

Abstract
The lateral habenula (LHb) is a small part of the epithalamus that projects to monoamine centers in the brain. Previously, neurotransmission onto the LHb was shown to be abnormally potentiated in animal models of depression. However, synaptic plasticity in this brain area and the effect of stressor exposure on synaptic plasticity of the LHb have not been investigated. Thus, we explored whether the LHb undergoes dynamic changes in synaptic efficacy or not. First, we observed that a moderate LTP occurs in a fraction of LHb neurons obtained from naive Sprague Dawley rats. Interestingly, a single exposure to acute stressors, such as inescapable foot shock or restraint plus tail shock (RTS), significantly enhances the magnitude of LTP in the LHb. We also observed an increased number of LHb neurons expressing phosphorylated cAMP response element-binding protein (pCREB) after exposure to stressors, which may contribute to determine the threshold for LTP induction. LTP induction in the LHb resulted in an additional increase in the number of pCREB-expressing neurons in stress-exposed animals but not in naive control animals. Together, we showed that LHb neurons have heterogeneous propensity for synaptic potentiation at rest; however, a single exposure to stressors greatly facilitates LTP induction in the LHb, suggesting that fundamental alterations in synaptic plasticity in the LHb may occur in animal models of depression or post-traumatic stress disorder.
SIGNIFICANCE STATEMENT Stress exposure is known to cause depression in human patients and animal models, although explanations at the cellular level remain to be elaborated. Here, we show that the lateral habenula (LHb) exhibits LTP after a pattern of brief strong stimulation. In addition, we show that stress exposure facilitates LTP in the LHb by lowering the threshold for LTP induction. We observed a selective increase in the number of neurons expressing pCREB in the LHb of animal models of depression. LTP induction results in a further increase in the density of pCREB-expressing neurons only after stress exposure. Our study provides the first evidence that animal models of depression exhibit altered synaptic plasticity of the LHb.
Keywords: depressive disorders, long-term potentiation, post-traumatic stress disorder, synaptic plasticity, the lateral habenula
Introduction
Several brain areas undergo synaptic changes in animal models of depression (Duman, 2002; Krishnan and Nestler, 2008; Duman and Aghajanian, 2012). One such region is the lateral habenula (LHb), a part of the epithalamus. The LHb receives inputs from areas of the forebrain, such as the ventral pallidum, globus palidus, and lateral preoptic area through the stria medularis, and projects to midbrain structures, such as the ventral tegmental area, rostromedial tegmental nucleus, and dorsal raphe nucleus (Herkenham and Nauta, 1977; Semba and Fibiger, 1992; Gonçalves et al., 2012; Shabel et al., 2012; Sego et al., 2014). Several lines of recent evidence suggest that the LHb is likely to play a critical role in depressive disorders (Sartorius and Henn, 2007; Sartorius et al., 2010; Li et al., 2011, 2013; Meng et al., 2011; Shabel et al., 2014). Importantly, metabolic activity in the LHb increases both in rodent models of depression and in human patients with major depressive disorder (Caldecott-Hazard et al., 1988; Shumake and Gonzalez-Lima, 2003). Excitatory synaptic inputs onto the LHb are abnormally potentiated in acute and congenital learned helpless animal models of depression. Thus, the potentiation of the LHb could likely mediate helplessness behaviors in rodents (Li et al., 2011, 2013), whereas lesions or deep brain stimulation of the LHb were reported to ameliorate depression-like symptoms in animal models and a treatment-resistant human patient (Yang et al., 2008; Sartorius et al., 2010; Li et al., 2011; Meng et al., 2011).
It has been suggested that stressful experiences disrupt synaptic plasticity in brain areas implicated in depressive disorders, including the hippocampus and PFC (Krishnan and Nestler, 2008; Duman and Aghajanian, 2012). Exposure to behavioral stressors, including repeated restraint stress and social defeat stress, was shown to impair LTP in the hippocampus (Foy et al., 1987; Garcia et al., 1997; Shors et al., 1997; Artola et al., 2006). Acute stress exposure has also been reported to facilitate LTP induction in the hippocampus-PFC pathway (Goto and Grace, 2006), whereas chronic stress exposure was shown to impair LTP induction in the PFC (Cerqueira et al., 2007).
It is well established that plastic changes in synapses accompany changes in the expression of cAMP response element-binding protein (CREB) (Dash et al., 1990; Pittenger et al., 2002; Ahmed and Frey, 2005; Benito and Barco, 2010; Gruart et al., 2012; Kandel, 2012). Classically, long-term facilitation of Aplysia neurons is impaired following microinjection of cAMP response element (CRE), which interacts with CREB and competes with endogenous CRE sites to bind to CREB (Dash et al., 1990). Certain types of LTP are impaired in the dorsal hippocampus of dominant-negative mutant CREB transgenic mice (Bourtchuladze et al., 1994; Pittenger et al., 2002), whereas hippocampal LTP is enhanced by the constitutively activated form of CREB (Du et al., 2000; Suzuki et al., 2011). Molecular studies have shown that patients with depression who are unmedicated have decreased CREB levels in the cerebral cortex (Dowlatshahi et al., 1998), whereas viral-mediated overexpression of CREB in the hippocampus ameliorates helpless behaviors in rodents (Chen et al., 2001). These studies suggest that restoring impaired synaptic plasticity may contribute to improvements in helpless behaviors in rodent models of depression.
Given that LHb activity is correlated with negative affective states in animals (Matsumoto and Hikosaka, 2007; Li et al., 2011), we hypothesized that synaptic efficacy in the LHb undergoes alterations depending on the emotional valence of experiences. We used two well-established acute stress paradigms: the inescapable, unpredictable foot shock (FS) paradigm, an animal model of learned helplessness (Maier, 1984; Li et al., 2011) and the restraint plus tail shock (RTS) paradigm, an animal model of post-traumatic stress disorder (Kim et al., 1996; Brennan et al., 2005). We investigated whether LTP occurs in the LHb as in other brain areas, and how exposure to stressful experiences may alter the capacity for synaptic plasticity in this brain area.
Materials and Methods
Subjects
Six- to 8-week-old male Sprague Dawley rats (Orient Bio, a branch of Charles River) were group-housed (4 rats per cage) under standard laboratory conditions on a 12 h light-dark cycle (lights on at 7:00 A.M., 45% humidity, 23 ± 1°C). Animals had free access to food and water. This study was performed in strict accordance with the recommendations in the National Institutes of Health Guide for the care and use of laboratory animals. All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee of Konkuk University, Seoul, Korea.
Behavioral procedures
Exposure to stressors.
Rats were subjected to either inescapable, unpredictable FS stress or RTS stress 24 h before behavioral tests or slice preparation. For the FS model, the animals were placed in inescapable chambers (12 width × 10 depth × 12 height, Coulbourn Instruments) and received unpredictable electrical FSs (120 unpredictable electric FSs at 0.8 mA over 40 min, controlled by precision adjustable shockers) (Li et al., 2011). For the RTS model, the animals were restrained in triangle-shaped plastic bags and received uncontrollable electrical shocks (60 unpredictable electric tail shocks at 1.2 mA, controlled by precision adjustable shockers) to the tail for an hour (Foy et al., 1987; Kim and Diamond, 2002; Kim et al., 2005). After exposure to the stressors, the rats were housed individually. Nonstressed animals remained group-housed until 24 h before behavioral testing.
