

Abstract
Tumorigenic and non-neoplastic tissue injury occurs via the ischemic microenvironment defined by low oxygen, pH, and nutrients due to blood supply malfunction. Ischemic conditions exist within regions of pseudopalisading necrosis, a pathological hallmark of glioblastoma (GBM), the most common primary malignant brain tumor in adults. To recapitulate the physiologic microenvironment found in GBM tumors and tissue injury, we developed an in vitro ischemic model and identified chromodomain helicase DNA binding protein 7 (CHD7) as a novel ischemia-regulated gene. Point mutations in the CHD7 gene are causal in CHARGE syndrome, a CNS developmental disorder, and interrupt the epigenetic functions of CHD7 in regulating neural stem cell maintenance and development. Using our ischemic system, we observed microenvironment-mediated decreases in CHD7 expression in brain tumor initiating cells and neural stem cells. Validating our approach, CHD7 was suppressed in the perinecrotic niche of GBM patient and xenograft sections, and an interrogation of patient gene expression datasets determined correlations between low CHD7, increasing glioma grade and worse patient outcomes. Segregation of GBM by molecular subtype revealed a novel observation that CHD7 expression is elevated in proneural vs mesenchymal GBM. Genetic targeting of CHD7 and subsequent gene ontology analysis of RNA sequencing data indicated angiogenesis as a primary biological function affected by CHD7 expression changes. We validated this finding in tube formation assays and vessel formation in orthotopic GBM models. Together, our data provide further understanding of molecular responses to ischemia and a novel function of CHD7 in regulating angiogenesis in both neoplastic and non-neoplastic systems.
Keywords: CHD7, ischemia, glioblastoma, tumor initiating cell, cancer stem cell, tumor microenvironment, hypoxia
Graphical Abstract
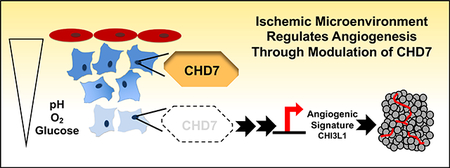
The ischemic microenvironment in both tumor and non-neoplastic tissue can drive new blood vessel formation to adapt to changes in pH, oxygen tension, and restricted nutrient availability. Using neural progenitor and glioblastoma cells in an in vitro ischemic model, we demonstrated that mRNA and protein expression of the epigenetic reader CHD7 was suppressed in the ischemic microenvironment. Reducing CHD7 altered the transcriptome to increase angiogenesis-related pathways that include CHI3L1 (YKL-40) and increased measures of angiogenesis in vitro and in vivo.
INTRODUCTION
Glioblastoma (GBM) is the most common primary malignant brain tumor in adult patients with a median survival rate of less than fifteen months after diagnosis, despite aggressive treatment(1–3). Treatment difficulties reflect the massive heterogeneity of GBMs. Molecular analysis of GBM tumors, including samples from different locations, and single cells demonstrates diverse genetic and epigenetic alterations(4–9). Transcriptomes from GBM patients can imitate neural lineages, indicating the need to further understand mechanisms through which tumors reacquire developmental programs. Novel treatments that address these complexities must be developed to improve the dismal outlook for GBM patients.
Heterogeneity in the tumor environment, particularly with regards to oxygen tension, is well recognized to partially mediate poor treatment response and therapeutic resistance(1). Solid tumor environments characterized by hypoxia, acidity, and nutrient restriction result from unconstrained growth and the altered metabolic profile of cancer cells. More extreme microenvironments may be present in regions of pseudopalisading necrosis, a pathologic marker of GBM where tumor cells surround necrotic foci. These perinecrotic niches are regions where Brain Tumor Initiating cells (BTICs) localize(10–16), and BTIC phenotypes--as measured through stem cell marker expression, neurosphere formation, and tumorigenic potential--are increased when in vivo microenvironments are modeled in vitro (10–12, 17–20). BTICs residing in perinecrotic niches contribute to highly vascular GBMs through elevated production of angiogenic factors including VEGF, which has autocrine effects on BTIC viability and paracrine effects on endothelial cells(21, 22). As VEGF antibody-based approaches to GBM treatment have failed in the clinic, exploring the molecular regulation of tumor environment may identify novel mechanisms leading to effective therapies targeting the resistant niche.
