

Abstract
Multiple studies have established that brain-derived neurotrophic factor (BDNF) plays a critical role in the regulation of synaptic plasticity via its receptor, TrkB. In addition to being phosphorylated, TrkB has also been demonstrated to be ubiquitinated. However, the mechanisms of TrkB ubiquitination and its biological functions remain poorly understood. In this study, we demonstrate that ubiquitin C-terminal hydrolase L1 (UCH-L1) promotes contextual fear conditioning learning and memory via the regulation of ubiquitination of TrkB. We provide evidence that UCH-L1 can deubiquitinate TrkB directly. K460 in the juxtamembane domain of TrkB is the primary ubiquitination site and is regulated by UCH-L1. By using a peptide that competitively inhibits the association between UCH-L1 and TrkB, we show that the blockade of UCH-L1-regulated TrkB deubiquitination leads to increased BDNF-induced TrkB internalization and consequently directs the internalized TrkB to the degradation pathway, resulting in increased degradation of surface TrkB and attenuation of TrkB activation and its downstream signaling pathways. Moreover, injection of the peptide into the DG region of mice impairs hippocampus-dependent memory. Together, our results suggest that the ubiquitination of TrkB is a mechanism that controls its downstream signaling pathways via the regulation of its endocytosis and postendocytic trafficking and that UCH-L1 mediates the deubiquitination of TrkB and could be a potential target for the modulation of hippocampus-dependent memory.
SIGNIFICANCE STATEMENT Ubiquitin C-terminal hydrolase L1 (UCH-L1) has been demonstrated to play important roles in the regulation of synaptic plasticity and learning and memory. TrkB, the receptor for brain-derived neurotrophic factor, has also been shown to be a potent regulator of synaptic plasticity. In this study, we demonstrate that UCH-L1 functions as a deubiquitinase for TrkB. The blockage of UCH-L1-regulated deubiquitination of TrkB eventually results in the increased degradation of surface TrkB and decreased activation of TrkB and its downstream signaling pathways. In vivo, UCH-L1-regulated TrkB deubiquitination is necessary for hippocampus-dependent memory. Overall, our study provides novel insights into the mechanisms of UCH-L1-mediated neurobiological functions and suggests that ubiquitination is an important regulatory signal for TrkB functions.
Keywords: endocytosis, learning and memory, recycling, TrkB, ubiquitination, UCHL1
Introduction
Ubiquitin C-terminal hydrolase L1 (UCH-L1), one of the UCH deubiquitinating enzymes (DUBs), is highly expressed in neurons and accounts for 1–2% of total soluble proteins in the brain (Wilkinson et al., 1989). As a DUB, UCH-L1 has been demonstrated to remove small adducts of ubiquitin (Larsen et al., 1998). Recently, more physiological substrates of UCH-L1 were suggested. For example, UCH-L1 has been demonstrated to deubiquitinate Hif-1α and NOXA, consequently promoting metastases and increasing sensitivity to genotoxic chemotherapy (Gérecová et al., 2013; Goto et al., 2015). In addition to its hydrolase activity, UCH-L1 has also been shown to exhibit a dimerization-dependent ubiquityl ligase activity, which can add ubiquitin chains to α-synuclein (Liu et al., 2002). It has also been reported that UCH-L1 stabilizes monoubiquitin, a process that is dependent on its affinity for monoubiquitin rather than hydrolase activity (Osaka et al., 2003). Given the multiple functions of UCH-L1, it is apparent that UCH-L1 plays a critical role in the CNS. Indeed, it has been demonstrated that UCH-L1 plays a significant role in maintaining normal synaptic structures and synaptic plasticity in hippocampal neurons (Cartier et al., 2009). Memory in passive avoidance learning and exploratory behavior are reduced in UCH-L1 deficit mice (Sakurai et al., 2008). Dysfunction or mutation of UCH-L1 has been reported to be associated with several neurodegenerative diseases. For example, a rare I93M mutation in UCH-L1 has been reported to cause Parkinson's disease (Leroy et al., 1998); recently, an E7A mutation was reported to lead to an early-onset progressive neurodegenerative syndrome (Bilguvar et al., 2013). In the brains of Alzheimer's disease (AD) patients, UCH-L1 was found to be downregulated (Choi et al., 2004). Overexpression of UCH-L1 has been shown to improve memory and rescue memory impairments caused by amyloid β (Aβ) (Gong et al., 2006; Zhang et al., 2014). However, how UCH-L1 mediates enhanced memory is not fully understood, and the in vivo substrates of UCH-L1 that are involved are still unknown.
Brain-derived neurotrophic factor (BDNF) is the most abundant neurotrophic factor in the brain. By activating its downstream receptor, TrkB, it regulates neuronal survival, development, synaptic plasticity, and learning and memory. By binding with its receptor, TrkB, BDNF activates TrkB, followed by endocytosis and intracellular postendocytic trafficking of the BDNF/TrkB complex, which is important for the strength and maintenance of the downstream signaling pathways (Sommerfeld et al., 2000; Huang et al., 2013). Ubiquitination has been identified as a mechanism that controls endocytosis and sorting of postendocytic receptors to degradative or recycling pathways (Haglund and Dikic, 2012). It has been reported BDNF induces the ubiquitination of TrkB (Makkerh et al., 2005; Arévalo et al., 2006) and a few studies suggest that TRAF6, c-Cbl, might be the E3 ligase responsible for TrkB ubiquitination (Moises et al., 2009; Pandya et al., 2014). However, the detailed mechanisms of the ubiquitination of TrkB and the biological functions associated with the ubiquitination of TrkB remain elusive.
In this study, we found that UCH-L1 binds specifically to the juxtamembrane 1 (JM1) motif of the TrkB receptor and regulates its ubiquitination as a deubiquitinase. As a result, UCH-L1-regulated TrkB deubiquitination affects TrkB endocytosis and postendocytic traffic, thus regulating the downstream signaling pathways. This in vitro result was also confirmed in in vivo experiments and we further demonstrated that UCH-L1 contributes to contextual fear conditioning (CFC) memory by regulating the deubiquitination of TrkB.
Materials and Methods
Animals
Male C57BL/6 mice, 8 weeks old and weighing 23–25 g, were housed at 22 ± 2°C on a 12 h light/dark cycle. Food and water were available ad libitum. All animal procedures were conducted on the basis of the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and were approved by the institutional animal care and use committee of Shandong University.
Reagents and antibodies
Human recombinant BDNF was purchased from PeproTech. Sulfo-NHS-S-S-biotin and Avidin beads were obtained from Thermo Fisher Scientific. We used the peptidomimetic method to block the interaction between UCH-L1 and TrkB. The peptides were synthesized and purified by GL Biochem. The peptides contains the 75–85 aa residues of UCH-L1 (VSPKVYFMKQT, referred to Tat-UCH-L175–85) fused to a Tat-like polyarginine membrane permeability sequence (GRRRRRRRRRRR) to promote its entrance into the cell and a biotin molecule for detection. A reversed sequence of the 75–85 aa residues of UCH-L1 was used as a control peptide with biotin and Tat at the N terminals (biotin-GRRRRRRRRRRR-TQKMFYVKPSV, named Tat-Con). The restriction enzymes were purchased from Fermentas.
Antibodies were purchased as follows: rabbit anti c-Myc antibody from Bethly; rabbit anti-Flag antibody from Thermo Fisher Scientific; rabbit anti-TrkB antibody from Millipore; mouse anti-ubiquitin (P4D1), mouse anti-UCH-L1 (13C4), mouse anti-Akt1 (B-1), and mouse anti-pY99 from Santa Cruz Biotechnology; mouse anti-polyubiquitinylated conjugates (FK1) from Enzo Life Sciences; mouse anti-phospho-p44/42 (ERK1/2; Thr202/Tyr204), rabbit anti-p42/44 (ERK1/2), rabbit anti-phospho-TrkA (pY490), rabbit anti-phospho-Akt (Ser473), and rabbit anti-K48 and k63 polyubiquitin antibodies from Cell Signaling Technology; mouse anti-FLAG (M1, M2) antibodies from Sigma-Aldrich; horseradish peroxidase-conjugated goat anti-mouse or rabbit IgG antibodies used for Western blot from Calbiochem; horseradish peroxidase-conjugated goat anti-mouse IgM antibody used for Western blot from Jackson ImmunoResearch Laboratories; and rabbit anti-GFP antibody and Alexa Fluor 488- or 594-conjugated goat anti-mouse or rabbit IgG heavy and light chains (H+L) used for immunofluorescent staining from Invitrogen. All the other reagents were from Sigma-Aldrich unless otherwise indicated.
Plasmid constructs and siRNA
The rat TrkB-FL, TrkB.T1, mutant TrkB constructs (JM1, JM0 and ΔJM1), IL2R, and Flag-tagged IL2R-JM1 were kept in our laboratory. The coding region of rat UCH-L1 was amplified from rat cDNA and subcloned into pCMV-3tag-7. TrkB and UCH-L1 site-directed mutations (K454R, K460R, K464R, K506R, K524R, K533R TrkB; C90S, D30A UCH-L1) and deletion mutation UCH-L1(Δ60–75, Δ75–85, Δ85–100, Δ110–120, Δ120–147 UCH-L1) were generated by means of two-step PCR,and subcloned into pcDNA3.1 or pCMV-3tag-7 expression vectors. The truncated mutation of UCH-L1 (UCH-L1 1–150, UCH-L1 60–223) was subcloned into pCMV3-tag7 expression vector (Agilent Technologies). HA-tagged rat UCH-L1 was subcloned into pCDNA3.1 expression vector (Invitrogen). GFP-tagged 75–85 of UCH-L1 was subcloned into pEGFPC2 expression vector (Invitrogen). All constructs were confirmed by DNA sequencing to exclude potential mutations introduced by PCR.
Three small interfering RNA (siRNA) against rat UCH-L1 was designed, synthesized, and purified by RiboBio. The target sequences were as follows: 5′-GGATGGATCAGTCCTGAAA-3′, 5′-GGTAGACGACAAAGTGAAT-3′, and 5′-CCAAGACAAGCTGGAATTT-3′. Scramble siRNAs were used as a negative control in all RNA interference experiments.
Lentivirus and adeno-associated virus serotype-9 (AAV9) preparation
UCH-L1 sequence was subcloned into the pUltra-hot vector and the empty pUltra-hot was used as a control. The lentivirus was produced by transient transfection of the UCH-L1 construct or control vector into HEK293T cells using nanofectin with pMDLg/pRR, pMDG-VSVG, and pRSV-Rev. After ∼48–72 h, the supernatant containing lentivirus was collected and filtered using a 0.45 μm filter. Then, 100 μl of supernatant containing lentivirus was added into Neurobasal medium to transfect 1 × 105 neurons. The culture medium was changed to Neurobasal medium containing 2% B27 supplement (Invitrogen) and l-glutamine (0.5 mm) after ∼8–12 h transfection.
The shRNA specific for mouse TrkB kinase domain and an shRNA-resistant TrkB FL used in previous studies were also used in our study (Jiang et al., 2015). The recombinant AAV9 carrying the shRNA, AAV9 encoding WT and C90S UCH-L1, AAV9 encoding shRNA-resistant TrkB, and empty AAV9 with a reporter gene were generated at Vigene Biosciences.
Hippocampal neuron culture and transfection
Culture of hippocampal neurons were prepared as described previously. At ∼7–9 d in vitro (DIV) cultured hippocampal neurons were transfected with plasmids or siRNA using Lipofectamine 2000 or lipofectamine RNAiMAX (Invitrogen) following the manufacturer's instructions. Two to 4 d after transfection, neurons were used for the indicated experiments.
Coimmunoprecipitation (Co-IP) assay
For Co-IP, HEK293 cells transfected with the indicated constructs, or cultured hippocampal neurons or mice hippocampi were lysed with TNE buffer (10 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 10% glycerol with protease inhibitor cocktail, Sigma-Aldrich). The lysates were centrifuged at 14,000 × g for 10 min at 4°C. The supernatant was incubated with indicated antibodies overnight at 4C, followed by 2 h of incubation at 4°C with protein A or G-Sepharose beads. Then, after five washes with TNE buffer, the beads were eluted by boiling in sample buffer for SDS-PAGE and further immunoblot analysis was conducted with the indicated antibodies. Each experiment was repeated at least three times.
Biotinylation assay to analyze surface TrkB level, surface TrkB degradation, internalization, and postendocytic recycling
To determine the effects of Tat-UCH-L175–85 on BDNF induced-degradation of surface TrkB, cell surface biotinylation and degradation assays were performed as described previously (Huang et al., 2009). Briefly, cultured hippocampal neurons (7–9 DIV) were serum starved overnight and, after a 30 min pretreatment of vehicle or the peptide, the surface proteins were labeled with sulfo-N-hydroxysuccinimide-biotin at 4°C followed by vehicle or BDNF (50 ng/ml) stimulation at 37°C for 60 min. Subsequently, neurons were chilled on ice and lysed by TNE buffer (containing 10 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 10% glycerol with protease inhibitors). The lysate was clarified by centrifugation at 12,000 × g for 10 min and the biotinylated proteins were isolated by streptavidin-conjugated Sepharose beads. TrkB-FL pulled down by streptavidin beads, which represents the undegraded TrkB, was separated by SDS-PAGE and detected using immunoblot.
