

Abstract
When activating muscles, motor neurons in the spinal cord also activate Renshaw cells, which provide recurrent inhibitory feedback to the motor neurons. The tight coupling with motor neurons suggests that Renshaw cells have an integral role in movement, a role that is yet to be elucidated. Here we used the selective expression of the nicotinic cholinergic receptor α2 (Chrna2) in mice to genetically target the vesicular inhibitory amino acid transporter (VIAAT) in Renshaw cells. Loss of VIAAT from Chrna2Cre-expressing Renshaw cells did not impact any aspect of drug-induced fictive locomotion in the neonatal mouse or change gait, motor coordination, or grip strength in adult mice of both sexes. However, motor neurons from neonatal mice lacking VIAAT in Renshaw cells received spontaneous inhibitory synaptic input with a reduced frequency, showed lower input resistance, and had an increased number of proprioceptive glutamatergic and calbindin-labeled putative Renshaw cell synapses on their soma and proximal dendrites. Concomitantly, Renshaw cells developed with increased excitability and a normal number of cholinergic motor neuron synapses, indicating a compensatory mechanism within the recurrent inhibitory feedback circuit. Our data suggest an integral role for Renshaw cell signaling in shaping the excitability and synaptic input to motor neurons.
SIGNIFICANCE STATEMENT We here provide a deeper understanding of spinal cord circuit formation and the repercussions for the possible role for Renshaw cells in speed and force control. Our results suggest that while Renshaw cells are not directly required as an integral part of the locomotor coordination machinery, the development of their electrophysiological character is dependent on vesicular inhibitory amino acid transporter-mediated signaling. Further, Renshaw cell signaling is closely associated with the molding of motor neuron character proposing the existence of a concerted maturation process, which seems to endow this particular spinal cord circuit with the plasticity to compensate for loss of the Renshaw cell in adult circuit function.
Keywords: ChAT, Chrna2, mouse, spinal cord, VIAAT
Introduction
The precise activation of muscles by motor neurons (MNs) is a core component of movement in vertebrates (Kullander, 2005). In parallel with sending the activating signal to muscles, motor neurons activate inhibitory interneurons called Renshaw cells (RCs) through axon collaterals. Renshaw cells in turn send a recurrent inhibitory signal to the motor neuron (Renshaw, 1946; Eccles et al., 1954). The close relationship of Renshaw cells with motor neurons through recurrent inhibitory circuits suggests an integral function of Renshaw cells for motor neuron activity, but their specific role for movement remains poorly understood (Windhorst, 1996; Alvarez and Fyffe, 2007).
In rodents and cats, Renshaw cells are rhythmically active during fictive locomotion (McCrea et al., 1980; Nishimaru et al., 2006). Motor neurons constitute the main source of excitatory inputs to Renshaw cells during locomotor activity, and blocking this input with the nicotinic receptor antagonist mecamylamine limits the spiking activity of Renshaw cells (Noga et al., 1987; Nishimaru et al., 2006). Under these conditions, a lowered fictive locomotor frequency in the mouse and an increased number of spikes generated by motor neurons per burst in the cat is observed (Noga et al., 1987; Nishimaru et al., 2006). A similarly reduced locomotor frequency was observed when motor neuron input to Renshaw cells was removed in choline acetyltransferase (ChAT) knock-out mice and when the so-called V1 neurons (a diverse population of spinal interneurons that include Renshaw cells) were ablated, both locomotor frequency and motor neuron spiking were affected (Myers et al., 2005; Gosgnach et al., 2006). These studies infer a role for Renshaw cells in the regulation of locomotor frequency and motor neuron spiking; however, the methodologies used affected additional spinal populations. Thus, firm conclusions regarding the specific role of Renshaw cells are therefore difficult to make (Alvarez and Fyffe, 2007; Goulding, 2009; Brownstone and Bui, 2010).
To address the particular role of Renshaw cells in the development and function of locomotor circuitry, we used a transgenic mouse that specifically targets Renshaw cells expressing the nicotinic cholinergic receptor α2 (Chrna2; Perry et al., 2015). We crossed the Chrna2Cre mouse with a mouse carrying a conditional allele for the transporter essential to load GABA and glycine into synaptic vesicles, the vesicular inhibitory amino acid transporter (VIAAT) Slc32a1 (Tong et al., 2008), and studied the phenotypic consequences and possible alterations in the motor neuron–Renshaw cell circuit.
Materials and Methods
Mice.
Chrna2Cre mice, Viaatlx/lx mice and R26.lsl.tdTomato mice have been described previously (Tong et al., 2008; Madisen et al., 2010; Leão et al., 2012; Perry et al., 2015). All animal procedures were approved by the appropriate local Swedish ethical committee (permit C248/11), and experiments were performed in accordance with the Swedish guidelines. Animals of either sex were maintained on a mixed 129/Sv;C57BL/6 background and housed under the approval of the animal care facility of Uppsala University. Chrna2Cre− littermates (Viaatlx/lx) were used as the control animals. Tail tips from newborn mice were routinely collected for DNA extraction followed by genotyping using PCR.
In situ hybridization and immunofluorescence.
In situ hybridization and immunohistochemistry were performed as previously described on lumbar (L) spinal cord tissue from 3-week-old mice (Enjin et al., 2010). The Viaat (SLC32A1) cRNA probe covered nucleotides 588–2072 (GeneID: 22348/NM_009508.2). The following antibodies were used in the immunohistochemistry procedures: polyclonal goat anti-ChAT (1:100; AB144P, Millipore); polyclonal goat anti-vesicular acetylcholine transporter (VAChT; 1:500; AB1578, Millipore); polyclonal rabbit and guinea pig anti-vesicular glutamate transporter 1 (VGluT; both at 1:300; Fujiyama et al., 2001); polyclonal rabbit anti-calbindin (1:5000; CB38, Swant); monoclonal mouse anti-VIAAT (1:500; 131 011, Synaptic Systems); and monoclonal mouse anti synaptophysin (1:500; Sigma-Aldrich). Images were captured on a Zeiss LSM Meta 510 confocal microscope and analyzed using ImageJ software.
Quantification of synapses was performed on 14 μm sections of lumbar spinal cord from 3-week-old mice. Only motor neurons located in the lateral motor column (LMC) were analyzed and putative gamma-motor neurons, defined by a small soma area and the absence of C-boutons (Kanning et al., 2010), were excluded from analysis. Synapses, defined by the expression of specific synapse markers coexpressed with synaptophysin, located on motor neuron soma and proximal dendrites (up to 20 μm from soma) were manually counted in a z-stack, captured with a 63×, 1.4 numerical aperture objective at a resolution of 1024 × 1024. Quantification of the number of calbindin-expressing cells and coexpression of Calbindin and Viaat mRNA was performed on 7 μm z-stacks from lumbar spinal cord.
Electrophysiology.
Spinal cord tissue from control (Viaatlx/lx) and Chrna2Cre; Viaatlx/lx mice [postnatal day 0 (P0) to P7] were prepared as previously described (Perry et al., 2015) with modifications to slicing thickness (270–300 μm). Slices were collected from the entire length of the lumbar region and incubated for 45 min to 1 h in artificial CSF (aCSF) containing (in mm) 128 NaCl, 4 KCl, 0.5 NaH2PO4, 21 NaHCO3, 30 d-glucose, 1.5 CaCl2, and 1 MgSO4, equilibrated with 95% O2 and 5% CO2, at 35°C and subsequently held at room temperature (22–24°C) during electrophysiological recordings. The spinal cord slices were placed into the recording chamber and superfused with oxygenated aCSF at a rate of 2–4 ml/min. Patch electrodes (3–9 MΩ) from borosilicate glass capillaries (GC150F-10, Harvard Apparatus) pulled on a PC-10 gravitational pipette puller (Narishige) contained a K+-based internal solution containing the following (in mm): 130 K-gluconate, 7 NaCl, 10 HEPES, 0.1 EGTA, 0.3 MgCl2, 2 ATP, and 0.5 GTP, with pH adjusted to 7.2 using KOH with an osmolarity between 280 and 300 mOsm/l. The liquid junction potential was calculated as 14.4 mV using Clampex software version 10.2.
Motor neurons, identified by their stereotypical morphology, and Renshaw cells, identified by RFP expression and ventral horn location, were visualized on an Olympus BX51WI Microscope fitted with infrared differential interference contrast optics and a Lambda LS Xenon Arc Lamp (Sutter Instruments) for fluorescent light. Ventral roots were positioned into glass suction electrodes, and Renshaw cell firing was confirmed through an antidromic response to ventral root stimulation, where stimulation was ∼1.5× threshold (A360 Stimulus Isolator, World Precision Instruments).
Whole-cell current-clamp recordings from identified motor neurons and Renshaw cells were made using a Multiclamp 700B or an Axopatch 200B amplifier (Molecular Devices) and digitalized with a data acquisition card (National Instruments), low-pass filtered at 4 or 5 kHz, digitized at 10 kHz, and acquired in WinWCP software (Dr. J. Dempster, University of Strathclyde, Glasgow, UK), AxoGraph X (Molecular Devices) and/or MATLAB (MathWorks). Electrophysiological data were analyzed in Axograph X or MATLAB.
A small hyperpolarizing bias current was used to maintain a resting membrane potential of −60 mV for motor neurons. Renshaw cells were voltage clamped at −60 mV. Motor neurons and Renshaw cells with a stable resting membrane potential lower than −45 mV were included in analysis. Action potentials (APs) elicited from depolarizing current pulses (5 pA increments, 20 ms) or a suprathreshold current injection (3 nA, 2 ms; Nakanishi and Whelan, 2010) from resting potential were analyzed for AP and afterhyperpolarization (AHP) parameters, as follows: amplitude, half-width (50% of spike amplitude or 50% of negative peak amplitude from onset baseline), rise (from 10% to 90% of peak), location (time at which peak amplitude occurs), and onset (at 5% of negative peak amplitude). The AHP time to peak was calculated as the location of the peak minus the AHP onset. Rheobase was noted as the minimum depolarizing injected current (motor neurons; 20 pA increments, 25 ms: Renshaw cells 5 pA increments, 20 ms) sufficient to evoke an action potential. The AP threshold potential was measured from the first AP fired and noted as the point when the increase in potential exceeds >50 mV/ms. Motor neuron input resistance was calculated from the average response to a hyperpolarizing current (−50 pA, 500 ms, 20 repetitions). Depolarizing current steps (−300 to +400 pA, 50 pA increments, 1 s duration) were used to record AP firing frequency (calculated from the last 500 ms of a 1 s current step) and initial doublet distance [400 pA (MN) and 100 pA (RC). The initial (maximal) firing frequency (in hertz) was defined as the inverse of the first three interspike intervals during a 50/100/250 pA current step. The steady-state firing frequency (in hertz) was defined as the average of the inverse of the last three interspike intervals in a 50/100/250 pA current step. The percentage increase or decrease in Renshaw cell and motor neuron properties from control was calculated by dividing the calculated difference between control and Chrna2Cre; Viaatlx/lx values by the control value for each parameter. For recordings of miniature IPSCs (mIPSCs), a cesium chloride-based internal solution (in mm: 120 CsCl, 4 NaCl, 4 MgCl2, 0.001 CaCl2, 10 HEPES, 3 Mg-ATP, 0.3 GTP-Tris, and 10 EGTA) was used, with the chloride reversal potential close to zero (Cl− 0.6 mV). Tetrodotoxin (TTX; 1 μm; Tocris Bioscience) was routinely added to the aCSF. Free-run traces of 60 s were collected using AxoGraph (Molecular Devices), and mIPCS were detected based on threshold and waveform using MATLAB (version 2013a, MathWorks). The instantaneous frequency (1/interspike interval), amplitude, and rise time of mIPSCs were analyzed before and after the addition of the GABAA receptor antagonist picrotoxin (PTX; 10 μm; Tocris Bioscience). Sample means are plotted as box plots and reported as the mean ± SEM. Data larger than q3 + 1.5 × (q3 − q1) or smaller than q1 − 1.5 × (q3 − q1), with q1 and q3 denoting the 25th and 75th percentiles (see box plots), was considered an outlier and discarded.