Alterations in the epigenetics of neoplastic cells and the neighboring cells composing the microenvironment occur in response to environmental pressures and may play an important role in therapeutic resistance. Understanding how the physicologic microenvironment modulates epigenetics is critical: the epigenetic state of GBM tracks with patient survival and therapeutic efficacy(23, 24). An epigenetic modifier which we suggest is regulated by the tumor environment is Chromodomain-helicase-DNA-binding protein 7 (CHD7). CHD7 is one of a family of CHD proteins involved in transcription, chromosomal stability, and DNA repair(25, 26), which binds methylated histone H3 lysine 4 (H3K4me)(27). It has been reported that CHD7 is capable of both enhancing and inhibiting embryonic stem cell genes, by co-localizing with Oct4, Sox2, and Nanog at enhancer regions(28), and a recent report indicates a requirement for CHD7 in oligodendrocyte precursor cell survival(29). CHD7 is mutated in a congenital disorder called CHARGE syndrome which stand for coloboma, heart defects, atresia choanae, growth retardation, genital abnormalities, and ear abnormalities (30, 31). However, very little is known about CHD7 and cancer(32–34), although one recent report indicated that a subset of medulloblastoma patients with low CHD7 and high BMI1 expression had poor prognosis(35). Another intriguing potential role for CHD7 is in angiogenesis. Two recently published studies showed CHD7 binding within 10kb of VEGF as well as increases in VEGF transcription upon loss of CHD7 in mice(28, 36). CHD7 in extracellular vesicles has also been proposed as a marker of endothelial dysfunction(37). Given that current knowledge regarding epigenetic control of tumor angiogenesis in GBM is limited, understanding how tumor microenvironments impact epigenetics to regulate angiogenesis is likely to provide future opportunities for therapeutic targeting of GBMs.
RESULTS AND DISCUSSION
While the effects of hypoxia on epigenetic factor regulation and chromatin marks has been previously explored(38–41), little is known about the contribution of tumor environments other than hypoxia to tumor epigenetic states. We therefore used a chromatin remodeling factor array to evaluate the expression of eighty-four genes important for epigenetic regulation when BTICs were treated with acidic stress or hypoxia (Figure 1A). Multiple genes were identified with 20 percent or greater repression by both acidic stress and hypoxia (Figure 1B, 1C). We next considered whether patient data suggested that repression of these genes by ischemic conditions was contributing to tumor biology by screening for correlations with poor patient outcomes. Using the Repository for Molecular Brain Neoplasia Data (REMBRANDT) and The Cancer Genome Atlas (TCGA), we determined that out of these genes, only lower mRNA levels of CHD7 significantly correlated with worse patient survival in both Rembrandt and TCGA (Figures 1B, 1D, S1).
Figure 1. Screening for Microenvironment-Regulated Epigenetic Related Genes in Glioblastoma.

Cells isolated from human GBM xenografts were cultured in normal, acidic (pH 6.5), or hypoxic conditions (2% O2) for 72 hrs. (A) An array measuring RNA expression of 84 epigenetic related genes from 1016 GBM PDX. (B) Genes sorted by at least a 20% reduction in expression in the presence of both hypoxia and acidic stress. Genes were interrogated to determine whether their downregulation correlated with worse patient prognosis in multiple GBM databases. (C) Validation of CHD family member mRNA expression across multiple GBM PDX in the context of acidic stress. (D) CHD7 mRNA expression was screened for correlations with patient outcomes using REMBRANDT and TCGA. In silico analysis demonstrates expression of CHD7 in (E) GBM patient specimens compared to non-transformed neural stem cells (Lee et al., Cancer Cell 2006), (F) GBMs, lower grade astrocytomas and other gliomas (Bredel et al., Cancer Research 2005, Freije et al., Cancer Research 2004, or Sun et al., Cancer Cell 2006), (G) G-CIMP vs non G-CIMP tumors (TCGA), and methylated vs unmethylated MGMT promoter (TCGA), or (H) primary and recurrent GBMs (Murat et al., J Clin Oncol 2008). (I) CHD7 mRNA expression does not change in GBM xenograft derived cells resistant to irradiation. *, p<0.05, **** p<0.0001 with t-test or ANOVA.
