

Abstract
Alzheimer's disease (AD) is characterized by progressive cognitive decline, increasingly attributed to neuronal dysfunction induced by amyloid-β oligomers (AβOs). Although the impact of AβOs on neurons has been extensively studied, only recently have the possible effects of AβOs on astrocytes begun to be investigated. Given the key roles of astrocytes in synapse formation, plasticity, and function, we sought to investigate the impact of AβOs on astrocytes, and to determine whether this impact is related to the deleterious actions of AβOs on synapses. We found that AβOs interact with astrocytes, cause astrocyte activation and trigger abnormal generation of reactive oxygen species, which is accompanied by impairment of astrocyte neuroprotective potential in vitro. We further show that both murine and human astrocyte conditioned media (CM) increase synapse density, reduce AβOs binding, and prevent AβO-induced synapse loss in cultured hippocampal neurons. Both a neutralizing anti-transforming growth factor-β1 (TGF-β1) antibody and siRNA-mediated knockdown of TGF-β1, previously identified as an important synaptogenic factor secreted by astrocytes, abrogated the protective action of astrocyte CM against AβO-induced synapse loss. Notably, TGF-β1 prevented hippocampal dendritic spine loss and memory impairment in mice that received an intracerebroventricular infusion of AβOs. Results suggest that astrocyte-derived TGF-β1 is part of an endogenous mechanism that protects synapses against AβOs. By demonstrating that AβOs decrease astrocyte ability to protect synapses, our results unravel a new mechanism underlying the synaptotoxic action of AβOs in AD.
SIGNIFICANCE STATEMENT Alzheimer's disease is characterized by progressive cognitive decline, mainly attributed to synaptotoxicity of the amyloid-β oligomers (AβOs). Here, we investigated the impact of AβOs in astrocytes, a less known subject. We show that astrocytes prevent synapse loss induced by AβOs, via production of transforming growth factor-β1 (TGF-β1). We found that AβOs trigger morphological and functional alterations in astrocytes, and impair their neuroprotective potential. Notably, TGF-β1 reduced hippocampal dendritic spine loss and memory impairment in mice that received intracerebroventricular infusions of AβOs. Our results describe a new mechanism underlying the toxicity of AβOs and indicate novel therapeutic targets for Alzheimer's disease, mainly focused on TGF-β1 and astrocytes.
Keywords: Alzheimer's disease, astrocyte, synapse loss, TGF-β1
Introduction
Alzheimer's disease (AD) is the most common form of dementia in the elderly, accounting for 50–80% of cases (Abbott, 2011), and is characterized by progressive decline in cognitive functions. The pathogenesis of AD involves initial synapse/neuronal dysfunction, followed by extensive neurodegeneration in the hippocampus and other brain regions (Palop and Mucke, 2010).
Soluble oligomers of the amyloid-β peptide (AβOs) accumulate in AD brains (Gong et al., 2003) and are increasingly considered major toxins leading to neuronal dysfunction in AD (Ferreira and Klein, 2011; Mucke and Selkoe, 2012; Hong et al., 2016). Among other deleterious actions, AβOs impair axonal transport, increase production of neuronal reactive oxygen species, and induce tau phosphorylation and mitochondrial damage, contributing to neuronal degeneration and death (De Felice et al., 2007, 2008; Alberdi et al., 2010; Decker et al., 2010). AβOs further induce synapse dysfunction and loss of dendritic spines (Lacor et al., 2004; Shankar et al., 2007), thus impairing synaptic plasticity (Lambert et al., 1998).
Emerging evidence indicates that astrocytes play important roles in synapse formation, maintenance and plasticity during brain development and adulthood (Araque et al., 1999; Eroglu and Barres, 2010; Han et al., 2013; Diniz et al., 2014a). Some of the molecules released by astrocytes that mediate these functions have been identified, and include cholesterol (Mauch et al., 2001), thrombospondin 1 (TSP-1; Christopherson et al., 2005), glypican (Allen et al., 2012), and hevin (Kucukdereli et al., 2011). On the other hand, there is limited information concerning the roles of astrocytes in synapse maintenance and function in neurodegenerative disorders, including AD.
Defective astroglial function and reactivity has been proposed to contribute to brain dysfunction in aging and in neurodegenerative diseases (Rodríguez-Arellano et al., 2016). Astroglial reactivity has been frequently found in close association with senile plaques and damaged neurons in postmortem analysis of AD brains (Beach and McGeer, 1988; Hippius and Neundörfer, 2003). Consistent with a role of astroglial cells in AD, astrocyte activation has been reported to be triggered by infusion of AβOs in the brains of mice (Ledo et al., 2013) and monkeys (Forny-Germano et al., 2014), as well as in the brains of transgenic mouse models of AD (Furman et al., 2012). In addition, a number of studies have shown that different aggregated forms of Aβ are internalized by astrocytes in vitro and in vivo (Matsunaga et al., 2003; Nagele et al., 2003; Wyss-Coray et al., 2003; Alarcón et al., 2005). Although those findings suggest that astrocytes are targets for AβOs, the impact of astrocyte dysfunction on the pathogenesis of AD has been subject of controversy.
We have shown that astrocytes control the balance between excitatory and inhibitory synapses in the cerebral cortex through secretion of transforming growth factor β 1 (TGF-β1) (Diniz et al., 2012, 2014b). TGF-β1 levels are reduced in the plasma of AD patients, which might contribute to neuronal death and exacerbation of neuroinflammation (Mocali et al., 2004; Juraskova et al., 2010). Additionally, deficits in TGF-β pathways have been reported in AD brains, in the brains of aged mice, and in mouse models of AD (Lee et al., 2006; Tesseur et al., 2006; Ueberham et al., 2006; Chalmers and Love, 2007; Tichauer et al., 2014; Caraci et al., 2015).
Given the increasing recognition of the roles played by astrocytes in synapse physiology, we hypothesized that synapse loss in AD might be, at least in part, a consequence of the impact of AβOs on astrocytes. We found that AβOs directly target astrocytes and impair their neuroprotective actions. Using purified human and murine astrocyte cultures, and mice that received an intracerebroventricular infusion of AβOs, we further demonstrated that astrocyte-derived TGF-β1 protects synapses and prevents memory deficits induced by AβOs. Results indicate that AβOs impact on astroglial cells renders synapses vulnerable to degeneration. Our work identifies a novel mechanism underlying cognitive impairment in AD, and provides insight into the role of glial cells in synapse pathology.
Materials and Methods
Animals.
Embryonic (E15–E16), newborn (P0–P2), and 3-month-old male Swiss mice were used. All animal-use protocols were approved by the Animal Use Ethics Committee of the Federal University of Rio de Janeiro (protocol no. 004/2016).
Preparation and characterization of AβOs.
AβOs were prepared weekly from synthetic Aβ1–42 (American Peptide Company) and were routinely characterized by size-exclusion chromatography and occasionally by Western immunoblots, as previously described (De Felice et al., 2007, 2008; Sebollela et al., 2012) AβOs ranged from dimers (∼9 kDa) to higher molecular weight oligomers (∼50–100 kDa). Oligomer preparations were aliquoted and kept at −70°C, and aliquots were defrosted at the time of use.
Murine astrocyte cultures.
Primary astrocyte cultures were prepared from neonatal Swiss mice (1- to 2-d-old) as previously described (Diniz et al., 2014b). Hippocampus were removed, stripped of meninges, and placed in DMEM/F12 (DMEM and nutrient mixture F12, Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen). Cultures were incubated at 37°C in a humidified 5% CO2, 95% air chamber for 7–10 d until confluence. After confluence, cells were subjected to passages to generate pure astrocytic cultures. This protocol yields an astrocyte-enriched culture consisting of >98% glial fibrillary acidic protein (GFAP)-positive cells.
Human astrocyte cultures.
Adult primary human astrocytes were isolated from surgically removed anterior temporal lobe tissue, from patients selected for surgical treatment of temporal-lobe epilepsy associated with hippocampal sclerosis (TLE-HS), as previously described (Diniz et al., 2012). All patients gave written consent to the study, and the procedures were reviewed and approved by the Brazilian Ministry of Health Ethics Committee. Astrocytes were grown in DMEM/F12 medium supplemented with 10% FCS, in a humidified 5% CO2, 95% air. New passages of cells were generated by harvesting confluent astrocyte cultures using trypsin-EDTA solution (0.25% trypsin with EDTA, Invitrogen). Human astrocytes up to the third passage were used in the study. Human astrocytes expressed the typical astrocyte markers GLAST (glutamate-aspartate transporter) in a typical punctate distribution pattern in their membranes, and human leukocyte antigen in a spread distribution pattern over the monolayer, attesting their human and astrocytic origin (Diniz et al., 2012).
Astrocyte conditioned medium.
