

Abstract
The atypical vesicular glutamate transporter type 3 (VGLUT3) is expressed by subpopulations of neurons using acetylcholine, GABA, or serotonin as neurotransmitters. In addition, VGLUT3 is expressed in the inner hair cells of the auditory system. A mutation (p.A211V) in the gene that encodes VGLUT3 is responsible for progressive deafness in two unrelated families. In this study, we investigated the consequences of the p.A211V mutation in cell cultures and in the CNS of a mutant mouse. The mutation substantially decreased VGLUT3 expression (−70%). We measured VGLUT3-p.A211V activity by vesicular uptake in BON cells, electrophysiological recording of isolated neurons, and its ability to stimulate serotonergic accumulation in cortical synaptic vesicles. Despite a marked loss of expression, the activity of the mutated isoform was only minimally altered. Furthermore, mutant mice displayed none of the behavioral alterations that have previously been reported in VGLUT3 knock-out mice. Finally, we used stimulated emission depletion microscopy to analyze how the mutation altered VGLUT3 distribution within the terminals of mice expressing the mutated isoform. The mutation appeared to reduce the expression of the VGLUT3 transporter by simultaneously decreasing the number of VGLUT3-positive synaptic vesicles and the amount of VGLUT3 per synapses. These observations suggested that VGLUT3 global activity is not linearly correlated with VGLUT3 expression. Furthermore, our data unraveled a nonuniform distribution of VGLUT3 in synaptic vesicles. Identifying the mechanisms responsible for this complex vesicular sorting will be critical to understand VGLUT's involvement in normal and pathological conditions.
SIGNIFICANCE STATEMENT VGLUT3 is an atypical member of the vesicular glutamate transporter family. A point mutation of VGLUT3 (VGLUT3-p.A211V) responsible for a progressive loss of hearing has been identified in humans. We observed that this mutation dramatically reduces VGLUT3 expression in terminals (∼70%) without altering its function. Furthermore, using stimulated emission depletion microscopy, we found that reducing the expression levels of VGLUT3 diminished the number of VGLUT3-positive vesicles at synapses. These unexpected findings challenge the vision of a uniform distribution of synaptic vesicles at synapses. Therefore, the overall activity of VGLUT3 is not proportional to the level of VGLUT3 expression. These data will be key in interpreting the role of VGLUTs in human pathologies.
Keywords: DFNA25, mutant mice VGLUT3A224V/A224V, point mutation (p.A211V), STED, synaptic vesicles, vesicular glutamate transporter 3 (VGLUT3)
Introduction
Glutamate accumulation within synaptic vesicles (SVs) is facilitated by the vesicular glutamate transporters VGLUT1, VGLUT2, and VGLUT3 (for review, see El Mestikawy et al., 2011). VGLUTs are crucial anatomical and functional markers of glutamatergic transmission. The amount of glutamate that is packaged into SVs and the amount of glutamate that is released are believed to be proportional to the density of VGLUT expression (Daniels et al., 2004, 2006, 2011; Wilson et al., 2005; Moechars et al., 2006). In addition to glutamate vesicular packaging, additional roles for the VGLUTs have recently been revealed. For example, VGLUTs influence the mobility of SVs in the axon and their stability in the synapse, as well as the intrinsic release probability of glutamatergic vesicles (Weston et al., 2011; Siksou et al., 2013; Herman et al., 2014).
The VGLUTs are structurally and functionally similar but are anatomically segregated. VGLUT1 and VGLUT 2 are used by cortical and subcortical excitatory terminals, respectively (Bellocchio et al., 2000; Takamori et al., 2000, 2001; Fremeau et al., 2001; Herzog et al., 2001; Varoqui et al., 2002). In contrast, VGLUT3 is localized in small populations of neurons using neurotransmitters other than glutamate, such as cholinergic interneurons in the striatum, subsets of GABAergic interneurons in the hippocampus and cortex, and serotonergic neurons in the dorsal and median raphe nuclei (Fremeau et al., 2002; Gras et al., 2002; Schäfer et al., 2002; Takamori et al., 2002; Herzog et al., 2004). At the cellular level, VGLUTs are present in terminals. However, unlike VGLUT1 and VGLUT2, VGLUT3 is also observed in somato-dendritic compartments (Herzog et al., 2004). VGLUT3 facilitates vesicular accumulation and release of acetylcholine, serotonin, and GABA via a mechanism called “vesicular synergy” (Gras et al., 2008; Amilhon et al., 2010; Zander et al., 2010). VGLUT3 confers on “non-glutamatergic” cells the ability to release glutamate (Varga et al., 2009; Higley et al., 2011; Nelson et al., 2014).
Mice that no longer express VGLUT3 (VGLUT3−/−) display hypersensitivity to pain, increased anxiety, and increased sensitivity to cocaine (Gras et al., 2008; Seal et al., 2009; Amilhon et al., 2010; Peirs et al., 2015; Sakae et al., 2015). Interestingly, heterozygous mice display no specific altered phenotype.
VGLUT3 is expressed by sensory inner hair cells in the auditory system. Therefore, VGLUT3−/− mice are profoundly deaf (Ruel et al., 2008; Seal et al., 2008). In humans, the p.A211V mutation of the gene that encodes VGLUT3 (Slc17a8) is responsible for a type of progressive deafness, DFNA25 (Ruel et al., 2008). This observation was the first evidence of an association between human pathology and a VGLUT mutation.
The mutated alanine is part of a peptide sequence that is highly conserved among the VGLUTs and their orthologs. The A211 alanine in human VGLUTs corresponds to position 224 in the mouse VGLUT3 sequence (A224). In this study, we generated a mutant mouse carrying the p.A211V mutation (VGLUT3A224V/A224V) and investigated its effects on the CNS. The VGLUT3A224V/A224V mice present the same progressive loss of hearing reported in humans (Ruel et al., 2008).
In the CNS, we observed that the amount of mutated VGLUT3-p.A224V protein was dramatically reduced in nerve endings (∼70%). Moreover, VGLUT3-dependent vesicular accumulation and synaptic release of glutamate were only minimally altered in VGLUT3A224V/A224V mice. Increased anxiety as well as increased basal or cocaine-induced locomotor activity have been observed in VGLUT3−/− mice. Despite substantial loss of VGLUT3, VGLUT3A224V/A224V mice displayed no such behavioral alterations. Therefore, the molecular, cellular, and behavioral functions of VGLUT3 are as efficiently fulfilled with 100% or with 30% expression of the transporter. This observation suggests that VGLUT3 global activity is not linearly correlated with the amounts of VGLUT3 present in terminals.
Finally, with stimulated emission depletion (STED) microscopy, we observed a decrease in VGLUT3-p.A224V-positive vesicles in the terminals. Together, these observations establish that the p.A211V mutation has complex effects on VGLUT3 expression and targeting.
Materials and Methods
Animals
Animal care and experiments were conducted in accordance with the European Communities Council Directive for the Care and the Use of Laboratory Animals (86/809/EEC) and in compliance with the Ministère de l'Agriculture et de la Forêt, Service Vétérinaire de la Santé et de la Protection Animale (authorization number 01482.01 from ethics committee Darwin #5). All efforts were made to minimize the number of animals used in the course of the study and to ensure their well-being. The animals were housed in a temperature-controlled room (21 ± 2°C) with ad libitum access to water and food under a 12 h light/dark cycle (lights on 7:30 A.M. to 7:30 P.M.).
Construction, genotyping, and breeding of VGLUT3A224V/A224V and VGLUT3A224V/− mice
The p.A211V mutation has been described in two unrelated human families (Ruel et al., 2008). The alanine at position 211 of human VGLUT3 is part of a KWAPPLER motif and is highly conserved in all three VGLUTs among different species (Ruel et al., 2008). In mouse VGLUT1 and VGLUT3, this alanine is at positions 198 and 224, respectively (Fig. 1A). A mouse line expressing the p.A224V mutation was generated at Phenomin–Institut Clinique de la Souris (Illkirch, France; http://www.phenomin.fr/) and was named VGLUT3A224V/A224V. A point mutation was introduced in exon 5 of the mouse Slc17a8 gene: a GCG (coding for an alanine) was exchanged for a GTG (coding for a valine; Fig. 1B). Mice were genotyped by PCR analysis of tail DNA with the following PCR primers: p1, 5′-CGGAGGGGAAGCCAGGAAAGGG-3′, and p2, 5′-GACAGCTCAGTGAGCTGTAGACCCAG-3′ for the WT and the mutated allele, yielding bands of 219 and 306 bp, respectively (Fig. 1C).