Extracellular electrophysiology.
P0–P2 (fictive locomotion) or P5–P6 (stretch reflex) mice were anesthetized with isoflurane, decapitated, eviscerated, and submerged in ice-cold dissection buffer containing the following (in nm): 128 NaCl, 4.69 KCl, 25 NaHCO3, 1.18 KH2PO4, 3.5 MgSO4, 0.25 CaCl2, and 22 d-glucose (equilibrated with 95% O2 and 5% CO2). The spinal cord was carefully dissected out of the spinal column and placed in a Sylgard-coated chamber. Spinal cords for stretch reflex experiments were hemisected along the midline using a tungsten needle. The spinal cord was then left for >15 min to recover while the chamber was superfused with aCSF containing the following (in nm): 128 NaCl, 4.69 KCl, 25 NaHCO3, 1.18 KH2PO4, 1.25 MgSO4, 2.5 CaCl2, and 22 d-glucose (equilibrated with 95% O2 and 5% CO2).
For stretch reflex experiments, dorsal and ventral L2 or L3 roots were placed into glass suction electrodes. Dorsal roots were stimulated using a stimulus isolator unit (A365, World Precision Instruments) with current pulses of increasing intensity (0–30 μA, 0.2 ms pulses) until maximal reflex amplitude was reached. During experiments, stimulus intensity was increased to 1.5× the intensity that generated the maximal response (7.5–45 μA), and stimulus pulses were given every 30 s. For synaptic depression experiments, 10 1.5× intensity pulses were delivered at 1 and 8 Hz. Recordings were made with alternating current filtered at 0.1–1000 Hz, amplified (1000×), and digitized at 10 kHz with a Digidata 1440A using pClamp version 10.2 software (Molecular Devices).
Fictive locomotion experiments were performed as previously described (Rabe et al., 2009; Andersson et al., 2012). Spinal cords were placed in the recording chamber and continually superfused with aCSF, and fictive locomotion was induced by the addition of NMDA (5 μm) plus serotonin (5-HT; 10 μm) to the perfusate (Wegmeyer et al., 2007). For experiments with increasing NMDA concentration, 50 μm dopamine was also included. Recorded compound action potentials were bandpass filtered (100–10,000 Hz) and digitized using AxoScope version 8.1 (Molecular Devices). For mecamylamine experiments, mecamylamine (50 μm) was added in combination with the neurochemicals after 20 min of stable locomotion had been recorded. The spinal cord was exposed to mecamylamine for 50 min before recording. After recording, the spinal cord was perfused with NMDA and 5-HT for 50 min before recording the washout traces. There was little or no recovery after washout, as has previously been described (Mentis et al., 2005).
Data analysis.
Recordings were analyzed using Clampfit version 10.2 (Molecular Devices). Stretch reflex experiments were analyzed off-line by averaging 18–20 traces (deviant spontaneous events were excluded from the averages). The latency was defined as the time between stimulus artifact to first deflection in potential (monosynaptic) and second deflection potential (polysynaptic). The amplitude was measured as the highest peak following the monosynaptic and polysynaptic deflections. The polysynaptic area was measured from the second deflection and the following 80 ms between the response and baseline. For synaptic depression experiments, the maximal monosynaptic amplitude was measured after each stimulus pulse. The monosynaptic amplitudes were normalized relative to the first amplitude.
Fictive locomotion data were analyzed using the MATLAB-based programs SpinalCore and NeuroData (Mor and Lev-Tov, 2007; Zhang et al., 2008). For analysis in SpinalCore, the data were downsampled to 250 Hz and rectified. A Morlet wavelet transform was used to extract the phase and frequency throughout 300 s of data. A continuous band of significant coherence was selected for analysis of mean phase, frequency, and analysis. For analysis in Neurodata, 600 s of data were low-pass filtered at 5 Hz, rectified, and high-pass filtered at 0.01 Hz. The resulting trace was analyzed for burst, interburst, and cycle period duration as described previously (Zhang et al., 2008).
Behavior.
All behaviors were tested on eight control mice (Viaatlx/lx) and nine Chrna2Cre; Viaatlx/lx 8-week-old mice, unless otherwise stated. The experiments were designed with a given maximum time for all experiments. If the maximum time was reached, that animal was excluded from the test. The experimenter was blind to genotype while performing and analyzing the experiments.
Treadmill locomotor experiments were performed and analyzed essentially as described previously (Andersson et al., 2012). Treadmill locomotion was conducted at 20 cm/s, and gait parameters were automatically analyzed using TreadScan software (CleverSys). Outliers and miscalculated steps were recognized by the TreadScan software and were manually removed before statistical comparison.
Accelerating rotarod experiments used a Rotorod device (IITC Life Science) in three trials, each of which was separated by at least 10 min. The rod accelerated from 0 to 45 rpm over 60 s and maintained rotation at maximum speed for another 60 s. In most experiments, the device semiautomatically recorded the speed at which the mouse fell from the rod. Steady-speed rotarod experiments were tested at 5, 10, 15, and 20 rpm. Mice were placed on a still rotarod, and the time they remained on the rotarod while it was moving was recorded. Occasionally, a mouse would fall without the sensors detecting it, in which case the experimenter would register the fall manually. The lag in these instances was not more than 1 s, corresponding to 0.75 rpm. In some instances, a mouse would cling to the rods, spin one rotation, and then fall. At this time, the experimenter manually recorded the time of the fall after one full rotation.
Beam walking, the ability to cross a 1 m rounded wooden beam, was tested on 8-week-old mice. First, the mice were given three test trials on a 25-mm-diameter rod. For the experiment, the mice were tested on a 12-mm-diameter rod. The mice were videotaped from behind, and the time to cross the rod was recorded manually. During analysis, the videotape was played in slow motion and the hindpaw foot faults of each mouse were counted. A maximum crossing time of 15 s was used to exclude mice that were not directly crossing the beam.
Ladder walking was performed on an elevated ladder with 30-cm-high plastic glass walls and 4 mm metal rungs separated by 1 cm, as described by Metz and Whishaw, 2009. Each mouse was given three trials to complete the task. For the second round, 21 of the rungs were removed, giving a random spacing (1 and 4 cm) between the rungs. The mice were videotaped from the side during experimentation, and foot slips were analyzed in slow motion.
Maximal grip strength was measured using the Bioseb Grip Strength test (Bio-Gs3, Bioseb). Using their forepaws, mice grasped a wire grid and were pulled back slowly by the experimenter. The force when the mouse released the grid was detected and recorded. Data are presented as the average grip strength across five trials.
Statistical analysis.
All statistical analysis was performed using Prism version 5.0a (GraphPad) or Matlab (R2013b, MathWorks). Normal distribution was assessed by the Shapiro–Wilk statistical test. The null hypothesis, stating that the population is normally distributed, is accepted when α is determined as ≥0.05. All behavioral data were analyzed using a Student's unpaired, two-tailed t test. All histology data were analyzed using a Mann–Whitney test. Circular statistics and locomotor burst parameters were analyzed as described in the studies by Mor and Lev-Tov (2007) and Zhang et al. (2008). The mecamylamine and increasing NMDA concentration experiments were analyzed using a two-way ANOVA, where the interaction and drug effects were considered to be significant at p < 0.05. Active and passive electrophysiological membrane properties, including AP parameters and firing frequency from control and Chrna2Cre; Viaatlx/lx motor neurons and Renshaw cells were analyzed using a Student's unpaired, two-tailed t test, testing whether the data were normally distributed, or the Mann–Whitney test, testing whether the data were not normally distributed. All histological, electrophysiological, and behavioral data represent the mean ± SEM, unless otherwise stated. Significance was determined and reported as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
Results
Chrna2Cre labels Renshaw cell-derived synapses on motor neurons
For all experiments, we used Chrna2Cre mice, which express Cre in defined interneuron populations in the cortex, hippocampus, and spinal cord (Leão et al., 2012; Perry et al., 2015). To visualize the genetically targeted Chrna2 spinal populations, we crossed Chrna2Cre mice with the R26.lsl.tdTomato line. In the resulting mouse, we found that the large majority (94%) of cells in the ventrolumbar spinal cord that expressed Chrna2Cre costained with an antibody for the known Renshaw cell marker calbindin (n = 52 cells; 3 mice, P10-P15; see also Perry et al., 2015). Chrna2Cre; R26.lsl.tdTomato+ boutons that costained with calbindin and the VIAAT protein were prominent in the motor neuron region (Fig. 1B). To corroborate that Chrna2Cre; R26.lsl.tdTomato+ expression was indeed in Renshaw cells, we used antidromic stimulation of ventral roots and simultaneous patching of tomato+ neurons. Such stimulation activated a synaptic current in the Chrna2Cre cells with a short latency (<2.2 ms), similar to the observed latency of Renshaw cells (Mentis et al., 2006; Perry et al., 2015). Application of the nicotinic antagonist mecamylamine (50 μm) reduced the synaptic current induced in Chrna2Cre; R26.lsl.tdTomato+ cells upon ventral root stimulation (Fig. 1C; n = 3 cells). Thus, Chrna2Cre cells in the spinal cord ventral horn have the hallmark properties of Renshaw cells.
Figure 1.

Chrna2Cre labels Renshaw cell-derived synapses on motor neurons. A, Schematic illustration of the recurrent inhibitory (RC–MN) circuit. Calbindin and Chrna2+ Renshaw cells (red) form inhibitory VIAAT synapses on motor neurons and are reciprocally innervated by cholinergic motor neuron axons (blue). B, Immunohistochemistry of calbindin+ (left) and VIAAT+ (right) contacts derived from Chrna2Cre; R26.lsl.tdTomato+ cells on motor neurons (ChAT) in the lumbar spinal cord of adult mice. Inset shows the overlap among VIAAT, calbindin, and Chrna2Cre; R26.lsl.tdTomato. C, A schematic illustration of a hemisected spinal cord slice detailing the antidromic ventral root setup (left) with example traces showing the response of a Chrna2Cre; R26.lsl.tdTomato+ cell to ventral root stimulation in the absence (red) and presence (black) of mecamylamine (right).