Our data suggested that CHD7 and two other CHD family members, CHD2 and CHD9, both within the top 15 downregulated genes by acidic stress alone, correlated with GBM outcomes by contributing to stress response. To further investigate this, we utilized multiple GBM patient derived xenografts (PDXs) of known subtype and mutational status for key oncogenes and tumor suppressors. Xenograft tumors were dissociated, and cells were cultured in BTIC media with standard or low pH. After 72 hours of treatment in either condition, we collected RNA from cells isolated from 7 GBM PDXs and the U87 cell line. While CHD2 and CHD9 were not consistently repressed by acidic stress, CHD7 mRNA was downregulated in 5 of 7 GBM PDXs as well as U87 cells (Figure 1C). When CHD7, CHD2, and CHD9 were interrogated for their significance in patient data, CHD7 was once again the only gene significantly correlating with worse survival in all four sample sets tested (Figures 1, S1). We also found that of CHD2, CHD7, or CHD9, only CHD7 had reduced mRNA expression in pseudopalisading cells in comparison to the cellular tumor (Figure S2A and data not shown), consistent with our findings of reduced CHD7 expression by ischemic environments in vitro.
We next sought to understand the significance of CHD7 expression in the overall context of glioma and the heterogeneous nature of GBM in particular. To that end, we queried multiple publicly available datasets and discovered that CHD7 expression was decreased in GBM compared to neural stem cells (Figure 1E) and reduced in GBM as compared to lower grade gliomas such as astrocytoma or oligodendroglioma (Figure 1F). Consistent with the notion that CHD7 was higher in less aggressive gliomas, G-CIMP positive GBMs had elevated CHD7 in comparison to non G-CIMP GBMs (Figure 1G) but CHD7 levels did not directly correlate with MGMT status. When comparing recurrent GBMs to primary tumors, CHD7 expression was also decreased (Figure 1H). However, CHD7 mRNA levels were unchanged in irradiation-resistant PDX derived cells generated by our laboratory (Figure 1I). Overall, these data suggest that CHD7 may have an important role to play in GBM, and that this role may be different in certain subsets of patients.
To improve upon attempts to mimic the in vivo microenvironment, we next determined whether similar results were obtained under ischemic conditions that included physiologic oxygen tension, glucose, and pH found in combination within GBMs. We confirmed that in vitro ischemia was able to suppress CHD7 mRNA (Figure 2A) and protein (Figure 2B) expression in GBM PDX cells. Interestingly, we also found that CHD7 was repressed by ischemia in immortalized neural progenitors (Figure 2A), suggesting that environmental repression of CHD7 could be broadly applicable. Validating this novel in vitro response, we also observed a markedly reduced expression of CHD7 in perinecrotic zones of PDXs (Figure 2C). Furthermore, staining of primary human GBM tissue sections determined a similar pattern of expression. In these sections, we noticed that CHD7 tends to be highly expressed adjacent to functional blood vessels, and lowest in the perinecrotic zones where ischemic conditions occur (Figure 2C and data not shown). This pattern of expression was also observed in images of GBM patient tumors obtained from the Human Protein Atlas(42) (Figure 2C) and similar, but not identical to, the mRNA expression pattern in the Ivy GAP dataset (Figure S2A). Overall, these data suggest that CHD7 levels are reduced under conditions found in the ischemic/perinecrotic microenvironment.
Figure 2. CHD7 is Repressed by Ischemic and Perinecrotic Conditions and is Differentially Expressed in GBM Molecular Subtypes.

(A) qPCR and (B) western blot confirm the decrease in CHD7 mRNA in GBM PDX and neural progenitors in ischemic conditions. (C) Representative images at 20x magnification of H&E and CHD7 expression in D456 GBM xenografts, human GBM patient specimens, and in the Human Protein Atlas (proteinatlas.org) IHC of normal brain section and high-grade glioma patient sections. (D) CHD7 expression in patient specimens from the TCGA database were ordered by their expression in GBM subtypes. (E) mRNA and (F) protein expression of CHD7 expression at baseline in multiple GBM PDX lines, human neural progenitors, and human astrocytes. ** p<0.01, *** p<0.001, **** p<0.0001 with t-test or ANOVA.