Figure 1.

Alignment of VGLUT protein sequences and the VGLUT3A224V/A224V mouse genomic construct. A, Alignment of mouse (mVGLUT3) and human (hVGLUT3) VGLUT3 and mouse (mVGLUT1) VGLUT1 amino acid sequences. The three peptide sequences are highly conserved (black letters indicate residues conserved in the sequences, and blue letters indicate residues that are different). The KWAPPLER motif (red boxed text) is conserved in all three sequences. The mutated alanine residue is in red. This alanine is at positions 224, 211, and 198 in the mVGLUT3, hVGLUT3, and mVGLUT1 amino acid sequences, respectively. B, Schematic representation of the targeting strategy. A targeting vector was constructed in which the GCG codon (encoding alanine 224) is replaced by a GTG codon in exon 5. A neomycin resistance selection cassette (flanked by two sites, Lox and LoxP_Neo_LoxP) was integrated downstream of exon 5. An auto-excision of the selection cassette in a male chimera germ line provided a targeting allele with a valine at position 224 in exon 5 and a LoxP site used for genotyping. C, Genotyping strategy of mouse VGLUT3 by PCR. Mice were genotyped with two primers (arrowheads P1 and P2 in B) flanking each side of the LoxP site. PCR amplification of the WT allele (+) yielded a 219 bp band, and the mutated allele (A224V) yielded a 306 bp band in an agarose gel. Asterisks represent the targeted site in exon 5.
Mice used in the study were age 10 days to 12 months. They were obtained by crossing heterozygous VGLUT3A224V/+ (C57BL/6N genetic background) or VGLUT3+/− (VGLUT3−/−; Sakae et al., 2015) mice with VGLUT3A224V/+ (C57BL/6N) mice (Table 1). Breeding provided mice expressing (1) two copies of the VGLUT3 WT allele (VGLUT3+/+ or WT), (2) one copy of mutated and one copy of VGLUT3 WT allele (VGLUT3A224V/+), (3) two copies of mutated alleles (VGLUT3A224V/A224V), or (4) only one copy of mutated alleles (VGLUT3A224V/−).
Table 1.
List of mutant mice used in this study
| Name | Number of copies of VGLUT3 allele |
||
|---|---|---|---|
| VGLUT3+ | VGLUT3A224V | VGLUT3− | |
| WT or VGLUT3+/+ | 2 | 0 | 0 |
| VGLUT3A224V/+ | 1 | 1 | 0 |
| VGLUT3A224V/A224V | 0 | 2 | 0 |
| VGLUT3A224V/− | 0 | 1 | 1 |
| VGLUT3−/ − | 0 | 0 | 2 |
Male littermates were used for the behavioral analysis, and females or males were used for the anatomical and biochemical experiments. The animals were randomly allocated to the experimental groups. Whenever possible, investigators were blinded to the genotypes during the experimental procedures. Animals were excluded from the experimental data analysis only when their results were detected as outliers using Grubb's test (GraphPad Prism, GraphPad Software).
Behavioral experiments
Spontaneous locomotor activity.
Basal locomotor activity was assessed as described previously (Gras et al., 2008). Mice were placed individually in activity boxes (20 × 15 × 25 cm), where their horizontal and vertical activities were measured by photocell beams located across the long axis, 15 mm (horizontal activity) and 30 mm (vertical activity) above the floor. Each box was connected by an interface to a computer (Imetronic). Spontaneous locomotor activity was measured in 15 min intervals over 5 h between 6:30 and 11:30 P.M.
Open field.
The open-field test was performed in a white Perspex arena (43 × 43 × 26 cm) located in a 10 lux illuminated room, as previously reported (Amilhon et al., 2010). The virtual central compartment square represented one-third of the total arena. Mice were introduced into the central area and allowed to freely explore the open field for 360 s. The durations, frequencies, and time courses of various behaviors (walking, rearing, stretching and grooming) were measured in different regions of the open field (central vs periphery zone). The time and number of entries in the center of the open field were evaluated as an index of an anxiety-related response.
Elevated plus maze.
The elevated plus maze (EPM) was used to measure unconditioned anxiety-like behavior (Amilhon et al., 2010). The EPM, which consisted of two open arms, two enclosed arms, and a central platform elevated 38.5 cm above the ground, was placed into 10 lux ambient light. After being allowed 1 h of habituation in the testing room, the animals were placed in the central area, facing one of the closed arms, and were tested for 360 s. The total time spent in each compartment (open vs closed arms) was recorded by video tracking (Viewpoint).
Cocaine-induced locomotor activity.
Cocaine-induced locomotor activity was measured in a cyclotron, which consisted of a circular corridor with four infrared beams placed at 90° angles (Imetronic). Activity was counted as the consecutive interruption of two adjacent infrared beams (one-fourth of a tour). To assess acute cocaine-induced locomotion, the animals were (1) placed in the cyclotron for 4 h for habituation, (2) given injections of saline (0.9% NaCl) and placed back in the cyclotron for 60 min, and (3) given injections of cocaine (10 mg/kg, i.p.). Locomotion was recorded for 95 min after cocaine injection.
Neuronal microculture and electrophysiological recording of autapses
Hippocampi were harvested at postnatal day 0 (P0) to P1 from VGLUT1−/− mice of either sex (Wojcik et al., 2004). Neurons were plated on island cultures at a density of 2000–3000 neurons per 35 mm dish. Recordings were performed from 14 to 18 days in vitro (DIV). VGLUT1−/− autaptic neurons were infected with either VGLUT3 or VGLUT3-p.A211V lentiviral vectors (10 and 40 ng of p24 per well, respectively). To normalize expression levels and to compare electrophysiological activity of the two isoforms, five times more VGLUT3-p.A211V than WT-expressing viral particles were used to rescue VGLUT1−/− hippocampal neurons.
The standard extracellular solution contained the following (in mm): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 MgCl2, and 2 CaCl2, pH 7.3. The internal solution contained the following (in mm): 135 KCl, 18 HEPES, 1 EGTA, 4.6 MgCl2, 4 ATP, 0.3 GTP, 15 creatine phosphate, and 20 U/ml phosphocreatine kinase. EPSCs were evoked by 2 ms of depolarization at 0 mV, resulting in an unclamped action potential. Readily releasable vesicle pool (RRP) size was assessed by pulsed (5 s) application of a hypertonic sucrose solution (500 mm sucrose added to extracellular solution) and by integrating the transient inward current component. Vesicular release probability was computed by dividing the EPSC charge by the RRP charge. Current traces were analyzed using Axograph X, Excel (Microsoft), and Prism (GraphPad Software). The mEPSCs were detected with a template function (Axograph; template: rise, 0.5 ms; decay, 3 ms; criteria range: rise, 0.15–1.5 m; decay, 0.5–5 ms).
In situ hybridization labeling of VGLUT3 mRNA
Regional in situ hybridization was performed as described previously (Gras et al., 2008; Amilhon et al., 2010; Vigneault et al., 2015). Mouse brains were rapidly dissected and frozen in isopentane at −30°C. Coronal brain sections (12 μm) were cut with a cryostat (Leica Biosystems) at −20°C, thaw-mounted on glass slides, fixed in 4% formaldehyde, washed with PBS, dehydrated in 50 and 70% ethanol, and air dried. The sections were incubated with a mixture of nine antisense oligonucleotides specific for mouse VGLUT3 (5′-TCAGAAGCTGTCATCCTCTCTCAACTCCAG-3′, 5′-GGCATCTTCCTCTTCATTGGTCCCATCGAT-3′, 5′-CCCTTCTCCTCTCGATCCAGAATCAACAAA-3′, 5′-ACTGCGCTTGCCCTGGAGGAACACTTGAAA-3′, 5′-GCCGTAATGCACCCTCGCCGCAGAAGGGATAAAC-3′, 5′-GGGCAGCCAAGCTCAGAATGAGCACAGCCTGTATCC-3′, 5′-AGTCACAGATGTACCGCTTGGGGATGCCGCAGCAG-3′, 5′-AGCCAGTTGTCCTCCGATGGGCACCACGATTGTC-3′, and 5′-CCACAATGGCCACTCCAAGGTTGCACCGAATCCC-3′). These oligonucleotides were labeled with [35S]-dATP (PerkinElmer) to a specific activity of 5 × 108 dpm/μg using terminal deoxynucleotidyl transferase (Promega). Sections were incubated for 18 h at 42°C, washed, and exposed to a BAS-SR Fujifilm imaging plate for 7 d. The plates were scanned with a Fuji Bioimaging Analyzer BAS-5000. Densitometry measurements were performed with MCID analysis software. Densitometric analysis of four to six sections for each region was averaged per mouse (eight mice per genotype).