Genetic elimination of VIAAT from Renshaw cells alters synaptic inputs on motor neuron soma and proximal dendrite
To explore the contribution of Renshaw cells to the development of motor neurons and motor circuits, we crossed the Chrna2Cre mouse to a mouse carrying an allele flanking the second exon of Viaat with lox sites for conditional deletion of the Viaat gene (Tong et al., 2008). In situ hybridization confirmed that Viaat mRNA was absent from all calbindin-immunoreactive ventral horn neurons in the lumbar spinal cord of 3-week-old Chrna2Cre; Viaatlx/lx mice, whereas in control mice all calbindin-expressing cells costained with Viaat mRNA (Fig. 2A; control mice: 22 cells/6 sections/2 cords; Chrna2Cre; Viaatlx/lx mice: 105 cells/23 sections/2 cords). The number of calbindin+ cells in the lumbar ventral horn was similar between control and Chrna2Cre; Viaatlx/lx mice, suggesting that the elimination of VIAAT has no effect on Renshaw cell survival (control mice: 3.7 ± 0.67 cells/section, n = 6 sections, Chrna2Cre; Viaatlx/lx mice: 4.6 ± 0.60 cells/section, n = 23 sections both from 2 mice, p = 0.70). Expression of the Cre protein in mice carrying the Chrna2Cre allele can be detected at embryonic day 12.5 (data not shown), suggesting that in Chrna2Cre; Viaatlx/lx mice the loss of VIAAT activity is initiated early during development. To examine how the absence of VIAAT-mediated Renshaw cell neurotransmission affects the development of recurrent inhibition circuits, we first quantified the number of synapses on both calbindin+ Renshaw cells and ChAT+ motor neurons (Fig. 2B,C). The number of cholinergic contacts (ChAT and VAChT) on calbindin+ Renshaw cells were unchanged in Chrna2Cre; Viaatlx/lx mice compared with control mice (Fig. 2B; ChAT+: control mice, 4.0 ± 0.81, n = 13 Renshaw cells; Chrna2Cre; Viaatlx/lx mice, 4.2 ± 1.0, n = 6 Renshaw cells both from two mice, p = 0.85; VAChT: control mice, 3.76 ± 0.51, n = 29 Renshaw cells; Chrna2Cre; Viaatlx/lx mice, 3.09 ± 0.51, n = 11 Renshaw cells both from two mice, p = 0.68). Motor neuron collaterals are the sole cholinergic input to Renshaw cells (Zagoraiou et al., 2009), suggesting that motor neurons do not require Renshaw cell-mediated inhibitory feedback (either via GABA or glycine) to develop synapses onto Renshaw cell soma.
Figure 2.

Chrna2Cre; Viaatlx/lx motor neurons develop with altered synaptic inputs. A, In situ hybridization for Viaat (red) combined with an immunohistochemistry for calbindin (green) confirmed that Renshaw cells in the lumbar spinal cord did not express Viaat mRNA in adult Chrna2Cre; Viaatlx/lx mice (right). All control (Viaatlx/lx) calbindin+ Renshaw cells expressed Viaat mRNA (left). B, Immunohistochemistry of calbindin+ Renshaw cells (green) and motor axon collaterals stained by VAChT or ChAT (red), colocalized with synaptophysin (blue, top). Inset and arrows show triple overlap (top) or VAChT+ and calbindin+ signals in close opposition (bottom). Motor neuron innervation of Renshaw cells was unchanged in Chrna2Cre; Viaatlx/lx mice. C, Immunohistochemistry of calbindin+ Renshaw cell synapses (green, arrows) and ChAT+ C-boutons (red) on motor neurons, colocalized with synaptophysin (blue). Inset shows triple overlap. Chrna2Cre; Viaatlx/lx motor neurons had an increased number of calbindin synapses. D, Immunohistochemistry of VGluT1+ contacts (green), colocalized with synaptophysin+ (blue), on ChAT+ motor neuron soma and proximal dendrites (red). Inset and arrows show triple overlap. The number of VGluT1+ synapses was increased in Chrna2Cre; Viaatlx/lx motor neurons. Data represent the mean ± SEM. Mann–Whitney U test, *p < 0.05, ***p < 0.001.
Next, we looked at the synaptic boutons on lumbar LMC motor neuron soma and proximal dendrites. Motor neurons had an increased number of calbindin+/synaptophysin+ contacts on their cell bodies (Fig. 2C; calbindin+: control mice, 2.30 ± 0.29, n = 43 motor neurons; Chrna2Cre; Viaatlx/lx mice, 3.73 ± 0.46, n = 30 motor neurons both from two mice, p = 0.014), which most likely originate from Renshaw cells or possibly interneurons in the dorsal spinal cord (Geiman et al., 2000). Additionally, glutamatergic input, labeled by the VGluT1 and labeling proprioceptive sensory Ia afferents (Betley et al., 2009), was more abundant on Chrna2Cre; Viaatlx/lx motor neurons when compared with motor neurons in control mice (Fig. 2D; control mice: 3.39 ± 0.72, n = 13 motor neurons; Chrna2Cre; Viaatlx/lx mice, 7.24 ± 1.06, n = 17 motor neurons both from two mice, p = 0.001). Last, presumable partition cell synapses immunopositive for ChAT (Zagoraiou et al., 2009) were unchanged in Chrna2Cre; Viaatlx/lx mice (Fig. 2C; ChAT+: control mice, 11.85 ± 0.97, n = 34 motor neurons; Chrna2Cre; Viaatlx/lx mice: 13.36 ± 1.26, n = 33 motor neurons both from two mice, p = 0.53). Thus, VIAAT-mediated Renshaw cell signaling influences the development of synapses to motor neurons.
Normal dorsal root-evoked potentials in Chrna2Cre; Viaatlx/lx mice
To test whether the increased number of excitatory VGluT1+ synaptic boutons on motor neurons translate into an altered motor circuit function, we exploited the sensory–motor circuit where dorsal root stimulation primarily activates VGluT1+ proprioceptive synapses in the monosynaptic time window (the stretch reflex). Lumbar level 2 or 3 dorsal roots were stimulated at 1.5× the intensity that generated a maximal response recorded from the corresponding ventral root (Fig. 3A). Despite the increased number of VGluT1+ synapses, the monosynaptic response in control (n = 12) and Chrna2Cre; Viaatlx/lx (n = 16) motor neurons had a similar peak amplitude (control: 1.70 ± 0. 54 mV; Chrna2Cre; Viaatlx/lx: 1.21 ± 0.24 mV; p = 0.38) and latency (control: 4.78 ± 0.21 ms; Chrna2Cre; Viaatlx/lx: 4.82 ± 0.19 ms; p = 0.90; Fig. 3B,C). Likewise, the activated polysynaptic currents (which are not specific to VGluT1 synapses) were of a similar amplitude (control: 0.39 ± 0.073 mV; Chrna2Cre; Viaatlx/lx: 0.45 ± 0.092 mV; p = 0.67), latency (control: 11.38 ± 1.13 ms; Chrna2Cre; Viaatlx/lx: 11.79 ± 0.72 ms; p = 0.76), and area (control: 13.13 ± 2.60 mV/ms; Chrna2Cre; Viaatlx/lx: 19.90 ± 5.73 mV/ms, p = 0.42) in both Chrna2Cre; Viaatlx/lx and control mice (Fig. 3B,C).
Figure 3.
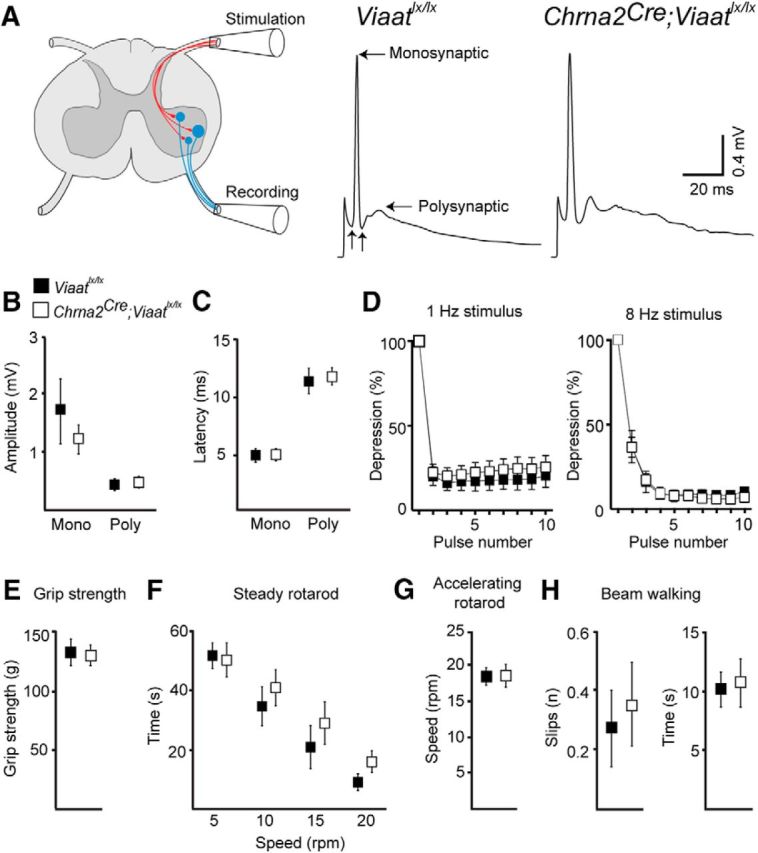
Chrna2Cre; Viaatlx/lx mice had normal stretch reflex parameters and motor behavior despite an increased number of proprioceptive synapses. A, Schematic of the spinal cord illustrating the stretch reflex experimental setup and examples of the averaged ventral root responses to dorsal root stimulation in control Viaatlx/lx (left) and Chrna2Cre; Viaatlx/lx (right) mice. Horizontal arrows indicate monosynaptic and polysynaptic peaks; vertical arrows indicate the onset of monosynaptic and polysynaptic episodes. B, C, The amplitudes (B) and latencies (C) of the monosynaptic and polysynaptic peaks were not altered in neonatal Chrna2Cre; Viaatlx/lx mice. D, Synaptic depression of the monosynaptic amplitude in response to 1 Hz (left) and 8 Hz (right) stimulus volley was not altered in Chrna2Cre; Viaatlx/lx mice. E–H, Chrna2Cre; Viaatlx/lx mice exhibit normal motor behavior across a range of tasks. E, Forepaw grip strength was similar between adult control and Chrna2Cre; Viaalx/lx mice. F, Time to fall on the steady speed rotarod at 5, 10, 15, and 20 rpm was unaffected in Chrna2Cre; Viaatlx/lx mice compared with controls. G, Chrna2Cre; Viaatlx/lx mice maintained the rotational speed of the accelerating rotarod and fell at a similar speed compared with control mice. H, The average number of hindfoot slips (left) and the time to cross (right) a wooden beam were similar between Chrna2Cre; Viaatlx/lx and control mice. Data represent the mean ± SEM. Two-tailed Student's t test.
Stimulating dorsal roots with a low frequency (<16 Hz) leads to significant synaptic depression that translates into a reduced evoked amplitude in motor neurons (Li and Burke, 2001). To test whether the increased number of VGluT1 synapses in Chrna2Cre; Viaatlx/lx mice might have affected synaptic depression, we stimulated dorsal roots at two low frequencies, 1 and 8 Hz. Both stimuli lead to significant depression of a similar magnitude in both mutant (n = 5) and control mice (n = 6; Fig. 3D; 1 Hz: stimulus number, F(9,81) = 259, p < 0.0001; interaction, F(9,81) = 0.42, p = 0.92; 8 Hz: stimulus number, F(9,81) = 288, p < 0.0001; interaction, F(9,81) = 0.19, p = 0.99). Thus, the increased number of VGluT1+ proprioceptive synapses onto motor neurons did not translate into altered motor neuron activation by dorsal root stimulation.
