

Abstract
α-Synuclein is a presynaptic protein the function of which has yet to be identified, but its neuronal content increases in patients of synucleinopathies including Parkinson's disease. Chronic overexpression of α-synuclein reportedly expresses various phenotypes of synaptic dysfunction, but the primary target of its toxicity has not been determined. To investigate this, we acutely loaded human recombinant α-synuclein or its pathological mutants in their monomeric forms into the calyces of Held presynaptic terminals in slices from auditorily mature and immature rats of either sex. Membrane capacitance measurements revealed significant and specific inhibitory effects of WT monomeric α-synuclein on vesicle endocytosis throughout development. However, the α-synuclein A53T mutant affected vesicle endocytosis only at immature calyces, whereas the A30P mutant had no effect throughout. The endocytic impairment by WT α-synuclein was rescued by intraterminal coloading of the microtubule (MT) polymerization blocker nocodazole. Furthermore, it was reversibly rescued by presynaptically loaded photostatin-1, a photoswitcheable inhibitor of MT polymerization, in a light-wavelength-dependent manner. In contrast, endocytic inhibition by the A53T mutant at immature calyces was not rescued by nocodazole. Functionally, presynaptically loaded WT α-synuclein had no effect on basal synaptic transmission evoked at a low frequency, but significantly attenuated exocytosis and impaired the fidelity of neurotransmission during prolonged high-frequency stimulation. We conclude that monomeric WT α-synuclein primarily inhibits vesicle endocytosis via MT overassembly, thereby impairing high-frequency neurotransmission.
SIGNIFICANCE STATEMENT Abnormal α-synuclein abundance is associated with synucleinopathies including Parkinson's disease, but neither the primary target of α-synuclein toxicity nor its mechanism is identified. Here, we loaded monomeric α-synuclein directly into mammalian glutamatergic nerve terminals and found that it primarily inhibits vesicle endocytosis and subsequently impairs exocytosis and neurotransmission fidelity during prolonged high-frequency stimulation. Such α-synuclein toxicity could be rescued by blocking microtubule polymerization, suggesting that microtubule overassembly underlies the toxicity of acutely elevated α-synuclein in the nerve terminal.
Keywords: alpha-synuclein, calyx of Held, microtubules, synaptic transmission, vesicle endocytosis
Introduction
Synucleins were originally cloned from Torpedo electric lobe, where they localize in cell nuclei and axon terminals (Maroteaux et al., 1988). The α isoform of synuclein, NACP, is enriched in mammalian presynaptic terminals (Jakes et al., 1994; Murphy et al., 2000). α-Synuclein interacts with a variety of proteins (Chung et al., 2017), including calmodulin (Zhou et al., 2004), actin (Jensen et al., 1999; Sousa et al., 2009), tubulin (Alim et al., 2002; Zhou et al., 2004), tau protein (Jensen et al., 1999; Zhou et al., 2004), septin 4 (Ihara et al., 2007), ubiquitin (Zhou et al., 2004), phospholipids (Stöckl et al., 2008), and ATP synthase (Ludtmann et al., 2016), but its functional role has yet to be established.
In synucleinopathies including Parkinson's disease (PD), aggregates of α-synuclein are found in neurons as Lewy bodies (Spillantini et al., 1998) initially in axon terminals (Marui et al., 2002). Missense mutations in the N-terminal α-helix region, such as A30P and A53T (Polymeropoulos et al., 1997; Kruger et al., 1998), and multiplication of the α-synuclein gene SNCA (Singleton et al., 2003; Ibáñez et al., 2004) are thought to underlie PD. In fact, α-synuclein is doubly expressed in SNCA triplication (Singleton et al., 2003; Farrer et al., 2004) and PD progression is reportedly proportional to the SNCA gene dosage (Chartier-Harlin et al., 2004). In the early stage of sporadic PD, α-synuclein is accumulated in brainstem presynaptic terminals and is subsequently propagated to upper CNS synapses (Braak et al., 2003), possibly by a prion-like protein-misfolding mechanism (Bendor et al., 2013).
Experimental overexpression of α-synuclein disrupts microtubule (MT)-dependent vesicle trafficking in cultured cells (Lee et al., 2006) and reduces dopamine release from secretory cells (Larsen et al., 2006; Choi et al., 2013) or from dopaminergic neurons in vivo (Lundblad et al., 2012). α-Synuclein overexpression also reduces glutamate release and the number of recycling vesicles in cultured hippocampal neurons (Nemani et al., 2010). In contrast, α-synuclein overexpression reportedly promotes vesicle endocytosis in neuronal cell lines (Ben Gedalya et al., 2009) and causes synaptic potentiation in cultured hippocampal neurons (Liu et al., 2004). Such diverse phenotypes of α-synuclein overexpression may include secondary effects, such as those mediated by α-synuclein binding partners. Therefore, we searched for a primary target of elevated α-synuclein toxicity by acutely loading it directly into glutamatergic presynaptic terminals, the calyces of Held, in slices from rat brainstem. Our results indicate that WT monomeric α-synuclein inhibits vesicle endocytosis, thereby impairing the fidelity of high-frequency neurotransmission. Our results also suggest a mechanism of excess MT assembly underlying the toxic effect of α-synuclein.
Materials and Methods
Experiments.
All experiments were performed in accordance with guidelines of the Physiological Society of Japan and animal experiment regulations at the Okinawa Institute of Science and Technology Graduate University.
Preparation.
After decapitation under halothane anesthesia, brainstems were isolated from Wistar rats of either sex at two different ages, one before at postnatal days 7 (P7)–P9 and the other after (P13–P14) the hearing onset. Transverse slices of the brainstem containing the medial nucleus of the trapezoid body (MNTB) were cut at a thickness of 175–200 μm using a microtome (VT1200S; Leica). Before recordings, slices were incubated for 1 h at 36–37°C in artificial CSF (aCSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, 3 myo-inositol, 2 sodium pyruvate, 0.5 sodium ascorbate, 1.25 NaH2PO4, and 26 NaHCO3 at 310–315 mOsm, pH 7.4, saturated with 95% O2 and 5% CO2. WT human recombinant α-synuclein was subcloned into pRK172 and expressed in E. coli BL21(DE3) (Masuda et al., 2006); monomeric WT α-synuclein was purified as described by Oikawa et al. (2016). Monomeric WT α-synuclein (tagged with histidine) was also obtained from Sigma-Aldrich. Monomeric α-synuclein mutants (A30P and A53T) were also purchased from Sigma-Aldrich. Lyophilized powders were dissolved at 36 μm in H2O and desalted and concentrated using centrifugal filter units (Amicon Ultra-2 ml 3K; Millipore). After adjusting the concentration to 360 μm, they were diluted with pipette internal solution to different concentrations (0.7–22 μm). Nocodazole (Calbiochem) and photostatin-1 (see below for details) were dissolved in internal solution containing 0.1% DMSO and infused into presynaptic terminals at a final concentration of 10 μm.
Electrophysiology.
Calyces of Held presynaptic terminals and postsynaptic MNTB principal cells were identified visually with a 40× water-immersion objective attached to an upright microscope (BX51WI; Olympus). Data were acquired at a sampling rate of 50 kHz using an EPC-10 USB double patch-clamp amplifier controlled by PatchMaster software (HEKA Elektroniks, RRID:SCR_000034) after online filtering at 5 kHz. Experiments were performed at room temperature (26–27°C). Resistances of patch electrodes were 4–8 and 1–3 MΩ for presynaptic and postsynaptic recording, respectively. Proteins and drugs were infused from whole-cell pipettes into calyceal terminals by diffusion. For whole-cell voltage-clamp recordings, presynaptic and postsynaptic series resistances were 8–14 and 4–7 MΩ and compensated by up to 60% for a final value of 7 and 3 MΩ, respectively. Care was taken to keep the presynaptic access resistance before compensation <14 MΩ. With these access resistances (8–14 MΩ), α-synuclein (20 kDa) diffuses into a calyx terminal having a volume of 960 μm3 (Vasileva et al., 2013) with a time constant of 11–20 s after whole-cell rupture (Pusch and Neher, 1988). Data were obtained 4–30 min after break-in.