Immunoautoradiographic labeling of VGLUT3
Immunoautoradiography experiments were performed on fresh frozen mouse brain sections (12 μm) as described previously (Amilhon et al., 2010; Vigneault et al., 2015). Brain slices were incubated with VGLUT3 rabbit polyclonal antiserum (1:20,000, Synaptic Systems) and then with anti-rabbit [125I]-IgG (PerkinElmer). The sections were then washed in PBS, rapidly rinsed in water, dried, and exposed to x-ray films (Biomax MR, Kodak) for 5 d. Standard radioactive microscales were exposed to each film to ensure that labeling densities were in the linear range. Densitometry measurements were performed with MCID analysis software on four to six sections for each region per mouse (eight mice per genotype).
Immunofluorescence
Immunofluorescence experiments were performed on BON cells, hippocampal primary neurons, and brain slices as described previously (Herzog et al., 2001, 2011; Gras et al., 2002). Cells or brain sections were incubated with anti-human VGLUT3 rabbit polyclonal antiserum (1:1000; Gras et al., 2002; Vigneault et al., 2015), anti-rodent VGLUT3 rabbit polyclonal antiserum (1:2000, Synaptic Systems), anti-rodent VGLUT1 rabbit polyclonal antiserum (1:2000; Herzog et al., 2001), anti-rodent MAP2 mouse monoclonal antiserum (1:1000, Sigma), anti-rodent bassoon mouse monoclonal antiserum (1:2000, Abcam), or PSD-95 mouse monoclonal antiserum (1:2000, Abcam). Immunolabeling was detected with anti-rabbit or anti-mouse secondary antisera coupled to Alexa Fluor 555 or Alexa Fluor 488 (1:2000, Invitrogen). Nuclei were labeled using DAPI (1:5000, Sigma). The cells and sections were observed with a fluorescence microscope equipped with an Apotome module (Axiovert 200 M, Zeiss) or a confocal laser-scanning microscope (Leica TCS SP5, Leica Microsystems). Fluorescence intensity in the synaptic boutons of hippocampal neuronal cultures was quantified using the MacBiophotonics plugins package for ImageJ software. In the brain slices, semiquantification of VGLUT3 in the soma and terminals was performed with ImageJ software (MacBiophotonics plugins). The contour of neuronal soma or brain areas containing terminals were delineated manually, and the integrated intensity within these regions of interest (ROIs) were measured using the ROI manager of the ImageJ software.
Electron microscopy immunogold detection of VGLUT3
Electron microscopy experiments were performed on WT and VGLUT3A224V/A224V mice as described previously (Bernard et al., 1999; Herzog et al., 2001). Briefly, the animals were deeply anesthetized and perfused transcardially with a mixture of 2% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, and 0.2% glutaraldehyde. Their brains were dissected, fixed overnight in 2% paraformaldehyde, and stored in PBS until use. Sections (70 μm) from the midbrain, including the striatum, were cut on a vibrating microtome (VT1000S, Leica Biosystems). Sections were successively incubated in anti-rodent VGLUT3 rabbit polyclonal antiserum (1:2000, Synaptic Systems), in goat anti-rabbit coupled to biotin (Vector Laboratories), and in streptavidin coupled to gold particles (1.4 nm in diameter; Nanoprobes; 1:100 in PBS/ BSA-c, negatively charged acetylated bovine serum albumin, Aurion). The signal of the gold immunoparticles was increased using a silver enhancement kit (HQ silver, Nanoprobes) for 2 min at room temperature in the dark. Finally, after treatment with 1% osmium, dehydration, and embedding in resin, ultrathin sections were cut, stained with lead citrate, and examined in a transmission electron microscope (EM 912 OMEGA, Zeiss) equipped with a LaB6 filament at 80 kV. Images were captured with a digital camera (SS-CCD, 2kx2k, Veleta).
VGLUT3 immunodetection by STED microscopy
STED microscopy experiments were performed on WT, VGLUT3A224V/+, VGLUT3A224V/A224V, VGLUT3A224V/−, and VGLUT3−/− mice. Briefly, the animals were deeply anesthetized and perfused transcardially with paraformaldehyde (2%). Their brains were dissected, fixed overnight in 2% paraformaldehyde, and stored in PBS until use. Sections (70 μm) from the midbrain, including the striatum, were cut on a vibrating microtome (VT1000S, Leica). VGLUT3 was detected in axonal varicosities in the mouse caudate–putamen. To ensure that VGLUT3 was detected in varicosities, synaptophysin I, a synaptic vesicular protein, was codetected with VGLUT3. Sections were successively incubated in a mixture of anti-rodent VGLUT3 rabbit polyclonal antiserum (1:2000, Synaptic Systems) and anti-synaptophysin I mouse antiserum (1:2000, Synaptic Systems), in anti-mouse biotinylated secondary antibody (1:100, Vector Laboratories), and in a mixture of streptavidin (1:100, BD Horizon V500, BD Biosciences) and Oregon Green 488 goat anti-rabbit IgG (Thermo Fisher Scientific) and mounted in ProLong Gold (Thermo Fisher Scientific).
Sections were observed using a SP8 gated-STED microscope (Leica Microsystems) equipped with a 592 nm depletion laser. BD Horizon V500 and Oregon Green 488 were excited at 470 and 514 nm, respectively. All acquisitions were performed using the same excitation laser power (50%). Alternatively, we compared the effect of increased laser power on the number of spots (50, 75, and 100%). Images were submitted to deconvolution (Huygens software, Scientific Volume Imaging), which permits the recovery of objects that are degraded by blurring and noise. Finally, the images were analyzed using ImageJ and Adobe Photoshop. The number of VGLUT3-positive puncta per varicosity surface was quantified in each genotype (80, 76, 76, 90, and 105 varicosities per animals were quantified in six WT, six VGLUT3A224V/+, six VGLUT3A224V/A224V, six VGLUT3A224V/−, and five VGLUT3−/−mice, respectively).
Fluorescence recovery after photobleaching
Fluorescence recovery after photobleaching (FRAP) experiments were performed to compare the mobility of VGLUT3-p.A211V with that of wild-type VGLUT3 at synapses. Primary culture of hippocampal neurons (as described below) were infected 48 h after plating with either VGLUT3 or VGLUT3-p.A211V lentiviral vectors (10 and 40 ng of p24 per well, respectively). To normalize expression levels of the two isoforms, five times more VGLUT3-p.A211V than WT-expressing viral particles were used to infect hippocampal primary neurons. Experiments were performed as described previously (Herzog et al., 2011) using a SP5 laser-scanning microscope (Leica) with a 63×/1.32 numerical aperture oil-immersion objective and a thermal incubator set to 37°C surrounding the setup (Leica Microsystems). The pinhole was opened to 2.5 Airy units to enhance signal detection. A bleaching protocol was used to prevent spontaneous recovery of venus fluorescence, as previously reported (McAnaney et al., 2005). Fluorescence recovery was monitored every 30 s during the first 5 min and then every 5 min for the next 70 min for FRAP and every 5 s for 10 min for fast FRAP. The entire FRAP and fast FRAP procedures were automated using SP5 live data mode software. Image processing was automated using ImageJ macro commands. Integrated fluorescence intensities were extracted from six bleached and eight control boutons, as well as one background area. The background signal was subtracted.