To obtain conditioned medium (CM) from AβO-exposed astrocytes (CM AβΟ), confluent astrocyte cultures (murine or human) were exposed to 500 nm AβOs or vehicle for 24 h (for CMAβ or CM, respectively). Cells were washed three times to eliminate AβOs and fresh serum-free DMEM/F12 medium was added. Medium was collected after 24 h and centrifuged at 1000 × g for 10 min to remove cellular debris. To guarantee that residual AβOs were not present in the CM, a dot immunoblot assay was performed using the NU4 anti-AβO antibody (Lambert et al., 2007). No residual AβO immunoreactivity was found under these conditions (data not shown).
TGF-β1 depletion from astrocyte conditioned medium.
For depletion of TGF-β1, CM was incubated with 1 μg/ml neutralizing antibody against TGF-β1 (Abcam) for 30 min at room temperature. According to information from the manufacturer, this concentration of antibody neutralizes 50% of the bioactivity of 0.25 ng/ml TGF-β1 in the HT 2 cell line. Neuronal cultures were maintained simultaneously in the presence of CM and the neutralizing antibody for an additional 3 h, followed by fixation and immunostaining of dendritic spine proteins.
Mature hippocampal neuronal cultures.
Primary hippocampal neuronal cultures were prepared as described previously (Diniz et al., 2012) using E15–E16 Swiss mice, and were used after 14 d in vitro. Cultures were prepared and maintained in Neurobasal medium (Invitrogen) supplemented with B-27, penicillin, streptomycin, l-glutamine, fungizone, and Ara-C (0.65 μm; Sigma-Aldrich). Cultures were previously treated at 37°C for 30 min with CM/CMAβ and exposed to 500 nm AβOs or an equivalent volume of vehicle (2% DMSO in PBS) for 3 h. For inhibition of TGF-β activity of the CM, neuronal cultures were preincubated with the TGF-β pathway inhibitor, SB-431542 (10 μm; Sigma-Aldrich), for 30 min before addition of conditioned medium. Cultures were then maintained in the presence of astrocyte conditioned medium and the inhibitor for 3 additional hours.
Immunoblotting.
Protein concentration in cell and tissue extracts was measured using the BCA protein assay kit (Cole-Parmer). Forty micrograms protein/lane were electrophoretically separated on a 10% SDS polyacrylamide gel. After separation, proteins were electrotransferred to a Hybond-P polyvinylidene difluoride membrane (Millipore) for 1.5 h. Nonspecific sites were blocked by membrane incubation in PBS containing 5% milk for 1 h. Primary antibodies were incubated in block solution overnight, followed by 1 h incubation with IRDye 680CW goat anti-mouse (LI-COR, RRID: AB_10715072) antibody and IRDye 800CW goat anti-rabbit antibody (LI-COR, RRID: AB_621848). Membranes were scanned and analyzed using Un-Scan-It gel v6.1 (Silk Scientific). Primary antibodies were mouse anti-synaptophysin (1:1000; Millipore Bioscience Research Reagents, RRID: AB_94947), rabbit anti-PSD-95 (1:1000; Abcam, RRID: AB_444362), rabbit anti-cyclophilin B (1:1000; Sigma-Aldrich, RRID: AB_10743624), rabbit anti-drebrin A/E (1:1000; Millipore, RRID: AB_1977159).
In-cell Western.
Astrocytes were grown in 96-well plates for 2 d in 50 μl of DMEM/F12 supplemented with 10% FCS. For fixation and permeabilization, a volume of 50 μl of 8% PFA was added to the culture and after 20 min, cells were washed three times with PBS-containing 0.1% Triton X-100. Blocking was done by incubating the cells in Odyssey blocking buffer (LI-COR) for 1.5 h at 24°C. Anti-TGF-β1 antibody was diluted in blocking buffer (1:100; Abcam, RRID: AB_1144265) and added to cells overnight at 4°C. Plates were washed with PBS-containing 0.1% Tween-20 three times, followed by 1 h incubation with IRDye 680CW goat anti-rabbit (1:800; LI-COR, RRID: AB_10706167) and IRDye 800CW goat anti-mouse antibodies (1:800; LI-COR, RRID: AB_621847). Plates were scanned with the Odyssey Infrared Imaging System and analyzed using the program Un-Scan-It gel v6.1 (Silk Scientific).
Immunocytochemistry.
After fixation with 4% paraformaldehyde for 15 min, cultures were permeabilized with 0.2% Triton X-100 for 5 min at room temperature, and nonspecific sites were blocked with 3% bovine serum albumin, 5% normal goat serum (Sigma-Aldrich) diluted in PBS for 1 h before immunoreaction with the following antibodies: rabbit anti-GFAP (1:500; DAKO Cytomation, RRID: AB_10013382), mouse anti-synaptophysin (1:1000; Millipore Bioscience Research Reagents, RRID: AB_94947); rabbit anti-PSD-95 (1:100; Cell Signaling Technology, RRID: AB_561221), rabbit anti-Spinophilin (1:500; Abcam, RRID: AB_444532), AβO-selective NU4 mouse monoclonal antibody (1 μg/ml; Lambert et al., 2007; RRID: AB_2313889), guinea pig anti-glutamate transporter (1:300; Millipore, AB_90949), mouse anti-TGF-β1 (1:100; Abcam, RRID: A_1144265). After primary antibody incubation, the cells were thoroughly washed with PBS and incubated with secondary antibodies for 2 h at room temperature. Secondary antibodies were AlexaFluor 546-conjugated goat anti-rabbit IgG (RRID: AB_10584649) or goat anti-mouse IgG (1:1000; RRID: AB_2534071, Invitrogen), or AlexaFluor 488-conjugated goat anti-rabbit IgG (RRID: AB_143165), goat anti-guinea pig IgG (RRID: AB_2534117), or goat anti-mouse IgG (1:300; RRID: AB_2534069, Invitrogen). Nuclei were counterstained with DAPI (Sigma-Aldrich).
For analysis of AβOs binding and quantification of excitatory synapses, hippocampal neurons that were at least two cell bodies away from the nearest neighboring neuron were imaged. The number of puncta for NU4 labeling was quantified using the Puncta Analyzer plug-in of ImageJ v1.29 (NIH; RRID: SCR_003070). Neuronal cell bodies were excluded from the analysis, resulting in the analysis of the NU4 immunoreactivity present only in dendritic processes. Cells were imaged on a TE 2000 Nikon microscope. Colocalization of synaptic proteins was analyzed as previously described (Diniz et al., 2012).
Immunohistochemistry.
Vibratome slices (40 μm thick) were incubated for 72 h with the following primary antibodies: rabbit anti-GFAP (1:1000; DAKO Cytomation, RRID: AB_10013382), mouse anti-synaptophysin (1:1000; Millipore Bioscience Research Reagents, RRID: AB_94947), rabbit anti-drebrin A/E (1:500; Millipore Bioscience Research Reagents, RRID: AB_1977159), mouse anti-TGF-β1 (1:1000; Abcam, RRID: AB_1144265). The slices were then washed three times with PBS and incubated with secondary antibodies for 2 h at room temperature. Secondary antibodies were AlexaFluor 546-conjugated goat anti-rabbit IgG (RRID: AB_10584649) or goat anti-mouse IgG (1:1000; RRID: AB_2534071, Invitrogen), or AlexaFluor 488-conjugated goat anti-rabbit IgG (RRID: AB_143165) or goat anti-mouse IgG (1:300; RRID: AB_2534069, Invitrogen). Nuclei were counterstained with DAPI (Sigma-Aldrich). Coverslips were mounted with DAKO Mounting Media and imaged on a confocal microscope (Leica TCS SPE).
Astrocyte morphometry and TGF-β1 levels.
Astrocytes in the CA1 stratum radiatum hippocampal region were immunostained for GFAP and TGF-β1. Images were acquired by confocal microscopy (Leica TCS SPE) with a 63× objective. For morphometric analysis, cells exhibiting a good delimitation relative to other astrocytes were randomly selected, and the number of processes was counted manually. Cell area and average fluorescence intensity per cell were analyzed with Fiji (NIH; RRID: SCR_002285) software. Levels of astrocyte TGF-β1 were quantified using the Puncta Analyzer plugin from ImageJ (NIH). Cells were randomly selected around their extremities based on GFAP staining, and the number of colocalized puncta of TGF-β1 and GFAP staining was quantified in each cell. Results represent the ratio between the number of colocalized puncta and the total area of each selected image. At least 30–35 cells were analyzed per experimental condition.
Reactive oxygen species measurements.
Dihydroethidium (DHE; Invitrogen) was freshly prepared immediately before each experiment. Astrocyte cultures incubated for 3 h at 37°C with 500 nm AβOs or vehicle were loaded with DHE at a final concentration of 10 μm for 40 min, and cells were imaged on the Nikon microscope. Images were collectively analyzed for DHE intensity using ImageJ (RRID:SCR_003070) software as described previously (De Felice et al., 2007).