For recording evoked EPSCs, simultaneous presynaptic and postsynaptic whole-cell recordings were made from a calyceal nerve terminal and postsynaptic cell. Throughout the experiments, presynaptic recordings were made in current-clamp mode, whereas postsynaptic recordings were made in voltage-clamp mode at a holding potential of −70 mV. Pipette solution for recording of presynaptic action potentials (APs) contained the following (in mm): 110 K-gluconate, 10 l-glutamate, 30 KCl, 10 HEPES, 0.5 EGTA, 12 Na2-phosphocreatine, 3 MgATP, 1 MgCl2, and 0.3 Na2GTP at 315 mOsm, pH 7.3, adjusted with KOH. Solution for postsynaptic recording contained the following (in mm): 110 CsF, 30 CsCl, 10 HEPES, 5 EGTA, 1 MgCl2, and 5 QX314-Cl at 300 mOsm, pH 7.3, adjusted with CsOH. EPSCs were evoked by depolarizing current injection (0.5–1 nA, 1 ms) into the presynaptic terminal via a recording glass electrode in the presence of bicuculline methiodide (10 μm) and strychnine hydrochloride (0.5 μm).
For recording postsynaptic APs, simultaneous presynaptic and postsynaptic whole-cell recordings were made from calyceal terminals and postsynaptic MNTB principal neurons, both in current-clamp mode, in the presence of bicuculline methiodide (10 μm) and strychnine hydrochloride (0.5 μm). Pipette solution for postsynaptic AP recording contained the following (in mm): 120 K-gluconate, 30 KCl, 5 EGTA, 12 Na2-phosphocreatine, 3 MgATP, 1 l-arginine, 1 MgCl2, and 0.3 Na2GTP at 315 mOsm, pH 7.3, adjusted with KOH. Presynaptic APs were elicited by a square pulse current injection into calyces in current-clamp mode via recording glass electrodes filled with K-gluconate-based internal solution (as above).
For membrane capacitance measurements, calyceal terminals were voltage clamped at a holding potential of −80 mV and a sinusoidal voltage command with a peak-to-peak voltage of 60 mV was applied at 1 kHz (Sun and Wu, 2001; Yamashita et al., 2005). To isolate presynaptic voltage-gated Ca2+ currents (ICa), the aCSF contained 10 mm tetraethylammonium chloride, 0.5 mm 4-aminopyridine, 1 μm tetrodotoxin, 10 μm bicuculline methiodide, and 0.5 μm strychnine hydrochloride. Intracellular solution for presynaptic terminals contained the following (in mm): 125 Cs-methanesulfonate, 30 CsCl, 10 HEPES, 0.5 EGTA, 12 Na2-phosphocreatine, 3 MgATP, 1 MgCl2, and 0.3 Na2GTP at 315–320 mOsm, pH 7.3, adjusted with CsOH. Tips of recording pipettes were coated with dental wax to reduce stray capacitance (4–6 pF). Single-pulse step depolarization (to +10 mV, 20 ms) was used to induce presynaptic ICa unless otherwise noted. Membrane capacitance (Cm) changes within 450 ms of square-pulse stimulation were excluded from analysis to avoid contamination with conductance-dependent capacitance artifacts. Rapid endocytosis was elicited by a 20 ms depolarizing pulse train (20 times, 1 Hz) and the endocytic rate was estimated from the slope fit with a linear regression line to Cm decay 0.45–1 s after each pulse. To avoid the influence of capacitance drift on baseline, we discarded data when the baseline drift measured 0–10 s before stimulation was >5 fF/s. When the capacitance baseline drift was 1–5 fF/s, for the baseline correction, we subtracted a linear regression line of the baseline from the data.
Native gel electrophoresis.
α-Synuclein proteins (3.6 μm) were resuspended in pipette internal solution before loading onto a Novex native gel (3–12% gradient; Invitrogen). Protein size markers (Native Mark; Invitrogen) were used as a standard and electrophoresis was performed according to the manufacturer's instructions. After the protein transfer, the nitrocellulose membrane was labeled using anti-α-synuclein primary antibody (SC-7011-R, RRID:AB_2192953; Santa Cruz Biotechnology) and secondary goat-anti-rabbit IgG-HRP (RRID:AB_11125142; Bio-Rad Laboratory). Bands were visualized using the Molecular Imager Chemi-Doc system (Bio-Rad).
Photostatin.
Photo-switchable microtubule inhibitor photostatin-1 (PST-1, 46 mm stock solution in DMSO; Borowiak et al., 2015) was diluted to 10 mm stock DMSO solution. Just before the experiments, stock solution was diluted to 10 μm with pipette internal solution (0.1% in final DMSO concentration) and sonicated to avoid precipitation. To minimize adhesion of PST-1 to plastic surfaces, glass syringes and vials were used and a metal spinal needle (TOP) was used for loading internal solution into patch pipettes. PST-1 loaded in presynaptic terminals was activated by illumination at 390 nm and inactivated at 510 nm supplied to slices through a 40× water-immersion objective lens with a Xenon lamp (70W, 100 ms pulses every 2 s) under the control of a shutter (Lambda 10-B; Sutter Instrument). When the shutter was opened, the whole area under the objective lens was illuminated. Cells illuminated at 390 nm showed inhibited exocytic capacitance change. This effect was prevented by replacing 20 mm methanesulfonate with equimolar glutathione in the presynaptic pipette solution (Hori and Takahashi, 2012).
Data analysis.
Data were analyzed using IGOR Pro 6 (RRID:SCR_000325; WaveMatrics) with Patcher's Power Tools (RRID:SCR_001950; Max-Planck-Institute), Microsoft Excel 2011/2016 for Mac, and StatPlus:mac (RRID:SCR_014635; AnalystSoft). Sample Cm records are shown as average values for each 50 data points (for 50 ms) plotted every 50 ms (for shorter time scales) or every 500 ms (for longer time scales). All values are given as mean ± SEM. For statistical tests, p < 0.05 (indicated by single asterisks) was taken as significant in Student's t test, paired t test, one-way ANOVA with the Tukey–Kramer post hoc test, or two-way ANOVA with the Scheffe post hoc test.