For assessing the effects of Tat-UCH-L175–85 on BDNF-induced internalization and recycling of TrkB receptors, cell surface receptor cleavable biotinylation, internalization, and recycling assays were conducted as described previously (Huang et al., 2009). Briefly, after serum starvation overnight, neurons (7–9 DIV) were pretreated with Tat-UCH-L175–85 for 30 min and labeled with sulfo-NHS-SS-biotin at 4°C. Then the neurons were treated with vehicle or BDNF (50 ng/ml) for 15 min. For internalization assay, the remaining surface biotin was removed using stripping solution (containing 50 mm glutathione, 0.01 g/ml BSA, 75 mm NaOH, 75 mm NaCl, 10 mm EDTA) for strip group and internalization group; the biotin was not removed from cell surface in the surface group in which the biotinylated proteins represents the total surface biotinylated receptors. Then, the neurons were lysed with TNE buffer, the lysates were centrifuged, and the biotinylated proteins in the supernatant, which represent the total surface TrkB (surface group) or internalized TrkB (internalization group), were isolated using streptavidin-conjugated Sepharose beads. The precipitated beads were then washed with TNE buffer and denatured in sample buffer at 98°C for 5 min. The biotinylated TrkB receptors from the various groups were separated by SDS-PAGE and then detected by immunoblot. The internalization rate was calculated as follows: (internalized TrkB)/(total surface TrkB) * 100%. For the recycling assay, neurons were pretreated with proteasome inhibitor MG132 (10 μm), lysosomal protease inhibitors leupeptin/pepstatin A (100 μg/ml) for 90 min, and Tat-UCH-L175–85 for 30 min. MG132 and leupeptin/pepstatin A were included in all subsequent steps to inhibit proteolysis. After being labeled with sulfo-NHS-SS-biotin as described above, neurons were treated with vehicle or BDNF treatment at 37°C for 15 min to induce internalization. Subsequently, for internalization and recycling groups, the remaining surface biotin was removed with stripping solution on ice, leaving internalized receptor proteins biotinylated. Then, the neurons of surface, strip and internalization group were lysed with TNE buffer and the neurons of recycling group were rewarmed in fresh medium at 37°C for 45 min to allow the recycling of the internalized TrkB back to surface. Then, the recycling neurons were stripped again to remove the biotin labeling from the recycled receptors. Afterward, neurons were lysed and the unrecycled receptors were pulled down with streptavidin beads as described above. The TrkB from the different groups were detected by anti-TrkB immunoblotting. The rate of unrecycled TrkB was calculated directly as follows: rewarming group/internalization group * 100%, which is inversely proportional to the recycling rate.
Fluorescence ratio microscopy analysis of the levels of TrkB receptor internalization and postendocytic recycling
The fluorescence images described below were acquired with an Eclipse TE2000-U (Nikon) using an HQ2 cooled CCD camera and a 60× objective lens. Both wild-type and mutants TrkB-FL were Flag tagged and only Flag-positive images of cultured hippocampal neurons were captured. The internalization of TrkB-FL was assessed as described previously (Liu et al., 2015). Cultured hippocampal neurons (7–9 DIV) were transfected with Flag-TrkB-FL construct (Flag epitope fused to the extracellular N-terminal) using Lipofectamine 2000 2 d before the experiment. Next, neurons were serum starved overnight. Then, the total pool of TrkB-FL initially present at the cell surface was labeled with Alexa Fluor 594-conjugated anti-Flag antibody (594-M1) at 20°C for 20 min. Afterward, neurons were stimulated with BDNF (50 ng/ml) at 37°C for 15 min to drive internalization of TrkB. Then, neurons were fixed with 4% paraformaldehyde in PBS (4% PFA), followed by labeling the receptors remaining at cell surface with Alexa Fluor 488-conjugated anti-mouse IgG secondary antibody under nonpermeabilizing conditions. Images were acquired and the ratio of 488/594 fluorescence was calculated. The mean 488/594 ratio of the internalized group (named A) represented the un-internalized TrkB-FL. For each experiment, there was a parallel control group in which neurons were fixed immediately after the 20 min incubation with 594-M1 and labeled with Alexa Fluor 488-conjugated anti-mouse IgG antibody and the mean 488/594 ratio of this group (named B) represented the 100% surface TrkB-FL. Therefore, the percentage of internalized TrkB receptors was calculated using the following formula: (1 − A/B) × 100.
The level of TrkB recycling was assessed as described previously (Zhao et al., 2009). In brief, cultured hippocampal neurons (7–9 DIV) transfected with the Flag-TrkB were serum starved overnight and then incubated with anti-Flag antibody (M1, calcium sensitive) conjugated to Alexa Fluor 594 (594-M1) at 20°C for 20 min to label surface TrkB receptor. Then, neurons were stimulated with BDNF (50 ng/ml) at 37°C for 15 min to drive TrkB internalization, followed by quick washes with cold PBS containing EDTA (1 mm) to dissociate the 594-M1 bound to un-internalized TrkB receptors. Next, neurons were incubated in fresh medium containing Alexa Fluor 488-conjugated anti-mouse IgG secondary antibody at 37°C for 45 min to label recycled receptors with 594-M1 that returned back to the cell surface. Then, neurons were immediately fixed. Images were acquired using epifluorescence microscopy. The fluo-488/fluo-594 fluorescence ratio was calculated using MetaMorph software. For each experiment, there were two parallel control groups: in the first, neurons were fixed immediately after the 20 min incubation with 594-M1 and subsequently labeled with Alexa Fluor-488-conjugated anti-mouse IgG antibody, the 100% surface control. In the second, neurons were fixed immediately after the EDTA-stripping step and then labeled with Alexa Fluor 488-conjugated anti-mouse IgG antibody, as the 0% recycled control. Therefore, the percentage of recycled TrkB receptors in individual cells was calculated from the (fluo-488/fluo-594) fluorescence ratio using the following formula: (E − Z)/(C − Z) × 100, where E is the mean ratio for the experimental coverslip, Z is the mean ratio for the zero recycle control, and C is the mean ratio for the 100% surface control.
In vitro deubiquitination assays
UCH-L1 with His tag was expressed in the BL21 Escherichia coli (DE3 strain; Novagen). Then, BL21 cells were lysed and UCH-L1 was purified according to the instruction of His Ni-Agarose tag protein purification kit (CoWin Biotech).
HEK293 cells were transfected with HA-Ub and Flag-TrkB. Forty-eight hours after transfection, cells were lysed with TNE buffer containing protease inhibitors and anti-Flag M2-conjugated beads (for TrkB) to immunoprecipitated Flag-TrkB, in which a large amount of Flag-TrkB was ubiquitinated. Immunoprecipitated complexes were washed and incubated with recombinant His-UCH-L1 in deubiquitylation assay buffer (50 mm HEPES/NaOH, pH 7.8, 0.5 mm EDTA, 1 mm DTT, 0.1 mg/ml ovalbumin) at 37°C for 1 h. The reaction was terminated by adding SDS sample buffer followed by 5 min heat denaturation at 95C. The ubiquitination of TrkB was detected by immunoblotting with the HA antibodies.
In vitro binding assay
HA-UCH-L1 and Flag-TrkB proteins were expressed with a TNT Quick Coupled Transcription/Translation System (Promega) according to the instructions of the manufacturer. HA-UCH-L1 and Flag-TrkB were mixed, followed by immunoprecipitation with Flag antibodies and immunoblot with HA antibodies.
Surgical procedure and microinjection
To microinject peptides, after being anesthetized with 5% chloral hydrate (0.6 ml/100 g, i.p.), the mice were implanted bilaterally with 26-gauge guide cannulas to the DG. The coordinates were as follows: anteroposterior (AP), −1.75 mm; lateral (L), ±1.3 mm; dorsoventral (V), −2.24 mm. A stylus was placed in the guide cannula to prevent clogging. After surgery, the animals were allowed to recover for 1 week and then TAT-scramble or Tat-UCH-L175–85 was injected. For AAV9 virus injection, after being anesthetized with 5% chloral hydrate (0.6 ml per 100 g, i.p.), the viruses were injected into bilateral DG without cannula implantation.
For the microinjection, the injection cannula was connected to a microsyringe driven by a microinfusion pump (KDS310; KD Scientific). Viruses were microinfused at a rate of 0.1 μl/min and an additional 5 min was allowed before withdrawal to ensure the infusions' diffusion. After AAV9 injection, the mice were put back in their cages for ∼7–10 d until subsequent experiments.
Drug administration
N-(2-((2-oxoazepan-3-yl)carbamoyl)phenyl)benzo[b]thiophene-2-carboxamide (ANA-12), an inhibitor of TrkB receptors, was dissolved in 1% dimethylsulfoxide in physiological saline. Mice were treated with ANA-12 (0.5 mg/kg) by intraperitoneal injection for 4 d before the acquisition trial and, on the last day, were treated with ANA-12 30 min before the acquisition trial.
Contextual fear conditioning
On the day of experiment, the mice were put into the conditioning chamber (25 × 25 × 25 cm) and allowed to habituate for 2 min without any stimulation; then they received 3 consecutive 0.4 or 0.7 mA 2 s foot shocks through a stainless steel grid floor with an interval of 60 s. After the last shock, the mice remained in the chamber for additional 60 s and were then placed back in their home cages. Short-term memory (STM) was tested 1 h after training. The animals were placed in the chamber in which the training occurred and the freezing behavior was recorded for 5 min without foot shock.
Locomotor activity test
The locomotor activity was assessed in the open-field test. The open field apparatus consists of a 40 × 40 cm area with 35-cm-high walls. A single mouse was placed in the center of the open-field arena and left free to explore for 10 min. After each trial, the apparatus was cleaned with a 50% ethanol solution to prevent olfactory cues. The total distance traveled in the arena was recorded to test the locomotor activity.
Experimental design and statistical analysis
Experiment 1: UCH-L1 promotes CFC memory by regulating TrkB.
To investigate whether UCH-L1 could promote CFC memory via regulation of TrkB, the following experiments were performed. First, 8-week-old male mice were separated into the following groups: control, WT or C90S UCH-L1 overexpression, TrkB overexpression, TrkB knockdown, and TrkB knockdown with UCH-L1 overexpression. AAV viruses (AAV9-UCH-L1 or AAV9-UCH-L1 C90S or AAV9-shTrkB or AAV9-OE TrkB, specified in Fig. 1D,E) to overexpress UCH-L1 with/without TrkB knockdown were stereotaxically injected into the DG region. AAV viruses expressing mCherry only or scramble RNA were used as control. n = ∼10–12 per group. Fifteen days later, the mice were subjected to CFC training and STM test. Second, in addition to using shRNA to knock down TrkB levels, we also applied the TrkB inhibitor-ANA12 to inhibit TrkB activity with or without UCH-L1 overexpression (see Fig. 1K,L, n = 8 per group). The inhibitor was intraperitoneally injected for 4 d before the acquisition trial and, on the last day, mice were treated with ANA-12 30 min before the CFC training. One hour later, the STM test was performed. After the STM test, the DG regions from those mice were separated and proteins were extracted. TrkB activation, ubiquitination of TrkB, and TrkB and UCH-L1 levels were examined by immunoprecipitation and Western blot (n = 4 per group).
Figure 1.