BON cell culture and transfection
Human carcinoid BON cells were maintained in 1:1 DMEM/F-12 medium supplemented with 10% fetal bovine serum (PAA Laboratories, GE Healthcare Life Sciences), 100 units/ml penicillin, and 100 μg/ml streptomycin (Life Technologies) at 37°C in a humidified 5% CO2 incubator as described previously (Herzog et al., 2001; Gras et al., 2002). BON cells were transfected using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions with the expression vector pcDNA3 (Invitrogen) containing the sequence coding for human VGLUT3 or VGLUT3-p.A211V coupled to green fluorescent protein (GFP) reporter (pcDNA3-VGLUT3-IRES-GFP or pcDNA3-VGLUT3-p.A211V-IRES-GFP). Stable clones were selected using G418 antibiotics (Merck, Millipore), and flow cytometry was used to select GFP- and VGLUT3-positive cells. Stable clones expressing VGLUT3 or VGLUT3-p.A211V were maintained in culture medium containing G418 (0.6 mg/ml).
Hippocampal neuronal culture
For immunofluorescence experiments, hippocampal cell cultures were prepared from newborn P0–P2 (P0 being the day of birth) C57BL/6 pups of either sex as described previously (Fasano et al., 2008). After 7 d in culture, neurons were transfected with linearized plasmid pcDNA3-VGLUT3-IRES-GFP or pcDNA3-VGLUT3-p.A211V-IRES-GFP with Lipofectamine 2000 (Thermo Fisher Scientific; 1 μg of DNA for 1 μl of lipofectamine). At 9 d in vitro (DIV), neurons were fixed and immunofluorescence experiments were performed. For FRAP experiments, hippocampal cell cultures were prepared as described previously (Siksou et al., 2013). After 2 d in culture, neurons were infected with lentiviral vectors containing either VGLUT3-venus or VGLUT3-p.A211V-venus inserts under the control of the synapsin promoter (10 and 40 ng of p24 per well, respectively). Viral particles expressing VGLUT3-venus or VGLUT3-p.A211V-venus were diluted 1:1000 in neurobasal medium (Life Technologies) containing Glutamax (Thermo Fisher Scientific), B27, and penicillin/streptomycin (Sigma-Aldrich). The diluted virus solutions (50 μl at and 300 μl for VGLUT3-venus and VGLUT3-p.A211V-venus-expressing lentivirus, respectively) were incubated for 15 d with the primary neuronal cultures. FRAP imaging of live dissociated neuron cultures was performed at 17 DIV.
Mutagenesis and construction of VGLUT3-p.A211V and VGLUT1-p.A198V
To introduce a point mutation in the WT alleles, we used the QuikChange II XL Site-Directed Mutagenesis kit (Stratagene) and a set of complementary primers as described previously (De Gois et al., 2015). All clones were sequenced in both directions, and the plasmids were purified using the Plasmid Maxi kit (Qiagen) before use.
Vesicular glutamate uptake assay with vesicles from stable BON clones
Synaptic vesicle preparations from the BON cells and [3H]l-glutamate uptake assays were performed as described previously (Herzog et al., 2001; Gras et al., 2002). Transport activity was triggered by the addition of 20 μl of vesicles (200 μg of protein) to 180 μl of uptake buffer containing ATP (2 mm, Sigma-Aldrich), l-glutamate (40 μm, pH 7.4, Sigma-Aldrich), and [3H] l-glutamate (6 μCi, PerkinElmer) with or without carbonyl cyanide m-chlorophenylhydrazone (50 μm, Sigma-Aldrich). After 10 min at 37°C, the uptake assays were terminated by dilution with 3 ml of ice-cold 0.15 m KCl, rapid filtration through a 0.45 μm pore size membrane filter (MF; Millipore), and three washes with 3 ml of ice-cold 0.15 m KCl. The radioactivity retained on the filters was measured by scintillation counting. Each uptake measurement was performed in triplicate. All experiments were performed independently three times on three independent BON–VGLUT3 clones.
Vesicular [3H]5-HT uptake assay in mouse brain synaptic vesicles
Synaptic vesicle isolation from mouse cortex and uptake assays of [3H]5-HT were performed as described previously (Amilhon et al., 2010). Transport reactions were initiated by adding 10 μl of cortical synaptic vesicles (25 μg of protein) to 90 μl of uptake buffer containing ATP (2 mm, Sigma-Aldrich) and [3H]5-HT (0.55 μCi, 50 nm, PerkinElmer) with or without 2 μm reserpine (Sigma-Aldrich) or l-glutamate (10 mm, Sigma-Aldrich). After 10 min at 37°C, vesicular uptake was stopped by dilution in 3 ml of ice-cold 0.15 m KCl, rapid filtration through mixed cellulose esters filters (MF; Millipore), and three washes with 3 ml of ice-cold 0.15 m KCl. Radioactivity retained on the filters was measured by scintillation counting. Each determination was performed in triplicate, and independent experiments were performed seven times using different synaptic vesicle preparations.
Western blotting
Western blot experiments were conducted on BON cell extracts or on homogenates from different brain regions (cortex, striatum, hippocampus), as described previously (Gras et al., 2008; Vigneault et al., 2015). Nitrocellulose membranes (0.4 μm pore size, Invitrogen) were incubated overnight with anti-human VGLUT3 rabbit polyclonal antiserum (1:1000; Vigneault et al., 2015), anti-rodent VGLUT3 rabbit polyclonal antiserum (1:2000, Synaptic Systems) or anti-rodent VGLUT1 rabbit polyclonal antiserum (1:5000; Herzog et al., 2001) and then with IRDye 800-conjugated secondary antibodies (1:5000, Invitrogen). α-Tubulin was used as the loading control (mouse monoclonal antiserum, 1:20,000, Sigma-Aldrich) detected with IRDye 700-conjugated secondary antibodies (1:5000, Invitrogen). The membranes were scanned using an Odyssey infrared imaging system (LI-COR). Integrated intensity was measured for each band and averaged for five to seven samples.
Quantitative RT-PCR analysis
The expression of VGLUT3 transcript in stable BON cells was estimated by quantitative RT-PCR as described previously (Gras et al., 2002). Nucleic acids were extracted from 65 × 104 BON cells (RNAeasy Mini kit, Qiagen). Reverse transcription was performed with the SuperScript Reverse Transcriptase kit (Life Technologies) using 2 μg of nucleic acid extract. cDNA amplification was performed with Taq DNA polymerase (Sigma-Aldrich) and the following primers: 5′-ACTCTGAACATGTTTATTCCC-3′ and 5′-CTTAGACTAACCACGTTGGC-3′ (3 min at 94°C, followed by 30 s at 94°C, 30 s at 55°C, and 40 s at 72°C for 40 cycles). The RT-PCR products were separated on a 1% agarose gel and viewed under UV light. Intensity quantification was performed with ImageJ software (MacBiophotonics plugins).
Homology modeling of VGLUT3
A putative 3D structure of VGLUT3 was established based on the x-ray crystal structure of the glycerol-3-phosphate transporter (GlpT) from Escherichia coli, which is a distant ortholog of vesicular glutamate carriers (Almqvist et al., 2007). Secondary structures were predicted using the membrane protein topology prediction method TransMembrane prediction using Hidden Markov Models and a Hidden Markov Model for Topology Prediction (Krogh et al., 2001; Tusnády and Simon, 2001). Sequence alignments were generated between human VGLUT3 (SWISS-PROT accession number Q8NDX2) and GlpT (P08194) using Clustal W (Thompson et al., 1994). Alignments were manually refined to avoid gaps in predicted (human VGLUT) and known (GlpT) secondary structure elements. VGLUT3 3D models were built from these alignments and from the crystallographic atomic coordinates of GlpT [Protein Data Bank (PDB) ID 1PW4] using the automated comparative modeling tool MODELER 9.0 (Discovery Studio 4.1, Accelrys Software). A three-dimensional model of the VGLUT3-p.A211V mutant was generated using a Build mutant protocol (Feyfant et al., 2007). The mutant model was minimized using the Adopted Basis Newton-Raphson (NR) algorithm, with a maximum step of 500 and a “generalized Born with Implicit Membrane” as an implicit solvent model. A 1-palmitoyl-2-oleoyl-phosphatidylcholine membrane of 100 × 80 Å was added using software from Visual Molecular Dynamic (VMD 1.9.2, http://www.ks.uiuc.edu/Research/vmd/vmd-1.9.2/). Proteins were solvated, and ions were added using the solvation and ionization package from VMD1.9.2. The structure was minimized using a NR algorithm, with a maximum step of 500 and an implicit solvent model. The system was then equilibrated using a short Nanoscale Molecular Dynamic software program (NAMD) of 1 ns.
