

Abstract
Neurons and glial cells exchange energy-rich metabolites and it has been suggested, originally based on in vitro data, that astrocytes provide lactate to glutamatergic synapses (“lactate shuttle”). Here, we have studied astrocytes that lack mitochondrial respiration in vitro and in vivo. A novel mouse mutant (GLASTCreERT2::Cox10flox/flox) was generated, in which the administration of tamoxifen causes mutant astrocytes to fail in the assembly of mitochondrial cytochrome c oxidase (COX). Focusing on cerebellar Bergmann glia (BG) cells, which exhibit the highest rate of Cre-mediated recombination, we found a normal density of viable astrocytes even 1 year after tamoxifen-induced Cox10 gene targeting. Our data show that BG cells, and presumably all astrocytes, can survive by aerobic glycolysis for an extended period of time in the absence of glial pathology or unspecific signs of neurodegeneration.
SIGNIFICANCE STATEMENT When astrocytes are placed into culture, they import glucose and release lactate, an energy-rich metabolite readily metabolized by neurons. This observation led to the “glia-to-neuron lactate shuttle hypothesis,” but in vivo evidence for this hypothesis is weak. To study astroglial energy metabolism and the directionality of lactate flux, we generated conditional Cox10 mouse mutants lacking mitochondrial respiration in astrocytes, which forces these cells to survive by aerobic glycolysis. Here, we report that these mice are fully viable in the absence of any signs of glial or neuronal loss, suggesting that astrocytes are naturally glycolytic cells.
Keywords: astrocytes, brain energy metabolism, glycolysis, lactate shuttle, mitochondria
Introduction
Maintaining brain function is dependent upon an uninterrupted supply of energy. The electrical activity of neurons and synaptic neurotransmission consume most of the cell's ATP supply (Attwell et al., 2010; Harris et al., 2012; Mergenthaler et al., 2013; Rangaraju et al., 2014). A cellular network comprising brain capillaries, astrocytes, and oligodendrocytes is thought to provide energy-rich metabolites to neurons and their axonal and dendritic processes (Bélanger et al., 2011; Pellerin and Magistretti, 2012; Bouzier-Sore and Pellerin, 2013; Morrison et al., 2013; Hirrlinger and Nave, 2014).
A specific role for astrocytes in the support of synaptic energy metabolism was proposed by the “astrocyte-to-neuron lactate shuttle” (ANLS) hypothesis (Pellerin and Magistretti, 1994). In this model, astrocytes provide lactate to the glutamatergic synapses that they engulf and astroglial glucose import is feed-back regulated by glutamate uptake at the synaptic cleft. Simplified, it was suggested that astrocytes perform aerobic glycolysis and release excess lactate into the synaptic compartment, thereby supporting neuronal energy metabolism as a function of synaptic activity (Pellerin and Magistretti, 1994, 2012; Pellerin et al., 1998; Magistretti and Allaman, 2015).
In agreement with the ANLS hypothesis were human PET studies showing that increases in cortical blood flow and glucose uptake, triggered by neuronal activity, only partly match rises in oxygen utilization (Fox and Raichle, 1986; Fox et al., 1988; Magistretti, 2006; Rasmussen et al., 2011). Using magnetic resonance spectroscopy (MRS) imaging, the utilization of plasma-derived 13C-labeled lactate in neurons could be visualized directly (Boumezbeur et al., 2010). Indeed, lactate can sustain neuronal activity in vivo and neurons exhibit a preference for lactate over glucose when both metabolites are available (Wyss et al., 2011). In culture, astrocytes metabolize glucose preferentially over lactate, which is released into the medium and used by neurons (Itoh et al., 2003; Bouzier-Sore et al., 2006). Purified neurons and astrocytes also differ in the expression of glycolytic pathway genes, which is higher in glia (Lovatt et al., 2007; Cahoy et al., 2008; Zhang et al., 2014).
However, proving the cellular origin of lactate in the intact CNS has been difficult. Glycogen granules are specific to astrocytes (Wender et al., 2000; Dienel and Cruz, 2015) and pharmacologically blocking glycogen breakdown in rats interferes with hippocampal LTP and also suppresses the increase of lactate normally associated with memory formation (Suzuki et al., 2011). Such behavioral data are compelling but remain indirect with respect to the in vivo source of lactate. Indeed, it has been argued that cortical astrocytes provide neurons with glucose and only take up lactate from glycolytic neuronal domains to dispose of excess lactate into the brain vasculature (Dienel, 2012; Hertz et al., 2014).
For neurons, glycolysis is insufficient to generate all of the ATP required for normal function and survival. Indeed, neurons are unable to upregulate glycolysis due to low levels of Pfkfb3, which generates the most potent activator of glycolysis (Herrero-Mendez et al., 2009). Cortical and hippocampal neurons degenerate when mitochondrial respiration is impaired genetically (Fukui et al., 2007; Diaz, 2010; Fünfschilling et al., 2012). In these in vivo experiments, the assembly of cytochrome c oxidase (COX) was disrupted by Cre-mediated targeting of the nuclear Cox10 gene (Diaz et al., 2005), which is essential for COX assembly and complex IV function. The same approach led to the surprising finding that oligodendrocytes readily survive as glycolytic cells, which are metabolically coupled to the axonal compartment that they feed with lactate (Fünfschilling et al., 2012). Indeed, loss of the lactate transporter MCT1/Slc16a1 from oligodendrocytes causes progressive axonal but not oligodendroglial degeneration (Lee et al., 2012).
Here, we generated GlastCreERT2:Cox10flox/flox mice in which a large fraction of astrocytes, and virtually all Bergmann glia (BG) cells of the cerebellum, lack mitochondrial respiration upon tamoxifen-induced deletion of Cox10 (Diaz et al., 2005; Mori et al., 2006). Strikingly, we observed fully viable but respiration-deficient astrocytes even 1 year after gene targeting and in the absence of signs of glial pathology, neurodegeneration, or inflammation.
Materials and Methods
Cell culture.
Primary astrocytes were isolated as described previously (Arai and Lo, 2010). Briefly, cortices dissected from postnatal day 2 (P2) Sprague Dawley rat pups were digested in minimum essential medium (MEM; Invitrogen) containing 2% cysteine (Sigma-Aldrich), 2% DNase (Roche), and 4% papaine (Worthington) for 1 h at 37°C and 5% CO2. Cells were plated onto 75 cm2 flasks previously coated with poly-l-Lysine (PLL, 10 μg/ml; Sigma-Aldrich) and grown in DMEM (Lonza) supplemented with 10% heat-inactivated fetal calf serum (FCS; Hyclone), 1% glutamine (Invitrogen), and 1% penicillin/streptomycin (Invitrogen) in a humidified incubator at 37°C and 5% CO2. After mixed glial cell cultures were confluent (12–14 d), nonastrocytic cells such as microglia and oligodendrocytes were detached from the flasks by manual shaking and removed by changing the medium. Astrocytes were dissociated by trypsinization and 20,000 cells were reseeded on PLL-coated (0.1 mg/ml) coverslips (13 mm). Astrocytic cultures were maintained in DMEM containing 10% FCS, 1% glutamine, and 1% penicillin/streptomycin at 37°C and 5% CO2. Human embryonic stem cell-derived astrocytes were cultured as described previously (Joannides et al., 2007).
