading to sarcopenia.12 Considering the critical role of TGF‐β1 signalling in muscle pathogenesis, the identification of novel proteins that regulate TGF‐β1 signalling could provide potential therapeutic targets for muscle‐wasting disorders.
One mechanism for specifically regulating TGF‐β signalling is to promote or inhibit ligand‐receptor binding by coreceptors.13, 14 Betaglycan, epidermal growth factor‐cripto‐1/FRL‐1/cryptic proteins, and the repulsive guidance molecule (RGM) family of glycosylphosphatidylinositol (GPI)‐anchored proteins represent three different types of coreceptors for TGF‐β signalling.14, 15, 16, 17, 18, 19 Among them, RGM family members (RGMa‐c) are regarded as the specific coreceptors for the BMP subfamily. For instance, both RGMa and DRAGON (RGMb) are expressed in the nervous system and enhance BMP signalling by binding with BMP ligands, BMP type I receptors, and BMP type II receptors.17, 19 However, a more recent study demonstrated that RGMa also facilitated TGFβ1/Smad2/3 signalling by interacting with TGF‐β1 receptor activin‐like kinase 5 and Smad2/3 during glial scar formation,20 suggesting an additional role for RGM family members in TGF‐β1 signalling. Unlike RGMa and RGMb, hemojuvelin (also known as HJV or RGMc) is not detected in the brain but is highly expressed in skeletal muscle, heart, and liver.21 Previous data have shown that Hjv (the murine homologue of HJV) functions as a coreceptor for BMPs in the liver and is required for BMP‐mediated induction of hepcidin, a key regulator of iron homeostasis.22, 23 Mutations in the HJV gene have been linked to the severe iron‐storage disease juvenile hemochromatosis, associated with decreased BMP signalling and lower hepcidin expression.18, 23, 24, 25, 26 Although Hjv expression is highest in skeletal muscle,21 only a few studies have explored its role in muscle. Comparative analyses of multiple mammalian species revealed two evolutionary conserved E‐boxes and an MEF2 site in the promoter region of the Hjv gene.27 Both myogenin and MEF2C are capable of increasing Hjv promoter activity.27 Hjv mRNA is quickly induced and parallels the rapid increase in expression of myogenin and MEF2C during myogenic differentiation.27, 28 These data prompted us to investigate whether Hjv is involved in muscle physiological and pathophysiological processes by serving as a coreceptor for TGF‐β1 signalling.
To characterize potential non‐iron‐related functions of Hjv, we investigated muscle‐specific functions of Hjv using conventional and conditional Hjv knockout mice and discovered a novel link between Hjv and muscle wasting. Notably, we found that HJV expression was significantly reduced in both aged muscles and Duchenne muscular dystrophic muscles. Hjv reconstitution in mdx and aged mice rescued the pathogenesis of muscles as well as dystrophic and age‐related TGF‐β1/Smad3 signalling activation. Moreover, we explored the underlying molecular mechanisms by which Hjv protects muscles from wasting. As the muscle‐specific function of Hjv is different from its liver function in regulating iron metabolism, there is potential to devise a novel drug‐development strategy to specifically target the muscle function of Hjv to treat DMD or age‐related muscle wasting.
Materials and methods
Human skeletal muscle samples
Quadriceps femoris from DMD patients (n = 3, mean age: 11.7 ± 5.7 years) and healthy individuals (n = 3, mean age: 13 ± 3 years) were obtained after patients had signed informed consent forms according to the requirement of Institutional Review Board of Chinese People's Liberation Army General Hospital. The diagnosis of DMD was genetically confirmed. The young (n = 10, 20% women, mean age: 26.9 ± 6.9 years) and aged (n = 10, 20% women, mean age: 75.1 ± 9.5 years) skeletal muscle samples were obtained after individuals had signed informed consent forms according to the requirements of Ethics Committee of No. 454 Hospital of People's Liberation Army. Detailed information of human subjects is listed in Supporting Information, Tables S1 and S2.
Mice
Heterozygote Hjv knockout mice (Hjv +/−) were crossed to yield homozygous Hjv knockout mice (Hjv −/−) and wild‐type (WT) littermates.29 Hjv flox/flox mice were crossed with human alpha‐skeletal actin‐Cre transgenic [B6.Cg‐Tg (ACTA1‐cre)79Jme/J strain; The Jackson Laboratory] mice to generate muscle‐specific Hjv knockout (MKO) mice30; Hjv flox/flox littermates were used as WT control mice. All mice were maintained on standard chow diet at a constant temperature (20 °C) under a 12 h/12 h artificial light/dark cycle. WT mice at the age of 2 months or 22–24 months were purchased from Vital River, Charles River China. Five‐week‐old mdx female mice and WT female control mice (C57BL/10ScSn) were purchased from the Institute of Model Organisms, Nanjing University, China. At the end of the experimental period, mice were deeply anaesthetized by intraperitoneal injection of pentobarbital sodium (80 mg/kg body weight). Gastrocnemius muscles from both legs and livers were collected using standardized dissection methods and cleaned of excess fat and connective tissue. All tissues were rapidly frozen in liquid nitrogen for subsequent use of RNA isolation and protein extraction, or embedded in tissue‐freezing medium and frozen in isopentane for sectioning and subsequent morphological and immunohistochemistry analyses. All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1996) and were approved by the Institutional Animal Care and Use Committee of the China Astronaut Research and Training Center.
Quantification of non‐heme iron
The concentration of non‐heme iron in the liver and skeletal muscles was measured by the ferrozine assay as previously described.31 Results were expressed as micrograms of iron per gram of wet tissue weight.
Evaluation of exercise performance
Twelve‐week‐old mice were adapted to moderate treadmill running (10 m/min for 15 min) every day for 1 week before the exercise endurance test. After acclimation, exercise endurance test was determined using a treadmill running test, with the speed 10 m/min for 5 min. After the initial warm‐up period, the running velocity was set to 10 m/min for 1 h and increased by 2 m/min increments at every 15 min until mice could not be prompted to continue running by moderate electric stimulation (less than 0.1 mA) and remained at the electrode for at least 10 continuous seconds.32 Mouse endurance capacity was estimated by running to exhaustion on an Exer4‐OxyMax motorized treadmill (Columbus Instruments, Columbus, OH, USA). Investigators who performed exercise endurance were blind to mice genotypes.
Muscle force assay
Gastrocnemius muscles from mice were excised and mounted on an isometric transducer (AD Instruments, Bella Vista, NSW, Australia) and bathed in an oxygenized (95% O2 and 5% CO2) Krebs–Henseleit solution (120.5 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 20.4 mM NaHCO3, 1.6 mM CaCl2, 10 mM glucose, and 1 mM pyruvate, pH 7.6) at 30 °C. The muscles were adjusted to the optimal length showing maximal isometric twitch tension and allowed to rest for 30 min. To measure skeletal muscle performance, the muscles were continuously stimulated at 100 Hz until the force output reached 10% of the initial force.
Vector construction
Mouse Hjv CDS was amplified from hepatic cDNA and then ligated into the pCMV‐3tag3a vector (Agilent Technologies, Santa Clara, CA, USA). The control was set as the pCMV‐3tag3a vector. The Hjv shRNA plasmids used in this research were constructed by Guangzhou Ribobio Co., Ltd. The sequences are shown in Supporting Information, Table S3.
Cell culture and transfection
C2C12 cells were purchased from China Infrastructure of Cell Line Resource and were cultured in growth medium comprising high‐glucose Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Thermo Scientific, Waltham, MA, USA), 100 units/mL penicillin, 0.1 mg/mL streptomycin, and 20 mM glutamine. Transfections were performed with the NeonTM Transfection System (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions. After 48 h, C2C12 cells were cultured in differentiation medium (high‐glucose DMEM supplemented with 2% horse serum, 100 units/mL penicillin, 0.1 mg/mL streptomycin, and 20 mM glutamine) for 120 h or treated with or without 2 ng/mL TGF‐β1 for 12 h before being harvested.
Luciferase assay
Smad‐responsive CAGA luciferase reporter (CAGA‐Luc) was purchased from Promega (Madison, WI, USA). C2C12 cells were transiently transfected with 2 μg CAGA‐Luc, in combination with 200 ng pRL‐TK vector to control for transfection efficiency, with co‐transfection with mouse Hjv CDS or Hjv shRNA. After 48 h, C2C12 cells were treated with or without 2 ng/mL TGF‐β1 for 12 h. Cells were then lysed and luciferase activity was analysed using the dual‐luciferase reporter assay system purchased from Promega according to the manufacturer's instructions.
Quantitative real‐time RT‐PCR
Total RNA was extracted with TRIzol according the manufacturer's protocol (Life Technologies, Carlsbad, CA, USA). The PrimeScript RT reagent Kit (TaKaRa, Kusatsa, Shiga, Japan) was used to reverse transcribe total RNA (2 μg) into complementary DNA with random hexamer primers, according to the manufacturer's instructions. Quantitative real‐time RT‐PCR (qRT‐PCR) was performed in triplicate using the Step One Plus Realtime PCR system (Applied Biosystems, Singapore). Each PCR mixture (final reaction volume, 50 μL) contained 21 μL sterile water, 25 μL SYBR Green (Applied Biosystems, 7 Kingsland Grange, Warrington, UK), 2 μL cDNA (50 ng/μL), 1 μL forward primer (10 pmol/μL), and 1 μL reverse primmer (10 pmol/μL). PCR cycling conditions were as follows: initial denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 10 s, annealing according to the melting temperature of the primers for 15 s, and elongation at 72 °C for 20 s, finally concluding with a melting curve step. The results were compared with a standard curve and normalized using housekeeping genes (18 s rRNA or GAPDH). The sequences of the primers used were listed in Supporting Information, Table S4.
Immunoblotting and immunoprecipitation
C2C12 cells or skeletal muscles were homogenized in RIPA‐based lysis buffer (Merck, Darmstadt, Germany) with Complete EDTA‐free protease and phosphatase inhibitor cocktails (Roche, Mannheim, Germany). Supernatants were collected and protein concentration was determined using the Bradford protein assay reagent (Bio‐Rad, Hercules, CA, USA). Equal amounts of extracted proteins (30 μg per lane) were denatured in sodium dodecyl sulfate loading buffer, boiled for 3 min, centrifuged briefly to remove insoluble material, and separated on sodium dodecyl sulfate‐polyacrylamide gels. The protein was then transferred onto a nitrocellulose membrane, which were blocked in 5% nonfat milk or bovine serum albumin diluted in Tris‐buffered saline‐Tween for 1 h and then incubated overnight at 4 °C with the respective following primary antibodies against Hjv (R&D Systems, Minneapolis, MN, USA), TGF‐β RII (TβRII) (Merck, Darmstadt, Germany), TGF‐β1, p‐Smad3, Smad3, p‐Smad2, Smad2, Smad1/5/8 (Abcam, Cambridge, MA, USA), p‐Smad1/5/8 (Cell Signaling, Danvers, MA, USA), Atrogin‐1, MuRF1 (ECM Biosciences, Versailles, KY, USA), and β‐actin (Santa Cruz Biotechnologies, Dallas, TX, USA). Signals were visualized by incubating with horseradish peroxidase‐coupled secondary antibodies and an enhanced chemiluminescence reagent (Thermo Scientific, Rockford, IL, USA). Blots were stripped using Stripping Reagent (Thermo Scientific, Rockford, IL, USA) according to the manufacturer's instructions and re‐probed if necessary. Protein bands were quantified using Image‐Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA); the strength of each protein signal was normalized to the corresponding β‐actin signal. Data are expressed as percent of the expression in the control group.
