

Abstract
Fever occurs upon binding of prostaglandin E2 (PGE2) to EP3 receptors in the median preoptic nucleus of the hypothalamus, but the origin of the pyrogenic PGE2 has not been clearly determined. Here, using mice of both sexes, we examined the role of local versus generalized PGE2 production in the brain for the febrile response. In wild-type mice and in mice with genetic deletion of the prostaglandin synthesizing enzyme cyclooxygenase-2 in the brain endothelium, generated with an inducible CreERT2 under the Slco1c1 promoter, PGE2 levels in the CSF were only weakly related to the magnitude of the febrile response, whereas the PGE2 synthesizing capacity in the hypothalamus, as reflected in the levels of cyclooxygenase-2 mRNA, showed strong correlation with the immune-induced fever. Histological analysis showed that the deletion of cyclooxygenase-2 in brain endothelial cells occurred preferentially in small- and medium-sized vessels deep in the brain parenchyma, such as in the hypothalamus, whereas larger vessels, and particularly those close to the neocortical surface and in the meninges, were left unaffected, hence leaving PGE2 synthesis largely intact in major parts of the brain while significantly reducing it in the region critical for the febrile response. Furthermore, injection of a virus vector expressing microsomal prostaglandin E synthase-1 (mPGES-1) into the median preoptic nucleus of fever-refractive mPGES-1 knock-out mice, resulted in a temperature elevation in response to LPS. We conclude that the febrile response is dependent on local release of PGE2 onto its target neurons and not on the overall PGE2 production in the brain.
SIGNIFICANCE STATEMENT By using mice with selective deletion of prostaglandin synthesis in brain endothelial cells, we demonstrate that local prostaglandin E2 (PGE2) production in deep brain areas, such as the hypothalamus, which is the site of thermoregulatory neurons, is critical for the febrile response to peripheral inflammation. In contrast, PGE2 production in other brain areas and the overall PGE2 level in the brain do not influence the febrile response. Furthermore, partly restoring the PGE2 synthesizing capacity in the anterior hypothalamus of mice lacking such capacity with a lentiviral vector resulted in a temperature elevation in response to LPS. These data imply that the febrile response is dependent on the local release of PGE2 onto its target neurons, possibly by a paracrine mechanism.
Keywords: cyclooxygenase-2, endothelial cells, fever, median preoptic nucleus, microsomal prostaglandin E synthase-1, prostaglandin E2
Introduction
It is now well established that prostaglandin E2 (PGE2) is the final mediator of inflammation-induced fever (Li et al., 1999; Engblom et al., 2003). Fever has long been associated with elevated brain levels of PGE2 (Splawinski, 1977); in response to a peripheral immune stimulus, PGE2 levels in the CSF increase concomitant with the febrile response (Inoue et al., 2002), and injection of PGE2 into the CSF elicits fever in a dose-dependent way (Nilsberth et al., 2009b). PGE2 is synthesized by brain endothelial cells through the concerted action of the inducible enzymes cyclooxygenase (Cox)-2 and microsomal prostaglandin E synthase-1 (mPGES-1; Ek et al., 2001; Yamagata et al., 2001). These correlational studies have later been complemented by functional genetic studies, which have shown that genetic deletion of Cox-2 or mPGES-1 in the brain endothelium strongly attenuates the febrile response (Wilhelms et al., 2014), hence demonstrating that the brain endothelium plays a critical role for the generation of the PGE2 that is seen in the brain during fever.
Because a peripheral immune stimulus elicits the induction of prostaglandin-synthesizing enzymes throughout the brain vasculature (Ek et al., 2001; Yamagata et al., 2001), suggesting a generalized PGE2 release, the specificity of the PGE2-elicited responses comes about through the distinct distribution of its receptor subtypes (Zhang and Rivest, 1999; Ek et al., 2000; Oka et al., 2000), as demonstrated by the attenuation of fever through the deletion of EP3 receptors in the median preoptic nucleus but not by deletion of these receptors at other sites (Lazarus et al., 2007). However, it is less clear whether the responses evoked by PGE2 binding to its receptors is the result of PGE2 release locally, in a paracrine fashion, or whether PGE2 released at other sites also can elicit fever under physiological conditions (Matsumura et al., 1997). Although studies using injections of PGE2 or cyclooxygenase inhibitors support the importance of local synthesis (Scammell et al., 1996, 1998), the critical site of prostaglandin synthesis has not been investigated using modern functional genetic techniques. Here we addressed this question by examining in normal mice and in mice with the deletion of Cox-2 in the brain endothelium the relationship between the magnitude of the febrile response to a peripheral immune stimulus and the level of induced PGE2 in the CSF, as seen in individual animals. We also examined the relationship between the magnitude of the febrile response and the level of Cox-2 mRNA in the hypothalamus, as well as the PGE2 levels in plasma, and we determined by using immunohistochemistry how Cox-2 expression in different types of vessels was related to the febrile response. Finally, we examined whether local endogenous immune-induced production of PGE2 in the anterior hypothalamus resulted in a temperature response. Our data show that local PGE2 release onto brain PGE2 receptors is the mechanism governing the febrile response to peripheral immune challenge.
Materials and Methods
Animals.
Mice with specific deletion of Cox-2 in brain endothelial cells were generated by crossing animals in which exons 4–5 of the Cox-2 gene (Ptgs2) are flanked by loxP-sites (Ishikawa and Herschman, 2006) with animals expressing a tamoxifen-inducible CreERT2 under the Slco1c1 promoter (expressed in the cerebrovascular endothelium; Ridder et al., 2011). The tamoxifen (Sigma-Aldrich; 1 mg diluted in a mixture of 10% ethanol and 90% sunflower seed oil) was injected (0.1 ml, i.p.) twice a day for 5 d, followed by a 5 week recovery period before any further experiments were performed. The inducible Cre line was also crossed with a Cre reporter line, which expresses a Gt(ROSA)26Sor locus with a loxP-flanked STOP cassette preventing transcription of a CAG promoter-driven red fluorescent protein variant (tdTomato; The Jackson Laboratory; RRID: IMSR_JAX:007914), and the offspring were treated with tamoxifen, as described above. Mice with deletion of the Ptges gene (Trebino et al., 2003), encoding mPGES-1, were from our own breeding and on a C57BL/6 background. All animal experiments were approved by the local animal care and use committee and followed international guidelines.
Telemetric temperature recordings.
The mice were briefly anesthetized with isoflurane (Abbot Scandinavia) and implanted intraperitoneally with a transponder that records core body temperature (Mini Mitter). Immediately after surgery, the mice were transferred to a room in which the ambient temperature was set to 29°C, providing near-thermoneutral conditions (Rudaya et al., 2005).
Injection of lentiviral vectors.
Mice were anesthetized with isoflurane and mounted onto a stereotaxic frame. The scalp was exposed, and two small holes were drilled at the level of the bregma on each side of the midline. A Hamilton syringe was lowered to a position that was 0.0 mm anteroposterior to bregma, 0.3 mm mediolateral to bregma, and 5.5 mm dorsoventral to bregma. A lentiviral (lenti) vector was then injected at a rate of 180 nl/min over 3 min. The syringe remained in place for at least 3 min after the infusion and was then slowly removed, after which the skin was closed. Mice were injected either with a lentiviral vector, in Dulbecco's PBS containing MgCl2 and CaCl2 (catalog #D8662, Sigma-Aldrich), expressing GFP [lenti-5-KP-pgk-GFP (5 × 108 to 1 × 109 transducing units/ml), produced as previously described (Zufferey et al., 1997; Georgievska et al., 2004; a gift from Johan Jakobsson, Lund University, Sweden] or a lentiviral vector expressing human mPGES-1 [suCMV promoter-human Ptges (NM_004878)-Rsv promoter-puromycin resistance, 108 infectious units/ml; AMS Biotechnology], to which was added 10% of the lenti-GFP virus to permit subsequent immunofluorescent identification of the injection site (we found no available antibody that could detect mPGES-1). During the same surgical session, a temperature transponder was implanted in the abdominal cavity, as described above.
