

Abstract
The complement system, typically associated with innate immunity, is emerging as a key controller of nonimmune systems including in development, with recent studies linking complement mutations with neurodevelopmental disease. A key effector of the complement response is the activation fragment C5a, which, through its receptor C5aR1, is a potent driver of inflammation. Surprisingly, C5aR1 is also expressed during early mammalian embryogenesis; however, no clearly defined function is ascribed to C5aR1 in development. Here we demonstrate polarized expression of C5aR1 on the apical surface of mouse embryonic neural progenitor cells in vivo and on human embryonic stem cell-derived neural progenitors. We also show that signaling of endogenous C5a during mouse embryogenesis drives proliferation of neural progenitor cells within the ventricular zone and is required for normal brain histogenesis. C5aR1 signaling in neural progenitors was dependent on atypical protein kinase C ζ, a mediator of stem cell polarity, with C5aR1 inhibition reducing proliferation and symmetric division of apical neural progenitors in human and mouse models. C5aR1 signaling was shown to promote the maintenance of cell polarity, with exogenous C5a increasing the retention of polarized rosette architecture in human neural progenitors after physical or chemical disruption. Transient inhibition of C5aR1 during neurogenesis in developing mice led to behavioral abnormalities in both sexes and MRI-detected brain microstructural alterations, in studied males, demonstrating a requirement of C5aR1 signaling for appropriate brain development. This study thus identifies a functional role for C5a–C5aR1 signaling in mammalian neurogenesis and provides mechanistic insight into recently identified complement gene mutations and brain disorders.
SIGNIFICANCE STATEMENT The complement system, traditionally known as a controller of innate immunity, now stands as a multifaceted signaling family with a broad range of physiological actions. These include roles in the brain, where complement activation is associated with diseases, including epilepsy and schizophrenia. This study has explored complement regulation of neurogenesis, identifying a novel relationship between the complement activation peptide C5a and the neural progenitor proliferation underpinning formation of the mammalian brain. C5a was identified as a regulator of cell polarity, with inhibition of C5a receptors during embryogenesis leading to abnormal brain development and behavioral deficits. This work demonstrates mechanisms through which dysregulation of complement causes developmental disease and highlights the potential risk of complement inhibition for therapeutic purposes in pregnancy.
Keywords: aPKC, C5a, C5aR1, complement, neurogenesis, polarity
Introduction
The key complement activation fragment, anaphylatoxin C5a and its primary receptor C5aR1, plays a pivotal role in inflammation and immune defense. However, it is increasingly recognized that this evolutionarily ancient system also possesses unexpected roles in development, such as in morphogenesis, neurogenesis, migration, and neuronal synapse pruning (Hawksworth et al., 2017; Gorelik et al., 2017). Defects in complement signaling have been associated with neurodevelopmental abnormalities such as autism, schizophrenia, and 3MC syndrome (Corbett et al., 2007; Rooryck et al., 2011; Sekar et al., 2016).
Despite these emerging developmental roles of complement, the functional role of C5aR1 in embryonic development remains poorly defined. During organogenesis of Xenopus embryos, C5 is expressed in the neural plate of the developing nervous system (McLin et al., 2008), and in mice and humans, C5 and C5aR1 are expressed in the developing neural tube (Denny et al., 2013). Postnatally, C5a–C5aR1 expression continues in neural stem cells in vitro, is expressed in migrating neuroblasts in response to ischemia (Rahpeymai et al., 2006), and promotes the proliferation of progenitor cells within the external granular layer of the postnatal rat cerebellum (Bénard et al., 2008). We also previously demonstrated that C5 and C5aR1 are expressed in human embryonic stem cells and can regulate pluripotency (Hawksworth et al., 2014). This early embryonic expression pattern of C5aR1 in the absence of other factors of the canonical pathogen-initiated complement cascade (Jeanes et al., 2015) suggests that C5aR1 signaling has adopted additional roles in mammalian development beyond innate immunity. However, despite this clear expression of C5a receptors during brain development, a neurodevelopmental role for C5a remains poorly defined.
To investigate the role of C5aR1 in neural progenitor cell (NPC) physiology, we used both mouse models and human embryonic stem (hES) cells differentiated to a stage resembling the ventricular zone of the developing brain. In these hES-derived cultures, neural rosettes are formed that display apical polarization and interkinetic nuclear migration of periluminal cells, similar to that seen during neurulation and in the cortical ventricular zone (Shi et al., 2012; Ziv et al., 2015). The signaling mechanisms in the control of rosette polarity are highly conserved, and the localization of the Par3/Par6/protein kinase C ζ (PKCζ) complex to the apical membrane is essential for self-renewal of neural progenitors through the orchestration of the balance between symmetric and asymmetric division (Fietz and Huttner, 2011). Here we show that C5aR1 is a regulator of the apicobasal polarity of neural stem cells and that the acute pharmacological blockade of C5aR1 signaling during neurogenesis results in reduced ventricular zone proliferation and cerebral disorganization, leading ultimately to behavioral alterations. Collectively, our data reveal that C5aR1 functions as a regulator of mammalian brain development under normal physiological conditions in both mice and humans. This work complements recent studies documenting mutations in complement activation pathways that contribute to an increased risk of neurodevelopmental disorders.
Materials and Methods
Reagents.
Mouse recombinant C5a (mC5a) was obtained from Sigma-Aldrich and reconstituted in 0.25% BSA in PBS. Human isolated C5a (hC5a) was obtained from CompTech. PMX53 was synthesized as described previously (Pavlovski et al., 2012), stored lyophilized, and reconstituted in purified water before use.
Tissue collection and processing.
All animal experiments in this study were performed with prior approval from the animal ethics committee of The University of Queensland. Animal housing and time-mating of mice was provided by University of Queensland Biological Resources. Tissues were collected after the mice were killed by cervical dislocation. Tissues preserved for RNA/protein analysis were snap frozen in liquid nitrogen and stored at −80°C until extraction. Protein was extracted using modified RIPA buffer prepared in house. Tissues used for histological analysis were incubated at 4°C overnight in freshly prepared 4% paraformaldehyde (PFA). Tissues were prepared for cryosection by sequential passaging through serial sucrose solutions (10, 20, and 30%), removed into OCT for freezing, and sectioned at 12 μm, unless otherwise stated.
Embryonic CSF was obtained from embryonic day 13.5 (E13.5) embryos using a pulled glass pipette attached to a vacuum. Pooled CSF from three litters was used in analysis of mC5a concentration through ELISA. CSF was treated with EDTA (5 mm final concentration) to prevent coagulation and extrinsic complement activation and stored at −80°C until analysis.
RT-PCR and qPCR.
RNA was extracted using RNeasy plus spin columns (QIAGEN) and treated for genomic DNA contamination using Turbo DNase (Invitrogen). All RNA was additionally checked for gDNA contamination by PCR analysis. Primer sequences and PCR conditions can be found in Table 1. qPCR was performed using SYBR green PCR mastermix (Ambion) and machine settings according to the manufacturer's instructions. The ΔΔCt method to assess fold change of gene expression was used, and all data points within an individual sample were referenced back to r18s expression levels.
Table 1.
PCR primer sequences and conditions
| Name | Species | Use | Forward (5′–3′) | Reverse (5′–3′) | Annealing temperature (°C) | Product size (bp) |
|---|---|---|---|---|---|---|
| mC5aR1 | Mouse | RT-PCR | ATGCTGATGCTGATGCTGATCG | ATGCTGATGGCTGATCGTCGGATGCTGAT | 60 | 562 |
| mActB | Mouse | RT-PCR | GTGGGCCGCCCTAGGCACCAG | CTCTTTGATGTCACGCACGATTTC | 60 | 103 |
| mC5aR1 | Mouse | qPCR | GGGATGTTGCAGCCCTTATCA | CGCCAGATTCAGAAACCAGATG | 60 | 131 |
| mSox2 | Mouse | qPCR | TAGAGCTAGACTCCGGGCGATGA | TTTCGTGGTCTTGTTTAAGGCAA | 60 | 296 |
| m18s | Mouse | qPCR | GATCCATTGGAGGGCAAGTCT | CCAAGATCCAACTACGAGCTT | 60 | 103 |
| hC5AR | Human | qPCR | TCCTTCAATTATACCACCCCTGA | GGAAGACGACTGCAAAGATGA | 60 | 139 |
| hC5 | Human | qPCR | ACTGAATTTGGTTGCTACTCCTC | GTATTACTGGGACTCCTCCTACC | 60 | 110 |
| hACTB | Human | qPCR | GCGGGAAATCGTGCGTGACATT | GATGGAGTTGAAGGTAGTTTGGTG | 60 | 232 |
| hCDH2 | Human | qPCR | ATCAACCCCATACACCAGCC | GTCGATTGGTTTGACCACGG | 60 | 128 |
Western blot.