Differences in the tumor microenvironments influence GBM heterogeneity which can include transcriptional signatures used for subtype classification. Considering the link between hypoxia and epithelial to mesenchymal transition, we evaluated whether lower levels of CHD7 were found in any particular GBM molecular subtype. We accessed microarray data from TCGA and discovered that CHD7 is differentially expressed in the GBM molecular subtypes, with the highest expression seen in the proneural subtype and lowest levels in the mesenchymal subtype (Figure 2D). Using PDXs of known GBM subtype and human neural progenitors and astrocytes, we also measured baseline expression of CHD7 RNA (Figure 2E) and protein in vitro (Figure 2F). These data largely corroborated data retrieved from patient databases in the above panel, with lowest levels in mesenchymal GBMs. Together, our findings suggest that CHD7 levels are reduced in GBM in comparison to neural progenitors and non-neoplastic brain, reduced in the mesenchymal GBM subtype, and repressed by ischemic microenvironments.
Given the limited reports of a role for CHD7 in cancer, we took an unbiased approach to evaluate the effects of CHD7 loss on the transcriptome. In neural progenitor cells and D456 GBM PDX that express relatively high CHD7 levels, we expressed shRNA directed against CHD7 or a non-targeting control shRNA to reduce CHD7 mRNA (Figure 3A) and protein (Figure 3B) levels. While it would have been beneficial to do the converse experiment and restore CHD7 to the low expressing GBM PDXs, the high molecular weight of CHD7 (approximately 336 kDa) presented a major obstacle which we could not overcome. We analyzed the GBM and neural progenitor samples with and without CHD7 targeting using RNAseq. Utilizing a list of genes that were altered in response to CHD7 KD in both D456 and NPCs, gene ontology analysis suggested multiple angiogenesis-related pathways in the top 5 diseases or functions predicted to be CHD7 regulated (Figure 3C). From this, we filtered the gene list by angiogenesis functions as well as whether the gene was altered in the same direction in both cell types (schematic in Figure 3D). The resulting four genes were decorin (DCN), platelet-derived growth factor delta (PDGFD), chitinase 3-like protein 1 (CHI3L1, also known as YKL-40), and tropomodulin 1 (TMOD1) (Figure 3D). To further confirm the importance of these genes, we sought to independently validate their expression in response to CHD7 knockdown via shRNA in both NPCs and D456. Our results indicated that CHI3lL1 was upregulated in both cell types when CHD7 was reduced (Figure 3E). This data supports previous studies showing that CHI3L1 is a biomarker for high-grade GBM and important in GBM angiogenesis (43–45). To further link CHD7 and CHI3L1, we determined whether the expression of these two genes correlated in patient specimens. Using the TCGA data via GlioVis, we found that CHD7 inversely correlated with CHI3L1 in glioma patient samples (Figure S2B). Connecting a CHD7/CHI3L1 pathway to angiogenesis and tumor microenvironmental responses, CHD7 inversely and CHI3L1 positively correlated with the hypoxia and acidic stress regulated angiogenesis modulators VEGFA and carbonic anhydrase 9 (Figure S2B). CHI3L1 was also elevated in pseudopalisading cells (Figure S2C) where CHD7 levels were lower. Together, these data suggest a common role for CHD7 in regulating angiogenesis in both non-neoplastic and neoplastic cells via regulation of the downstream target CHI3L1.
Figure 3. Transcriptomic Effects of CHD7 Knockdown with Gene Ontology Analysis.

(A, B) Stable knockdown of CHD7 using shRNA in neural progenitor cells (NPCs) and D456 GBM xenografts shows reduced CHD7 transcript and protein. (C) Gene ontology analysis using Ingenuity Pathway Analysis (IPA) identified enrichment in angiogenesis pathways following CHD7 knockdown. D) Schematic of the logic used to identify candidate downstream genes. (E) Independent validation of the candidate genes from RNAseq was performed in separate experiments using mRNA from CHD7 KD NPC and D456 cells in comparison to NT control identified CHI3L1 as a common gene upregulated in response to CHD7 suppression. *, p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 with ANOVA.