For immunoprecipitation (IP), the gastrocnemius muscles of WT mice were homogenized in a lysis buffer containing 50 mM Tris, pH 7.6, 100 mM NaCl, 1 mM dithiothreitol, 0.5% Triton X‐100, and protease inhibitor cocktail. Simultaneously, 5 μg of the mouse monoclonal antibody to Hjv or TβRII, or non‐specific mouse IgG diluted in 200 μL phosphate buffer saline (PBS) with 0.01% Tween‐20 was incubated with 50 μL Dynabeads Protein A (Life Technologies, Carlsbad, CA, USA) for 10 min at room temperature. The beads were then washed twice followed by incubation with approximately 1 mg total protein overnight at 4 °C with rotation. After washing three times with PBS, the protein‐bound beads were finally resuspended in 20 μL Elution Buffer and denatured in premixed NuPAGE LDS Sample Buffer (Life Technologies, Carlsbad, CA, USA). The samples were boiled at 95 °C for 5 min, and the supernatant was loaded on the gel for immunoblotting.
Single myofibre isolation
Single myofibre isolation was performed as described in Shefer et al.33 Briefly, WT mice were deeply anaesthetized by intraperitoneal injection of pentobarbital sodium (80 mg/kg body weight). The skin over the gastrocnemius muscle and around the ankle was torn back to expose the Achilles tendon, which was cut and used to pull up the gastrocnemius muscle while trimming the fascia and connective tissue as much as possible on either side. After rinsing the muscle in PBS, the excised muscles were immediately transferred to 15 mL tubes containing digestion medium (DMEM supplemented with 35 μg/mL glucose, 100 units/mL penicillin G, 100 μg/mL streptomycin, and 400 units/mL collagenase type I) that was pre‐warmed for at least 10 min. The digest mixture was rocked for 1.5 h at 37 °C using a rotator and then 3–4 mL was transferred to a series of 10 cm plates containing warm RV medium (DMEM supplemented with 10% fetal bovine serum, 35 μg/mL glucose, 100 units/mL penicillin G, and 100 μg/mL streptomycin). Individual fibres were extracted using a pipette and gently dispensed into the centre of a Matrigel (BD Biosciences, Billerica, MA, USA) coated confocal dish. After culturing in RV medium for 24 h, the myofibres were fixed by 2% formaldehyde for 5 min and then stained as described in the succeeding text.
Immunostaining and trichrome staining
For immunostaining, C2C12 cells were fixed with 4% formaldehyde for 30 at 4 °C and then treated with 0.5% Triton‐X 100 in PBS for 5 min at room temperature. Then, cells were incubated with a primary antibody against myosin (Hybridoma Bank, IA, USA) or Flag (Abcam, Cambridge, MA, USA) at 4 °C overnight, followed by incubation with the Alexa Fluor‐594 or Alexa Fluor‐488 (Life Technologies, Carlsbad, CA, USA) fluorescent dye conjugated to an anti‐mouse secondary antibody, respectively. For each sample, more than eight fields per dish were picked. Myofibres isolated were stained with primary antibodies against Hjv (R&D Systems, Minneapolis, MN, USA) and TβRII (Merck, Darmstadt, Germany) at 4 °C overnight, followed by incubation with the Alexa Fluor‐488 (Life Technologies, Carlsbad, CA, USA) fluorescent dye conjugated to an anti‐goat secondary antibody and Alexa Fluor‐594 (Life Technologies, Carlsbad, CA, USA) fluorescent dye conjugated to an anti‐rabbit secondary antibody. Cryosections of gastrocnemius muscles were fixed in 4% paraformaldehyde in PBS for 10 min at 4 °C and washed in PBS. After incubating for 30 min with 0.3% Triton X‐100 in PBS, sections were then blocked for 1 h in 5% goat serum in PBS. The cross‐sectional area of the gastrocnemius muscles was determined by immunostaining with an anti‐laminin antibody (Abcam, Cambridge, MA, USA). The primary antibody was detected by Alexa Fluor‐594 (Life Technologies, Carlsbad, CA, USA) fluorescent dye conjugated to an anti‐rabbit secondary antibody. Photo capture was performed using a Nikon laser microscope (Eclipse E600, Nikon Instruments, Inc., Japan). Trichrome staining was performed following the protocol of trichrome stain (Masson) kit from Sigma‐Aldrich (St. Louis, MO, USA). To measure the fibre cross‐sectional area and fibrosis area, images were captured manually to ensure that adjacent images had slightly overlapping areas. Using Adobe Photoshop CS6 (Adobe Systems Inc.), these images were aligned so that identical areas overlapped and they could then be assembled into one picture covering the entire muscle. The merged pictures were directly analysed using Image‐Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA). By performing Count/Size/Select Colors/Segmentation, Image‐Pro Plus 6.0 automatically traces the fibre or fibrosis outlines based on a pixel classification algorithm. In some cases, manual identification of the boundaries is necessary to delete false boundaries and draw missing ones. The ‘Area’ parameter in the ‘Measurement’ property function was selected to calculate the fibre cross‐sectional area and the fibrotic area.
In vivo transfection
Plasmid containing mouse Hjv CDS or control plasmid was ethanol precipitated and then resuspended in a 25% sucrose‐1 × PBS solution at 4 °C overnight. Prior to plasmid injection, old WT mice at the age of 22–24 months were anaesthetized with pentobarbital sodium (40 mg/kg). A syringe containing 20 μg/10 μL control or Hjv plasmid was inserted near the distal myotendinous junction and pushed along the longitudinal axis of the left or right gastrocnemius muscle, respectively. Young WT mice at the age of 2 months were used as control mice. Plasmid was injected evenly throughout the longitudinal axis of the muscle during syringe withdrawal. Electric pulses (50 Volts/cm, 5 pulses, 200 ms intervals) were then applied by two stainless steel spatula electrodes placed on each side of the gastrocnemius muscle belly by using an ECM 830 electroporator (BTX, Holliston, MA, USA). Two weeks after injections, the gastrocnemius muscles were removed and rapidly frozen in liquid nitrogen for subsequent use for RNA isolation and protein extraction or embedded in tissue‐freezing medium and frozen in isopentane for sectioning and subsequent immunohistochemistry analyses. The same procedures were conducted on 5‐week‐old mdx (C57BL/10ScSn‐Dmdmdx/J) female mice and WT female control mice (C57BL/10ScSn).
Statistical analysis
Data are presented as mean ± SD. The two‐tailed t‐test was used for comparison between two groups, and multi‐group comparisons were performed with one‐way analysis of variance (ANOVA) test followed by Bonferroni post hoc test using (GraphPad PRISM version 5.0, La Jolla, CA, USA). P values of less than or equal to 0.05 were considered to be statistically significant.
Results
Hjv deficiency results in myofibre atrophy, fibrosis, and reduced exercise capacity
To characterize potential non‐iron‐related functions of Hjv in skeletal muscle, we first performed systemic studies on Hjv global knockout mice compared with their WT littermates. Consistent with our previous reports,34, 35 Hjv global knockout resulted in iron accumulation in the liver but not in skeletal muscles (Supporting Information, Figure S1). Although Hjv −/− mice were indistinguishable in appearance from their WT littermates 12 weeks after birth (Figure 1A), immunostaining for laminin in gastrocnemius cross sections clearly showed decreased myofibre size in Hjv −/− mice (Figure 1B), indicative of myofibre atrophy. Two well‐known muscle‐specific ubiquitin ligases, MAFbx (Atrogin‐1) and muscle RING finger protein 1 (MuRF1), were up‐regulated at both mRNA and protein levels in Hjv −/− mice (Figure 1C and D). In addition, the gastrocnemius muscle of Hjv −/− mice showed increased fibrotic connective tissue in the interstitial spaces of the muscle (Figure 1E). The mRNA levels of collagen type 1a1 (Col1a1), collagen type 1a2 (Col1a2), and collagen type 3a1 (Col3a1), typical markers of fibrosis, were significantly higher in the muscles of Hjv −/− mice than in those of WT mice (Figure 1F). These structural changes in Hjv −/− muscles led to the decreased exercise capacity of Hjv −/− mice, as demonstrated by reduced running time and distance. On average, Hjv −/− and WT mice ran to exhaustion for 126 and 144 min (P = 0.01), corresponding to distances of 1541 and 1901 m, respectively (P = 0.01) (Figure 1G). Maximal titanic force was also reduced in Hjv −/− mice (P = 0.04) (Figure 1H). Taken together, Hjv appeared to be important for preventing muscle atrophy, which was reflected at both structural and functional levels.
Figure 1.
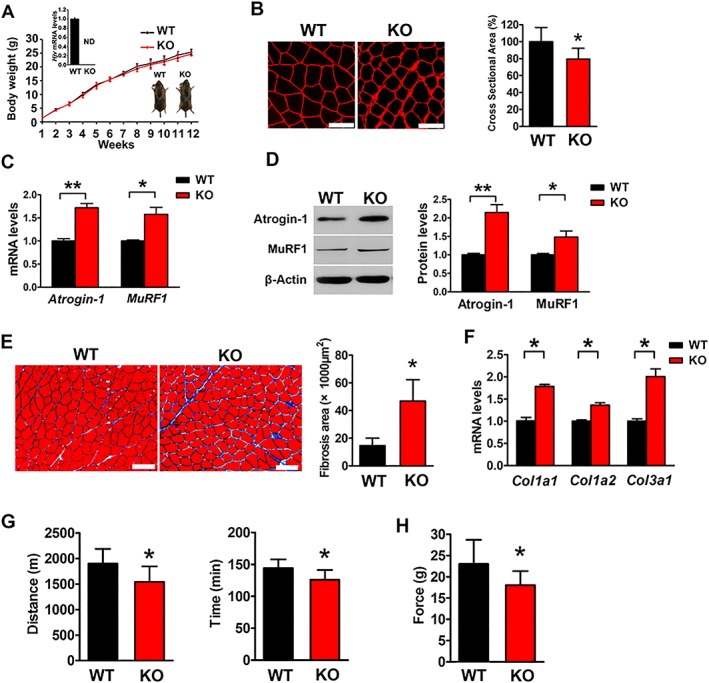
Hjv‐deficient mice display myofibre atrophy, fibrosis, and impaired muscle performance. (A) Gross morphology and mean body weights of WT and KO mice during the first 12 weeks after birth (n = 10). ND, not detected. (B) Immunostaining for laminin in muscle cross sections of WT and KO mice and quantification of cross‐sectional area (n = 5). Scale bars: 75 μm. (C) qRT‐PCR analyses of Atrogin‐1 and MuRF1 expression in muscles of WT and KO mice (n = 5). (D) Western blotting of Atrogin‐1 and MuRF1 in muscles of WT and KO mice (n = 5). (E) Masson's trichrome staining and quantification of fibrosis area in muscle cross sections of WT and KO mice (n = 4). Scale bars: 100 μm. (F) qRT‐PCR analysis of Col1a1, Col1a2, and Col3a1 mRNA expression in muscles of WT and KO mice (n = 3). (G) Exercise endurance of WT and KO mice was determined by running on a treadmill until exhaustion; running distance (left) and time (right) are shown (n = 10). (H) Maximal tetanic forces of muscles isolated from WT and KO mice were measured during continuous electrical stimulation (n = 10). All data are shown as mean ± SD. * P < 0.05, ** P < 0.01 by t‐test. Hjv, hemojuvelin; KO, knockout; WT, wild‐type.