Nitrite measurement.
NO production was determined indirectly through the assay of nitrite (NO2−), a stable metabolite of NO, based on the Griess reaction (Ding et al., 1998). Briefly, a 50 μl aliquot of conditioned medium was mixed with an equal volume of Griess reagent [0.1% N-(1-naphthyl) ethylenediamine dihydrochloride, 1% sulfanilamide, and 2.5% phosphoric acid], incubated for 10 min at 22°C, and the absorbance was measured at 540 nm. Nitrite concentrations were calculated from a standard curve of NaNO2 (Sigma-Aldrich) ranging from 0 to 100 μm. Background NO2− was subtracted from the experimental values.
Small interfering RNA assays.
Hippocampal astrocyte purified cultures were transfected using the Lipofectamine 2000 (Invitrogen) protocol according to manufacturer's instructions. Cells were transfected for 4 h with 50 nm small interfering RNA (siRNA) for TGF-β1 (which specifically silences TGF-β1 α gene through 3–5 targeted siRNA of 19–25 nt; Santa Cruz Biotechnology, SC-37192). Previous results from our group demonstrated that this protocol leads to a 60% decrease in TGF-β1 expression in a purified culture of midbrain astrocytes (data not shown). Culture treatments were conducted 24 h after transfection.
Animals and intracerebroventricular injections.
Three-month-old Swiss mice were kept in groups of five animals per cage in standard housing conditions (12 h light/dark cycle with controlled room temperature and humidity) with ad libitum access to food and water. All mice experiments were performed in accordance with Principles of Laboratory Animal Care from the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of the Federal University of Rio de Janeiro (protocol no. IBqM 041/2011). Intracerebroventricular (i.c.v.) injections were performed as previously described (Figueiredo et al., 2013). Briefly, mice were anesthetized using isoflurane 2.5% (Cristália) in a vaporizer system and were gently restrained during the intracerebroventricular procedure. Mice received injections of 10 ng TGF-β1, 10 pmol AβOs, or vehicle (final volume: 3 μl, i.c.v.) through a 2.5-mm-long needle. The needle was inserted unilaterally 1 mm to the right of the midline point equidistant from each eye and 1 mm posterior to a line drawn through the anterior base of the eye, as previously described (Figueiredo et al., 2013). Mice presenting hemorrhages were removed from the analysis.
Novel object recognition test.
The behavioral analyzes were performed according to a previously described protocol (Figueiredo et al., 2013). Mice were initially placed in an open-field arena and were submitted to a 5 min habituation session. The number of crossings and rearings was determined. Mice were then exposed to a training phase, in which animals explored two identical objects during 5 min and the time spent exploring each object was determined. One hour after the training section, mice were again placed in the arena for the test session, and one of the two familiar objects used in the training session was replaced by a novel one. Again, the time spent exploring familiar and novel objects was determined and all data analyzed. The arena and objects were cleaned thoroughly between trials with 40% ethanol to eliminate olfactory cues. Results were expressed as percentage of time exploring each object during the training or test session and were analyzed using a one-sample Student's t test comparing the mean exploration time for each object with the fixed value of 50%. By definition, animals that recognize the familiar object as such (i.e., learn the task) spend more time exploring the novel object.
Statistical analysis.
GraphPad software v5.0 (RRID:SCR_002798), was used for statistical analysis. Because all statistical tests involved multiple conditions, ANOVA was applied in all comparisons, followed by Tukey's post-test when statistical significance was achieved. A confidence interval of 95% was used, and a p value <0.05 was considered statistically significant. Densitometry of blotted gels was performed using Un-Scan-It gel v6.1 (Silk Scientific). Data are reported as means ± SEM and error bars in the graphs represent SEM.
Results
AβOs trigger functional and morphological changes in astrocytes
To investigate the impact of AβOs on astrocytes, we initially exposed purified mouse hippocampal astrocyte cultures to vehicle (control) or 500 nm AβOs for 24 h. Immunofluorescence confocal microscopy analysis showed that AβOs mostly localize to the astrocyte plasma membrane but are also present in the cytoplasm, suggesting that AβOs interact with the membrane and are internalized by astrocytes (Fig. 1A–D).
Figure 1.
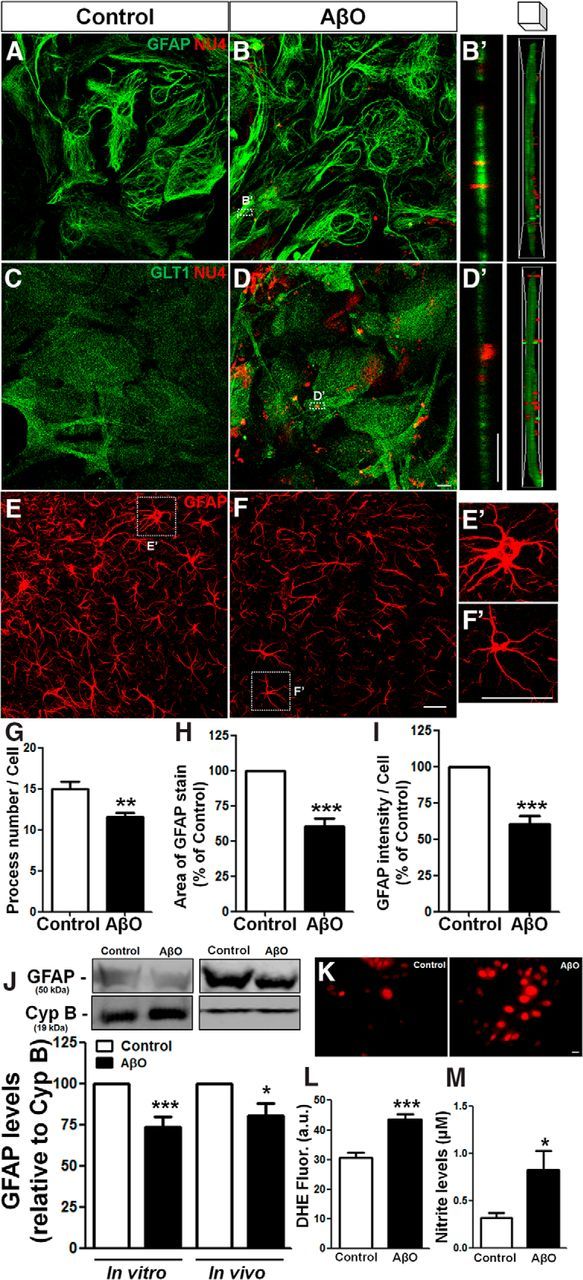
AβOs bind to astrocyte membranes, are internalized, and trigger astrocyte activation. Mouse hippocampal astrocyte cultures were exposed to vehicle or 500 nm AβOs for 24 h. Cells were then analyzed by immunocytochemistry for GFAP, GLT1, and AβOs (NU4 antibody immunoreactivity; A–D); representative z-stack orthogonal cut images of GFAP/NU4 (B′) and GLT1/NU4 (D′); levels of ROS (K, L) and nitrite production (M). E–I, AβO or vehicle was infused intracerebroventricularly in mice and GFAP immunostaining was analyzed in the hippocampus (E, F). Astrocyte morphology was evaluated by analyzing the number of processes (G), cellular area (H), and intensity of GFAP immunoreactivity per cell (I). GFAP levels were analyzed by Western blotting of cultured astrocytes or hippocampal tissue homogenates (J). Exposure to AβOs triggered astrocyte activation in vitro and induced astrocytic atrophy in vivo. Scale bars: D, D′, K, 10 μm; F, F′, 30 μm. *p < 0.050, **p < 0.010, and ***p < 0.001; n = 3–6 experiments with independent astrocyte cultures and three animals per experimental group; 30–45 cells were analyzed per experimental condition. Student's t test.
We next aimed to investigate the effects of AβOs on astrocytes in vivo. To this end, Swiss mice received an intracerebroventricular infusion of AβOs and morphological analysis of astrocytes present in the CA1 stratum radiatum hippocampal region was performed. This revealed that AβOs induced significant morphological changes in astrocytes, with a 23% decrease in the number of process (Fig. 1E–G), a 40% decrease in cell area (Fig. 1H) and a 40% decrease in GFAP immunoreactivity per cell (Fig. 1I) in astrocytes from AβO-infused mice. Immunoblotting analysis revealed that AβOs induced comparable decreases in GFAP levels in cell culture homogenates and in hippocampal extracts (Fig. 1J). We further found that cultured astrocytes exposed to AβOs presented increased levels of reactive oxygen species (ROS; Fig. 1K,L) and extracellular nitrite (NO2−), a stable metabolite of NO (Fig. 1M). These results suggest that AβOs directly interact with astrocyte membranes, are internalized, and impact astrocyte physiology and morphology.