Statistics
All statistical comparisons were performed with Prism 5 (GraphPad Software). Each statistical test was appropriately chosen for the relevant experimental design. To compare two groups, a nonparametric Mann–Whitney U test was performed. One-way ANOVA, Kruskal–Wallis test, or repeated-measures ANOVA was used for multiple group comparisons. All results are expressed as the mean ± SEM. Differences were considered significant at p < 0.05.
Results
The VGLUT3-p.A211V mutation reduces the expression level of VGLUT3 in vitro
To compare the expression of native VGLUT3 and VGLUT3-p.A211V, both alleles were expressed in cultures of stably transfected BON cells or hippocampal neurons. The transcripts coding for both isoforms were expressed at similar levels in BON cells when detected by RT-PCR (Fig. 2A; Mann–Whitney U test, p > 0.05, n = 6). In contrast, VGLUT3-p.A211V protein levels were markedly reduced relative to the WT isoform when measured by Western blotting in BON cells (Fig. 2B; −64%, Mann–Whitney U test, p = 0.0079, n = 5) or in hippocampal neuronal cultures (Fig. 2C; −66%, Mann–Whitney U test, p = 0.028, n = 4). This decrease was also confirmed by immunofluorescence detection of VGLUT3 and VGLUT3-p.A211V in BON cells (Fig. 2D,G,H; −79%, Mann–Whitney U test, p = 0.028, n = 4) or in hippocampal neuronal cultures (Fig. 2E,I,J; −86%, Mann–Whitney U test, p = 0.008, n = 5). The same point mutation was then introduced into the coding sequence of VGLUT1 (VGLUT1-p.A198V). Interestingly, VGLUT1-p.A198V expression was dramatically reduced in BON cells (Fig. 2F,K,L; −90%, Mann–Whitney U test, p = 0.005, n = 6). Therefore, exchanging the alanine of the KWAPPLER motif for a valine was sufficient to markedly reduce the expression of vesicular glutamate transporters.
Figure 2.

Expression of VGLUT3-p.A211V in cell cultures. A, Gel image of RT-PCR products (negative image) showing no significant difference between WT and VGLUT3-p.A211V transcript from BON cells stably expressing each allele. B, C, Western blot (WB) quantification of VGLUT3 and VGLUT3-p.A211V in BON cell extracts (B) and in primary cultures of hippocampal neurons transfected with a VGLUT3 or VGLUT3-p.A211V expression plasmid (C). VGLUT3-p.A211V expression was reduced by 64% in BON cells (B) and by 66% in neurons (C). D, E, Quantification of fluorescence intensity of WT and VGLUT3-p.A211V in stable BON cells (D) or in synaptic boutons in hippocampal neurons in culture (E). F, Western blot detection and quantification of WT and VGLUT1-p.A198V in BON cell extracts. G, H, Immunofluorescence microphotographs of VGLUT3 (green) in BON cells stably transfected with a VGLUT3 (G) or VGLUT3-p.A211V (H) expression plasmid. I, J, Hippocampal neurons transiently transfected with a VGLUT3 (I) or VGLUT3-p.A211V (J) expression plasmid. Nuclei were labeled with DAPI (blue). K, L, Immunofluorescence of the WT isoform of VGLUT1 (K, red) or VGLUT1-p.A198V (L, red) in BON cell cultures that were transiently transfected with expression plasmid. M–R, Colocalizations of VGLUT3 WT (M, O, Q, green) or VGLUT3-p.A211V (N, P, R, green) with MAP2 (M, N, red), bassoon (O, P, red), and PSD-95 (Q, R, red) in primary hippocampal neuronal cultures transfected with a VGLUT3 or VGLUT3-p.A211V plasmid. Areas surrounded by a dashed line are enlarged in the insets. Scale bars: R, 5 μm; G–L, 10 μm; M–R, insets, 1 μm. * p < 0.05; ** p < 0.01.
We then assessed whether VGLUT3-p.A211V was expressed in synaptic boutons in neuronal hippocampal cultures. Both VGLUT3 and VGLUT3-p.A211V were expressed in punctiform structures apposed to MAP2-positive dendritic processes (Fig. 2M,N). These VGLUT3- and VGLUT3-p.A211V-positive puncta colocalized with presynaptic markers, such as bassoon, and were apposed to PSD-95-positive elements (Fig. 2O–R). Thus, the p.A211V mutation does not qualitatively alter the targeting of VGLUT3 to synaptic boutons.
Glutamate vesicular uptake and release are minimally altered by the p.A211V mutation
We then wondered whether p.A211V altered the 3D structure of VGLUT3. Theoretical models (Fig. 3A–H) were obtained using the crystal structure of the GlpT from E. coli (PDB ID 1PW4, 19% identity, 39% similarity with VGLUT3) as a template (Almqvist et al., 2007). As shown in Figure 3, in these putative 3D models, alanine 211 is strategically located on a small cytoplasmic loop [cytoplasmic loop 4 (CL4)] and at the top of the transporter pore. Switching a valine for an alanine in this position only minimally modified the 3D structure of the loop. In particular, in the WT isoform, A211 interacted with the N-terminal domain through cysteine 67 hydrophobic binding (Fig. 3C,D), as well as with leucine 214 in CL4. These interactions were potentially altered with the p.A211V mutation (Fig. 3G,H). Both WT and mutant isoforms conserved a short-distance interaction pattern with CL4 (K209, W210, P212, P213, L214, and E215). However, in the N-terminal domain of the mutant, the interaction of A211 with C67 was abolished. In parallel, A211 from the WT isoform interacted with T289 of the cytoplasmic loop 6. This interaction was lost in the VGLUT3 p.A211V mutated isoform but was partially compensated by interactions with residues of CL6: H276 and I287 (Fig. 3H).
Figure 3.
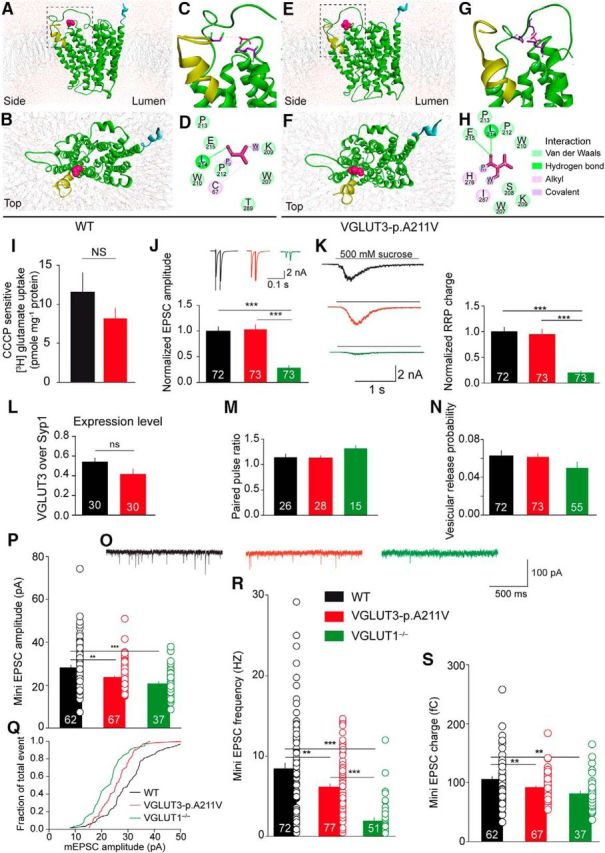
Effect of the p.A211V mutation on 3D structure, glutamate vesicular accumulation, and release. A–H, Three- and two-dimensional model of human VGLUT3 (A–D) and VGLUT3-p.A211V (E–H) using crystallographic MFS transporter structures as a template. Packing of the helices viewed from the side (A, C, E, G) or the top (B, F) is shown. The alanine residue (in position 211) of VGLUT3 (A, B) and the valine residue of VGLUT3-p.A211V (E, F) that are exposed to the pore are shown in pink. Regions boxed in A and E are enlarged in C and G, respectively. D, H, Close-up view and two-dimensional interactions diagram of alanine 211 (D) or valine 211 (H). I, Effect of the p.A211V mutation on H+ ionophore carbonyl cyanide m-chlorophenylhydrazone (CCCP)-sensitive [3H]l-glutamate uptake by synaptic vesicles from BON cells stably expressing VGLUT3 (black bars) or VGLUT3-p.A211V (red bars). The small difference observed between the two populations of vesicles is not significant (NS). J–S, Electrophysiological recordings of VGLUT1−/− hippocampal autaptic neurons infected with lentivirus expressing VGLUT3 (WT) or VGLUT3-p.A211V isoforms. The number of recorded cells is indicated in bar graphs. Data are pooled from two independent cultures in M and from four independent cultures in J–L and N–S. J, Top, Representative traces of current responses after two unclamped action potentials with an interstimulus interval of 25 ms in VGLUT1−/− autaptic neurons (green bars and traces) expressing VGLUT3 (black bars and traces) or VGLUT3-p.A211V (red bars and traces). Artifacts and action potentials are blanked. Bottom, Plot of average EPSC amplitude size (first pulse) normalized to WT. K, Left, Representative traces of current responses after application of sucrose (500 mm) for 5 s. Right, Plot of the average readily releasable pool (RRP) charge normalized to WT. L, Comparison of the expression levels of WT and VGLUT3-p.A211V, determined by measuring immunofluorescence intensities and normalizing to the intensities of synaptic marker synaptophysin I (Syp1). M, Plot of average paired-pulse ratios with an interstimulus interval of 25 ms. Data are pooled from two independent cultures. N, Plot of average vesicular release probability. Data are pooled from four independent cultures. O, Example of mEPSC traces in autaptic neurons. P–S, Scattered points and bar graphs of average amplitude (P), fraction of events (Q), frequency (R), and EPSC charge (S). Data are pooled from four independent cultures.