We further investigated a role for CHD7 in angiogenesis by co-culturing CHD7-knockdown cells with human brain microvascular endothelial cells (HBMEC) (Figure 4A) or human umbilical vein endothelial cells (data not shown) and measuring tube formation. The loss of CHD7 in both GBM and non-neoplastic neural stem cells contributed to an increase in endothelial tube length and number of segments (Figure 4A, 4B), which was not due to changes in GBM cell proliferation (data not shown). In an orthotopic model of GBM, staining of sections taken from intracranial tumors formed by D456 GBM PDX expressing CHD7 shRNA showed dramatic increases in collapsed and malformed blood vessels and increases in areas of thrombosis, marked by expression of CD31 (arrowhead), within the brain tumors (Figure 4C). Targeting of CHD7 contributed to an increase in vessel formation in vivo (Figure 4C, 4D), but knockdown of CHD7 alone was not sufficient to significantly decrease survival (Figure S3). In these GBM xenograft models, it remains possible that the relatively low levels of CHD7 and high levels of proliferation and angiogenesis preclude any increases in angiogenesis from acting as a dominant regulator of overall animal survival. Alternatively, the human/mouse cell microenvironmental interactions in PDX models may not be optimal for observing CHD7 impacts on GBM development or progression and transgenic mouse models may be required. However, our data support the hypothesis that CHD7 is a relevant determinant of human patient outcomes that modulates angiogenesis. We stratified CHD7 expression with both CA9, a hypoxia marker, and VEGFA, a master regulator of angiogenesis in TCGA data, and correlated these expression statuses with patient survival. Interestingly, lower levels of CA9 and VEGFA only correlate with better prognosis if CHD7 levels are high (Figure S4). Ultimately, these data further suggest the importance of the link between CHD7 levels and angiogenesis and hypoxia for patient outcomes.
Figure 4. Angiogenic Effects of in vitro and in vivo Modulation of CHD7 Expression.

(A) Representative images of tube formation after D456 GBM PDX or neural progenitor cells expressing CHD7 shRNA or NT control were co-cultured with human brain microvascular endothelial cells. (B) Quantification of branch length and number of segments in the tube formation assay. (C) Representative images at 10x magnification of sections obtained from orthotopic xenografts formed by intracranial injection of D456 cells expressing shRNA specific for CHD7. Immunohistochemistry shown represents CHD7 expression and blood vessel formation by CD31 staining. Abnormal vasculature and thrombosis marked by arrowheads. (D) Quantification of CD31 staining as a percentage of total slide micrograph area from three tumor sections. *, p<0.05 with ANOVA.
CONCLUSION
Tumors can be considered wounds that do not heal and employ developmental pathways to facilitate progression(46). We have determined that ischemic conditions commonly found in the tumor environment that can also occur during damage to non-tumor tissue have a novel molecular consequence: repression of CHD7. Loss of CHD7 is well recognized as the cause of CHARGE syndrome, but ischemia-related dysregulation of CHD7 has not been previously reported to our knowledge. The consistency of the repression of CHD7 under ischemia in neural progenitors and GBM PDX cells suggests the pathway is important for multiple brain pathologies. We believe it could be interesting to investigate whether changes in CHD7 levels were associated with perinatal asphyxia and resulting developmental delays. Our use of cells derived from GBM PDXs under the BTIC/cancer stem cell maintenance condition and the known role for CHD7 in neuronal differentiation also suggests that there may be implications for brain lineages that have not yet been investigated.
Cells in perinecrotic/ischemic microenvironments can attempt to respond to the lack of nutrients and oxygen by sending signals for neovascularization(46). We found that angiogenesis was elevated when CHD7 was reduced, suggesting that repression of CHD7 may be important for adaptation to the ischemic microenvironment. This molecular alteration could provide a new link between angiogenesis and epigenetics, as CHD7 is a chromatin remodeler known to “fine-tune” gene expression(28). Prior reports have demonstrated that histone demethylase Jumonji domain-containing proteins are regulated by hypoxia and can modulate angiogenesis in cancers including GBM(47). However, hypoxia-independent microenvironment-mediated mechanisms to regulate angiogenesis via epigenetic alterations are virtually unknown. Hypoxia in vivo will often accompany lower pH and glucose levels, but the potential for isolated microenvironmental conditions exists(16). Our finding that CHD7 can be repressed by acidic stress alone or ischemia is therefore unique and highlights the importance of further defining the molecular targets of CHD7, including CHI3L1, that mediate the angiogenic effect. Novel understanding of how ischemia affects angiogenesis is critical for developing new treatments for highly necrotic tumor types such as GBM and for recovery from injury, such as stroke or aneurysm.
MATERIALS AND METHODS
Cell Culture/Patient Specimens
Glioblastoma tumor initiating cells were isolated via enzymatic dissociation of patient-derived xenografts (PDX) and maintained in culture as neurospheres in Dulbecco’s Modified Eagle Medium (DMEM)/F12 supplemented with Gem21 without Vitamin A, 1% penicillin/streptomycin, 1% sodium pyruvate, and 10 μg of recombinant human epidermal growth factor (EGF) and fibroblast growth factor (FGF) basic. Cells were maintained in an incubator at 37°C with or without modulation of oxygen tensions and pH as indicated. De-identified glioma tissue samples from human patients were provided as paraffin-embedded slides.