To confirm that the muscle atrophy observed in Hjv global knockout mice was due, at least in part, to the non‐iron regulatory function of Hjv, we generated muscle‐specific Hjv knockout mice by crossing human alpha‐skeletal actin‐Cre transgenic mice with mice bearing Hjv floxed alleles (Hjv flox/flox). We refer to these mice with deletion of Hjv in skeletal muscle as MKO mice, and their littermate Hjv flox/flox mice were used as the WT control. qRT‐PCR confirmed the absence of Hjv mRNA in the skeletal muscles of MKO mice (Figure 2A), whereas Hjv expression was unchanged in the other tissues examined, including heart, kidney, and liver (Supporting Information, Figure S2).
Figure 2.
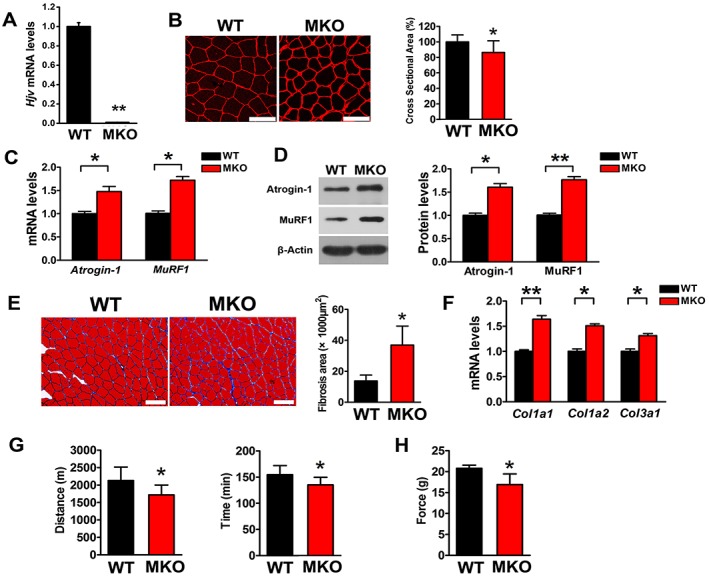
Muscle‐specific deletion of Hjv results in myofibre atrophy, fibrosis, and impaired muscle performance. (A) qRT‐PCR analysis of Hjv expression in muscles from WT and MKO mice (n = 3). (B) Immunostaining for laminin in muscle cross sections of WT and MKO mice and quantification of cross‐sectional area (n = 5). Scale bars: 75 μm. (C) qRT‐PCR analyses of Atrogin‐1 and MuRF1 expression in muscles of WT and MKO mice (n = 3). (D) Western blotting of Atrogin‐1 and MuRF1 in muscles of WT and MKO mice (n = 3). (E) Masson's trichrome staining and quantification of fibrosis area in muscle cross sections of WT and MKO mice (n = 4). Scale bars: 100 μm. (F) qRT‐PCR analysis of Col1a1, Col1a2, and Col3a1 mRNA expression in muscles of WT and MKO mice (n = 3). (G) Exercise endurance of WT and MKO mice was determined by running on a treadmill until exhaustion; running distance (left) and time (right) are shown (n = 10). (H) Maximal tetanic forces of gastrocnemius muscles isolated from WT and MKO mice were measured during continuous electrical stimulation (n = 5). All data are shown as mean ± SD. * P < 0.05, ** P < 0.01 by t‐test. Hjv, hemojuvelin; MKO, muscle‐specific Hjv knockout; WT, wild‐type.
Muscle‐specific Hjv knockout mice were viable, fertile, and of normal size and weight compared with their WT littermates, and no mobility or behavioural deficits were observed. However, like the Hjv −/− mice, the gastrocnemius muscle of MKO mice exhibited atrophic myofibres and fibrosis (Figure 2B and E), as well as increased expression of atrophy and fibrosis markers (Figure 2C, D, and F). Consistent with the structural changes, the running distance (P = 0.02), running time (P = 0.02), and maximal tetanic force (P = 0.04) were reduced in MKO mice (Figure 2G and H). Notably, the specific knockout of Hjv in skeletal muscle has no effect on whole‐body iron homeostasis.24, 30 Therefore, these data demonstrate that the structural and functional deficiency observed in Hjv −/− and MKO mice resulted from the deletion of Hjv in muscle and not the iron overload.
Hemojuvelin is significantly reduced in Duchenne muscular dystrophy and aged muscles
To address whether HJV is involved in certain muscle‐wasting disorders, we searched the Gene Expression Omnibus (GEO) database for the gene expression profiles of quadriceps muscle biopsies from 10 DMD patients (mean age: 2.6 ± 1.9 years) and 11 unaffected controls (36.4% women, mean age: 8.7 ± 10.4 years) (GEO ID: 4809984) (Figure 3A). We found that in addition to several TGF‐β/Smad pathway‐related genes, HJV was significantly reduced in DMD muscles (Figure 3A). Moreover, muscular gene expression in young (10 weeks) and aged (24 months) WT mice extrapolated from the public database (GEO ID: 107050532) indicated that Hjv expression was significantly reduced in aged muscles (Figure 3B). To validate these gene expression results, we examined the HJV mRNA levels in muscles from DMD patients (n = 3, mean age: 11.7 ± 5.7 years) and unaffected controls (n = 3, mean age: 13 ± 3 years) as well as in gastrocnemius muscles from WT and mdx (DMD mouse model) mice. Furthermore, muscle samples from young (n = 10, 20% women, mean age: 26.9 ± 6.9 years) and old (n = 10, 20% women, mean age: 75.1 ± 9.5 years) people as well as young (2 months) and old (22–24 months) mice were examined. qRT‐PCR confirmed that HJV mRNA levels were significantly reduced in dystrophic (Figure 3C and D) and aged muscles (Figure 3F and G), and western blotting analyses demonstrated that Hjv protein levels were also significantly decreased in muscles of mdx and aged mice (Figure 3E and H).
Figure 3.
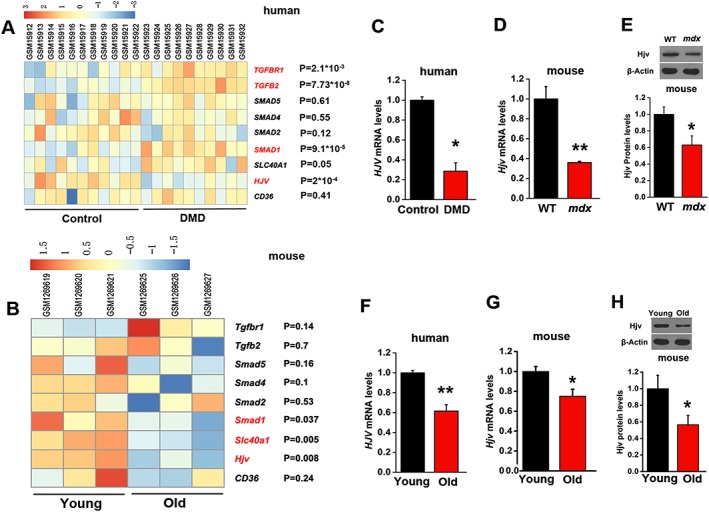
Decreased expression of HJV in DMD and aged muscles. (A) Heatmap of the gene expression of TGFBR1, TGFB2, SMAD5, SMAD4, SMAD2, SMAD1, SLC40A1, HJV, and CD36 was created based on published gene expression profiles of quadriceps muscles from patients with DMD (n = 10) and unaffected controls (n = 11). (B) Heatmap of the gene expression of Tgfbr1, Tgfb2, Smad5, Smad4, Smad2, Smad1, Slc40a1, Hjv, and CD36 was created based on published gene expression profiles of skeletal muscles from young and aged mice (n = 3). (C) qRT‐PCR analysis of HJV expression in muscles from patients with DMD and unaffected controls (n = 3). (D) qRT‐PCR analyses of Hjv in muscles of WT and mdx mice (n = 3). (E) Western blotting of Hjv in muscles of WT and mdx mice (n = 4). (F) qRT‐PCR analysis of HJV expression in muscles of young and aged humans (n = 10). (G) qRT‐PCR analyses of Hjv expression in muscles of young and aged WT mice (n = 3). (H) Western blotting of Hjv in muscles of young and aged WT mice (n = 4). All data are shown as mean ± SD. * P < 0.05, ** P < 0.01 by t‐test. DMD, Duchenne muscular dystrophy; HJV, hemojuvelin.
Hemojuvelin overexpression rescues dystrophic and age‐related muscle wasting
To address whether the loss of Hjv contributes to dystrophic or aged muscle wasting, we carried out a set of rescue experiments to determine whether the overexpression of exogenous Hjv reverses DMD and age‐related muscle wasting. The Hjv overexpression plasmid was injected and electroporated into the gastrocnemius muscles of mdx and aged (22–24 months) mice. Fourteen days after electroporation, muscles were harvested for analyses. Western blotting confirmed the overexpression of exogenous Hjv in mdx and aged muscles (Supporting Information, Figure S3A and B; Tables S5 and S6); mdx mice displayed a higher variation in muscle fibre size, as indicated by a higher number of small and large fibres compared with WT mice. However, the mdx muscles transfected with Hjv displayed a lower variation in fibre size and had fewer small and large fibres compared with mdx alone (Figure 4A). In addition, muscles from mdx mice typically showed obvious fibrosis in the interstitial spaces, but overexpression of Hjv significantly suppressed fibrosis (Figure 4B, Supporting Information, Table S5). Consistently, the up‐regulated expression of fibrosis genes (Col1a1, Col1a2, and Col3a1) in mdx muscles was inhibited by overexpression of Hjv (Figure 4C, Supporting Information, Table S5). Dystrophic muscles are more vulnerable to being infiltrated by adipocytes.36 To examine the effects of Hjv on adipogenesis in mdx muscles, we measured the mRNA levels of fatty acid binding protein 4 (Fabp4), adiponectin, and Pparγ1. As expected, muscles of mdx mice expressed higher levels of Fabp4, adiponectin, and Pparγ1 mRNA in comparison with WT muscles, while overexpression of Hjv in mdx muscles significantly reduced the expression of these three genes (Supporting Information, Figure S4). A similar protective effect of Hjv was also observed in aged mice. As shown in Figure 5 and Supporting Information, Table S6, the myofibre atrophy and fibrosis in aged muscles were alleviated by Hjv overexpression. Together, these data suggested that Hjv sufficiently suppresses DMD and age‐related muscle wasting.