Results
WT α-synuclein inhibits vesicle endocytosis, but the A30P and A53T mutants do not
It has been reported recently that WT α-synuclein and its A53T mutant inhibit endocytosis of synaptic vesicles at the calyx of Held of immature rodents before hearing onset (P7–P10; Xu et al., 2016). Because endocytic properties at the calyx of Held undergo significant changes after hearing (Renden and von Gersdorff, 2007; Yamashita et al., 2010; Eguchi et al., 2012), we revisited this issue using posthearing rats (P13–P14). In contrast to the report by Xu et al. (2016) showing oligomeric bands for the commercially available α-synuclein or its mutants, our WT α-synuclein, purified or purchased (His tag; Sigma-Aldrich), as well as α-synuclein mutants A30P and A53T (Sigma-Aldrich), showed single bands in native PAGE gel-electrophoresis (Fig. 1A). Because α-synuclein purified from brain tissue is predominantly monomeric (Burré et al., 2013) and it takes several days to convert it to oligomer (Krishnan et al., 2003; Kim et al., 2008; Burré et al., 2013), it should remain monomeric in the terminal within the time of our experiments. WT α-synuclein monomer (3.6 μm) with or without His tag loaded into presynaptic terminals via a recording pipette significantly inhibited endocytic capacitance change (Fig. 1B), prolonging its half decay time (T0.5) by ∼2-fold without affecting presynaptic Ca2+ current (QCa) or exocytic capacitance change (ΔCm) (Fig. 1B2). As reported in prehearing immature calyces (Xu et al., 2016), the A30P mutant (3.6 μm) had no effect, but unlike that report, A53T also had no effect at more mature calyces (Fig. 1B1,B2). The results were also essentially the same at physiological temperature (35°C) except that endocytic kinetics were faster (T0.5, 6.5 ± 0.6 s in control, n = 5; 12.1 ± 2.0 s with α-synuclein, n = 6). The endocytic inhibitory effect of WT synuclein was concentration dependent, approaching a maximum at ∼3.6 μm (Fig. 1C; see also Xu et al., 2016). WT α-synuclein also inhibited fast endocytosis of subsecond time constants that are known to accelerate during a train of 1 Hz stimulation (Wu et al., 2005; Yamashita et al., 2010) by strongly attenuating the maximal rate of endocytosis (Fig. 1D). Neither the A30P nor A53T mutant attenuated this fast endocytosis (Fig. 1D). Therefore, WT α-synuclein, but neither of these pathological mutants, inhibited vesicle endocytosis at this auditory relay synapse after hearing onset. Although neither WT nor mutant α-synuclein affected exocytosis evoked by a single pulse depolarization (Fig. 1B), prolonged repetitive stimulation revealed, in cumulative plots, a significant inhibition of exocytosis by WT α-synuclein, and enhancement of exocytosis by A30P mutant (p < 0.05; Fig. 1D).
Figure 1.
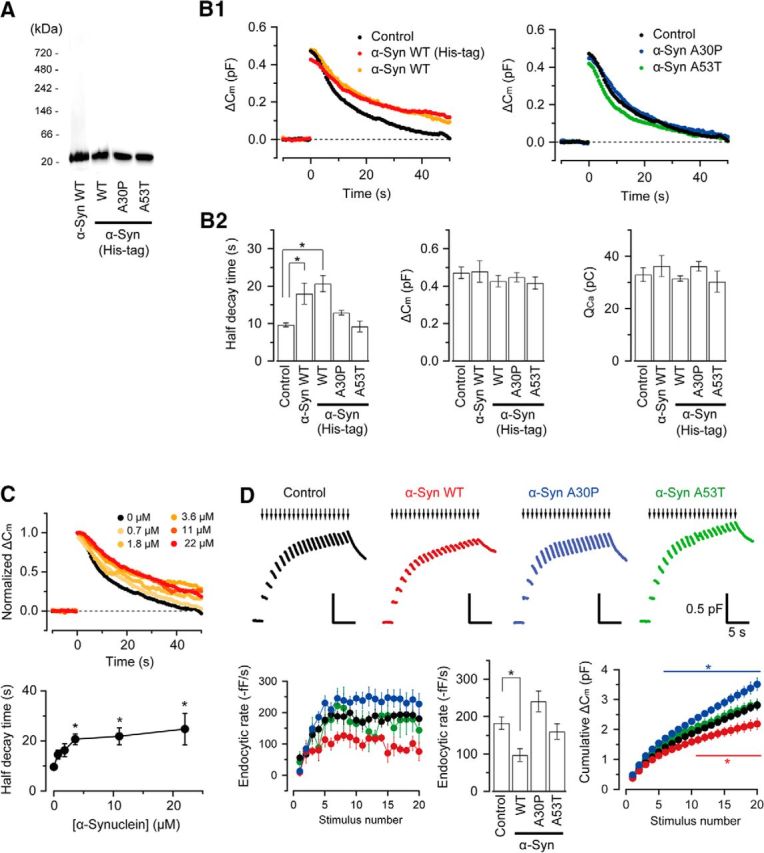
WT monomeric α-synuclein inhibits slow and fast vesicle endocytosis at calyces of Held in slices from posthearing rats, but its mutants do not. A, Native PAGE (from left to right); monomeric human WT α-synuclein (purified by M.H.), that obtained from Sigma-Aldrich (His tag), and monomeric A30P and A53T α-synuclein mutants from Sigma-Aldrich, all dissolved in pipette solution. B1, Exo–endocytic Cm changes of calyceal presynaptic terminals evoked by ICa (induced by a 20 ms pulse stepping from −80 mV to +10 mV). Left, Intraterminal loading of monomeric WT α-synucleins (3.6 μm) from different sources similarly slowed endocytic Cm changes without affecting exocytic Cm changes or ICa (red and yellow traces superimposed with control black traces, each averaged from records in seven to eight terminals). Right, A30P or A53T α-synuclein mutants (3.6 μm), loaded into presynaptic terminals, had no significant effect (superimposed blue or green traces, averaged from records in four to six terminals). B2, Bar graphs showing the effects of WT α-synucleins, His tag WT α-synuclein, and A30P and A53T mutants on the 50% decay time of endocytic Cm (left), the magnitudes of exocytic Cm jump (middle), and presynaptic Ca2+ current charge (QCa, right). C, Intraterminal loading of α-synuclein (His tag) at different concentrations (0.7–22 μm) slowed vesicle endocytosis in a concentration-dependent manner. Top, Cm traces; data from six to seven calyces were averaged, normalized at the peak, and superimposed. Bottom, Half endocytic decay time at different α-synuclein concentrations (n = 6–7 for each data point). Asterisks indicate a significant difference from the control (with no α-synuclein). D, Top, Cm changes induced by 20 ms depolarizing pulses (arrows indicate stimulations) repeated at 1 Hz for 20 s at calyceal terminals loaded with α-synuclein (His tag, red), A30P (blue), A53T mutant (green), or without loading (black, control). Traces represent average Cm changes from four to seven calyces. Bottom left, Endocytic rate during the train (fF/s, ordinate) estimated from a linear regression line fit to the Cm decay 0.45–1 s after each pulse versus stimulus number (abscissa) in calyceal terminals with α-synuclein (red, n = 5), A30P (blue, n = 6), A53T (green, n = 4), or without them (control: black, n = 7). The endocytic Cm decay after each pulse (initial rate: 56.1 ± 12 fF/s) became gradually faster and reached a maximal rate of 183 ± 14 fF/s at around the 10th pulse (mean value of 10th–20th events, n = 7). α-Synuclein decreased the maximal rate to 96.8 ± 17 fF/s (n = 5, p < 0.05). Bottom center, Bar graphs indicating the maximal endocytic rate in the presence of α-synuclein, A30P, A53T mutants, or in their absence. Bottom right, Cumulative ΔCm during the 20 s train (ordinate) versus stimulus number (abscissa) in calyceal terminals loaded with WT α-synuclein (red, n = 5), A30P (blue, n = 6), A53T (green, n = 4), or without loading (control, black, n = 7). WT α-synuclein significantly reduced the cumulative ΔCm (p < 0.05 from 11th stimulus), whereas A30P mutant significantly increased the cumulative ΔCm (p < 0.05 from sixth stimulus) compared with controls. A53T mutant had no effect.