Viral Infection and Vectors
293T cells were plated 24 hours before transfection at 80% confluency. Using Fugene HD, cells were transfected using pGIPZ (Dharmacon) lentiviral vectors encoding CHD7 shRNA along with PAX2 (Addgene #12260) and VSVG (Addgene #8454) vectors in penicillin/streptomycin free media. 24 hours after transfection, media was replaced with fresh media containing antibiotics. Virus was harvested 72 hours after transfection, filtered with .45 micron syringe filters, and titered. To infect, viral media was added to cells that had been plated 24 hours earlier, and replaced with fresh media 24 hours after adding viral media. Cells were grown for 72 hours after infection before adding puromycin for selection.
RNA and Protein Expression
RNA was extracted from glioma cells using TRIzol reagent (ThermoFisher). To generate cDNA from extracted RNA, a reverse transcriptase kit (Bio-Rad) using random primers was employed. CHD7 was detected via quantitative Polymerase Chain Reaction (qPCR) using a set of primers specific for CHD7 (Bio-Rad qHsaCID0013442; Amplicon Context Sequence: GACAGAGTTGAACGTGGTTGTGTATCATGGGAGTCAAGCTAGTCGTCGGACCATTCAGTTGTATGAAATGTACTTCAAAGATCCCCAGGGTCGAGTGATAAAGGGGTCCTATAAGTTTCATGCCATCATCACTACATTTGAGATGATTTTGACTGATTGT). To measure protein expression of CHD7, cells were lysed, and cytoplasmic and nuclear compartment cell fractions were collected using NE-PER kit (ThermoFisher). Cell lysate was quantified using DC Protein Assay Kit (Bio-Rad) and run on SDS-PAGE gradient gel, and the blot was probed with antibody specific for CHD7 (Novus) or Lamin-B1 (Cell Signaling).
Epigenetics RNA Expression Array
GBM cells from the 1016 PDX line were grown in atmospheric oxygen tension (21% O2), hypoxia (5% O2), or acidic stress (6.5) for 72 hours with media changed once every 24 hours. RNA was extracted, cDNA generated using illustra RNAspin Mini Kit (GE) and iScript cDNA Synthesis Kit (Bio-Rad) and quantified using a Human Epigenetic Chromatin Remodeling Factor RNA expression array (Qiagen).
RNA sequencing
Human neural progenitor cells (Millipore Sigma) and D456 GBM PDX cells were transduced with lentivirus encoding either for a control shRNA or shRNA targeting CHD7 mRNA. Following selection for 72 hours with puromycin, cells were collected and RNA was extracted using Norgen Total RNA Purification Kit including DNAse treatment (cat #37500). RNA-sequencing libraries were constructed from polyA mRNA using the NEBNext polyA isolation module (cat# E7490). Libraries were constructed from 800 ng of polyA RNA using the NEBNext Ultra RNA library kit for Illumina (cat# E7530). Each of the twelve libraries was barcoded using the Illumina Multiplex oligos (cat # E7335) and pooled before sequencing on one lane of HiSeq 2500. Raw reads were aligned to hg37 using STAR with default settings implemented in our published primary analysis pipeline(48). Raw counts were analyzed using DESeq2(49) to identify genes differentially expressed in all CHD7 knockdown experiments compared to the non-targeting controls in NPCs and D456 individually (Supplemental Table 1). Raw data and processed count tables are available at GEO GSE119688. Ingenuity Pathway Analysis (IPA) (Qiagen) was used for gene ontology analysis.
In Silico Analysis
Correlations between patient survival and CHD7 gene expression were plotted in Kaplan-Meier survival curves (top vs. bottom quartile) using data from the National Cancer Institute’s Repository for Molecular Brain Neoplasia Data (REMBRANDT). CHD7 RNA expression was also plotted in GBM vs lower grade gliomas, primary vs recurrent GBM, and GBM vs Neural Stem Cells using the Lee Brain, Bredel Brain, Freije Brain, Sun Brain, and Murat Brain and downloaded via the Oncomine database (Thermo Fisher, www.oncomine.org). Additional datasets were analyzed and downloaded using Gliovis (http://gliovis.bioinfo.cnio.es/)(50). Sample sections with IHC for CHD7 were downloaded via the Human Protein Atlas (http://www.proteinatlas.org/).