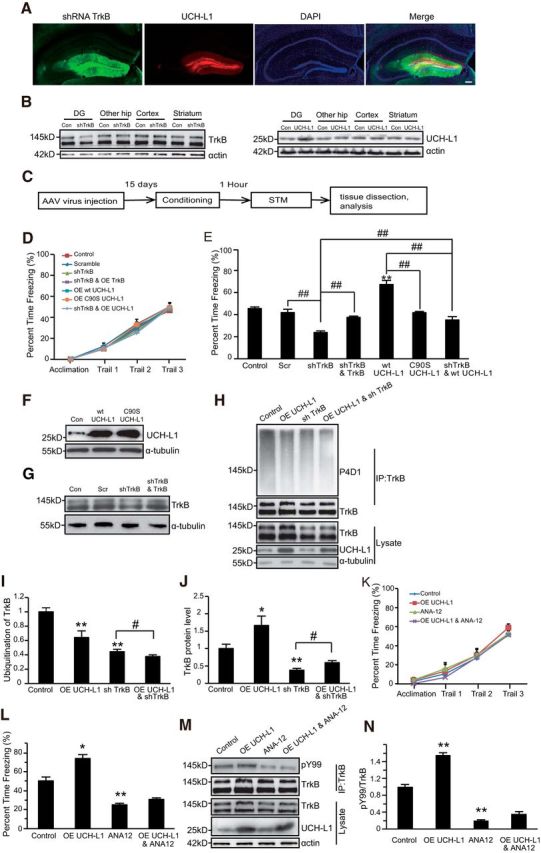
UCH-L1 promotes hippocampus-dependent learning and memory via regulating TrkB. A, Representative images of the DG region injected with AAV9 viruses. The immunofluorescence of GFP or mCherry represents AAV9-infected cells. 200 μm. B, Overexpression of UCH-L1 and the knockdown of TrkB in the DG of hippocampus, other parts of hippocampus, cortex, and striatum were examined by Western blot. C, Schematic representation of the experimental schedule to investigate the effects of UCH-L1 and TrkB on STM. D, E, CFC training and STM test were performed in the indicated mice. **p < 0.01 versus control or scramble group, ##p < 0.01, indicated groups, one-way ANOVA. All values are presented as mean ± SEM, n = 10–12 per group. F, G, Representative immunoblot images of the overexpression of WT UCH-L1 or C90S UCH-L1 (F) and the knockdown of TrkB levels (G). H–J, Representative immunoblot images (H) and densitometric analysis of TrkB ubiquitination (I) and protein levels (J) in the DG of mice. The control group consisted of naive mice injected with vehicle. n = 4 per group. **p < 0.01 versus control, #p < 0.05, indicated groups, one-way ANOVA. K, L, CFC training and STM test were performed in mice with TrkB inhibition (ANA12) or/and UCH-L1 overexpression. **p < 0.01 versus control group. #p < 0.05, indicated groups, one-way ANOVA. All values are presented as mean ± SEM, n = 8 per group. M, N, Representative immunoblot images (M) and densitometric analysis of TrkB activation (N) in the DG of mice. The control group consisted of naive mice injected with vehicle. n = 4 per group. **p < 0.01 versus control, one-way ANOVA.
Experiment 2: UCH-L1 interacts with TrkB.
This experiment was to verify whether UCHL1 interacts with TrkB. Co-IPs were performed to examine the interaction between exogenous or endogenous expressed UCH-L1 and TrkB. For the endogenous Co-IP assay, the DG regions from male C57 mice were separated and proteins were extracted. Anti-TrkB antibody was used to enrich the protein and anti-rabbit IgG was used as a negative control. In vitro binding assay was performed to confirm the interaction between UCH-L1 and TrkB. All of the experiments were performed at least three times.
Experiment 3: Mapping the TrkB/UCH-L1 interaction domain.
To find the specific interaction domains in UCH-L1 and TrkB, several truncated mutants or deletion mutants of UCH-L1 and TrkB were constructed and Co-IP assays were performed. More detailed information can be found in the Results section describing Figures 3 and 4. All of the experiments were performed at least three times.
Figure 3.

JM1 of TrkB is both necessary and sufficient for the association between TrkB and UCH-L1. A, Schematic presentation of TrkB-FL, TrkB-T1, JM1, JM0, and ΔJM1 used in this study. B, Co-IP was performed in HEK293 cell lysates expressing Myc-UCH-L1 and Flag-TrkB FL or other TrkB mutants: JM1, JM0, or ΔJM1. Cell lysates were immunoprecipitated (IP) using anti-Flag antibodies. C, Schematic presentation of IL-2R and IL-2R JM1 used in this study. D, Co-IP was performed in HEK293 cell lysates expressing Myc-UCH-L1 and Flag-TrkB FL or IL-2R or IL-2R JM1. Cell lysates were immunoprecipitated using anti-Flag antibodies.
Figure 4.

The 75–85 aa region of UCH-L1 is necessary and sufficient for the interaction between UCH-L1 and TrkB. A, Schematic presentation of UCH-L1-FL, UCH-L1 1–150, and UCH-L1 60–223 used in this study. B, Co-IP was performed in HEK293 cell lysates expressing Myc UCH-L1 or UCH-L1 1–150 or UCH-L1 60–223 and Flag-TrkB FL. Cell lysates were immunoprecipitated using anti-Myc antibodies and analyzed by immunoblotting with Flag antibodies. C, Schematic presentation of UCH-L1-FL and its deletion mutats. D, Co-IP was performed in HEK293 cell lysates expressing Myc UCH-L1 or its deletion mutants and Flag-TrkB. Cell lysates were immunoprecipitated (IP) using anti-Myc antibodies and analyzed by immunoblotting with Flag antibodies. E, Co-IP was performed in HEK293 cell lysates expressing pEGFPC2 (GFP) or pEGFPC2-UCH-L175–85 (GFP-75–85) and Flag-TrkB FL. Cell lysates were IP using an anti-Flag antibodies and analyzed by immunoblotting with GFP antibodies. F, Co-IP was performed in HEK293 cell lysates expressing Myc UCH-L1 or its mutants and Flag-TrkB. Cell lysates were IP using an anti-Flag antibodies and analyzed by immunoblotting with Myc antibodies. G, Quantification of the affinity between TrkB FL and UCH-L1 or mutants of UCH-L1. *p < 0.05 versus WT UCH-L1, one-way ANOVA.
Experiment 4: UCH-L1 regulates TrkB ubiquitination as a deubiquitinating enzyme.
To investigate whether UCHL1 could regulate TrkB deubiquitination via its deubiquitinating enzyme activity, we cotransfected HEK293 cells with HA-Ub, Flag-TrkB together with WT UCH-L1, or mutant UCH-L1 lacking deubiquitinating activity (C90S or D30A UCH-L1) and examined the ubiquitination of TrkB. To exclude artifact caused by exogenous expression of UCH-L1 and TrkB, we used the siRNA to knock down the UCH-L1 (3 siRNAs against UCH-L1 were used and scramble siRNA was used as control) or overexpress WT UCH-L1 or C90S/D30A UCH-L1 with lentivirus in cultured primary neurons and detected the endogenous TrkB ubiquitination level. To further validate the above results, we performed in vitro deubiquitination assays. More detailed information can be found in the Results section describing Figure 5. All of the experiments were performed at least three times.
Figure 5.

UCH-L1 regulates TrkB deubiquitination. A, TrkB undergoes multimonoubiquitination in response to BDNF. Primary neurons were serum starved for 8–12 h before stimulation with BDNF (50 ng/ml) for 15 min. The ubiquitination of TrkB was examined by immunoprecipitation with anti-TrkB antibodies followed by immunoblotting with P4D1 antibodies, which recognize both multimonoubiquitinated and polyubiquitinated substrates, and FK1 antibodies, which recognize only polyubiquitinated substrates. B, β-catenin was polyubiquitinated. Primary neurons were serum starved for 8–12 h and the ubiquitination of β-catenin was examined by both P4D1 and FK1 antibodies. C, TrkB is not ubiquitinated by K48 or K63 polyubiquitin chains. Primary neurons were serum starved for 8–12 h before stimulation with BDNF (50 ng/ml) for 15 min. The ubiquitination of TrkB was examined by immunoprecipitation with anti TrkB antibodies followed by immunoblotting with P4D1, anti-K48, and anti-K63 antibodies. D, UCH-L1 was knocked down with three siRNAs in primary neurons and the ubiqutination of TrkB was examined. E, UCH-L1 was overexpressed with lenti-WT UCH-L1 or C90S UCH-L1 in primary neurons and the ubiquitination of TrkB and the binding between UCH-L1 and TrkB were examined. F, Quantification of UCH-L1 and TrkB in cultured primary neurons before and after BDNF application. *Significant difference with p < 0.05, Student's t test. G, UCH-L1 deubiquitinates TrkB dependent on its hydrolase activity. HEK293 cells were transfected with Myc-UCH-L1, C90S, or D30A mutants together with Flag-TrkB FL and HA-Ub. Cell lysates were immunoprecipitated using anti-Flag antibodies and analyzed by immunoblotting with HA antibodies. H, UCH-L1 deubiquitinates TrkB in an in vitro deubiquitination assay. HEK293 cells were transiently transfected with Flag-TrkB and HA-Ub and ubiquitinated TrkB was enriched with anti-Flag antibodies. Purified UCH-L1 was incubated together with ubiquitinated TrkB in deubiquitylation assay buffer at 37°C for 1 h. The ubiquitination of TrkB was analyzed by immunoblotting.
Experiment 5: Study of the ubiquitination site in TrkB regulated by UCH-L1.
To investigate the ubiquitination sites in TrkB regulated by UCH-L1, we constructed several point mutants of TrkB, the mutation sites of which were near the UCHL1/TrkB interaction area, and the amino acid sites were mutated from lysine (K) to arginine (R): K454R, K460R, K464R, K506R, K524R, K533R, KJMR (all of the lysine in the JM region of TrkB mutated to arginine) and R460K (all of the lysine in the JM region of TrkB but K460 mutated to arginine) TrkB. We cotransfected HA-Ub, Flag-WT TrkB, or mutant TrkB together with UCHL1 into HEK293 cells and the ubiquitination of TrkB was examined. All of the experiments were performed at least three times.
Experiment 6: Effects of Tat-UCH-L175-85 on ubiquitination of TrkB, TrkB degradation, and intracellular trafficking.
To explore the effects of the peptide on TrkB ubiquitination, the cultured primary hippocampal neurons (DIV = 7) were pretreated with Tat-con or Tat-UCHL175–85 for 30 min followed by BDNF stimulation. The ubiquitination of TrkB was examined. The effects of the peptides on surface TrkB degradation were measured by surface biotin assay. More detailed information can be found in the Results section describing Figure 7.
Figure 7.

Ubiquitination of TrkB-FL promotes surface TrkB-FL degradation. A, Tat-UCH-L175–85 inhibits the interaction between UCH-L1 and TrkB. HEK293 cells were transfected with Flag-TrkB FL and Myc-UCH-L1. Cells were treated with Tat-Con or Tat-UCH-L175–85 before lysis. The interaction between UCH-L1 and TrkB was analyzed by immunoprecipitation (IP) using anti-Flag antibodies followed by immunoblotting with myc antibodies. B, Tat-UCH-L175–85 inhibits UCH-L1-regulated TrkB-FL deubiquitination in HKE293 cells. HEK293 cells were transfected with Flag-TrkB-FL together with Myc UCH-L1 and HA-Ub and then treated with Tat-Con or Tat-UCH-L175–85. TrkB ubiquitination was examined by immunoprecipitation using anti-Flag antibodies followed by immunoblotting with HA antibodies. C, Tat-UCH-L175–85 inhibits UCH-L1-regulated TrkB deubiquitination in neurons. Primary neurons were pretreated with Tat-UCH-L175–85 followed by stimulation with BDNF (50 ng/ml) for 15 min. The ubiquitination of endogenous TrkB was assessed by immunoprecipitation with TrkB antibodies followed by immunoblotting with P4D1 antibodies. D, Tat-UCH-L175–85 could not block UCH-L1-mediated deubiquitination of Hif-1α. ΗΕK293 cells transfected with Hif-1α, together with UCH-L1 and HA-Ub, were pretreated with Tat-UCH-L175–85 and the ubiquitination of Hif-1α was examined. E, Representative immunoblot images from the cleavable surface biotinylation assay (see Materials and Methods for details). Neuronal surface TrkB receptors were biotinylated, followed by BDNF stimulating for 60 min. The remaining biotinylated surface TrkB under different conditions were examined. F, Statistical results of surface TrkB level. **p < 0.01 versus no BDNF treatment group. ##p < 0.01 versus BDNF group treated with Tat-Con, one-way ANOVA. G, Representative immunoblot images from the cleavable surface biotinylation assay. HEK293 cells were transfected with WT or K460R TrkB FL. Then, surface TrkB receptors were biotinylated, followed by BDNF stimulating for 60 min. The remaining biotinylated TrkB-FL under different conditions were examined. H, Statistical results of surface TrkB-FL levels. *p < 0.05, **p < 0.01 versus control group or the indicated group, one-way ANOVA. All data are presented as mean ± SEM from three independent experiments.
To study the effects of Tat-UCH-L175–85 on TrkB endocytosis and postendocytic recycling, fluorescence-based internalization and recycling assays were performed. For each experiment at least 30 successfully transfected neurons per group were selected. At the same time, surface biotinylation internalization and recycling assays were also performed. More detailed information can be found in the Results section. All of the experiments were performed at least three times.
Experiment 7: Effects of Tat-UCH-L175-85 on TrkB and its downstream signaling pathways.
To determine whether Tat-UCH-L175–85 has any effects on TrkB signaling pathways, cultured primary hippocampal neurons were pretreated with Tat-Con or Tat-UCH-L175–85 for 30 min, followed by BDNF stimulation for the indicated times. Then, pTrkB, pERK1/2, and pAkt were examined by Western blot. All of the experiments were performed at least three times.
Experiment 8: Effects of Tat-UCH-L175-85 on CFC learning and memory.
This experiment was to investigate the effects of the Tat-UCH-L175–85 on CFC learning and memory. The Tat-scramble or Tat-UCH-L175–85 was injected into the DG region of mice (for more details, plese see the “Surgical procedure and microinjection” section) Thirty minutes later, the mice were subjected to CFC training followed by the STM test (n = 12 for each group). Then, the mice were killed and the DG regions were separated and proteins were extracted to detect the indicated protein levels. More detailed information can be found in the Results section describing Figure 10.