Chrna2Cre;Viaatlx/lx mice performed normally in sensory motor tests
Next, we performed a series of motor behavior tests on 8-week-old Chrna2Cre; Viaatlx/lx (n = 9) and control (n = 8) mice. We found no measureable deficiencies in the forepaw grip strength of Chrna2Cre; Viaatlx/lx mice (Fig. 3E; control mice: 133.2 ± 10.5 g; Chrna2Cre; Viaatlx/lx mice: 130.9 ± 8.1 g; p = 0.84). Further, we investigated the ability of the mice to maintain balance at different speeds (steady rotarod), their ability to maintain balance as speed increases (accelerating rotarod), and their ability for correct foot placement during a balance act (beam and ladder walking). Motor coordination, both balance and foot placement, was not affected in Chrna2Cre; Viaatlx/lx mice when compared with controls (Fig. 3F–H: steady rotarod at 5 rpm (control 51.0 ± 4.4 s, Chrna2Cre; Viaatlx/lx 49.3 ± 6.0 s, p = 0.82), 10 rpm (control mice: 34.2 ± 6.5 s; Chrna2Cre; Viaatlx/lx mice: 40.6 ± 5.7 s, p = 0.47), 15 rpm (control mice: 20.6 ± 7.1 s; Chrna2Cre; Viaatlx/lx mice: 28.4 ± 7.4 s, p = 0.46), and 20 rpm (control mice: 9.3 ± 2.5 s; Chrna2Cre; Viaatlx/lx mice: 15.6 ± 3.7 s, p = 0.19); accelerating rotarod: control mice, 18.4 ± 1.1 rpm, Chrna2Cre; Viaatlx/lx mice, 18.5 ± 1.4 rpm, p = 0.93; beam walk foot slips: control mice, 0.3 ± 0.1 slips; Chrna2Cre; Viaatlx/lx mice, 0.4 ± 0.1 slips, p = 0.67; time control: control mice, 10.1 ± 1.4 s; Chrna2Cre; Viaatlx/lx mice, 10.7 ± 2.0 s, p = 0.82). The number of foot slips on (control mice, 0.9 ± 0.3 slips; Chrna2Cre; Viaatlx/lx mice, 2.2 ± 0.7 slips; p = 0.13) and the time to cross (control mice, 37.1 ± 10.5 s; Chrna2Cre; Viaatlx/lx mice, 51.3 ± 11.4 s; p = 0.39) a 1 m ladder with random distances between rungs were similar between Chrna2Cre; Viaatlx/lx and control mice (Fig. 3H). Thus, VIAAT-mediated Renshaw cell signaling appears dispensable for the development of normal coordination and maximal strength.
The effects of VIAAT-mediated Renshaw cell signaling on motor behavior
To determine whether the absence of VIAAT-mediated Renshaw cell signaling during development alters locomotor behavior in these mice, we performed a detailed analysis of gait. Chrna2Cre; Viaatlx/lx (n = 9) and control (n = 8) mice were placed to run on a clear treadmill at 20 cm/s while being videotaped from underneath. Paw contact with the treadmill and the subsequent gait parameters (stride length, stride frequency, swing, and stance) were semiautomatically analyzed for locomotor behavior (Table 1). All gait parameters were normal in both control and Chrna2Cre; Viaatlx/lx mice, suggesting that the absence of Viaat-mediated Renshaw cell signaling did not affect the development of the neural circuitry required for the maintenance of normal gait.
Table 1.
Locomotor behavior is unaffected in Chrna2Cre;Viaatlx/lx mice
| Forelimbs |
Hindlimbs |
|||||
|---|---|---|---|---|---|---|
| Control | Chrna2Cre; Viaatlx/lx | p Value | Control | Chrna2Cre; Viaatlx/lx | p Value | |
| Stride time (ms) | 264.8 ± 5.9 | 252.5 ± 5.8 | 0.18 | 266.0 ± 5.4 | 269.0 ± 8.2 | 0.77 |
| Stance time (ms) | 149.7 ± 7.6 | 140.9 ± 9.2 | 0.47 | 153.9 ± 5.3 | 152.0 ± 4.8 | 0.80 |
| Swing time (ms) | 115.1 ± 5.3 | 114.1 ± 4.2 | 0.93 | 109.8 ± 4.3 | 112.1 ± 6.1 | 0.64 |
| Break time (ms) | 83.9 ± 2.6 | 83.2 ± 3.4 | 0.34 | 89.2 ± 2.8 | 91.2 ± 3.1 | 0.98 |
| Propulsion (ms) | 66.8 ± 6.9 | 57.6 ± 6.1 | 0.88 | 66.5 ± 6.3 | 66.4 ± 4.0 | 0.76 |
Data represent the mean ± SEM, unless otherwise indicated. Control, n = 9; Chrna2Cre;Viaatlx/lx, n = 8. Removal of Viaat from the recurrent inhibitory circuit does not affect adult locomotor behavior.
Fictive locomotion is normal in the absence of VIAAT-mediated Renshaw cell signaling
The activation of isolated spinal cord central pattern generator (CPG) networks induces a stereotypical pattern of motor activation that generates rhythmic locomotor-like activity in the absence of sensory and supraspinal inputs. To determine whether the intrinsic pattern of locomotion was affected by a loss of Renshaw cell VIAAT-mediated inhibition, we measured locomotor-like activity through the spinal cord ventral roots at L2 (flexion) and L5 (extension). Drug-induced locomotion (5 μm NMDA and 10 μm 5-HT) induced a stable rhythm in both control and Chrna2Cre; Viaatlx/lx spinal cords (Fig. 4A; control, n = 6; Chrna2Cre; Viaatlx/lx, n = 5). The average frequency of fictive locomotor activity was similar in control and Chrna2Cre; Viaatlx/lx mice where a coherency region emerged at 0.4 Hz for both genotypes (Fig. 4B–E; frequency: left (l)/right (r): control, 0.44 ± 0.028 Hz; Chrna2Cre; Viaatlx/lx, 0.41 ± 0.027 Hz, p = 0.63; flexion (f)/extension (e): control, 0.48 ± 0.052 Hz; Chrna2Cre; Viaatlx/lx, 0.43 ± 0.087 Hz, p = 0.61; coherence: l/r: control, 0.87 ± 0.012; Chrna2Cre; Viaatlx/lx, 0.88 ± 0.0098, p = 0.47; f/e: control, 0.86 ± 0.019; Chrna2Cre; Viaatlx/lx, 0.84 ± 0.014, p = 0.41). At this frequency, normal L2 left/right alternation activity and flexion–extension (L2 vs L5) coordination was observed in both control and Chrna2Cre; Viaatlx/lx cords (Fig. 4C). To extend our analysis of the locomotor cycle, we applied a time-series algorithm to look at the individual bursts from the L2 segment. In accordance with the other parameters of fictive locomotion, there was no difference in the average cycle period (control, 2.44 ± 0.16 s; Chrna2Cre; Viaatlx/lx, 2.56 ± 0.19 s; p = 0.66), burst duration (control, 1.16 ± 0.063 s; Chrna2Cre; Viaatlx/lx, 1.19 ± 0.064 s; p = 0.73), or interburst duration (control, 1.28 ± 0.097 s; Chrna2Cre; Viaatlx/lx, 1.34 ± 0.11 s; p = 0.69) between control and Chrna2Cre; Viaatlx/lx mice (Fig. 4F). Additionally, the stability of these parameters was consistent cycle to cycle as the coefficient of variation (CoV) was unchanged in the Chrna2Cre; Viaatlx/lx mutant (Fig. 4G; cycle period: control, 0.065 ± 0.0065; Chrna2Cre; Viaatlx/lx, 0.11 ± 0.020, p = 0.10; burst duration: control, 0.052 ± 0.0052; Chrna2Cre; Viaatlx/lx, 0.066 ± 0.0086, p = 0.22; interburst duration: control, 0.10 ± 0.012; Chrna2Cre; Viaatlx/lx, 0.15 ± 0.033, p = 0.22). Altogether, this suggests that CPG circuits remain activated in Chrna2Cre; Viaatlx/lx mice and maintain a rhythmic bursting similar to that of controls.
Figure 4.

Chrna2Cre; Viaatlx/lx mice exhibit normal locomotor-like activity. A, Schematic drawing of an isolated spinal cord with recording electrodes placed on the l and r lumbar L2 and L5 ventral roots. Raw traces of locomotor-like activity from ventral root recordings from control (Viaatlx/lx) and Chrna2Cre; Viaatlx/lx spinal cords during fictive locomotion. The rectified, smoothened trace (blue) defined the burst, interburst, and cycle periods. B, Coherence power spectra of l/r alternation activity recorded from lL2 vs rL2 ventral roots and f/e alternation from L2 vs L5 ventral roots from control and Chrna2Cre; Viaatlx/lx spinal cords. The coherence power was normalized to a color-graded scale (right). The white boxed region denotes the area used for coherence and frequency analysis; the black line delineates the cone of influence. Calibration: 40 s. C, Circular statistic analysis confirmed an unchanged locomotor pattern in Chrna2Cre; Viaatlx/lx spinal cords; control and Chrna2Cre; Viaatlx/lx mice displayed normal left/right and flexor/extensor alternation. Arrows denote the mean phase value given by the coherence band, where a value near 0° represents synchrony and a value near 180° represents alternation; dashed inner line denotes significance (0.8, Rayleigh test). D, E, The mean locomotor frequency (D) and coherence (E) for both left/right and flexion/extension coordination was unchanged between control and Chrna2Cre; Viaatlx/lx spinal cords. F, The mean cycle period, burst duration, and interburst duration were normal in Chrna2Cre; Viaatlx/lx mice. G, The coefficient of variation (CoV) for each burst parameter; the cycle period, burst duration, and interburst duration were normal in Chrna2Cre; Viaatlx/lx mice. Data represent the mean ± SEM. Two-tailed Student's t test.
The effect of mecamylamine on locomotor frequency during fictive locomotion is not attributable to Renshaw cells
Even if the baseline parameters of fictive locomotion were normal in Chrna2Cre; Viaatlx/lx mice, it remained possible that provocation of the CPG circuit would uncover a contribution from Renshaw cells to fictive locomotion. Since the nicotinic antagonist mecamylamine lowers the fictive locomotor frequency and blocks the activity of Renshaw cells in fictive locomotion (Nishimaru et al., 2006), we hypothesized that the addition of mecamylamine should not affect the frequency of the locomotor rhythm in Chrna2Cre; Viaatlx/lx mice. Surprisingly, the application of mecamylamine reduced the locomotor frequency in spinal cords of Chrna2Cre; Viaatlx/lx mice (n = 9) by a magnitude similar to that of spinal cords in control (n = 8; Fig. 5A–D; frequency: mecamylamine, F(1,15) = 29, p < 0.0001; interaction, F(1,15) = 1.1, p = 0.31; coherence: mecamylamine, F(1,15) = 5.4, p = 0.03; interaction, F(1,15) = 0.020, p = 0.89). This slower frequency was attributed to the burst duration (mecamylamine, F(1,15) = 27, p = 0.0001; interaction, F(1,15) = 0.28, p = 0.60), interburst duration (mecamylamine, F(1,15) = 18, p = 0.0008; interaction, F(1,15) = 0.0028, p = 0.96), and cycle period (mecamylamine, F(1,15) = 21, p = 0.0003; interaction, F(1,15) = 0.037, p = 0.85) of the locomotor pattern being similarly increased during mecamylamine application in both mutant and control spinal cords (Fig. 5E). The coefficient of variation of these variables was similarly influenced by the application of mecamylamine [Fig. 5E; burst duration (mecamylamine, F(1,15) = 11, p = 0.0051; interaction, F(1,15) = 0.74, p = 0.40), interburst duration (mecamylamine, F(1,15) = 9.3, p = 0.0080; interaction, F(1,15) = 0.0025, p = 0.96), and cycle period (mecamylamine, F(1,15) = 11, p = 0.0041; interaction, F(1,15) = 0.083, p = 0.78). Thus, the effect of mecamylamine on fictive locomotion appears to be independent of Renshaw cell function.