Tube Formation Assay
D456 human GBM PDX or human neural progenitor cells (Millipore Sigma) were stably infected with shRNAs targeting CHD7 or non-targeting controls and validated for decreased CHD7 RNA and protein expression. Cells were plated on Geltrex matrix in co-culture with equal numbers of human brain microvascular endothelial cells (HBMEC; ATCC) and allowed to grow for 24 hours in HBMEC media. Cells were imaged for tube formation using EVOS FL Auto (ThermoFisher) microscope and tubes were quantified using ImageJ software angiogenesis analyzer plugin.
In Vivo Tumor Initiation
All tumor initiation studies were performed in accordance with UAB IACUC guidelines and requirements and were initiated in athymic BALB/c nu/nu mice housed in a UAB animal facility. Subcutaneous tumor formation including xenograft propagation was initiated using GBM PDX cell lines that were dissociated into single cells and counted. 1 × 106 cells were injected subcutaneously in the right flank. Intracranial tumors were initiated after 72 hours of infection with lentiviral vectors followed by 72 hours of selection with 1 μg/mL puromycin. After dissociation and counting, 500 and 5,000 cells were implanted in the left frontal lobe of the mice. Mice were maintained up to 9 weeks or until the development of neurological symptoms of a tumor. Brains were collected from euthanized mice and fixed in 10% formalin for 18 hours, and paraffin embedded.
Immunohistochemistry
Specific antibodies for CHD7 (Novus) and CD31 (Abcam) were used for immunohistochemical detection in tissue specimens. Sections were developed using Vector ImmPACT NovaRED chromogen reagent.
Statistical Analysis
All statistics were performed with GraphPad Prism Version 7 (GraphPad Software Inc., La Jolla, CA) unless otherwise noted. One-way ANOVA and multiple t-tests were performed with Dunnett correction for multiple comparisons, and p values were adjusted to account for multiple comparisons with a confidence level of 95% and significance of 0.05.
Supplementary Material
SIGNIFICANCE STATEMENT.
Inactivating mutations in the epigenetic modifier chromodomain helicase DNA binding protein 7 (CHD7) are associated with CHARGE syndrome, a disorder causing Coloboma, Heart defects, Atresia choanae, growth Retardation, Genital abnormalities, and Ear abnormalities. We find for the first time that ischemic microenvironments repress CHD7 levels in neural progenitors and brain tumor initiating cells/cancer stem cells. Our novel results also demonstrate that reduced levels of CHD7 in neural progenitors or brain tumor initiating cells increase angiogenesis. We believe our data may have important implications for development, stem cells, and cancer.
Acknowledgments
Financial Support: This work was supported by National Institutes of Health grant R21NS096531, R01NS104339 (ABH), F31CA200085 (NHB), the UAB Brain Tumor SPORE Career Development Award, a pilot award from the UAB-HudsonAlpha Center for Genomic Medicine, and startup funds from the University of Alabama at Birmingham. These startup funds include contributions from the Department of Cell, Developmental and Integrative Biology, the Comprehensive Cancer Center, the Civitan International Research Center for Glial Biology in Medicine, the Center for Free Radical Biology, and the Neuro-Oncology Brain SPORE. Additional support is provided by P30 NS47466 (UAB Neuroscience Molecular Detection Core) and P30 AI027767 (Center for AIDS Research Flow Cytometry Core).