Protein samples (20 μg) were subjected to electrophoresis at 100 V on a 10% polyacrylamide gel until good separation was achieved. Primary antibodies directed against C5aR1 (1:500, Hycult Biotech, clone 10/92, RRID:AB_10130226), phospho-Erk (1:1000, CST, catalog #9106, RRID:AB_331768), total-Erk (1:1000, Cell Signaling Technology, catalog #9102, RRID:AB_330744), and β-tubulin (1:2000, Sigma Aldrich, clone TUB2.1, AB_10679259) were diluted in 0.5× odyssey blocking buffer (LiCor) and incubated with the membrane rocking overnight at 4°C. Incubation with specific LiCor odyssey secondary antibodies was performed according to the manufacturer's instructions. Blots were imaged using the LiCor odyssey system and software. Optical densitometry values were derived from analysis of the image in ImageJ (NIH, RRID:SCR_003070).
Immunofluorescence.
Tissues or cells were blocked using 0.1% Triton X-100/4% goat serum in PBS for 1 h. In the case of live staining for NE-4C cultures, a primary antibody was added to unfixed cells on ice for 30 min before fixation. Primary antibodies raised against mC5aR1 (HBT, clone 10/92, 1:200, RRID:AB_10130226), Pax6 (R&D Systems, MAB1260, 1:500, RRID:AB_2159696), phosphohistone H3 (CST, catalog #9706, 1:1000, RRID:AB_331749), doublecortin (DCX; CST, catalog #4604, 1:500, RRID:AB_561007), Sox2 (CST, catalog #3728, 1:500, RRID:AB_2194037), acetylated α-tubulin (Sigma Aldrich, catalog #T7451, 1:500, RRID:AB_609894), Zo-1 (Invitrogen, catalog #402300, 1:500, RRID:AB_2533457), hC5aR1 (BD Biosciences, catalog #550733, 3 μg/ml, RRID:AB_393854), C5 (HBT, clone bb5.1, 1:250, RRID:AB_10992443), Tubb3 (Millipore, MAB1637, 1:1000, RRID:AB_2210524), NCAD (Sigma Aldrich, C3865, 1:2000, RRID:AB_262097), Arl13b (NeuroMab, 73-287, 1:200, RRID:AB_11000053), PKCζ (Abcam, ab59412, 1:250, RRID:AB_946308), or isotype control antibodies were incubated overnight at 4°C. Appropriate Alexa Fluor secondary antibodies (Invitrogen, 1:1000) were incubated with the samples for 2 h at room temperature before counterstaining (1 μg/ml DAPI, 5 min) and mounting. For C5aR/PKCζ costaining, the issue of using two rabbit antibodies was circumvented with the use of conjugated Fab fragments and intermediate blocking with an unconjugated Fab fragment (Jackson ImmunoResearch Laboratories). All immunofluorescence images were acquired via confocal microscopy (DMi8, Leica Microsystems) and processed with ImageJ software. Further image analysis was performed with either ImageJ or CellProfiler software (Broad Institute, RRID:SCR_007358), as stated in relevant sections.
mC5a ELISA.
Maternal and embryonic brain sample concentrations were determined by BCA assay (Thermo Fisher Scientific). Aliquots of each sample were measured in technical triplicate for mC5a concentration by ELISA (R&D Systems, RRID:AB_2067297) according to the manufacturer's instructions. mC5a concentrations were normalized to protein concentration (nanograms per milligram, brain samples) or volume (nanograms per milliliter, CSF).
Neurosphere culture.
Telencephalon from litters of E14.5 C57BL6/J mice (RRID:IMSR_JAX:000664) were isolated and mechanically dissociated. Cells were maintained in DMEM/F-12 medium supplemented with 1× B27 supplement, l-glutamine, 10 ng/ml bFGF, 10 ng/ml EGF, and penicillin/streptomycin. Isolated human rosettes were maintained in N2B27 throughout the neurosphere assay. To assess the effect of C5aR1 modulation on neurosphere growth, 103 cells at passage 3 were seeded into each well of a 96-well plate in the presence or absence of 10 nm mC5a. Wells were imaged after 1 week in culture, and the number and diameter of neurospheres were assessed. Medium was replaced every 48 h. Before treatment with 10 nm mC5a, cells were deprived of growth factors (bFGF/EGF) for 6 h.
NE-4C culture.
NE-4C cells were acquired from the American Type Culture Collection (catalog #CRL-2925, RRID:CVCL_B063) and expanded in MEM (Sigma-Aldrich) supplemented with 10% FCS (Lonza), l-glutamine, and nonessential amino acids (Invitrogen). For transwell culture, NE-4C cells were plated on poly-l-lysine-coated 0.2 μm transwell membranes in a 24-well plate. Cells were maintained in medium as described above, and treatment (mC5a or vehicle) was added to the upper compartment 12 h before fixation for immunofluorescence.
Human embryonic stem cell culture and neuronal differentiation.
H9 hES cells (RRID:CVCL_9773) were maintained on Matrigel (BD Biosciences)-coated dishes in mouse embryonic fibroblast-conditioned KSR medium [DMEM/F-12 supplemented with 20% KnockOut Serum Replacement (Invitrogen) and 0.1 mm nonessential amino acids, 1 mm l-glutamine, 0.1 mm β-mercaptoethanol, and 10 ng/ml human basic fibroblast growth factor (FGF2), all from Thermo Fisher Scientific] as described previously (Briggs et al., 2013). Neuronal induction was performed using an adapted method of dual SMAD inhibition (Chambers et al., 2009). Briefly, high-density H9 cells were incubated for 10 d in N2B27 medium (1:1 mixture of DMEM/F-12 supplemented with N2 and Neurobasal medium supplemented with B27, 0.1 mm nonessential amino acids, 1 mm l-glutamine, 0.1 mm β-mercaptoethanol, 50 U/ml penicillin, and 50 mg/ml streptomycin) supplemented with 1 μm Dorsomorphin and 10 μm SB431542. At day 10, cultures were bulk passaged using 1 mg/ml dispase solution and seeded on Matrigel-coated plates in N2B27 medium without Dorsomorphin and SB431542. The cultures passaged again using a similar technique at day 15, maintaining bulk and high-density culture. Cells were supplemented with 2 ng/ml FGF2 from day 16 to day 22 to promote emergence and proliferation of rosettes. Using this method, rosettes began to emerge around days 16–18, reaching maturity 7–10 d later. To ensure purity of neuronal cultures, rosettes were manually harvested around day 24.
Human neural rosette experiments.
Rosette cultures were single cell dissociated with Accutase (Thermo Fisher Scientific) and seeded at either high density (3.2 × 105 cells/cm2) or low density (1.2 × 105 cells/cm2) for subsequent experiments. At high density, 100% of cells re-formed rosettes, allowing for experimentation on a pure culture of uniformly sized rosettes. At low density, the ability of rosette re-formation was limited (see Results), allowing for interrogation of the ability of C5aR1 signaling to promote reestablishment of rosettes over differentiation.
Low-density single cells were grown for 7 d in the presence of 10 nm hC5a, 1 μm PMX53 plus hC5a, or vehicle control, after which cells were fixed in 4% paraformaldehyde and stained for NCAD using the above immunocytochemistry methods protocol. Acquired images were analyzed using CellProfiler (RRID:SCR_007358) to quantify rosette number through the measurement and quantification of NCAD-positive rosette lumens (Kamentsky et al., 2011).
High-density single cells were cultured for 5 d to allow reestablishment of rosettes, after which cells were tested in DAPT treatment and cell-cycle progression experiments. For DAPT treatment, cells were pretreated with 10 nm hC5a or vehicle control for 1 h, after which 1 μm DAPT was added. Cells were incubated overnight, after which they were fixed, stained for NCAD, imaged, and analyzed with CellProfiler to quantify rosette number or RNA collected for qRT-PCR. For cell-cycle testing, cells were serum starved overnight in DMEM/F-12 plus 0.25% BSA, after which 10 nm hC5a or vehicle control was added to the cells. The cells were then incubated overnight, after which they were fixed and stained with anti-phosphorylated histone H3 (pHH3) antibody and DAPI using the above immunocytochemistry methods protocol. The ratio of pHH3-positive nuclei was calculated using analysis with CellProfiler.