The putative 3D model suggests also that the alanine residue of the WT and the valine residue of VGLUT3-p.A211V faced the pore and hence could affect the entry of glutamate into the lumen of synaptic vesicles. Therefore, [3H]l-glutamate accumulation was measured in vesicular fractions from BON cells stably expressing VGLUT3 or VGLUT3-p.A211V. A small but nonsignificant reduction in vesicular glutamate uptake was observed with the VGLUT3-p.A211V isoform (Fig. 3I; Mann–Whitney U test, p > 0.05, n = 10). Hence, despite a 60–80% reduction in expression, the mutated VGLUT3 appeared to be as efficient as the WT allele at translocating glutamate into vesicles.
The capacity for glutamate release from neurons expressing the VGLUT3-p.A211V isoform was then evaluated by electrophysiological recordings. Autaptic hippocampal cultured neurons obtained from newborn VGLUT1−/− mice were rescued by lentiviral-driven expression of either VGLUT3 (WT; Fig. 3J, black bars) or VGLUT3-p.A211V (Fig. 3J, red bars). Uninfected neurons are shown with green bars. In these experiments, to determine the intrinsic properties of the VGLUT3-p.A211V isoform, expression levels of the mutant were purposely raised to equal WT. After 2 weeks in culture, synaptic responses were evoked by 2 ms depolarization of the cell at 0 mV. This resulted in an unclamped action potential and release of glutamate, which in turn generated EPSCs. The mean normalized EPSC peak amplitude was not different between the WT (n = 72) and the VGLUT3-p.A211V (n = 73) neurons (Fig. 3J). This was in contrast to the EPSC responses from VGLUT1−/− neurons, which showed a severe reduction in synaptic response, demonstrating that the WT and the VGLUT3-p.A211V mutant rescued responses equally. We next determined the RRP size of VGLUT3, VGLUT3-p.A211V, and VGLUT1−/− neurons by applying hypertonic sucrose (500 mm for 5 s; Rosenmund and Stevens, 1996). The charge in VGLUT1−/− neurons was reduced by 80%, as expected (Fig. 3K; n = 73, p < 0.01; Wojcik et al., 2004). VGLUT3 and VGLUT3-p.A211V neurons had a similar RRP charge (Fig. 3K). The expression levels of WT and VGLUT3-p.A211V as determined by immunofluorescence were not different (Fig. 3L). Moreover, the facilitation of responses that were evoked in pairs of action potential stimulation (25 ms interval; Fig. 3M) and the vesicular release probability were not different between VGLUT3 (n = 72) and VGLUT3-p.A211V (n = 73) neurons (Fig. 3N). The release probability in the VGLUT1−/− neurons was reduced, as expected (Wojcik et al., 2004; Herman et al., 2014).
Analysis of spontaneous release activity (Fig. 3O, trace sample) demonstrated that mean mEPSC amplitude was slightly reduced in VGLUT3-p.A211V neurons compared with WT neurons (Fig. 3P,Q; WT, n = 62; VGLUT3-p.A211V, n = 67; p < 0.005). The mean frequency of mEPSCs was significantly different among all three groups (Fig. 3R). In VGLUT1−/− neurons, mEPSC frequency was reduced by 80% (Fig. 3R; 1.8 Hz, n = 51, p < 0.0005), a finding consistent with the reduced RRP size (Fig. 3K) and the results of a previous publication (Wojcik et al., 2004). In VGLUT3-p.A211V expressing hippocampal isolated neurons, mEPSC frequency was reduced by almost 30% (Fig. 3R; WT, 8.5 Hz, n = 72; VGLUT3-p.A211V, 6 Hz, n = 77, p < 0.005). The mEPSC charge was also slightly decreased in VGLUT3-p.A211V neurons compared with WT neurons (Fig. 3S; WT, n = 62; VGLUT3-p.A211V, n = 67; p < 0.005). Together, the results suggested that the p.A211V mutation does not alter the quantity of glutamate release, the RRP size or the release probability. In contrast, a small reduction in spontaneous release activity was observed.
The expression of VGLUT3-p.A224V is dramatically reduced in the terminals of the mouse CNS
The p.A211V mutation is responsible for DFNA25 progressive deafness (Ruel et al., 2008). To gain insight into the underlying mechanisms, we used newly generated mutant mice in which the corresponding amino acid was mutated in the VGLUT3 mouse genome (VGLUT3A224V/A224V; Fig. 1). The expression levels of VGLUT3 mRNA and protein were measured by in situ hybridization, immunoautoradiography, and Western blotting at different ages (P10 and 3, 6, and 12 months) in WT, heterozygous, and homozygous mice (Fig. 4).
Figure 4.
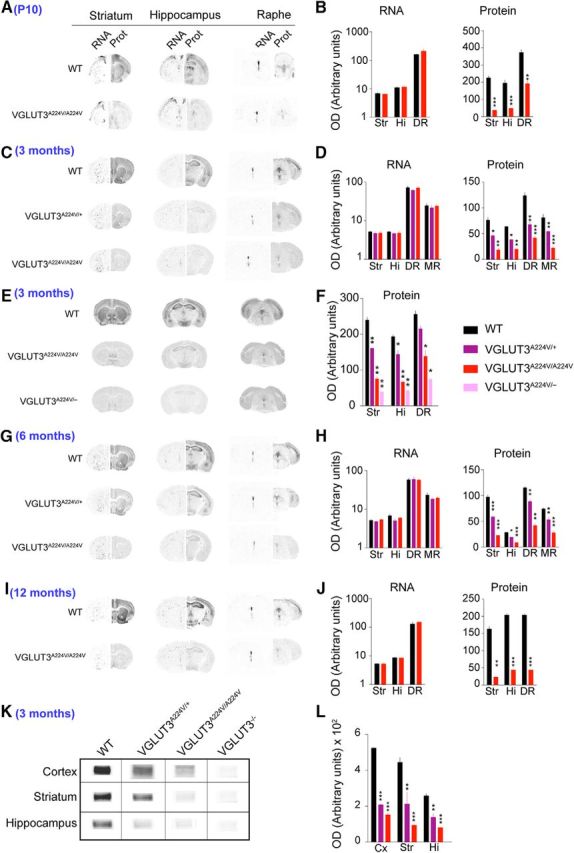
Regional expression of VGLUT3, VGLUT3A224V/+, VGLUT3A224V/A224V, and VGLUT3A224V/− in the CNS of mice at different ages. A–J, Detection and quantification of VGLUT3, VGLUT3A224V/+, VGLUT3A224V/A224V, and VGLUT3A224V/− mRNA and protein (Prot) expression by in situ hybridization or immunoautoradiography on coronal mouse brain sections taken at P10 (A, B) and at 3 months (C–F), 6 months (G, H), or 12 months (I, J) in the striatum (Str), hippocampus (Hi), dorsal raphe (DR), and median raphe (MR). WT, VGLUT3A224V/+, and VGLUT3A224V/A224V mice expressed a similar level of transcripts (B, D, H, J, left; Mann–Whitney U test for B, J; Kruskal–Wallis test for D, H; p > 0.05, n = 8). B, D, H, right, F, Loss of the protein in all areas from P10 until 1 year in VGLUT3A224V/A224V mice (n = 8 for each genotype, Mann–Whitney U test or Kruskal–Wallis test, *p < 0.05, **p < 0.01, ***p < 0.001). In VGLUT3A224V/+ mice, there is a 34% decrease in VGLUT3 in all areas at 3 and 6 months (D, H). E, F, In this experiment, the protein expression of VGLUT3 was compared in WT, VGLUT3A224V/A224V, and VGLUT3A224V/− mice (n = 8) in the striatum, hippocampus, and dorsal raphe. VGLUT3 expression decreased by 70% in VGLUT3A224V/A224V mice and further decreased by 84% in VGLUT3A224V/− mice in the striatum (Kruskal–Wallis test; in the striatum, WT vs VGLUT3A224V/+, p = 0.0043; WT vs VGLUT3A224V/A224V, p = 0.0043; WT vs VGLUT3A224V/−, p = 0.0012; in the hippocampus, WT vs VGLUT3A224V/+, p = 0.01; WT vs VGLUT3A224V/A224V, p = 0.0043; WT vs VGLUT3A224V/−, p = 0.0012; in raphe nuclei, WT vs VGLUT3A224V/+, p = 0.0571; WT vs VGLUT3A224V/A224V, p = 0.0159; WT vs VGLUT3A224V/−, p = 0.0286). K, L, Western blot detection (K) and quantification (L) of VGLUT3 in the cortex, striatum, and hippocampus of VGLUT3A224V/+ and VGLUT3A224V/A224V mice at 3 months (n = 5; Kruskal–Wallis test, **p < 0.01).