Primary neurons were isolated as described previously (Meberg and Miller, 2003; Sinnamon et al., 2012). Briefly, cortices dissected from embryonic day 17 Sprague Dawley rats were collected in ice-cold HBSS without Mg2+ or Ca2+ with 5 mm HEPES and digested with 2.5% trypsin (Invitrogen) in HBSS for 20 min at 37°C. Trypsinization was stopped by adding 10% FCS in DMEM. Cortical homogenates were incubated with 0.2 mg/ml DNase in DMEM for 5 min at room temperature and subsequently dissociated carefully by 10 passes through a fire-polished Pasteur pipette. The suspension was filtered through a 40 μm cell strainer to remove debris. Then, 20,000 cells were plated on 13 mm coverslips previously coated with PLL (10 μg/ml) in a 24-well format. Cells were cultivated in DMEM supplemented with 10% heat-inactivated FCS, 1% glutamine, 1% penicillin/streptomycin, and 2% B27 supplement (Invitrogen) and maintained at 37°C and 5% CO2 in a humidified incubator. The next day, the medium was changed to Neurobasal medium (Invitrogen) containing 1% glutamine, 1% penicillin/streptomycin, and 2% B27 supplement. From there on, every 3–4 d, a half medium change was performed. To obtain pure neuronal cultures, cytosine arabinofuranoside was added (to a final concentration of 5 μm) at d in vitro 4 (DIV4) and DIV8 to kill postmitotic glial cells.
Pharmacological inhibition of mitochondrial respiratory chain complex I and complex IV and genetic disruption of complex IV in vitro.
Rotenone and potassium cyanide (KCN) were used to inhibit mitochondrial chain complex I and complex IV, respectively. Rotenone treatments were conducted for 24 h and initiated in astrocytes at DIV25 and neurons at DIV10. Fresh rotenone powder (Sigma-Aldrich) was dissolved in dimethyl sulfoxide to obtain a 10 mm solution. The final rotenone concentrations used in each experiment are indicated in the figures. At DIV21, the human astrocyte monolayer was exposed to KCN, dissolved in DMSO, for 24 h at varying concentrations (50 μm, 100 μm, and 1 mm). To eliminate COX10 expression and to generate astrocytes lacking complex IV in vitro, astrocytes were cultured from whole-brain homogenates from Cox10flox/flox mice. Cultured astrocytes were then exposed to lentiviral Cre (generated by Pamela Brown, University of Edinburgh) for 48 h and maintained for 8 weeks in vitro. Resuspended cells were then used to assess oxygen consumption by respirometry, as described previously (Ziabreva et al., 2010).
Trypan blue exclusion.
Cells were cultured as stated above and treated in the presence or absence of rotenone. Subsequently, they were harvested by light trypsinization (5 min, 0.05% trypsin) and suspended in defined medium. Next, cells were incubated in 0.4% trypan blue (Sigma-Aldrich) solution for 5 min at room temperature (RT) and the number of trypan blue-negative cells was counted using a hemocytometer to determine viable cells per 13 mm coverslip.
Immunocytochemistry.
Cells were washed with 1× PBS, pH 7.4, fixed with 4% (w/v) paraformaldehyde (PFA) for 15 min at 37°C, washed with 1× PBS, and permeabilized with 0.2% Triton X-100 and 10% horse serum in 1× PBS for 1 h at RT. Primary antibodies were applied in the same solution overnight at 4°C. Dilutions were as follows: for GFAP (1:200, mouse; Novocastra), NeuN (1:100, mouse; Millipore), Tuj-1 (1:200, rabbit; Covance), Vimentin (1:200, mouse; Novocastra NCL-VIM-V9), and Aquaporin residue 249–323 (1:100, rabbit; Sigma-Aldrich). Secondary antibodies to mouse and rabbit conjugated to Alexa Fluor 488 and Alexa Fluor 555 (both 1:2000; Invitrogen) were incubated in 2% horse serum in 1× PBS or PBS/BSA [1% (w/v) BSA] for 1 h at RT. For nuclei labeling, DAPI (0.025 μg/ml final concentration) was added to the secondary antibody. Coverslips were mounted using Aqua-Polymount (Eppelheim).
Animals.
All animal experiments were performed in compliance with animal policies of the State of Lower Saxony, Germany. Animals were maintained on a C57BL/6 genetic background. Cox10flox/flox mice, CKIIα-Cre mice, GLASTCreERT2 mice, and R26R-EYFP mice were genotyped as described previously (Minichiello et al., 1999; Srinivas et al., 2001; Diaz et al., 2005; Mori et al., 2006). All tamoxifen-sensitive mutant and control mice were treated with tamoxifen (Sigma-Aldrich) or the vehicle oil only. Control mice received the vehicle or tamoxifen when GlastCreERT2 was present or not, respectively. Tamoxifen was dissolved in corn oil to obtain a 10 mg/ml solution. Mice were injected intraperitoneally with a tamoxifen dosage of 100 mg/kg body weight for 5 consecutive days starting at P30. For the proliferation assay, mice were exposed to 0.2 mg/ml 5-ethynyl-2-deoxyuridine (EdU; Invitrogen) in the drinking water at the age of 2 months for 14 d. EdU solution was freshly prepared every 2 d.
PCR on genomic DNA.
A total of 30 μg of total cerebellar and brain DNA was used in a PCR to detect the recombined Cox10 allele with the primers TGAGTAGAATGGCTTCCGGAAGGG and AGCAGCAAAGAGGGCTCACTTCTTGC, generating a template of 465 bp. For each genotype, three female mice were analyzed.
Immunohistochemistry.
Mice were anesthetized with Avertin [2,2,2-tribromoethanol [2% (v/v)] and amyl alcohol [2% (w/v)], 0.2 ml per 10 g body weight] and perfused intracardially with 15 ml of HBSS and subsequently with 4% (w/v) PFA in 0.1 m sodium phosphate buffer. Brains were kept in the same fixative overnight at 4°C and then either embedded in paraffin or cut using a vibratome.
For reporter studies, 30-μm-thick cerebellum sections were cut sagittally with a vibratome. Sections were washed with 1× PBS and permeabilized with 0.2% Triton X-100 and 5% horse serum in 1× PBS for 1 h at RT. Primary antibodies against GFP (1:1000, goat; Rockland) and S100β (1:200, rabbit; Abcam 52642) were applied in the same solution overnight at 4°C. For each genotype, four mice of either sex were analyzed.
For fluorescent immunolabeling, 5 μm paraffin sections were cut using a microtome. After deparaffinization and rehydration, sections were boiled for 10 min in 0.1 m citrate buffer, pH 6.0, in a microwave (650 W). After cooling down to RT, sections were rinsed with 0.05 m Tris buffer, pH 7.6, containing 2% (w/v) fat-free milk powder and incubated in blocking solution containing 20% goat serum in 1× PBS for 30 min at RT. Primary antibodies GFAP (1:200, mouse; Novocastra) and S100β (1:200, rabbit; Abcam 52642) were diluted in BSA/PBS and incubated overnight at 4°C. For each genotype, three to six mice of either sex were analyzed.