Figure 4.
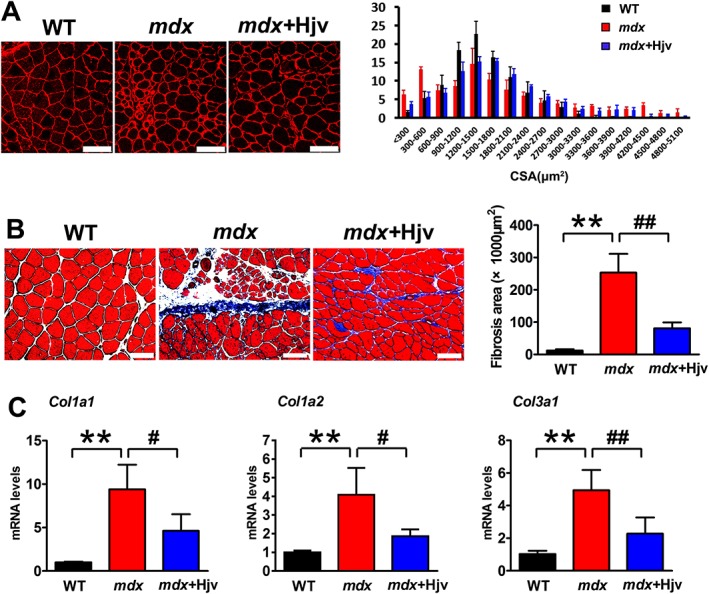
Hjv overexpression ameliorates dystrophic muscle wasting. (A) Immunostaining for laminin in muscle cross sections of WT and mdx mice transfected with control (mdx) or Hjv overexpression (mdx+Hjv) plasmids and the distribution plot of myofibre cross‐sectional area (n = 3). Scale bars: 100 μm. (B) Masson's trichrome staining and quantification of fibrosis area in muscle cross sections from the transfected muscles as shown in (A) (n = 4). Scale bars: 100 μm. (C) qRT‐PCR analysis of Col1a1, Col1a2, and Col3a1 mRNA levels expression in the transfected muscles shown in (A) (n = 4). All data are shown as mean ± SD. ** P < 0.01, # P < 0.05, ## P < 0.01 by one‐way analysis of variance. Hjv, hemojuvelin; WT, wild‐type.
Figure 5.
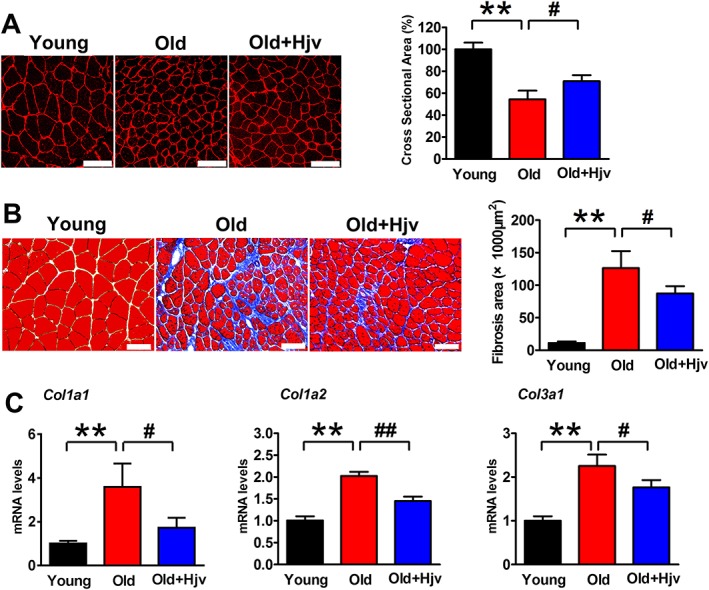
Hjv overexpression attenuates age‐related muscle wasting. (A) Immunostaining for laminin in muscle cross sections of young and old mice transfected with control (Old) or Hjv overexpression (Old+Hjv) plasmids and quantification of cross‐sectional area (n = 3). Scale bars: 100 μm. (B) Masson's trichrome staining and quantification of fibrosis area in muscle cross sections from the transfected muscles as shown in (A) (n = 4). Scale bars: 100 μm. (C) qRT‐PCR analysis of Col1a1, Col1a2, and Col3a1 mRNA expression in the transfected muscles shown in (A) (n = 4). All data are shown as mean ± SD. ** P < 0.01, # P < 0.05, ## P < 0.01 by one‐way analysis of variance. Hjv, hemojuvelin.
Lack of hemojuvelin enhances the transforming growth factor‐β1 signalling pathway in vivo and in vitro
Previous studies identified Hjv as a BMP coreceptor required for BMP‐mediated p‐Smad1/5/8 activation. We therefore investigated whether the effect of Hjv on muscle was associated with altered BMP signalling. Gastrocnemius muscles from WT and Hjv −/−mice were isolated and analysed by western blotting. Unexpectedly, Hjv deficiency did not affect p‐Smad1/5/8 expression but up‐regulated TGF‐β1, TβRII, and p‐Smad2/3 expression compared with that in WT littermates (Figure 6A). We next constructed short hairpin RNA (shRNA) plasmids specifically against Hjv, of which the highest knockdown efficiency was selected for subsequent experiments (Supporting Information, Figure S5). As shown in Figure 6B, Hjv knockdown in C2C12 cells resulted in up‐regulation of TGF‐β1, TβRII, and p‐Smad2/3 expression, while no changes in the expression of p‐Smad1/5/8 were observed. Moreover, C2C12 cells transfected with Hjv shRNAs formed fewer and smaller myotubes than those transfected with control shRNA, with concomitant higher expression of atrophy genes (Atrogin‐1 and MuRF1) (Supporting Information, Figure S6).
Figure 6.
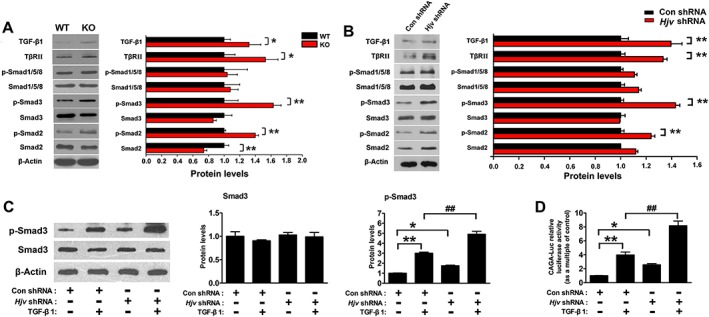
Hjv deficiency promotes activation of the TGF‐β1/Smad3 signalling pathway in muscle. (A) Western blotting of the indicated proteins in muscles of WT and KO mice (n = 4). (B) Western blotting of the indicated proteins in C2C12 myotubes transfected with either Hjv shRNA or control shRNA (n = 4). (C) C2C12 cells were transfected with either Hjv shRNA or control shRNA. Forty‐eight hours post‐transfection, C2C12 cells were incubated in the absence or presence of 2 ng/mL TGF‐β1 for 12 h. Smad3 and p‐Smad3 were then detected by western blotting (n = 3). (D) C2C12 cells were transfected with a CAGA‐Luc and a control pRL‐TK, together with either Hjv shRNA or control shRNA. Forty‐eight hours post‐transfection, cells were incubated in the absence or presence of 2 ng/mL TGF‐β1 for 12 h before cell lysates were analysed for luciferase activity. Relative luciferase activity was calculated as the ratio of firefly to Renilla luciferase activity (n = 6). All data are shown as mean ± SD. * P < 0.05, ** P < 0.01, ## P < 0.01 by t‐test (A, B) or one‐way analysis of variance (C, D). Hjv, hemojuvelin; KO, knockout; TGF‐β1, transforming growth factor‐β1; WT, wild‐type.
The up‐regulation of TGF‐β1, TβRII, and p‐Smad2/3 in Hjv‐deficient muscle raises the question of whether loss of Hjv aggravates the activation of downstream signalling by exogenous TGF‐β1. We therefore transfected C2C12 cells with control or Hjv shRNA, in combination with a Smad‐responsive CAGA luciferase reporter (CAGA‐Luc). The transfected cells were then incubated with or without TGF‐β1. As expected, Hjv knockdown increased the basal levels of p‐Smad3 and CAGA luciferase activity in the absence of TGF‐β1 (Figure 6C and D). p‐Smad3 was quickly activated by TGF‐β1, and this activation was further enhanced in C2C12 cells transfected with Hjv shRNA (Figure 6C). In agreement with western blotting results, TGF‐β1 increased CAGA luciferase activity, and the increase in CAGA luciferase activity induced by TGF‐β1 was more evident in C2C12 cells transfected with Hjv shRNA (Figure 6D). These data clearly demonstrate that loss of Hjv enhances TGF‐β1 signalling in skeletal muscle.
Hemojuvelin overexpression inhibits transforming growth factor‐β1 signalling in vivo and in vitro
To examine whether Hjv overexpression was sufficient to inhibit the activation of TGF‐β1 signalling in muscle, we transfected C2C12 cells with CAGA‐Luc in combination with a plasmid encoding the mouse Hjv coding sequence or empty plasmid (Supporting Information, Figure S7). Transfected cells were then treated with or without TGF‐β1. As shown in Figure 7A, Hjv overexpression reduced the basal levels of p‐Smad3 and CAGA luciferase activity in the absence of TGF‐β1. Of note, the up‐regulation of p‐Smad3 and CAGA luciferase activity induced by TGF‐β1 were also significantly attenuated in C2C12 cells transfected with Hjv (Figure 7B).
Figure 7.