In utero injections.
Time-mated dams at E13.5 were anesthetized under 1% isofluorane for surgery. One microliter of 100 nm mC5a, 10 μm PMX53, or respective vehicle control was injected into the ventricular system of the embryos. Abdominal incisions were closed with sutures, and dams were administered 0.1 mg/kg buprenorphine for analgesia after surgery. Dams were killed and tissues were collected at 24 h after surgery for tissue analysis.
For analysis of proliferation, the embryonic telencephalon was sectioned coronally, and sections at the level of the preoptic area were used for histological analysis. M-phase cells, as determined by phosphohistone H3 staining, were counted using ImageJ at the apical surface of the telencephalic ventricular zone. Phosphohistone H3-positive cells per 100 μm were calculated for each individual embryo, and differences between treatment groups were analyzed by Students t test.
Treatment of animals for behavioral experiments and magnetic resonance imaging.
Time-mated dams were acquired from UQBR and housed under standard conditions under the care of animal house staff. Mice were administered 1 mg/kg PMX53 or sterile water vehicle control (n = 6 per group) in a 100 μl volume via intraperitoneal injection over 3 d (E12.5–E14.5). Dams were allowed to litter down in individual cages. Gestational age at birth was defined as the number of days after discovery of the vaginal plug (E0.5). Litter number, weight, crown-rump length, and snout-occiput length were taken at birth. In addition, pup weight was tracked over the first 5 weeks of life to determine whether any differences existed in growth parameters.
At 8 weeks of age, male and female mice from the litters were randomly selected for participation in behavioral experiments (n = 8 per group). After behavioral experiments, mice were anesthetized with a zylazine/xoletil mixture and perfused with PBS, followed by 4% PFA via an intracardiac cannula. Whole heads were incubated in 4% PFA for an additional 3 d before washes with PBS and careful removal of the brain. Brains were stored in fresh PBS until magnetic resonance imaging (MRI) analysis.
Grip strength.
Mice were assessed for motor weakness using the grip strength test. Briefly, mice gripped a bar attached to a force transducer. The experimenter gently pulled backward on the base of the tail until the mouse dislodged from the bar. The maximum force recorded over three trials was designated as the grip strength. Both forelimb and hindlimb grip strength was assessed.
Balance beam.
Mice were assessed for higher motor coordination using the balance beam test. The apparatus consisted of a 70 cm (length) × 3 mm (width) beam suspended 1 m above a surface. The beam was held in a room with bright overhead lights kept at a constant output of 150 lumens, and a covered platform was set at the end of the beam. Mice were trained, through four training attempts, to move toward the covered platform through the use of a training beam of 80 mm width. After training, mice were exposed to the test apparatus. Time taken to cross the beam and footfall errors were recorded. A footfall error was deemed to have occurred if the paw of the animal moved from a position on the beam and crossed a threshold 10 mm beneath the beam.
Open-field test.
The open-field test used 50 × 50 cm infrared photobeam tracking arenas (MED Associates) to measure activity in a novel environment. Mice were placed in the center of the arena, and after a 30 s initiation period, movement in the x, y, and z planes was tracked for the following 30 min. Arenas were cleaned with 70% ethanol and allowed to dry between experiments. Thigmotaxis over the initial 5 min was used as a measure of anxiety in the new environment and was assessed as beam breaks within the center (25 × 25 cm) square of the arena.
Y-maze.
The Y-maze consisted of a Y-shaped maze of opaque white plastic with three identical arms set at 120° angles. The arms consisted of a home arm of plain design and two exploratory arms where the walls were decorated with different repetitive geometric patterns. For the exploratory task, one exploratory arm was blocked from the maze by use of a plastic divider. A subject was placed in the home arm and allowed to explore the home arm and remaining exploratory arm for 5 min. The subject was then reintroduced to the maze after a 30 min period with the arm divider removed, allowing for entry into the second novel, exploratory arm. The movement of the mouse around the maze was tracked with EthoVision video tracking software (Noldus). Frequency of entry into the novel arm was used as a measure of short-term memory.
MRI analysis of brain regions.
Brains stored for MRI analysis were washed extensively in PBS, followed by a 48 h incubation in gadolinium contrast agent [0.2% Magnevist (Bayer Healthcare Pharmaceuticals) in PBS]. Brains were imaged on 16.4T small-animal vertical wide bore NMR spectrometer (Bruker BioSpin) at the Centre for Advanced Imaging, The University of Queensland. Brains were immersed in fomblin oil (Solvay Solexis) inside a glass test tube of 10 mm diameter and fitted inside a quadrature birdcage coil (M2M Imaging). T1-weighted images and multishell diffusion-weighted images (DWIs) were obtained within a total scan time of 18 h as described previously (Sepehrband et al., 2015). Briefly, DWI datasets were composed of three B0 images and 60 diffusion-weighted images for each shell. Optimally ordered gradient directions with electrostatic energy minimization were obtained using the Camino software package (Jones et al., 1999; Cook et al., 2007).
Volumetric analysis of the obtained T1 images was achieved using Advanced Normalization Tools software. Briefly, all T1 images were warped to produce a common template image. Warp fields containing Jacobian values for the individual images were subjected to a modified t test using the randomize function of FSL (Oxford Center for Functional MRI of the Brain Software Library) to determine significantly different Jacobian value voxels between the vehicle- and PMX53-treated groups. Inverse warp fields were applied to anatomical area mapping of the common template to generate volumetric values for regions of sample brains. T1 images and generated anatomical masks were visualized and refined in ITKsnap software (University of Pennsylvania). Differences in brain region volume were tested using Student's t test.
Images were registered using the FSL linear registration tool to compare anatomically similar voxels between samples. Comparison of each of the diffusion parameters was achieved using the randomize function of FSL to generate a probability map of differences between vehicle- and PMX53-treated samples. Probability maps were thresholded to significance (p ≤ 0.05) and displayed on a generated template image.
Statistical analyses.
Graphing and statistics were performed using GraphPad Prism software (version 6.0c, GraphPad Software) using Student's t test and one-way ANOVA with Dunnett's post-test for the relevant statistical analysis.
Results
C5aR1 is expressed in murine neural progenitor cells and is localized to the apical ventricular zone
We have previously reported the neuroepithelial expression of C5aR1 during mouse neurulation (E7.5–10.5; Denny et al., 2013), but it is unknown whether C5aR1 expression continues during the period of neurogenesis. We therefore examined the temporal expression of C5aR1 in embryos 12.5–18.5 dpc. RT-PCR analysis of whole-brain RNA extracts revealed C5ar1 expression during this key period of brain formation (Fig. 1A). Immunohistochemistry analysis indicated that C5aR1 protein was distinctly localized to the apical surface of the ventricular zone (Fig. 1A). The ligand of C5aR1, mC5a, was detected at very low levels in embryonic and adult brain tissue; however, it was found at much higher levels in CSF sampled from E14.5 embryonic ventricles, but not adult mouse CSF (Fig. 1D). Human concordance with these results has recently been reported, with hC5a detected in the CSF of newborn infants (Pataky et al., 2016). Importantly, these embryonic mC5a CSF concentrations equated to ∼1.4 nm, a functionally active concentration for this potent signaling peptide (Hawksworth et al., 2014).
Figure 1.

Localization of C5aR1 and ligands. A, C5aR1 (red) is expressed in the developing neocortex at the apical surface of the ventricular zone at E14.5 (top row). Shown is counterstain with Pax6 (green) and DAPI (blue). Scale bar, 50 μm. Merged images of the ventricular zone at E12.5, E16.5, and E18.5 are shown in the second row. RT-PCR demonstrates C5aR1 expression in embryonic brain tissue, neurosphere, and NE-4C culture. B, C5aR1 expression (red) within sectioned neurosphere. Neg, Secondary-only negative control. Scale bar, 50 μm. C, C5aR1 expression (red) on NE-4C cells grown in monolayer. Neg, Secondary-only negative control. Scale bar, 20 μm. D, Embryonic CSF contains mC5a at significantly greater concentrations than brain tissue or maternal CSF. E, C5aR1 is detected within NE-4C cultures by Western blot at the predicted molecular weight (50 kDa) and decreases at stage VI of differentiation. Stages indicate morphologically distinct progression of NE-4C differentiation (see Materials and Methods). F, Expression of C5aR1 mRNA decreases with differentiation of NE-4C cells. Progenitor marker Sox2 is assayed as a comparison. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
We next demonstrated that C5aR1 expression is maintained ex vivo in neurospheres derived from the telencephalon of E14.5 mice, and the immortalized neural progenitor cell line NE-4C, as determined by RT-PCR analysis (Fig. 1A) and immunocytochemistry (Fig. 1B,C). Interestingly, differentiation of the NE-4C line with retinoic acid, confirmed by downregulation of Sox2, caused a statistically significant reduction in C5aR1 mRNA between stages II and IV (Fig. 1F), a period that corresponds to the beginning of neurogenesis and migration in the cultures (Schlett and Madarász, 1997). No reduction of C5aR1 protein was detected until stage VI, a period that marks the onset of gliogenesis (Fig. 1E). The discordance in timing between mRNA and protein signal loss reflects previous measurements of protein half-life in culture (Schwanhäusser et al., 2011). Overall, this indicates that C5aR1 retains a polarized localization in neural progenitor cells ex vivo and that expression of the receptor reduces as cells differentiate.