Secondary antibodies to mouse, goat, and rabbit conjugated to Alexa Fluor 488 (1:2000; Invitrogen) and Alexa Fluor 555 (1:2000; Invitrogen) in 2% horse serum in 1× PBS or PBS/BSA [1% (w/v) BSA] were incubated for 2 h at RT. For nuclei labeling, DAPI (0.025 μg/ml final concentration) was added to the secondary antibody. Sections were mounted using Aqua Polymount.
For chromogenic staining, paraffin sections (5 μm) were immunolabeled with antibodies against APP (1:750, mouse; Millipore), CD3 (1:150, rat; Serotec), Mac3 (1:400, rat; Millipore), and GFAP (1:200, mouse; Novocastra) using the LSAB2 kit (Dako), the Vector Elite ABC kit (Vector Laboratories), and the Zytomed kit (Zytomed Systems) according to the manufacturer's instructions. For each genotype, three to six mice were analyzed. For the quantification of CD3-positive T cells and Mac3-positive microglia, nuclei surrounded by DAB-positive deposits were quantified using the Cell Counter plug-in of ImageJ. For semiautomated analysis of the GFAP-positive area, our own ImageJ plug-in (http://www1.em.mpg.de/Goebbels2016_GFAP) was used. For each genotype, four to six mice of either sex were analyzed.
For hematoxylin staining, sections were deparaffinized, rehydrated, and incubated in 0.1% hematoxylin for 3–5 min. For each genotype, three to six mice of either sex were analyzed.
For TUNEL staining, paraffin sections were treated with DeadEnd Colorimetric TUNEL System kit (Promega) following the manufacturer's instructions. For each genotype, three to six mice of either sex were analyzed.
The proliferation assay was performed on paraffin sections using the Click-it EdU Alexa Fluor 555 Imaging kit (Invitrogen) after immunolabeling. The tissue was permeabilized with 0.5% Triton X-100 and 3% horse serum in 1× PBS for 1 h at RT and the following steps were performed according to the manufacturer's instructions. For each genotype, three to four female mice were assessed.
COX/SDH histochemistry.
Sequential histochemistry was performed to detect COX and succinate dehydrogenase (SDH, also known as mitochondrial complex II) activity as described previously (Campbell et al., 2011). Briefly, 8-mm-thick brain sections were cut using a cryostat and air dried for 30 min at RT. Astrocytes were removed from medium and the coverslips were air dried for 30 min at RT. Cryosections and astrocytes on coverslips were exposed to COX medium containing 500 μM cytochrome c, 5 mm diaminobenzidine tetrahydrochloride, and 20 μg/ml catalase at 37°C for 40 min. Sections were washed 3 times with 1× PBS, followed by application of SDH medium containing 130 mm sodium succinate, 200 mm phenazine methosulphate, 1 mm sodium azide, and 1.5 mm nitroblue tetrazolium in 0.1 m phosphate buffer, pH 7.0, for 30 min at 37°C. For each genotype, three mice of either sex were analyzed.
Electron microscopy.
Mice were anesthetized with Avertin, perfused intracardially with 15 ml of HBSS and subsequently with 4% (w/v) PFA and 2.5% (v/v) glutaraldehyde in 0.1 m sodium phosphate buffer. Brains were kept in the same fixative for postfixation overnight. Cerebellar lobe 3 was punched out from 300-μm-thick sagittal cerebellar sections, dehydrated in an ascending series of ethanol, and then embedded in Epon. Ultrathin sections (50 nm) were cut using an ultramicrotome (Ultracut S) and contrasted with 4% uranyl acetate, followed by application of lead citrate (Reynolds, 1963). EM images from the upper third of the molecular layer were acquired at 5000× with an electron microscope (Leo 912; Zeiss). For each genotype, four to six female mice were analyzed and, for each animal, 10 random photographs were taken. Synapses between parallel fiber (PF) axons and Purkinje cell (PC) dendrites were identified by morphological characteristics (Yamada and Watanabe, 2002) and counted using the Cell Counter plug-in of ImageJ. BG processes were identified by their irregular appearance and characteristic bundles of intermediate filaments in a relatively clear cytoplasm (Palay and Chan-Palay, 1974). The area of BG processes was analyzed with the point-hit method with a defined grid area of 2.0 μm2 using ImageJ. EM images from the PC layer were acquired at 3150×. For each genotype, three female mice were analyzed. BG were identified by their localization and morphological features (Palay and Chan-Palay, 1974). The area of BG cytoplasm was measured and the number and size of BG mitochondria were determined using ImageJ.
Localized proton MRS.
Mice [Cox10fl/fl-GLAST-Cre (n = 11) and Cox10fl/fl controls] were initially anesthetized with 4% isoflurane and subsequently intubated and kept under anesthesia with 1.75% isoflurane in ambient air enriched with 20% oxygen (positive pressure ventilation, constant respiratory frequency of 85/min).
Localized proton MRS was performed at a field strength of 9.4 T (Bruker Biospin) by using a STEAM sequence (TR/TE/TM = 6000/10/10 ms, outer volume suppression, water suppression with CHEmical Shift Selective (CHESS) pulses). T2-weighted images (2D SE, TR/TE = 2500/20 ms, spatial resolution 130 × 130 μm2, slice thickness 300 μm, axial and sagittal orientation) were used for positioning of the volumes of interest. Spectra were obtained in the midfrontal cortex (3.9 × 0.7 × 3.2 mm3, number of averages 128) and the cerebellum (4 × 1 × 2 mm3, number of averages 128). Metabolite quantification involved spectral evaluation by LCModel (Version 6.3–1L) and calibration with a brain water concentration of 43.7 mol/L. Values with Cramer–Rao lower bounds >20% were excluded from further analyses. For each genotype, four to five female mice were analyzed.
Statistics.
Statistical significance was evaluated using unpaired Student's t test. Values are represented as mean ± SEM. p < 0.05 was considered statistically significant.
Results
Cultured astrocytes resist mitochondrial respiratory chain complex I and complex IV inhibitors that are toxic for neurons
To compare the ability of cultured astrocytes and neurons to survive by glycolysis in the presence of oxygen, purified cells were treated with rotenone, a plant-derived inhibitor of the mitochondrial respiratory chain complex I (Lindahl and Oberg, 1961), and potassium cyanide, a COX inhibitor (Campbell et al., 2011). As expected, cortical neurons were very sensitive to the pharmacological inhibition. Addition of as little as 10 nm rotenone led to a striking reduction of Tuj1-positive neuronal processes 24 h later and apoptotic cells with pyknotic nuclei were obvious (Fig. 1A). At higher concentrations of rotenone, we observed complete neuronal loss within 24 h. In contrast, the exposure of astrocytes to 10–40 nm rotenone for 24 h caused no obvious alteration of morphology (Fig. 1B) and no signs of cell death when quantified by trypan blue exclusion (Fig. 1C); this was expected because astrocytes are resistant to anoxia (Kelleher et al., 1993). Likewise, the addition of KCN at doses lethal to neurons (Campbell et al., 2011) was also well tolerated by both human astrocytes (Fig. 1D) and mouse astrocytes (data not shown) despite previously reported species differences in cultured astrocytes (Ahlemeyer et al., 2013). We therefore aimed to target mitochondrial complex IV in vivo, the functional loss of which is only associated with a delayed generation of ROS (Diaz et al., 2012), using a long-term in vivo approach in mutant mice.
Figure 1.