Soluble factors released by murine and human astrocyte cultures reduce AβO binding to neurons and protect synapses
Astrocytes are a major source of soluble factors that regulate cell survival and synaptogenesis (Araque et al., 1999; Clarke and Barres, 2013; Diniz et al., 2014a). Thus, we next assessed whether soluble factors released to the CM by healthy astrocytes could interfere with the binding of AβOs to hippocampal neurons in purified cultures. Cultured neurons treated with CM presented a 53% decrease in AβO binding to their membranes (Fig. 2A,B,D), whereas CM from AβO-exposed astrocyte cultures (CM AβO) reduced binding to a significantly lesser extent (34%; Fig. 2C,D).
Figure 2.

Astrocyte CM protects neurons against the deleterious effects of AβOs. Mature hippocampal neurons (14 DIV) were maintained for 30 min in the presence of vehicle, CM derived from astrocyte cultures (CM), or CM derived from astrocytes previously primed by AβOs (CM AβO), and were then exposed for 3 h to 500 nm AβOs. After this period, the binding of AβOs (NU4 immunoreactivity; A–D), density of dendritic spines (spinophilin immunoreactivity; E–H), and density of synapses (synaptophysin/PSD-95 immunoreactivity; I–L) Twelve to 15 images were acquired from duplicate coverslips in each experimental condition, 90–150 cells were analyzed per experimental condition. Scale bars, 10 μm. *p < 0.050, **p < 0.010, and ***p < 0.001; comparisons between multiple groups were analyzed using a one-way ANOVA followed by Tukey's post hoc tests, n = 3–4 experiments with independent neuronal cultures.
We next asked whether the reduction in AβOs binding to neurons promoted by astrocyte CM was associated with protection of synapses and dendritic spines. In line with previous studies (Brito-Moreira et al., 2017), neuronal exposure to AβOs for 3 h decreased synapse density by 33% (Fig. 2L). Interestingly, CM from healthy astrocytes not only completely prevented the decrease in spine density induced by AβOs (Fig. 2E–H) but potently induced synaptogenesis (Fig. 2I–L), even in the presence of AβOs. We further note that CM derived from astrocytes exposed to AβOs afforded significantly less protection against AβO-induced synaptotoxicity (Fig. 2H,L).
Although their mechanism of action has been less investigated, human astrocytes are important modulators of synapse formation and function (Diniz et al., 2012, 2014b; Han et al., 2013). Thus, we next asked whether soluble factors released by ex vivo human astrocytes could also protect neurons from the toxic impact of AβOs. We found that human astrocyte conditioned medium (HCM) completely abolished AβO binding to murine neurons (Fig. 3A,B,D), whereas a 59% decrease in binding was observed in neurons treated with CM from human astrocytes previously exposed to AβOs (HCM AβO; Fig. 3C,D). Additionally, both HCM and HCM AβO prevented AβO-induced damage to synapses (Fig. 3E,F). Similar to the observations described above with murine CM, we found that, even in the presence of AβOs, HCM induced a very significant increase in the number of synapses in hippocampal neurons. Additionally, exposure of human astrocytes to AβOs impaired HCM synaptogenic potential (Fig. 3F).
Figure 3.

HCM abolishes AβOs binding to hippocampal neurons and synapse loss. Mature mouse hippocampal neurons (19–21 DIV) were maintained for 30 min in the absence (A) or presence (B) of HCM, or CM from human astrocytes previously primed by AβO (HCM AβO; C), and were then exposed for 3 h to 500 nm AβOs. After this period, AβO binding (A–D) and density of synapses (E, F) were analyzed by quantification of the number of NU4 and synaptophysin/PSD-95 puncta, respectively. Fifteen images were acquired from duplicate coverslips for each experimental condition. Scale bar, 10 μm. *p < 0.050, ***p < 0.001, one-way ANOVA followed by Tukey's post hoc tests; n = 3 experiments with independent neuronal cultures; 90–100 cells were analyzed per experimental condition.
Altogether, these results demonstrate that soluble factors released by murine and, notably, human astrocytes prevent AβOs binding to neurons and protect synapses in vitro. Further, we show that prior exposure to AβOs partially impairs the synaptogenic potential of murine and human astrocytes.
Astrocyte-derived TGF-β1 protects neurons against AβO-induced synapse loss
We have previously reported that TGF-β1 secreted by astrocytes regulates excitatory synapse formation (Diniz et al., 2012). To determine whether TGF-β1 was involved in the synaptoprotective actions of astrocytes against AβOs, we downregulated TGF-β1 activity/signaling by three different approaches: 1) pharmacological inhibition, 2) use of a neutralizing antibody, and 3) RNAi-mediated knock-down of TGF-β expression.
First, we added SB-431542, a TGF-β receptor antagonist, to CM or CM AβO before their addition to hippocampal neuronal cultures. Although SB-431542 had no effect on the protective action of CM AβO on synapses, we found that the TGF-β antagonist blocked the protective effect of CM on synapses by 35%, which we evaluated by quantification of the number of colocalized synaptophysin/PSD-95 puncta (Fig. 4A–F).
Figure 4.
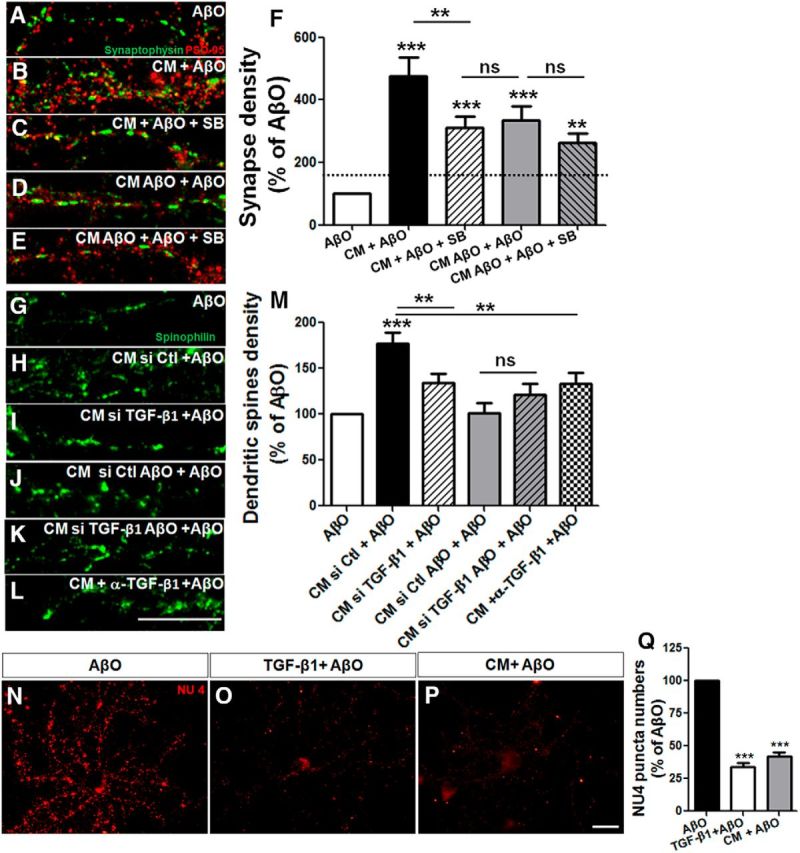
Astrocyte protection against AβOs is mediated by TGF-β1. Mature hippocampal neurons (14 DIV) were maintained for 30 min in the absence or presence of CM derived from astrocyte cultures (CM) or CM derived from astrocyte cultures previously primed by AβO (CM AβO), and were then exposed for 3 h to 500 nm AβOs. When present, 10 μm SB-431542 was added 30 min before CM addition. Synapse density was evaluated by double immunocytochemistry for PSD-95 and synaptophysin and determination of juxtaposed puncta (A–F). A–E, Representative images of dendritic segments under different experimental conditions. Density of dendritic spines were analyzed by spinophilin labeling in neurons cultured for 12 DIV and maintained in different CM plus AβOs. Neurons were treated with CM siRNA control (CM si Ctl), CM siRNA for TGF-β1 (CM siTGF-β1), or in the presence of neutralizing antibody against TGF-β1 (CM + α-TGF-β1). Representative dendrites of spinophilin labeling (G–L) and quantification of the density of dendritic spines (M). Prior addition of 10 ng/ml purified TGF-β1 to AβO-exposed neurons mimics the effect of CM and impairs AβO binding to neurons (measured by the quantification of the number of NU4 puncta; N–Q). Scale bars: 10 μm. **p < 0.010 and ***p < 0.001, one-way ANOVA followed by Tukey's post hoc tests. F, M, Q, n = 3–4 experiments with independent neuronal cultures. A total of 90–100 cells (F), 37–40 cells (M), and 60–95 cells were analyzed per experimental condition (Q).