Figure 10.

TAT-UCH-L175–85 decreases hippocampus-dependent memory of C57 mice. A, Representative immunoblot images of TrkB ubiquitination, TrkB-FL, and the interaction between UCH-L1 and TrkB-FL in the DG of mice with or without CFC test. n = 4–6 per group. B–D, Quantification of TrkB ubiquitination (B), TrkB levels (C), and the interaction between UCH-L1 and TrkB (D). **p < 0.01 versus Tat-Con (CFC) group; #p < 0.05, ## p < 0.01, indicated groups, one-way ANOVA. *p < 0.05, Student's t test. n = 4/group. E, CFC test was performed to test the effects of Tat-UCH-L1 on hippocampus-dependent learning and memory. The mice were given a DG injection of Tat-Con or Tat-UCH-L175–85. CFC training (E) was performed 30 min later followed by the STM 1 h later (F). n = 12 per group; **p < 0.01, Student's t test. All data are presented as mean ± SEM.
Statistical analysis
All statistical analyses were performed using SPSS software. Comparisons of multiple groups were performed using one-way ANOVA followed by post hoc tests. When equal variances were assumed, an LSD test was used to compare the differences between groups. If not, a Dunnett's T3 test was used. Comparisons of two groups were performed using a two-tailed t test. Data are presented as the mean ± SEM and the significance was set at p < 0.05.
Results
UCH-L1 promotes hippocampus-dependent memory by regulating TrkB
It has been demonstrated that UCH-L1 plays an important role in maintaining learning and memory abilities (Sakurai et al., 2008; Zhang et al., 2014). BDNF–TrkB signaling pathways have also been shown to be critical in regulating learning and memory (Minichiello et al., 1999). First, an AAV virus that expressed UCH-L1 (AAV9-UCH-L1) or knocked down TrkB (AAV9-shTrkB) was stereotaxically injected into the DG region of mice. Many GFP- or mCherry-positive cells were observed in the DG region of AAV-virus infected mice (Fig. 1A), which demonstrated that the viruses successfully infected the cells in vivo. The efficacy of UCH-L1 overexpression and TrkB knockdown in the DG region of the hippocampus was also examined by Western blot. As shown in Figure 1B, the overexpression of UCH-L1 or the knockdown of TrkB was only limited to the DG region of the hippocampus. Ten days after the virus injection, CFC training and STM test (1 h after training) were performed. To avoid a ceiling effect, a lower current of 0.4 mA was used. We found that neither the overexpression of UCH-L1 nor the knockdown of TrkB could interfere with the CFC training process (Fig. 1D). Overexpression of WT UCH-L1 but not C90S UCH-L1 (Fig. 1F), which lacks deubiquitinating activity, significantly increased the freezing time (from 45.46 ± 1.45% to 67.26 ± 3.68%, Fig. 1E, F(6,76) = 35.842, p < 0.001; p < 0.001, WT UCH-L1 vs control; p = 0.588, C90S UCH-L1 vs control, one-way ANOVA), indicating that UCH-L1 promotes STM and this effect is dependent on its deubiquitinating activity. Decreased TrkB expression with shRNA led to a significant reduction in freezing time (from 41.85 ± 2.97% to 23.79 ± 1.41%), and overexpression of shRNA-resistant TrkB-FL in shRNA expressing brains to restore the level of TrkB-FL (Fig. 1G) could lead to a significant increase in the freezing time compared to shRNA TrkB group (from 23.79 ± 1.41% to 37.48 ± 1.15%, Fig. 1E, F(6,76) = 35.842, p < 0.001; p = 0.007, shTrkB vs Scr; p = 0.002, shTrkB and TrkB vs shTrkB, one-way ANOVA). These results consistently suggested that TrkB is critical in regulating hippocampus-dependent memory. Next, we investigated the relationship between UCH-L1 and TrkB. To find out, AAV9-UCH-L1 and AAV9-shTrkB viruses were simultaneously injected into the DG of mice and STM was performed. It is interesting that the freezing time of mice in the UCH-L1+ shTrkB group increased compared with the mice in the TrkB knockdown group (35.2 ± 2.93% vs 23.79 ± 1.41%), but the freezing time was still lower than in the vehicle control group and even much lower than in the UCH-L1 overexpression group (67.26 ± 3.68% vs 35.2 ± 2.93%, Fig. 1E, F(6,76) = 35.842, p < 0.001; p = 0.004, UCH-L1+ shTrkB vs shTrkB; p < 0.001, UCH-L1+ shTrkB vs WT UCH-L1, one-way ANOVA). These results indicated that the knockdown of TrkB prevents UCH-L1 induced improvements in STM. Therefore, TrkB is likely downstream of UCH-L1 in the regulation of STM. To further confirm this hypothesis, we applied the TrkB inhibitor ANA-12 and examined its effects on STM. ANA12 dramatically decreased the activation of TrkB in the DG of mice, as shown in Figure 1, M and N (F(3,8) = 331.963, p < 0.001; p < 0.001, control vs ANA-12, one-way ANOVA). In addition, the results of the behavior test were consistent with the knockdown of TrkB by shRNA. The inhibition of TrkB also led to a significant decrease in freezing time and impairing their STM (from 50.33 ± 4.28% to 24.82 ± 1.66%). This effect could not be reversed by the overexpression of UCH-L1 (24.82 ± 1.66% vs 31.05 ± 0.99%, Fig. 1L, F(3,20) = 40.450, p < 0.001; p = 0.005, control vs ANA-12; p = 0.181, ANA-12 vs OE UCH-L1 and ANA-12, one-way ANOVA). All of these results suggest that UCH-L1 regulates hippocampus-dependent memory via TrkB signaling pathways. Because UCH-L1 is a member of the deubiquitinase family and promotes STM dependent on its hydrolase activity, we speculated that it could regulate the ubiquitination of TrkB. Indeed, we found that the overexpression of UCH-L1 in the DG led to a decrease in the ubiquitination of TrkB (Fig. 1H,I, F(3,13) = 18.082, p < 0.001; p = 0.002, control vs UCH-L1 OE, one-way ANOVA) and the content of TrkB significantly increased compared with controls (Fig. 1H,J, F(3,13) = 39.103, p < 0.001; p = 0.020, control vs UCH-L1 OE, one-way ANOVA), which suggests that UCH-L1 may regulate TrkB ubiquitination as a deubiquitinase during the CFC process and thus alter TrkB protein levels.
UCH-L1 interacts with TrkB independent of TrkB kinase activity
To further investigate whether UCH-L1 could regulate TrkB ubiquitination as a deubiquitinase, we first tested whether UCH-L1 could associate with TrkB in an exogenous Co-IP assay. Flag-tagged TrkB-FL or TrkB.T1 with Myc-tagged UCH-L1 was transfected into HEK293 cells. After the immunoprecipitation of TrkB with Flag antibodies, an association between TrkB and UCH-L1 was observed by immunoblotting with myc antibodies (Fig. 2A). The complex that formed between TrkB-FL and UCH-L1 was also confirmed after immunoprecipitation with myc antibodies and immunoblotting with Flag antibodies (Fig. 2B). To exclude any artifacts caused by overexpression and to determine whether this interaction occurs in physiological conditions, we performed an endogenous Co-IP assay with rat brain lysate. The lysate was immunoprecipitated with anti-TrkB antibodies and immunoblotted with anti-UCH-L1 antibodies. We found an association between endogenous UCH-L1 and TrkB (Fig. 2C). Therefore, we determined that TrkB and UCH-L1 could interact with each other and form a complex both in vitro and in vivo. To investigate whether UCH-L1 interacts directly with TrkB, we performed the in vitro binding assay with purified UCHL1 and TrkB and the result demonstrated that UCHL1 associates with TrkB directly (Fig. 2D).
Figure 2.

UCH-L1 associates with TrkB-FL independently of TrkB kinase activity. A, Co-IP was performed in lysates of HEK293 cells expressing Flag-tagged TrkB-FL (FL) or TrkB.T1 (T1) and Myc-tagged UCH-L1. Cell lysates were immunoprecipitated (IP) with anti-Flag antibodies. B, Co-IP was performed in lysates of HEK293 cells expressing Flag-tagged TrkB-FL and Myc-tagged UCH-L1. Cell lysates were immunoprecipitated with anti-Myc antibodies. C, Endogenous association of UCH-L1 and TrkB receptors was detected by Co-IP in C57 mice brain tissues. Brain lysates were immunoprecipitated with anti-TrkB antibodies. A preimmunized IgG (rabbit) was used as a negative control. D, In vitro binding assay to investigate the interaction between TrkB and UCH-L1. Flag-TrkB FL and HA-UCH-L1 were obtained by in vitro transcription and translation. Interaction between TrkB and UCH-L1 was assayed by mixing Flag-TrkB FL and HA-UCH-L1 together, followed by immunoprecipitation with Flag antibodies. E, Co-IP was performed in lysates of HEK293 cells expressing Myc-tagged UCH-L1 and Flag-tagged TrkB-FL or TrkB KD. Cell lysates were immunoprecipitated with anti-Flag antibodies. F, Quantification of the affinity between UCH-L1 and TrkB FL or TrkB KD. Data are shown as the mean ± SEM. p > 0.05, Student's t test. Each Co-IP experiment was repeated three times and representative data are shown.
We have shown that both TrkB-FL and TrkB.T1 interact with UCH-L1 with similar affinities (Fig. 1A) and TrkB.T1 is a truncated TrkB with no intracellular kinase domain (Fig. 3A). It is likely that the association between TrkB and UCH-L1 is independent of TrkB kinase activity. To further confirm this, wild-type Flag-tagged TrkB-FL or TrkB-Kinase dead construct (TrkB-KD) (Zhao et al., 2009) were transfected into HEK293 cells with Myc-tagged UCH-L1. We found that UCH-L1 interacted with both TrkB-FL and TrkB-KD at comparable levels (Fig. 2E,F, t′ = 0.294, p = 0.807, two-tailed t test), indicating that the association is independent of the kinase activity of TrkB.
Mapping the TrkB/UCH-L1 interaction domain
Because TrkB and UCH-L1 form a complex, we next mapped the domain involved in their interaction. Our results showed that both TrkB-FL and TrkB.T1 could interact with UCH-L1 (Fig. 2A). As shown in Figure 3A, TrkB.T1 shares with TrkB-FL a consensus 12 aa (K454–G465, JM1) in the intracellular JM domain. To address whether these 12 aa play an important role in their interaction, several TrkB mutant constructs were used as previously reported: TrkB-JM1 that retained the 12 aa in the TrkB intracellular domain, TrkB-JM0 with the entire TrkB intracellular domain deletion, and TrkBΔJM1 with the 12 aa deletion in the TrkB intracellular domain (Fig. 3A; Huang et al., 2011). The Co-IP assay revealed that TrkB-JM1, but not TrkBΔJM1 or TrkB-JM0, could still interact with UCH-L1 (Fig. 3B), suggesting that the JM1 domain is necessary for TrkB/UCH-L1 interaction. To further investigate whether the JM1 domain is sufficient for the TrkB/UCH-L1 interaction, we transplanted JM1 to the C-terminal of IL2R, which has a short cytoplasmic tail (Fig. 3C). We found that IL2R itself could not interact with UCH-L1 and the JM1 transplantation could confer its binding ability with UCH-L1 (Fig. 3D), suggesting that the JM1 domain in TrkB is not only necessary but also sufficient for the TrkB/UCH-L1 interaction.
To verify which region in UCH-L1 is involved in the UCH-L1/TrkB interaction, we constructed several deletion mutants of UCH-L1 with N-terminal myc tags (Fig. 4A) and performed Co-IP assays. TrkB was found to interact with aa 1–150 and 60–223 in UCH-L1 (Fig. 4B), indicating that TrkB interacts with a region within aa 60–150 of UCH-L1. To define more precisely the region required for TrkB binding, a series of mutants of UCH-L1 with corresponding amino acid deletions were constructed based on its tertiary structure: Δ60–75, Δ75–85, Δ85–100, Δ110–125, and Δ125–147 (Fig. 4C). The Co-IP was performed and the result showed that TrkB interacts with Δ60–75, Δ85–100, Δ110–125, and Δ125–147 UCH-L1, but not with Δ75–85 UCH-L1 (Fig. 4D), which indicated that aa 75–85 of UCH-L1 are necessary for the association between UCH-L1 and TrkB. To further investigate whether aa 75–85 of UCH-L1 are sufficient for the TrkB/UCH-L1 interaction, we transplanted aa 75–85 of UCH-L1 to a pEGFP-C2 vector. We found that pEGFP-C2 itself could not interact with TrkB and aa 75–85 transplantation confers its binding ability with TrkB (Fig. 4E). The above results suggest that aa 75–85 in UCH-L1 are not only necessary but also sufficient for the TrkB/UCH-L1 interaction.