Cultured astrocytes resist mitochondrial respiratory chain complex I and complex IV inhibitors that are toxic for neurons. A, Cultures of rat cortical neurons were treated with rotenone for 24 h (concentrations as indicated) and immuostained for TuJ1 (red) and NeuN (green). Toxicity is indicated by the loss of staining and pyknotic neuronal nuclei (DAPI). Insets show condensed neuronal nuclei at higher magnification. Scale bars, 50 and 10 μm. B, Cultured astrocytes were treated with rotenone for 24 h (concentrations as indicated) and immunostained for GFAP (green). C, Quantified by a trypan blue exclusion assay, rotenone treatment did not affect astrocyte survival. Data are shown as mean ± SEM. D, Cultured human astrocytes, immunostained for vimentin (green) and aquaporin-4 (red), were resistant to KCN administered at low (50 μm) and high (1 mm) concentrations. Scale bar, 50 μm. n.s., Not significant.
Astrocyte-specific recombination of Cox10 and loss of cytochrome c oxidase
To determine whether adult astrocytes in the living brain are dependent on oxidative phosphorylation, we crossbred Cox10flox/flox mice (Diaz et al., 2005) with a tamoxifen-sensitive GLASTCreERT2 driver line that targets astrocytes selectively in a temporally controlled fashion (Mori et al., 2006; Buffo et al., 2008). In the absence of the farnesyl-transferase Cox10, functional COX cannot be assembled, as shown in other mouse models in which mutant cells either die or survive by aerobic glycolysis (Fukui et al., 2007; Fünfschilling et al., 2012).
Mice received daily tamoxifen injections for 5 consecutive days beginning at P30. Age-matched control mice were homozygous for the floxed Cox10 allele, but either lacked GLASTCreERT2 (when injected with tamoxifen) or expressed GLASTCreERT2 but received vehicle only. In the adult rodent brain, the half-life of mitochondria is 24–26 d (Beattie et al., 1967; Menzies and Gold, 1971). First histological and electron microscopy analyses were therefore performed at 3 months of age (Fig. 2A), when we expected a significant loss of mitochondrial respiration, and neuronal Cox10 mutants are clearly affected (data not shown). Later, we investigated long-term effects in Cox10 mutant mice at ∼1 year of age. All mutant mice were obtained at the expected Mendelian ratio, long-lived and indistinguishable in the cage from control littermates.
Figure 2.
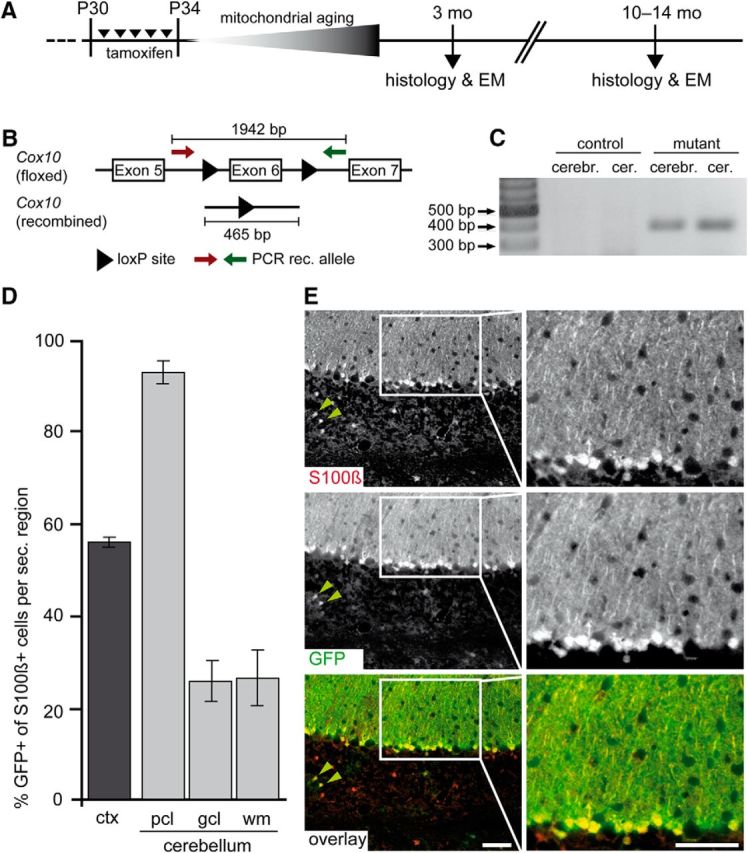
Inducible deletion of Cox10 and specific reporter gene recombination. A, GLASTCreERT2:Coxflox/flox mice (age P30) were tamoxifen injected for 5 consecutive days and analyzed months later as indicated. B, Schema of the floxed Cox10 gene with the excision of exon 6 (not drawn to scale). Arrows indicate the location of PCR primers. C, Verification of Cox10 genomic recombination in mutant brains and cerebelli at the age of 3 months. D, Estimated recombination efficiency of GLASTCreERT2 mice in the cortex (ctx) and cerebellar subregions (pcl, PC layer; gcl, granule cell layer; wm, white matter) by analysis of R26R-EYFP reporter gene expression. Astrocytes, identified by S100β colabeling, were up to 93% recombinant (mean ± SEM). E, Virtually all BG marked with S100β (top: red channel) expressed EYFP (middle: green channel, immunostained). Black and white images have been pseudocolored in the bottom panel. Scale bars, 50 μm.
Recombination of the floxed Cox10 locus was confirmed by PCR analysis of genomic DNA (Fig. 2B) purified from the cerebellum and the cerebrum. For both brain regions, we could determine the loss of exon 6 by the emergence of a 465 bp DNA fragment (Fig. 2C). From genomic DNA of control mice, no corresponding PCR product could be amplified.
Specificity of Cre recombination was addressed by crossbreeding Cox10flox/flox::GLASTCreERT2 mice to the reporter strain R26R-EYFP (Srinivas et al., 2001), leading to the excision of a floxed “stop” sequence and expression of yellow fluorescent protein (YFP). Positive identification of astrocytes was achieved by colabeling YFP+ cells for S100β, a calcium-binding protein and widely used astrocyte marker (Donato, 1991).
As expected, inducible GLASTCreERT2 expression was mosaic for cortical astrocytes (Mori et al., 2006). In the somatosensory cortex 55% of S100β-marked astrocytes were Cre recombined. In the cerebellum, on average, 75% of all S100β-positive astrocytes were Cre recombined, including 25% EYFP-labeled astrocytes in the granule cell layer and underlying white matter (Fig. 2D). However, cerebellar BG cells, a subgroup of astrocytes localized in the PC layer (Yamada and Watanabe, 2002), revealed a very high degree (93 ± 2.4%) of recombination (Fig. 2D,E). Therefore, further analyses focused on these cells.
To demonstrate functional loss of mitochondrial respiration in BG, we performed sequential COX and SDH histochemistry (Fig. 3A,B), a widely used technique to identify mitochondrial dysfunction (Campbell et al., 2011; Fünfschilling et al., 2012). This technique revealed the absence of cytochrome c oxidase activity from Cox10-deficient astrocytes by virtue of a characteristic blue precipitate that cannot form in wild-type cells and their processes (Fig. 3C,D). Here, cytochrome c oxidase generates a brown precipitate that prevents the subsequent formation of a blue SDH reaction product. To demonstrate the efficient loss of COX activity on the single-cell level, we cultured primary astrocytes obtained from Cox10-floxed mice for 8 weeks (allowing for mitochondrial protein turnover), transduced them with a Cre-expressing lentivirus, and applied COX/SDH histochemistry. The emergence of the blue and the absence of the brown precipitate when only COX medium was added to Cox10-floxed astrocytes upon lentiviral-mediated Cox10 depletion indicates the reliable inactivation of cytochrome c oxidase in all astrocytes (Fig. 3E).