Sucrose preference test.
A two-bottle sucrose preference test was used to test the animals' anhedonic behavior. Rats were singly caged during this test and deprived of water for 4 h before the test. Both bottles with tap water and 1% sucrose solution were presented to the single caged rat for 1 h at the beginning of the dark cycle for 2 consecutive days. The amount of consumption for each solution was measured by comparing the weight of the bottle before and after the test. Sucrose preference was quantified as the amount of sucrose consumption divided by total amount of liquid consumption on day 2.
Elevated plus maze.
To test the level of anxiety, we conducted an elevated plus maze task. This apparatus consisted of a central platform (10 cm × 10 cm), 50 cm off the ground with four branching arms (50 cm × 10 cm), where two of the arms facing each other are open and the other two arms are enclosed by Plexiglas walls (40 cm high). Rats were placed at the center of the plus maze and allowed to freely explore the maze for 5 min. The entire sessions were recorded by a video camera mounted at the top of the maze. The time spent and the number of entries in open arms and close arms were measured manually in offline. The percentage of time an animal spent in the open arms was quantified as the amount of open arm retention time divided by the total test time (5 min).
Forced swim test (FST).
To test despair behavior in the rat, we measured immobility within an inescapable water bath (25 × 50 cm). A cylinder-shaped water bath was filled with 26 ± 1°C water to height of 35 cm. Rats remained in the water for 15 min, and we measured their immobility during this time. The animals were then removed from the cylinder and dried using a towel. On the next day, the same protocol was repeated. The FST sessions were recorded by a video camera from the side of the cylinder, and the amount of time that the animal spent immobile, passively floating in the water was scored manually by a trained scorer. The scorer was blinded to the condition of behavioral manipulation. Immobility was quantified as the length of immobile time during the last 5 min of the FST on the second test day (Li et al., 2011).
Slice preparation and electrophysiology
Animals were killed for electrophysiological recordings 24 h after FS or RTS. Sagittal brain slices containing the LHb (350 μm thick) were prepared using a Leica VT 1000S with ice-cold choline dissection buffer (in mm as follows: 110 choline chloride, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 11.6 Na-ascorbic acid, 3.1 pyruvic acid, 7 MgCl2, 0.5 CaCl2, 25 glucose) bubbled with 95% O2/5% CO2 mixed gas. Brain slices were placed in a recovery chamber containing aCSF (in mm as follows: 1 NaH2PO4, 26.2 NaHCO3, 118 NaCl, and 2.5 KCl, 11 glucose, 2 CaCl2, and 1 MgCl2 with bubbling), kept for 45 min in a 35°C water bath, and then stored at room temperature. LHb neurons were voltage clamped at −60 mV using Axopatch 200B and Clampex 10.3 (Molecular Devices), filtered at 5 kHz, and sampled at 10 kHz. Recording pipettes with resistances of 2–6 mΩ were filled with internal solution (in mm as follows: 115 Cs-methanesulphonate, 20 CsCl, 10 HEPES, 2.5 MgCl2, 0.6 EGTA, 5 QX314, 4 Na2-ATP, 0.4 Na2-GTP, and 10 Na-phosphocreatine). To isolate EPSCs, picrotoxin (50 μm in DMSO) was added to the aCSF. Evoked EPSCs (eEPSCs) were elicited by placing a bipolar stimulator (FHC) on the stria medullaris. All electrophysiological recordings were obtained at a holding potential of −60 mV at 30°C-32°C. Recordings that showed a >20% change in the amplitude of eEPSCs during the baseline in either pathway were discarded from further analysis. The magnitude of LTP was determined by the averaged eEPSC amplitudes during the 25–30 min after LTP induction compared with those during the 5 min baseline.
Immunohistochemistry
For c-fos immunoreactivity experiments, 45 min after the end of exposure to the stressor, the animals were deeply anesthetized using isoflurane and transcardially perfused with 0.01 m PBS, followed by cold 4% PFA in 0.01 m PBS. Brains were postfixed with 4% PFA for 24 h and transferred to a 30% sucrose solution until they sank for cryoprotection. Free floating brain sections (thickness: 50 μm, 8 sections/animal, 6 or 7 animals/group) were incubated with 3% FBS/0.3% BSA/0.2% Triton X-100 in 0.1 m phosphate buffer for an hour at room temperature. They were then incubated with anti-c-fos antibody (1:1000, Santa Cruz Biotechnology) overnight. After wash, sections were incubated with fluorescence-conjugated secondary antibody (1:500, AlexaFluor-488 or -594, Invitrogen) in 0.3% BSA/0.3% Triton X-100/0.1 m phosphate buffer for 1 h. After 5 times of washes, sections were mounted on slides and protected using ProLong Gold antifade reagent (Invitrogen). Images were obtained using an FV1000 spectral microscope (Olympus) and analyzed with ImageJ software.
For projection-specific c-fos analysis, a retrograde tracer, cholera toxin subunit B conjugated with AlexaFluor-488 (1 μl, Invitrogen) was injected into the ventral tegmental area (VTA, anteroposterior: −5.6, mediolateral: ±1, dorsoventral: −8.55), rostromedial tegmental nucleus (RMTg, anteroposterior: −5.6, mediolateral: ±1, dorsoventral: −8.55), or dorsal raphe nucleus (DRN, anteroposterior: −7.44, mediolateral: ±3.8, dorsoventral: −6.6, θ = 303). Three days after the injection, the animals were exposed to stress and prepared for c-fos immunohistochemistry experiments.
For phosphorylated CREB immunoreactivity experiments, 1 d after application of either the stress or no behavior paradigm, rats were anesthetized with isoflurane and whole brains were removed for preparation of acute brain slices. After 1 h of recovery, LTP was induced by theta burst stimulation (TBS) on stria medullaris. Habenular-containing brain slices were fixed immediately with 4% PFA for 2 d and sectioned at 50 μm using a microtome. Endogenous peroxidase activity was quenched by incubating in 3% H2O2/10% MeOH in PBS for 30 min. After incubation in a blocking solution containing 10% FBS/0.3% Triton X-100/0.01 m PBS for an hour, brain sections (5 sections/animal, 4 animals/group) were then incubated with phosphorylated CREB antibody (pCREB, 1:1000, Millipore) for an hour at room temperature and overnight at 4°C. After washing, sections were incubated for 1 h with biotinylated anti-rabbit IgG (1:1000, Vector Laboratories) and then for 1 h in ExtrAvidin-peroxidase conjugate (1:1000, Sigma-Aldrich). A diaminobenzidine peroxidase substrate kit (Vector Laboratories) was used for staining. After mounting, sections were dried for at least 5 d and protected with Permount reagent (Fisher Scientific). Images were obtained using a microscope (BX51, Olympus) with an attached digital microscope camera (DP72, Olympus) and analyzed using ImageJ software.