Our previous studies have shown that the TrkB/UCH-L1 interaction does not depend on TrkB kinase activity and we further investigated whether the interaction depends on the hydrolase activity of UCH-L1. We cotransfected HEK293 cells with a Flag-tagged TrkB-FL and a Myc-tagged WT or C90S or D30A UCH-L1. The C90S mutation abolishes its enzymatic hydrolase activity and maintains ubiquitin binding affinity, whereas the D30A mutation causes a loss of ubiquitin-binding ability (Larsen et al., 1996; Osaka et al., 2003). The Co-IP results showed that both WT and mutant UCH-L1 interacted with TrkB-FL; the C90S mutation results in a weaker association (Fig. 4G), but D30A did not (Fig. 4F,G, F(2,6) = 10.981, p = 0.010; p = 0.015, C90S vs WT; p = 0.294, D30A vs WT, one-way ANOVA), suggesting that UCH-L1 hydrolase activity plays an important role in the regulation of their interaction.
UCH-L1 regulates TrkB ubiquitination as a deubiquitinating enzyme
Because UCH-L1 and TrkB interact with each other and the overexpression of UCH-L1 led to a reduction in TrkB ubiquitination in vivo, we hypothesized that UCH-L1 regulates TrkB ubiquitination as a deubiquitinase. To study our hypothesis, first we examined TrkB ubiquitination. Primary hippocampal neurons at DIV7 were stimulated with BDNF (50 ng/ml) for 15 min and the cell extracts were subjected to immunoprecipitation with TrkB antibodies and blotted in parallel with antibodies that recognize both multimonoubiquitination and polyubiquitination (P4D1) or only polyubiquitination (FK1) as described previously (Arévalo et al., 2006). Consistent with the results of previous study (Arévalo et al., 2006), BDNF induced a rapid and significant increase in the ubiquitination of TrkB, as detected with the P4D1, but not with FK1 (Fig. 5A). To confirm the fidelity of the FK1 antibody, the state of ubiquitination of β-catenin, which has been reported to be polyubiquitinated (Dimitrova et al., 2010), was also analyzed in neurons. The polyubiquitinated β-catenin was effectively detected by both FK1 and P4D1 antibodies (Fig. 5B). We also found that anti-K48 or K63 polyubiquitin antibodies could not detect BDNF-induced ubiquitination of TrkB in cultured primary neurons (Fig. 5C). These data suggested that TrkB is likely to be multimonoubiquitinated in response to BDNF.
In contrast with TrkB.T1, the full-length TrkB (TrkB FL), which contains the kinase activity, plays a more important role in modulation of neural activity, synaptic structures, and functions (Zhao et al., 2009; Huang et al., 2013) and we focused on studying the effects of UCH-L1 on TrkB FL in the following studies. To further confirm that UCH-L1 regulates the ubiquitination of TrkB, we examined endogenous BDNF-induced TrkB ubiquitination in primary hippocampal neurons via the overexpression or knockdown of UCH-L1. As shown in Figure 5D, the reduction of UCH-L1 by three different siRNAs led to increased ubiquitination of TrkB. In addition, the ubiquitination level of TrkB decreased with the overexpression of WT UCH-L1, but C90S UCH-L1 (Fig. 5E), which lacks hydrolase activity, could not reduce the ubiquitination of TrkB. These results suggested that UCH-L1 could regulate the deubiquitination of TrkB and this regulation is dependent on its hydrolase activity. This also occurred in HEK293 cells with TrkB and WT or mutant UCH-L1 expression. In HEK293 cells, the level of TrkB ubiquitination decreased significantly with WT UCH-L1 overexpression and C90S or the D30A mutant UCH-L1, both of which lack the hydrolase activity, failed to decrease the ubiquitination of TrkB (Fig. 5G), also suggesting that UCH-L1 regulates TrkB ubiquitination mainly via its hydrolase activity. In addition, an in vitro deubiquitination assay was performed. As expected, recombinant UCH-L1 protein was able to potently deubiquitinate TrkB (Fig. 5H), strongly suggesting UCH-L1 as a deubiquitinating enzyme that regulates TrkB ubiquitination. We also found that binding between UCH-L1 and TrkB increased after BDNF treatment (Fig. 5E,F), suggesting that the affinity between UCH-L1 and TrkB could be regulated by BDNF.
UCH-L1 mainly deubiquitinates TrkB at K460
There are ∼20 potential ubiquitin lysine loci in the intracellular domain of TrkB-FL and we next investigated which sites could be the major ubiquitination sites regulated by UCH-L1. Considering that the JM1 region of TrkB is important for the interaction between TrkB and UCH-L1, we speculated that the TrkB ubiquitination sites regulated by UCH-L 1 were mainly located near the JM area. To test the hypothesis, several TrkB mutants were constructed in which each of the lysines in JM region was mutated to arginine (K454R, K460R, K464R, K506R, K524R, and K533R). We cotransfected Flag-tagged TrkB-FL or TrkB mutants with or without UCH-L1 in HEK293 cells and the ubiquitination of TrkB was examined. As shown in Figure 6, A and B, the K460R mutation led to a significant reduction in TrkB ubiquitination (F(13,28) = 7.869, p < 0.001; p = 0.003, K460R TrkB vs WT TrkB FL, one-way ANOVA) and the ubiquitination level of other mutant TrkB receptors remained at a comparable level as WT TrkB-FL; UCH-L1 overexpression failed to deubiquitinate TrkB when K460 was changed to arginine, however, UCH-L1 could still deubiquitinate the other mutant TrkB receptors. These results suggest that K460 may be the major ubiquitination site of TrkB and may also be the major ubiquitination site regulated by UCH-L1. However, this could not exclude the possibility that the ubiquitination of K460R TrkB has reached the lowest level, so that overexpression of UCH-L1 could not lead to further reduction of ubiquitination. To confirm that UCH-L1 deubiquitinates TrkB at K460, we constructed two other constructs: in one construct, all lysines in the JM region were mutated to arginine (KJMR) and, in the other construct, all lysines but K460 in the JM region were mutated to arginine (R460K). The ubiquitination level of these two constructs was detected with or without UCH-L1 overexpression. We found that the ubiquitination level of KJMR TrkB decreased significantly, as expected, and UCH-L1 had no effects on its ubiquitination; however, the ubiquitination of R460K TrkB was at a comparable level as WT TrkB and UCH-L1 could still deubiquitinate R460K TrkB (Fig. 6C,D, F(5,12) = 46.461, p < 0.001; p = 0.004 KJMR TrkB vs TrkB FL; p = 0.667, KJMR+UCH-L1 vs KJMR, p = 0.670, R460K TrkB vs TrkB FL, p = 0.001, R460K TrkB vs R460K TrkB+UCH-L1, one-way ANOVA). All of the above data suggested that K460 in TrkB is the major site of ubiquitination and also the primary site regulated by UCH-L1.
Figure 6.

UCH-L1 mainly deubiquitinates TrkB at K460 site. A, K460 is the major site of ubiquitination of TrkB. HEK293 cells were transiently transfected with Flag-TrkB FL or its site mutants together with Myc-UCH-L1 and HA-Ub. Cell lysates were immunoprecipitated using anti-Flag antibodies and the ubiquitination of TrkB was analyzed by immunoblotting with HA antibodies. B, Quantification of the ubiquitination level of WT or the site mutant TrkB FL. *p < 0.05, **p < 0.01 versus no UCH-L1 control, ##p < 0.01 versus WT TrkB-FL control, one-way ANOVA. C, UCH-L1 regulates TrkB ubiquitination at the K460 site. HEK293 cells were transfected with Flag-TrkB, Flag-TrkB KJMR, or Flag-TrkB R460K together with Myc-UCH-L1 and HA-Ub. Cell lysates were immunoprecipitated (IP) using anti-Flag antibodies and the level of ubiquitination of WT or mutant TrkB was analyzed by immunoblotting with HA antibodies. Statistical results are shown in D. **p < 0.01 versus no UCH-L1-expressing control, ##p < 0.01 versus WT TrkB-FL control, one-way ANOVA. All data are presented as mean ± SEM.
Blockade of UCH-L1-regulated TrkB deubiquitination by Tat-UCH-L175–85 promotes TrkB degradation
Ubiquitination of surface receptors has been demonstrated to regulate their biological functions, such as intracellular sorting and degradation (Haglund et al., 2003; Widagdo et al., 2015), so we further explored the effects of UCH-L1-regulated TrkB deubiquitination on TrkB degradation and intracellular trafficking. To this end, a specific peptide, aa 75–85 of UCH-L1 fused with biotin and the membrane permeability domain (Tat) at the N-terminal (Tat-UCH-L175–85), which specifically blocks UCH-L1/TrkB interaction in a competitive manner, was used instead of the UCH-L1 inhibitor LDN57444 or the knockdown of UCH-L1. In addition to inhibiting the activity of UCH-L1, the inhibitor also alters proteasome activity (Tan et al., 2008). UCH-L1 is a multifunctional proteins (Larsen et al., 1996; Liu et al., 2002; Osaka et al., 2003; Kabuta et al., 2013) and the knockdown of UCH-L1 has broader effects in addition to its effects on TrkB. Therefore, the peptide could be a better tool with which to investigate the specific effects of UCH-L1-regulated TrkB. We first tested the efficiency of Tat-UCH-L175–85. HEK293 cells transfected with TrkB and UCH-L1 were treated with Tat-UCH-L175–85 (10 μm) for 30 min and a Co-IP assay was performed. As shown in Figure 7A, the association between UCH-L1 and TrkB was efficiently inhibited by the peptide. We next investigated whether this inhibition affects ubiquitination of TrkB. As expected, the deubiquitination of TrkB regulated by UCH-L1 was significantly inhibited by Tat-UCH-L175–85 (Fig. 7B). A similar result was also observed in cultured primary hippocampal neurons, in which the addition of Tat-UCH-L175–85 efficiently increased BDNF-induced TrkB ubiquitination (Fig. 7C). These results suggest that the inhibition of the association between UCH-L1 and TrkB by Tat-UCH-L175–85 promotes the ubiquitination of TrkB. It has been demonstrated that UCH-L1 could also deubiquitinate other proteins such as Hif-1α and NOXA (Brinkmann et al., 2013; Goto et al., 2015). To confirm the specificity of the peptide, we investigated whether Tat-UCH-L1 had any effects on UCH-L1-mediated deubiquitination of Hif-1a. As shown in Figure 7D, UCH-L1 successfully reduced the ubiquitination of Hif-1α; however, Tat-UCH-L175–85 failed to block UCH-L1-mediated deubiquitination of Hif-1α. All of these resutls suggest that Tat-UCH-L175–85 could specifically inhibit UCH-L1-mediated TrkB deubiquitination.
The Tat-UCH-L175–85 was used to investigate the effects of UCH-L1-regulated deubiquitination of TrkB-FL. First, we performed a surface biotin assay to study its effects on BDNF-mediated surface TrkB-FL degradation. Cultured hippocampal neurons were pretreated with 10 μm Tat-Con or Tat-UCH-L175–85 for 30 min and then the surface proteins were biotinylated, followed by BDNF (50 ng/ml) exposure for 60 min to allow for the degradation of endocytic TrkB-FL. The biotin-labeled TrkB-FL, which represented the undegraded TrkB-FL, was pulled down by avidin-agarose and detected by immunoblot with TrkB antibodies. The quantification of data revealed that, in the Tat-Con group, after BDNF stimulation, 54.4 ± 0.68% of surface TrkB-FL was degraded, in contrast to 70.9 ± 2.40% of surface TrkB-FL degraded in Tat-UCH-L175–85-treated neurons (Fig. 7E,F, F(3,8) = 46.057, p = 0.001; p = 0.001, Tat-Con + BDNF vs Tat UCH-L175–85 + BDNF, one-way ANOVA). These results suggest that the blockage of the interaction of UCH-L1 and TrkB with Tat-UCH-L175–85 increases the ubiquitination of TrkB and promotes BDNF-induced surface TrkB-FL degradation. The peptide caused an increase of ubiquitination of TrkB-FL and the K460R mutation caused a reduction of ubiquitination of TrkB-FL. Therefore, to better understand the effects of TrkB-FL ubiquitination on its degradation, we also investigated the BDNF-induced degradation of surface K460R TrkB. As shown in Figure 7, G and H, BDNF led to a significant reduction of WT TrkB-FL; however, the K460R mutation made TrkB-FL more resistant to degradation (Fig. 7G,H, F(3,8) = 10.186, p = 0.024; p = 0.011, TrkB-FL vs TrkB-FL + BDNF, p = 0.565, TrkB K460R vs TrkB K460R + BDNF, one-way ANOVA). These results indicate that the ubiquitination of TrkB-FL could promote degradation of surface TrkB-FL receptors.