We next knocked down TGF-β1 expression in astrocytes using siRNA (Fig. 4G–J,M) and immunodepleted TGF-β1 from the astrocyte conditioned medium using a neutralizing antibody (Fig. 4K–M). Both approaches severely impaired the capacity of astrocyte CM to protect against AβO toxicity (Fig. 4G–M).
We further tested the effect of exogenous purified TGF-β1 (10 ng/ml) applied to neuronal cultures 30 min before exposure to AβOs. Purified TGF-β1 decreased AβO binding to neurons to a similar extent as CM, suggesting that boosting TGF-β1 signaling protects neurons from Aβ oligomers (Fig. 4N–Q).
We next verified whether hippocampal astrocytes produce TGF-β1 in vitro (Fig. 5A,B) and in vivo (Fig. 5D,E). TGF-β1 immunostaining was found in the intracellular compartments of both cultured hippocampal astrocytes (Fig. 5A,B) and in astrocytes in the CA1 hippocampal subfield (Fig. 5D,E). Exposure of cultured astrocytes to AβOs slightly decreased the levels of TGF-β1 in these cells (Fig. 5C). Levels of TGF-β1 were also reduced in the hippocampus of AβO-injected mice (Fig. 5F).
Figure 5.

Exposure to AβOs reduces levels of astrocyte TGF-β1 in vitro and in vivo. TGF-β1 immunoreactivity in hippocampal astrocytes in vitro (A–C) and in vivo (D–I). Intracerebroventricular infusion of AβOs in mice and exposure of astrocyte cultures to AβOs reduced TGF-β1 levels in vitro (C, in-cell Western), and in vivo (F, Western blotting; G–I, immunohistochemistry). Scale bars: A, B, 10 μm; D, E, H, 30 μm. **p < 0.010 and ***p < 0.001, Student's t test (C, F, I); n = 5 experiments with independent astrocyte cultures, and three animals per experimental group; 30–35 cells were analyzed per experimental condition (I).
To fully correlate AβOs effects with astrocytic TGF-β1 production, we quantified levels of TGF-β1 by immunohistochemistry in hippocampal astrocytes from mice intracerebroventricularly injected with AβOs. We found a decrease in levels of astrocyte TGF-β1 in AβO-injected mice, indicating that AβOs impact TGF-β1 production by astrocytes in vivo (Fig. 5G–I).
Collectively, results suggest that AβO-induced decrease in TGF-β1 levels in (and its secretion by) astrocytes renders neurons more vulnerable to the synaptotoxicity of AβOs.
TGF-β1 protects against synapse loss and memory impairment triggered by AβOs
Previous studies have shown that intracerebroventricular infusion of AβOs reduces synaptophysin and PSD-95 levels in the hippocampi of mice (Figueiredo et al., 2013; Lourenco et al., 2013) and monkeys (Forny-Germano et al., 2014). To determine the ability of TGF-β1 to protect synapses from the deleterious impact of AβOs in vivo, we assessed the effect of AβO infusion on the levels of drebrin, a marker of dendritic spines, in the mouse brain. Intracerebroventricular infusion of 10 pmol AβOs caused a 63% decrease in drebrin immunoreactivity in the hippocampal CA1 region of mice (Fig. 6B,D). Intracerebroventricular infusion of 10 ng TGF-β1 30 min before the injection of AβOs fully prevented AβO-induced decrease in drebrin levels (Fig. 6A–D). Western blotting analyses further showed that, whereas AβOs decreased the levels of the synaptic proteins, drebrin, PSD-95 and synaptophysin, TGF-β1 restored the levels of these proteins in vivo (Fig. 6E,F). Further, TGF-β1 rescued the atrophy of astrocyte processes induced by AβOs in vivo (Fig. 6G–J). These results establish that TGF-β1 protects astrocytes and neuronal synapses from the deleterious effects of AβOs.
Figure 6.

TGF-β1 prevents AβO-induced synapse loss in vivo. Vehicle (A), AβO (B), or TGF-β1+AβO (C) were infused intracerebroventricular in mice. After 48 h, animals were submitted to histological evaluation of dendritic spines (Drebrin staining; D) in the CA1 region of the hippocampus, and Western blot analysis of the levels of the synaptic proteins, Drebrin, synaptophysin, and PSD-95 (E, F). Astrocytes from hippocampus were morphologically analyzed by GFAP staining followed by process counting (G–J). AβOs induce loss of dendritic spines and astrocytic process atrophy, whereas TGF-β1 prevents these deficits. Scale bars, 30 μm. *p < 0.050, **p < 0.010, and ***p < 0.001; n = 3, one-way ANOVA followed by Tukey's post hoc tests.
Synaptic and memory loss is well characterized in several animal models of AD (LaFerla and Green, 2012). Thus, we sought to determine the effects of TGF-β1 on memory impairment caused by AβOs in mice. To do that, 10 ng TGF-β1 was administered intracerebroventricularly 30 min before the infusion of 10 pmol AβOs (i.c.v.). We initially evaluated locomotor and exploratory activities of mice in different experimental conditions. Mice were allowed to freely explore an empty arena in a 5-min-long habituation session, and the numbers of rearings (elevations on rear paws) and of lines crossed in the arena floor were recorded. No differences were found in rearings or crossings between experimental groups (Fig. 7A,B), indicating that AβO infusion or treatment with TGF-β1 had no effect on locomotor/exploratory activities. We next (48 h after AβOs infusion) performed the novel object recognition test, a nonaversive declarative memory test, in mice treated or not with TGF-β1. In the training session, animals were placed at the center of the arena in the presence of two objects for 5 min and the amount of time spent exploring each object was determined. Results indicated that the mice had no preference for any of the objects (Fig. 7C). In the test session, one of the objects was replaced by a new one. As expected, vehicle-injected mice learned the task and spent more time exploring the novel object. In contrast, and in line with our previous studies (Figueiredo et al., 2013; Lourenco et al., 2013) mice injected with 10 pmol AβOs failed to acquire the OR memory. Remarkably, prior treatment with 10 ng TGF-β1 blocked AβO-induced cognitive impairment in mice (Fig. 7D).
Figure 7.

TGF-β1 prevents AβO-induced memory impairment. Vehicle (A), AβO (B), or TGF-β1+AβO (C) were infused intracerebroventricularly in mice. After 48 h, animals were submitted to behavior assays, crossing (A), rearing (B), and recognition memory of objects (C, D). TGF-β1 significantly prevented the cognitive impairment triggered by AβO. *p < 0.001; n = 7–9, Student's t test comparing the mean exploration time for each object with the fixed value of 50%.
Discussion
In the current study, we have identified a molecular mechanism by which astrocytes protect synapses and cognitive function against AβOs, soluble neurotoxins thought to be responsible for synaptic deterioration underlying Alzheimer's memory loss. We showed that AβOs trigger astrocyte activation and oxidative stress, decrease production of TGF-β1 and impair the synapse-protective function of astrocytes against AβOs. Additionally, we showed that TGF-β1 prevents AβOs damage in vivo, suggesting that boosting astroglial TGF-β1 pathway may provide a useful strategy for treatment of the early stages of AD.
The role of astrocytes in Aβ processing and AD pathogenesis has been a controversial matter. Although reactive astrocytes have been suggested to participate in the clearance and degradation of β-amyloid in vivo and in vitro (Matsunaga et al., 2003; Nagele et al., 2003; Wyss-Coray et al., 2003; Alarcón et al., 2005; Allaman et al., 2010; Verkhratsky et al., 2010), the effects of Aβ peptides, specifically AβOs, and their interaction with astrocytes, remain uncertain. Here, we demonstrated that AβOs interact with astrocyte membranes, trigger oxidative stress and impact astrocyte function. Multiple receptors and neuronal membrane proteins have been implicated in AβOs binding to the excitatory synaptic terminal, including the NMDA (De Felice et al., 2007), insulin receptors (Zhao et al., 2008; De Felice et al., 2009), AMPA (Zhao et al., 2010), Wnt receptors (Magdesian et al., 2008), and PrPC (Laurén et al., 2009; Beraldo et al., 2016). Although astrocytes express most of these molecules, potential AβOs binding sites in astrocytes remain to be determined.
We also found that intracerebroventricular injection of AβOs induces atrophy of astrocytes in the mouse hippocampus. Reduction of astroglial volume, surface area and astrocyte morphological complexity has been described in two transgenic mouse models of AD (3xTg-AD and PDAPP-J20 mice; Olabarria et al., 2010; Yeh et al., 2011; Kulijewicz-Nawrot et al., 2012; Beauquis et al., 2013). In early stages of AD, astrocytes undergo degeneration and loss of cellular processes, which appear to contribute to the progression of synapse loss and early cognitive deficits (Rodríguez-Arellano et al., 2016). In later stages of AD, however, there is a chronic neuroinflammation and reactive astrocytes are found associated with neuritic plaques, a feature commonly found in animal models and in diseased human tissue. These results are in agreement with the notion that astrocytes surrounding neuritic plaques undergo robust hypertrophy and proliferation, forming the glial scar, whereas astrocytes more distal from the local plaque injury not necessarily undergo such alterations, although they present metabolic defects (Rodríguez-Arellano et al., 2016).