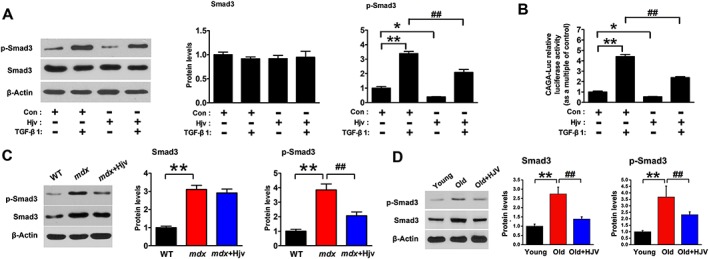
Hjv overexpression inhibits the activation of TGF‐β1/Smad3 signalling in muscle. (A) C2C12 cells were transfected with either a control or Hjv overexpression plasmid. Forty‐eight hours post‐transfection, C2C12 cells were incubated in the absence or presence of 2 ng/mL TGF‐β1 for 12 h. Smad3 and p‐Smad3 were then detected by western blotting (n = 3). (B) C2C12 cells were transfected with a CAGA‐Luc and a control pRL‐TK, together with either a control or Hjv overexpression plasmid. Forty‐eight hours post‐transfection, cells were incubated in the absence or presence of 2 ng/mL TGF‐β1 for 12 h before cell lysates were analysed for luciferase activity. Relative luciferase activity was calculated as the ratio of firefly to Renilla luciferase activity (n = 6). (C) Western blotting of p‐Smad3 and Smad3 in the muscles of WT and mdx mice transfected with a control (mdx) or Hjv overexpression (mdx+Hjv) plasmids (n = 4). (D) Western blotting of p‐Smad3 and Smad3 in the muscles of young and old mice transfected with a control (Old) or Hjv overexpression (Old+Hjv) plasmid (n = 3). All data are shown as mean ± SD. * P < 0.05, ** P < 0.01, ## P < 0.01 by one‐way analysis of variance. Hjv, hemojuvelin; TGF‐β1, transforming growth factor‐β1; WT, wild‐type.
Activation of TGF‐β1 signalling is closely associated with structural and functional changes in dystrophic and aged muscles. Thus, we determined whether the protective effect of Hjv overexpression on dystrophic and aged muscles could be attributed to the inhibition of TGF‐β1 signalling. As shown in Figure 7C and D, p‐Smad3 was activated in dystrophic and aged gastrocnemius muscles compared with control muscles. When Hjv was overexpressed in mdx and aged muscles, p‐Smad3 activation was eliminated (Figure 7C and D). Together, these results indicate that Hjv overexpression is sufficient to inhibit TGF‐β1 signalling in muscles.
Hemojuvelin acts as a coreceptor for transforming growth factor‐β receptor II in skeletal muscle
Transforming growth factor‐β receptor II resides on the cell membrane and is critical for mediating intracellular signalling transduction by TGF‐β family members. To determine whether the Hjv protein interacts with TGF‐β receptors, we performed immunofluorescence using isolated myofibres from WT mouse gastrocnemius muscles. Our results showed the co‐localization of Hjv and TβRII on the myofibre membrane (Figure 8A). In addition, co‐IP experiments further confirmed that Hjv directly interacts with TβRII in murine skeletal muscle (Figure 8B). Thus, we concluded that Hjv inhibits TGF‐β1 signalling by interacting with TβRII.
Figure 8.
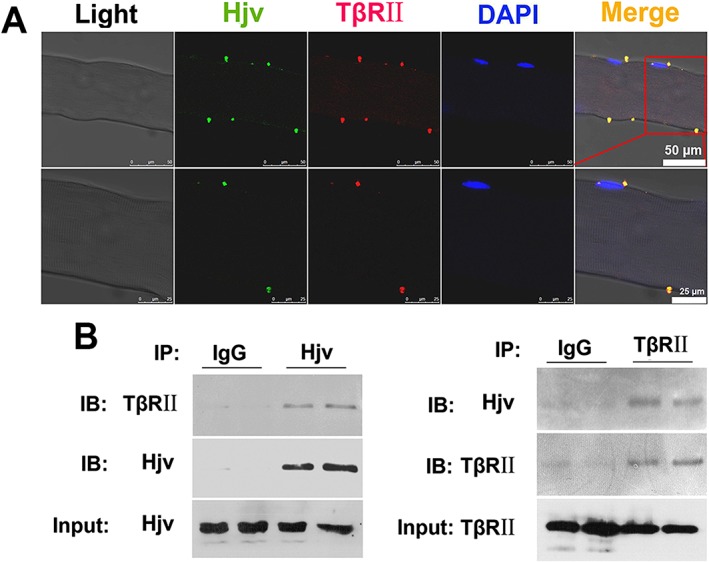
Hjv directly interacts with TβRII on the muscle membrane. (A) Representative immunofluorescence micrographs demonstrating co‐localization of Hjv and TβRII on myofibre membranes isolated from the muscles of WT mice (n = 4). The lower panel (scale bar: 25 μm) shows magnified images of the upper panel (scale bar: 50 μm). (B) Co‐immunoprecipitation analysis of Hjv and TβRII in the muscles of WT mice (n = 4). Hjv, hemojuvelin; IB, immunoblotting; IP, immunoprecipitation; TβRII; TGF‐β receptor II; WT, wild‐type.
Discussion
Hemojuvelin (HJV) is a member of the RGM family that localizes on the cell membrane. As a coreceptor for BMP, Hjv activates hepcidin expression in the liver via the BMP‐SMAD signalling pathway.18, 22 In the clinic, disease‐associated HJV mutations result in juvenile hemochromatosis.25, 37, 38 Previous studies have shown that Hjv is expressed in skeletal muscle, heart, and liver, with the highest expression being observed in skeletal muscle.21 Hjv deficiency in murine skeletal muscle reportedly does not affect systemic iron homeostasis.24, 30 However, whether Hjv plays a role in skeletal muscle remains largely unclear.
In this study, we found that Hjv deficiency in skeletal muscle resulted in myofibre atrophy, fibrosis, and reduced muscle force and exercise capacity. Muscle fibrosis in Hjv −/− or MKO mice may compensate for the loss of myofibre size hence the reason muscle mass was not decreased in Hjv −/− or MKO mice (data not shown). Our findings seem to contradict the fact that patients with HJV mutations rarely develop skeletal muscle complaints. As suggested by a recent study reporting variable expressivity of HJV‐related hemochromatosis in patients, the symptoms associated with mild and/or late‐onset HJV hemochromatosis may have been overlooked because of the relatively small number of cases analysed, mixed genotypes, and/or missing data in patients' medical history.39 Therefore, the clinical presentation of HJV hemochromatosis could change significantly, and the currently recognized common complications may be encountered less frequently. For example, a 30‐year‐old female patient with compound heterozygous G320V and C321W HJV mutations was diagnosed when she presented with progressive fatigue and early onset menopause.40 Although excessive iron accumulated in the liver of Hjv −/− mice, we can rule out an association between iron overload and the structural changes in Hjv‐deficient muscles based on several key observations. First, we detected the ex vivo maximal titanic force of muscles from Hjv −/− mice and their WT littermates, revealing reduced maximal titanic force in Hjv −/− mouse muscles. Notably, the iron content was unchanged in Hjv −/− muscles.24 Second, MKO mice presented structural and functional deficiencies similar to those observed in Hjv −/− mice, with no alterations in serum iron content or skeletal muscle.24, 30 Finally, C2C12 myoblasts transfected with Hjv shRNAs also formed fewer and smaller myotubes than those transfected with a control shRNA, with higher expression of Atrogin‐1 and MuRF1 being observed. Therefore, for the first time, we reveal a previously unrecognized non‐iron regulatory function of Hjv in maintaining muscle structure and function.
Hjv was third RGM family member to be characterized and is thus also known as RGMc. Previous data showed that Hjv functions as a coreceptor for BMPs in the liver and enhances classical BMP signalling.18, 22, 23 The absence of Hjv results in the reduced expression of phosphorylated Smad1/5/8 in the Hjv −/− liver.22 These data led us to examine whether the muscle wasting and dysfunction observed in Hjv −/− mice was because of impaired BMP signalling in Hjv −/− muscles. Unexpectedly, the lack of Hjv did not affect p‐Smad1/5/8 expression in muscles but up‐regulated TGF‐β1, TβRII, and p‐Smad2/3 expression. Our in vitro experiments confirmed that loss of Hjv not only increased basal levels of p‐Smad3 and CAGA luciferase activity in C2C12 cells but also augmented the activation of Smad3 signalling induced by TGF‐β1. In contrast, Hjv overexpression was sufficient to inhibit TGF‐β1/Smad3 signalling in vivo and in vitro. Thus, it is plausible to conclude that Hjv functions as an inhibitor for TGF‐β1 signalling in skeletal muscle. In DMD and aged muscles, TGF‐β1 was up‐regulated, with a concomitant decrease in Hjv. The loss of Hjv may increase the sensitivity of muscle cells to TGF‐β1, leading to greater activation of downstream Smad3 signalling and thereby promoting the pathogenesis of these muscle‐wasting disorders.
Mammals express over 40 TGF‐β family members but only five type I receptors and seven type II receptors.14, 41 The receptors, especially type II receptors, determine the specificity of binding with TGF‐β family members, which require association with some accessory coreceptors.13, 14, 41 Thus, the identification of coreceptors for TGF‐β signalling is critical for specifically regulating this pathway. Betaglycan, epidermal growth factor‐cripto‐1/FRL‐1/cryptic proteins, and the RGM family of GPI‐anchored proteins represent three different coreceptors for TGF‐β signalling.14, 15, 16, 17, 18, 19 Interestingly, some coreceptors induce totally different effects by mediating interactions between different TGF‐β ligands and their respective receptors. For example, betaglycan serves as a TGF‐β RIII and facilitates the interaction of TβRII with TGF‐β2, thus maximizing TGF‐β signalling.42 Betaglycan also acts as a coreceptor for inhibin, a molecule known as an activin antagonist.43, 44 Given that both TGF‐β and activin initiate the Smad2/3 pathway, betaglycan can exert opposing effects on Smad2/3 activation. The loss of Hjv results in the alteration of distinct downstream signalling pathways in the liver and muscle; we speculate that Hjv possesses functional properties similar to those of betaglycan by acting as a coreceptor for distinct receptors. This notion is supported by our immunostaining and IP experiments, which show that Hjv and TβRII are co‐localized and interact at the myofibre membrane. It had been reported that Hjv could interact with several types of receptors. For example, Hjv activates BMP signalling by binding with BMP type I receptor.22 In hepatoma‐derived cells, Hjv forms a multi‐protein membrane complex with the hemochromatosis protein and transferrin receptor 2.45 Recently, we found that Hjv can interact with interleukin 12 receptor subunit beta 1.46 In this study, our results provide the first evidence that Hjv serves as a coreceptor for TβRII in skeletal muscle, conferring an additional layer of regulation to control TGF‐β1 signalling. Further work is needed to determine the precise mechanism by which Hjv inhibits TGF‐β1 signalling in skeletal muscle. Hjv likely interferes with the interaction between TGF‐β ligands and TβRII. Alternatively, Hjv may competitively interact with TβRII, impairing the recruitment and activation of type I receptors.