Immune stimulation.
Mice were injected intraperitoneally with bacterial wall lipopolysaccharide (LPS) from Escherichia coli (catalog #O111:B4, Sigma-Aldrich; 120 μg/kg body weight, diluted in 100 μl), 1 or 3 weeks (virus-injected mice) following implantation of the temperature transponder.
Tissue collection.
Mice injected with LPS only were killed 5 h after injection (this time point was selected since it corresponds to the time of peak fever in this experimental paradigm; Hamzic et al., 2013). Blood was drawn from the right atrium, transferred to EDTA-coated tubes (Sarstedt) to which was added indomethacin (10 μm; Sigma-Aldrich), and centrifuged at 7000 × g for 7 min at 4°C. The plasma was immediately frozen on dry ice and kept at −70°C. The animals were then placed in a stereotaxic frame, the atlanto-occipital membrane was exposed, and CSF was withdrawn from the cisterna magna using a Hamilton syringe mounted on a micromanipulator and immediately frozen. Samples that contained traces of blood were discarded. The whole procedure from when the animals were killed until CSF was withdrawn took <10 min. A hypothalamic block was then dissected and placed in RNAlater stabilization reagent (Qiagen) and stored at −70°C until analysis. This block was first isolated by two coronal cuts, one placed 0.5 mm rostral to the apex of the optic chiasm and the other at the caudal margin of the mammillary bodies. The resulting slab was then trimmed by sagittal cuts on each side through the sulcus between the hypothalamus and the temporal lobe. Finally, a horizontal cut was placed slightly above the anterior commissure. Mice injected with viral vectors were killed the day after the immune challenge with LPS. After asphyxiation with CO2, one group of mice was fixed by transcardial perfusion with a phosphate-buffered (0.1 m) paraformaldehyde solution (4%). The brains were removed and post-fixed for 3 h in the same fixative and then cryoprotected with 25% sucrose in PBS. In another group of mice, the brain was immediately removed. The hypothalamus was dissected and stored in RNAlater (Qiagen) at −70°C.
Immunohistochemistry.
Brains were cut in the frontal plane at 30 μm on a freezing microtome. The immunohistochemical procedures were performed according to standardized protocols (Engström et al., 2012). In brief, sections were incubated in a blocking solution [PBS containing 3% normal donkey serum (Jackson ImmunoResearch), 1% bovine serum albumin (Sigma-Aldrich), and 0.3% Triton X (Merck)] for 45 min, followed by incubation overnight at room temperature with rabbit anti-Cox-2 antibody (1:500; sc-1747 M-19, Santa Cruz Biotechnology; RRID: AB_2084976) and goat anti-lipocalin-2 antibody (1:500; AF1857, R&D Systems; RRID: AB_355022), rinsed in PBS and then incubated with Alexa Fluor 555 donkey anti-rabbit antibody and Alexa Fluor 488 donkey anti-goat antibody (both 1:500; Life Technologies). Sections from lentivector-injected brains were incubated with chicken anti-GFP antibody (1:10,000; ab13970, Abcam; RRID: AB_300798), followed by Alexa Fluor 488 goat anti-chicken IgG (heavy chain and light chain) antibody (Life Technologies). The sections were finally mounted on SuperFrost Plus glasses (ThermoFisher Scientific) with Prolong Gold Anti-Fade Reagent (Life Technologies).
Assays for PGE2 levels in CSF and plasma.
The concentration of PGE2 in CSF (diluted 1:100) was determined using a High Sensitivity Prostaglandin E2 Enzyme Immunoassay Kit (Assay Designs). The values were calculated using a standard curve ranging from 7.81 to 1000 pg/ml (R2 = 1). The kit antiserum shows the following cross-reactivity, according to the manufacturer: PGE2, 100%; PGE1, 70%; PGE3, 16.3%; PGF1α, 1.4%; PGF2α, 0.7%; 6-keto-PGF1α, 0.6%; PGA2, 0.1%; PGB1, 0.1%; and <0.1% for 13,14-dihydro-15-keto-PGF2α, 6, 15-keto, 13, 4-dihydro-PGF1α, thromboxane B2, 2 arachidonoylglycerol, anandamide, PGD2, and arachidonic acid. The concentration of PGE2 metabolites in plasma (diluted 1:20) was determined with a Prostaglandin E Metabolite EIA Kit (Cayman Chemical). The values were calculated using a standard curve ranging from 0.2 to 50 pg/ml (R2 = 0.999). The kit antiserum recognizes derivatized 13,14-dihydro-15-ketoPGE1, 13,14-dihydro-15-ketoPGE2, and bicycloPGE1, but has <0.01% cross-reactivity with arachidonic acid, leukotriene B4, tetranor-PGEM, tetranor-PGFM, PGD2, PGE1, 6-keto PGE1, PGE2, PGF1α, 6-keto PGF1α, PGF2α and thromboxane B2.
Real-time quantitative PCR analyses.
RNA was extracted with RNeasy Universal Plus kit or RNeasy Micro Kit (Qiagen), and reverse transcription was performed with High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Real-time quantitative PCR (qPCR) was then performed using Gene Expression Master Mix (Applied Biosystems) on a 96-well plate (7900HT Fast RT-PCR System, Applied Biosystems). The following assays (all from Applied Biosystems) were used: Ptgs2: Mm00478374_m1; human Ptges (Hs01115r610_m1); and Gapdh (Mm99999915_g1).
Experimental design and statistical analysis.
For all comparisons between genotypes, littermates were used. Mice were of both sexes, and experimental groups were balanced with respect to sex and age. Sample size is reported in the figure legends. All statistical analyses were performed in GraphPad Prism (GraphPad Software). Analysis of differences in body temperature at 5 h between LPS-treated WT mice and mice with endothelial-specific deletion of Cox-2 was performed with a one-way ANOVA followed by Sidak's multiple-comparisons test. The same analysis was used for differences between groups with respect to PGE2 in CSF and qPCR data. For PGE2 metabolites in plasma, nonparametric statistics were used (Kruskal–Wallis test followed by Dunn's multiple-comparisons test). Temperature responses after injections of viral vectors were analyzed with a two-way repeated-measures ANOVA followed by Tukey's post hoc test. Regression analysis was performed with F statistics. Qualitative data were analyzed with Fisher's exact test. Results were considered significant at p < 0.05.
Results
Deletion of Cox-2 in brain endothelial cells results in attenuated fever response to LPS
As reported previously (Wilhelms et al., 2014), mice with deletion of Cox-2 in brain endothelial cells (Cox-2ΔSlco1c1) show attenuated fever following intraperitoneal injection of LPS (Fig. 1a). At 5 h after injection, when the animals were killed and tissue was collected for analysis, LPS-treated Cox-2ΔSlco1c1 mice displayed significantly lower body temperatures than their wild-type (WT) Cox-2fl/fl littermates (F(3,37) = 6.283, p = 0.0015; LPS Cox-2ΔSlco1c1 vs LPS WT: p = 0.0219; Fig. 1b).
Figure 1.