Astrocytes are an important source of soluble factors that actively participate in synapse formation, maintenance and elimination (Clarke and Barres, 2013; Diniz et al., 2014a). Here, we showed that treatment of neurons with astrocyte CM reduces AβOs binding to neurons and synapse loss induced by AβOs. We also found that CM from healthy astrocytes is more effective in maintaining synapses than CM from astrocytes previously exposed to AβOs, indicating that AβOs impair the synaptogenic potentials of both murine and human astrocytes.
Our results show that AβOs reduce levels of astrocyte TGF-β1 in vitro and in vivo, suggesting that AβOs downregulate astrocyte production of factors that attenuate oligomer binding to neurons and neurotoxicity. Likewise, exposure of cultured astrocytes to Aβ has been shown to decrease the release of the synaptogenic molecule, TSP-1, resulting in decreased levels of synaptic proteins in hippocampal neurons (Rama Rao et al., 2013). Although we cannot rule out production of TGF-β1 by other cell types in vivo, our results showing that hippocampal astrocytes produce TGF-β1 in vivo and that AβOs decrease the levels of TGF-β1 in vitro and in vivo suggest that decreased levels of TGF-β1 and TGF-β1 signaling in astrocytes might be one of the mechanisms of AβO-induced synaptotoxicity.
In addition to decreased production and release of synaptogenic molecules, such as TSP and TGF-β1, we cannot completely rule out that secretion of toxic molecules by astrocytes may also contribute to AβO-induced impairment of synaptogenic potential of astrocytes. Such toxic molecules could comprise, among others, soluble Aβ40 and Aβ42 peptides (Qiao et al., 2016), the inhibitory neurotransmitter, GABA (Jo et al., 2014), and oxidative stress molecules (shown here) secreted by astrocytes in response to AβOs or in AD transgenic models.
In agreement with our results, a study using a transgenic AD mouse model indicated that astrocytes display reduced expression of neuronal support genes and genes involved in neuronal communication (Orre et al., 2014). They argued that Aβ-induced astrocyte reactivity might compromise normal astrocyte phenotype and function, leading to a less favorable support for neurons, thus contributing to the neuronal dysfunction and cognitive decline in AD.
Early stages of the neurodegenerative process of AD are associated with astroglial dysfunction, including deficits in glutamate and K+ buffering, glutamate-glutamine cycling, and oscillations in cytosolic calcium concentrations, leading to disruption in synaptic connectivity, and, ultimately, to neuronal death (Osborn et al., 2016; Rodríguez-Arellano et al., 2016). Data presented here extend this scenario, suggesting that astrocyte dysfunction may contribute to synaptic deficit in early phases of AD, when Aβ plaques and neuronal degeneration are not yet present.
We found that inhibition of TGF-β1 signaling partially abrogates the protection of synapses conferred by astrocytes, suggesting that TGF-β1 is a key factor in astrocyte protection against AβOs. It is interesting to note that astrocyte CM, which contains TGF-β1, not only protects synapses from AβOs toxicity but also increases the density of synapses beyond the control level. This can be probably attributed to the synaptogenic activity of this cytokine, as observed here and in other works (Bae et al., 2011; Diniz et al., 2012, 2014b; Caraci et al., 2015).
TGF-β1 is a pleiotropic molecule, which performs critical functions in nervous system repair and development (Diniz et al., 2014a). More recently, TGF-β1 has been strongly implicated in synapse formation, transmission and plasticity (Aberle et al., 2002; Packard et al., 2003; Sanyal et al., 2004; Fukushima et al., 2007; Lacmann et al., 2007; Heupel et al., 2008; Fong et al., 2010; Sun et al., 2010; Diniz et al., 2012, 2014b; Caraci et al., 2015). Deregulation of TGF-β signaling has been associated with a broad spectrum of behavioral abnormalities, including cognitive impairment, affective disorders, and deficits in sensorimotor gating (Vivien and Ali, 2006; Graciarena et al., 2010; Sun et al., 2010; Krieglstein et al., 2011). Our data showing that AβOs decrease the levels of TGF-β1 in vitro and in vivo are in agreement with the observation that TGF-β1 levels are reduced in the plasma of AD patients, which might contribute to neuronal death and exacerbation of the neuroinflammatory process (Mocali et al., 2004; Juraskova et al., 2010). Additionally, deficiency in TGF-β1 signaling, including reduced expression of neuronal TβRII and Smad3 and defects in subcellular localization and nuclear translocation of phosphorylated Smad2/3 (von Bernhardi et al., 2015), has been reported in postmortem AD brain and in AD animal models and aged mice (Lee et al., 2006; Tesseur et al., 2006; Ueberham et al., 2006; Chalmers and Love, 2007; Tichauer et al., 2014; Caraci et al., 2015).
We showed here that an intracerebroventricular administration of TGF-β1 prevented the deleterious effects of AβOs in memory impairment and synapse loss in mice. Our data thus extend two recent works that showed that TGF-β1 prevents retinal and brain damage elicited by AβOs (Chen et al., 2015; Fisichella et al., 2016). Intracerebroventricular administration of TGF-β1 before Aβ1–42 injection has been shown to ameliorate Aβ1–42-induced neurodegeneration and to prevent Aβ1–42-induced increases in glia-derived proinflammatory mediators, as well as T-cell-derived proinflammatory cytokines, in the hypothalamus, serum and CSF of an AD rat model (Chen et al., 2015). Importantly, pretreatment with TGF-β1 also prevented Aβ1–42-induced decreases in levels of neurotrophic factors, including IGF-1, GDNF, and BDNF. Here, we found that TGF-β1 rescues AβO-induced alterations in astrocyte morphology in mice. This result is corroborated by the observation that astrocytes from transgenic animals that overexpress TGF-β1 present increased number and complexity of processes (Bae et al., 2011). Together, these data suggest that at least part of the deficits observed in AβO-injected mice might be due to astrocyte dysfunction and correlate to impairment of TGF-β1 signaling.
TGF-β1 secreted by astrocytes regulates microglia-mediated synaptic pruning through the complement cascade (Bialas and Stevens, 2013). The recent demonstration that microglia mediates early synapse loss in AD mouse models through the complement cascade (Hong et al., 2016) suggests that deficits in TGF-β1 pathway reported in AD patients and here may contribute to impact astrocyte-microglia-synapse interactions in AD. Altogether, these data suggest a double mechanism underlying TGF-β1 action against Aβ toxins: a direct reduction in AβOs binding and synapse protection and/or strengthening, and an indirect effect via modulation of the synthesis and secretion of other factors involved in control of the neuroinflammation process.
Recently, two types of reactive astrocytes, called A1 and A2, have been suggested to play key roles in the progression of CNS diseases (Liddelow et al., 2017). A1 astrocytes are activated by microglia and produce large amounts of inflammatory molecules and complement cascade proteins, previously shown to be destructive to synapses (Bialas and Stevens, 2013). A2 astrocytes, on the other hand, exhibit increased release of neurotrophic factors and, therefore, aid in neuronal survival. In human AD, the presence of A1 astrocytes is abundant, which supports our hypothesis of the impairment of synaptogenic and protective astrocytic function, and the release of neurotoxic mediators (Liddelow et al., 2017), such as NO and ROS, verified here.
In conclusion, our results suggest a novel mechanism underlying astrocyte function in AD. We propose that, by enhancing TGF-β1 signaling, astrocytes may protect neurons against AβO-induced synaptotoxicity. Additionally, we show that astrocytes are directly affected by AβOs, which inhibit their ability to protect neurons. These findings open a new perspective to guide the search for novel therapeutic targets for AD, potentially focused on TGF-β signaling and astrocyte biology.
Footnotes
This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Ministério da Saúde, Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ). We thank Marcelo Meloni and Grasiela Ventura for technical assistance.
The authors declare no competing financial interests.