Mechanism underlying the inhibitory effect of α-synuclein on vesicle endocytosis
Although the inhibitory effect of α-synuclein on vesicle endocytosis has been suggested (Boassa et al., 2013; Busch et al., 2014) and demonstrated (Xu et al., 2016; Fig. 1), little is known about the underlying mechanism. Because α-synuclein binds to PIP2 (Stöckl et al., 2008), which operates for accelerating endocytosis (Eguchi et al., 2012; Taoufiq et al., 2013), we first tested whether intraterminal PIP2 loading might rescue α-synuclein-induced endocytic inhibition. PIP2 (50 μm) loaded with WT α-synuclein showed a tendency to rescue the inhibitory effect of α-synuclein on endocytosis, but the difference was statistically insignificant (Fig. 2A, p = 0.20). Because α-synuclein can bind to actin (Jensen et al., 1999; Sousa et al., 2009), we also tested whether the actin depolymerizer latrunculin A might rescue α-synuclein-induced endocytic inhibition. As shown in Figure 2B, intraterminal loading of latrunculin A (25 μm) had no rescuing effect on α-synuclein-induced endocytic inhibition. Among various binding partners, α-synuclein binds to tubulins (Alim et al., 2002; Zhou et al., 2004) and reportedly polymerizes them into MTs in vitro (Alim et al., 2004, but see Oikawa et al., 2016). It can also bind to tau protein (Jensen et al., 1999; Zhou et al., 2004) and promotes its phosphorylation (Jensen et al., 1999). We therefore investigated whether pharmacological block of tubulin polymerization might rescue the inhibitory effect of α-synuclein on vesicle endocytosis. We first tested nocodazole, which specifically blocks MT assembly. Nocodazole loaded alone into presynaptic terminals (10 μm) had no effect on presynaptic membrane capacitance change, but when coloaded with α-synuclein, completely rescued the inhibitory effect of α-synuclein on vesicle endocytosis (Fig. 2C). Likewise, nocodazole (10 μm) rescued the inhibitory effect of α-synuclein on the accelerating fast endocytosis during the stimulus train and slow endocytosis thereafter (Fig. 2D).
Figure 2.
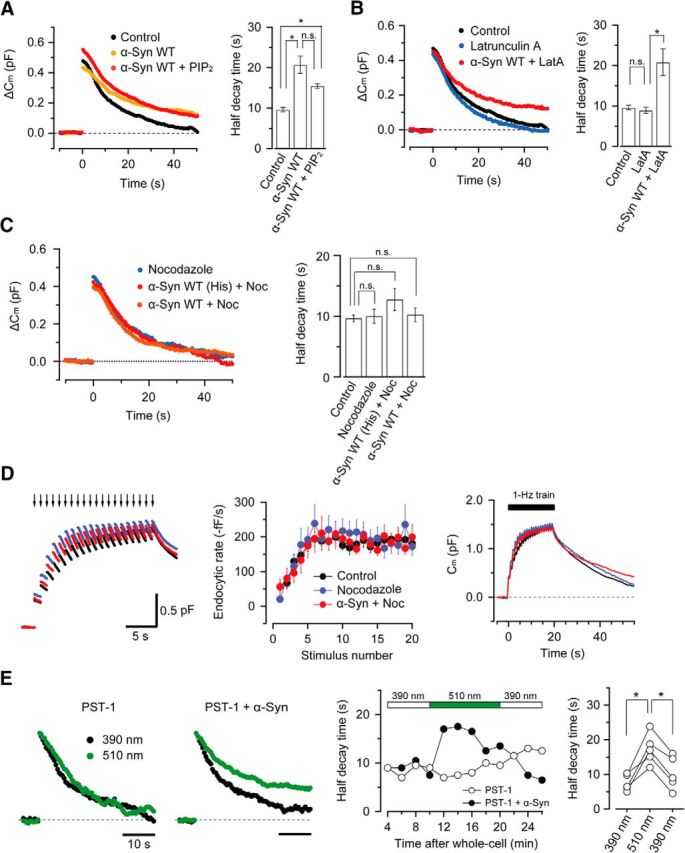
The tubulin depolymerizers nocodazole and photostatin-1 can rescue the inhibitory effects of α-synuclein on vesicle endocytosis. A, B, Coloading of PIP2 (50 μm, n = 5, red in A) or latrunculin A (25 μm, n = 6, red in B) together with WT α-synuclein had no significant rescuing effect (n.s. in A, p = 0.20) for the inhibitory effect of α-synuclein on vesicle endocytosis. Yellow trace in A indicates the endocytic inhibitory effect of WT α-synuclein for comparison. Bar graphs indicate endocytic 50% decay time, averaged from data in five to seven calyces. Latrunculin A loaded alone in calyceal terminals had no effect on vesicle endocytosis (n = 5, blue trace in B). C, Intraterminal loading of nocodazole (10 μm) together with WT α-synuclein with (red) or without (orange) His tag (3.6 μm) rescued the inhibitory effect of α-synuclein on vesicle endocytosis induced by a single 20 ms pulse. Nocodazole alone (blue trace) had no effect. Bar graphs indicate endocytic 50% decay time averaged from data in five to seven calyces. D, Nocodazole coloading (10 μm) rescued the inhibitory effects of α-synuclein (His tag, 3.6 μm) on accelerating fast endocytosis induced by a train of 20 ms pulse at 1 Hz. Left, Traces of control (black), nocodazole alone (blue), and WT α-synuclein coloaded with nocodazole (red, superimposed). Center and right, Endocytic rates were similar in control (black), nocodazole alone (blue), and α-synuclein + nocodazole (red), as shown in time plots and bar graphs (n = 5–7). E, PST-1 reversibly rescued endocytic inhibitory effects of α-synuclein. Intraterminal loading of PST-1 (10 μm) alone had no effect under 390 nm (black trace) or 510 nm (green trace) illumination (left sample traces), but, when loaded with α-synuclein (His tag, 3.6 μm, right traces), rescued its inhibitory effect on vesicle endocytosis under 390 nm illumination (black trace), but not under 510 nm light (right sample traces). In sample traces, ΔCm amplitude was normalized at the peak to compare endocytic time courses. Time plots (middle panel) indicate endocytic 50% decay time under 390 or 510 nm illumination in the presence of PST-1 and α-synuclein (filled circles) and PST-1 alone (open circles). In the presence of PST-1 and α-synuclein, the endocytic rate was slowed when PST-1 was inactivated by 510 nm light, but became faster when returned to 390 nm illumination. Right panel summarizes the mean half endocytic decay time from five terminals at three epochs in the time plot showing significant and reversible rescue by PST-1 from α-synuclein-induced endocytic inhibition under 390 nm illumination. Asterisks indicate a significant difference (*p < 0.05).
To confirm the specificity of the nocodazole inhibition of tubulin polymerization, we used the recently introduced photo-switchable tubulin polymerization inhibitor PST-1 (Borowiak et al., 2015), a colchicine derivative activated under short-wavelength light, but inactivated under long-wavelength light. When PST-1 (10 μm) was loaded alone into the calyx of Held terminal, it had no effect on exo–endocytic capacitance changes under both short-wavelength (390 nm) and long-wavelength (510 nm) light (Fig. 2E). When it was coloaded with α-synuclein under 390 nm light, α-synuclein no longer inhibited endocytosis, whereas switching the light wavelength to 510 nm revealed its inhibitory effect on endocytosis. When the light was switched back to 390 nm, the endocytic inhibitory effect of α-synuclein was again cancelled. Therefore, PST-1 reversibly rescued the inhibitory effect of α-synuclein on vesicle endocytosis. These pharmacological results using nocodazole and PST-1 strongly suggest that MT assembly is involved in the inhibitory effect of α-synuclein on vesicle endocytosis.