Ubiquitination of TrkB-FL results in the enhanced internalization of the TrkB-FL receptor
We hypothesized that the increased degradation of surface TrkB-FL is caused by the altered intracellular trafficking steps after BDNF stimulation, which includes the initial internalization process and the postendocytic recycling or degradation pathways. To determine whether the endocytosis of TrkB-FL could be influenced by Tat-UCH-L175–85, a live-cell ratiometric fluorescence-based internalization assay was performed (see detailed protocol in Material and Methods). We found that, after stimulation with BDNF, significantly fewer TrkB-FL receptors remained on the cell surface in Tat-UCH-L175–85-pretreated neurons compared with the control group (Fig. 8A), suggesting that blocking the UCH-L1/TrkB interaction with Tat-UCH-L175–85 facilitates the endocytosis of surface TrkB (from 40.62 ± 2.99% to 60.04 ± 1.96%, Fig. 8B, n = 30, t = −5.803, p < 0.001, two-tailed t test). To eliminate any artifacts caused by overexpression, a cleavable surface biotinylation internalization assay was applied to measure endogenous surface TrkB internalization. We also found that more surface TrkB-FL was internalized in respond to BDNF in the Tat-UCH-L175–85-treated neurons (from 41.89 ± 5.22% to 63.80 ± 1.89%, Fig. 8C,D, t = −3.944, p = 0.017, two-tailed t test). We also investigated BDNF-induced internalization of K460R TrkB-FL, which shows less ubiquitination, with a live-cell ratiometric fluorescence-based internalization assay. The results revealed that the K460R mutation inhibits the endocytosis of surface TrkB (from 38.547 ± 2.98% to 25.04 ± 1.96%, Fig. 8E, n = 30, t = 3.362, p < 0.001, two-tailed t test). These results suggest that the ubiquitination of TrkB promotes BDNF-mediated surface TrkB-FL endocytosis.
Figure 8.
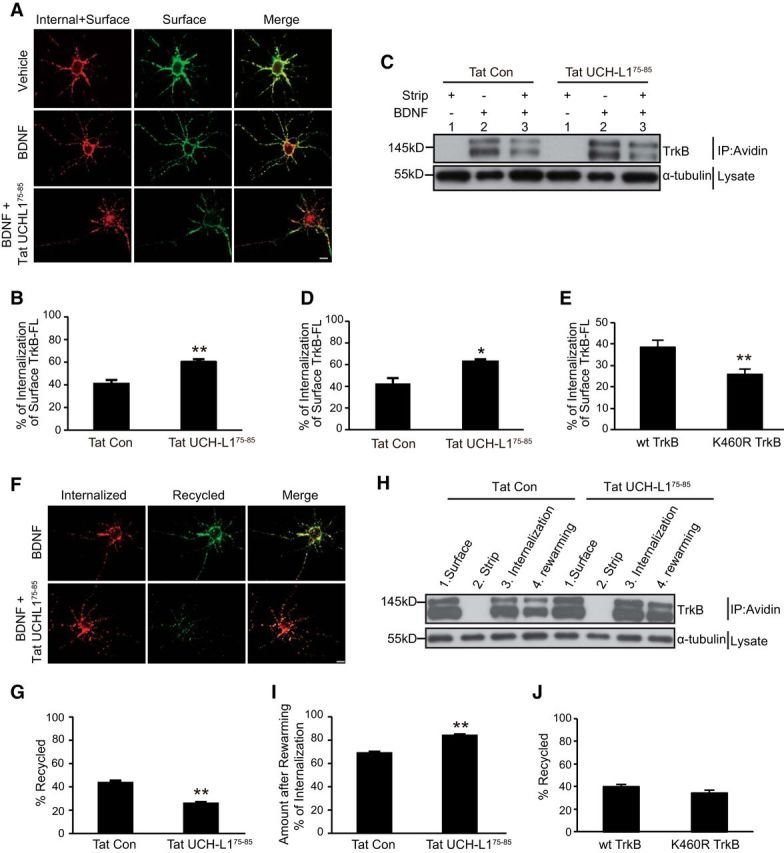
Ubiquitination of TrkB facilitates BDNF-induced TrkB-FL internalization. A, Representative epifluorescence images from the TrkB-FL internalization assay. Scale bar, 20 μm. B, Quantification of internalized TrkB levels in A was performed as described in the Materials and Methods. All data are presented as mean ± SEM determined from analysis of more than three independent experiments (n = 30 cells for each condition per independent experiment). **p < 0.01, Student's t test. C, Representative images from the cleavable surface biotinylation assay (see Materials and Methods for details). Lane 1 shows the efficiency of the stripping procedure; lane 2 shows the total biotinylated TrkB-FL receptors at the neuronal surface; lane 3 shows the internalized biotinylated TrkB receptors. D, Quantification of internalized TrkB levels in C was performed as described in the Materials and Methods. All data are presented as mean ± SEM determined from analysis of more than three independent experiments. *p < 0.05, Student's t test. E, Quantification of internalized K460R TrkB-FL levels performed by epifluorescence internalization assay. All data are presented as mean ± SEM from three independent experiments (n = 30 cells for each condition per independent experiment). **p < 0.01, Student's t test. F, Representative epifluorescence images from the TrkB-FL postendocytic recycling assay. Scale bar, 20 μm. G, Quantitative analysis of recycled TrkB receptors as described in E. Graphs represent mean ± SEM determined from results of more than three independent experiments (n = 30 cells for each condition per independent experiment). **p < 0.01, Student's t test. H, Representative data from the cleavable surface biotinylation assay (see Materials and Methods for details). Lane 1 shows the total biotinylated TrkB receptors at neuronal surface; lane 2 shows the efficiency of the stripping procedure; lane 3 shows the internalized biotinylated TrkB receptors; lane 4 shows the un-recycled TrkB receptors during the rewarming period. I, Densitometric quantitation of un-recycled biotinylated TrkB receptor levels in G. Bar graphs represent mean ± SEM (n > 3). **p < 0.01, Student's t test. J, Quantitative analysis of recycled K460R TrkB receptors as performed by live-cell epifluorescence recycling assay. Graphs represent mean ± SEM from three independent experiments (n = 30 cells for each condition per independent experiment).
Next, we investigated the effects of Tat-UCH-L175–85 on the postendocytic recycling of TrkB-FL. A live-cell ratiometric fluorescence-based recycling assay was applied to measure the recycling level of TrkB (see detailed protocol in Materials and Methods), which has been used in our previous studies (Zhao et al., 2009; Huang et al., 2013). Quantification of these results by ratiometric fluorescence analysis revealed that, when the UCH-L1/TrkB interaction was blocked by Tat-UCH-L175–85, the levels of BDNF-dependent TrkB-FL recycling were significantly reduced (from 43.7 ± 1.68% to 25.9 ± 0.99%, Fig. 8F,G, n = 30, t′ = 9.055, p < 0.001, two-tailed t test), suggesting that the blockade of UCH-L1/TrkB association by Tat-UCH-L175–85 induces less internalized TrkB-FL to be recycled back to the cell surface. A cleavable surface biotinylation recycling assay was also applied to measure endogenous BDNF-dependent postendocytic recycling of TrkB. We found a significant increase in the remaining TrkB after rewarming upon treatment with Tat-UCH-L175–85 (from 69.07 ± 1.07% to 84.07 ± 0.95%, Fig. 8H,I, t = −10.418, p < 0.001, two-tailed t test), suggesting Tat-UCH-L175–85 causes less internalized TrkB to be recycled back to the cell surface to be cleaved. Therefore, these results indicate that, by inhibiting the UCH-L1/TrkB interaction, Tat-UCH-L175–85 promotes TrkB ubiquitination and reduces postendocytic recycling of TrkB-FL. We further investigated whether the K460R TrkB-FL increased recycling as K460R TrkB has decreased ubiquitination with a ratiometric fluorescence-based recycling assay. However, we found the BDNF-dependent postendocytic recycling of K460R TrkB was at comparable levels as WT TrkB (Fig. 8J, n = 30, t = 0.942, p = 0.354, two-tailed t test), indicating that the ubiquitination of TrkB-FL may be not a major factor in the regulation of the recycling of TrkB. All of these results suggest that the ubiquitination of TrkB could facilitate BDNF-dependent TrkB-FL internalization and direct more internalized TrkB-FL to degradation pathways.
TAT-UCHL75–85 attenuates TrkB and its downstream signaling pathways
To further investigate the functional consequences of UCHL-regulated TrkB ubiquitination, we examined BDNF-induced downstream signaling in cultured hippocampal neurons pretreated with Tat-UCH-L175–85. We found that, with Tat-UCH-L175–85 pretreatment, the level of phosphorylated TrkB decreased significantly after 15 min of exposure to BDNF (Fig. 9A,B, F(7,24) = 16.704, p < 0.001; p = 0.001, Tat-Con + BDNF vs Tat-UCH-L175–85 + BDNF at 15 min, one-way ANOVA), indicating that TrkB activation was attenuated as early as 15 min. The activation of Akt and ERK1/2, which are well established downstream signaling molecules of TrkB, was monitored by immunoblot. To our surprise, phosphorylated Akt was increased by ∼2-fold at 15 min (Fig. 9A,C, F(7,24) = 18.536, p < 0.001; p < 0.001, Tat-Con + BDNF vs Tat-UCH-L175–85 + BDNF at 15 min, one-way ANOVA), suggesting that Akt is activated at 15 min. The activation of Akt at 15 min is most likely due to the enhanced endocytosis caused by Tat-UCH-L175–85 (Fig. 8)because previous studies have demonstrated the activation of Akt signaling downstream of TrkB is dependent on ligand-receptor endocytosis (Zheng et al., 2008). After 60 min of exposure to BDNF, significantly decreased activities of both ERK1/2 and Akt pathways (Fig. 9A–D) were revealed by decreased phosphorylation of Akt (Fig. 9C, F(7,24) = 18.536, p < 0.001; p = 0.027, Tat-Con + BDNF vs Tat-UCH-L175–85 + BDNF at 60 min, one-way ANOVA) and ERK1/2 (Fig. 9D, F(7,24) = 20.328, p < 0.001; p = 0.001, Tat-Con + BDNF vs Tat-UCH-L175–85 + BDNF at 60 min, one-way ANOVA). Together, in the long term, the increased ubiquitination of TrkB induced by Tat-UCH-L175–85 causes decreased activity of TrkB and its downstream signaling pathways. We also investigated the effects of K460R mutation on TrkB activation. WT or K460R TrkB was transfected into HEK293 cells, followed by BDNF application for the indicated times. As shown in Figure 9, E and F, the activation of K460R TrkB-FL was stronger and persisted for a longer amount of time compared with WT TrkB. All of these data suggest that the ubiquitination of TrkB could reversibly regulate the activation of TrkB.
Figure 9.

Tat-UCH-L175–85 causes decreased activation of TrkB and its downstream signaling pathways. A, Representative images of immunoblots showing BDNF-induced phosphorylation of TrkB, Akt, and ERK1/2 in cultured hippocampal neurons under the indicated conditions. Neurons were pretreated with Tat-Con (10 μm, 30 min) or Tat-UCH-L175–85(10 μm, 30 min) before BDNF (50 ng/ml) stimulation. B–D, Quantitative analysis of TrkB (B), Akt (C), and ERK1/2 (D) activation in neurons under Tat-UCH-L175–85 and Tat-Con incubation. p-TrkB, p-ERK1/2, and p-Akt levels were normalized to the phosphorylation levels of BDNF alone treatment group detected at the 15 min time point. Graphs represent means ± SEM. n = 4, *p < 0.05, **p < 0.01, one-way ANOVA. E, Representative images of immunoblots showing BDNF-induced phosphorylation of TrkB in HEK293 cells transfected with WT or K460R TrkB for the indicated times. F, Quantitative analysis of TrkB activation. Graphs represent means ± SEM. n = 3, *p < 0.05, **p < 0.01, one-way ANOVA.