References
- Abbott A. (2011) Dementia: a problem for our age. Nature 475:S2–S4. 10.1038/475S2a [DOI] [PubMed] [Google Scholar]
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS (2002) Wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron 33:545–558. 10.1016/S0896-6273(02)00589-5 [DOI] [PubMed] [Google Scholar]
- Alarcón R, Fuenzalida C, Santibáñez M, von Bernhardi R (2005) Expression of scavenger receptors in glial cells: comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J Biol Chem 280:30406–30415. 10.1074/jbc.M414686200 [DOI] [PubMed] [Google Scholar]
- Alberdi E, Sánchez-Gómez MV, Cavaliere F, Pérez-Samartín A, Zugaza JL, Trullas R, Domercq M, Matute C (2010) Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 47:264–272. 10.1016/j.ceca.2009.12.010 [DOI] [PubMed] [Google Scholar]
- Allaman I, Gavillet M, Bélanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ (2010) Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci 30:3326–3338. 10.1523/JNEUROSCI.5098-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA (2012) Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 486:410–414. 10.1038/nature11059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215. 10.1016/S0166-2236(98)01349-6 [DOI] [PubMed] [Google Scholar]
- Bae JJ, Xiang YY, Martinez-Canabal A, Frankland PW, Yang BB, Lu WY (2011) Increased transforming growth factor-beta1 modulates glutamate receptor expression in the hippocampus. Int J Physiol Pathophysiol Pharmacol 3:9–20. [PMC free article] [PubMed] [Google Scholar]
- Beach TG, McGeer EG (1988) Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer's disease visual cortex. Brain Res 463:357–361. 10.1016/0006-8993(88)90410-6 [DOI] [PubMed] [Google Scholar]
- Beauquis J, Pavía P, Pomilio C, Vinuesa A, Podlutskaya N, Galvan V, Saravia F (2013) Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer's disease. Exp Neurol 239:28–37. 10.1016/j.expneurol.2012.09.009 [DOI] [PubMed] [Google Scholar]
- Beraldo FH, Ostapchenko VG, Caetano FA, Guimaraes AL, Ferretti GD, Daude N, Bertram L, Nogueira KO, Silva JL, Westaway D, Cashman NR, Martins VR, Prado VF, Prado MA (2016) Regulation of amyloid beta oligomer binding to neurons and neurotoxicity by the prion protein-mGluR5 complex. J Biol Chem 291:21945–21955. 10.1074/jbc.M116.738286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas AR, Stevens B (2013) TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16:1773–1782. 10.1038/nn.3560 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Brito-Moreira J, Lourenco MV, Oliveira MM, Ribeiro FC, Ledo JH, Diniz LP, Vital JFS, Magdesian MH, Melo HM, Barros-Aragao F, de Souza JM, Alves-Leon SV, Gomes FCA, Clarke JR, Figueiredo CP, De Felice FG, Ferreira ST (2017) Interaction of amyloid-beta (Abeta) oligomers with neurexin 2alpha and neuroligin 1 mediates synapse damage and memory loss in mice. J Biol Chem 292:7327–7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraci F, Gulisano W, Guida CA, Impellizzeri AA, Drago F, Puzzo D, Palmeri A (2015) A key role for TGF-beta1 in hippocampal synaptic plasticity and memory. Sci Rep 5:11252. 10.1038/srep11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers KA, Love S (2007) Neurofibrillary tangles may interfere with Smad 2/3 signaling in neurons. J Neuropathol Exp Neurol 66:158–167. 10.1097/nen.0b013e3180303b93 [DOI] [PubMed] [Google Scholar]
- Chen JH, Ke KF, Lu JH, Qiu YH, Peng YP (2015) Protection of TGF-beta1 against neuroinflammation and neurodegeneration in Aβ1–42-induced Alzheimer's disease model rats. PLoS One 10:e0116549. 10.1371/journal.pone.0116549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433. 10.1016/j.cell.2004.12.020 [DOI] [PubMed] [Google Scholar]
- Clarke LE, Barres BA (2013) Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci 14:311–321. 10.1038/nrn3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA (2010) Amyloid-β peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3β in primary cultured hippocampal neurons. J Neurosci 30:9166–9171. 10.1523/JNEUROSCI.1074-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL (2007) Aβ oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 282:11590–11601. 10.1074/jbc.M607483200 [DOI] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL (2008) Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol Aging 29:1334–1347. 10.1016/j.neurobiolaging.2007.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL (2009) Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc Natl Acad Sci U S A 106:1971–1976. 10.1073/pnas.0809158106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M, Zhang M, Wong JL, Rogers NE, Ignarro LJ, Voskuhl RR (1998) Antisense knockdown of inducible nitric oxide synthase inhibits induction of experimental autoimmune encephalomyelitis in SJL/J mice. J Immunol 160:2560–2564. [PubMed] [Google Scholar]
- Diniz LP, Almeida JC, Tortelli V, Vargas Lopes C, Setti-Perdigão P, Stipursky J, Kahn SA, Romão LF, de Miranda J, Alves-Leon SV, de Souza JM, Castro NG, Panizzutti R, Gomes FC (2012) Astrocyte-induced synaptogenesis is mediated by transforming growth factor beta signaling through modulation of D-serine levels in cerebral cortex neurons. J Biol Chem 287:41432–41445. 10.1074/jbc.M112.380824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz LP, Matias IC, Garcia MN, Gomes FC (2014a) Astrocytic control of neural circuit formation: highlights on TGF-beta signaling. Neurochem Int 78:18–27. 10.1016/j.neuint.2014.07.008 [DOI] [PubMed] [Google Scholar]
- Diniz LP, Tortelli V, Garcia MN, Araújo AP, Melo HM, Seixas da Silva GS, De Felice FG, Alves-Leon SV, de Souza JM, Romão LF, Castro NG, Gomes FC (2014c) Astrocyte transforming growth factor beta 1 promotes inhibitory synapse formation via CaM kinase II signaling. Glia 62:1917–1931. 10.1002/glia.22713 [DOI] [PubMed] [Google Scholar]
- Eroglu C, Barres BA (2010) Regulation of synaptic connectivity by glia. Nature 468:223–231. 10.1038/nature09612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ST, Klein WL (2011) The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol Learn Mem 96:529–543. 10.1016/j.nlm.2011.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM, Mota-Sales AP, Saraiva LM, Klein WL, Sebollela A, De Felice FG, Ferreira ST (2013) Memantine rescues transient cognitive impairment caused by high-molecular-weight Aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci 33:9626–9634. 10.1523/JNEUROSCI.0482-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisichella V, Giurdanella G, Platania CB, Romano GL, Leggio GM, Salomone S, Drago F, Caraci F, Bucolo C (2016) TGF-β1 prevents rat retinal insult induced by amyloid-β (1–42) oligomers. Eur J Pharmacol 787:72–77. 10.1016/j.ejphar.2016.02.002 [DOI] [PubMed] [Google Scholar]
- Fong SW, McLennan IS, McIntyre A, Reid J, Shennan KI, Bewick GS (2010) TGF-β2 alters the characteristics of the neuromuscular junction by regulating presynaptic quantal size. Proc Natl Acad Sci U S A 107:13515–13519. 10.1073/pnas.1001695107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forny-Germano L, Lyra e Silva NM, Batista AF, Brito-Moreira J, Gralle M, Boehnke SE, Coe BC, Lablans A, Marques SA, Martinez AM, Klein WL, Houzel JC, Ferreira ST, Munoz DP, De Felice FG (2014) Alzheimer's disease-like pathology induced by amyloid-β oligomers in nonhuman primates. J Neurosci 34:13629–13643. 10.1523/JNEUROSCI.1353-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima T, Liu RY, Byrne JH (2007) Transforming growth factor-β2 modulates synaptic efficacy and plasticity and induces phosphorylation of CREB in hippocampal neurons. Hippocampus 17:5–9. 10.1002/hipo.20243 [DOI] [PubMed] [Google Scholar]
- Furman JL, Sama DM, Gant JC, Beckett TL, Murphy MP, Bachstetter AD, Van Eldik LJ, Norris CM (2012) Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer's disease. J Neurosci 32:16129–16140. 10.1523/JNEUROSCI.2323-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL (2003) Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A 100:10417–10422. 10.1073/pnas.1834302100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graciarena M, Depino AM, Pitossi FJ (2010) Prenatal inflammation impairs adult neurogenesis and memory related behavior through persistent hippocampal TGFβ1 downregulation. Brain Behav Immun 24:1301–1309. 10.1016/j.bbi.2010.06.005 [DOI] [PubMed] [Google Scholar]