C5aR1 is apically localized in human embryonic stem cell-derived neural progenitors
To explore the role of C5aR1 in a human setting, we first assessed whether the mouse apical neural localization of C5aR1 was conserved in human tissue. We have previously reported C5aR1 expression in a similar localization to mouse, with staining for C5aR1 restricted to the cortical ventricular zone of Carnegie stage 13 human neural tissue (Denny et al., 2013). To further assess C5aR1 localization and function in human development, we used hES-derived neuronal progenitors as an in vitro model system. To this end, hES cells were differentiated into cortical neural rosettes, a stage with characteristics analogous to the cortical ventricular zone (Shi et al., 2012; Ziv et al., 2015). The cells in these human neural rosettes expressed the neural markers TUBB3 and NCAD, with strong expression of the tight junction marker ZO-1 on the apical luminal surface of each rosette, indicative of the distinct apicobasal polarity that defines the rosette architecture (Fig. 2A). C5aR1 expression was robustly detected at the apical surface of rosettes, where it colocalized strongly with the apical membrane marker atypical PKCζ but not with markers of cell–cell junctions (NCAD) or cilia (Arl13b), suggesting that the protein is confined to the apical plasma membrane (Fig. 2A). In neural progenitor cells, the apical membrane attachment acts as an anchor for determinants of polarity such as the Par3/Par6/PKCζ complex, which together with apical NCAD-based adherens junctions maintains tissue architecture and apicobasal polarity (Götz and Huttner, 2005). Interestingly, we observed a close correlation between NCAD and C5aR1 expression levels during the neural differentiation of hES cells. Upregulation of NCAD after neural induction peaked at the rosette stage and decreased after further maturation of the cultures. Both C5aR1 and C5 closely followed this pattern of expression (Fig. 2B). C5 protein expression appeared diffuse and punctate throughout the rosettes (Fig. 2A), and the processed form of C5, hC5a, was detectable in the lysate of rosette cells at a similar time point (Fig. 2C). High lysate levels of C5a may reflect intracellular stores of C5a, as has been observed and discussed previously for the complement anaphylatoxins (Pavlovski et al., 2012; Hawksworth et al., 2014; Elvington et al., 2017). These results indicate that human subcellular and temporal C5aR1 expression during development closely mirrors that observed in developing mouse brain.
Figure 2.
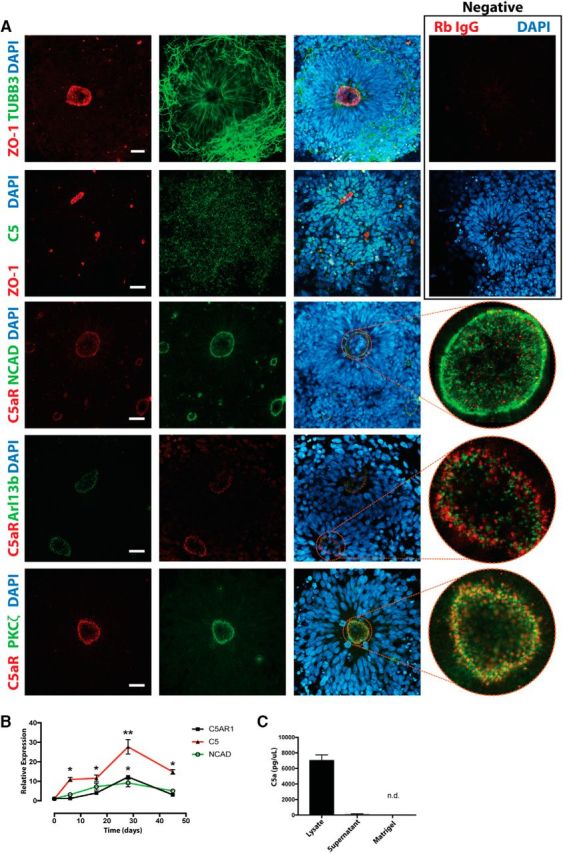
Expression of C5aR1 in human embryonic stem cell-derived rosettes. A, Immunocytochemistry of human neural rosettes showing staining for neural marker TUBB3, tight junction marker ZO-1, and C5. C5aR1 localizes to the apical membrane, colocalizing with PKCζ, but not markers of cilia (Arl13b) or tight junctions (NCAD). Negative controls are shown at top right. Scale bar, 20 μm. B, Transcript expression as hES cells (day 0) are differentiated through the cortical rosette stage (day 28) to a mature neuronal lineage. C5AR1 expression is highest at the rosette stage of neuronal differentiation. C, hC5a is detected through ELISA within the lysate of rosette cultures and is not derived from the exogenous extracellular matrix (Matrigel). *p ≤ 0.05, **p ≤ 0.01.
C5aR1 signals via PKCζ and Erk in mouse and human neural progenitors to promote polarization and proliferation
Intrigued by the unique apical localization of C5aR1 in vivo and in vitro, and the well described importance of the apical membrane and PKCζ in controlling progenitor pool proliferation, we next assessed the functional role of C5aR1 in these processes. In mouse neurosphere cultures, mC5a addition caused an increase in p42/44 (ERK) phosphorylation (Fig. 3A), which could be effectively inhibited through pretreatment with a specific PKCζ pseudosubstrate inhibitor (Fig. 3B). Similarly, in human rosette cultures, treatment with human C5a caused an increase in p42/44 phosphorylation that was attenuated through pretreatment with either a selective C5aR1 antagonist (C5aR1-A, PMX53) or with PKCζ inhibition (Fig. 3C), indicating that C5aR1 signaling is conserved between human and mouse. Given the association between ERK signaling and mitogenic activity, we investigated the ability of C5aR1 signaling to modulate neural progenitor proliferation. Daily addition of species-specific C5a to neurosphere cultures resulted in an increase in both number and diameter of mouse and human neurospheres over a 7 d period (Fig. 3D–G). Additionally, in human neural rosette cultures, the addition of C5a led to a 59% increase in pHH3-positive nuclei, indicative of increased mitotic activity within rosettes (Fig. 3H).
Figure 3.

C5aR1 signals through PKCζ to maintain cell polarity in vitro. A, Mouse neurosphere cultures demonstrate C5a concentration-dependent p42/44 phosphorylation. B, The response to 100 nm C5a is prevented by PKCζ inhibition. C, Human rosette cultures demonstrate time-dependent p42/44 phosphorylation to 10 nm hC5a. The response is prevented through pretreatment with C5aR1-A or PKCζ inhibition. D–G, Mouse and human neurosphere cultures dissociated and grown over a 7 d period demonstrate an increase in number (D, F) and diameter (E, G) in response to C5a. H–K, Treatment of human rosettes with 10 nm C5a. H, C5a increases M-phase-positive cells in neural rosettes as determined by pHH3 immunocytochemical analysis. I, DAPT (gray bars) treatment induced loss of rosettes and a decrease in mRNA of NCAD, C5AR1, and C5 compared with vehicle (black bars) treatment. J, K, Maintenance of rosette architecture after single-cell dissociation (K) or DAPT treatment (J) was promoted by exogenous C5a addition. Adjacent images are representative of DAPT-treated rosettes in the presence or absence of C5a. NCAD (white) and computational outlines (green) of rosette apical lumens are shown. L, M, NE-4C cells grown on transwell membranes demonstrate reorganization of the mitotic spindle (L), as determined by acetylated tubulin staining (green), and reduction in apical surface area (M), outlined by ZO-1 (red) in response to C5a. White arrows are representative distances from mitotic spindle to calculated cell center as shown in L. Scale bar, 20 μm. Veh., Vehicle. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
The observation that C5aR1 signaling is mediated by PKCζ, a component of the Par3/Par6/PKCζ cell polarity complex, combined with its localization at the apical membrane of neural rosettes strongly supports a role for C5aR1 in controlling NPC polarity. We therefore examined the role of C5aR1 in NPC polarity signaling pathways. Previous studies have shown that a loss of paracrine Notch signaling results in downregulation of NCAD and the loss of apicobasal polarity (Main et al., 2013). Treatment of human neural rosettes with the Notch signaling inhibitor DAPT significantly decreased the expression of NCAD, leading to a disruption of cell–cell contacts and inducing a loss of rosette architecture (Fig. 3I). Interestingly, both C5aR1 and C5 expression also decreased with DAPT treatment, which may indicate polarity-dependent expression of these complement factors (Fig. 3I). Single-cell dissociation of rosettes also interferes with paracrine signaling and cell–cell contact, resulting in loss of cell polarity and impaired rosette formation. Exogenous addition of hC5a maintained rosette architecture in the presence of DAPT (Fig. 3J) and promoted reestablishment of rosette architecture after dissociation, an effect that was blocked after C5aR1 antagonism (Fig. 3K). We conclude that C5aR1 is not only dependent on but also actively promotes cell polarity.