As shown in Figure 4, the levels of VGLUT3 transcript were unaltered in the striatum, hippocampus, and raphe area at all ages assessed. In contrast, protein levels were markedly altered at various ages in the striatum, hippocampus, and raphe of VGLUT3A224V/A224V mice (Fig. 4, Table 2). In the striatum of VGLUT3A224V/A224V mice, the level of the mutant protein, as measured by immunoautoradiography, was reduced by 76–85% compared with that in WT mice at all ages (Fig. 4A–J, Table 2; Mann–Whitney U test for P10 and 12 months, Kruskal–Wallis test for 3 and 6 months; *p < 0.05, **p < 0.01, ***p < 0.001). A similar decrease in protein levels was observed in the hippocampus as well as the raphe nuclei with 69–76% and 50–74% reductions measured in the respective tissues at the same time points. Hence, the mean decrease across age and brain areas in VGLUT3A224V/A224V mice was 70.3% (SEM, ±2.7; n = 12). Reduced VGLUT3-p.A224V levels were also measured in mice expressing only one copy of the mutated gene and one copy of the WT gene (3- and 6-month-old heterozygous VGLUT3A224V/+ mice; Fig. 4, Table 2). In heterozygous mice, the mean decrease was 34% (SEM, ±4.1; n = 8). These results were further confirmed by Western blotting experiments performed in the cortex, striatum, and hippocampus in 3-month-old VGLUT3A224V/+ and VGLUT3A224V/A224V mice (Fig. 4K,L; Tables 2, 3; Kruskal–Wallis test, *p < 0.05, **p < 0.01, ***p < 0.001). In summary, VGLUT3 expression was globally reduced by 70% in the brains of VGLUT3A224V/A224V mice of all ages.
Table 2.
Percentage of decrease in VGLUT3 expression as determined by immunoautoradiography in the striatum, hippocampus, and raphe nuclei of VGLUT3A224V/+, VGLUT3A224V/A224V, and VGLUT3A224V/− at P10 and 3, 6, or 12 months of age
| Brain area | Genotype | % VGLUT3 decrease |
|||
|---|---|---|---|---|---|
| P10 | 3 months | 6 months | 12 months | ||
| Striatum | VGLUT3A224V/+ | N.A. | 34 | 50 | N.A. |
| VGLUT3A224V/A224V | 83 | 76 | 77 | 85 | |
| VGLUT3A224V/− | N.A. | 84 | N.A. | N.A. | |
| Hippocampus | VGLUT3A224V/+ | N.A. | 26 | 38 | N.A. |
| VGLUT3A224V/A224V | 76 | 69 | 70 | 78 | |
| VGLUT3A224V/− | N.A. | 88 | N.A. | N.A. | |
| Dorsal raphe | VGLUT3A224V/+ | N.A. | 16 | 24 | N.A. |
| VGLUT3A224V/A224V | 50 | 46 | 61 | 77 | |
| VGLUT3A224V/− | N.A. | 71 | N.A. | N.A. | |
| Median raphe | VGLUT3A224V/+ | N.A. | 20 | 29 | N.A. |
| VGLUT3A224V/A224V | N.A. | 74 | 63 | N.A. | |
N.A., Not applicable.
Table 3.
Percentage decrease in VGLUT3 expression as determined by Western blot analysis of the cortex, striatum, and hippocampus of heterozygous and homozygous VGLUT3-p.A224V-expressing mice at 3 months of age
| Brain area | Genotype | % VGLUT3 decrease |
|---|---|---|
| Cortex | VGLUT3A224V/+ | 63 |
| VGLUT3A224V/A224V | 72 | |
| Striatum | VGLUT3A224V/+ | 55 |
| VGLUT3A224V/A224V | 85 | |
| Hippocampus | VGLUT3A224V/+ | 47 |
| VGLUT3A224V/A224V | 70 |
To obtain mice with only one copy of the VGLUT3-p.A224V isoform, we crossed heterozygous VGLUT3+/− mice with heterozygous VGLUT3A224V/+ mice. As shown in Figure 4, E and F, the decrease in VGLUT3 expression was more pronounced in VGLUT3A224V/− than in VGLUT3A224V/A224V mice. In the striatum of 3-month-old VGLUT3A224V/− mice, VGLUT3 expression was reduced by 84% (Mann–Whitney U test, p = 0.012 relative to WT, n = 8). Moreover, 88% of the protein was lost in the hippocampus (Mann–Whitney U test, p = 0.012 relative to WT, n = 8) and 71% was lost in the raphe nuclei (Mann–Whitney U test, p = 0.028 relative to WT, n = 4). Therefore, mice with one copy of the VGLUT3-p.A224V isoform demonstrate a larger decrease in VGLUT3 expression than mice expressing two mutant copies.
Unlike VGLUT1 and VGLUT2, which are almost exclusively present in nerve endings, VGLUT3 is present in both terminals and neuronal cell bodies (Gras et al., 2002; Somogyi et al., 2004). We therefore studied the cellular and subcellular distribution of VGLUT3-p.A224V in these cellular compartments (Fig. 5). The expression of VGLUT3-p.A224V was dramatically reduced in terminals from the striatum or hippocampus (CA1) compared with WT levels (Fig. 5A–C; striatum: −69%, Mann–Whitney U test, p < 0.0001, n = 19; Fig. 5E–G, hippocampus: −72%, Mann–Whitney U test, p < 0.0001, n = 20; eight animals per genotype). However, surprisingly, VGLUT3 levels were similar in the soma of tonically active cholinergic interneurons (TANs) and the hippocampal basket cells from WT and VGLUT3A224V/A224V mice (Fig. 5D,H; Mann–Whitney U test, p > 0.05). Furthermore, using electron microscopy, we observed that in TANs, the WT as well as the VGLUT3-p.A224V isoforms were distributed over similar subcellular organelles, including the endoplasmic reticulum (Fig. 5I,J; n = 5 for each genotype). Together, these data suggest that the VGLUT3-p.A224V isoform is not abnormally accumulated in the soma of neurons and that its expression is markedly reduced in terminals.
Figure 5.