Tat-UCH-L175–85 impairs hippocampus-dependent memory
Our in vitro experiments have demonstrated that, by regulating TrkB deubiquitination, UCH-L1 is involved in the regulation of BDNF-dependent endocytosis, postendocytic recycling of TrkB, and, subsequently, TrkB downstream signaling pathways. Given that UCH-L1 promotes hippocampus-dependent memory via TrkB (Fig. 1), we hypothesized that UCH-L1-regulated TrkB deubiquitination contributes to the increased memory induced by UCH-L1 in vivo. To test this hypothesis, we first investigated whether Tat-UCH-L175–85 could inhibit TrkB ubiquitination in vivo. We used the stereotaxic injection of drugs into the brain DG regions of ∼6- to 8-week-old mice. After a week of recuperation, Tat control or Tat-UCH-L175–85 was injected into mice brain DG regions 30 min before CFC training and, 1 h later, the DG regions of the brains were collected and the interaction of UCH-L1/TrkB and the ubiquitination level of TrkB was examined. As expected, the interaction between UCH-L1 and TrkB was significantly inhibited with Tat-UCH-L175–85 treatment (Fig. 10A). Under basic conditions (without CFC training), Tat-UCH-L175–85 had no impact on the ubiquitination level (Fig. 10B, F(3,12) = 12.388, p < 0.001; p = 0.870, Tat-Con without CFC vs Tat-UCH-L175–85 without CFC, one-way ANOVA) or protein level of TrkB (Fig. 10A,C, F(3,12) = 50.202, p < 0.001; p = 0.181, Tat-Con without CFC vs Tat-UCH-L175–85 without CFC, one-way ANOVA); however, after CFC training, there was a robust increase in the ubiquitination level of TrkB (Fig. 10A,B, F(3,12) = 12.388, p < 0.001; p = 0.044, Tat-Con without CFC vs Tat-Con after CFC, one-way ANOVA), a significant reduction in TrkB content (Fig. 10C, F(3,12) = 50.202, p < 0.001; p < 0.001, Tat-Con without CFC vs Tat-Con after CFC, one-way ANOVA), and the binding of UCH-L1 and TrkB increased significantly (Fig. 10D, n = 4, t = −3.754, p = 0.023, two tailed t test). Tat-UCH-L175–85 treatment led to a further increase in the ubiquitination of TrkB and a decrease in the expression of TrkB in the mice brains (Fig. 10A–C, F(3,12) = 50.202, p < 0.001; p = 0.005, Tat-Con after CFC vs Tat-UCH-L175–85 after CFC, one-way ANOVA). These results suggested that CFC training may induce BDNF release and lead to an increased ubiquitination of TrkB; the inhibition of UCH-L1/TrkB interaction by Tat-UCH-L175–85 promotes CFC training-induced TrkB ubiquitination in vivo.
Next, we examined the effects of Tat-UCH-L175–85 on hippocampus-dependent learning and memory with CFC test. Mice were injected with Tat-Con or Tat-UCH-L175–85 for 30 min and CFC was applied to test STM. We found that the two groups of mice exhibited intact freezing responses during the training process (Fig. 10E). We then tested the contextual fear STM of these two groups of mice. The mice injected with Tat-UCH-L175–85 exhibited significant decreased freezing time (from 65.26 ± 4.45% to 11.4 ± 2.34%, Fig. 10F, t′ = 10.710, n = 12, p < 0.001, two-tailed t test) and this effect was not caused by differences in the locomotor activity (data not shown), suggesting that Tat-UCH-L175–85 impaired contextual fear memory. The above results indicate the blockade of UCH-L1-regulated deubiquitination of TrkB by Tat-UCH-L175–85 impairs hippocampus-dependent STM.
Discussion
UCH-L1 has been demonstrated to be required for synaptic function and the maintenance of memory in passive avoidance learning through the regulation of the transcription factor cAMP response element-binding protein (CREB) (Gong et al., 2006; Sakurai et al., 2008). In this study, we demonstrate that UCH-L1 could facilitate hippocampus-dependent memory via the regulation of TrkB ubiquitination. Moreover, we found that UCH-L1 functions as a deubiquitinating enzyme that regulates TrkB ubiquitination level, thus altering BDNF-dependent TrkB endocytosis and postendocytic traffic. Finally, the UCH-L1-mediated TrkB ubiquitination contributes to BDNF-mediated downstream signaling pathways, which suggests that UCH-L1-regulated TrkB deubiquitination plays important roles in BDNF-associated functions.
Our study provides several novel insights into the mechanisms and significance of UCH-L1 and TrkB ubiquitination. First, we provide direct evidence that UCH-L1 is responsible for the deubiquitination of TrkB. Limited studies were conducted to study the ubiquitination of TrkB and c-Cbl and TRAF6 have been demonstrated to be the E3 ligases for TrkB ubiquitination (Jadhav et al., 2008; Pandya et al., 2014). However, the detailed mechanisms and biological functions are still not clear. Here, we demonstrate that UCH-L1 regulates the deubiquitination of TrkB (Fig. 4). The ubiquitination of TrkB is precisely regulated by the E3 ligase and DUB. We have demonstrated the TrkB KD could associate with UCH-L1 with a similar affinity as WT TrkB and the loss of hydrolase activity of UCH-L1 attenuates the association between UCH-L1 and TrkB (Figs. 2, 3), which suggests that the interaction between UCH-L1 and TrkB is dependent on UCH-L1 hydrolase activity rather than TrkB kinase activity. It has been reported that BDNF could upregulate deubiquitinating activity of UCH-L1 in hippocampal synaptoneurosomes (Santos et al., 2015). Therefore, it is possible UCH-L1 and TrkB associate constitutively with each other. Neuronal activity such as LTP and learning and memory processes, which could trigger the local secretion of BDNF (Greenberg et al., 2009; Ma et al., 2011), causes a rapid increase in the ubiquitination of TrkB (Fig. 1). At the synapse, BDNF also upregulates the hydrolase activity of UCH-L1 to deubiquitinate TrkB. How E3 ligase and UCH-L1 coordinate to regulate the ubiquitination of TrkB needs further investigation. Impairment of the regulation, such as the inhibition of UCH-L1 regulated deubiquitination of TrkB by Tat-UCH-L175–85, could lead to attenuated TrkB signaling pathways, which underlies the deficit in learning and memory.
Second, we showed that UCH-L1-regulated deubiquitination of TrkB plays an important role in the regulation of BDNF-mediated endocytosis and postendocytic trafficking of surface TrkB. Monoubiquitination of receptors has been demonstrated to be responsible for receptors movement from the plasma membrane to the lysosome (Haglund et al., 2003). We demonstrated that TrkB is more likely to be multimonoubiquitinated in response to BDNF (Fig. 5), which agrees with the ubiquitination of TrkA, another Trk family member (Arévalo et al., 2006). Many studies have been conducted to study the ubiquitination of TrkA. Nedd4–2 is one of the E3 ligases for the multimonoubiquitination of TrkA and mediates its intracellular recycling and degradation, thus controlling its downstream signaling pathways and functions (Arévalo et al., 2006; Kiris et al., 2014; Yu et al., 2014). The common effects of the ubiquitination of both TrkA and TrkB are that it provides signals for TrkA or TrkB to be sorted to degradation pathways. In contrast to the effects of ubiquitination of TrkA, we demonstrate that the ubiquitination of TrkB promotes BDNF-mediated internalization; however, the Nedd4-2-mediated ubiquitination of TrkA has no effects on endocytosis (Arévalo et al., 2006; Yu et al., 2011). This is likely due to the differential timing of ubiquitination; the Nedd4-2-mediated ubiquitination of TrkA occurs after internalization, like the agonist-induced ubiquitination of the AMPA receptor (Widagdo et al., 2015), whereas the ubiquitination of TrkB occurs on the plasma membrane right after activation. The ubiquitination of TrkA has been associated with its long-distance transport. Nedd4-2 depletion enhances the retrograde transport of TrkA in DRG neurons (Yu et al., 2011). It has been demonstrated UCH-L1 is important for supporting TrkB retrograde transport (Poon et al., 2013), which may be due to UCH-L1-regulated deubiquitination of TrkB. In addition to multimonoubiquitination, TrkA has also been demonstrated to be polyubiquitinated, which controls its internalization and signaling (Geetha et al., 2005). Therefore, polyubiquitination of TrkB may also occur under certain circumstances to control its functions.
Among all of the potential ubiquitination sites of TrkB, K460 is the key site for TrkB ubiquitination and is also the major deubiquitination site regulated by UCH-L1. It is interesting that K460 residues in the KFG motif are conserved in all Trk family members (Peng et al., 1995), which has also been demonstrated to be important for the ubiquitination of TrkA and its functions, such as neurite differentiation and sensitivity to thermal stimuli (Kiris et al., 2014). The mutation of K460 to R alone leads to a dramatic decrease in the ubiquitination level of TrkB (Fig. 5). It is likely that K460 is the prime site for the progressive ubiquitination of other lysines, which may cause sequential interactions with other proteins, thus directing TrkB toward the vesicular bodies. The KFG motif is in the JM domain of TrkB, which has been proposed to be a recycling signal for both TrkA and TrkB (Chen et al., 2005; Zhao et al., 2009). We found that the Tat-UCH-L175–85 led to increased ubiquitination of TrkB and decreased recycling of TrkB (Fig. 8F–I); however, the K460R mutation, which result in decreased ubiquitination of TrkB, had no effects on BDNF-dependent recycling of TrkB (Fig. 8J). Therefore, these data indicate that the ubiquitination of lysine at KFG motif of TrkB is not likely to be the mechanism responsible for regulating the recycling of the internalized TrkB. The ubiquitination of TrkB is responsible for surface TrkB internalization and directs more internalized TrkB to the degradation pathway. The JM1 has also been demonstrated to play an important role in the regulation of TrkB anterograde transport by association with JIP3 (Huang et al., 2011). Taking into account the proximity of the ubiquitination site and the interaction motif, it is likely the ubiquitination of TrkB at K460 could affect the association between TrkB and the adaptor proteins, thus affecting the intracellular transport of TrkB.
The UCH-L1-regutated deubiquitination of TrkB therefore controls the activation of TrkB and its downstream signaling pathways. In the long term (1 h or longer), increased ubiquitination of TrkB caused by Tat-UCH-L175–85 leads to increased TrkB degradation and decreased activity of TrkB and downstream signaling pathways. This phenomenon was also confirmed with K460R TrkB, in which the BDNF-induced ubiquitination is inhibited and results in increased activation of TrkB (Fig. 9). To our surprise, we found that the ubiquitination of TrkB could affect the phosphorylation of TrkB as early as 15 min, when there is no obvious degradation of TrkB receptor (Zhao et al., 2009). It has been proposed that ubiquitination is a mechanism to control the recruitment and interaction of proteins to activate Trk (Kiris et al., 2014). Furthermore, ubiquitination of TrkA is suggested to favor kalirin displacement to facilitate TrkA activation (Kiris et al., 2014). Kalirin has also been reported to be involved in the regulation of TrkB activation (Yan et al., 2016). Whether the ubiquitination of TrkB is involved needs further investigation.
Finally, the UCH-L1-regulated deubiquitination of TrkB is required for the maintenance of hippocampus-dependent memory (Fig. 10). It has been reported that the UCH-L1-deficient gracile axonal dystrophy mouse exhibits impaired memory in passive avoidance learning at young ages (Sakurai et al., 2008), which could be due to the altered CREB activation and synaptic plasticity (Hegde et al., 1997; Gong et al., 2006). CREB has been shown to be an important regulator of BDNF–TrkB induced gene expression (Finkbeiner et al., 1997), so it is plausible that UCH-L1 upregulates the cAMP–CREB pathway via the regulation of deubiquitination of TrkB.
AD is one the most common neurodegenerative diseases, causing learning and memory deficit at early stages, and several lines of evidence indicate that UCH-L1 is involved in AD pathogenesis. Decreased UCH-L1 expression has been observed in the brains of AD mice and AD patients (Choi et al., 2004; Poon et al., 2013) and the overexpression of UCH-L1 reduces the number of Aβ plaques and improves memory deficits in AD mice (Gong et al., 2006; Zhang et al., 2014). UCH-L1-mediated APP and BACE1 degradation has been proposed to have a protective effect in AD cell and mouse models (Zhang et al., 2012; Zhang et al., 2014). BDNF–TrkB signaling pathways have also been demonstrated to improve hippocampus-dependent memory independently of Aβ metabolism (Nagahara et al., 2009; Massa et al., 2010). Our study revealed that UCH-L1-regulated deubiquitination of TrkB, which is necessary for the maintenance of normal hippocampus-dependent memory, may contribute to its protective effects on AD. In addition, the regulation of TrkB by UCH-L1 could be a potential drug target for the treatment of AD.
In conclusion, we have demonstrated here that the UCH-L1-regulated deubiquitination of TrkB is necessary for the activation of TrkB and its downstream signaling pathways and for UCH-L1-promoted hippocampus-dependent memory. Modulation of the interaction between UCH-L1 and TrkB could be a novel target for drugs to treat dementias. In addition to learning and memory, BDNF–TrkB pathways have been shown to be involved in emotion and other advanced behaviors (Chen et al., 2006), in which the UCH-L1-regulated ubiquitination of TrkB may also participate. Future studies are needed to better understand the role of UCH-L1 in vivo.
Footnotes
This work was supported by National 973 Basic Research Program of China (Grant 2012CB911004), the National Natural Science Foundation of China (Grants 31571449 and 31401219), and the China Postdoctoral Science Foundation (Grant 2016M600534).
The authors declare no competing financial interests.