Attenuated fever in mice with gene deletion of Cox-2 in brain endothelial cells. a, Temperature recordings from WT and Cox-2ΔSlco1c1 mice immune challenged by intraperitoneal injection of LPS (120 μg/kg). The initial temperature peak is due to the handling stress in conjunction with the injection procedure. It is prostaglandin independent (Saha et al., 2005) and does not differ between genotypes. b, Bar graph showing mean fever 5 h after LPS injection in WT type and Cox-2ΔSlco1c1 mice. *p < 0.05. In a and b: n = 15 for WT LPS; n = 11 for Cox-2ΔSlco1c1 LPS; n = 9 for WT NaCl; and n = 6 for Cox-2ΔSlco1c1 NaCl. Error bars = SEM.
No difference in PGE2 levels in CSF between Cox-2ΔSlco1c1 mice and WT mice
We next examined whether the difference in the febrile response to LPS between Cox-2ΔSlco1c1 mice and WT mice was associated with a difference in CSF levels of PGE2. LPS treatment resulted in elevated levels of PGE2 in CSF at 5 h after injection (F(3,37) = 8.003, p < 0.003; LPS WT vs NaCl WT: p = 0.0005; LPS Cox-2ΔSlco1c1 vs NaCl Cox-2ΔSlco1c1: p = 0.0198), but there was no difference between genotypes (Fig. 2a).
Figure 2.
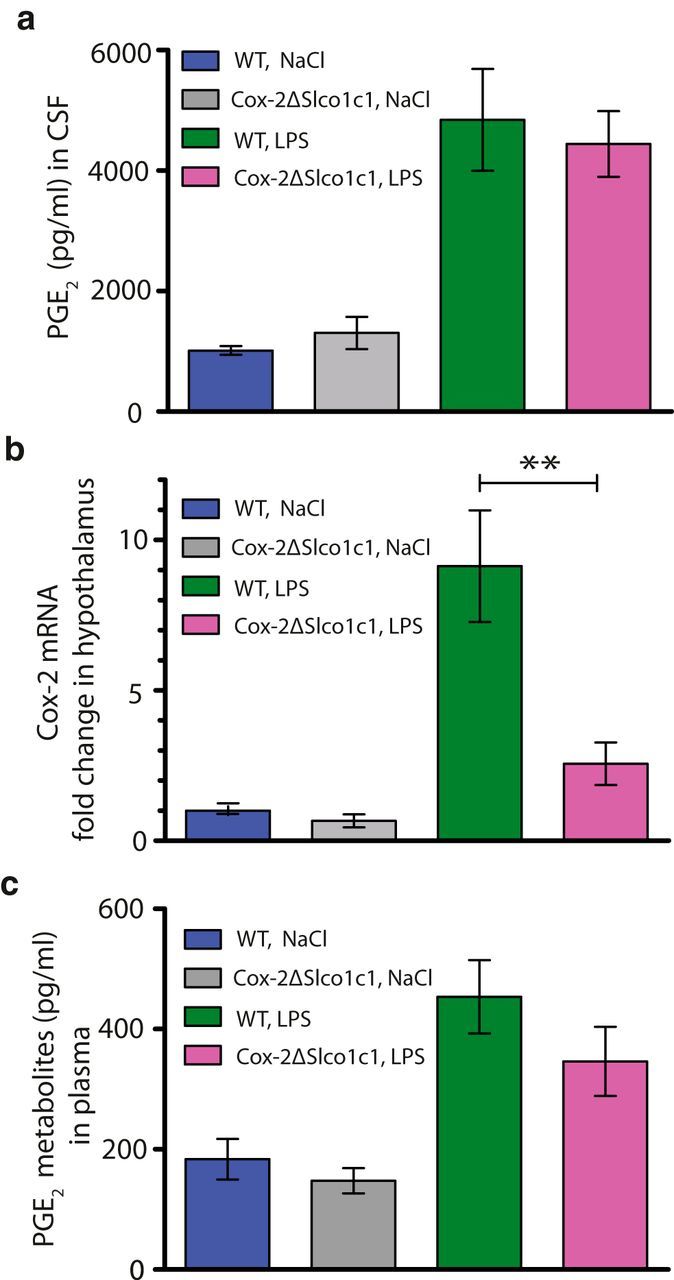
PGE2 levels in brain and plasma and Cox-2 mRNA expression in the hypothalamus in WT mice and in mice with gene deletion of Cox-2 in brain endothelial cells. a, Immune stimulation with LPS (120 μg/kg, i.p.) increases the PGE2 concentration in the CSF to similar levels in both WT and Cox-2ΔSlco1c1 mice. b, The immune-induced Cox-2 mRNA induction in the hypothalamus is significantly attenuated in Cox-2ΔSlco1c1 mice. **p < 0.01. c, The immune-induced levels of PGE2 metabolites in plasma do not differ between WT and Cox-2ΔSlco1c1 mice. In all graphs: n = 15 for WT LPS; n = 10–11 for Cox-2ΔSlco1c1 LPS; n = 9–10 for WT NaCl; and n = 5–8 for Cox-2ΔSlco1c1 NaCl. Error bars = SEM.
Lower levels of induced Cox-2 mRNA in the hypothalamus of Cox-2ΔSlco1c1 mice
qPCR analysis of the levels of Cox-2 mRNA in the hypothalamus of Cox-2ΔSlco1c1 mice and WT mice showed significantly lower induction following LPS administration in the gene-deleted mice than in the WT mice (F(3,39) = 21.78, p < 0.0001; LPS WT vs LPS Cox-2ΔSlco1c1: p = 0.0021; Fig. 2b). As expected, there was also a small (but statistically not significant) reduction of Cox-2 mRNA in the NaCl-treated Cox-2ΔSlco1c1 mice compared with the WT mice (Fig. 2b).
No difference in PGE2 metabolite levels in plasma between Cox-2ΔSlco1c1 mice and WT mice
To examine whether the gene deletion in the Cox-2ΔSlco1c1 mice, which should occur only in brain endothelial cells (Ridder et al., 2011), had influenced prostaglandin synthesis peripherally, the levels of PGE2 metabolites in plasma were analyzed. This assay was chosen instead of the direct measurement of PGE2 because PGE2 in plasma is difficult to measure reliably (Samuelsson et al., 1975) since it is rapidly converted in vivo to its 13,14-dihydro-15-keto metabolite, with >90% of circulating PGE2 being cleared by a single passage through the lungs (Hamberg and Samuelsson, 1971). The levels of PGE2 metabolites in plasma were elevated following immune challenge with LPS (Kruskal-Wallis statistic = 19.84, p = 0.0002; LPS WT vs NaCl WT: p = 0.0022; LPS Cox-2ΔSlco1c1 vs NaCl Cox-2ΔSlco1c1: p = 0.0327), but there was no difference between the genotypes (Fig. 2c).
Body temperature following immune stimulation correlates with Cox-2 mRNA levels in the hypothalamus but not with PGE2 levels in CSF
To further examine the relationship between body temperature and central prostaglandin synthesis, we next performed a regression analysis of the individual temperatures and the levels of PGE2 in CSF in Cox-2ΔSlco1c1 mice and WT mice 5 h after LPS injection. As shown (Fig. 3a), there was only weak relationship between these two parameters (R2 = 0.157, F(1,23) = 4.292, p = 0.0497; Fig. 3a). In contrast, there was a strong correlation between temperature and Cox-2 mRNA in the hypothalamus (R2 = 0.593, F(1,22) = 32.01, p < 0.0001; Fig. 3b). There was also a moderately strong relationship between body temperature and PGE2 metabolites in plasma (R2 = 0.4310, F(1,23) = 17.42, p = 0.0004; Fig. 3c).
Figure 3.