Figure 3.
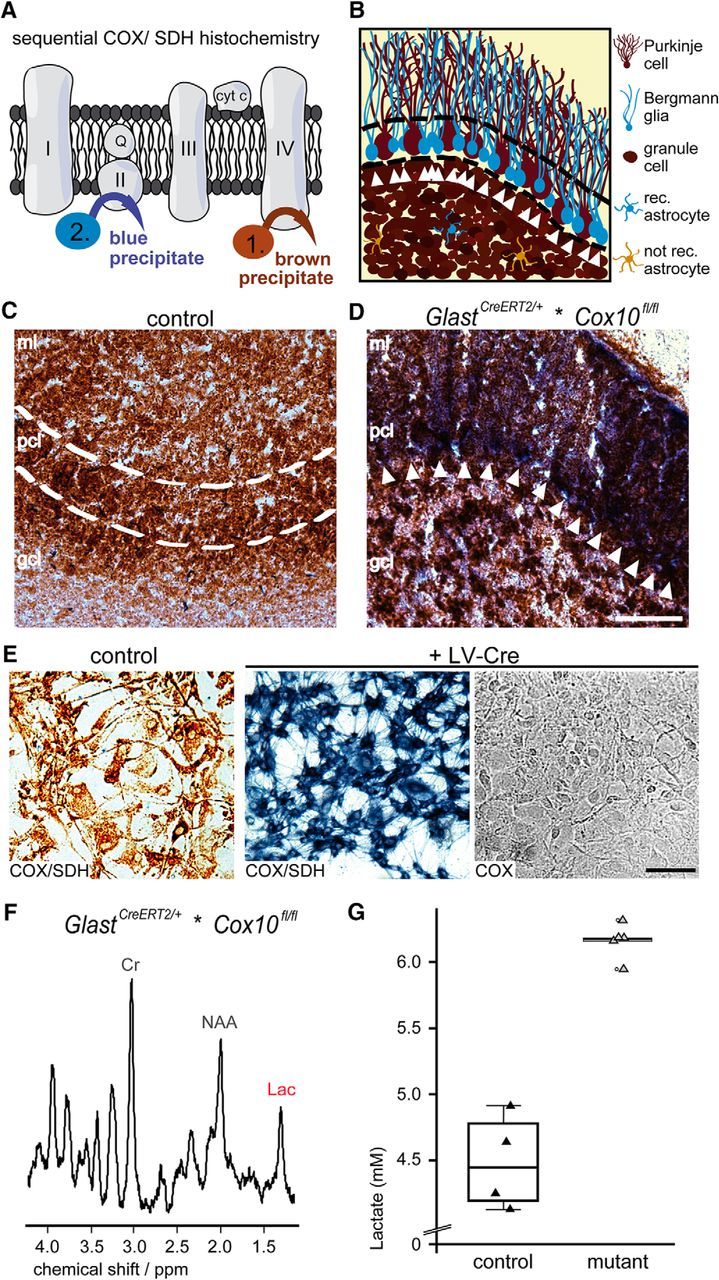
Sequential COX/SDH histochemistry proving the loss of COX activity in BG cells. A, Identification of respiration deficient (COX neg.) cells by sequential COX/SDH histochemistry. In control cells, COX activity generates a brown precipitate and prevents the emergence of a blue precipitate by the SDH reaction, which therefore marks mutant cells. B, Scheme illustrating mutant BG in the PC layer (blue, arrowheads) next to PCs (brown, COX active) as shown below. C, In control mice, mitochondrial COX activity leads to a brown precipitate in all cells of the cerebellum (age 14 months). D, In age-matched mutant mice, the blue precipitate marks BG cells (white arrows). Note also blue spots in the granule cell layer indicating astrocytes. Scale bar, 50 μm, E, Lentiviral-mediated deletion of Cox10 in cultured astrocytes maintained for 8 weeks in vitro leads to inactive cytochrome c oxidase in all astrocytes, as detectable by the emergence of a blue precipitate and the absence of a brown precipitate when only COX medium was applied. Scale bar, 100 μm. F, Localized proton MR spectra obtained from the cerebellum (voxel size: 4 × 1 × 2 mm3) revealed increased lactate concentrations in GlastCreERT2/+*COX10fl/fl mice. G, When quantified, mutant mice revealed a concentration of 6.2 ± 0.2 mm (controls 4.5 ± 0.4 mm). Lac, lactate; Cr, creatine; NAA, N-acetylaspartate; rec., Cre recombined; ml, molecular layer; pcl, PC layer; gcl, granule cell layer.
The fact that we observed Cox10-deficient BG in 1-year-old mutants implies that these astrocytes must have survived in the absence of mitochondrial respiration as glycolytic cells. Therefore, we hypothesized that astrocytes survived by increasing ATP production by aerobic glycolysis and predicted that elevated lactate levels would be detected in vivo in Cox10 mutant brains. Localized proton MRS detecting levels of different metabolites, among them lactate, was performed in aged mice (20–25 months). Indeed, under isoflurane anesthesia, Cox10 mutant mice showed increased brain lactate concentrations (Fig. 3F). When quantified, we determined in the cerebellum >6 mm in mutant and >4 mm in control mice (in the cortex, >4 mm in mutant and >3 mm in control, data not shown) (Fig. 3G).
Normal cerebellar organization and survival of BG cells
By histological staining (hematoxylin) at the age of 3 months and 14 months, loss of mitochondrial respiration in astrocytes did not affect the gross morphology of the cerebellum (or any other brain area). At both ages, cerebellar size and cytoarchitecture were unaffected in mutant mice (Fig. 4A and data not shown). When stained for S100β and coimmunolabeled for GFAP, the density and morphology of BG cells and their many processes were indistinguishable from wild-type (Fig. 4B,C). Therefore, respiration-deficient BG survive well in vivo. As shown by electron microscopy, these cells exhibited glycogen granules of normal size and their mitochondria were unaltered in size and number (Fig. 4D,E).
Figure 4.

Cerebellum and BG cell morphology in Cox10 conditional mutant mice. A, Hematoxylin staining showing that the cerebellum appears normal in mutant mice (14 months). Scale bar, 500 μm. B, Single BG cells colabeled for S100β (red) and GFAP (green) were morphologically indistinguishable in 14-month-old mutant mice and controls. Asterisks mark PCs (unstained). Scale bar, 100 μm. C, Quantification of BG cell numbers at the age of 3 and 9–10 months. D, At the electron microscope level, mutant BG exhibit glycogen inclusion of normal size and mitochondria are morphologically unaffected (age 10 months). Scale bar, 500 nm. E, Quantitation of mitochondrial data. Numbers are mean ± SEM. G, Glycogen; N, nucleus; M, mitochondria; n.s., not significant.