Statistical analysis
The data were analyzed using Student's t tests, ANOVA tests with Bonferroni correction, and F tests unless specified otherwise. p values <0.05 were considered significant. The data are shown as mean ± SEM.
Results
Long-term synaptic potentiation occurs in a subset of LHb neurons
The LHb has been shown to be highly potentiated during negative affective states, such as the absence of expected reward (Matsumoto and Hikosaka, 2007) or chronic depression (Li et al., 2011; Proulx et al., 2014). To investigate whether synaptic plasticity occurs in the LHb, we recorded eEPSCs from LHb neurons in sagittal brain slices while stimulating the stria medullaris (Fig. 1A). Upon TBS (5 stimuli at 100 Hz, repeated 20 times with 200 ms intervals), the amplitude of the eEPSCs of the LHb showed only a very moderate potentiation of ∼ 20% (n = 16, p < 0.05; Fig. 1C,D). The magnitudes of LTP exhibited a bimodal distribution, suggesting that there are two populations of cells with different capacities for LTP induction (Fig. 1E). The median of the LTP magnitude was 115.9% of baseline (n = 16, all recordings included). Eight of 16 cells exhibited no potentiation upon TBS (Fig. 1B, right, representative recording of a cell with no potentiation, 94.9 ± 4.5% of baseline), whereas the remaining cells exhibited significant potentiation upon TBS (Fig. 1B, left, representative recording of a cell with potentiation, 140.5 ± 5.6% of baseline). The magnitude of LTP was significantly different between the potentiated and nonpotentiated neurons (p < 0.001). The peak amplitudes of eEPSCs do not determine whether potentiation occurs upon TBS. Baseline eEPSC amplitudes between control versus stimulated pathway, as well as between potentiated versus nonpotentiated neurons were not different (p > 0.1). Our observations suggest that there is a heterogeneous propensity for induction of synaptic plasticity in the LHb (Fig. 1E): moderate LTP occurs in a subset of LHb neurons (Fig. 1B, left), whereas no LTP occurs in another subpopulation of LHb neurons (Fig. 1B, right). Overall, TBS induces mild synaptic potentiation in LHb neurons (Fig. 1C,D).
Figure 1.

Mild long-term potentiation occurs in the LHb. A, Recording configuration. B, Examples of TBS-induced (5 stimuli at 100 Hz, repeated 20 times with 200 ms intervals) synaptic potentiation in the LHb obtained from naive animals. Left, Example of a potentiated LHb neuron. Right, Nonpotentiated LHb neuron. Calibration: 20 ms, 100 pA. C, Summary of compiled data for normalized amplitudes of eEPSCs before and after TBS (117.7 ± 6.9% of baseline for stimulated pathway, n = 16, p < 0.05 compared with before TBS or the control pathway). D, Averaged magnitude of LTP after TBS in naive animals (CTRL: unstimulated control pathway; TBS: stimulated pathway). E, The distribution of probability plot for the magnitude of LTP in naive animals. *p < 0.05.
Stress exposure activates LHb neurons with limited specificity
A growing body of literature indicates that stressful experiences may alter synaptic efficacy or synaptic plasticity in many brain areas that are reportedly involved in stress regulation or reward processing (Pittenger and Duman, 2008). We therefore investigated whether stressor exposure may shift the propensity for LTP induction in the LHb. We used two behavioral paradigms: inescapable FS stress and uncontrollable RTS stress. It has been shown that the FS paradigm induces helpless behaviors in rodents (Li et al., 2011). To examine whether a single exposure to FS or RTS induces depressive-like behaviors, either inescapable and unpredictable FSs were administered to the animals (120 shocks, 0.8 mA for 10 s each) or unpredictable tail shocks were given for 1 h (60 shocks, 1.2 mA for 1 s each) while the animals were restrained (Kim et al., 1996). A series of behavioral tests performed 24 h after exposure to FS or RTS (Fig. 2A) showed that the animals exhibited a significantly reduced preference for sucrose (F(2,29) = 8.07, p < 0.01 by one-way ANOVA with Bonferroni correction; Fig. 2B), time on the open arm of elevated plus maze (F(2,29) = 5.44, p < 0.05; Fig. 2C, left), and open arm entry (F(2,29) = 7.05, p < 0.01; Fig. 2C, right). Moreover, increased immobility during the FST was observed (F(2,29) = 7.71, p < 0.05; Fig. 2D). These data suggest that both FS and RTS paradigms induce helpless behaviors in rodents. RTS-exposed animals spent notably less time in the open arms of the elevated plus maze (p < 0.01 compared with naive animals, p < 0.05 compared with FS animals; Fig. 2C, left, by Bonferroni post hoc analysis). Time spent in the open arms in FS-exposed animals was comparable with that in unstressed naive animals (p > 0.9; Fig. 2C, left). The FS group showed a tendency for a decreased number of entries to the open arms (p = 0.078; Fig. 2C, right), whereas the RTS group had a significantly decreased number of entries to the open arms (p < 0.01; Fig. 2C, right). These observations suggest that both types of stressors induce anxiety-like behavior with helplessness, although FS exposure may induce milder anxiety compared with RTS exposure.
Figure 2.