Inhibitory effect of A53T mutants on vesicle endocytosis in prehearing animals
Because the α-synuclein A53T mutant inhibits vesicle endocytosis at calyces of Held in prehearing rats (Xu et al., 2016), but not in posthearing rats (Fig. 1B), we reexamined the effect of A53T mutant on endocytosis at calyces from prehearing (P7–P9) rats (Fig. 3). As reported previously (Xu et al., 2016), both WT and A53T mutant α-synucleins inhibited endocytosis, whereas A30P mutant had no effect (Fig. 3A). As in posthearing rats (Fig. 2C), nocodazole (10 μm) rescued the inhibitory effect of WT α-synuclein (Fig. 3B). Like calyces in posthearing rats (Fig. 2C), nocodazole alone had no effect on endocytosis in prehearing rats (Fig. 3B,C). However, unlike in posthearing rats, nocodazole failed to rescue the endocytic inhibitory effect of the A53T mutant in prehearing rat calyces (Fig. 3C), suggesting that a distinct MT-independent mechanism may underlie the endocytic inhibitory effect of A53T mutant in prehearing rodents.
Figure 3.
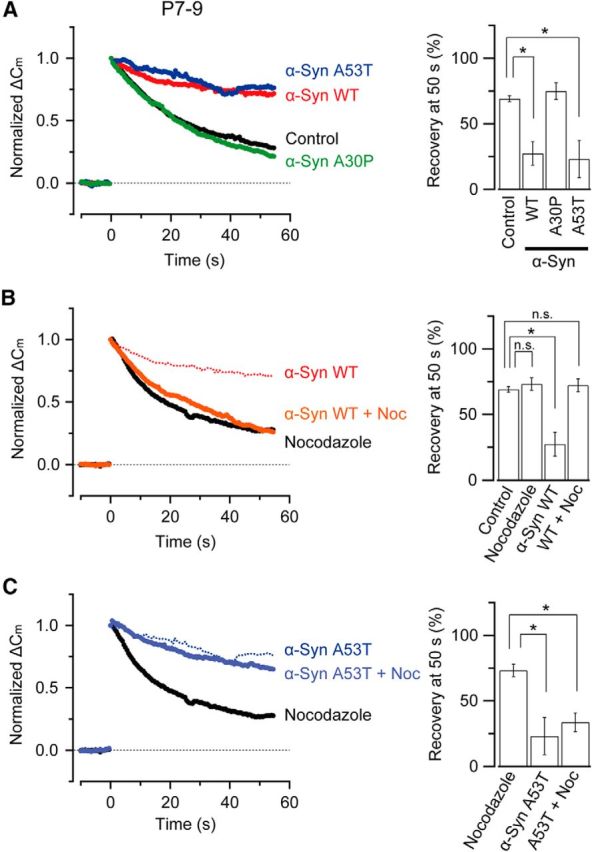
α-Synuclein A53T mutant inhibits vesicle endocytosis at calyces of Held in prehearing rats in a MT-independent manner. A, In rat calyces of Held from prehearing rats (P7–P9), both WT α-synuclein (His tag, 3.6 μm, n = 5, red) and A53T (3.6 μm, n = 5, blue) inhibited endocytic capacitance change elicited by a 20 ms depolarizing pulse (p < 0.05), whereas the A30P mutant (3.6 μm, n = 5, green) had no effect (p = 0.42). Bar graphs indicate mean endocytic recovery (%) from exocytic ΔCm 50 s after stimulation. B, Endocytic inhibition by WT α-synuclein (His tag) rescued by nocodazole (10 μm, n = 4–5, no significant difference between nocodazole alone and WT α-synuclein + nocodazole, p = 0.89). C, Endocytic inhibition by the A53T mutant could not be rescued by nocodazole (10 μm, n = 4–5, significant difference between nocodazole alone and A53T + nocodazole, p < 0.05). Asterisks indicate a significant difference (*p < 0.05).
Impact of endocytic inhibition by α-synuclein on neurotransmission
The inhibitory effect of a-synuclein on vesicle endocytosis has been inferred from expanded presynaptic membrane after its overexpression or injection (Boassa et al., 2013; Busch et al., 2014) and was shown directly by Xu et al. (2016) and here (Fig. 1) at rodent calyces of Held. However, it remains unexplained how a-synuclein impairs neurotransmission. In simultaneous presynaptic and postsynaptic whole-cell patch-clamp recordings at the calyx of Held, we loaded WT α-synuclein (3.6 μm) into presynaptic terminals. α-Synuclein had no effect on basal glutamatergic neurotransmission; the mean amplitude or shape of EPSCs evoked at 0.03 Hz remained unchanged (Fig. 4A). We then tested its effect on high-frequency neurotransmission. A train of 30 stimulations at 100 Hz induces short-term depression (STD) of EPSC amplitude (Fig. 4B). Cumulative EPSC amplitude during the train was unchanged by α-synuclein loading (p = 0.91). The STD is followed by a recovery with a biexponential time course (Fig. 4C), reflecting fast and slow replenishment of synaptic vesicles to release sites (Wang and Kaczmarek, 1998). In these experiments, at room temperature, the overall recovery rate is twice as slow as that at physiological temperature (Kushmerick et al., 2006). The slow recovery component reflects the time course of vesicle endocytosis (Hosoi et al., 2009), whereas the fast recovery is caused by Ca2+-dependent vesicle replenishment to release sites (Wang and Kaczmarek, 1998). α-Synuclein selectively prolonged the slow component without affecting the fast component of recovery (Fig. 4C). Nocodazole alone (10 μm) loaded into presynaptic terminals had no effect on the recovery rate, but when coloaded with α-synuclein (3.6 μm), canceled its inhibitory effect on the recovery rate (Fig. 4D). Therefore, nocodazole rescued the inhibitory effect of α-synuclein on the slow replenishment of synaptic vesicles.
Figure 4.

Toxic effects of WT α-synuclein on high-frequency neurotransmission. A, WΤ α-synuclein (His tag) has no effect on basal transmission at 0.03 Hz. EPSCs (bottom traces) evoked by presynaptic action potentials (top traces) at 0.03 Hz in the presence (red) or absence (black) of WT α-synuclein (3.6 μm, superimposed). In bar graphs, the mean amplitude of EPSCs was 6.8 ± 1.3 nA (n = 6) and 6.1 ± 1.4 nA (n = 6) in the absence and presence of α-synuclein, respectively (p = 0.72, Student's t test). The decay time constant of EPSCs was 0.96 ± 0.09 ms (n = 6) and 1.1 ± 0.1 ms (n = 6) in the presence and absence of α-synuclein, respectively (p = 0.29). B, Left, EPSCs evoked by a train of 30 stimuli at 100 Hz in the presence (red) or absence (control, black) of α-synuclein (His tag, 3.6 μm) in calyceal terminals. Right, Cumulative amplitudes of EPSCs during a 100 Hz train with (red, n = 6) or without (black, n = 6) WT α-synuclein. WT α-synuclein had no effect on cumulative amplitude of EPSCs (p = 0.91). C, Recovery of EPSCs from STD in the presence (red) and absence (black) of α-synuclein (His tag, 3.6 μm) in calyceal terminals. The 100 Hz stimulation was followed by test stimuli at different interstimulus intervals (0.02–20 s, protocol on the top panel). Sample traces in insets indicate EPSCs at different intervals after STD (superimposed). Right bar graphs indicate mean and SEMs of fast and slow recovery time constants (τ) from five to six synapses showing a significant prolongation (*) by α-synuclein of the slow recovery time constant in double exponential fits (solid lines in time plots); 6.2 ± 0.8 s in control, 12 ± 2 s with α-synuclein. There was no significant difference in fast recovery time constants. D, Nocodazole (10 μm) rescued the inhibitory effect of α-synuclein (His tag) on recovery of EPSCs from STD (n = 7). Recovery in the presence of α-synuclein alone (Fig. 4B) is indicated by a dashed line. Loading nocodazole alone (10 μm, blue, n = 6) had a similar recovery time to control (black). Bar graphs in the right panel indicate no significant effect of α-synuclein on the recovery time after STD in the presence of nocodazole. E, Left panels show postsynaptic APs (output) in response to presynaptic APs (input) elicited at 100 Hz in the control (left) in the presence of α-synuclein (middle) and α-synuclein + nocodazole (right) at different time periods during sustained stimulation. Center panels indicate the inhibitory effect of α-synuclein on synaptic fidelity (in output/input ratio; filled red circles, n = 4; left) compared with control (open black circles, n = 5). Right panel shows rescuing effect of coloaded nocodazole (10 μm, right panel, open red circles, n = 4). Nocodazole alone (10 μm) had no effect (blue-filled circles, n = 4). The dashed line indicates impaired synaptic fidelity by α-synuclein for comparison.