Analysis of glial cell death, neurodegeneration, and regeneration
CNS integrity could theoretically be preserved when Cox10 mutant astrocytes are replaced by newly generated astrocytes that have escaped Cre recombination. We explored the proliferation index of astrocytes by a 2-week daily treatment with EdU beginning 50 d after tamoxifen-induced deletion of Cox10. By costaining for S100β, we only detected sporadic EdU-labeled astrocytes. Similarly, the proliferation of EdU+ cells in the cerebella of Cox10 mutants and controls was the same (Fig. 5A,B).
Figure 5.

Survival of BG cells in conditional Cox10 mutant mice. A, As shown by EdU labeling, mitotic cells (in red) and S100β-positive astrocytes (in green) were not costained in the cerebellum of Cox10 mutant and control mice (arrowhead indicates a mitotic nonastrocytic cell). Scale bar, 20 μm. B, When quantified, the total number of EdU-positive cells is not different in the mutant cerebellum. Data are shown as mean ± SEM. cer. sec., Cerebellar section. C, D, No signs of cell death in the cerebellum were observed by TUNEL staining (age 3 months; C), in contrast to a positive control (D), where Cox10 mutant neurons undergo apoptosis in the forebrain of CKIIα-Cre*Cox10fl/fl mice (age 4 months). Scale bar, 100 μm. n.s., Not significant.
To detect apoptotic mutant cells directly, we performed a TUNEL assay. No evidence for increased apoptotic cell death was found in the brains of mutant or age-matched control mice at the ages of 3 and 10 months (Fig. 5C and data not shown). For comparison, when using CKIIα-Cre mice to delete Cox10 in cortical neurons (beginning at age P5–P10), we observed massive apoptosis of projection neurons in the forebrain at 4 months (Fig. 5D). As shown previously, these mice suffer from severe neurodegeneration, confirming that neuronal survival requires oxidative phosphorylation (Fukui et al., 2007; Diaz, 2010), a striking difference from the survival of mutant glial cells.
Lack of secondary neuroinflammation
Neuroinflammation is a sensitive response to perturbations of CNS integrity and metabolic distress (Xanthos and Sandkühler, 2014). However, we found no evidence for astrogliosis by GFAP immunostaining of brains from 14-month-old Cox10 mutant mice (Fig. 6A). Immunostaining for Mac3, an inducible marker of activated microglia, revealed no alterations compared with age-matched controls (Fig. 6B). We detected a few CD3+ T cells in brain sections, but they were comparable in number to control brains (Fig. 6C). The accumulation of APP-positive endosomes in axonal spheroids is a sign of disturbed axonal transport and a key feature of respiration-deficient cortical projection neurons. However, conditional Cox10flox/flox::GLASTCreERT2 mutants did not exhibit any APP accumulation at the two ages tested (Fig. 6D and data not shown).
Figure 6.

No signs of inflammation and neurodegeneration were seen in Glast-Cre::Cox10 mutant mice. A, Immunostaining of sagittal cerebellar sections for GFAP revealed no signs of gliosis in mutant mice (age 14 months). B, Activated Mac3-positive microglia (arrowheads) were not more frequent in mutant than in control mice. C, Invading T-cells, immunostained for CD3 (arrowheads), were not significantly increased in mutants compared with age-matched controls. D, As shown by APP immunostaining, axonal swellings were not observed in control and Glast-Cre::Cox10fl/fl mutant mice, in contrast to the forebrain of CKIIα-Cre::Cox10fl/fl mice (positive control, right). All sections are counterstained with hematoxylin (blue). Scale bars, 100 μm. Numbers are mean ± SEM. n.s., Not significant.
BG morphology and synapse density
Astrocytes contribute to the formation and maintenance of functional synapses. To explore whether respiration-deficient BG cells and their processes cause morphological changes of synapses between PF axons and PF dendritic spines, we performed an electron microscopy study. Quantification of PF–PC synapse density in the upper part of the cerebellar molecular layer revealed no significant difference between Cox10 mutant mice and controls (Fig. 7A,B). We then investigated whether BG processes surrounding the PF–PC synapses are morphologically intact. Swollen processes could indicate lactate accumulation (Rehncrona, 1985), whereas retracted processes would suggest synaptic dysfunction. However, we observed no morphological differences and the calculation of BG “process area” in mutants and controls was the same at both 3 and 10 months of age (Fig. 7A,C). Moreover, mitochondrial density in BG processes was not affected in mutants compared with age-matched controls (Fig. 7D). This is in agreement with our conclusion that glycolytic astrocytes are healthy and maintain normal brain functions in vivo.
Figure 7.

BG cell processes covering PC synapses. A, Electron micrographs showing BG processes (false-colored in pink) at PF–PC synapses, marked with asterisks, in the upper third of the molecular layer. Images from mutant and control mice are indistinguishable (age 9–10 months). Scale bar, 1 μm. B, PF–PC synapse density in the upper part of the molecular layer was unaltered in mutant mice at the indicated ages. C, PC synapses are normally covered by BG processes in mutant mice at both time points. D, Calculated mitochondrial density of BG processes was unaltered in mutant mice compared with controls. Data shown as mean ± SEM. n.s., Not significant.
Discussion
The cellular origin and metabolic fate of brain lactate has been difficult to study in vivo, which has led to diverse hypotheses, often based on conflicting data obtained in vitro (Pellerin and Magistretti, 1994; DiNuzzo et al., 2010; Dienel, 2012; Bouzier-Sore and Pellerin, 2013; Hertz et al., 2014; Patel et al., 2014; Magistretti and Allaman, 2015; Mächler et al., 2016). Direct in vivo approaches to determine the origin of endogenously generated brain lactate are technically difficult to implement. To address the question of whether astrocytes in the living brain are, at least in principle, a continuous source of lactate for neighboring cells, we generated a new mouse model in which astrocytes that could not survive by aerobic glycolysis alone would simply die. This was possible by genetically targeting Cox10 and inactivating the mitochondrial complex IV (COX) in a large subset of adult astrocytes. Successful recombination of the tamoxifen-inducible GLASTCreERT2 mouse line (Mori et al., 2006) was shown by reporter gene expression (EYFP) of nearly all BG cells in the cerebellum (Srinivas et al., 2001). The extent of recombination in cortical astrocytes may have been underestimated in the R26R-EYFP reporter mice. Successful elimination of mitochondrial respiration in BG was shown by the absence of COX activity (by sequential COX/SDH histochemistry), even in aged mutant mice, and the efficient inactivation of COX on a single-cell level was demonstrated in cultured Cox10-floxed astrocytes. COX/SDH histochemistry is a very specific technique in which the complete loss of mitochondrial cytochrome c oxidase activity is detected reliably. If anything, the opposite poses a problem: that COX deficiencies are overlooked in mitochondrial diseases because the reaction products saturate despite significantly reduced enzymatic activity (Mahad et al., 2009; Rocha et al., 2015)
Cox10 mutant cells are forced to live glycolytically, so lactate is a necessary by-product generated to maintain the NAD+/NADH ratio required for continuous glycolysis (as shown in Cnp1-Cre::Coxflox/flox mice by Fünfschilling et al., 2012). As shown by localized proton MRS, elevated lactate concentrations in the cerebellum of Cox10 mutant mice could be observed in vivo under isoflurane anesthesia (from 4.5 mm to >6 mm in mutant cerebella), demonstrating that mutant astrocytes survive by sufficient ATP supply from enhanced aerobic glycolysis. Given that the these measurements include a large background of genetically wild-type cells (mostly cerebellar granule cells), the data demonstrate that mutant astrocytes are a source of elevated lactate levels and this lactate is neither stored as glycogen nor metabolized by OXPHOS in these astrocytes, but rather shuttled out of mutant astrocytes. Indeed, in this experimental situation, purely glycolytic astrocytes will use more of the available glucose than normal, which may increase the lactate utilization by “glucose-starved” neurons. Therefore, the flow rate of lactate (from astrocytes to neurons) may be higher in mutant compared with wild-type brains.