Figure 5.
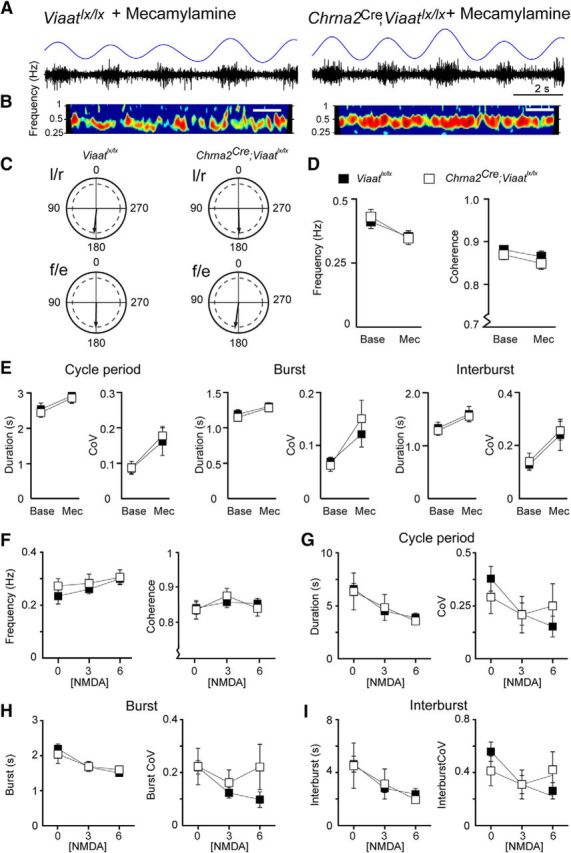
Mecamylamine and NMDA had similar effects on the fictive locomotor rhythm. A, Example raw traces of locomotor-like activity recorded from ventral roots of control (Viaatlx/lx) and Chrna2Cre; Viaatlx/lx spinal cords after the application of mecamylamine (50 μm) to the perfusate. The rectified, smoothened trace is shown in blue. B, Coherence power spectra of l/r alternation activity after exposure to mecamylamine for 50 min. Calibration: 40 s. C, The application of mecamylamine did not alter the locomotor pattern in control or Chrna2Cre; Viaatlx/lx spinal cords; normal left/right and flexor/extensor alternation was maintained. Arrows denote the mean phase value given by the coherence band; the dashed inner line denotes significance. D, Mecamylamine induced a similar reduction in the frequency (left) and coherence (right) in Chrna2Cre; Viaatlx/lx and control spinal cords. E, Mecamylamine (Mec) increased the cycle period, burst, and interburst duration as well as the CoV (right) for these parameters during locomotor-like activity in both control and Chrna2Cre; Viaatlx/lx cords compared with baseline (Base). F, Increasing NMDA concentrations increased the locomotor frequency and coherence in both control and Chrna2Cre; Viaatlx/lx spinal cords. G–I, Increasing NMDA concentrations shortened the locomotor cycle period (G), burst (H), and interburst (I) duration but did not influence the coefficient of variation for these parameters during locomotor-like activity in both control and Chrna2Cre; Viaatlx/lx spinal cords. Data represent the mean ± SEM. Two-tailed Student's t test and two-way ANOVA.
To further probe for a possible function of Renshaw cells during fictive locomotion, we tested increasing excitation levels to mimic increased locomotor speed. To achieve this, we exposed the isolated spinal cords from Chrna2Cre; Viaatlx/lx (n = 6) and age-matched control (n = 5) mice to 10 μm 5-HT and 50 μm dopamine followed by increasing concentrations of NMDA (3 and 6 μm). The addition of NMDA similarly increased the frequency of the locomotor cycle in both genotypes (Fig. 5F; frequency: concentration, F(2,16) = 5.0, p = 0.02; interaction, F(2,16) = 0.53, p = 0.60; coherence: concentration, F(2,16) = 4.2, p = 0.04; interaction, F(2,16) = 1.31, p = 0.30). This faster frequency in response to increasing NMDA concentrations was caused by a shortening of the burst duration, interburst duration, and cycle period in control and Chrna2Cre; Viaatlx/lx mice, while the coefficient of variation for each parameter remained unaffected (Fig. 5G–I; burst duration: concentration, F(2,16) = 15.7, p = 0.0002; interaction, F(2,16) = 0.69, p = 0.52; burst duration CoV: concentration, F(2,16) = 1.4, p = 0.27; interaction, F(2,16) = 0.78, p = 0.48; interburst duration: concentration, F(2,16) = 9.3, p = 0.002; interaction, F(2,16) = 0.21, p = 0.81; interburst duration CoV: concentration, F(2,16) = 3.0, p = 0.08; interaction, F(2,16) = 1.60, p = 0.23; cycle period: concentration, F(2,16) = 13.2, p = 0.0004; interaction, F(2,16) = 0.23, p = 0.80; cycle period CoV: concentration, F(2,16) = 2.9, p = 0.09; interaction, F(2,16) = 1.08, p = 0.36). Thus, we found no evidence that the formation of a functional CPG, including the induction, coordination, and stability of the basic fictive locomotor rhythm, required VIAAT-mediated Renshaw cell signaling.
Motor neurons receive a lower frequency of mIPCSs during the first postnatal days in Chrna2Cre; Viaatlx/lx mice
To examine whether the lack of VIAAT-mediated Renshaw cell signaling onto motor neurons was noticeable in the general spontaneous inhibitory release, we recorded mIPSCs using whole-cell patch clamp in slices from control mice (Viaatlx/lx; postnatal age, 2.4 ± 0.4 d; n = 7) and Chrna2Cre; Viaatlx/lx mice (postnatal age, 1.7 ± 0.6 d; n = 7) = in the presence of TTX (1 μm; Fig. 6). Traces containing miniature synaptic currents were analyzed pairwise before and after the addition of PTX (10 μm). We measured a significantly lower instantaneous mIPSC frequency from motor neurons in slices where Renshaw cells lacked VIAAT compared with control littermates (Fig. 6B,C; control mice, 8.85 ± 1.15 Hz; Chrna2Cre; Viaatlx/lx, 5.56 ± 0.72 Hz; p = 0.032). No statistical difference in instantaneous mIPSC frequency was seen following application of the GABAA receptor antagonist PTX (Fig. 6B,C; control, 8.33 ± 1.39 Hz; Chrna2Cre; Viaatlx/lx, 5.09 ± 1.95 Hz; p = 0.21), indicating that the difference in frequency relies on the glycinergic component of mIPSCs. We did not detect a difference in the average amplitude of mIPSCs (control. 78.29 ± 10.92 pA; Chrna2Cre; Viaatlx/lx, 61,44 ± 7.02 pA; p = 0.22) or after application of PTX (Fig. 6D,E; control, 52.11 ± 9.61 pA; Chrna2Cre; Viaatlx/lx, 63.97 ± 6.68 pA; p = 0.3354). However, the mean mIPSC rise time was slower in Chrna2Cre; Viaatlx/lx mice compared with control littermates (Fig. 6F; control, 3.53 ± 0.12 ms; Chrna2Cre; Viaatlx/lx, 4.06 ± 0.14 ms; p = 0.013). The application of PTX removed this difference in average rise time (control, 3.32 ± 0.02 ms; Chrna2Cre; Viaatlx/lx, 3.34 ± 0.02 ms; p = 0.36), indicating that the remaining glycinergic component of mIPSCs, with a faster rise time (compared with the average rise time when GABAergic and glycinergic mIPSCs are mixed), is not different between control and animals lacking VIAAT in Renshaw cells (Fig. 6F). Therefore, the loss of Renshaw cell inhibition onto motor neurons (P0–P3) resulted in a reduced frequency of inhibitory spontaneous release, and with a component of GABAA receptor-mediated signaling affecting rise time.
Figure 6.

Neonatal lumbar motor neurons receive lower-frequency inhibitory spontaneous release in Chrna2Cre; Viaatlx/lx mice. A, Schematic drawing depicting the lost RC VIAAT-mediated inhibition onto MNs (IaIN, Ia inhibitory interneurons) in Chrna2Cre; Viaatlx/lx mice. B, Example traces of lumbar motor neuron mIPSCs before (left) and following PTX application (right) for control mice (Viaatlx/lx, top) and Chrna2Cre; Viaatlx/lx mice (bottom). C, Box plot of mIPSC frequencies for control (n = 8), control + PTX (n = 7), Chrna2Cre; Viaatlx/lx (n = 7) and Chrna2Cre; Viaatlx/lx +PTX (n = 7). D, Example mIPSCs (50 overlaps in gray and mean in black) are shown before (left) and during (right) PTX application for control mice (top) and Chrna2Cre; Viaatlx/lx mice (bottom). E, Box plot of mIPSC amplitudes. F, Box plot of mIPSC rise times. Data represent the mean ± SEM. Two-tailed Student's t test: *p < 0.05, **p < 0.01, ***p < 0.001.
Impact of Renshaw cells on the development of motor neuron electrical properties
To examine whether VIAAT-mediated Renshaw cell signaling affects the development of motor neuron intrinsic properties, we performed whole-cell patch-clamp experiments from motor neurons in transverse slices from P0–P3 Chrna2Cre; Viaatlx/lx mice (n = 13) and compared the results to age-matched control mice (Viaatlx/lx, n = 9). There was no difference in the membrane potential of motor neurons between control mice (−54.9 ± 1.9 mV) and Chrna2Cre; Viaatlx/lx mice (−54.6 ± 3.4 mV), but there was a significant difference in input resistance (control mice, 119.5 ± 17.4 MΩ; Chrna2Cre; Viaatlx/lx mice, 60.1 ± 5.3 MΩ; p = 0.012). Motor neurons from Chrna2Cre; Viaatlx/lx mice were notably less excitable, although there was no significant difference in the average action potential threshold (control mice, −22.2 ± 2.5 mV; Chrna2Cre; Viaatlx/lx mice, 17.3 ± 3.3 mV; p = 0.247). Further, we found no significant differences in average rheobase current, action potential amplitude, or half-width (Table 2). The membrane AHP was altered, where Chrna2Cre; Viaatlx/lx motor neurons showed a faster AHP onset and shorter duration compared with controls (Fig. 7A, Table 2; AHP rise: control mice, 20.9 ± 1.9 ms; Chrna2Cre; Viaatlx/lx mice, 13.6 ± 0.5 ms; p = 0.013; AHP location: control mice, 134.6 ± 10.0 ms; Chrna2Cre; Viaatlx/lx mice, 97.2 ± 3.8 ms; p = 0.016; AHP onset: control mice, 97.0 ± 7.3 ms; Chrna2Cre; Viaatlx/lx mice, 72.0 ± 2.8; p = 0.027; AHP time to peak: control mice, 37.7 ± 4.0 ms; Chrna2Cre; Viaatlx/lx mice, 25.2 ± 1.5 ms; p = 0.040). Other AHP parameters including amplitude and half-width, were not different between genotypes (Table 2). Thus, due to the lower input resistance, Chrna2Cre; Viaatlx/lx motor neurons required more current input to generate sufficient depolarization to reach action potential threshold.