This promotion of polarity was also observed in mouse-derived NE-4C cells cultured on transwell plates. The addition of exogenous mC5a led to restriction of the apical surface area by ∼50%, as measured by ZO-1 staining, and microtubule-organizing center localization in these cells was significantly closer to the apical center, indicative of the induction of cell polarization (Fig. 3L,M). Collectively, these data confirmed the polarity-dependent expression of C5aR1, an involvement of C5aR1 signaling in the maintenance of neural progenitor polarity and, subsequently, proliferation.
C5aR1 signaling increases the proliferation of neural progenitor cells of the embryonic ventricular zone in vivo
Having defined a role for C5aR1 in NPC proliferation and establishment of cell polarity in vitro, we next wished to assess its role in mouse brain development by in utero delivery of mC5a or C5aR1-A (PMX53) into the embryonic ventricle of E13.5 mouse embryos (Fig. 4A). We observed that 24 h after a single injection of mC5a, there was a twofold increase in the number of apical progenitors in the M phase of the cell cycle, as indicated by pHH3 staining (Fig. 4B). In contrast, blockade of C5aR1 signaling resulted in a decrease in the number of M-phase apical progenitors after 24 h (Fig. 4B). Analysis of the cleavage plane of actively dividing cells in these samples demonstrated a significant shift from symmetric to asymmetric division after C5aR1-A injection (Fig. 4C). We conclude that C5aR1 signaling promotes NPC proliferation in vivo and affects the balance between symmetric and asymmetric division, in concordance with in vitro observations. Combined with the robust expression of mC5a in embryonic CSF (Fig. 1D), this supports an endogenous physiological role for mC5a–C5aR1 signaling during embryonic neurogenesis.
Figure 4.
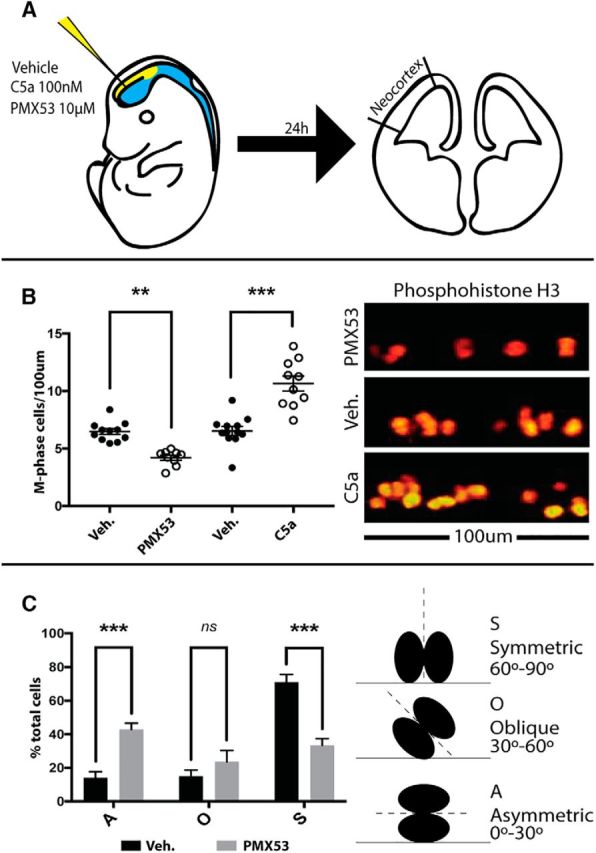
C5aR1 signaling alters neural progenitor division planes and proliferation in vivo. A, Schema of the in utero injection process. Briefly, 1 μl of 100 nm mC5a, 1 μm PMX53, or vehicle was delivered to the embryonic ventricle in utero. After 24 h, brains were processed for immunohistochemistry. M-phase cells, as determined by pHH3 staining, were counted along the ventricular surface of the neocortex. B, In utero injection of mC5a to the embryonic ventricle increases, whereas blockade of C5aR1 signaling using PMX53 decreases, the number of M-phase apical progenitor cells. C, Cleavage plan analysis demonstrates a shift from symmetric division toward asymmetric division after treatment with C5aR1 antagonist. S, Symmetric division; A, asymmetric division; O, oblique division; Veh., vehicle. **p ≤ 0.01, ***p ≤ 0.001.
Blockade of C5aR1 from E12.5 to E14.5 results in behavioral abnormalities in the adult mouse
Given that acute disruption of C5a–C5aR1 signaling affects proliferation of apical progenitors and the balance between symmetric and asymmetric cell division, we next determined whether acute pharmacological blockade of C5aR1 during neurogenesis translates into behavioral abnormalities later in life. Time-mated dams were intraperitoneally injected daily with 1 mg/kg C5aR1-A during the critical neurodevelopmental window (E12.5–E14.5), and resultant litters were subjected to behavioral testing between 6 and 8 weeks of age (Fig. 5A). We first confirmed that intraperitoneal delivery of C5aR1-A over the 3 d embryonic window resulted in impaired neurogenesis. Compared with vehicle controls, C5aR1-A-treated embryos displayed a reduction in the size of the ventricular zone, as measured by Sox2 staining, and an increase in the thickness of the maturing cortex, as demonstrated by the postmitotic maturing neuron marker DCX at E16.5. This collectively resulted in a significant decrease of the Sox2/DCX ratio (Fig. 5B). We next assessed whether C5aR1-A treatment resulted in any gross developmental or pregnancy complications. C5aR1-A-treated pups showed no change in postnatal growth (Fig. 5C), litter size (Fig. 5D), and snout-occiput length (Fig. 5F); however, a minor, but significant, reduction in crown-rump length was observed (Fig. 5E).
Figure 5.