Mutant GLASTCreERT2::Coxflox/flox mice, observed up to 14 months of age, were phenotypically indistinguishable from controls. The cerebellum and all other CNS areas investigated were histologically normal, including the morphology and density of BG cells themselves, and this was also consistent with the normal survival of rotenone- and potassium cyanide-treated astrocytes in vitro. Importantly, no change in mitochondrial numbers or mitochondrial morphology was detected. The possibility that mitochondrial respiration might persist due to fusion or fission events is highly unlikely because Cox10 is a nuclear gene and its recombination leads to the gradual loss of complex IV from all mitochondria. We note that swellings have been observed as signs of “unphysiological” mitochondrial dysfunction, for example, in Cox10-deficient skeletal muscle cells, hepatocytes, and Schwann cells (Diaz et al., 2005, 2008; Fünfschilling et al., 2012). Our findings here suggest that a physiological adaptation has taken place, possibly as a result of normal astroglial differentiation, which could include a developmental “metabolic switch,” but this possibility needs to be further investigated. We note that a similar metabolic switch has been associated with the differentiation of myelinating oligodendrocytes (Fünfschilling et al., 2012) and is reminiscent of the “Warburg effect” in many tumor cells (Gatenby and Gillies, 2004).
Axonal swellings, APP accumulation, and neuroinflammation comprise a sensitive response to neurodegenerative processes. Their absence at any age tested suggests that mutant astrocytes (and their neighbors) are not distressed by the lack of mitochondrial respiration. BG cells play an important role in maintaining the physiology and plasticity of synapses between PFs and PF dendrites (Saab et al., 2012). The densities of cerebellar PF–PC synapses and the area occupied by astrocytic processes enwrapping these synapses was the same in mutant and control mice.
Our data do not suggest that astrocytes do not require mitochondrial respiration. In postnatal development, during cellular growth, and after CNS injury, it is even likely that normal astrocyte behavior requires mitochondrial ATP generation. However, our work provides in vivo evidence that in the adult steady-state respiration-deficient BG cells survive normally, which necessitates that they generate ATP merely by aerobic glycolysis. The data also imply that these astrocytes at least are a continuous source (not a sink) of lactate; that is, not for short time, but rather for 1 year or more. In the cerebellum, this applies to >90% of all BG cells in the absence of any sign of pathology or cerebellar dysfunction. Although it remains a theoretical possibility that lactate fluxes are very different in wild-type mice, we take our phenotypical data as strong supportive in vivo evidence for a model of astrocytes as naturally glycolytic cells.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Grants SPP1757 and CNMPB) to K.-A.N. and Progressive MS Alliance Planning Grant to D.M. K.-A.N. holds a European Research Council Advanced Investigator Grant. We thank W. Möbius, T. Ruhwedel, and A. Fahrenholz for support with electron microscopy and histology; T. Pawelz and C. Casper for mice husbandry; P. Brown (Biomolecular Core, University of Edinburgh) for providing Cre expressing lentivirus; and A. Mot for critical reading of the manuscript.
The authors declare no competing financial interests.
References
- Ahlemeyer B, Kehr K, Richter E, Hirz M, Baumgart-Vogt E, Herden C (2013) Phenotype, differentiation, and function differ in rat and mouse neocortical astrocytes cultured under the same conditions. J Neurosci Methods 212:156–164. 10.1016/j.jneumeth.2012.09.016 [DOI] [PubMed] [Google Scholar]
- Arai K, Lo EH (2010) Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J Neurosci Res 88:758–763. 10.1002/jnr.22256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA (2010) Glial and neuronal control of brain blood flow. Nature 468:232–243. 10.1038/nature09613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie DS, Basford RE, Koritz SB (1967) The turnover of the protein components of mitochondria from rat liver, kidney, and brain. J Biol Chem 242:4584–4586. [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14:724–738. 10.1016/j.cmet.2011.08.016 [DOI] [PubMed] [Google Scholar]
- Boumezbeur F, Petersen KF, Cline GW, Mason GF, Behar KL, Shulman GI, Rothman DL (2010) The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J Neurosci 30:13983–13991. 10.1523/JNEUROSCI.2040-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Pellerin L (2013) Unraveling the complex metabolic nature of astrocytes. Front Cell Neurosci 7:179. 10.3389/fncel.2013.00179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Voisin P, Bouchaud V, Bezancon E, Franconi JM, Pellerin L (2006) Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur J Neurosci 24:1687–1694. 10.1111/j.1460-9568.2006.05056.x [DOI] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M (2008) Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A 105:3581–3586. 10.1073/pnas.0709002105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. 10.1523/JNEUROSCI.4178-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O, Lassmann H, Turnbull DM, Mahad DJ (2011) Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 69:481–492. 10.1002/ana.22109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz F. (2010) Cytochrome c oxidase deficiency: patients and animal models. Biochim Biophys Acta 1802:100–110. 10.1016/j.bbadis.2009.07.013 [DOI] [PubMed] [Google Scholar]
- Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT (2005) Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet 14:2737–2748. 10.1093/hmg/ddi307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz F, Garcia S, Hernandez D, Regev A, Rebelo A, Oca-Cossio J, Moraes CT (2008) Pathophysiology and fate of hepatocytes in a mouse model of mitochondrial hepatopathies. Gut 57:232–242. 10.1136/gut.2006.119180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz F, Garcia S, Padgett KR, Moraes CT (2012) A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum Mol Genet 21:5066–5077. 10.1093/hmg/dds350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA. (2012) Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab 32:1107–1138. 10.1038/jcbfm.2011.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA, Cruz NF (2015) Contributions of glycogen to astrocytic energetics during brain activation. Metab Brain Dis 30:281–298. 10.1007/s11011-014-9493-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M, Mangia S, Maraviglia B, Giove F (2010) Changes in glucose uptake rather than lactate shuttle take center stage in subserving neuroenergetics: evidence from mathematical modeling. J Cereb Blood Flow Metab 30:586–602. 10.1038/jcbfm.2009.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R. (1991) Perspectives in S-100 protein biology. Cell Calcium 12:713–726. 10.1016/0143-4160(91)90040-L [DOI] [PubMed] [Google Scholar]
- Fox PT, Raichle ME (1986) Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc Natl Acad Sci U S A 83:1140–1144. 10.1073/pnas.83.4.1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PT, Raichle ME, Mintun MA, Dence C (1988) Nonoxidative glucose consumption during focal physiologic neural activity. Science 241:462–464. 10.1126/science.3260686 [DOI] [PubMed] [Google Scholar]