Table 2.
Electrical properties of the recurrent inhibition circuit are altered in Chrna2Cre;Viaatlx/lx mice
| Motor neurons |
Renshaw cells |
|||
|---|---|---|---|---|
| Control | Chrna2Cre; Viaatlx/lx | Control | Chrna2Cre; Viaatlx/lx | |
| RMP (mV) | −54.9 ± 1.9 | −54.6 ± 3.4 | −51.6 ± 0.8 | −48.2 ± 1.3** |
| Input resistance (Ω) | 119.5 ± 17.4 | 60.1 ± 5.3* | 282.0 ± 31.2 | 423.7 ± 36.6** |
| Rheobase (pA) | 735.4 ± 151.5 | 1205 ± 315.9 | 56.1 ± 4.9 | 39.6 ± 6.7* |
| AP Threshold (mV) | −22.2 ± 2.5 | −17.3 ± 3.3 | −30.3 ± 1.3 | −38.4 ± 1.3*** |
| AP Amplitude (mV) | 54.2 ± 3.3 | 49.3 ± 4.6 | 62.5 ± 3.53 | 64.90 ± 5.31 |
| AP half-width (ms) | 1.2 ± 0.09 | 1.3 ± 0.1 | 2.2 ± 0.2 | 3.0 ± 0.3* |
| AHP amplitude (mV) | −5.9 ± 1.4 | −5.7 ± 1.0 | −9.4 ± 0.8 | −9.4 ± 0.7 |
| Location of peak (ms) | 134.6 ± 10.0 | 97.2 ± 3.8* | 148.8 ± 2.5 | 159.1 ± 4.2* |
| Onset (ms) | 97.0 ± 7.3 | 72.0 ± 2.8* | 127.18 ± 2.1 | 129.34 ± 1.2 |
| AHP rise (ms) | 20.9 ± 1.9 | 13.6 ± 0.5* | 12.3 ± 0.9 | 17.8 ± 1.9** |
| AHP half-width (ms) | 112.0 ± 12.2 | 89.0 ± 6.0 | 63.1 ± 5.7 | 97.8 ± 13.4* |
Data represent the mean ± SEM. Motor neurons: control, n = 13; Chrna2Cre;Viaatlx/lx, n = 9; Renshaw cells: control, n = 13; Chrna2Cre;Viaatlx/lx, n = 13. Removal of Viaat from the recurrent inhibitory circuit alters motor neuron and Renshaw cell electrical properties.
*p < 0.05.
**p < 0.01.
***p < 0.001.
Figure 7.

VIAAT-mediated Renshaw cell signaling is required for the development of motor neuron and Renshaw cell electrical properties. A, Example action potential traces from control (black) and Chrna2Cre; Viaatlx/lx (gray) motor neurons (left) and Renshaw cells (right). Arrows indicate the AP threshold, and dashed gray lines highlight the AHP rise and half-width for the Renshaw cells, while motor neurons required a suprathreshold current injection to compare AHP between genotypes (see inset on right). B, Control (black) and Chrna2Cre; Viaatlx/lx (gray) motor neurons have a similar spike frequency in response to depolarizing injected current. Inset, Average spike frequency (F) for control and Chrna2Cre; Viaatlx/lx mice per injected current block: block A, 0–800 pA; block B, 0–1250 pA; block C, 850–1900 pA; block D, 1300–3500 pA. C, Chrna2Cre; Viaatlx/lx Renshaw cells (gray) had a slower firing frequency than control Renshaw cells (black) in response to increasing depolarizing current steps. D, The percentage change, either increase or decrease, in action potential properties from both Chrna2Cre; Viaatlx/lx motor neurons (dark gray) and Chrna2Cre; Viaatlx/lx Renshaw cells (light gray) compared with control animals. Data represent the mean ± SEM. Two-tailed Student's t test, Mann–Whitney U test: *p < 0.05, **p < 0.01, ***p < 0.001.
Although requiring more depolarizing current to achieve action potentials, a faster repolarization from hyperpolarized potentials (as seen with the AHP quantification) may allow Chrna2Cre; Viaatlx/lx motor neurons to readily return to resting membrane potential and thus still be able to follow high firing frequencies when excited by sufficient input to reach threshold. We sought to elucidate the steady-state firing frequency of motor neurons in response to depolarization; however, due to the variability in the low input resistance of Chrna2Cre; Viaatlx/lx motor neurons, a variable amount of current needed to be injected to generate spike trains. To initially elucidate the motor neuron average firing frequency, we grouped their firing responses over two defined current blocks for control and Chrna2Cre; Viaatlx/lx mice (Fig. 7B, inset; control mice: block A, 0–800 pA; block C, 850–1900 pA; Chrna2Cre; Viaatlx/lx mice: block B, 0–1250 pA; block D, 1300–3500 pA). Motor neuron firing frequency was similar in response to low and high current injection for both genotypes (control mice: block A, 12.5 ± 1.0 Hz; block C, 12.2 ± 0.8 Hz; Chrna2Cre; Viaatlx/lx mice: block B, 9.3 ± 0.4 Hz; block D, 10.3 ± 0.5 Hz), but Chrna2Cre; Viaatlx/lx motor neurons fired significantly slower than control neurons at lower current inputs (Fig. 7B; control mice: block A, 12.5 ± 1.0 Hz; Chrna2Cre; Viaatlx/lx mice: block B, 9.3 ± 0.4 Hz, p = 0.003). As such, we compared the slope of the frequency–current gain (in hertz per picoampere) in response to increased current injections instead of a comparison of the firing frequency at a specific current step. We found no significant difference in slope between control and Chrna2Cre; Viaatlx/lx motor neurons (Fig. 7B; control: 0.0195 ± 0.003 Hz/pA, n = 12; Chrna2Cre; Viaatlx/lx: 0.0125 ± 0.0029 Hz/pA; n = 6; p = 0.151).
Impact of the loss of Renshaw cell VIAAT-mediated signaling on the Renshaw cell itself
To determine whether VIAAT-mediated Renshaw cell signaling affected the electrophysiological development of the Renshaw cell population, we performed patch-clamp experiments from Renshaw cells in transverse slices from P1–P6 Chrna2Cre; Viaatlx/lx mice (n = 13) and compared them to age-matched control mice (n = 13). Passive and active firing properties were examined in a whole-cell current-clamp configuration at rest. The Chrna2Cre; Viaatlx/lx Renshaw cells had a more depolarized resting membrane potential (control, −51.6 ± 0.8 mV; Chrna2Cre; Viaatlx/lx, −48.2 ± 1.3 mV; p = 0.002) and markedly higher input resistances than control neurons (control, 282.0 ± 31.2 MΩ; Chrna2Cre; Viaatlx/lx, 423.7 ± 36.6 MΩ; p = 0.0071). Stepwise injections of depolarizing current (20 ms, 5 pA increments) generated action potentials in Renshaw cells from both Chrna2Cre; Viaatlx/lx and control mice (Fig. 7A), where differences in the action potential shape were apparent. The action potential threshold was more hyperpolarized (control, −30.3 ± 1.3 mV; Chrna2Cre; Viaatlx/lx, −38. 4 ± 1.3 mV; p = 0.0003), which coincided with a significantly reduced rheobase current (control, 56.1 ± 4.9 pA; Chrna2Cre; Viaatlx/lx, 39.6 ± 6.7 pA; p = 0.05) in Chrna2Cre; Viaatlx/lx Renshaw cells (Fig. 7A; Table 2). Like the motor neurons, the AP amplitude was similar between genotypes; however, the AP half-width was significantly wider in Chrna2Cre; Viaatlx/lx Renshaw cells (Fig. 7A; Table 2; control, 2.2 ± 0.2 ms; Chrna2Cre; Viaatlx/lx, 3.0 ± 0.3 ms; p = 0.023). Action potentials from both control and Chrna2Cre; Viaatlx/lx Renshaw cells repolarized with afterhyperpolarization potentials of similar amplitudes (control, −9.4 ± 0.8 mV; Chrna2Cre; Viaatlx/lx, −9.4 ± 0.7 mV; p = 0.98); however, Chrna2Cre; Viaatlx/lx Renshaw cell AHPs had a slower onset and longer duration compared with control neurons (Fig. 7A; Table 2; AHP rise: control, 12.3 ± 0.9 ms; Chrna2Cre; Viaatlx/lx, 17.8 ± 1.9 ms; p = 0.014; AHP half-width: control, 63.1 ± 5.7 ms; Chrna2Cre; Viaatlx/lx, 97.8 ± 13.4 ms; p = 0.022; AHP location: control, 148.8 ± 2.5 ms; Chrna2Cre; Viaatlx/lx, 159.1 ± 4.2 ms; p = 0.046). Thus, Chrna2Cre; Viaatlx/lx Renshaw cells are hyperexcitable and require less depolarizing input to reach the threshold for generating an action potential.
Renshaw cells elicited tonically firing trains of action potentials, triggered by depolarizing current steps (0–250 pA, 50 pA increments, 2 s duration). Loss of VIAAT-mediated signaling affected the frequency at which Renshaw cells responded to increasing depolarization as Chrna2Cre; Viaatlx/lx Renshaw cells fired action potentials at a slower frequency than controls (Fig. 7C). This slower firing frequency was maintained across all depolarizing steps (control vs Chrna2Cre; Viaatlx/lx, respectively: at 50 pA: 13.6 ± 0.6 Hz, 11.0 ± 0.9 Hz, p = 0.03; at 100 pA: 19.1 ± 1.3 Hz, 14.8 ± 1.5 Hz, p = 0.02; at 150 pA: 24.7 ± 0.7 Hz, 17.6 ± 1.5 Hz, p = 0.0003; at 200 pA: 26.5 ± 1.3 Hz, 18.3 ± 2.2 Hz, p = 0.004; at 250 pA: 29.6 ± 1.5 Hz, 21.4 ± 3.0 Hz, p = 0.02). This slower firing in Chrna2Cre; Viaatlx/lx mice could be attributed to the slower AHP in Chrna2Cre; Viaatlx/lx Renshaw cells. To further quantify Renshaw cell firing across increasing depolarizing current steps, the firing frequency was extrapolated from the interspike intervals of the first three (maximal) and the last three (steady state) action potentials in the spike train. A comparison of steady-state and maximal firing frequencies revealed that Renshaw cells from both control and Chrna2Cre; Viaatlx/lx mice adapt their firing frequencies similarly over the course of depolarization (data not shown).
To examine the changes within the recurrent inhibition circuit, we compared motor neurons and Renshaw cells and their relative changes when VIAAT was deleted (Fig. 7D). Interestingly, in many electrophysiological properties the percentage change of Chrna2Cre; Viaatlx/lx motor neurons was matched by an opposite change in Chrna2Cre; Viaatlx/lx Renshaw cells. Most notably, Chrna2Cre; Viaatlx/lx motor neurons had a 49% decrease in input resistance compared with control motor neurons, whereas Chrna2Cre; Viaatlx/lx Renshaw cells had a 50% increase in input resistance. Moreover, Chrna2Cre; Viaatlx/lx motor neurons had a 22% and 35% decreases in action potential threshold and AHP rise, respectively, whereas Chrna2Cre; Viaatlx/lx Renshaw cells had 26% and 44% increases, respectively. Thus, significant cellular changes were observed in both motor neurons and Renshaw cells as a consequence of the VIAAT deletion, and in the majority of altered parameters the changes were reciprocal.