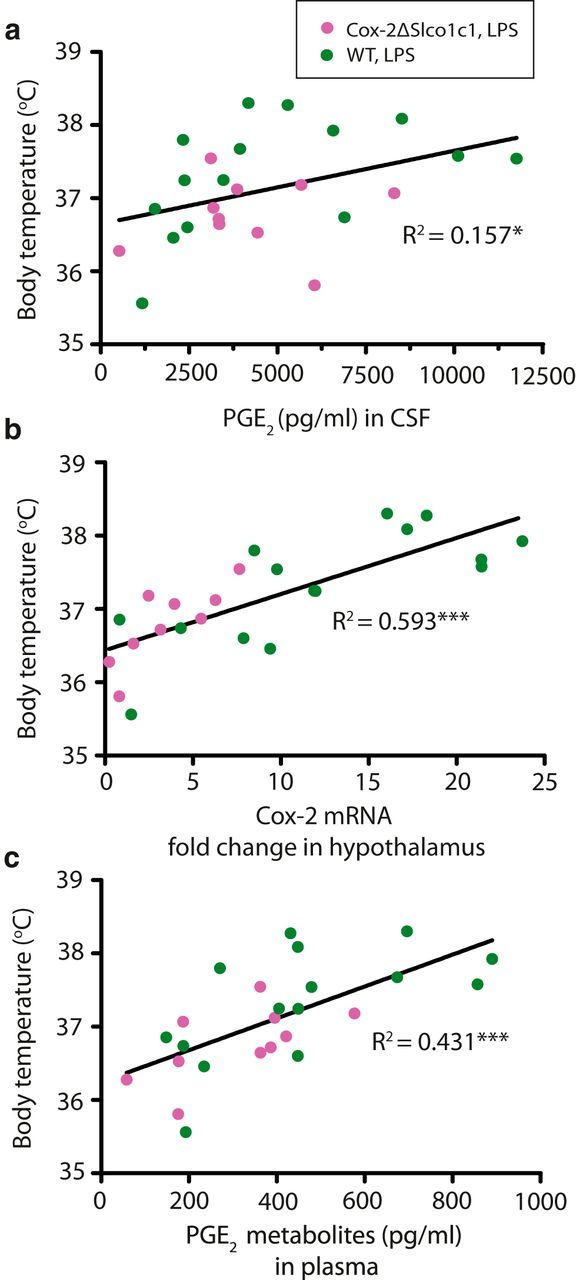
Relationship among body temperature and PGE2 in the CSF, Cox-2 mRNA in the hypothalamus, and PGE2 metabolites in plasma, respectively, following immune stimulation with LPS (120 μg/kg, i.p.). a, Weak relationship between body temperature and PGE2 in the CSF. b, Strong relationship between body temperature and Cox-2 mRNA in the hypothalamus. c, Moderately strong relationship between body temperature and PGE2 metabolites in plasma. *p < 0.05, ***p < 0.001.
The levels of PGE2 in CSF correlate only weakly with Cox-2 mRNA expression in the hypothalamus and not with levels of PGE2 metabolites in plasma
To examine what affects the PGE2 levels in CSF, regression analysis was performed for the relationship between PGE2 in CSF and Cox-2 mRNA expression in the hypothalamus of LPS-treated mice (Fig. 4a) and PGE2 metabolites in plasma in these mice (Fig. 4b), respectively. PGE2 levels in CSF showed only a weak relationship with the Cox-2 mRNA expression in the hypothalamus (R2 = 0.1783, F(1,25) = 5.424, p = 0.0282; Fig. 4a). In the same vein, there was no significant relationship between PGE2 levels in CSF and the levels of PGE2 metabolites in plasma (R2 = 0.1321, F(1,24) = 3.654, p = 0.0679; Fig. 4b), indicating that they have different sources of origin.
Figure 4.
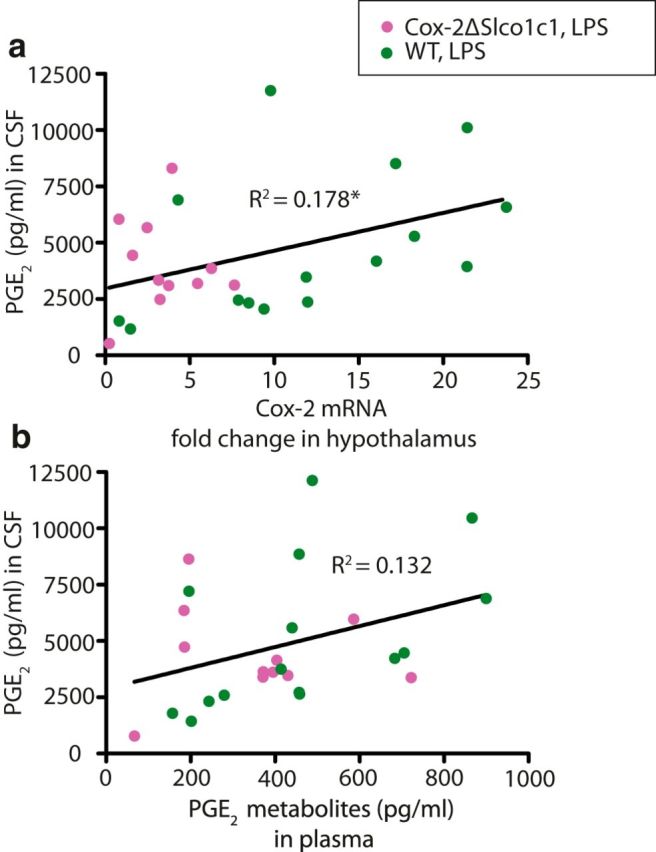
Relationship between PGE2 levels in the CSF and Cox-2 mRNA and PGE2 metabolites in plasma, respectively. a, Weak relationship between PGE2 in the CSF and Cox-2 mRNA in the hypothalamus. b, No significant relationship between PGE2 in the CSF and PGE2 metabolites in plasma. *p < 0.05.
The gene deletion in Cox-2ΔSlco1c1 mice mainly occurs in smaller vessels in the brain parenchyma but not in large vessels
Since the above data indicated that the gene deletion in Cox-2ΔSlco1c1 mice preferentially affected induced prostaglandin synthesis in the hypothalamus, but not generally in the brain, we examined histologically where the gene deletion took place. We crossed the mice with the tamoxifen-inducible CreERT2 under the Slco1c1 promoter with a Cre reporter line expressing a Gt(ROSA)26Sor locus with a loxP-flanked STOP cassette, which prevents transcription of a CAG promoter-driven red fluorescent protein variant (tdTomato). Following tamoxifen treatment of the offspring, we subjected it to immune challenge with LPS. We found that Cre-induced recombination occurred preferentially in small- and medium-sized blood vessels; in the latter, tdTomato staining was found to be colocalized with induced Cox-2 immunoreactivity in endothelial cells (Fig. 5a,b). In contrast, Cox-2-positive cells in larger blood vessels rarely expressed tdTomato staining (Fig. 5b,c). We also examined brains from Cox-2ΔSlco1c1 mice and WT mice for the coexpression of Cox-2 and lipocalin-2 immunoreactivity to evaluate whether the endothelial cells had responded to LPS since lipocalin-2 is expressed in endothelial cells in response to LPS together with, but independently of, Cox-2 (Hamzic et al., 2013; Vasilache et al., 2015). We found that lipocalin-2-expressing cells in larger vessels, and in particular in vessels close to the surface of the neocortex, coexpressed Cox-2 extensively in both genotypes (Fig. 6a,b). Strong Cox-2 labeling was also seen among lipocalin-2-expressing cells in meningeal vessels in both genotypes (Fig. 6c,d). However, smaller vessels in Cox-2ΔSlco1c1 mice, preferentially in the more medial parts of the brain such as in the hypothalamic region, more rarely expressed Cox-2 while displaying extensive labeling for lipocalin-2 (Fig. 6f). This was in stark contrast to what was seen in WT mice, in which there was extensive coexpression of both proteins also in small vessels (Fig. 6e). Although there was some variation across animals, when a blinded investigator qualitatively determined the genotype of the animals by examining the degree of colocalization of Cox-2 and lipocalin-2 in differently sized vessels, the correct genotype was determined in 83% of the animals (p = 0.0073; n = 13 for Cox-2ΔSlco1c1 mice, and n = 9 for WT mice).