References
- Arévalo JC, Waite J, Rajagopal R, Beyna M, Chen ZY, Lee FS, Chao MV (2006) Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron 50:549–559. 10.1016/j.neuron.2006.03.044 [DOI] [PubMed] [Google Scholar]
- Bilguvar K, et al. (2013) Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci U S A 110:3489–3494. 10.1073/pnas.1222732110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann K, Zigrino P, Witt A, Schell M, Ackermann L, Broxtermann P, Schüll S, Andree M, Coutelle O, Yazdanpanah B, Seeger JM, Klubertz D, Drebber U, Hacker UT, Krönke M, Mauch C, Hoppe T, Kashkar H (2013) Ubiquitin C-terminal hydrolase-L1 potentiates cancer chemosensitivity by stabilizing NOXA. Cell Rep 3:881–891. 10.1016/j.celrep.2013.02.014 [DOI] [PubMed] [Google Scholar]
- Cartier AE, Djakovic SN, Salehi A, Wilson SM, Masliah E, Patrick GN (2009) Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J Neurosci 29:7857–7868. 10.1523/JNEUROSCI.1817-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Ieraci A, Tanowitz M, Lee FS (2005) A novel endocytic recycling signal distinguishes biological responses of Trk neurotrophin receptors. Mol Biol Cell 16:5761–5772. 10.1091/mbc.E05-07-0651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS (2006) Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314:140–143. 10.1126/science.1129663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L (2004) Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson's and Alzheimer's diseases. J Biol Chem 279:13256–13264. 10.1074/jbc.M314124200 [DOI] [PubMed] [Google Scholar]
- Dimitrova YN, Li J, Lee YT, Rios-Esteves J, Friedman DB, Choi HJ, Weis WI, Wang CY, Chazin WJ (2010) Direct ubiquitination of beta-catenin by Siah-1 and regulation by the exchange factor TBL1. J Biol Chem 285:13507–13516. 10.1074/jbc.M109.049411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19:1031–1047. 10.1016/S0896-6273(00)80395-5 [DOI] [PubMed] [Google Scholar]
- Geetha T, Jiang J, Wooten MW (2005) Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol Cell 20:301–312. 10.1016/j.molcel.2005.09.014 [DOI] [PubMed] [Google Scholar]
- Gérecová G, Kopanicová J, Jaká P, Běhalová L, Juhásová B, Bhatia-Kiššová I, Forte M, Polčic P, Mentel M (2013) BH3-only proteins Noxa, Bik, Bmf, and Bid activate Bax and Bak indirectly when studied in yeast model. FEMS Yeast Res 13:747–754. 10.1111/1567-1364.12074 [DOI] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O (2006) Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell 126:775–788. 10.1016/j.cell.2006.06.046 [DOI] [PubMed] [Google Scholar]
- Goto Y, et al. (2015) UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF-1alpha. Nat Commun 6:6153. 10.1038/ncomms7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Xu B, Lu B, Hempstead BL (2009) New insights in the biology of BDNF synthesis and release: implications in CNS function. J Neurosci 29:12764–12767. 10.1523/JNEUROSCI.3566-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K, Dikic I (2012) The role of ubiquitylation in receptor endocytosis and endosomal sorting. J Cell Sci 125:265–275. 10.1242/jcs.091280 [DOI] [PubMed] [Google Scholar]
- Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I (2003) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol 5:461–466. 10.1038/ncb983 [DOI] [PubMed] [Google Scholar]
- Hegde AN, Inokuchi K, Pei W, Casadio A, Ghirardi M, Chain DG, Martin KC, Kandel ER, Schwartz JH (1997) Ubiquitin C-terminal hydrolase is an immediate-early gene essential for long-term facilitation in Aplysia. Cell 89:115–126. 10.1016/S0092-8674(00)80188-9 [DOI] [PubMed] [Google Scholar]
- Huang SH, Zhao L, Sun ZP, Li XZ, Geng Z, Zhang KD, Chao MV, Chen ZY (2009) Essential role of Hrs in endocytic recycling of full-length TrkB receptor but not its isoform TrkB.T1. J Biol Chem 284:15126–15136. 10.1074/jbc.M809763200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SH, Duan S, Sun T, Wang J, Zhao L, Geng Z, Yan J, Sun HJ, Chen ZY (2011) JIP3 mediates TrkB axonal anterograde transport and enhances BDNF signaling by directly bridging TrkB with kinesin-1. J Neurosci 31:10602–10614. 10.1523/JNEUROSCI.0436-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SH, Wang J, Sui WH, Chen B, Zhang XY, Yan J, Geng Z, Chen ZY (2013) BDNF-dependent recycling facilitates TrkB translocation to postsynaptic density during LTP via a Rab11-dependent pathway. J Neurosci 33:9214–9230. 10.1523/JNEUROSCI.3256-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav T, Geetha T, Jiang J, Wooten MW (2008) Identification of a consensus site for TRAF6/p62 polyubiquitination. Biochem Biophys Res Commun 371:521–524. 10.1016/j.bbrc.2008.04.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Huang S, Li X, Li X, Zhang Y, Chen ZY (2015) Tyrosine kinase receptor B protects against coronary artery disease and promotes adult vasculature integrity by regulating Ets1-mediated VE-cadherin expression. Arterioscler Thromb Vasc Biol 35:580–588. 10.1161/ATVBAHA.114.304405 [DOI] [PubMed] [Google Scholar]
- Kabuta T, Mitsui T, Takahashi M, Fujiwara Y, Kabuta C, Konya C, Tsuchiya Y, Hatanaka Y, Uchida K, Hohjoh H, Wada K (2013) Ubiquitin C-terminal hydrolase L1 (UCH-L1) acts as a novel potentiator of cyclin-dependent kinases to enhance cell proliferation independently of its hydrolase activity. J Biol Chem 288:12615–12626. 10.1074/jbc.M112.435701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiris E, Wang T, Yanpallewar S, Dorsey SG, Becker J, Bavari S, Palko ME, Coppola V, Tessarollo L (2014) TrkA in vivo function is negatively regulated by ubiquitination. J Neurosci 34:4090–4098. 10.1523/JNEUROSCI.4294-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen CN, Price JS, Wilkinson KD (1996) Substrate binding and catalysis by ubiquitin C-terminal hydrolases: identification of two active site residues. Biochemistry 35:6735–6744. 10.1021/bi960099f [DOI] [PubMed] [Google Scholar]
- Larsen CN, Krantz BA, Wilkinson KD (1998) Substrate specificity of deubiquitinating enzymes: ubiquitin C-terminal hydrolases. Biochemistry 37:3358–3368. 10.1021/bi972274d [DOI] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH (1998) The ubiquitin pathway in Parkinson's disease. Nature 395:451–452. 10.1038/26652 [DOI] [PubMed] [Google Scholar]
- Liu XH, Geng Z, Yan J, Li T, Chen Q, Zhang QY, Chen ZY (2015) Blocking GSK3beta-mediated dynamin1 phosphorylation enhances BDNF-dependent TrkB endocytosis and the protective effects of BDNF in neuronal and mouse models of Alzheimer's disease. Neurobiol Dis 74:377–391. 10.1016/j.nbd.2014.11.020 [DOI] [PubMed] [Google Scholar]
- Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT Jr (2002) The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell 111:209–218. 10.1016/S0092-8674(02)01012-7 [DOI] [PubMed] [Google Scholar]
- Ma L, Wang DD, Zhang TY, Yu H, Wang Y, Huang SH, Lee FS, Chen ZY (2011) Region-specific involvement of BDNF secretion and synthesis in conditioned taste aversion memory formation. J Neurosci 31:2079–2090. 10.1523/JNEUROSCI.5348-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkerh JP, Ceni C, Auld DS, Vaillancourt F, Dorval G, Barker PA (2005) p75 neurotrophin receptor reduces ligand-induced Trk receptor ubiquitination and delays Trk receptor internalization and degradation. EMBO Rep 6:936–941. 10.1038/sj.embor.7400503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, Longo FM (2010) Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest 120:1774–1785. 10.1172/JCI41356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R (1999) Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24:401–414. 10.1016/S0896-6273(00)80853-3 [DOI] [PubMed] [Google Scholar]
- Moises T, Wüller S, Saxena S, Senderek J, Weis J, Kruttgen A (2009) Proteasomal inhibition alters the trafficking of the neurotrophin receptor TrkA. Biochem Biophys Res Commun 387:360–364. 10.1016/j.bbrc.2009.07.032 [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med 15:331–337. 10.1038/nm.1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaka H, Wang YL, Takada K, Takizawa S, Setsuie R, Li H, Sato Y, Nishikawa K, Sun YJ, Sakurai M, Harada T, Hara Y, Kimura I, Chiba S, Namikawa K, Kiyama H, Noda M, Aoki S, Wada K (2003) Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet 12:1945–1958. 10.1093/hmg/ddg211 [DOI] [PubMed] [Google Scholar]
- Pandya C, Kutiyanawalla A, Turecki G, Pillai A (2014) Glucocorticoid regulates TrkB protein levels via c-Cbl dependent ubiquitination: a decrease in c-Cbl mRNA in the prefrontal cortex of suicide subjects. Psychoneuroendocrinology 45:108–118. 10.1016/j.psyneuen.2014.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Greene LA, Kaplan DR, Stephens RM (1995) Deletion of a conserved juxtamembrane sequence in Trk abolishes NGF-promoted neuritogenesis. Neuron 15:395–406. 10.1016/0896-6273(95)90043-8 [DOI] [PubMed] [Google Scholar]
- Poon WW, Carlos AJ, Aguilar BL, Berchtold NC, Kawano CK, Zograbyan V, Yaopruke T, Shelanski M, Cotman CW (2013) beta-Amyloid (Abeta) oligomers impair brain-derived neurotrophic factor retrograde trafficking by down-regulating ubiquitin C-terminal hydrolase, UCH-L1. J Biol Chem 288:16937–16948. 10.1074/jbc.M113.463711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai M, Sekiguchi M, Zushida K, Yamada K, Nagamine S, Kabuta T, Wada K (2008) Reduction in memory in passive avoidance learning, exploratory behaviour and synaptic plasticity in mice with a spontaneous deletion in the ubiquitin C-terminal hydrolase L1 gene. Eur J Neurosci 27:691–701. 10.1111/j.1460-9568.2008.06047.x [DOI] [PubMed] [Google Scholar]
- Santos AR, Mele M, Vaz SH, Kellermayer B, Grimaldi M, Colino-Oliveira M, Rombo DM, Comprido D, Sebastião AM, Duarte CB (2015) Differential role of the proteasome in the early and late phases of BDNF-induced facilitation of LTP. J Neurosci 35:3319–3329. 10.1523/JNEUROSCI.4521-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerfeld MT, Schweigreiter R, Barde YA, Hoppe E (2000) Down-regulation of the neurotrophin receptor TrkB following ligand binding. Evidence for an involvement of the proteasome and differential regulation of TrkA and TrkB. J Biol Chem 275:8982–8990. 10.1074/jbc.275.12.8982 [DOI] [PubMed] [Google Scholar]
- Tan YY, Zhou HY, Wang ZQ, Chen SD (2008) Endoplasmic reticulum stress contributes to the cell death induced by UCH-L1 inhibitor. Mol Cell Biochem 318:109–115. 10.1007/s11010-008-9862-x [DOI] [PubMed] [Google Scholar]
- Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, Anggono V (2015) Activity-dependent ubiquitination of GluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep 10:783–795. 10.1016/j.celrep.2015.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J (1989) The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 246:670–673. 10.1126/science.2530630 [DOI] [PubMed] [Google Scholar]
- Yan Y, Eipper BA, Mains RE (2016) Kalirin is required for BDNF–TrkB stimulated neurite outgrowth and branching. Neuropharmacology 107:227–238. 10.1016/j.neuropharm.2016.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Calvo L, Anta B, López-Benito S, Southon E, Chao MV, Tessarollo L, Arévalo JC (2011) Regulation of trafficking of activated TrkA is critical for NGF-mediated functions. Traffic 12:521–534. 10.1111/j.1600-0854.2010.01156.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Calvo L, Anta B, López-Benito S, López-Bellido R, Vicente-García C, Tessarollo L, Rodriguez RE, Arévalo JC (2014) In vivo regulation of NGF-mediated functions by Nedd4–2 ubiquitination of TrkA. J Neurosci 34:6098–6106. 10.1523/JNEUROSCI.4271-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Deng Y, Luo Y, Zhang S, Zou H, Cai F, Wada K, Song W (2012) control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal hydrolase L1. J Neurochem 120:1129–1138. 10.1111/j.1471-4159.2011.07644.x [DOI] [PubMed] [Google Scholar]
- Zhang M, Cai F, Zhang S, Zhang S, Song W (2014) Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer's progression in vivo. Sci Rep 4:7298. 10.1038/srep07298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Sheng AL, Huang SH, Yin YX, Chen B, Li XZ, Zhang Y, Chen ZY (2009) Mechanism underlying activity-dependent insertion of TrkB into the neuronal surface. J Cell Sci 122:3123–3136. 10.1242/jcs.047712 [DOI] [PubMed] [Google Scholar]
- Zheng J, Shen WH, Lu TJ, Zhou Y, Chen Q, Wang Z, Xiang T, Zhu YC, Zhang C, Duan S, Xiong ZQ (2008) Clathrin-dependent endocytosis is required for TrkB-dependent Akt-mediated neuronal protection and dendritic growth. J Biol Chem 283:13280–13288. 10.1074/jbc.M709930200 [DOI] [PubMed] [Google Scholar]