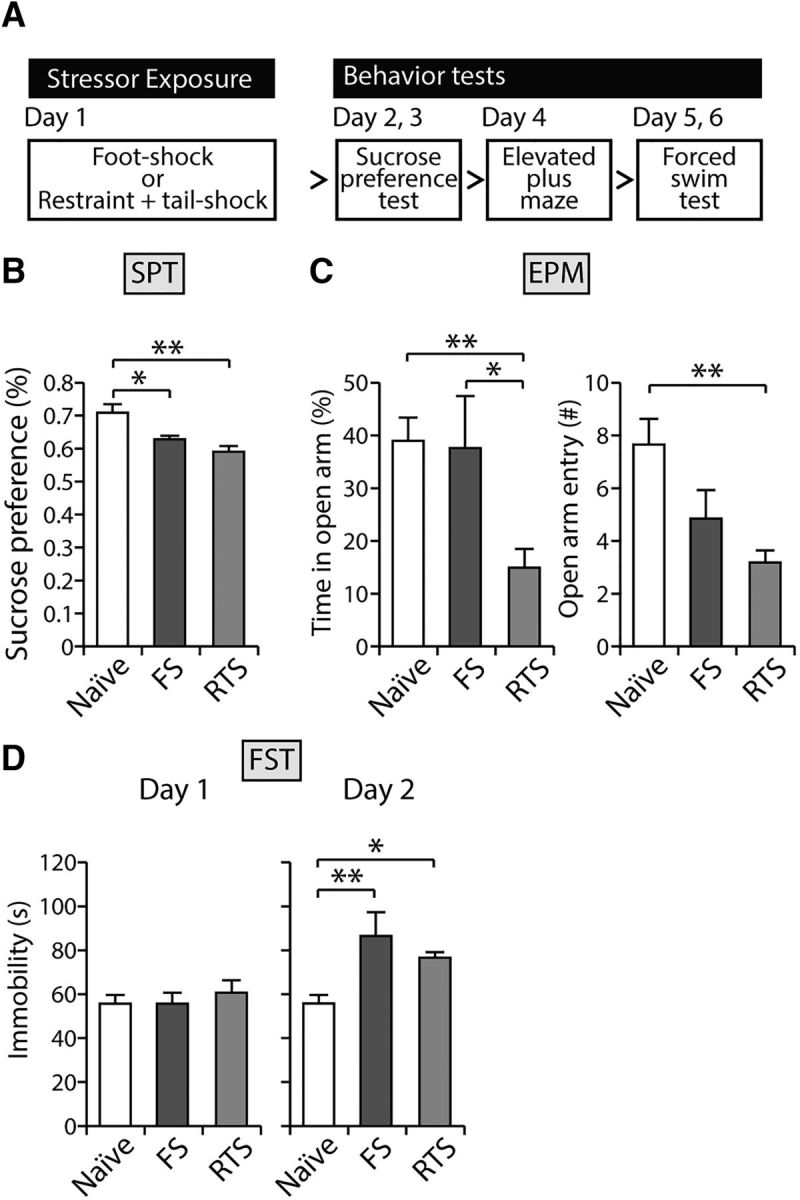
Both stress paradigms induce depression-like behaviors in rodents. A, Experimental scheme. B, FS and RTS animals exhibited significantly reduced preference for 1% sucrose solution compared with naive animals in the sucrose preference test (n = 8–14, F(2,29) = 8.07, p < 0.01, one-way ANOVA with Bonferroni post hoc analysis; p < 0.05 compared with naive and FS animals; p < 0.01 compared with naive and RTS animals). C, The amount of time spent in the open arm in elevated plus maze was significantly decreased in RTS animals (F(2,29) = 5.44, p < 0.01). However, this was comparable in the naive and FS groups (p > 0.9). RTS animals entered the open arms less frequently than naive animals (F(2,29) = 7.05, p < 0.01). The FS group had a tendency for less open arms entries (p < 0.1). D, FS and RTS animals showed comparable immobility times on the first day of the FST (F(2,29) = 0.33, p > 0.7). However, both groups of stressed animals spent significantly prolonged immobility times on day 2 in the FST compared with naive animals (F(2,29) = 7.71, p < 0.01, one-way ANOVA; p < 0.01, Bonferroni post hoc analysis compared with naive and FS animals; p < 0.05 compared with naive and RTS animals). *p < 0.05. **p < 0.01.
In another set of animals, we obtained brain samples 45 min after each stress paradigm and quantitatively analyzed c-fos immunoreactivity in the habenula complex (Fig. 3). The density of c-fos-positive neurons was selectively increased after FS or RTS in the LHb but not in the medial habenula (mHb), compared with unstressed naive animals (6 or 7 animals per group, 8 sections averaged per animal, F(2,17) = 2.3, p > 0.1 for mHb; F(2,17) = 31.40, p < 0.001 for LHb; Fig. 3B; one-way ANOVA with Bonferroni correction). In addition, there were significantly more c-fos-expressing neurons in the LHb after RTS than after FS (p < 0.01 by Bonferroni post hoc analysis). These observations suggest that exposure to stressors activates a subpopulation of LHb neurons and that the activation of the LHb may be correlated with the intensity of the stressor.
Figure 3.
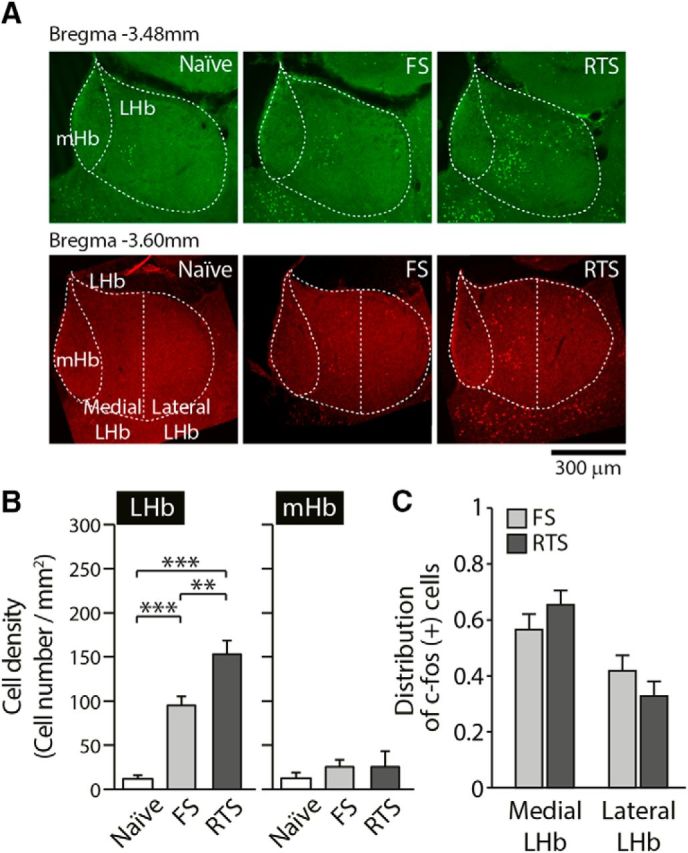
Exposure to stressors activates LHb neurons. A, Representative images of c-fos immunoreactivity in each group. B, The density of c-fos-positive cells in the LHb (right) was significantly increased after exposure to FS or RTS but not in the mHb (left) (one-way ANOVA, F(2,17) = 2.3, p > 0.1 for the mHb; F(2,19) = 38.26, p < 0.001 for the LHb, p < 0.001 when comparing naive vs FS animals, p < 0.001 when comparing naive vs RTS animals, Bonferroni post hoc analysis). In addition, the LHb showed significantly more c-fos-positive cells following RTS than after FS exposure (p < 0.01, Bonferroni post hoc analysis). C, Densities of c-fos-positive cells in the medial part of the LHb (medial LHb) and the lateral part of the LHb (lateral LHb) were analyzed 45 min after exposure to FS and RTS. The distributions of c-fos-positive cells do not differ in FS versus RTS animals (5 animals per group, 10 sections averaged per animal, two-way ANOVA, F(1,16) = 2.41, p > 0.1). **p < 0.01. ***p < 0.001.
To examine whether there is any specificity in the LHb neurons responding to stress, we quantified the density of c-fos-expressing neurons in the medial part of the LHb and the lateral part of the LHb, separately (Fig. 3). Even though the total numbers of c-fos-expressing neurons following FS and RTS are significantly different, the distribution of c-fos-positive neurons remained comparable following the two stress paradigms used (F(1,16) = 2.41, p > 0.1 two-way ANOVA; Fig. 3C). After FS and RTS, 57.5% and 66.6% of the c-fos-expressing neurons were found in the medial part of the LHb, respectively. Both stressors seem to recruit more neurons in the medial part than the lateral part of the LHb, although this difference was not statistically significant (p = 0.09 for FS, p = 0.12 for RTS).