Finally, we tested the effects of α-synuclein on the fidelity of high-frequency neurotransmission by recording postsynaptic APs (output) in response to presynaptic APs (input) elicited by 100 Hz stimulation (Eguchi et al., 2012). During a sustained 100 Hz stimulation, postsynaptic AP firings remained stable at least for 30 s in controls, whereas at synapses loaded with α-synuclein, APs started to fail within ∼10 s after stimulation (Fig. 4E). Fifty seconds after the sustained stimulation, synaptic fidelity, defined as a ratio of postsynaptic APs relative to presynaptic APs per second, dropped down to ∼50% at synapses loaded with α-synuclein, but remained at ∼75% in control (p < 0.05 in 2-way ANOVA with Scheffe post hoc test, n = 4–5). Therefore, presynaptically loaded α-synuclein significantly impaired fidelity of high-frequency neurotransmission.
We next investigated whether the toxic effect of α-synuclein on high-frequency transmission can be rescued by nocodazole. When nocodazole (10 μm) was coloaded with α-synuclein (3.6 μm) into calyceal terminals, synaptic fidelity during 100 Hz stimulation remained similar to controls without α-synuclein (Fig. 4E). Therefore, nocodazole rescued high-frequency neurotransmission impaired by α-synuclein.
Discussion
In the present study, we have shown that acute loading of WT monomeric α-synuclein into the calyx of Held presynaptic terminals inhibits endocytosis of synaptic vesicles, whereas the α-synuclein mutants A30P and A53T had no such effect. Although α-synuclein mutants enhance α-synuclein aggregation (Kruger et al., 1998; Li et al., 2001), acute loading or overexpression of the A30P mutant has no inhibitory effect on exo–endocytosis or vesicle recycling (Nemani et al., 2010; Busch et al., 2014; Xu et al., 2016). Nevertheless, at the calyx of Held, the A30P mutant significantly augmented exocytosis after a train of 1 Hz stimulation (Fig. 1D). Mechanisms underlying this effect of A30P mutant remain to be investigated. The A53T mutant had a similar effect to that of WT α-synuclein in overexpression studies at cultured hippocampal synapses (Nemani et al., 2010) and calyces of Held (Xu et al., 2016). In addition, in acute loading experiments, the A53T mutant inhibited endocytosis at prehearing rat calyces of Held (Xu et al., 2016; Fig. 3A), but this effect disappeared after hearing onset (Fig. 1A). At the calyx of Held, vesicle endocytic mechanisms undergo substantial changes during the second postnatal week, when rodents start to hear sound (Takahashi, 2015). For example, GTP-independent component disappears and calmodulin/calcineurin-dependent endocytosis becomes nonfunctional (Yamashita et al., 2010), whereas an activity/PKG-dependent endocytic facilitation mechanism becomes functional (Eguchi et al., 2012). Given that PD is associated with aged individuals, pathological significance of the endocytic inhibitory effect of α-synuclein A53T mutant, observed only transiently in immature animals (Xu et al., 2016), is unclear.
The α-synuclein preparations used by Xu et al. (2016) for intraterminal loading contained both monomers and oligomers. In the brain, native α-synuclein exists predominantly in a monomeric state (Burré et al., 2013), possibly in equilibrium with its oligomers (Bartes et al., 2011). When the α-synuclein concentration increases, it can slowly aggregate into fibrils with a time course of 20–60 h (Kim et al., 2008). It also takes several days before purified monomeric α-synuclein is converted to oligomers (Krishnan et al., 2003; Burré et al., 2013). Furthermore, in whole-cell recordings, soluble molecules in patch pipettes predominate in the presynaptic terminal cytosolic concentration (Pusch and Neher, 1988). Therefore, under our experimental conditions, WT α-synuclein should remain monomeric throughout the experimental period. In previous loss-of-function studies, αβγ-synuclein triple knock-out reduces mitochondrial ATP synthesis (Ludtmann et al., 2016) and inhibits vesicle endocytosis (Vargas et al., 2014), suggesting that α-synuclein, together with β-synuclein and γ-synuclein, may normally support ATP supply that is indispensable for vesicle endocytosis (Rangaraju et al., 2014). Despite the fact that α-synuclein is essential for vesicle endocytic function, its abnormal upregulation may induce toxic functions, like many other indispensable molecules. A simple increase of α-synuclein in cells may be sufficient for its misfolding and transcellular propagation (Bendor et al., 2013).
The inhibitory effect of α-synuclein on vesicle endocytosis was suggested previously (Boassa et al., 2013; Busch et al., 2014) and demonstrated directly at calyces of Held of prehearing (Xu et al., 2016) and posthearing (Fig. 1) rats. However, little has been investigated on the underlying mechanism. We have shown here that the MT-depolymerizing reagents nocodazole and PST-1, when loaded into calyceal terminals, completely rescued the inhibitory effect of α-synuclein on vesicle endocytosis (Fig. 2C–E). Convincingly, the photo-switchable PST-1 rescued endocytic inhibition by α-synuclein in a reversible manner (Fig. 2E). These results strongly suggest that overassembly of MTs by α-synuclein underlies its toxic effect on endocytosis.
It has been reported that α-synuclein assembles tubulins directly into MTs in vitro (Alim et al., 2002, 2004). Electron microscopic examination of the reaction products indicates MT formation by WT-α-synuclein, whereas A30P and A53T formed only amorphous aggregates that are distinct from MTs (Alim et al., 2004). These reports may explain how nocodazole rescues endocytic inhibition by α-synuclein, but not that by A53T in prehearing rat calyces (Fig. 3C). However, direct assembly of MTs by α-synuclein in vitro was reported recently to be unreproducible (Oikawa et al., 2016). Because WT α-synuclein promotes phosphorylation of tau protein by GSK3β (Duka et al., 2009), it may increase the soluble tau concentration (Ballatore et al., 2007), thereby promoting new MT assembly. At later stages of synucleinopathies, aberrant MTs, together with the tubulin aggregates promoted by the α-synuclein mutant, may induce traffic jams in intracellular organelle transport (Ebneth et al., 1998), thereby causing neuronal degeneration. The detailed mechanisms by which endocytosis is blocked by newly formed MTs remain to be investigated.
With respect to synaptic functions, presynaptically loaded α-synuclein had no effect on basal transmission at low frequency or during a short train of repetitive stimulation (Fig. 4A,B). However, it inhibited exocytosis during prolonged stimulation (Fig. 1D) and significantly impaired the fidelity of high-frequency neurotransmission (Fig. 4E). These results suggest that the primary target of α-synuclein toxicity is endocytosis. When endocytosis is inhibited for a prolonged period, exocytosis is secondarily impaired because of a slowing of recycling vesicle replenishment (Fig. 4C; see also Yamashita et al., 2005). This mechanism can explain results from previous α-synuclein overexpression studies reporting disrupted vesicle trafficking (Lee et al., 2006; Scott and Roy, 2012; Wang et al., 2014), reduced vesicle pool size (Outeiro and Lindquist, 2003; Nemani et al., 2010; Scott and Roy, 2012), and diminished transmitter release (Larsen et al., 2006; Nemani et al., 2010; Lundblad et al., 2012; Janezic et al., 2013). Therefore, even moderate inhibition of vesicle endocytosis can impair synaptic fidelity during high-frequency neurotransmission, as reported previously for protein kinase G inhibitors (Eguchi et al., 2012). Fast precise neurotransmission is essential for sensory cognition and memory formation (Sabatini and Regehr, 1999), as well as for motor coordination (Sugihara et al., 1993). Dopaminergic neurons, which are involved in PD, express type 3 vesicular glutamate transporters and form glutamatergic synapses that provide precisely timed signaling (Chuhma et al., 2004). Therefore, elevated presynaptic α-synuclein likely impairs fast neurotransmission in dopaminergic–glutamatergic synapses, thereby possibly generating early symptoms of synucleinopathies.