Discussion
Here we report that the developmental removal of VIAAT-mediated Renshaw cell signaling influences the components, but not the output, of the recurrent inhibition circuit. Renshaw cell function has been tightly linked to motor neuron activity for 60 years, but the precise contribution of Renshaw cells to motor control remains obscure. Our analysis of mice deficient in VIAAT-mediated Renshaw cell signaling suggests that Renshaw cells may be involved in the development of an appropriate response to the synaptic input of motor neurons. Interestingly, the absence of VIAAT-mediated Renshaw cell signaling does not translate to deficient motor behavior or physiological activity in spinal circuits.
Genetic elimination of VIAAT-mediated signaling in Renshaw cells
The subtle phenotypic consequences in Chrna2Cre; Viaatlx/lx mice raised concerns about the efficiency of the VIAAT deletion. We validated that the cre–lox combination used in this study resulted in the removal of Viaat mRNA (Fig. 2) and was restricted to Chrna2-expressing ventral horn populations. VIAAT is the primary vesicular transport protein for inhibitory transmitters, and no alternative uptake mechanism for GABA and/or glycine into vesicles is available (Rahman et al., 2015; Aubrey, 2016). Moreover, previous reports demonstrated that genetic deletion of VIAAT resulted in the absence of IPSCs in autaptic cultures (Wojcik et al., 2006) and that Chrna2+ hippocampal interneurons lost their optogenetically induced activity (Leão et al., 2012). When measuring spontaneous inhibitory neurotransmitter release (Fig. 6), we found a lower instantaneous frequency of mIPSCs onto Chrna2Cre; Viaatlx/lx motor neurons, which is indicative of a reduced spontaneous Renshaw cell-mediated inhibition.
Motor circuit and motor behavior development in the absence of Renshaw cell signaling
Due to their tight physiological coupling to motor neurons, Renshaw cells have long been thought to play an integral role in the control of locomotion. Here we demonstrate that genetically inactivating Renshaw cell inhibitory neurotransmitter transport did not result in any abnormalities in fictive locomotion or motor behavior. Fictive locomotion parameters including coordination, frequency, and burst activity, and the cycle-to-cycle variation in these parameters was not changed in Chrna2Cre; Viaatlx/lx mice, suggesting that neonatal locomotion is not modulated by Renshaw cells. Furthermore, if Renshaw cells have an integral role in fictive locomotion, the addition of mecamylamine, which depletes the spiking activity of Renshaw cells (Nishimaru et al., 2006), should have a limited impact on the locomotor rhythm in Chrna2Cre; Viaatlx/lx cords. A comparable decrease in locomotor frequency was observed in both control and Chrna2Cre; Viaatlx/lx mice, suggesting that the mecamylamine effect on fictive locomotion is likely due to the inhibition of nicotinic receptors on cells other than Renshaw cells.
When the NMDA concentration increased, the frequency of the locomotor rhythm increased due to a shortening of motor neuron bursting. Similar burst shortening was seen in Chrna2Cre; Viaatlx/lx ventral roots, suggesting that Renshaw cells do not limit motor neuron bursting during fictive locomotion. Moreover, adult Chrna2Cre; Viaatlx/lx mice behaved normally in all motor tasks, even when challenged, suggesting that Renshaw cells are not required for speed control, adaptation, or motor coordination under these circumstances. We propose these results can be explained either by a redundancy in Renshaw cell function for fictive and physical locomotion or by compensatory mechanisms in the mutant mice.
Compensation in the Renshaw cell–motor neuron circuit
The absence of behavioral deficiencies in Chrna2Cre; Viaatlx/lx mice suggests that the physiological role of Renshaw cells is potentially compensated for during network development. Network connectivity, function, synaptic strength, and excitability are shaped by spontaneous activity in developing circuits throughout spinal cord maturation (Blankenship and Feller, 2010). Moreover, spontaneous activity in embryonic mice, mediated by cholinergic, glycinergic, and GABAergic pathways, shapes the development and characteristics of spinal motor circuits (Hanson and Landmesser, 2003). Neural circuits have impressive capacities to generate “normal” output activity even if circuit components, such as VIAAT-mediated neurotransmission, are removed (for review, see Blankenship and Feller, 2010). Neurons maintain network activity levels within a defined physiological range by adjusting cellular excitability, rhythmogenic capabilities, and the strength of excitatory and inhibitory synaptic inputs (De Zeeuw et al., 2003; Leão et al., 2004; Sapir et al., 2004; Harley et al., 2015). In ovo application of the GABAA receptor antagonist gabazine, initially reduced spontaneous activity that later recovered with alterations to motor neuron excitability (Wilhelm and Wenner, 2008; Wilhelm et al., 2009). Further, a study of a mouse model of amyotrophic lateral sclerosis found that decreased GABA inhibition in the motor cortex was associated to hyperexcitable layer 5 pyramidal neurons and overall cortical excitability (Nieto-Gonzalez et al., 2011). These regulatory mechanisms could support the presumable compensatory changes in the recurrent inhibitory circuit found here; Chrna2Cre; Viaatlx/lx Renshaw cells became hyperexcitable, whereas motor neurons have a lower input resistance, requiring a larger current input to fire; therefore, this may reflect the processes that operate to ensure a robust and stable motor output. This presumable compensation seemed surprisingly complementary; motor neurons and Renshaw cells had relatively comparable and opposite trends in changes to some neural properties that contribute to cellular excitability.
The observed alterations could be explained partly by the significant changes to motor neuron and Renshaw cell input resistance; however, changes to ion channel protein levels and the magnitude and contribution of ionic conductances also contribute to recovering inherent activity (Chub and O'Donovan, 1998; Desai et al., 1999; De Zeeuw et al., 2003; MacLean et al., 2003; Swensen and Bean, 2005; Leão et al., 2010; Wenner, 2014). Although identifying possible ionic changes within the recurrent inhibition circuit was outside the scope of this study, there were notable changes to the composition of the spontaneous inhibitory neurotransmitter release on Chrna2Cre; Viaatlx/lx motor neurons (Fig. 6). This supports the further examination of ion channel contributions and possible modifications in future studies addressing compensation.
Compensatory adaptations in synaptic input
The modified cellular membrane properties within the Chrna2Cre; Viaatlx/lx recurrent inhibitory circuit were coupled to a significant increase in the number of synaptic connections to motor neurons, which could be part of the compensation mechanism to balance the lower input resistance of motor neurons. Chrna2Cre; Viaatlx/lx motor neurons had an increased number of presumable inhibitory calbindin+ contacts and excitatory VGluT1 contacts. Interestingly, despite the increased number of VGluT1 inputs, specific activation of these synapses by dorsal root stimulation generated a motor neuron response of normal amplitude (monosynaptic stretch reflex). This observation lends support to a model where the hypoexcitable motor neurons develop more synapses (presumably stronger synaptic input) to generate normal motor output.
Since Renshaw cells are the predominant source of calbindin+ contacts in the ventral horn (Carr et al., 1998), we find it likely that the increased calbindin+ contacts on motor neurons arise from Renshaw cells. This phenomenon has been observed during disease progression in the Sod1G93A mouse model, where Renshaw cell axonal sprouting gave rise to an increased number of calbindin+ Renshaw cell contacts on motor neurons (Wootz et al., 2013). The number of Renshaw cells appeared unaltered in Chrna2Cre; Viaatlx/lx mice, suggesting that Chrna2Cre; Viaatlx/lx Renshaw cells could have increased calbindin+ axonal connections to strengthen recurrent inhibition. In another mouse model of altered neuronal development, engrailed−/− mice, in which Renshaw cells and Ia inhibitory interneurons fail to develop, had a 50% reduction in the number of calbindin+ Renshaw cell–motor neuron contacts, whereas the total number of inhibitory synapses remained constant (Sapir et al., 2004). Locomotor behavior in engrailed−/− mice was unaffected, similar to Chrna2Cre; Viaatlx/lx mice, illustrating how synaptic changes can be associated with the compensation and maintenance of normal behavior.
Concluding remarks
In recent years, suggestions for the role of Renshaw cells have covered their involvement in speed and force regulation (Kiehn, 2006; Alvarez and Fyffe, 2007) through to sculpting the spiking activity of motor neurons (Pratt and Jordan, 1987; Brownstone and Bui, 2010). Our results support a role for Renshaw cells in modifying motor neuron activity but, in the absence of a locomotor phenotype, do not support a direct role in speed or force regulation. However, we found a presumable complementary compensation between motor neurons and Renshaw cells, which, despite a role for Renshaw cells in locomotor circuits, could underlie the undisturbed locomotor output in Chrna2Cre; Viaatlx/lx mice. In any case, our results highlight that the spinal cord recurrent inhibitory circuit is endowed with several mechanisms to maintain a purposeful output and indicate that further analysis of Renshaw cell function should be addressed using acute silencing.
Footnotes
This work was financed by grants from the Swedish Medical Research Council, Hållsten, Ländells, the Swedish Brain Foundation, the Swedish Foundation for Cooperation in Research and Higher Education, and CNPq-Brasil.
The authors declare no competing financial interests.