- Han X, Chen M, Wang F, Windrem M, Wang S, Shanz S, Xu Q, Oberheim NA, Bekar L, Betstadt S, Silva AJ, Takano T, Goldman SA, Nedergaard M (2013) Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12:342–353. 10.1016/j.stem.2012.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heupel K, Sargsyan V, Plomp JJ, Rickmann M, Varoqueaux F, Zhang W, Krieglstein K (2008) Loss of transforming growth factor-beta 2 leads to impairment of central synapse function. Neural Dev 3:25. 10.1186/1749-8104-3-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippius H, Neundörfer G (2003) The discovery of Alzheimer's disease. Dialogues Clin Neurosci 5:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee DY, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, et al. (2014) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nat Med 20:886–896. 10.1038/nm.3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juraskova B, Andrys C, Holmerova I, Solichova D, Hrnciarikova D, Vankova H, Vasatko T, Krejsek J (2010) Transforming growth factor beta and soluble endoglin in the healthy senior and in Alzheimer's disease patients. J Nutr Health Aging 14:758–761. 10.1007/s12603-010-0325-1 [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Zheng F, Unsicker K, Alzheimer C (2011) More than being protective: functional roles for TGF-beta/activin signaling pathways at central synapses. Trends Neurosci 34:421–429. 10.1016/j.tins.2011.06.002 [DOI] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C (2011) Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A 108:E440–E449. 10.1073/pnas.1104977108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulijewicz-Nawrot M, Verkhratsky A, Chvátal A, Syková E, Rodríguez JJ (2012) Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer's disease. J Anat 221:252–262. 10.1111/j.1469-7580.2012.01536.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacmann A, Hess D, Gohla G, Roussa E, Krieglstein K (2007) Activity-dependent release of transforming growth factor-beta in a neuronal network in vitro. Neuroscience 150:647–657. 10.1016/j.neuroscience.2007.09.046 [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL (2004) Synaptic targeting by Alzheimer's-related amyloid β oligomers. J Neurosci 24:10191–10200. 10.1523/JNEUROSCI.3432-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Green KN (2012) Animal models of Alzheimer disease. Cold Spring Harb Perspect Med 2:a006320. 10.1101/cshperspect.a006320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95:6448–6453. 10.1073/pnas.95.11.6448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL (2007) Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem 100:23–35. 10.1111/j.1471-4159.2006.04157.x [DOI] [PubMed] [Google Scholar]
- Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457:1128–1132. 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledo JH, Azevedo EP, Clarke JR, Ribeiro FC, Figueiredo CP, Foguel D, De Felice FG, Ferreira ST (2013) Amyloid-beta oligomers link depressive-like behavior and cognitive deficits in mice. Mol Psychiatry 18:1053–1054. 10.1038/mp.2012.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HG, Ueda M, Zhu X, Perry G, Smith MA (2006) Ectopic expression of phospho-Smad2 in Alzheimer's disease: uncoupling of the transforming growth factor-beta pathway? J Neurosci Res 84:1856–1861. 10.1002/jnr.21072 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, Sathler LB, Brito-Moreira J, Amaral OB, Silva CA, Freitas-Correa L, Espírito-Santo S, Campello-Costa P, Houzel JC, Klein WL, Holscher C, Carvalheira JB, Silva AM, Velloso LA, Munoz DP, et al. (2013) TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer's beta-amyloid oligomers in mice and monkeys. Cell Metab 18:831–843. 10.1016/j.cmet.2013.11.002 [DOI] [PubMed] [Google Scholar]
- Magdesian MH, Carvalho MM, Mendes FA, Saraiva LM, Juliano MA, Juliano L, Garcia-Abreu J, Ferreira ST (2008) Amyloid-beta binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/beta-catenin signaling. J Biol Chem 283:9359–9368. 10.1074/jbc.M707108200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga W, Shirokawa T, Isobe K (2003) Specific uptake of Aβ1–40 in rat brain occurs in astrocyte, but not in microglia. Neurosci Lett 342:129–131. 10.1016/S0304-3940(03)00240-4 [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW (2001) CNS synaptogenesis promoted by glia-derived cholesterol. Science 294:1354–1357. 10.1126/science.294.5545.1354 [DOI] [PubMed] [Google Scholar]
- Mocali A, Cedrola S, Della Malva N, Bontempelli M, Mitidieri VA, Bavazzano A, Comolli R, Paoletti F, La Porta CA (2004) Increased plasma levels of soluble CD40, together with the decrease of TGF beta 1, as possible differential markers of Alzheimer disease. Exp Gerontol 39:1555–1561. 10.1016/j.exger.2004.07.007 [DOI] [PubMed] [Google Scholar]
- Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2:a006338. 10.1101/cshperspect.a006338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagele RG, D'Andrea MR, Lee H, Venkataraman V, Wang HY (2003) Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971:197–209. 10.1016/S0006-8993(03)02361-8 [DOI] [PubMed] [Google Scholar]
- Olabarria M, Noristani HN, Verkhratsky A, Rodríguez JJ (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia 58:831–838. [DOI] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Osborn LM, Jansen AH, Kooijman L, Bossers K, Hol EM (2014) Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol Aging 35:2746–2760. 10.1016/j.neurobiolaging.2014.06.004 [DOI] [PubMed] [Google Scholar]
- Osborn LM, Kamphuis W, Wadman WJ, Hol EM (2016) Astrogliosis: an integral player in the pathogenesis of Alzheimer's disease. Prog Neurobiol 144:121–141. 10.1016/j.pneurobio.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Packard M, Mathew D, Budnik V (2003) Wnts and TGF beta in synaptogenesis: old friends signalling at new places. Nat Rev Neurosci 4:113–120. 10.1038/nrn1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L (2010) Amyloid-β–induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 13:812–818. 10.1038/nn.2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J, Wang J, Wang H, Zhang Y, Zhu S, Adilijiang A, Guo H, Zhang R, Guo W, Luo G, Qiu Y, Xu H, Kong J, Huang Q, Li XM (2016) Regulation of astrocyte pathology by fluoxetine prevents the deterioration of Alzheimer phenotypes in an APP/PS1 mouse model. Glia 64:240–254. 10.1002/glia.22926 [DOI] [PubMed] [Google Scholar]
- Rama Rao KV, Curtis KM, Johnstone JT, Norenberg MD (2013) Amyloid-beta inhibits thrombospondin 1 release from cultured astrocytes: effects on synaptic protein expression. J Neuropathol Exp Neurol 72:735–744. 10.1097/NEN.0b013e31829bd082 [DOI] [PubMed] [Google Scholar]
- Rodríguez-Arellano JJ, Parpura V, Zorec R, Verkhratsky A (2016) Astrocytes in physiological aging and Alzheimer's disease. Neuroscience 323:170–182. 10.1016/j.neuroscience.2015.01.007 [DOI] [PubMed] [Google Scholar]
- Sanyal S, Kim SM, Ramaswami M (2004) Retrograde regulation in the CNS; neuron-specific interpretations of TGF-beta signaling. Neuron 41:845–848. 10.1016/S0896-6273(04)00152-7 [DOI] [PubMed] [Google Scholar]
- Sebollela A, Freitas-Correa L, Oliveira FF, Paula-Lima AC, Saraiva LM, Martins SM, Mota LD, Torres C, Alves-Leon S, de Souza JM, Carraro DM, Brentani H, De Felice FG, Ferreira ST (2012) Amyloid-beta oligomers induce differential gene expression in adult human brain slices. J Biol Chem 287:7436–7445. 10.1074/jbc.M111.298471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL (2007) Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27:2866–2875. 10.1523/JNEUROSCI.4970-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Gewirtz JC, Bofenkamp L, Wickham RJ, Ge H, O'Connor MB (2010) Canonical TGF-β signaling is required for the balance of excitatory/inhibitory transmission within the hippocampus and prepulse inhibition of acoustic startle. J Neurosci 30:6025–6035. 10.1523/JNEUROSCI.0789-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, Lin AH, Crews L, Tremblay P, Mathews P, Mucke L, Masliah E, Wyss-Coray T (2006) Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest 116:3060–3069. 10.1172/JCI27341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichauer JE, Flores B, Soler B, Eugenín-von Bernhardi L, Ramírez G, von Bernhardi R (2014) Age-dependent changes on TGFbeta1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav Immun 37:187–196. 10.1016/j.bbi.2013.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueberham U, Ueberham E, Gruschka H, Arendt T (2006) Altered subcellular location of phosphorylated Smads in Alzheimer's disease. Eur J Neurosci 24:2327–2334. 10.1111/j.1460-9568.2006.05109.x [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ (2010) Astrocytes in Alzheimer's disease. Neurotherapeutics 7:399–412. 10.1016/j.nurt.2010.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivien D, Ali C (2006) Transforming growth factor-beta signalling in brain disorders. Cytokine Growth Factor Rev 17:121–128. 10.1016/j.cytogfr.2005.09.011 [DOI] [PubMed] [Google Scholar]
- von Bernhardi R, Cornejo F, Parada GE, Eugenín J (2015) Role of TGFbeta signaling in the pathogenesis of Alzheimer's disease. Front Cell Neurosci 9:426. 10.3389/fncel.2015.00426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9:453–457. 10.1038/nm838 [DOI] [PubMed] [Google Scholar]
- Yeh CY, Vadhwana B, Verkhratsky A, Rodríguez JJ (2011) Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer's disease. ASN Neuro 3:271–279. 10.1042/AN20110025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL (2008) Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J 22:246–260. 10.1096/fj.06-7703com [DOI] [PubMed] [Google Scholar]
- Zhao WQ, Santini F, Breese R, Ross D, Zhang XD, Stone DJ, Ferrer M, Townsend M, Wolfe AL, Seager MA, Kinney GG, Shughrue PJ, Ray WJ (2010) Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J Biol Chem 285:7619–7632. 10.1074/jbc.M109.057182 [DOI] [PMC free article] [PubMed] [Google Scholar]