Footnotes
This work was supported by the Okinawa Institute of Science and Technology, the Core Research for Evolutionary Science and Technology of the Japan Science and Technology Agency (T.T.), and the Japan Society for the Promotion of Science (Grant-in-Aid for Young Scientists to K.E.). We thank Yasuo Ihara and Takeshi Sakaba for comments and Steven Aird for editing this manuscript.
The authors declare no competing financial interests.
References
- Alim MA, Hossain MS, Arima K, Takeda K, Izumiyama Y, Nakamura M, Kaji H, Shinoda T, Hisanaga S, Ueda K (2002) Tubulin seeds α-synuclein fibril formation. J Biol Chem 277:2112–2117. 10.1074/jbc.M102981200 [DOI] [PubMed] [Google Scholar]
- Alim MA, Ma QL, Takeda K, Aizawa T, Matsubara M, Nakamura M, Asada A, Saito T, Kaji H, Yoshii M, Hisanaga S, Uéda K (2004) Demonstration of a role for α-synuclein as a functional microtubule-associated protein. J Alzheimers Dis 6:435–442; discussion 443–449. 10.3233/JAD-2004-6412 [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 8:663–672. [DOI] [PubMed] [Google Scholar]
- Bartes T, Choi JG, Selkoe DJ (2011) α-Synuclein occurs physiologically as helically folded tetramer that resists aggregation. Nature 477:107–110. 10.1038/nature10324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Gedalya T, Loeb V, Israeli E, Altschuler Y, Selkoe DJ, Sharon R (2009) α-Synuclein and polyunsaturated fatty acids promote clathrin-mediated endocytosis and synaptic vesicle recycling. Traffic 10:218–234. 10.1111/j.1600-0854.2008.00853.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendor JT, Logan TP, Edwards RH (2013) The function of α-synuclein. Neuron 79:1044–1066. 10.1016/j.neuron.2013.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boassa D, Berlanga ML, Yang MA, Terada M, Hu J, Bushong EA, Hwang M, Masliah E, George JM, Ellisman MH (2013) Mapping the subcellular distribution of α-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: Implications for Parkinson's disease pathogenesis. J Neurosci 33:2605–2615. 10.1523/JNEUROSCI.2898-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowiak M, Nahaboo W, Reynders M, Nekolla K, Jalinot P, Hasserodt J, Rehberg M, Delattre M, Zahler S, Vollmar A, Trauner D, Thorn-Seshold O (2015) Photo-switchable inhibitors of microtubule dynamics optically control mitosis and cell death. Cell 162:403–411. 10.1016/j.cell.2015.06.049 [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. 10.1016/S0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- Burré J, Vivona S, Diao J, Sharma M, Brunger AT, Südhof TC (2013) Properties of native brain α-synuclein. Nature 498:E4–E6; discussion E6–E7. 10.1038/nature12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch DJ, Oliphint PA, Walsh RB, Banks SM, Woods WS, George JM, Morgan JR (2014) Acute increase of α-synuclein inhibits synaptic vesicle recycling evoked during intense stimulation. Mol Biol Cell 25:3926–3941. 10.1091/mbc.E14-02-0708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A (2004) α-Synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364:1167–1169. 10.1016/S0140-6736(04)17103-1 [DOI] [PubMed] [Google Scholar]
- Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, Lee NK, Shin YK (2013) Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc Natl Acad Sci U S A 110:4087–4092. 10.1073/pnas.1218424110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Zhang H, Masson J, Zhuang X, Sulzer D, Hen R, Rayport S (2004) Dopamine neurons mediate a fast excitatory signal via their glutamatergic synapses. J Neurosci 24:972–981. 10.1523/JNEUROSCI.4317-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Khurana V, Yi S, Sahni N, Loh KH, Auluck PK, Baru V, Udeshi ND, Freyzon Y, Carr SA, Hill DE, Vidal M, Ting AY, Lindquist S (2017) In situ peroxidase labeling and mass-spectrometry connects alpha-synuclein directly to endocytic trafficking and mRNA metabolism in neurons. Cell Systems 4:242–250.e4. 10.1016/j.cels.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duka T, Duka V, Joyce JN, Sidhu A (2009) α-Synuclein contributes to GSK-3β-catalyzed tau phosphorylation in Parkinson's disease models. FASEB J 23:2820–2830. 10.1096/fj.08-120410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E (1998) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol 143:777–794. 10.1083/jcb.143.3.777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K, Nakanishi S, Takagi H, Taoufiq Z, Takahashi T (2012) Maturation of a PKG-dependent retrograde mechanism for exoendocytic coupling of synaptic vesicles. Neuron 74:517–529. 10.1016/j.neuron.2012.03.028 [DOI] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW (2004) Comparison of kindreds with Parkinsonism and α-synuclein genomic multiplications. Ann Neurol 55:174–179. 10.1002/ana.10846 [DOI] [PubMed] [Google Scholar]
- Hori T, Takahashi T (2012) Kinetics of synaptic vesicle refilling with neurotransmitter glutamate. Neuron 76:511–517. 10.1016/j.neuron.2012.08.013 [DOI] [PubMed] [Google Scholar]
- Hosoi N, Holt M, Sakaba T (2009) Calcium dependence of exo- and endocytotic coupling at a glutamatergic synapse. Neuron 63:216–229. 10.1016/j.neuron.2009.06.010 [DOI] [PubMed] [Google Scholar]
- Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A (2004) Causal relation between α-synuclein gene duplication and familial Parkinson's deisease. Lancet 364:1169–1171. 10.1016/S0140-6736(04)17104-3 [DOI] [PubMed] [Google Scholar]
- Ihara M, Yamasaki N, Hagiwara A, Tanigaki A, Kitano A, Hikawa R, Tomimoto H, Noda M, Takanashi M, Mori H, Hattori N, Miyakawa T, Kinoshita M (2007) Sept4, a component of presynaptic scaffold and Lewy bodies, is required for the suppression of α-synuclein neurotoxicity. Neuron 53:519–533. 10.1016/j.neuron.2007.01.019 [DOI] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M (1994) Identification of two distinct synucleins from human brain. FEBS Lett 345:27–32. 10.1016/0014-5793(94)00395-5 [DOI] [PubMed] [Google Scholar]
- Janezic S, Threlfell S, Dodson PD, Dowie MJ, Taylor TN, Potgieter D, Parkkinen L, Senior SL, Anwar S, Ryan B, Deltheil T, Kosillo P, Cioroch M, Wagner K, Ansorge O, Bannerman DM, Bolam JP, Magill PJ, Cragg SJ, Wade-Martins R (2013) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc Natl Acad Sci U S A 110:E4016–E4025. 10.1073/pnas.1309143110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R (1999) α-Synuclein binds to tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J Biol Chem 274:25481–25489. 10.1074/jbc.274.36.25481 [DOI] [PubMed] [Google Scholar]
- Kim M, Jung W, Lee IH, Bhak G, Paik SR, Hahn JS (2008) Impairment of microtubule system increases α-synuclein aggregation and toxicity. Biochem Biophys Res Commun 365:628–635. 10.1016/j.bbrc.2007.11.020 [DOI] [PubMed] [Google Scholar]
- Krishnan S, Chi EY, Wood SJ, Kendrick BS, Li C, Garzon-Rodriguez W, Wypych J, Randolph TW, Narhi LO, Biere AL, Citron M, Carpenter JF (2003) Oxidative dimer formation is the critical rate-limiting step for Parkinson's disease α-synuclein fibrillogenesis. Biochemistry 42:829–837. 10.1021/bi026528t [DOI] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O (1998) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson's disease. Nat Genet 18:106–108. 10.1038/ng0298-106 [DOI] [PubMed] [Google Scholar]