- Fukui H, Diaz F, Garcia S, Moraes CT (2007) Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 104:14163–14168. 10.1073/pnas.0705738104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, Nave KA (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485:517–521. 10.1038/nature11007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899. 10.1038/nrc1478 [DOI] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75:762–777. 10.1016/j.neuron.2012.08.019 [DOI] [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP (2009) The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11:747–752. 10.1038/ncb1881 [DOI] [PubMed] [Google Scholar]
- Hertz L, Gibbs ME, Dienel GA (2014) Fluxes of lactate into, from, and among gap junction-coupled astrocytes and their interaction with noradrenaline. Front Neurosci 8:261. 10.3389/fnins.2014.00261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirrlinger J, Nave KA (2014) Adapting brain metabolism to myelination and long-range signal transduction. Glia 62:1749–1761. 10.1002/glia.22737 [DOI] [PubMed] [Google Scholar]
- Itoh Y, Esaki T, Shimoji K, Cook M, Law MJ, Kaufman E, Sokoloff L (2003) Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc Natl Acad Sci U S A 100:4879–4884. 10.1073/pnas.0831078100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joannides AJ, Fiore-Hériché C, Battersby AA, Athauda-Arachchi P, Bouhon IA, Williams L, Westmore K, Kemp PJ, Compston A, Allen ND, Chandran S (2007) A scaleable and defined system for generating neural stem cells from human embryonic stem cells. Stem Cells 25:731–737. [DOI] [PubMed] [Google Scholar]
- Kelleher JA, Chan PH, Chan TY, Gregory GA (1993) Modification of hypoxia-induced injury in cultured rat astrocytes by high levels of glucose. Stroke 24:855–863. 10.1161/01.STR.24.6.855 [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448. 10.1038/nature11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl PE, Oberg KE (1961) The effect of rotenone on respiration and its point of attack. Exp Cell Res 23:228–237. 10.1016/0014-4827(61)90033-7 [DOI] [PubMed] [Google Scholar]
- Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M (2007) The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci 27:12255–12266. 10.1523/JNEUROSCI.3404-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martín A, Romero-Gómez I, Baeza-Lehnert F, Lengacher S, Schneider BL, Aebischer P, Magistretti PJ, Barros LF, Weber B (2016) In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab 23:94–102. 10.1016/j.cmet.2015.10.010 [DOI] [PubMed] [Google Scholar]
- Magistretti PJ. (2006) Neuron-glia metabolic coupling and plasticity. J Exp Biol 209:2304–2311. 10.1242/jeb.02208 [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Allaman I (2015) A cellular perspective on brain energy metabolism and functional imaging. Neuron 86:883–901. 10.1016/j.neuron.2015.03.035 [DOI] [PubMed] [Google Scholar]
- Mahad DJ, Ziabreva I, Campbell G, Laulund F, Murphy JL, Reeve AK, Greaves L, Smith KJ, Turnbull DM (2009) Detection of cytochrome c oxidase activity and mitochondrial proteins in single cells. J Neurosci Methods 184:310–319. 10.1016/j.jneumeth.2009.08.020 [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Miller MW (2003) Culturing hippocampal and cortical neurons. Methods Cell Biol 71:111–127. 10.1016/S0091-679X(03)01007-0 [DOI] [PubMed] [Google Scholar]
- Menzies RA, Gold PH (1971) The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem 246:2425–2429. [PubMed] [Google Scholar]
- Mergenthaler P, Lindauer U, Dienel GA, Meisel A (2013) Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36:587–597. 10.1016/j.tins.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R (1999) Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24:401–414. 10.1016/S0896-6273(00)80853-3 [DOI] [PubMed] [Google Scholar]
- Mori T, Tanaka K, Buffo A, Wurst W, Kühn R, Götz M (2006) Inducible gene deletion in astroglia and radial glia: a valuable tool for functional and lineage analysis. Glia 54:21–34. 10.1002/glia.20350 [DOI] [PubMed] [Google Scholar]
- Morrison BM, Lee Y, Rothstein JD (2013) Oligodendroglia: metabolic supporters of axons. Trends Cell Biol 23:644–651. 10.1016/j.tcb.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V (1974) The neuroglial cells of the cerebellar cortex. In: Cerebellar cortex (Palay SL, Chan-Pay V, eds), pp. 288–321. Berling: Springer. [Google Scholar]
- Patel AB, Lai JC, Chowdhury GM, Hyder F, Rothman DL, Shulman RG, Behar KL (2014) Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc Natl Acad Sci U S A 111:5385–5390. 10.1073/pnas.1403576111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ (1994) Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 91:10625–10629. 10.1073/pnas.91.22.10625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ (2012) Sweet sixteen for ANLS. J Cereb Blood Flow Metab 32:1152–1166. 10.1038/jcbfm.2011.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, Stella N, Magistretti PJ (1998) Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci 20:291–299. 10.1159/000017324 [DOI] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA (2014) Activity-driven local ATP synthesis is required for synaptic function. Cell 156:825–835. 10.1016/j.cell.2013.12.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen P, Wyss MT, Lundby C (2011) Cerebral glucose and lactate consumption during cerebral activation by physical activity in humans. FASEB J 25:2865–2873. 10.1096/fj.11-183822 [DOI] [PubMed] [Google Scholar]
- Rehncrona S. (1985) Brain acidosis. Ann Emerg Med 14:770–776. 10.1016/S0196-0644(85)80055-X [DOI] [PubMed] [Google Scholar]
- Reynolds ES. (1963) The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol 17:208–212. 10.1083/jcb.17.1.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha MC, Grady JP, Grünewald A, Vincent A, Dobson PF, Taylor RW, Turnbull DM, Rygiel KA (2015) A novel immunofluorescent assay to investigate oxidative phosphorylation deficiency in mitochondrial myopathy: understanding mechanisms and improving diagnosis. Sci Rep 5:15037. 10.1038/srep15037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab AS, Neumeyer A, Jahn HM, Cupido A, Šimek AA, Boele HJ, Scheller A, Le Meur K, Götz M, Monyer H, Sprengel R, Rubio ME, Deitmer JW, De Zeeuw CI, Kirchhoff F (2012) Bergmann glial AMPA receptors are required for fine motor coordination. Science 337:749–753. 10.1126/science.1221140 [DOI] [PubMed] [Google Scholar]
- Sinnamon JR, Waddell CB, Nik S, Chen EI, Czaplinski K (2012) Hnrpab regulates neural development and neuron cell survival after glutamate stimulation. RNA 18:704–719. 10.1261/rna.030742.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1:4. 10.1186/1471-213X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144:810–823. 10.1016/j.cell.2011.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR (2000) Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J Neurosci 20:6804–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B (2011) In vivo evidence for lactate as a neuronal energy source. J Neurosci 31:7477–7485. 10.1523/JNEUROSCI.0415-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthos DN, Sandkühler J (2014) Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci 15:43–53. 10.1038/nrn3617 [DOI] [PubMed] [Google Scholar]
- Yamada K, Watanabe M (2002) Cytodifferentiation of Bergmann glia and its relationship with Purkinje cells. Anat Sci Int 77:94–108. 10.1046/j.0022-7722.2002.00021.x [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziabreva I, Campbell G, Rist J, Zambonin J, Rorbach J, Wydro MM, Lassmann H, Franklin RJ, Mahad D (2010) Injury and differentiation following inhibition of mitochondrial respiratory chain complex IV in rat oligodendrocytes. Glia 58:1827–1837. 10.1002/glia.21052 [DOI] [PMC free article] [PubMed] [Google Scholar]