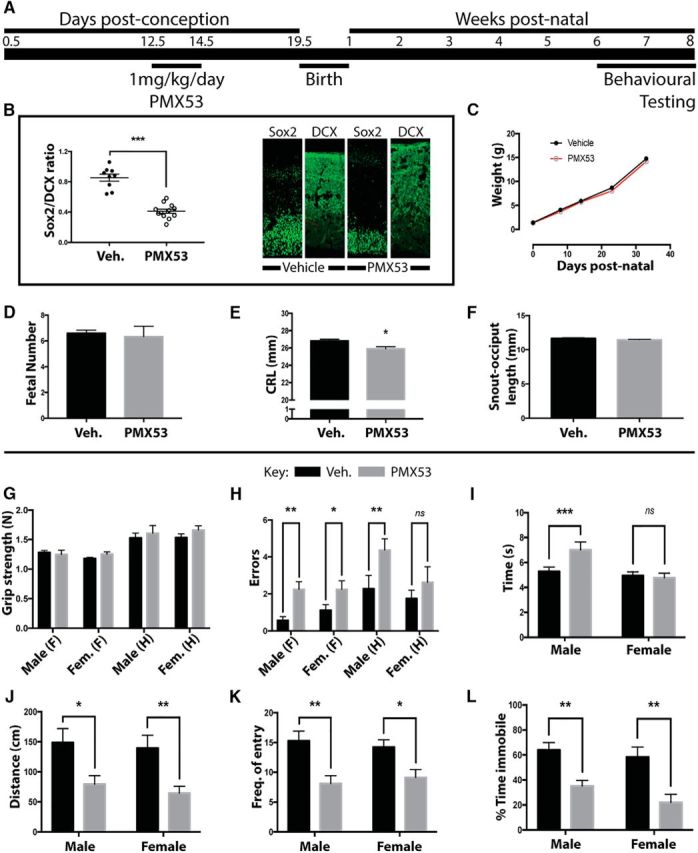
Blockade of C5aR1 signaling at E12.5–E14.5 causes behavioral changes in adult mice. A, Schema of the experimental process. Briefly, 1 mg · kg−1 · d−1 of the C5aR1 antagonist PMX53 was delivered by intraperitoneal injection to pregnant dams at E12.5–E14.5. Resultant litters were taken through behavioral testing from 6 to 8 weeks and killed, and brains were prepared for ex vivo MRI. Data show results for vehicle (V, black bars)- and PMX53 (P, gray bars)-treated mice. B, Sox2/DCX ratio of the ventricular zone of E16.5 embryos. C, D, F, No change in postnatal growth (C), litter size (D), and snout-occiput length (F) was seen. E, A significant reduction in crown-rump length was observed. G, No difference between treatment groups was found in grip strength for both forelimb (F) and hindlimb (H). H, Time to cross balance beam was increased in male animals from PMX53-treated litters. I, Footfall errors crossing balance beam. J–L, Distance moved in center of open-field arena (J), frequency of entry to novel arm Y-maze (K), and time spent immobile during the forced swim test (L) were significantly different in PMX53-treated litters. Veh., Vehicle; Fem., female. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Adult mice were next subjected to a series of behavioral tests to assess neuromotor and cognitive function. In utero C5aR1-A-treated mice demonstrated significant behavioral abnormalities in adulthood. In motor control tasks, there was no difference in grip strength (Fig. 5G); however balance beam testing of centrally controlled motor coordination tasks showed an increase in both footfall errors and time taken to cross the beam for C5aR1-A-treated mice (Fig. 5H,I). In an open-field test, C5aR1-A-treated animals of both genders demonstrated a decrease in distance traveled in the center areas of the cage (Fig. 5J), suggesting heightened anxiety in a novel environment. The Y-maze was used to assess short-term memory, and frequency of entry into the novel arm was reduced in C5aR1-A-treated animals (Fig. 5K). Finally, the forced swim test was used to assess depressive-like symptoms, where C5aR1-A-treated animals spent significantly reduced time being immobile compared with nontreated animals (Fig. 5L). These results clearly demonstrated that acute in utero blockade of C5aR1 signaling over a relatively short time frame led to a range of behavioral discrepancies that involved several anatomically distinct systems such as memory, coordination, and anxiety.
In utero blockade of C5aR1 between E12.5 and E14.5 results in microstructural differences on MRI analysis
Given the broad behavioral deficits induced by transient in utero C5aR1 antagonism between E12.5 and E14.5 of gestation, we next used ex vivo 16.4T MRI to identify structural alterations in the brain that could underlie the observed phenotypes. Firstly, a Jacobian map was used to measure the relative change of each brain structure required to fit to a template image, therefore identifying any volumetric change in distinct regions between sample groups (Leporé et al., 2007). C5aR1-A-treated animals showed significantly increased clusters of Jacobian values throughout the cortex and striatum (Fig. 6), indicating that spatial expansion was required to fit individual images to the template in these areas (Lepore et al., 2008). However, volumetric analysis of segmented brain regions (Ma et al., 2005) failed to show significant differences between groups (Table 2). These areas of volume difference correlated with microstructural difference as shown by fractional anisotropy (FA). FA values were higher in the frontal cortex, striatum, and hypothalamus of C5aR1-A-treated animals (Fig. 6), alluding to increased myelination, increased axonal density, or a reduction in fiber dispersion in these areas (Sepehrband et al., 2015), which may account for the behavioral differences seen between these two experimental groups (Soares et al., 2013). Overall, these results demonstrate multiple microstructural differences induced by C5aR1 inhibition in embryos, which are concordant with the behavioral deficits seen in these mice.
Figure 6.

Blockade of C5aR1 signaling at E12.5–E14.5 results in microstructural changes in adult brains. Top row, Significance map of Jacobian warping projected onto T1-weighted template average. Middle row, Significance map of FA values projected onto template average. For both maps: blue/purple, vehicle>PMX53 (C5aR1-A) treatment; red/yellow, PMX53>vehicle. Color-coded p values are shown at the bottom. Bottom row, Anatomical areas are labeled. Mot., Motor cortex; Orb., orbital cortex; OB, olfactory bulb; SS, somatosensory cortex; Gust., gustatory cortex; Pir., piriformis; CP, caudate/putamen; NA, nucleus accumbens; P., pallidum; H., hypothalamus; Veh, vehicle.
Table 2.
MRI volume comparison of brain regions for C5aR1-antagonist or vehicle-treated mice
| Area | Vehicle treatment |
C5aR1-A treatment |
p value | ||||
|---|---|---|---|---|---|---|---|
| Volume (mm3) | SE | n | Volume (mm3) | SE | n | ||
| Amygdala | 10.24 | 0.1425 | 7 | 10.18 | 0.2929 | 9 | 0.8764 |
| Caudate/putamen | 23.25 | 0.4326 | 7 | 22.85 | 0.5933 | 9 | 0.6175 |
| Corpus callosum and external capsule | 8.213 | 0.102 | 7 | 7.891 | 0.331 | 9 | 0.4203 |
| Central gray matter | 4.476 | 0.05 | 7 | 4.339 | 0.07 | 9 | 0.1762 |
| Cerebellum | 46.96 | 0.3563 | 7 | 42.83 | 1.764 | 9 | 0.0624 |
| Fimbria | 2.007 | 0.028 | 7 | 1.928 | 0.05576 | 9 | 0.2702 |
| Globus pallidus | 2.485 | 0.055 | 7 | 2.368 | 0.046 | 9 | 0.1209 |
| Hippocampus | 24.71 | 0.287 | 7 | 24.31 | 0.5846 | 9 | 0.5832 |
| Hypothalamus | 11.61 | 0.162 | 7 | 11.17 | 0.284 | 9 | 0.234 |
| Neocortex | 125.5 | 1.84 | 7 | 121.5 | 3.45 | 9 | 0.3668 |
| Olfactory bulbs | 17.88 | 0.2216 | 6 | 16.53 | 0.7673 | 9 | 0.1858 |
| Thalamus | 25.19 | 0.095 | 7 | 24.69 | 0.732 | 8 | 0.5444 |
Discussion
Proteins of the complement system are present during embryogenesis, playing novel roles in development (McLin et al., 2008; Denny et al., 2013; Jeanes et al., 2015). Our laboratory has previously demonstrated their presence and function on human embryonic stem cells and mouse neuroepithelial cells (Denny et al., 2013; Hawksworth et al., 2014). Additionally, we have previously shown C5aR1 expression on the apical neuroepithelium at a similar developmental stage to this study, in nonpathological human embryos (Denny et al., 2013), raising the question of roles for complement in normal embryonic development. Here we show that in both human and mouse models, the key complement effector system, C5a–C5aR1 signaling, functions to control progenitor cell polarity, proliferation, and the symmetry of the cell division. Ultimately, this loss of C5aR1 signaling manifests as altered cerebral organization and behavioral deficits.
In neural progenitor cells of the ventricular zone, the loss of apical attachment where C5aR1 resides is associated with decreased expression of factors responsible for the maintenance of stemness. Asymmetric inheritance of this attachment during mitosis is a catalyst for differentiation toward a postmitotic state, with loss of attachment initiating exit from the ventricular zone pool, and subsequent maturation into neuronal subtypes (Götz and Huttner, 2005; Miyamoto et al., 2015). Conversely, symmetric division of the apical membrane maintains both daughter cells within the ventricular zone progenitor pool, secondary to continued signaling from the apical membrane. The apically localized PKCζ is an essential second messenger in the promotion of symmetric division and the maintenance of neuroepithelial architecture (Ghosh et al., 2008). However, less is known about the receptors that trigger PKCζ activation. Here we have identified C5aR1 as a novel prime candidate, with polarized apical expression, for controlling endogenous PKCζ signaling during mammalian corticogenesis.
In addition, we have shown a plausible biological source for C5aR1 stimulation within the CSF of the developing embryo, which were at higher concentrations than both embryonic brain tissue and adult CSF. This may suggest that C5a is actively secreted into the CSF to stimulate proliferation of progenitor cells, given that concentrations are higher during development than in adulthood. The source of this C5a will require further delineation; however, we demonstrated C5 expression within neural rosette cultures (Fig. 2A). As the CSF was assayed before the advent of ependymal cell differentiation, this strongly suggests an autocrine production of C5a by neural progenitor cells.