Next, we analyzed the origins of the stress-activated LHb neurons using c-fos immunoreactivity analysis combined with a retrograde tracer injection. We injected a retrograde tracer, cholera toxin subunit B, conjugated with Alexa-488 into the VTA, RMTg, and DRN. This was followed by staining using c-fos antibody with or without stress exposure (Fig. 4). We found that the LHb sends stronger projections to the VTA than the RMTg or the DRN (Fig. 4A,B; two-way ANOVA, F(2,63) = 89.34, p < 0.001). Acute stress exposure did not alter the ratios of neurons projecting to these areas (Fig. 4B; two-way ANOVA, F(2,63) = 2.11, p > 0.1). We repeatedly observed an increased number of c-fos-expressing neurons after stress exposure (Fig. 4A,C; one-way ANOVA, F(2,63) = 65.182, p < 0.001). When we quantified the colabeled neurons, we failed to observe any significant preference for activation depending on the projection sites (Fig. 4D; two-way ANOVA, F(2,63) = 1.12, p > 0.3). Therefore, stress-induced LHb activation does not seem to be specific to either any subnucleus (Fig. 3) or projection site (Fig. 4).
Figure 4.
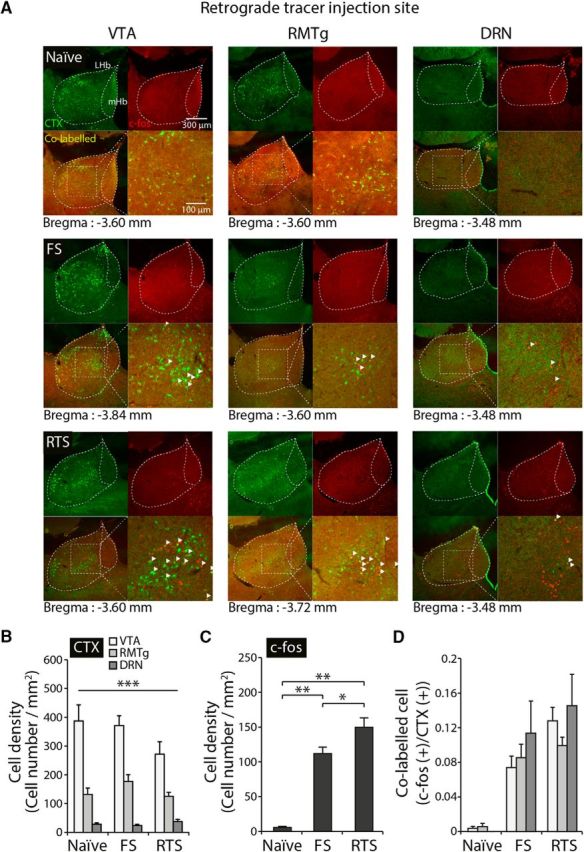
The activation of LHb neurons following stress exposure is independent of where they project. A, Representative images showing cholera toxin subunit B (CTX) and c-fos-colabeled cells in the habenular complex of naive, FS-exposed, and RTS-exposed animals. B, The density of CTX-positive cells in the LHb was significantly different depending on the LHb projection site (two-way ANOVA, F(2,63) = 89.41, p < 0.001). The strongest projection to the LHb was found to originate at the VTA, whereas the weakest projection was from the DRN. Stress exposure did not alter CTX-positive cell density depending on projection site (two-way ANOVA, F(2,63) = 2.11, p > 0.1). C, Density of c-fos-expressing neurons in the LHb. FS- and RTS-exposed animals exhibited significantly higher levels of c-fos (one-way ANOVA, F(2,69) = 3.13, p < 0.001). D, The numbers of colabeled cells were increased after exposure to stress (two-way ANOVA, F(2,63) = 30.31, p < 0.001). However, there was no significant difference in the ratio of colabeled LHb neurons depending on projection site after stress exposure compared with naive controls (two-way ANOVA, F(2,63) = 1.12, p > 0.3). There was no interaction effect (two-way ANOVA, F(2,63) = 0.69, p > 0.6). *p < 0.05. **p < 0.01. ***p < 0.001.
Exposure to stressors facilitates the induction of long-term synaptic potentiation in the LHb
Next, we investigated whether the stress-induced activation of LHb neurons altered the induction of LTP. Surprisingly, upon TBS onto the stria medullaris, the amplitudes of eEPSCs of the LHb obtained from FS- or RTS-exposed animals was greatly potentiated compared with that obtained from naive animals (n = 8–13, p < 0.05; Fig. 5A,B,D,E). A one-way ANOVA of the magnitude of LTP showed that the between-group effects (naive, FS, and RTS) were significant (F(2,36) = 5.44, p < 0.01). As shown in Figure 5C, F, there is an increased propensity for potentiation after exposure to stressors compared with the nonstressed naive group (shown in Fig. 1E). Moreover, exposure to stressors reduced the frequency with which no potentiation was observed. The magnitude of potentiation in those recordings showing potentiation higher than the median of naive group (115.9%) was larger following stressor exposure than when there were no stressors (40.5% potentiation in naive group vs 80.3% and 106.4% potentiation in the FS and RTS groups, respectively) (Fig. 5C,F). The median LTP magnitudes were 129.2% and 197.7% of baseline for the FS- and RTS-exposed groups, respectively. In addition, the magnitude of the LTP plot shows that there is a significantly higher variance in the stressed groups compared with the naive group (p < 0.001 for naive vs FS group, p < 0.01 for naive vs RTS group, F test; Fig. 5G). When we plotted the averaged magnitudes of LTP for each group against the average numbers of c-fos-positive cells in the LHb, we found a strong correlation between LTP magnitude and the number of c-fos-expressing neurons (R2 = 0.987; Fig. 5H). This observation suggests that the activation of the LHb may correlate with the propensity for LTP induction in the LHb.
Figure 5.

Exposure to stressors facilitates the induction of LTP in the LHb and alters the propensity for potentiation in the LHb. A, Summary of normalized amplitudes of eEPSCs before and after TBS in FS-exposed animals. Calibration: 20 ms, 100 pA. B, Averaged magnitude of LTP measured 30 min after TBS. After TBS, the amplitude of eEPSC was further potentiated in FS animals compared with naive animals (154.7 ± 20% of baseline, n = 13, p < 0.05). C, The probability plot for the magnitude of LTP in FS-exposed animals compared with naive animals. D, Summary of normalized amplitudes of eEPSCs before and after TBS in RTS-exposed animals. Calibration: 20 ms, 100 pA. E, Averaged magnitude of LTP measured 30 min after TBS. After TBS, the amplitude of eEPSC was further potentiated in RTS animals compared with naive animals (191.1 ± 19% of baseline, n = 8, p < 0.05). F, The probability plot for the magnitude of LTP in RTS-exposed animals compared with naive animals. G, Comparison of the magnitude of LTP in the stimulated pathway in individual experiments between naive, FS-exposed, and RTS-exposed groups (one-way ANOVA test, F(2,36) = 5.44, p < 0.01). The sample variances were 0.08, 0.52, and 0.29 for the naive, FS-, and RTS-exposed groups, respectively (F test, p < 0.01). H, Correlation between c-fos-positive cell density and magnitude of LTP (R2 = 0.987). *p < 0.05. **p < 0.01. ***p < 0.001.