References
- Alvarez FJ, Fyffe RE (2007) The continuing case for the Renshaw cell. J Physiol 584:31–45. 10.1113/jphysiol.2007.136200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson LS, Larhammar M, Memic F, Wootz H, Schwochow D, Rubin CJ, Patra K, Arnason T, Wellbring L, Hjälm G, Imsland F, Petersen JL, McCue ME, Mickelson JR, Cothran G, Ahituv N, Roepstorff L, Mikko S, Vallstedt A, Lindgren G, et al. (2012) Mutations in DMRT3 affect locomotion in horses and spinal circuit function in mice. Nature 488:642–646. 10.1038/nature11399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubrey KR. (2016) Presynaptic control of inhibitory neurotransmitter content in VIAAT containing synaptic vesicles. Neurochem Int 98:94–102. 10.1016/j.neuint.2016.06.002 [DOI] [PubMed] [Google Scholar]
- Betley JN, Wright CV, Kawaguchi Y, Erdélyi F, Szabó G, Jessell TM, Kaltschmidt JA (2009) Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell 139:161–174. 10.1016/j.cell.2009.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB (2010) Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci 11:18–29. 10.1038/nrn2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstone RM, Bui TV (2010) Spinal interneurons providing input to the final common path during locomotion. Prog Brain Res 187:81–95. 10.1016/B978-0-444-53613-6.00006-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr PA, Alvarez FJ, Leman EA, Fyffe RE (1998) Calbindin D28k expression in immunohistochemically identified Renshaw cells. Neuroreport 9:2657–2661. 10.1097/00001756-199808030-00043 [DOI] [PubMed] [Google Scholar]
- Chub N, O'Donovan MJ (1998) Blockade and recovery of spontaneous rhythmic activity after application of neurotransmitter antagonists to spinal networks of the chick embryo. J Neurosci 18:294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG (1999) Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci 2:515–520. 10.1038/9165 [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Chorev E, Devor A, Manor Y, Van Der Giessen RS, De Jeu MT, Hoogenraad CC, Bijman J, Ruigrok TJ, French P, Jaarsma D, Kistler WM, Meier C, Petrasch-Parwez E, Dermietzel R, Sohl G, Gueldenagel M, Willecke K, Yarom Y (2003) Deformation of network connectivity in the inferior olive of connexin 36-deficient mice is compensated by morphological and electrophysiological changes at the single neuron level. J Neurosci 23:4700–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Fatt P, Koketsu K (1954) Cholinergic and inhibitory synapses in a pathway from motor-axon collaterals to motoneurones. J Physiol 126:524–562. 10.1113/jphysiol.1954.sp005226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enjin A, Rabe N, Nakanishi ST, Vallstedt A, Gezelius H, Memic F, Lind M, Hjalt T, Tourtellotte WG, Bruder C, Eichele G, Whelan PJ, Kullander K (2010) Identification of novel spinal cholinergic genetic subtypes disclose Chodl and Pitx2 as markers for fast motor neurons and partition cells. J Comp Neurol 518:2284–2304. 10.1002/cne.22332 [DOI] [PubMed] [Google Scholar]
- Fujiyama F, Furuta T, Kaneko T (2001) Immunocytochemical localization of candidates for vesicular glutamate transporters in the rat cerebral cortex. J Comp Neurol 435:379–387. 10.1002/cne.1037 [DOI] [PubMed] [Google Scholar]
- Geiman EJ, Knox MC, Alvarez FJ (2000) Postnatal maturation of gephyrin/glycine receptor clusters on developing Renshaw cells. J Comp Neurol 426:130–142. [DOI] [PubMed] [Google Scholar]
- Gosgnach S, Lanuza GM, Butt SJ, Saueressig H, Zhang Y, Velasquez T, Riethmacher D, Callaway EM, Kiehn O, Goulding M (2006) V1 spinal neurons regulate the speed of vertebrate locomotor outputs. Nature 440:215–219. 10.1038/nature04545 [DOI] [PubMed] [Google Scholar]
- Goulding M. (2009) Circuits controlling vertebrate locomotion: moving in a new direction. Nat Rev Neurosci 10:507–518. 10.1038/nrn2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT (2003) Characterization of the circuits that generate spontaneous episodes of activity in the early embryonic mouse spinal cord. J Neurosci 23:587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CM, Reilly MG, Stewart C, Schlegel C, Morley E, Puhl JG, Nagel C, Crisp KM, Mesce KA (2015) Compensatory plasticity restores locomotion after chronic removal of descending projections. J Neurophysiol 113:3610–3622. 10.1152/jn.00135.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, Henderson CE (2010) Motor neuron diversity in development and disease. Annu Rev Neurosci 33:409–440. 10.1146/annurev.neuro.051508.135722 [DOI] [PubMed] [Google Scholar]
- Kiehn O. (2006) Locomotor circuits in the mammalian spinal cord. Annu Rev Neurosci 29:279–306. 10.1146/annurev.neuro.29.051605.112910 [DOI] [PubMed] [Google Scholar]
- Kullander K. (2005) Genetics moving to neuronal networks. Trends Neurosci 28:239–247. 10.1016/j.tins.2005.03.001 [DOI] [PubMed] [Google Scholar]
- Leão KE, Leão RN, Deardorff AS, Garrett A, Fyffe R, Walmsley B (2010) Sound stimulation modulates high-threshold K(+) currents in mouse auditory brainstem neurons. Eur J Neurosci 32:1658–1667. 10.1111/j.1460-9568.2010.07437.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão RN, Oleskevich S, Sun H, Bautista M, Fyffe RE, Walmsley B (2004) Differences in glycinergic mIPSCs in the auditory brain stem of normal and congenitally deaf neonatal mice. J Neurophysiol 91:1006–1012. [DOI] [PubMed] [Google Scholar]
- Leão RN, Mikulovic S, Leão KE, Munguba H, Gezelius H, Enjin A, Patra K, Eriksson A, Loew LM, Tort AB, Kullander K (2012) OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat Neurosci 15:1524–1530. 10.1038/nn.3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Burke RE (2001) Short-term synaptic depression in the neonatal mouse spinal cord: effects of calcium and temperature. J Neurophysiol 85:2047–2062. [DOI] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM (2003) Activity-independent homeostasis in rhythmically active neurons. Neuron 37:109–120. 10.1016/S0896-6273(02)01104-2 [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H (2010) A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13:133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea DA, Pratt CA, Jordan LM (1980) Renshaw cell activity and recurrent effects on motoneurons during fictive locomotion. J Neurophysiol 44:475–488. [DOI] [PubMed] [Google Scholar]
- Mentis GZ, Alvarez FJ, Bonnot A, Richards DS, Gonzalez-Forero D, Zerda R, O'Donovan MJ (2005) Noncholinergic excitatory actions of motoneurons in the neonatal mammalian spinal cord. Proc Natl Acad Sci U S A 102:7344–7349. 10.1073/pnas.0502788102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Siembab VC, Zerda R, O'Donovan MJ, Alvarez FJ (2006) Primary afferent synapses on developing and adult Renshaw cells. J Neurosci 26:13297–13310. 10.1523/JNEUROSCI.2945-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz GA, Whishaw IQ (2009) The ladder rung walking task: a scoring system and its practical application. J Vis Exp (28):e1204. 10.3791/1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor Y, Lev-Tov A (2007) Analysis of rhythmic patterns produced by spinal neural networks. J Neurophysiol 98:2807–2817. 10.1152/jn.00740.2007 [DOI] [PubMed] [Google Scholar]
- Myers CP, Lewcock JW, Hanson MG, Gosgnach S, Aimone JB, Gage FH, Lee KF, Landmesser LT, Pfaff SL (2005) Cholinergic input is required during embryonic development to mediate proper assembly of spinal locomotor circuits. Neuron 46:37–49. 10.1016/j.neuron.2005.02.022 [DOI] [PubMed] [Google Scholar]
- Nakanishi ST, Whelan PJ (2010) Diversification of intrinsic motoneuron electrical properties during normal development and botulinum toxin-induced muscle paralysis in early postnatal mice. J Neurophysiol 103:2833–2845. 10.1152/jn.00022.2010 [DOI] [PubMed] [Google Scholar]
- Nieto-Gonzalez JL, Moser J, Lauritzen M, Schmitt-John T, Jensen K (2011) Reduced GABAergic inhibition explains cortical hyperexcitability in the wobbler mouse model of ALS. Cereb Cortex 21:625–635. 10.1093/cercor/bhq134 [DOI] [PubMed] [Google Scholar]
- Nishimaru H, Restrepo CE, Kiehn O (2006) Activity of Renshaw cells during locomotor-like rhythmic activity in the isolated spinal cord of neonatal mice. J Neurosci 26:5320–5328. 10.1523/JNEUROSCI.5127-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noga BR, Shefchyk SJ, Jamal J, Jordan LM (1987) The role of Renshaw cells in locomotion: antagonism of their excitation from motor axon collaterals with intravenous mecamylamine. Exp Brain Res 66:99–105. [DOI] [PubMed] [Google Scholar]
- Perry S, Gezelius H, Larhammar M, Hilscher MM, Lamotte d'Incamps B, Leao KE, Kullander K (2015) Firing properties of Renshaw cells defined by Chrna2 are modulated by hyperpolarizing and small conductance ion currents Ih and ISK. Eur J Neurosci 41:889–900. 10.1111/ejn.12852 [DOI] [PubMed] [Google Scholar]
- Pratt CA, Jordan LM (1987) Ia inhibitory interneurons and Renshaw cells as contributors to the spinal mechanisms of fictive locomotion. J Neurophysiol 57:56–71. [DOI] [PubMed] [Google Scholar]
- Rabe N, Gezelius H, Vallstedt A, Memic F, Kullander K (2009) Netrin-1-dependent spinal interneuron subtypes are required for the formation of left-right alternating locomotor circuitry. J Neurosci 29:15642–15649. 10.1523/JNEUROSCI.5096-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman J, Besser S, Schnell C, Eulenburg V, Hirrlinger J, Wojcik SM, Hülsmann S (2015) Genetic ablation of VIAAT in glycinergic neurons causes a severe respiratory phenotype and perinatal death. Brain Struct Funct 220:2835–2849. 10.1007/s00429-014-0829-2 [DOI] [PubMed] [Google Scholar]
- Renshaw B. (1946) Observations on interaction of nerve impulses in the gray matter and on the nature of central inhibition. Am J Physiol 146:443–448. [DOI] [PubMed] [Google Scholar]
- Sapir T, Geiman EJ, Wang Z, Velasquez T, Mitsui S, Yoshihara Y, Frank E, Alvarez FJ, Goulding M (2004) Pax6 and engrailed 1 regulate two distinct aspects of renshaw cell development. J Neurosci 24:1255–1264. 10.1523/JNEUROSCI.3187-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Bean BP (2005) Robustness of burst firing in dissociated purkinje neurons with acute or long-term reductions in sodium conductance. J Neurosci 25:3509–3520. 10.1523/JNEUROSCI.3929-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB (2008) Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11:998–1000. 10.1038/nn.2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmeyer H, Egea J, Rabe N, Gezelius H, Filosa A, Enjin A, Varoqueaux F, Deininger K, Schnütgen F, Brose N, Klein R, Kullander K, Betz A (2007) EphA4-dependent axon guidance is mediated by the RacGAP alpha2-chimaerin. Neuron 55:756–767. 10.1016/j.neuron.2007.07.038 [DOI] [PubMed] [Google Scholar]
- Wenner P. (2014) Homeostatic synaptic plasticity in developing spinal networks driven by excitatory GABAergic currents. Neuropharmacology 78:55–62. 10.1016/j.neuropharm.2013.04.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm JC, Wenner P (2008) GABAA transmission is a critical step in the process of triggering homeostatic increases in quantal amplitude. Proc Natl Acad Sci U S A 105:11412–11417. 10.1073/pnas.0806037105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm JC, Rich MM, Wenner P (2009) Compensatory changes in cellular excitability, not synaptic scaling, contribute to homeostatic recovery of embryonic network activity. Proc Natl Acad Sci U S A 106:6760–6765. 10.1073/pnas.0813058106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windhorst U. (1996) On the role of recurrent inhibitory feedback in motor control. Prog Neurobiol 49:517–587. 10.1016/0301-0082(96)00023-8 [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Katsurabayashi S, Guillemin I, Friauf E, Rosenmund C, Brose N, Rhee JS (2006) A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron 50:575–587. 10.1016/j.neuron.2006.04.016 [DOI] [PubMed] [Google Scholar]
- Wootz H, Fitzsimons-Kantamneni E, Larhammar M, Rotterman TM, Enjin A, Patra K, André E, Van Zundert B, Kullander K, Alvarez FJ (2013) Alterations in the motor neuron-renshaw cell circuit in the Sod1(G93A) mouse model. J Comp Neurol 521:1449–1469. 10.1002/cne.23266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagoraiou L, Akay T, Martin JF, Brownstone RM, Jessell TM, Miles GB (2009) A cluster of cholinergic premotor interneurons modulates mouse locomotor activity. Neuron 64:645–662. 10.1016/j.neuron.2009.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Narayan S, Geiman E, Lanuza GM, Velasquez T, Shanks B, Akay T, Dyck J, Pearson K, Gosgnach S, Fan CM, Goulding M (2008) V3 spinal neurons establish a robust and balanced locomotor rhythm during walking. Neuron 60:84–96. 10.1016/j.neuron.2008.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]