Figure 5.
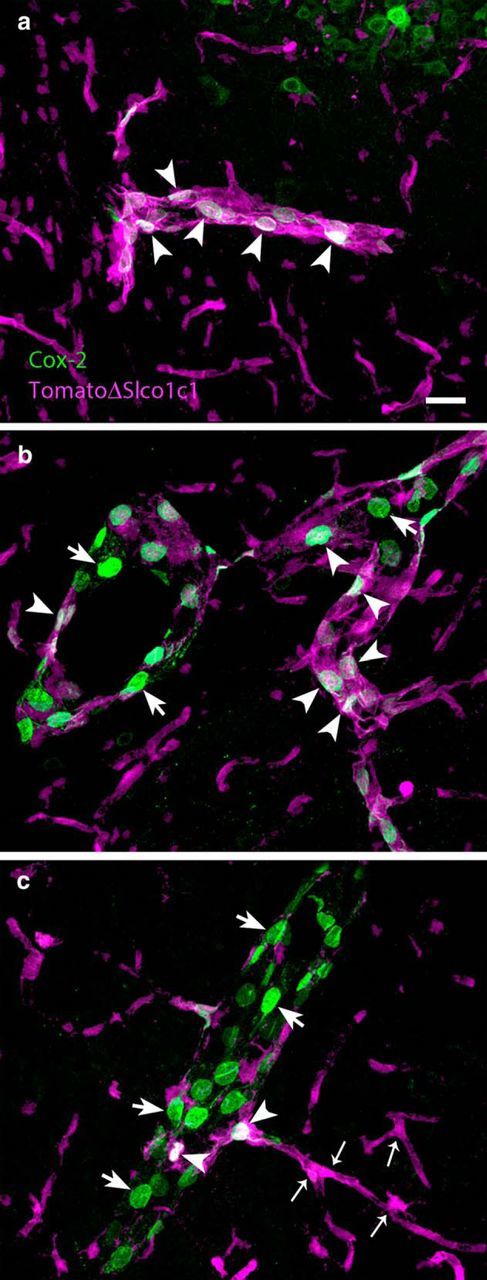
Gene deletion with Slco1c1-Cre targets mainly endothelial cells in small- and medium-sized vessels in the brain. a, Dual labeling (arrowheads) of the Cre reporter protein tdTomato and Cox-2 in a small vessel in the brain. b, Large vessel with smaller-sized branch. Most of the Cox-2-immunoreactive cells (green) in the large vessel do not express tdTomato (arrows), whereas those in the smaller sized branch do (arrowheads). c, Numerous Cox-2-expressing cells (arrows) in a large vessel are shown, but few of those cells also express tdTomato (arrowheads). All micrographs are from immune-challenged mice. Note that endothelial cells in capillaries express tdTomato (small arrows) but not Cox-2. Scale bar, 20 μm.
Figure 6.

Cox-2 and lipocalin-2 expression following immune challenge. a, b, Abundant expression of Cox-2 among lipocalin-2 (LCN2)-positive cells in a large vessel in both WT (a) and Cox-2ΔSlco1c1 mice (b). c, d, No difference in Cox-2 expression in the leptomeninges between WT (c) and Cox-2ΔSlco1c1 mice (d). e, f, Reduced Cox-2 expression in small lipocalin-2-stained vessel in a Cox-2ΔSlco1c1 mouse (f) and abundant expression in a WT mouse (e). Arrowheads point at dual-labeled cells, and arrows point at single-labeled cells. Scale bar, 20 μm.
Restoration of PGE2 synthesis in the preoptic hypothalamus of mPGES-1 knock-out mice results in a temperature response to LPS
To determine whether local PGE2 production in the hypothalamus results in a temperature response to LPS, we used intracerebral injection of a viral vector to restore the PGE2 synthesizing capacity in the median preoptic regions of mice with global deletion of mPGES-1, the inducible terminal PGE2-synthesizing enzyme (Jakobsson et al., 1999). We chose to use mPGES-1 knock-out (KO) mice, which previously have been shown to be unable to mount a temperature rise upon peripheral immune challenge (Engblom et al., 2003; Nilsberth et al., 2009a) instead of Cox-2 KO mice, because the latter are difficult to breed and suffer from various health problems, including chronic inflammation (Langenbach et al., 1999). mPGES-1 KO mice with injections of viral vectors expressing GFP aimed at the region of the median preoptic nucleus, the structure critical for the fever response to PGE2 (Scammell et al., 1996; Lazarus et al., 2007), displayed the same hypothermic response to immune challenge with LPS as shown previously for these mice (Engblom et al., 2003; Nilsberth et al., 2009a; Engström et al., 2012). In contrast, mPGES-1 KO mice injected with a vector expressing mPGES-1 showed no sustained hypothermia after the initial temperature drop following the handling stress-induced temperature peak but displayed a body temperature elevation compared with mice injected with a vector expressing GFP (Fig. 7a). Mean body temperature during the period of 60–480 min after LPS injection (i.e., after the handling stress-induced temperature peak) was significantly different between treatments (F(2,29) = 11.17, p = 0.0003; lenti-mPGES-1 vs lenti-GFP, p = 0.0042). While injection sites were determined by immunofluorescence to GFP (GFP lentiviral vector was added at 10% to the mPGES-1 vector; Fig. 7b,c), transcription of mPGES-1 was assured by qPCR analysis. In a total of eight mice injected with the viral vector expressing mPGES-1, all displayed mPGES-1 mRNA, whereas none of four mice injected with the GFP vector did so, as expected.
Figure 7.