The number of neurons expressing phosphorylated CREB is increased in the LHb of RTS-exposed animals after LTP induction
LTP induction is known to be accompanied by an increase in the expression levels of CREB and/or the phosphorylation of CREB (Pittenger and Kandel, 1998). To examine whether TBS at the LHb increases the levels of previously known synaptic plasticity-related molecules, we measured the levels of pCREB in the LHb. One day after exposure to the stressors, acute brain slices containing the LHb were obtained from naive and RTS animals. Forty-five minutes after TBS, control or LTP-induced LHb slices from naive, FS-, or RTS-exposed animals were fixed for immunohistochemical analysis of pCREB (Fig. 6). We observed that stress exposure per se increased the density of pCREB-positive neurons in the LHb (4 animals per group, 5 sections averaged per animal, F(2,114) = 9.37, p < 0.001 two-way ANOVA; F(2,57) = 3.85, p < 0.05 one-way ANOVA with Bonferroni correction; Fig. 6B). The densities of pCREB-expressing neurons after TBS are also different after stress exposure (p < 0.01 one-way ANOVA with Bonferroni correction; Fig. 6B). In addition, the magnitude of LTP was strongly correlated with the density of pCREB-expressing neurons before TBS (R2 = 0.969) and after TBS (R2 = 0.997). These analyses suggest that stress exposure lowers the threshold for LTP, thus facilitating LTP induction in the LHb. The density of pCREB-positive neurons in the mHb remained unchanged upon exposure to stressors (F(2,114) = 0.42, p > 0.7 two-way ANOVA) or TBS (F(1,114) = 0.45, p > 0.5, two-way ANOVA; Fig. 6C). Our observations suggest that exposure to stressful events may facilitate phosphorylation of CREB, accompanied with further long-term synaptic potentiation of LHb synapses.
Figure 6.

Stress exposure increased the density of neurons expressing phosphorylated CREB in the LHb. A, Representative images showing pCREB immunoreactivity in the habenular complex of naive, FS-, and RTS-exposed animals. B, The density of pCREB-expressing cells in the LHb was significantly increased upon exposure to stressors (two-way ANOVA, F(2,114) = 9.37, p < 0.001). Stress exposure per se increased pCREB-positive cell density in the LHb in the absence of any stimulation (F(2,57) = 3.85, p < 0.05, one-way ANOVA with Bonferroni post hoc analysis). C, No significant differences in the density of pCREB-positive cells were observed in the mHb upon stress exposure (two-way ANOVA, F(2,114) = 0.42, p > 0.7) or TBS (two-way ANOVA, F(1,114) = 0.45, p > 0.5). *p < 0.05. **p < 0.01.
Discussion
Here we investigated the principles of synaptic plasticity in the LHb and stress-induced alterations in synaptic plasticity using electrophysiological and molecular approaches. First, we observed a heterogeneous propensity for synaptic plasticity in the LHb: LTP occurs only in a subpopulation of LHb neurons, whereas the majority of neurons in the LHb exhibit little potentiation, if any, even after TBS. To our knowledge, our observation provides the first evidence showing that long-lasting synaptic potentiation can occur in LHb neurons following a brief, strong stimulation. We also examined how exposure to stressors may acutely alter the efficacy of long-term synaptic plasticity in this particular brain area. Interestingly, a single exposure to an inescapable FS or a restrain plus tail shock significantly facilitates LTP induction and increases the number of neurons expressing phosphorylated CREB protein in the LHb. Our observations suggest that stressor exposure may have lowered the threshold for induction of LTP in the LHb and shifts the propensity of LHb neurons toward more potentiation.
A previous study reported that synaptic activity of the subpopulation of LHb neurons was abnormally potentiated in animal models of depression (Li et al., 2011). Given that stressors can lead to increased transmission onto the LHb, we expected that synaptic plasticity might be reduced following stress; that is, if in vivo stressors would increase transmission via a limited amount of plasticity factors, it is expected to occlude subsequent in vitro plasticity. However, we found increased synaptic potentiation following stress. This suggests that stressors change the states of synapses, making them more sensitive to plasticity. Thus, a stimulus that activates an input to the LHb, which normally would not be potentiated, will become potentiated if activated following stress. Our observations of increased pCREB expression after stress exposure even without TBS (Fig. 6) may provide further support for this idea.
The LHb may encode different valences of affective states in distinct circuitry. The activity of the LHb is known to be altered by addictive drugs or reward prediction errors (Matsumoto and Hikosaka, 2007; Hikosaka, 2010; Maroteaux and Mameli, 2012; Proulx et al., 2014; Meye et al., 2015). While neurons in the LHb projecting to the VTA are selectively potentiated in animal models of depression, cocaine potentiates the activity of LHb neurons projecting to the RMTg, an inhibitory area of the dopaminergic system, but not to the VTA (Maroteaux and Mameli, 2012). Therefore, synaptic plasticity in the LHb may be distinctively controlled depending on affective state as well as on the targeted projection sites.
We showed that stress exposure directly activates LHb neurons (Fig. 3). However, stress exposure seems to activate LHb neurons with limited specificity. Even though the densities of c-fos-expressing neurons are significantly different following FS and RTS, there is no topographic difference in the distribution of activated neurons following the use of these stress paradigms (Fig. 3). In addition, stress-responsive LHb neurons were not projection-specific (Fig. 4). Despite previous studies reporting distinctive roles for LHb neurons depending on their target sites (Maroteaux and Mameli, 2012), we found no significant differences in c-fos-expressing neurons depending on projection site (Fig. 4, two-way ANOVA, F(2,69) = 1.63, p > 0.2). We cannot rule out the possibility that the principles of synaptic plasticity in the LHb differ depending on the projection site. Future studies may investigate input-specific modulation of synaptic plasticity.
To induce synaptic potentiation, a number of synaptic proteins need to be activated. These include PKC and CaMKII (Malinow et al., 1988; Malinow et al., 1989; Wang and Kelly, 1995; Sweatt, 1999; Ahmed and Frey, 2005). It is well established that phosphorylation of CREB via the signaling cascades of PKC or CaMKII is critical to maintaining potentiated synaptic transmission (Bourtchuladze et al., 1994; Sweatt, 1999). A previous study reported that congenitally learned helpless animals express increased levels of βCaMKII in the LHb and that LHb-specific knockdown of βCaMKII is sufficient to reverse helpless behaviors in congenitally learned helpless animals (Li et al., 2013). Therefore, it is possible that exposure to stressors may have led to increased βCaMKII levels, thereby contributing to the facilitation of LTP induction in the LHb, although this proposal requires further examination.