Muscle wasting resulting from physiological (ageing) and pathological (DMD) conditions has been recognized as a rising major risk factor for adverse health outcomes, warranting detailed studies on the molecular pathogenesis of metabolic and contractile dysregulation in the aged or dystrophic muscular system.2 The TGF‐β1/Smad signalling pathway is known as a central pathway associated with multiple types of muscle disorders, including dystrophic and age‐related muscle wasting.7, 9 TGF‐β1 was remarkably up‐regulated in the muscles of DMD patients and mdx mice and contributed to disease progression by promoting fibrosis formation.9, 11 In aged muscles, excessive TGF‐β1 is produced, inducing unusually high levels of TGF‐β1/p‐Smad3 in resident satellite cells and impairing their regenerative capacity.12 Therefore, the inhibition of TGF‐β1 signalling has been investigated as a treatment option for muscle‐wasting disorders.47, 48 For example, the inhibition of TGF‐β1 signalling by losartan treatment had dramatically beneficial effects on sarcopenic muscle by improving the regeneration after injury.49 Inhibition of TGF‐β1 ligands in mouse models of muscular dystrophy has been shown to variously reduce muscle fibrosis and increase mass and strength.50 However, TGF‐β1 signalling is found ubiquitously throughout the body, and disruption of this pathway is thus associated with several disease conditions, such as pathological cardiac diseases, bone, and connective tissue disorders.7, 10, 41, 51 Precise manipulation of this pathway is therefore necessary to avoid unexpected side effects. Given its relatively high expression in skeletal muscle coupled with its inhibitory effect on TGF‐β1 signalling, HJV may serve as an ideal target for regulating TGF‐β1 signalling in skeletal muscle with high spatiotemporal resolution. In this study, we found that Hjv expression was reduced in both aged and dystrophic muscles. Moreover, overexpressing Hjv in the muscles of aged and mdx mice reversed the up‐regulation of TGF‐β1/p‐Smad3 and significantly reduced both fibrosis and muscle wasting. These data provide compelling evidence in support of HJV as a potential therapeutic target for treating muscle‐wasting disorders, especially DMD. Although various gene‐based therapies (such as exon‐skipping, gene transfer, or gene editing) have been developed to restore a functional dystrophin in DMD muscles, the fibrosis induced by TGF‐β1 can severely compromise the efficacy of these therapies by limiting access of the therapeutic vectors to dystrophic muscles.52, 53 To overcome this limitation, ‘combined therapies’ including both a therapy to correct the genetic defect and an additional one to improve the status of recipient muscle have been proposed recently.54 The protective effects of Hjv on dystrophic muscles indicate that pretreating dystrophic muscles with a HJV expression vector may maximize the efficacy of gene‐based approaches by reducing the fibrosis, thereby allowing the use of lower and safer vector doses to achieve a therapeutic expression level of dystrophin. This would ultimately lower the costs and potential toxicity. Future studies are warranted to better understand the precise mechanism of HJV in the maintenance of muscle function and to develop potent and safe HJV agonists against muscle dystrophy and atrophy.
In summary, we revealed an unrecognized function of HJV in the maintenance of skeletal muscle health. As shown in Figure 9, under normal physiological conditions, HJV inhibits TGF‐β1 signalling in skeletal muscle by interacting with TβRII. In dystrophic and aged muscles, excessive TGF‐β1 is produced while HJV is decreased. More TGF‐β1 binds with TβRII, leading to greater activation of downstream Smad3 signalling and exacerbating the pathogenesis of these muscle‐wasting disorders. Taken together, our data functionally define the HJV‐TβRII‐Smad3 signalling cascade as a novel therapeutic target to improve the quality of life in both aged populations and patients with muscle dystrophy, such as DMD.
Figure 9.
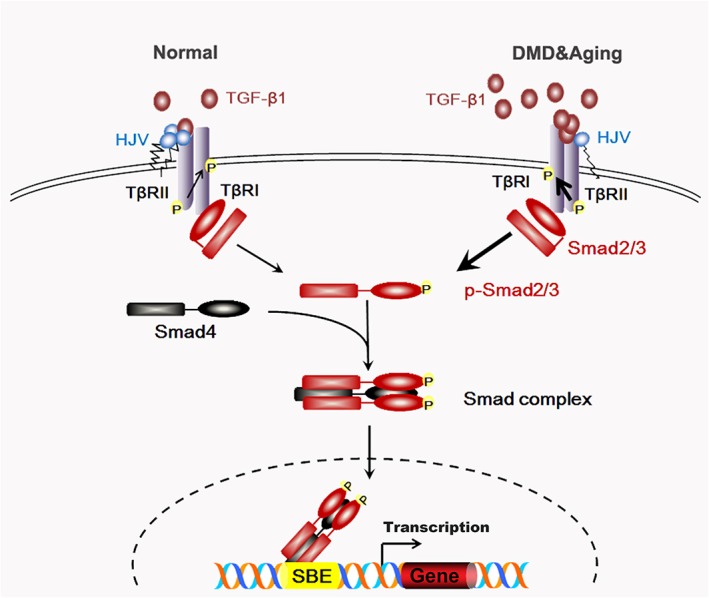
Schematic diagram depicting the role of HJV in TGF‐β1/Smad3 signalling in skeletal muscle. TGF‐β1 signals through binding with TβRII. The type II receptor then transphosphorylates the type I receptor (TβRІ), which subsequently transduces the signal by phosphorylating Smad2 and Smad3, allowing them to form a heterooligomeric complex with a common partner (co)‐Smad (Smad4). The complex translocates to the nucleus, where it interacts directly with the Smad binding element (SBE) and regulates expression of target genes. Under normal physiological conditions, HJV interferes with the interaction between TGF‐β1 and TβRII or impairs the activation of TβRІ by competitive binding with TβRII to avoid over‐activation of TGF‐β1 signalling. In dystrophic or aged muscles, excessive TGF‐β1 is produced while HJV is decreased. More TGF‐β1 binds with TβRII, thereby leading to greater activation of downstream Smad3 signalling and exacerbating the pathogenesis of these muscle‐wasting disorders. Arrow thickness represents different degrees of activation of TGF‐β1 signalling. HJV, hemojuvelin; TGF‐β1, transforming growth factor‐β1.
Conflict of interest
The authors declare no competing financial interests.
Supporting information
Figure S1. Quantification of non‐heme iron concentration in tissues of WT and Hjv −/− mice. Non‐heme iron concentration in liver and skeletal muscle of WT (n = 9) and Hjv −/− (n = 4) mice was measured by the ferrozine assay and results were expressed as micrograms of iron per gram of wet tissue weight. All data are shown as mean ± s.d. **P < 0.01 by t‐test.
{kind=link}
Figure S2. Validation of skeletal muscle‐specific Hjv knockout mice. Hjv mRNA levels was measured in heart, kidney and liver from WT and MKO mice by qRT‐PCR (n = 3). All data are shown as mean ± s.d.
{kind=link}
Figure S3. Overexpression of Hjv in mdx and aged muscles. (A) Western blotting of Hjv expression in the muscles of WT and mdx mice transfected with a control (mdx) or Hjv overexpression (mdx + Hjv) plasmid (n = 4). (B) Western blotting of Hjv expression in the muscles of young and old mice transfected with a control (Old) or Hjv overexpression (Old+ Hjv) plasmid (n = 4). All data are shown as mean ± s.d. **P < 0.01, # P < 0.05, ## P < 0.01 by one‐way ANOVA.
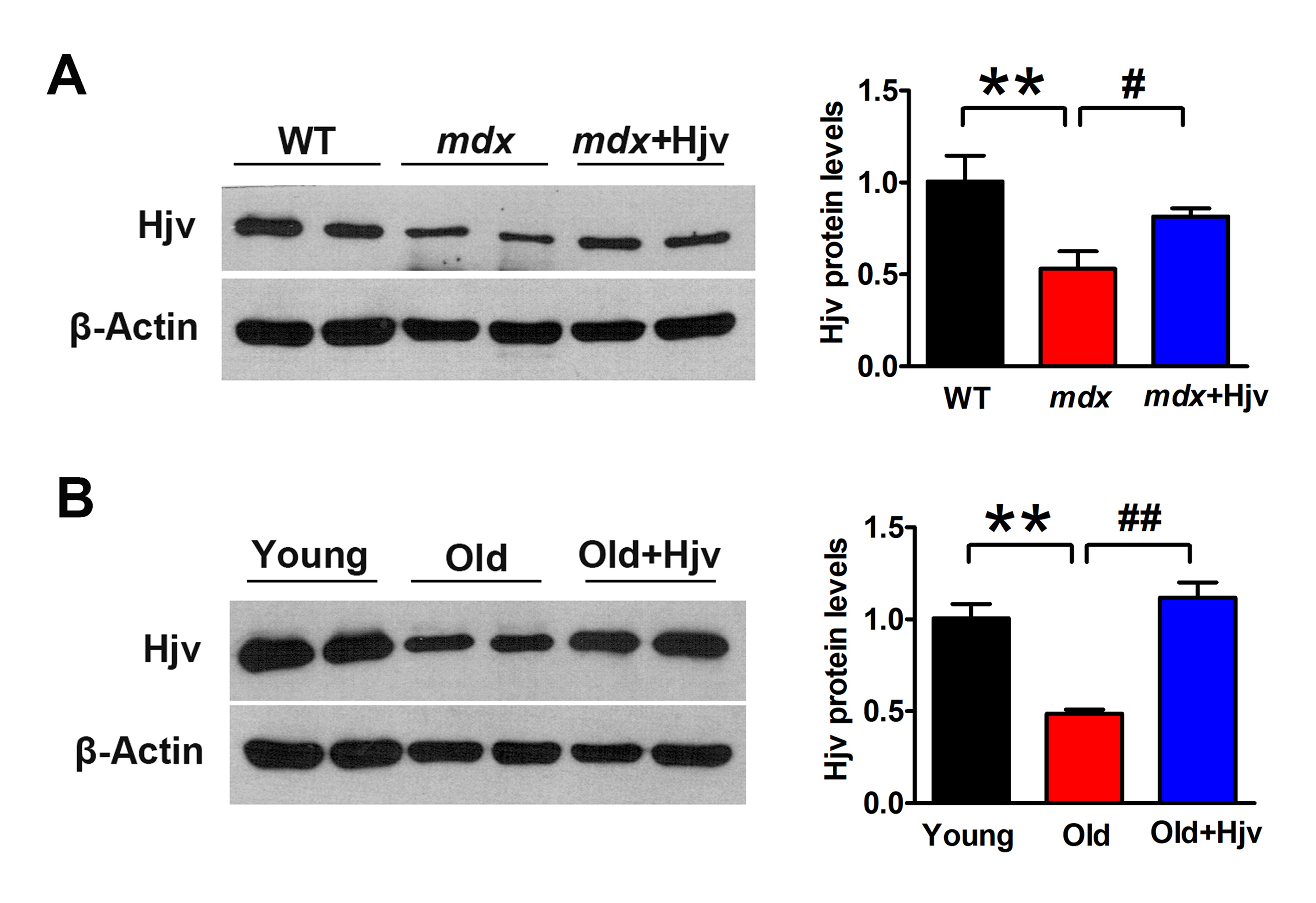{kind=link}
Figure S4. Hjv overexpression inhibits adipogenesis in dystrophic muscles. qRT‐PCR analysis of Fabp4, adiponectin, and Pparγ1 mRNA levels in muscles of WT and mdx mice transfected with control (mdx) or Hjv overexpression (mdx + Hjv) plasmids (n = 4). All data are shown as mean ± s.d. **P < 0.01, ## P < 0.01 by one‐way ANOVA.