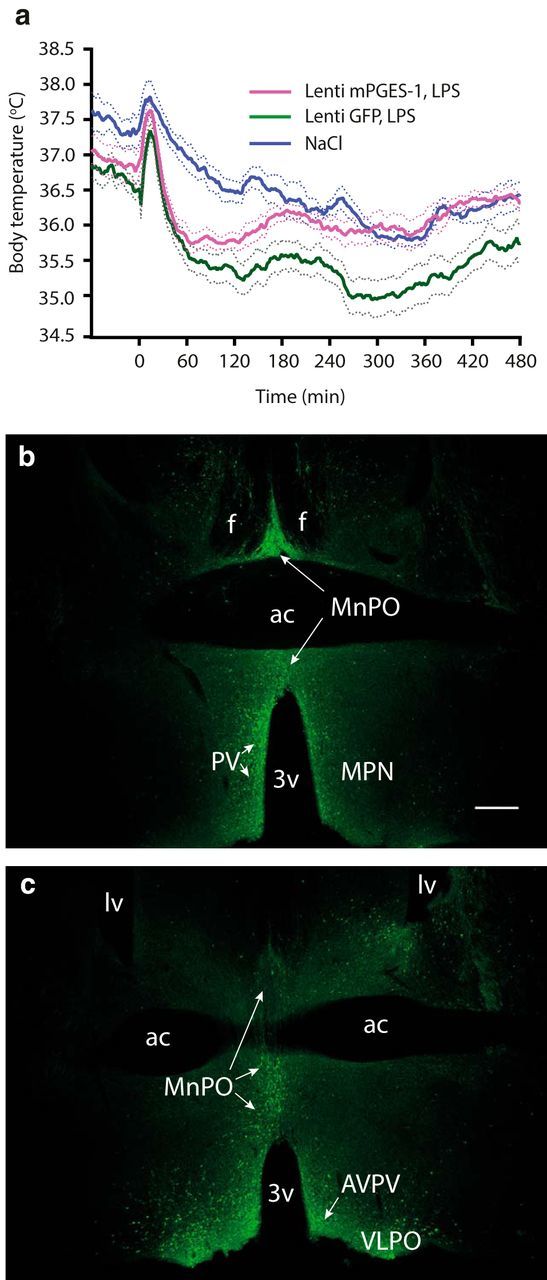
Mice injected into the preoptic hypothalamus with lentiviral vector encoding the terminal prostaglandin E2-synthesizing enzyme mPGES-1 display higher body temperature in response to LPS injection than mice injected with control vector. a, Temperature recordings of immune-challenged mPGES-1 knock-out mice injected with a viral vector encoding mPGES-1 (magenta trace) or given control injection of lentiviral vector encoding GFP (green trace). n = 12 for lenti-mPGES-1 LPS; n = 10 for lenti-GFP LPS; and n = 10 for NaCl (mixed group of lenti-mPGES-1 and lenti-GFP-injected mice). Error bars = SEM. b, c, Micrographs showing immunofluorescent staining for GFP in the preoptic hypothalamus after injection with viral vector. b and c are from different animals; the plane of the chosen sections corresponds approximately to bregma +0.14/+0.145 mm (b), and bregma +0.26/+0.245 mm (c) in the atlas of Paxinos and Franklin (2001) and the Allen Reference Atlas (Dong, 2008), respectively. 3v, Third ventricle; ac, anterior commissure; AVPV, anteroventral periventricular nucleus; f, fornix; lv, lateral ventricle; MnPO, median preoptic nucleus; MPN, medial preoptic nucleus; PV, periventricular nucleus; VLPO, ventrolateral preoptic nucleus. Scale bar, 500 μm.
Discussion
The febrile response is dependent on PGE2 synthesis by small- to medium-sized vessels in the hypothalamus but independent of global PGE2 synthesis in brain
This study shows that the magnitude of the febrile response was strongly correlated with the PGE2-synthesizing capacity in the hypothalamus, as reflected in the levels of Cox-2 mRNA, but was only weakly related to the PGE2 levels in CSF. These findings were corroborated by the histological demonstration that genetic deletion of Cox-2 in brain endothelial cells using a tamoxifen-inducible CreERT2 under the Slco1c1 promoter (Ridder et al., 2011) occurred preferentially in small- to medium-sized vessels deep in the brain parenchyma, such as in the hypothalamus, whereas larger vessels, particularly those close to the neocortical surface, and vessels in the meninges were left unaffected. Accordingly, whereas the gene deletion attenuated the PGE2-synthesizing capacity in the hypothalamus, in which the EP3 receptor-expressing neurons that are critical for the febrile response are located (Lazarus et al., 2007), it left the PGE2 synthesis intact in large parts of the brain. Together, these data imply that the febrile response is dependent on the local, possibly paracrine, release of PGE2 onto the preoptic EP3 receptors, whereas the overall PGE2 level in the brain, as reflected in the levels measured in the CSF, is not involved. This conclusion was further supported by the finding that local restoration of induced PGE2 synthesis in febrile-resistant mPGES-1 KO mice at loci involving the median preoptic nucleus, the critical site for immune-induced fever, resulted in a temperature elevation in response to intraperitoneal LPS.
Previous findings that the injection of PGE2 into the cerebral ventricles causes fever in a dose-dependent manner (Engblom et al., 2003; Lazarus et al., 2007; Nilsberth et al., 2009b) would seem to indicate that PGE2 synthesized at some sites in the brain other than in the immediate vicinity of the preoptic EP3 receptor-expressing neurons that are critical for the febrile response also could influence the firing properties of these neurons and, hence, elicit fever. However, as noted previously (Nilsberth et al., 2009b), the concentration in the CSF of exogenously administered PGE2 that is required for eliciting fever is on the order of 1000-fold higher than that seen in CSF during immune-induced fever. This observation suggests that the concentration of PGE2 at its target neurons that is needed for eliciting fever in response to a peripheral immune stimulus is much higher than that measured in the CSF. It seems likely that such high concentrations of PGE2 during physiological conditions could be achieved only by paracrine release. This idea is supported by the demonstration that intracerebral injection of a threshold dose of PGE2 causes fever when localized to or in the immediate vicinity of the median preoptic nucleus but not when localized to more distant sites (Scammell et al., 1996). Similarly, microinjections of a cyclooxygenase inhibitor into the same area attenuated LPS-induced fever, whereas microinjections at other sites did not (Scammell et al., 1998).
Relationship between fever and hypothermia
While injection of a viral vector encoding mPGES-1 resulted in a significantly higher body temperature after peripheral immune challenge than that displayed by mice subjected to control virus injections, the body temperature did not reach levels that could be classified as fever. The temperature response to immune challenge with LPS is likely the central compilation of pyrogenic and hypothermic signaling, with the neuronal substrate being reciprocally interconnected cell groups that generate temperature-elevating and temperature-lowering signals, respectively (Zhao et al., 2017). In the absence of induced PGE2 synthesis, animals immune challenged with LPS display hypothermia (Fig. 7; see also Engström et al., 2012), and such hypothermia also occurs in mice lacking EP3 receptors (Oishi et al., 2015). Hypothermia, which is elicited by an as yet unidentified cryogen (Almeida et al., 2006), is thus the response to LPS when no pyrogenic PGE2–EP3 signaling is present. While mPGES-1 KO mice, similar to WT mice, are fully responsive to intracerebroventricularly injected PGE2, hence demonstrating intact EP3 signaling (Engblom et al., 2003), it should be noted that the response of mice to the intracerebroventricularly injected PGE2 is graded. Low doses give rise to only a slight temperature elevation, whereas high doses elicit a body temperature on the order of 40°C (Nilsberth et al., 2009b). The inability to fully restore the febrile response by virus vector injection in the present study is therefore likely explained by only partly restored PGE2 synthesis, which in turn may be due to incomplete mPGES-1 expression and/or deficient mPGES-1 protein coupling to Cox-2. The latter is induced in brain endothelial cells (Engström et al., 2012); however, structures that had incorporated the virus (and hence were shown to express GFP) were preferentially of neuron/glial cell type and were only rarely suggestive of endothelial cells (data not shown). While speculative, mPGES-1 expressed by other cells than endothelial cells could perhaps couple with Cox-1 that is more ubiquitously expressed, hence resulting in PGE2 synthesis, although at levels that did not permit a full-fledged restoration of the febrile response. Nevertheless, the finding of a temperature elevation in response to LPS injection, although modest, in animals in which mPGES-1 was re-expressed in the preoptic region demonstrates the critical role for localized PGE2 synthesis for heat production in response to peripheral immune challenge.