Recent molecular studies have suggested that CREB-expressing neurons are preferentially recruited during fear memory formation (Han et al., 2007) and that CREB contributes to increase intrinsic neuronal excitability and synaptic plasticity (Dash et al., 1990; Benito and Barco, 2010; Suzuki et al., 2011; Gruart et al., 2012). Interestingly, we observed that exposure to stressors per se selectively increased the density of pCREB-positive neurons in the LHb. Given that neuronal excitability before training and CREB expression levels determine whether given neurons will be used for memory formation or not (Yiu et al., 2014), stressor exposure may increase neuronal excitability and thereby lower the threshold for synaptic plasticity following TBS. The magnitude of LTP correlates with the number of c-fos-positive cells (Fig. 5H). Notably, we observed a very strong correlation between the magnitude of LTP and the density of pCREB-expressing neurons before as well as after TBS, suggesting that the neuronal excitability before TBS may contribute to the magnitude of synaptic potentiation by lowering the threshold for LTP. These results may provide explanations for long-standing clinical observations that stressor exposure facilitates or causes the onset of depressive disorders at the cellular level (Cassem, 1995; Kendler et al., 1999; Hammen, 2005; Schneiderman et al., 2005).
Together, these data indicate that the LHb exhibits increased expression of synaptic potentiation-related molecules and facilitated long-term synaptic potentiation in helpless animals. Future investigation will uncover the underlying mechanisms and molecular mediators that may enhance synaptic efficacy in the LHb upon exposure to stressful experiences.
Footnotes
This work was supported by Korean Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea HI14C2136 and HI14C2137 to C.C., and Brain Research Program (through the National Research Foundation of Korea funded by the Ministry of Science, ICT, and Future Planning NRF-2015M3C7A1031395).
The authors declare no competing financial interests.
References
- Ahmed T, Frey JU (2005) Plasticity-specific phosphorylation of CaMKII, MAP-kinases and CREB during late-LTP in rat hippocampal slices in vitro. Neuropharmacology 49:477–492. 10.1016/j.neuropharm.2005.04.018 [DOI] [PubMed] [Google Scholar]
- Artola A, von Frijtag JC, Fermont PC, Gispen WH, Schrama LH, Kamal A, Spruijt BM (2006) Long-lasting modulation of the induction of LTD and LTP in rat hippocampal CA1 by behavioural stress and environmental enrichment. Eur J Neurosci 23:261–272. 10.1111/j.1460-9568.2005.04552.x [DOI] [PubMed] [Google Scholar]
- Benito E, Barco A (2010) CREB's control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci 33:230–240. 10.1016/j.tins.2010.02.001 [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79:59–68. 10.1016/0092-8674(94)90400-6 [DOI] [PubMed] [Google Scholar]
- Brennan FX, Beck KD, Ross RJ, Servatius RJ (2005) Stress-induced increases in avoidance responding: an animal model of post-traumatic stress disorder behavior? Neuropsychiatr Dis Treat 1:69–72. 10.2147/nedt.1.1.69.52292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott-Hazard S, Mazziotta J, Phelps M (1988) Cerebral correlates of depressed behavior in rats, visualized using 14C-2-deoxyglucose autoradiography. J Neurosci 8:1951–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassem EH. (1995) Depressive disorders in the medically ill: an overview. Psychosomatics 36:S2–S10. 10.1016/S0033-3182(95)71698-X [DOI] [PubMed] [Google Scholar]
- Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N (2007) The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci 27:2781–2787. 10.1523/JNEUROSCI.4372-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AC, Shirayama Y, Shin KH, Neve RL, Duman RS (2001) Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biol Psychiatry 49:753–762. 10.1016/S0006-3223(00)01114-8 [DOI] [PubMed] [Google Scholar]
- Dash PK, Hochner B, Kandel ER (1990) Injection of the cAMP-responsive element into the nucleus of Aplysia sensory neurons blocks long-term facilitation. Nature 345:718–721. 10.1038/345718a0 [DOI] [PubMed] [Google Scholar]
- Dowlatshahi D, MacQueen GM, Wang JF, Young LT (1998) Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet 352:1754–1755. 10.1016/S0140-6736(05)79827-5 [DOI] [PubMed] [Google Scholar]
- Du K, Asahara H, Jhala US, Wagner BL, Montminy M (2000) Characterization of a CREB gain-of-function mutant with constitutive transcriptional activity in vivo. Mol Cell Biol 20:4320–4327. 10.1128/MCB.20.12.4320-4327.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS. (2002) Pathophysiology of depression: the concept of synaptic plasticity. Eur Psychiatry 17 [Suppl 3]:306–310. [DOI] [PubMed] [Google Scholar]
- Duman RS, Aghajanian GK (2012) Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72. 10.1126/science.1222939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy MR, Stanton ME, Levine S, Thompson RF (1987) Behavioral stress impairs long-term potentiation in rodent hippocampus. Behav Neural Biol 48:138–149. 10.1016/S0163-1047(87)90664-9 [DOI] [PubMed] [Google Scholar]
- Garcia R, Musleh W, Tocco G, Thompson RF, Baudry M (1997) Time-dependent blockade of STP and LTP in hippocampal slices following acute stress in mice. Neurosci Lett 233:41–44. 10.1016/S0304-3940(97)00621-6 [DOI] [PubMed] [Google Scholar]
- Gonçalves L, Sego C, Metzger M (2012) Differential projections from the lateral habenula to the rostromedial tegmental nucleus and ventral tegmental area in the rat. J Comp Neurol 520:1278–1300. 10.1002/cne.22787 [DOI] [PubMed] [Google Scholar]
- Goto Y, Grace AA (2006) Alterations in medial prefrontal cortical activity and plasticity in rats with disruption of cortical development. Biol Psychiatry 60:1259–1267. 10.1016/j.biopsych.2006.05.046 [DOI] [PubMed] [Google Scholar]
- Gruart A, Benito E, Delgado-García JM, Barco A (2012) Enhanced cAMP response element-binding protein activity increases neuronal excitability, hippocampal long-term potentiation, and classical eyeblink conditioning in alert behaving mice. J Neurosci 32:17431–17441. 10.1523/JNEUROSCI.4339-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammen C. (2005) Stress and depression. Annu Rev Clin Psychol 1:293–319. 10.1146/annurev.clinpsy.1.102803.143938 [DOI] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, Neve RL, Guzowski JF, Silva AJ, Josselyn SA (2007) Neuronal competition and selection during memory formation. Science 316:457–460. 10.1126/science.1139438 [DOI] [PubMed] [Google Scholar]
- Herkenham M, Nauta WJ (1977) Afferent connections of the habenular nuclei in the rat: a horseradish peroxidase study, with a note on the fiber-of-passage problem. J Comp Neurol 173:123–146. 10.1002/cne.901730107 [DOI] [PubMed] [Google Scholar]