- Kushmerick C, Renden R, von Gersforff H (2006) Physiological temperature reduce the rate of vesicle pool depletion and short-term depression via an acceleration of vesicle recruitment. J Neurosci 26:1366–1377. 10.1523/JNEUROSCI.3889-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D (2006) α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci 26:11915–11922. 10.1523/JNEUROSCI.3821-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Lee S, Lee SJ (2006) Impairment of microtubule-dependent trafficking by overexpression of α-synuclein. Eur J Neurosci 24:3153–3162. 10.1111/j.1460-9568.2006.05210.x [DOI] [PubMed] [Google Scholar]
- Li J, Uversky VN, Fink AL (2001) Effect of familial Parkinson's disease point mutations A30P and A53T on the structure properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 40:11604–11613. 10.1021/bi010616g [DOI] [PubMed] [Google Scholar]
- Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins RD, Arancio O (2004) α-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J 23:4506–4516. 10.1038/sj.emboj.7600451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtmann MH, Angelova PR, Ninkina NN, Gandhi S, Buchman VL, Abramov AY (2016) Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J Neurosci 36:10510–10521. 10.1523/JNEUROSCI.1659-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad M, Decressac M, Mattsson B, Björklund A (2012) Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A 109:3213–3219. 10.1073/pnas.1200575109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH (1988) Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8:2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marui W, Iseki E, Nakai T, Miura S, Kato M, Uéda K, Kosaka K (2002) Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci 195:153–159. 10.1016/S0022-510X(02)00006-0 [DOI] [PubMed] [Google Scholar]
- Masuda M, Dohmae M, Nonaka T, Oikawa T, Hisanaga S, Goedert M, Hasegawa M (2006) Cysteine misincorporation in bacterially expresssed α-synuclein filament assembly. FEBS Lett 580:1775–1779. 10.1016/j.febslet.2006.02.032 [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM (2000) Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20:3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH (2010) Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65:66–79. 10.1016/j.neuron.2009.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Nonaka T, Terada M, Tamaoka A, Hisanaga S, Hasegawa M (2016) α-Synuclein fibrils exhibit gain of toxic function, promoting tau aggregation and inhibiting microtubule assembly. J Biol Chem 291:15046–15056. 10.1074/jbc.M116.736355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S (2003) Yeast cells provide insight into alpha-synuclein biology. Science 302:1772–1775. 10.1126/science.1090439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. 10.1126/science.276.5321.2045 [DOI] [PubMed] [Google Scholar]
- Pusch M, Neher E (1988) Rates of diffusional exchange between small cells and a measuring patch pipette. Pflugers Arch 411:204–211. 10.1007/BF00582316 [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA (2014) Activity-driven local ATP synthesis is required for synaptic function. Cell 156:825–835. 10.1016/j.cell.2013.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renden R, von Gersdorff H (2007) Synaptic vesicle endocytosis at a CNS nerve terminal: Faster kinetics at physiological temperatures and increased endocytic capacity during maturation. J Neurophysiol 98:3349–3359. 10.1152/jn.00898.2007 [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG (1999) Timing of synaptic transmission. Annu Rev Physiol 61:521–542. 10.1146/annurev.physiol.61.1.521 [DOI] [PubMed] [Google Scholar]
- Scott DA, Roy S (2012) α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci 32:10129–10135. 10.1523/JNEUROSCI.0535-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) α-Synuclein locus triplication causes Parkinson's disease. Science 302:841. 10.1126/science.1090278 [DOI] [PubMed] [Google Scholar]
- Sousa VL, Bellani S, Giannandrea M, Yousuf M, Valtorta F, Meldolesi J, Chieregatti E (2009) α-Synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Molec Biol Cell 20:3725–3739. 10.1091/mbc.E08-03-0302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 95:6469–6473. 10.1073/pnas.95.11.6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöckl M, Fischer P, Wanker E, Herrmann A (2008) α-Synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J Molec Biol 375:1394–1404. 10.1016/j.jmb.2007.11.051 [DOI] [PubMed] [Google Scholar]
- Sugihara I, Lang EJ, Llinás R (1993) Uniform olivocerebellar conduction time underlies Purkinje cell complex spike synchronicity in the rat cerebellum. J Physiol 470:243–271. 10.1113/jphysiol.1993.sp019857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JY, Wu LG (2001) Fast kinetics of exocytosis revealed by simultaneous measurements of presynaptic capacitance and postsynaptic currents at a central synapse. Neuron 30:171–182. 10.1016/S0896-6273(01)00271-9 [DOI] [PubMed] [Google Scholar]
- Takahashi T. (2015) Strength and precision of neurotransmission at mammalian presynaptic terminals. Proc Jpn Acad Ser B Physiol Biol Sci 91:305–320. 10.2183/pjab.91.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taoufiq Z, Eguchi K, Takahashi T (2013) Rho-kinase accelerates synaptic vesicle endocytosis by linking cyclic GMP-dependent protein kinase activity to phosphatidyl inositol-4,5-bisphosphate synthesis. J Neurosci 33:12099–12104. 10.1523/JNEUROSCI.0730-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS (2014) Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci 34:9364–9376. 10.1523/JNEUROSCI.4787-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasileva M, Renden R, Horstmann H, Gilter D, Kuner T (2013) Overexpression of synapsin 1a in the calyx of Held accelerates short-term plasticity and decreases synaptic vesicle volume and active zone area. Front Cell Neurosci 7:A270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK (1998) High frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature 394:384–388. 10.1038/28645 [DOI] [PubMed] [Google Scholar]
- Wang L, Das U, Scott DA, Tang Y, McLean PJ, Roy S (2014) α-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol 24:2319–2326. 10.1016/j.cub.2014.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Xu J, Wu XS, Wu LG (2005) Activity-dependent acceleration of endocytosis at a central synapse. J Neurosci 25:11676–11683. 10.1523/JNEUROSCI.2972-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu XS, Sheng J, Zhang Z, Yue HY, Sun L, Sgobio C, Lin X, Peng S, Jin Y, Gan L, Cai H, Wu LG (2016) α-Synuclein mutation inhibits endocytosis at mammalian central nerve terminals. J Neurosci 36:4408–4414. 10.1523/JNEUROSCI.3627-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Hige T, Takahashi T (2005) Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science 307:124–127. 10.1126/science.1103631 [DOI] [PubMed] [Google Scholar]
- Yamashita T, Eguchi K, Saitoh N, von Gersdorff H, Takahashi T (2010) Developmental shift to a mechanism of synaptic vesicle endocytosis requiring nanodomain Ca2+. Nat Neurosci 13:838–844. 10.1038/nn.2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Gu G, Goodlett DR, Zhang T, Pan C, Montine TJ, Montine KS, Aebersold RH, Zhang J (2004) Analysis of α-synuclein-associated proteins by quantitative proteomics. J Biol Chem 279:39155–39164. 10.1074/jbc.M405456200 [DOI] [PubMed] [Google Scholar]