Role of peripherally produced PGE2 for the febrile response
The present data confirm that prostaglandin production in brain endothelial cells is important for the febrile response (Engström et al., 2012; Eskilsson et al., 2014a; Wilhelms et al., 2014). However, it has been suggested that peripherally produced circulating PGE2 also is involved (Steiner et al., 2006), but data from mice chimeric for mPGES-1 imply that the role of PGE2 produced by hematopoietically derived cells is, at most, very small (Engström et al., 2012). Here we found no significant relationship between the levels of PGE2 metabolites in plasma and PGE2 levels in the CSF, suggesting that peripherally and centrally produced PGE2 are of distinct sources of origin, at least at the time point examined. Accordingly, although there was a moderately strong relationship in the present study between the levels of PGE2 metabolites in plasma and the magnitude of fever, there is most likely no causality between these events; in the case of a strong peripheral immune response, there is also a strong central immune response, and, conversely, a weak peripheral immune response is associated with a weak central immune response. This conclusion is supported by observations in the mice chimeric for mPGES-1. Thus, whereas WT mice carrying mPGES-1 KO hematopoietic cells displayed normal LPS-induced fever, mPGES-1 KO mice carrying WT hematopoietic cells did not mount a febrile response, despite showing strong induction of PGE2 metabolites in plasma (Engström et al., 2012). Furthermore, in those mice, central PGE2 levels were not significantly increased, providing additional support for the distinct origin of centrally and peripherally produced PGE2.
Heterogeneous distribution of transporter proteins among brain endothelial cells
We observed in the genetically modified mice that Cre recombinase expressed under the control of the Slco1c1 promoter produced recombination (and hence gene deletion) in brain endothelial cells in small- and medium-sized vessels but not in larger vessels. The Slco1c1 gene encodes the organic anion transporter 14 (Oatp14), which has been shown to be expressed selectively in endothelial cells of the brain (Ridder et al., 2011). The organic anion transporter family transports hormones and other organic molecules to and from the brain (Westholm et al., 2008), and it has been demonstrated that other members of the Oatp family also show heterogeneous distributions among the vessels in the brain similar to Oatp14, with the main expression being in smaller vessels (Daneman et al., 2010). Furthermore, it has been shown that transporter proteins overall have a more prominent expression in capillaries than in venules (Macdonald et al., 2010), which is consistent with the idea that the exchange of molecules and nutrients between blood and tissue occurs preferentially in the capillaries. However, induced prostaglandin synthesis, as reflected by induced expression of Cox-2, seems to occur in large-, medium-, and small-sized vessels but not in capillaries (Fig. 5; Eskilsson et al., 2014b), and it is not yet known which transporter is responsible for the transfer of PGE2 into the brain. As shown here, PGE2 synthesis in small- and medium-sized vessels is critical for the febrile response. The functional role of the PGE2 that is synthesized in the larger vessels, including vessels in meninges, which seems to account for most of the PGE2 that is seen in the CSF, remains to be clarified. It has been reported recently that the excitation of pyramidal cells in the cerebral cortex results in increased local cerebral blood flow via neuronal release of PGE2 and its binding to vasodilatory EP2 and EP4 receptors on vascular smooth muscle cells or pericytes (Lacroix et al., 2015). Whether PGE2 produced by endothelial cells in response to peripheral immune challenge also takes part in the vasodilation of cerebral vessels is not known, and neither is what functional role such vasodilation, if present, would subserve.
Footnotes
This study was supported by the Swedish Medical Research Council (A.B. and D.E.), The Swedish Cancer Foundation (A.B.), The European Research Council (starting grant to D.E.), The Knut and Alice Wallenberg Foundation (D.E.), The Swedish Brain foundation (A.B. and D.E.), and the County Council of Östergötland (A.B. and D.E). We thank Dr. Harvey Herschman for the gift of Cox-2 conditional knock-out mice.
The authors declare no competing financial interests.
References
- Almeida MC, Steiner AA, Branco LG, Romanovsky AA (2006) Neural substrate of cold-seeking behavior in endotoxin shock. PLoS One 1:e1. 10.1371/journal.pone.0000001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA (2010) The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One 5:e13741. 10.1371/journal.pone.0013741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HW. (2008) The Allen reference atlas: a digital color brain atlas of the C57BL/6J male mouse. Hoboken, NJ: Wiley. [Google Scholar]
- Ek M, Arias C, Sawchenko P, Ericsson-Dahlstrand A (2000) Distribution of the EP3 prostaglandin E(2) receptor subtype in the rat brain: relationship to sites of interleukin-1-induced cellular responsiveness. J Comp Neurol 428:5–20. [DOI] [PubMed] [Google Scholar]
- Ek M, Engblom D, Saha S, Blomqvist A, Jakobsson PJ, Ericsson-Dahlstrand A (2001) Inflammatory response: pathway across the blood-brain barrier. Nature 410:430–431. 10.1038/35068632 [DOI] [PubMed] [Google Scholar]
- Engblom D, Saha S, Engström L, Westman M, Audoly LP, Jakobsson PJ, Blomqvist A (2003) Microsomal prostaglandin E synthase-1 is the central switch during immune-induced pyresis. Nat Neurosci 6:1137–1138. 10.1038/nn1137 [DOI] [PubMed] [Google Scholar]
- Engström L, Ruud J, Eskilsson A, Larsson A, Mackerlova L, Kugelberg U, Qian H, Vasilache AM, Larsson P, Engblom D, Sigvardsson M, Jönsson JI, Blomqvist A (2012) Lipopolysaccharide-induced fever depends on prostaglandin E2 production specifically in brain endothelial cells. Endocrinology 153:4849–4861. 10.1210/en.2012-1375 [DOI] [PubMed] [Google Scholar]
- Eskilsson A, Mirrasekhian E, Dufour S, Schwaninger M, Engblom D, Blomqvist A (2014a) Immune-induced fever is mediated by IL-6 receptors on brain endothelial cells coupled to STAT3-dependent induction of brain endothelial prostaglandin synthesis. J Neurosci 34:15957–15961. 10.1523/JNEUROSCI.3520-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskilsson A, Tachikawa M, Hosoya K, Blomqvist A (2014b) The distribution of microsomal prostaglandin E synthase-1 in the mouse brain. J Comp Neurol 522:3229–3244. 10.1002/cne.23593 [DOI] [PubMed] [Google Scholar]
- Georgievska B, Jakobsson J, Persson E, Ericson C, Kirik D, Lundberg C (2004) Regulated delivery of glial cell line-derived neurotrophic factor into rat striatum, using a tetracycline-dependent lentiviral vector. Hum Gene Ther 15:934–944. 10.1089/hum.2004.15.934 [DOI] [PubMed] [Google Scholar]
- Hamberg M, Samuelsson B (1971) On the metabolism of prostaglandins E 1 and E 2 in man. J Biol Chem 246:6713–6721. [PubMed] [Google Scholar]
- Hamzic N, Blomqvist A, Nilsberth C (2013) Immune-induced expression of lipocalin-2 in brain endothelial cells: relationship with interleukin-6, cyclooxygenase-2 and the febrile response. J Neuroendocrinol 25:271–280. 10.1111/jne.12000 [DOI] [PubMed] [Google Scholar]