These findings are particularly of interest in light of previous reports demonstrating that, in adult mice, C5aR1 does not contribute to basal neurogenesis (Bogestål et al., 2007). This appears to contrast with our study, which identifies a role for C5aR1 in neurogenesis, albeit at an earlier stage of life than investigated by Bogestål et al. (2007). However, the receptor has also been demonstrated on migrating neuroblasts in models of cerebral ischemia (Rahpeymai et al., 2006), leading to some question of its role on these cells, given an apparent noncontributory role to neurogenesis. Although not directly tested, it could be argued that C5aR1 is responsive on neuroblasts to the higher C5a concentrations in the ischemic brain but has a dormant role in the nonpathological and, therefore, low-C5a setting.
The present discovery adds to previous studies that have identified neurodevelopmental deficits associated with aberrant complement activity, including in disorders such as autism, schizophrenia, and epilepsy (Hawksworth et al., 2017). For example, the fetal neurocognitive injury associated with maternal malaria infection has been shown to be mediated by C5aR1 (McDonald et al., 2015). Furthermore, allele variations leading to increased complement factor 4A (C4A) expression have been correlated to increased schizophrenia risk (Sekar et al., 2016). It is interesting to speculate, given our findings, on whether the behavioral deficits demonstrated in both diseases are the result of a direct effect of C5aR1 signaling on neural progenitors, altering corticogenesis, rather than the alternate hypothesis of a generalized inflammation and altered synaptic pruning. Within this hypothesis, classical complement cascade activation would inescapably lead to the formation of C5 convertases, and activation of C5. Given our identification of a mechanistic role for C5aR1 in ventricular zone progenitors and cerebral organization, it could equally be hypothesized that the increased C4A and classical complement cascade activation increases cerebral C5aR1 signaling, with consequent alterations in progenitor migration driving the complex cortical pathology associated with schizophrenia risk. Additionally, it is interesting that the short window of C5aR1 blockade used in our studies was also not compensated for later in development, as demonstrated by the behavioral and brain microstructural differences in adult mice. In contrast, there is a demonstrated functional compensation of impaired synaptic pruning resulting from other models of complement deficiency (Perez-Alcazar et al., 2014). This theory is supported by the finding of impaired short-term memory in C5aR1 knock-out animals (Gong et al., 2013), indicating that disruption of the receptor signaling, but not the cascade, is enough to alter neuronal circuitry.
Given the potential disparity between animal and human cognitive development, one focus of this study was to validate observations made in the mouse model in a human environment. We have previously shown expression of C5aR1 in human embryos, at Carnegie stage 13, where it is also localized to the apical neuroepithelium (Denny et al., 2013). The combination of this in vivo localization with the results presented in this study, demonstrating human neural progenitors signaling through C5aR1 in a similar manner to the mouse, strongly suggests conservation of C5aR1 function between mouse and human. C5aR1 is already a strong candidate target for direct therapeutics against inflammatory diseases of pregnancy. Maternal complement dysregulation is a factor in the pathogenesis of preeclampsia and infection-related preterm birth (Lokki et al., 2014; Denny et al., 2015). Interestingly, the humanized IgG2/4 monoclonal antibody directed against C5, eculizimab, has already been in use in pregnant women affected by paroxysmal nocturnal hemoglobinuria. Given the rarity of the disease, there have only been a few reports on the safety of this drug in pregnancy, with no obvious complications at birth or in early childhood development (Kelly et al., 2015). The effect of eculizimab is fortuitously confined to the maternal circulation by the poor transfer of IgG2 through the placenta, and comparison of maternal and cord blood from these pregnancies has shown no effect on the complement system of the fetus (Hallstensen et al., 2015). However, with the clinical development of small-molecule C5aR1 inhibitors, such as CCX-168 (Woodruff et al., 2011), it would be prudent to be cautious in the clinical use of C5aR1-directed therapeutics during pregnancy without prior consideration to fetal transfer and potential developmental implications of C5aR1 inhibition as highlighted in this study.
This study also adds to emerging work demonstrating a wide developmental role for complement components in mammalian development. These include roles for complement factors in developmental processes such as radial intercalation, migration, and synaptic pruning (Stevens et al., 2007; Carmona-Fontaine et al., 2011; Szabó et al., 2016; Gorelik et al., 2017). In this context, it is interesting to speculate what niche these proteins first filled. The origins of the evolutionarily ancient system of complement proteins may be as a controller of tissue organization and development, with utilization of complement in the context of innate immunity following later in evolution (Hawksworth et al., 2017).
In conclusion, here we show a novel role for C5aR1 as a modulator of apicobasal polarity in neural progenitor cells that is highly conserved between mice and humans. Inhibition of C5aR1 signaling during neurogenesis has deleterious consequences for cerebral organization, resulting in behavioral abnormalities in adult mice. Our data suggest that the development and use of C5aR1 antagonists as potential treatments for pregnancy-related inflammatory disease should be approached with extreme caution.
Footnotes
This work was supported by ARC Centre of Excellence “Stem Cells Australia” Grant SR110001002 (E.W.), National Health and Medical Research Council (NHMRC) Project Grant APP1082271 (T.M.W.), and NHMRC Career Development Fellowship APP1105420 (T.M.W.).
The authors declare no competing financial interests.
References
- Bénard M, Raoult E, Vaudry D, Leprince J, Falluel-Morel A, Gonzalez BJ, Galas L, Vaudry H, Fontaine M (2008) Role of complement anaphylatoxin receptors (C3aR, C5aR) in the development of the rat cerebellum. Mol Immunol 45:3767–3774. 10.1016/j.molimm.2008.05.027 [DOI] [PubMed] [Google Scholar]
- Bogestål YR, Barnum SR, Smith PL, Mattisson V, Pekny M, Pekna M (2007) Signaling through C5aR is not involved in basal neurogenesis. J Neurosci Res 85:2892–2897. 10.1002/jnr.21401 [DOI] [PubMed] [Google Scholar]
- Briggs JA, Sun J, Shepherd J, Ovchinnikov DA, Chung TL, Nayler SP, Kao LP, Morrow CA, Thakar NY, Soo SY, Peura T, Grimmond S, Wolvetang EJ (2013) Integration-free induced pluripotent stem cells model genetic and neural developmental features of Down syndrome etiology. Stem Cells 31:467–478. 10.1002/stem.1297 [DOI] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Theveneau E, Tzekou A, Tada M, Woods M, Page KM, Parsons M, Lambris JD, Mayor R (2011) Complement fragment C3a controls mutual cell attraction during collective cell migration. Dev Cell 21:1026–1037. 10.1016/j.devcel.2011.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280. 10.1038/nbt.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PA, Symms M, Boulby PA (2007) Optimal acquisition orders of diffusion-weighted MRI measurements. J Magn Reson Imaging 25:1051–1058. 10.1002/jmri.20905 [DOI] [PubMed] [Google Scholar]
- Corbett BA, Kantor AB, Schulman H, Walker WL, Lit L, Ashwood P, Rocke DM, Sharp FR (2007) A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol Psychiatry 12:292–306. 10.1038/sj.mp.4001943 [DOI] [PubMed] [Google Scholar]
- Denny KJ, Coulthard LG, Jeanes A, Lisgo S, Simmons DG, Callaway LK, Wlodarczyk B, Finnell RH, Woodruff TM, Taylor SM (2013) C5a receptor signaling prevents folate deficiency-induced neural tube defects in mice. J Immunol 190:3493–3499. 10.4049/jimmunol.1203072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny KJ, Coulthard LG, Mantovani S, Simmons D, Taylor SM, Woodruff TM (2015) The role of C5a receptor signaling in endotoxin-induced miscarriage and preterm birth. Am J Reprod Immunol 74:148–155. 10.1111/aji.12386 [DOI] [PubMed] [Google Scholar]