- Hikosaka O. (2010) The habenula: from stress evasion to value-based decision-making. Nat Rev Neurosci 11:503–513. 10.1038/nrn2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. (2012) The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5:14. 10.1186/1756-6606-5-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Karkowski LM, Prescott CA (1999) Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry 156:837–841. 10.1176/ajp.156.6.837 [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM (2002) The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci 3:453–462. 10.1038/nrn849 [DOI] [PubMed] [Google Scholar]
- Kim JJ, Foy MR, Thompson RF (1996) Behavioral stress modifies hippocampal plasticity through N-methyl-D-aspartate receptor activation. Proc Natl Acad Sci U S A 93:4750–4753. 10.1073/pnas.93.10.4750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Koo JW, Lee HJ, Han JS (2005) Amygdalar inactivation blocks stress-induced impairments in hippocampal long-term potentiation and spatial memory. J Neurosci 25:1532–1539. 10.1523/JNEUROSCI.4623-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ (2008) The molecular neurobiology of depression. Nature 455:894–902. 10.1038/nature07455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, Malinow R (2011) Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature 470:535–539. 10.1038/nature09742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Zhou T, Liao L, Yang Z, Wong C, Henn F, Malinow R, Yates JR 3rd, Hu H (2013) betaCaMKII in lateral habenula mediates core symptoms of depression. Science 341:1016–1020. 10.1126/science.1240729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF. (1984) Learned helplessness and animal models of depression. Prog Neuropsychopharmacol Biol Psychiatry 8:435–446. 10.1016/S0278-5846(84)80032-9 [DOI] [PubMed] [Google Scholar]
- Malinow R, Madison DV, Tsien RW (1988) Persistent protein kinase activity underlying long-term potentiation. Nature 335:820–824. 10.1038/335820a0 [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW (1989) Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245:862–866. 10.1126/science.2549638 [DOI] [PubMed] [Google Scholar]
- Maroteaux M, Mameli M (2012) Cocaine evokes projection-specific synaptic plasticity of lateral habenula neurons. J Neurosci 32:12641–12646. 10.1523/JNEUROSCI.2405-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O (2007) Lateral habenula as a source of negative reward signals in dopamine neurons. Nature 447:1111–1115. 10.1038/nature05860 [DOI] [PubMed] [Google Scholar]
- Meng H, Wang Y, Huang M, Lin W, Wang S, Zhang B (2011) Chronic deep brain stimulation of the lateral habenula nucleus in a rat model of depression. Brain Res 1422:32–38. 10.1016/j.brainres.2011.08.041 [DOI] [PubMed] [Google Scholar]
- Meye FJ, Valentinova K, Lecca S, Marion-Poll L, Maroteaux MJ, Musardo S, Moutkine I, Gardoni F, Huganir RL, Georges F, Mameli M (2015) Cocaine-evoked negative symptoms require AMPA receptor trafficking in the lateral habenula. Nat Neurosci 18:376–378. 10.1038/nn.3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, Duman RS (2008) Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33:88–109. 10.1038/sj.npp.1301574 [DOI] [PubMed] [Google Scholar]
- Pittenger C, Kandel E (1998) A genetic switch for long-term memory. C R Acad Sci III 321:91–96. 10.1016/S0764-4469(97)89807-1 [DOI] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER (2002) Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron 34:447–462. 10.1016/S0896-6273(02)00684-0 [DOI] [PubMed] [Google Scholar]
- Proulx CD, Hikosaka O, Malinow R (2014) Reward processing by the lateral habenula in normal and depressive behaviors. Nat Neurosci 17:1146–1152. 10.1038/nn.3779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorius A, Henn FA (2007) Deep brain stimulation of the lateral habenula in treatment resistant major depression. Med Hypotheses 69:1305–1308. 10.1016/j.mehy.2007.03.021 [DOI] [PubMed] [Google Scholar]
- Sartorius A, Kiening KL, Kirsch P, von Gall CC, Haberkorn U, Unterberg AW, Henn FA, Meyer-Lindenberg A (2010) Remission of major depression under deep brain stimulation of the lateral habenula in a therapy-refractory patient. Biol Psychiatry 67:e9–e11. 10.1016/j.biopsych.2009.08.027 [DOI] [PubMed] [Google Scholar]
- Schneiderman N, Ironson G, Siegel SD (2005) Stress and health: psychological, behavioral, and biological determinants. Annu Rev Clin Psychol 1:607–628. 10.1146/annurev.clinpsy.1.102803.144141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sego C, Gonçalves L, Lima L, Furigo IC, Donato J Jr, Metzger M (2014) Lateral habenula and the rostromedial tegmental nucleus innervate neurochemically distinct subdivisions of the dorsal raphe nucleus in the rat. J Comp Neurol 522:1454–1484. 10.1002/cne.23533 [DOI] [PubMed] [Google Scholar]
- Semba K, Fibiger HC (1992) Afferent connections of the laterodorsal and the pedunculopontine tegmental nuclei in the rat: a retro- and antero-grade transport and immunohistochemical study. J Comp Neurol 323:387–410. 10.1002/cne.903230307 [DOI] [PubMed] [Google Scholar]
- Shabel SJ, Proulx CD, Trias A, Murphy RT, Malinow R (2012) Input to the lateral habenula from the basal ganglia is excitatory, aversive, and suppressed by serotonin. Neuron 74:475–481. 10.1016/j.neuron.2012.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabel SJ, Proulx CD, Piriz J, Malinow R (2014) Mood regulation: GABA/glutamate co-release controls habenula output and is modified by antidepressant treatment. Science 345:1494–1498. 10.1126/science.1250469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, Gallegos RA, Breindl A (1997) Transient and persistent consequences of acute stress on long-term potentiation (LTP), synaptic efficacy, theta rhythms and bursts in area CA1 of the hippocampus. Synapse 26:209–217. [DOI] [PubMed] [Google Scholar]
- Shumake J, Gonzalez-Lima F (2003) Brain systems underlying susceptibility to helplessness and depression. Behav Cogn Neurosci Rev 2:198–221. 10.1177/1534582303259057 [DOI] [PubMed] [Google Scholar]
- Suzuki A, Fukushima H, Mukawa T, Toyoda H, Wu LJ, Zhao MG, Xu H, Shang Y, Endoh K, Iwamoto T, Mamiya N, Okano E, Hasegawa S, Mercaldo V, Zhang Y, Maeda R, Ohta M, Josselyn SA, Zhuo M, Kida S (2011) Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J Neurosci 31:8786–8802. 10.1523/JNEUROSCI.3257-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD. (1999) Toward a molecular explanation for long-term potentiation. Learn Mem 6:399–416. 10.1101/lm.6.5.399 [DOI] [PubMed] [Google Scholar]
- Wang JH, Kelly PT (1995) Postsynaptic injection of CA2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron 15:443–452. 10.1016/0896-6273(95)90048-9 [DOI] [PubMed] [Google Scholar]
- Yang LM, Hu B, Xia YH, Zhang BL, Zhao H (2008) Lateral habenula lesions improve the behavioral response in depressed rats via increasing the serotonin level in dorsal raphe nucleus. Behav Brain Res 188:84–90. 10.1016/j.bbr.2007.10.022 [DOI] [PubMed] [Google Scholar]
- Yiu AP, Mercaldo V, Yan C, Richards B, Rashid AJ, Hsiang HL, Pressey J, Mahadevan V, Tran MM, Kushner SA, Woodin MA, Frankland PW, Josselyn SA (2014) Neurons are recruited to a memory trace based on relative neuronal excitability immediately before training. Neuron 83:722–735. 10.1016/j.neuron.2014.07.017 [DOI] [PubMed] [Google Scholar]