- Inoue W, Matsumura K, Yamagata K, Takemiya T, Shiraki T, Kobayashi S (2002) Brain-specific endothelial induction of prostaglandin E(2) synthesis enzymes and its temporal relation to fever. Neurosci Res 44:51–61. 10.1016/S0168-0102(02)00083-4 [DOI] [PubMed] [Google Scholar]
- Ishikawa TO, Herschman HR (2006) Conditional knockout mouse for tissue-specific disruption of the cyclooxygenase-2 (Cox-2) gene. Genesis 44:143–149. 10.1002/gene.20192 [DOI] [PubMed] [Google Scholar]
- Jakobsson PJ, Thorén S, Morgenstern R, Samuelsson B (1999) Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci U S A 96:7220–7225. 10.1073/pnas.96.13.7220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix A, Toussay X, Anenberg E, Lecrux C, Ferreirós N, Karagiannis A, Plaisier F, Chausson P, Jarlier F, Burgess SA, Hillman EM, Tegeder I, Murphy TH, Hamel E, Cauli B (2015) COX-2-derived prostaglandin E2 produced by pyramidal neurons contributes to neurovascular coupling in the rodent cerebral cortex. J Neurosci 35:11791–11810. 10.1523/JNEUROSCI.0651-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenbach R, Loftin CD, Lee C, Tiano H (1999) Cyclooxygenase-deficient mice: a summary of their characteristics and susceptibilities to inflammation and carcinogenesis. Ann N Y Acad Sci 889:52–61. 10.1111/j.1749-6632.1999.tb08723.x [DOI] [PubMed] [Google Scholar]
- Lazarus M, Yoshida K, Coppari R, Bass CE, Mochizuki T, Lowell BB, Saper CB (2007) EP3 prostaglandin receptors in the median preoptic nucleus are critical for fever responses. Nat Neurosci 10:1131–1133. 10.1038/nn1949 [DOI] [PubMed] [Google Scholar]
- Li S, Wang Y, Matsumura K, Ballou LR, Morham SG, Blatteis CM (1999) The febrile response to lipopolysaccharide is blocked in cyclooxygenase-2(-/-), but not in cyclooxygenase-1(-/-) mice. Brain Res 825:86–94. 10.1016/S0006-8993(99)01225-1 [DOI] [PubMed] [Google Scholar]
- Macdonald JA, Murugesan N, Pachter JS (2010) Endothelial cell heterogeneity of blood-brain barrier gene expression along the cerebral microvasculature. J Neurosci Res 88:1457–1474. 10.1002/jnr.22316 [DOI] [PubMed] [Google Scholar]
- Matsumura K, Cao C, Watanabe Y (1997) Possible role of cyclooxygenase-2 in the brain vasculature in febrile response. Ann N Y Acad Sci 813:302–306. 10.1111/j.1749-6632.1997.tb51709.x [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Hamzic N, Norell M, Blomqvist A (2009a) Peripheral lipopolysaccharide administration induces cytokine mRNA expression in the viscera and brain of fever-refractory mice lacking microsomal prostaglandin E synthase-1. J Neuroendocrinol 21:715–721. 10.1111/j.1365-2826.2009.01888.x [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Elander L, Hamzic N, Norell M, Lönn J, Engström L, Blomqvist A (2009b) The role of interleukin-6 in lipopolysaccharide-induced fever by mechanisms independent of prostaglandin E2. Endocrinology 150:1850–1860. 10.1210/en.2008-0806 [DOI] [PubMed] [Google Scholar]
- Oishi Y, Yoshida K, Scammell TE, Urade Y, Lazarus M, Saper CB (2015) The roles of prostaglandin E2 and D2 in lipopolysaccharide-mediated changes in sleep. Brain Behav Immun 47:172–177. 10.1016/j.bbi.2014.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Oka K, Scammell TE, Lee C, Kelly JF, Nantel F, Elmquist JK, Saper CB (2000) Relationship of EP(1–4) prostaglandin receptors with rat hypothalamic cell groups involved in lipopolysaccharide fever responses. J Comp Neurol 428:20–32. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates, Ed 2. London: Academic. [Google Scholar]
- Ridder DA, Lang MF, Salinin S, Röderer JP, Struss M, Maser-Gluth C, Schwaninger M (2011) TAK1 in brain endothelial cells mediates fever and lethargy. J Exp Med 208:2615–2623. 10.1084/jem.20110398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudaya AY, Steiner AA, Robbins JR, Dragic AS, Romanovsky AA (2005) Thermoregulatory responses to lipopolysaccharide in the mouse: dependence on the dose and ambient temperature. Am J Physiol Regul Integr Comp Physiol 289:R1244–R1252. 10.1152/ajpregu.00370.2005 [DOI] [PubMed] [Google Scholar]
- Saha S, Engström L, Mackerlova L, Jakobsson PJ, Blomqvist A (2005) Impaired febrile responses to immune challenge in mice deficient in microsomal prostaglandin E synthase-1. Am J Physiol Regul Integr Comp Physiol 288:R1100–R1107. 10.1152/ajpregu.00872.2004 [DOI] [PubMed] [Google Scholar]
- Samuelsson B, Granström E, Green K, Hamberg M, Hammarström S (1975) Prostaglandins. Annu Rev Biochem 44:669–695. 10.1146/annurev.bi.44.070175.003321 [DOI] [PubMed] [Google Scholar]
- Scammell TE, Elmquist JK, Griffin JD, Saper CB (1996) Ventromedial preoptic prostaglandin E2 activates fever-producing autonomic pathways. J Neurosci 16:6246–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammell TE, Griffin JD, Elmquist JK, Saper CB (1998) Microinjection of a cyclooxygenase inhibitor into the anteroventral preoptic region attenuates LPS fever. Am J Physiol 274:R783–R789. [DOI] [PubMed] [Google Scholar]
- Splawinski JA. (1977) Mediation of hyperthermia by prostaglandin E2: a new hypothesis. Naunyn Schmiedebergs Arch Pharmacol 297 [Suppl 1]:S95–S97. 10.1007/BF00587791 [DOI] [PubMed] [Google Scholar]
- Steiner AA, Ivanov AI, Serrats J, Hosokawa H, Phayre AN, Robbins JR, Roberts JL, Kobayashi S, Matsumura K, Sawchenko PE, Romanovsky AA (2006) Cellular and molecular bases of the initiation of fever. PLoS Biol 4:e284. 10.1371/journal.pbio.0040284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP (2003) Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci U S A 100:9044–9049. 10.1073/pnas.1332766100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilache AM, Qian H, Blomqvist A (2015) Immune challenge by intraperitoneal administration of lipopolysaccharide directs gene expression in distinct blood–brain barrier cells toward enhanced prostaglandin E2 signaling. Brain Behav Immun 48:31–41. 10.1016/j.bbi.2015.02.003 [DOI] [PubMed] [Google Scholar]
- Westholm DE, Rumbley JN, Salo DR, Rich TP, Anderson GW (2008) Organic anion-transporting polypeptides at the blood-brain and blood-cerebrospinal fluid barriers. Curr Top Dev Biol 80:135–170. 10.1016/S0070-2153(07)80004-4 [DOI] [PubMed] [Google Scholar]
- Wilhelms DB, Kirilov M, Mirrasekhian E, Eskilsson A, Kugelberg UÖ, Klar C, Ridder DA, Herschman HR, Schwaninger M, Blomqvist A, Engblom D (2014) Deletion of prostaglandin E2 synthesizing enzymes in brain endothelial cells attenuates inflammatory fever. J Neurosci 34:11684–11690. 10.1523/JNEUROSCI.1838-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, Sugiura H, Cao C, Watanabe Y, Kobayashi S (2001) Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin-induced fever. J Neurosci 21:2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Rivest S (1999) Distribution, regulation and colocalization of the genes encoding the EP2- and EP4-PGE2 receptors in the rat brain and neuronal responses to systemic inflammation. Eur J Neurosci 11:2651–2668. 10.1046/j.1460-9568.1999.00682.x [DOI] [PubMed] [Google Scholar]
- Zhao ZD, Yang WZ, Gao C, Fu X, Zhang W, Zhou Q, Chen W, Ni X, Lin JK, Yang J, Xu XH, Shen WL (2017) A hypothalamic circuit that controls body temperature. Proc Natl Acad Sci U S A 114:2042–2047. 10.1073/pnas.1616255114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15:871–875. 10.1038/nbt0997-871 [DOI] [PubMed] [Google Scholar]