REFERENCES
- 1.Wen PY, Kesari S, Malignant gliomas in adults. The New England Journal of Medicine 359, 492–507 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Fuller GN, Scheithauer BW, The 2007 Revised World Health Organization (WHO) Classification of Tumours of the Central Nervous System: newly codified entities. Brain pathology 17, 304–307 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R et al. , Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10, 459–466 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Noushmehr H et al. , Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turcan S et al. , IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brennan CW et al. , The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips HS et al. , Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9, 157–173 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Verhaak RG et al. , Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel AP et al. , Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z et al. , Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flavahan WA et al. , Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci 16, 1373–1382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hjelmeland AB et al. , Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ 18, 829–840 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filatova A, Acker T, Garvalov BK, The cancer stem cell niche(s): the crosstalk between glioma stem cells and their microenvironment. Biochim Biophys Acta 1830, 2496–2508 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Calabrese C et al. , A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Heddleston JM et al. , Glioma stem cell maintenance: the role of the microenvironment. Curr Pharm Des 17, 2386–2401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukumura D et al. , Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res 61, 6020–6024 (2001). [PubMed] [Google Scholar]
- 17.Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN, The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8, 3274–3284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li P, Zhou C, Xu L, Xiao H, Hypoxia enhances stemness of cancer stem cells in glioblastoma: an in vitro study. Int J Med Sci 10, 399–407 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG, Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol 177, 1491–1502 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soeda A et al. , Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 28, 3949–3959 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Bao S et al. , Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 66, 7843–7848 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Hamerlik P et al. , Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. The Journal of Experimental Medicine 209, 507–520 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venneti S, Thompson CB, Metabolic modulation of epigenetics in gliomas. Brain Pathol 23, 217–221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malzkorn B, Wolter M, Riemenschneider MJ, Reifenberger G, Unraveling the glioma epigenome: from molecular mechanisms to novel biomarkers and therapeutic targets. Brain Pathol 21, 619–632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall JA, Georgel PT, CHD proteins: a diverse family with strong ties. Biochem Cell Biol 85, 463–476 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Marfella CG, Imbalzano AN, The Chd family of chromatin remodelers. Mutat Res 618, 30–40 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schnetz MP et al. , Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res 19, 590–601 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schnetz MP et al. , CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet 6, e1001023 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marie C et al. , Oligodendrocyte precursor survival and differentiation requires chromatin remodeling by Chd7 and Chd8. Proc Natl Acad Sci U S A 115, E8246–E8255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergman JE et al. , CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. Journal of medical genetics 48, 334–342 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Hsu P et al. , CHARGE syndrome: a review. J Paediatr Child Health 50, 504–511 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Colbert LE et al. , CHD7 expression predicts survival outcomes in patients with resected pancreatic cancer. Cancer Res 74, 2677–2687 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim MS, Chung NG, Kang MR, Yoo NJ, Lee SH, Genetic and expressional alterations of CHD genes in gastric and colorectal cancers. Histopathology 58, 660–668 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Tahara T et al. , Colorectal carcinomas with CpG island methylator phenotype 1 frequently contain mutations in chromatin regulators. Gastroenterology 146, 530–538 e535 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Badodi S et al. , Convergence of BMI1 and CHD7 on ERK Signaling in Medulloblastoma. Cell Rep 21, 2772–2784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Engelen E et al. , Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet 43, 607–611 (2011). [DOI] [PubMed] [Google Scholar]
- 37.de la Cuesta F et al. , Kalirin and CHD7: novel endothelial dysfunction indicators in circulating extracellular vesicles from hypertensive patients with albuminuria. Oncotarget 8, 15553–15562 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez-Perri JI, Acevedo JM, Wappner P, Epigenetics: new questions on the response to hypoxia. Int J Mol Sci 12, 4705–4721 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia X et al. , Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U S A 106, 4260–4265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pollard PJ et al. , Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem J 416, 387–394 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Yang J et al. , Role of hypoxia-inducible factors in epigenetic regulation via histone demethylases. Ann N Y Acad Sci 1177, 185–197 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Uhlen M et al. , Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Francescone RA et al. , Role of YKL-40 in the angiogenesis, radioresistance, and progression of glioblastoma. J Biol Chem 286, 15332–15343 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iwamoto FM et al. , Serum YKL-40 is a marker of prognosis and disease status in high-grade gliomas. Neuro Oncol 13, 1244–1251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanwar MK, Gilbert MR, Holland EC, Gene expression microarray analysis reveals YKL-40 to be a potential serum marker for malignant character in human glioma. Cancer Research 62, 4364–4368 (2002). [PubMed] [Google Scholar]
- 46.Dvorak HF, Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. The New England Journal of Medicine 315, 1650–1659 (1986). [DOI] [PubMed] [Google Scholar]
- 47.Ueda J et al. , The hypoxia-inducible epigenetic regulators Jmjd1a and G9a provide a mechanistic link between angiogenesis and tumor growth. Mol Cell Biol 34, 3702–3720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alonso A et al. , aRNApipe: a balanced, efficient and distributed pipeline for processing RNA-seq data in high-performance computing environments. Bioinformatics 33, 1727–1729 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M, GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol 19, 139–141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.