{kind=link}
Figure S5. RNAi‐mediated Hjv knockdown in C2C12 cells. C2C12 cells were transfected with either control shRNA or Hjv shRNA1, 2, 3. Seventy‐two hours post‐transfection, total proteins were extracted and Hjv protein levels were then detected by Western blotting (n = 3). All data are shown as mean ± s.d. **P < 0.01 by one‐way ANOVA.
{kind=link}
Figure S6. RNAi‐mediated knockdown of Hjv results in myotube atrophy. (A) C2C12 cells were transfected with either Hjv shRNA or control shRNA. Twenty‐four hours post‐transfection, C2C12 myotubes were induced in differentiation medium for 120 h and were then fixed with 4% formaldehyde and immunostained with an anti‐myosin antibody. Scale bars: 250 μm. Quantitative analysis of the myosin‐positive area was performed using Image‐Pro Plus 6.0 software (n = 4). (B) qRT‐PCR analysis of Atrogin‐1 and MuRF1 mRNA expression in C2C12 myotubes treated as described in (A). All data are shown as mean ± s.d. *P < 0.05,**P < 0.01 by t‐test.
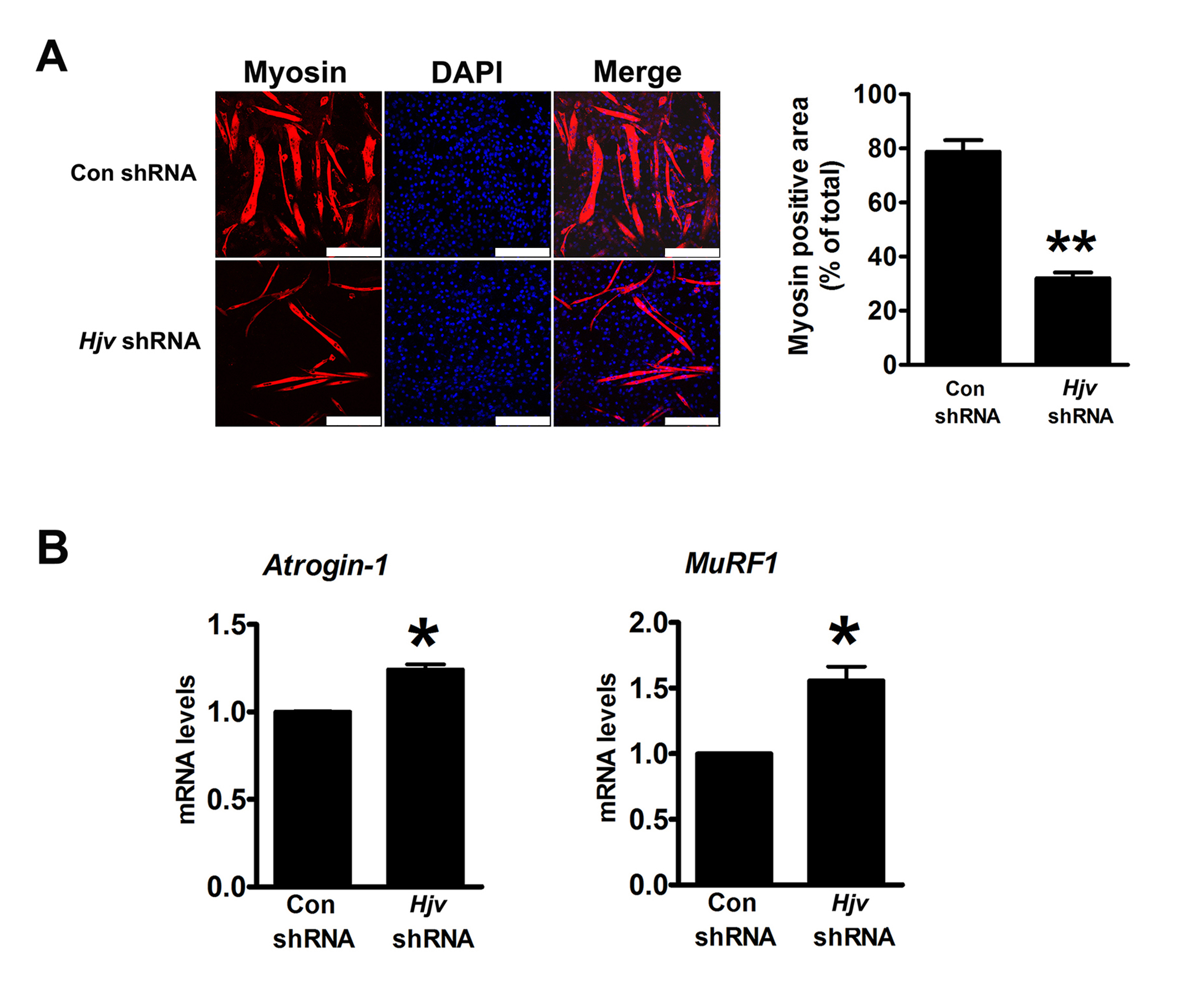{kind=link}
Figure S7. Transfection of Hjv expression vector and control vector in C2C12 cells. C2C12 cells were transfected with either a control or Hjv overexpression plasmid. Forty‐eight hours post‐transfection, C2C12 cells were immunostained with anti‐flag primary antibody and a secondary antibody conjugated with Alexa Fluor‐488 fluorescent dye to show transfection efficiency. Scale bars: 100 μm.
{kind=link}
Table S1. Detailed information for control and DMD patients
Table S2. Detailed information for young and aged patients
Table S3. Sequences used in plasmid construction
Table S4. Primers used in Real‐time PCR analysis
Table S5. Raw data for Figure 4 and Figure S3
Table S6. Raw data for Figure 5 and Figure S3
Acknowledgements
We thank Dr Nancy Andrews at Duke University for providing us with Hjv +/− and Hjv flox/flox mice. This work was supported by grants from the National Natural Science Foundation of China (81772016, 11727813, 31171144, and 81272177 to X.C.; 81871522 to P.Z.; 31330036; and 3150034 to F.D.W.) and the 1226 major project (AWS16J018 to X.C.).
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.55
Zhang P., He J., Wang F., Gong J., Wang L., Wu Q., Li W., Liu H., Wang J., Zhang K., Li M., Huang X., Pu C., Li Y., Jiang F., Wang F., Min J., and Chen X. (2019) Hemojuvelin is a novel suppressor for Duchenne muscular dystrophy and age‐related muscle wasting, Journal of Cachexia, Sarcopenia and Muscle, 10: 557–573. 10.1002/jcsm.12414.
Contributor Information
Fudi Wang, Email: fwang@zju.edu.cn.
Junxia Min, Email: junxiamin@zju.edu.cn.
Xiaoping Chen, Email: xpchen2009@163.com.