- Elvington M, Liszewski MK, Bertram P, Kulkarni HS, Atkinson JP (2017) A C3(H20) recycling pathway is a component of the intracellular complement system. J Clin Invest 127:970–981. 10.1172/JCI89412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fietz SA, Huttner WB (2011) Cortical progenitor expansion, self-renewal and neurogenesis–a polarized perspective. Curr Opin Neurobiol 21:23–35. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Marquardt T, Thaler JP, Carter N, Andrews SE, Pfaff SL, Hunter T (2008) Instructive role of aPKCzeta subcellular localization in the assembly of adherens junctions in neural progenitors. Proc Natl Acad Sci U S A 105:335–340. 10.1073/pnas.0705713105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Pan Y, Zhao W, Knable L, Vempati P, Begum S, Ho L, Wang J, Yemul S, Barnum S, Bilski A, Gong BY, Pasinetti GM (2013) IVIG immunotherapy protects against synaptic dysfunction in Alzheimer's disease through complement anaphylatoxin C5a-mediated AMPA-CREB-C/EBP signaling pathway. Mol Immunol 56:619–629. 10.1016/j.molimm.2013.06.016 [DOI] [PubMed] [Google Scholar]
- Gorelik A, Sapir T, Haffner-Krausz R, Olender T, Woodruff TM, Reiner O (2017) Developmental activities of the complement pathway in migrating neurons. Nat Commun, 8:15096 10.1038/ncomms15096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz M, Huttner WB (2005) The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6:777–788. 10.1038/nrm1739 [DOI] [PubMed] [Google Scholar]
- Hallstensen RF, Bergseth G, Foss S, Jæger S, Gedde-Dahl T, Holt J, Christiansen D, Lau C, Brekke O-L, Armstrong E, Stefanovic V, Andersen JT, Sandlie I, Mollnes TE (2015) Eculizumab treatment during pregnancy does not affect the complement system activity of the newborn. Immunobiology 220:452–459. 10.1016/j.imbio.2014.11.003 [DOI] [PubMed] [Google Scholar]
- Hawksworth OA, Coulthard LG, Taylor SM, Wolvetang EJ, Woodruff TM (2014) Brief report: complement C5a promotes human embryonic stem cell pluripotency in the absence of FGF2. Stem Cells 32:3278–3284. 10.1002/stem.1801 [DOI] [PubMed] [Google Scholar]
- Hawksworth OA, Coulthard LG, Woodruff TM (2017) Complement in the fundamental processes of the cell. Mol Immunol 84:17–25. 10.1016/j.molimm.2016.11.010 [DOI] [PubMed] [Google Scholar]
- Jeanes A, Coulthard LG, Mantovani S, Markham K, Woodruff TM (2015) Co-ordinated expression of innate immune molecules during mouse neurulation. Mol Immunol 68:253–260. 10.1016/j.molimm.2015.09.004 [DOI] [PubMed] [Google Scholar]
- Jones DK, Horsfield MA, Simmons A (1999) Optimal strategies for measuring diffusion in anisotropic systems by magnetic resonance imaging. Magn Reson Med 42:515–525. [DOI] [PubMed] [Google Scholar]
- Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE (2011) Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27:1179–1180. 10.1093/bioinformatics/btr095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RJ, Höchsmann B, Szer J, Kulasekararaj A, de Guibert S, Röth A, Weitz IC, Armstrong E, Risitano AM, Patriquin CJ, Terriou L, Muus P, Hill A, Turner MP, Schrezenmeier H, Peffault de Latour R (2015) Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 373:1032–1039. [DOI] [PubMed] [Google Scholar]
- Leporé N, Brun C, Pennec X, Chou Y-Y, Lopez OL, Aizenstein HJ, Becker JT, Toga AW, Thompson PM (2007) Mean template for tensor-based morphometry using deformation tensors. In: Medical image computing and computer-assisted intervention MICCAI, Lecture notes in computer science, Vol 4792, pp 826–833. Berlin: Springer. [DOI] [PubMed] [Google Scholar]
- Lepore N, Brun C, Chou YY, Chiang MC, Dutton RA, Hayashi KM, Luders E, Lopez OL, Aizenstein HJ, Toga AW, Becker JT, Thompson PM (2008) Generalized tensor-based morphometry of HIV/AIDS using multivariate statistics on deformation tensors. IEEE Trans Med Imaging 27:129–141. 10.1109/TMI.2007.906091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokki AI, Heikkinen-Eloranta J, Jarva H, Saisto T, Lokki ML, Laivuori H, Meri S (2014) Complement activation and regulation in preeclamptic placenta. Front Immunol 5:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hof PR, Grant SC, Blackband SJ, Bennett R, Slatest L, McGuigan MD, Benveniste H (2005) A three-dimensional digital atlas database of the adult C57BL/6J mouse brain by magnetic resonance microscopy. Neuroscience 135:1203–1215. 10.1016/j.neuroscience.2005.07.014 [DOI] [PubMed] [Google Scholar]
- Main H, Radenkovic J, Jin SB, Lendahl U, Andersson ER (2013) Notch signaling maintains neural rosette polarity. PLoS One 8:e62959. 10.1371/journal.pone.0062959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald CR, Cahill LS, Ho KT, Yang J, Kim H, Silver KL, Ward PA, Mount HT, Liles WC, Sled JG, Kain KC (2015) Experimental malaria in pregnancy induces neurocognitive injury in uninfected offspring via a C5a-C5a receptor dependent pathway. PLoS Pathog 11:e1005140. 10.1371/journal.ppat.1005140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLin VA, Hu CH, Shah R, Jamrich M (2008) Expression of complement components coincides with early patterning and organogenesis in Xenopus laevis. Int J Dev Biol 52:1123–1133. 10.1387/ijdb.072465v [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Sakane F, Hashimoto K (2015) N-cadherin-based adherens junction regulates the maintenance, proliferation, and differentiation of neural progenitor cells during development. Cell Adh Migr 9:183–192. 10.1080/19336918.2015.1005466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pataky R, Howie FA, Girardi G, Boardman JP (2016) Complement C5a is present in CSF of human newborns and is elevated in association with preterm birth. J Matern Fetal Neonatal Med 29:1–4. 10.1080/14767058.2016.1251896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovski D, Thundyil J, Monk PN, Wetsel RA, Taylor SM, Woodruff TM (2012) Generation of complement component C5a by ischemic neurons promotes neuronal apoptosis. FASEB J 26:3680–3690. 10.1096/fj.11-202382 [DOI] [PubMed] [Google Scholar]
- Perez-Alcazar M, Daborg J, Stokowska A, Wasling P, Björefeldt A, Kalm M, Zetterberg H, Carlström KE, Blomgren K, Ekdahl CT, Hanse E, Pekna M (2014) Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp Neurol 253:154–164. 10.1016/j.expneurol.2013.12.013 [DOI] [PubMed] [Google Scholar]
- Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, Zwirner J, Wetsel RA, Gerard C, Pekny M, Pekna M (2006) Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J 25:1364–1374. 10.1038/sj.emboj.7601004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooryck C, Diaz-Font A, Osborn DP, Chabchoub E, Hernandez-Hernandez V, Shamseldin H, Kenny J, Waters A, Jenkins D, Kaissi AA, Leal GF, Dallapiccola B, Carnevale F, Bitner-Glindzicz M, Lees M, Hennekam R, Stanier P, Burns AJ, Peeters H, Alkuraya FS, et al. (2011) Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat Genet 43:197–203. 10.1038/ng.757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlett K, Madarász E (1997) Retinoic acid induced neural differentiation in a neuroectodermal cell line immortalized by p53 deficiency. J Neurosci Res 47:405–415. [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473:337–342. 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, Genovese G, Rose SA, Handsaker RE, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Daly MJ, Carroll MC, Stevens B, McCarroll SA (2016) Schizophrenia risk from complex variation of complement component 4. Nature 530:177–183. 10.1038/nature16549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepehrband F, Clark KA, Ullmann JF, Kurniawan ND, Leanage G, Reutens DC, Yang Z (2015) Brain tissue compartment density estimated using diffusion-weighted MRI yields tissue parameters consistent with histology. Hum Brain Mapp 36:3687–3702. 10.1002/hbm.22872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ (2012) Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci 15:477–486. 10.1038/nn.3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares JM, Marques P, Alves V (2013) A hitchhiker's guide to diffusion tensor imaging. Front Neurosci 12:7–31. 10.3389/fnins.2013.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178. 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Szabó A, Cobo I, Omara S, McLachlan S, Keller R, Mayor R (2016) The molecular basis of radial intercalation during tissue spreading in early development. Dev Cell 37:213–225. 10.1016/j.devcel.2016.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff TM, Nandakumar KS, Tedesco F (2011) Inhibiting the C5–C5a receptor axis. Mol Immunol 48:1631–1642. 10.1016/j.molimm.2011.04.014 [DOI] [PubMed] [Google Scholar]
- Ziv O, Zaritsky A, Yaffe Y, Mutukula N, Edri R, Elkabetz Y (2015) Quantitative live imaging of human embryonic stem cell derived neural rosettes reveals structure-function dynamics coupled to cortical development. PLoS Comp Biol 11:e1004453 10.1371/journal.pcbi.1004453 [DOI] [PMC free article] [PubMed] [Google Scholar]