References
- 1. Frontera WR, Ochala J. Skeletal muscle: a brief review of structure and function. Calcif Tissue Int 2015;96:183–195. [DOI] [PubMed] [Google Scholar]
- 2. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015;14:58–74. [DOI] [PubMed] [Google Scholar]
- 3. Liguori I, Russo G, Aran L, Bulli G, Curcio F, Della‐Morte D, et al. Sarcopenia: assessment of disease burden and strategies to improve outcomes. Clin Interv Aging 2018;13:913–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Han A, Bokshan SL, Marcaccio SE, DePasse JM, Daniels AH. Diagnostic criteria and clinical outcomes in sarcopenia research: a literature review. J Clin Forensic Med 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee AJ, Buckingham ET, Kauer AJ, Mathews KD. Descriptive phenotype of obsessive compulsive symptoms in males with Duchenne muscular dystrophy. J Child Neurol 2018; 883073818774439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carter JC, Sheehan DW, Prochoroff A, Birnkrant DJ. Muscular dystrophies. Clin Chest Med 2018;39:377–389. [DOI] [PubMed] [Google Scholar]
- 7. Chen JL, Colgan TD, Walton KL, Gregorevic P, Harrison CA. The TGF‐β signalling network in muscle development, adaptation and disease. Adv Exp Med Biol 2016;900:97–131. [DOI] [PubMed] [Google Scholar]
- 8. Kollias HD, McDermott JC. Transforming growth factor‐beta and myostatin signaling in skeletal muscle. J Appl Physiol 1985;2008:104,579–104,587. [DOI] [PubMed] [Google Scholar]
- 9. Burks TN, Cohn RD. Role of TGF‐β signaling in inherited and acquired myopathies. Skeletal Muscle 2011;1:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sartori R, Gregorevic P, Sandri M. TGFβ and BMP signaling in skeletal muscle: potential significance for muscle‐related disease. Trends Endocrinol Metab 2014;25:464–471. [DOI] [PubMed] [Google Scholar]
- 11. Kharraz Y, Guerra J, Pessina P, Serrano AL, Munoz‐Canoves P. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed Res Int 2014;2014,965631:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008;454:528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feng XH, Derynck R. Specificity and versatility in TGF‐β signaling through Smads. Annu Rev Cell Dev Biol 2005;21:659–693. [DOI] [PubMed] [Google Scholar]
- 14. de Caestecker M. The transforming growth factor‐β superfamily of receptors. Cytokine Growth Factor Rev 2004;15:1–11. [DOI] [PubMed] [Google Scholar]
- 15. Bilandzic M, Stenvers KL. Betaglycan: a multifunctional accessory. Mol Cell Endocrinol 2011;339:180–189. [DOI] [PubMed] [Google Scholar]
- 16. Cheng SK, Olale F, Bennett JT, Brivanlou AH, Schier AF. EGF‐CFC proteins are essential coreceptors for the TGF‐β signals Vg1 and GDF1. Genes Dev 2003;17:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Samad TA, Rebbapragada A, Bell E, Zhang Y, Sidis Y, Jeong SJ, et al. DRAGON, a bone morphogenetic protein co‐receptor. J Biol Chem 2005;280:14122–14129. [DOI] [PubMed] [Google Scholar]
- 18. Core AB, Canali S, Babitt JL. Hemojuvelin and bone morphogenetic protein (BMP) signaling in iron homeostasis. Front Pharmacol 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Babitt JL, Zhang Y, Samad TA, Xia Y, Tang J, Campagna JA, et al. Repulsive guidance molecule (RGMa), a DRAGON homologue, is a bone morphogenetic protein co‐receptor. J Biol Chem 2005;280:29820–29827. [DOI] [PubMed] [Google Scholar]
- 20. Zhang R, Wu Y, Xie F, Zhong Y, Wang Y, Xu M, et al. RGMa mediates reactive astrogliosis and glial scar formation through TGFβ1/Smad2/3 signaling after stroke. Cell Death Differ 2018;25:1503–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez A, Pan P, Parkkila S. Expression studies of neogenin and its ligand hemojuvelin in mouse tissues. J Histochem Cytochem 2006;55:85–96. [DOI] [PubMed] [Google Scholar]
- 22. Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet 2006;38:531–539. [DOI] [PubMed] [Google Scholar]
- 23. Xia Y, Babitt JL, Sidis Y, Chung RT, Lin HY. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood 2008;111:5195–5204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gkouvatsos K, Wagner J, Papanikolaou G, Sebastiani G, Pantopoulos K. Conditional disruption of mouse HFE2 gene: maintenance of systemic iron homeostasis requires hepatic but not skeletal muscle hemojuvelin. Hepatology 2011;54:1800–1807. [DOI] [PubMed] [Google Scholar]
- 25. Wallace DF, Dixon JL, Ramm GA, Anderson GJ, Powell LW, Subramaniam N. Hemojuvelin (HJV)‐associated hemochromatosis: analysis of HJV and HFE mutations and iron overload in three families. Haematologica 2005;90:254–255. [PubMed] [Google Scholar]
- 26. Kuns‐Hashimoto R, Kuninger D, Nili M, Rotwein P. Selective binding of RGMc/hemojuvelin, a key protein in systemic iron metabolism, to BMP‐2 and neogenin. Am J Physiol Cell Physiol 2008;294:C994–C1003. [DOI] [PubMed] [Google Scholar]
- 27. Severyn CJ, Rotwein P. Conserved proximal promoter elements control repulsive guidance molecule c/hemojuvelin (Hfe2) gene transcription in skeletal muscle. Genomics 2010;96:342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuninger D, Kuns‐Hashimoto R, Kuzmickas R, Rotwein P. Complex biosynthesis of the muscle‐enriched iron regulator RGMc. J Cell Sci 2006;119:3273–3283. [DOI] [PubMed] [Google Scholar]
- 29. Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest 2005;115:2187–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen W, Huang FW, de Renshaw TB, Andrews NC. Skeletal muscle hemojuvelin is dispensable for systemic iron homeostasis. Blood 2011;117:6319–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang AS, Gao J, Koeberl DD, Enns CA. The role of hepatocyte hemojuvelin in the regulation of bone morphogenic protein‐6 and hepcidin expression in vivo. J Biol Chem 2010;285:16416–16423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, et al. Total skeletal muscle PGC‐1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab 2010;12:633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shefer G, Yablonka‐Reuveni Z. Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol Biol 2005;290:281–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu Q, Wang H, An P, Tao Y, Deng J, Zhang Z, et al. HJV and HFE play distinct roles in regulating hepcidin. Antioxid Redox Signal 2015;22:1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017;66:449–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cordani N, Pisa V, Pozzi L, Sciorati C, Clementi E. Nitric oxide controls fat deposition in dystrophic skeletal muscle by regulating fibro‐adipogenic precursor differentiation. Stem Cells 2014;32:874–885. [DOI] [PubMed] [Google Scholar]
- 37. Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest 2005;115:2180–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lanzara C, Roetto A, Daraio F, Rivard S, Ficarella R, Simard H, et al. Spectrum of hemojuvelin gene mutations in 1q‐linked juvenile hemochromatosis. Blood 2004;103:4317–4321. [DOI] [PubMed] [Google Scholar]
- 39. Hamdi‐Rozé H, Ben Ali Z, Ropert M, Detivaud L, Aggoune S, Simon D, et al. Variable expressivity of HJV related hemochromatosis: “Juvenile” hemochromatosis? Blood Cells Mol Dis 2019;74:30–33. [DOI] [PubMed] [Google Scholar]
- 40. Lee PL, Barton JC, Brandhagen D, Beutler E. Hemojuvelin (HJV) mutations in persons of European, African‐American and Asian ancestry with adult onset haemochromatosis. Br J Haematol 2004;127:224–229. [DOI] [PubMed] [Google Scholar]
- 41. Santibanez JF, Quintanilla M, Bernabeu C. TGF‐β/TGF‐β receptor system and its role in physiological and pathological conditions. Clin Sci (Lond) 2011;121:233–251. [DOI] [PubMed] [Google Scholar]
- 42. Sankar S, Mahooti‐Brooks N, Centrella M, McCarthy TL, Madri JA. Expression of transforming growth factor type III receptor in vascular endothelial cells increases their responsiveness to transforming growth factor β 2. J Biol Chem 1995;270:13567–13572. [DOI] [PubMed] [Google Scholar]
- 43. Lewis KA, Gray PC, Blount AL, MacConell LA, Wiater E, Bilezikjian LM, et al. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 2000;404:411–414. [DOI] [PubMed] [Google Scholar]
- 44. Wiater E, Harrison CA, Lewis KA, Gray PC, Vale WW. Identification of distinct inhibin and transforming growth factor β‐binding sites on betaglycan: functional separation of betaglycan co‐receptor actions. J Biol Chem 2006;281:17011–17022. [DOI] [PubMed] [Google Scholar]
- 45. D'Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane‐associated protein complex for hepcidin regulation. J Hepatol 2012;57:1052–1060. [DOI] [PubMed] [Google Scholar]
- 46. Wu Q, Shen Y, Tao Y, Wei J, Wang H, An P, et al. Hemojuvelin regulates the innate immune response to peritoneal bacterial infection in mice. Cell discovery 2017;3:17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Walton KL, Johnson KE, Harrison CA. Targeting TGF‐β mediated SMAD signaling for the prevention of fibrosis. Front Pharmacol 2017;8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov 2012;11:790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burks TN, Andres‐Mateos E, Marx R, Mejias R, Van Erp C, Simmers JL, et al. Losartan restores skeletal muscle remodeling and protects against disuse atrophy in sarcopenia. Sci Transl Med 2011;3:82ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen JL, Walton KL, Hagg A, Colgan TD, Johnson K, Qian H, et al. Specific targeting of TGF‐β family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc Natl Acad Sci U S A 2017;114:E5266–E5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gordon KJ, Blobe GC. Role of transforming growth factor‐β superfamily signaling pathways in human disease. Biochim Biophys Acta 2008;1782:197–228. [DOI] [PubMed] [Google Scholar]
- 52. Trollet C, Athanasopoulos T, Popplewell L, Malerba A, Dickson G. Gene therapy for muscular dystrophy: current progress and future prospects. Expert Opin Biol Ther 2009;9:849–866. [DOI] [PubMed] [Google Scholar]
- 53. Zhou L, Lu H. Targeting fibrosis in Duchenne muscular dystrophy. J Neuropathol Exp Neurol 2010;69:771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cordova G, Negroni E, Cabello‐Verrugio C, Mouly V, Trollet C. Combined therapies for Duchenne muscular dystrophy to optimize treatment efficacy. Front Genet 2018;9:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Quantification of non‐heme iron concentration in tissues of WT and Hjv −/− mice. Non‐heme iron concentration in liver and skeletal muscle of WT (n = 9) and Hjv −/− (n = 4) mice was measured by the ferrozine assay and results were expressed as micrograms of iron per gram of wet tissue weight. All data are shown as mean ± s.d. **P < 0.01 by t‐test.
Figure S2. Validation of skeletal muscle‐specific Hjv knockout mice. Hjv mRNA levels was measured in heart, kidney and liver from WT and MKO mice by qRT‐PCR (n = 3). All data are shown as mean ± s.d.
Figure S3. Overexpression of Hjv in mdx and aged muscles. (A) Western blotting of Hjv expression in the muscles of WT and mdx mice transfected with a control (mdx) or Hjv overexpression (mdx + Hjv) plasmid (n = 4). (B) Western blotting of Hjv expression in the muscles of young and old mice transfected with a control (Old) or Hjv overexpression (Old+ Hjv) plasmid (n = 4). All data are shown as mean ± s.d. **P < 0.01, # P < 0.05, ## P < 0.01 by one‐way ANOVA.
Figure S4. Hjv overexpression inhibits adipogenesis in dystrophic muscles. qRT‐PCR analysis of Fabp4, adiponectin, and Pparγ1 mRNA levels in muscles of WT and mdx mice transfected with control (mdx) or Hjv overexpression (mdx + Hjv) plasmids (n = 4). All data are shown as mean ± s.d. **P < 0.01, ## P < 0.01 by one‐way ANOVA.
Figure S5. RNAi‐mediated Hjv knockdown in C2C12 cells. C2C12 cells were transfected with either control shRNA or Hjv shRNA1, 2, 3. Seventy‐two hours post‐transfection, total proteins were extracted and Hjv protein levels were then detected by Western blotting (n = 3). All data are shown as mean ± s.d. **P < 0.01 by one‐way ANOVA.
Figure S6. RNAi‐mediated knockdown of Hjv results in myotube atrophy. (A) C2C12 cells were transfected with either Hjv shRNA or control shRNA. Twenty‐four hours post‐transfection, C2C12 myotubes were induced in differentiation medium for 120 h and were then fixed with 4% formaldehyde and immunostained with an anti‐myosin antibody. Scale bars: 250 μm. Quantitative analysis of the myosin‐positive area was performed using Image‐Pro Plus 6.0 software (n = 4). (B) qRT‐PCR analysis of Atrogin‐1 and MuRF1 mRNA expression in C2C12 myotubes treated as described in (A). All data are shown as mean ± s.d. *P < 0.05,**P < 0.01 by t‐test.
Figure S7. Transfection of Hjv expression vector and control vector in C2C12 cells. C2C12 cells were transfected with either a control or Hjv overexpression plasmid. Forty‐eight hours post‐transfection, C2C12 cells were immunostained with anti‐flag primary antibody and a secondary antibody conjugated with Alexa Fluor‐488 fluorescent dye to show transfection efficiency. Scale bars: 100 μm.
Table S1. Detailed information for control and DMD patients
Table S2. Detailed information for young and aged patients
Table S3. Sequences used in plasmid construction
Table S4. Primers used in Real‐time PCR analysis
Table S5. Raw data for Figure 4 and Figure S3
Table S6. Raw data for Figure 5 and Figure S3