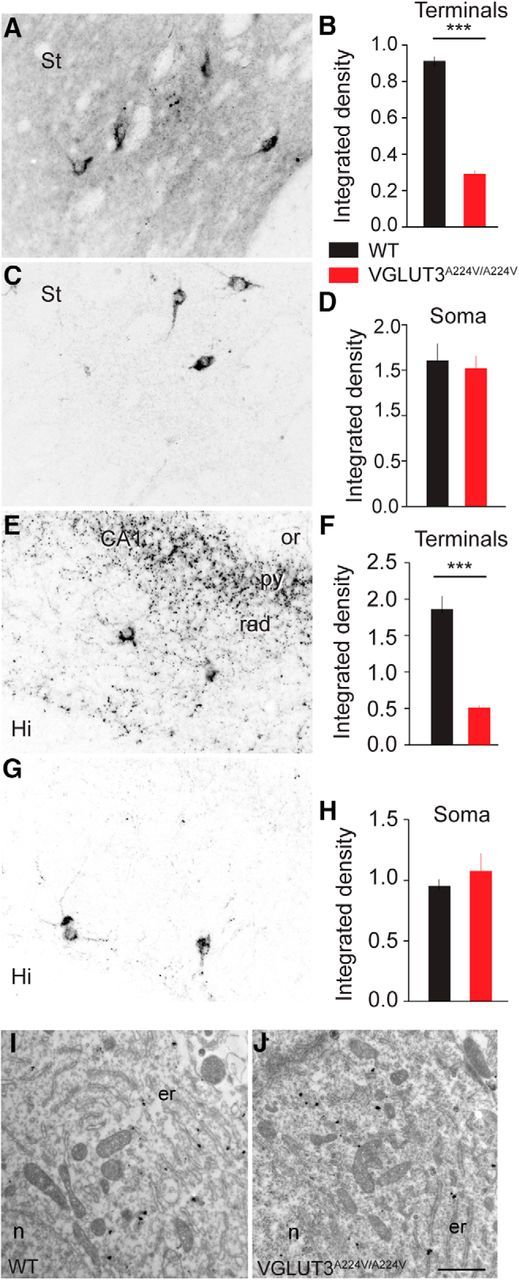
Expression of VGLUT3-p.A224V in soma and terminals of VGLUT3-positive neurons in the brain of WT and VGLUT3A224V/A224V mice. A–H, Immunofluorescence visualization and quantification of VGLUT3 in the striatum (A, C) and in the CA1 pyramidal field of the hippocampus (E, G) in WT (A, E) and VGLUT3A224V/A224V (C, G) mice. VGLUT3 expression is substantially reduced in terminals of the striatum (B, −69%) and hippocampus (F, −72%), whereas its expression is unchanged in the soma of TANs and basket cells (D, H). I, J, Electron microphotographs of the soma of TANs in striatal sections of WT (I) and VGLUT3A224V/A224V (J) mice. VGLUT3 is labeled with gold particles. The distribution of VGLUT3 labeling is similar in WT (I) and VGLUT3A224V/A224V (J) mice. St, Striatum; Hi, hippocampus; er, endoplasmic reticulum; n, nucleus; or, stratum oriens of the hippocampus; py, stratum pyramidale of the hippocampus; rad, stratum radiatum. Scale bar, A, C, E, G, 35 μm; I, J, 1 μm. *** p < 0.001.
The mobility of VGLUT3-p.A211V is minimally modified
The aforementioned results suggested that VGLUT3-p.A211V was either not properly trafficked or was degraded in the nerve endings. To test the first hypothesis, we used FRAP to assess the mobility of VGLUT3 WT or VGLUT3-p.A211V in synaptic boutons, as described previously (Herzog et al., 2011). Hippocampal neurons were transduced at 2 DIV with a lentiviral vector expressing either the WT isoform of VGLUT3 or VGLUT3-p.A211V; both were tagged with the YFP derivative venus (Fig. 6A). VGLUT3-p.A211V transduction was adjusted to match the expression level of WT VGLUT3. Transduction of both isoforms yielded a punctate distribution of venus fluorescence that was reminiscent of presynaptic localization. A bleaching light pulse was applied to ROIs surrounding individual boutons in mature neurons after 17 d in culture (Fig. 6A). The exchange of bleached SVs and fluorescently labeled SVs from neurites surrounding the ROI was monitored during the next 75 min (Fig. 6A). VGLUT3-p.A211V fluorescence recovered to levels similar to those of the wild-type protein (Fig. 6A,B). An additional FRAP experiment with faster imaging rates in the first 5 min was performed to reveal possible differences in the initial recovery phase (Fig. 6B, fast FRAP inset). The kinetics of recovery seemed slightly different, but these differences were not statistically significant. However, the amplitude of recovery at 1 h was similar for the mutant and wild-type experiments (Fig. 6C; Mann–Whitney U test, p = 0.1129). Therefore, although we cannot rule out that the mobility of the overexpressed venus-tagged VGLUT3-p.A211V mutant isoform was altered, we conclude that the p.A211V mutation powerfully reduces VGLUT3 expression in terminals without apparently altering its mobility.
Figure 6.

The p.A211V mutation does not alter VGLUT3 mobility. A, Representative image sequence depicting the initial fluorescence in the chosen boutons (white arrow), the loss of fluorescence after bleaching (time point, 2:10 min), and the gradual recovery of fluorescent material in the bleached region in hippocampal cultures expressing either VGLUT3-venus or VGLUT3-p.A211V-venus. B, Kinetics of FRAP over 75 min in neurons expressing VGLUT3-venus or VGLUT3-p.A211V-venus. Average recovery curves (error bars represent SEM) are shown in black for VGLUT3-venus and in red for VGLUT3-p.A211V-venus. The inset shows FRAP curves during the first 10 min, with images taken every 5 s. C, Amplitude of recovery for VGLUT3-venus or VGLUT3-p.A211V-venus in bleached boutons 60 min after recovery from bleaching. Data are pooled from five independent cultures for FRAP and three independent cultures for fast FRAP (n = 25 boutons per condition).
The p.A224V mutation does not alter the behavior of mutant mice
Mice lacking VGLUT3 (VGLUT3−/−) demonstrate increased anxiety behavior, as well as augmented basal and cocaine-induced locomotor activity (Gras et al., 2008; Amilhon et al., 2010; Sakae et al., 2015). We thus decided to characterize these behaviors in adult (3- to 4-month-old) male VGLUT3A224V/A224V mice with only 30% VGLUT3 expression. We first assessed the anxiety levels of WT and mutant animals in an open field and an elevated plus maze (Fig. 7A,B). Spontaneous as well as cocaine-induced locomotor activities were also measured (Fig. 7C,D). No significant differences between WT and VGLUT3A224V/A224V mice were observed in any paradigm. Therefore, the p.A211V mutation does not modify the VGLUT3-dependent mood or locomotor phenotypes.
Figure 7.
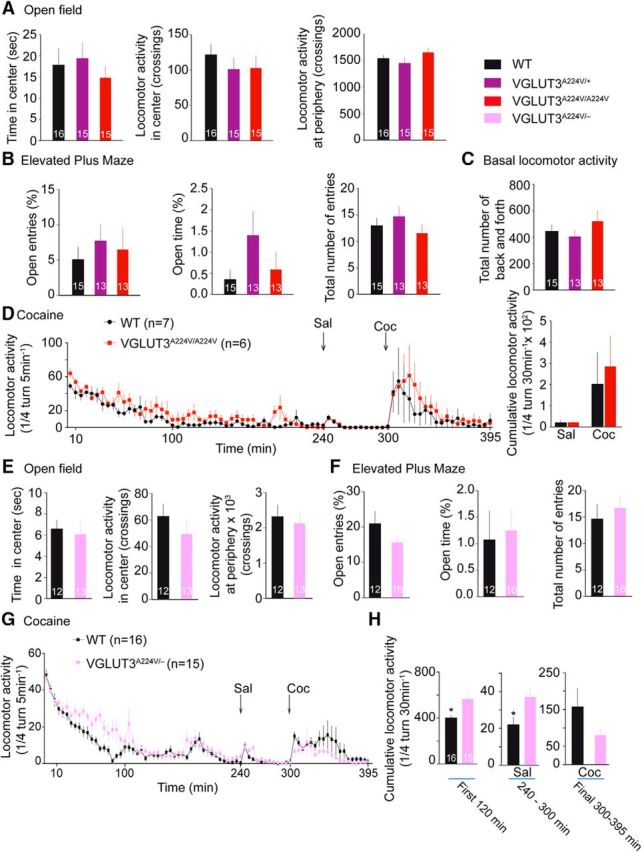
Behavioral analysis of VGLUT3A224V/A224V mice. A, In the open field, WT littermates and VGLUT3A224V/+ and VGLUT3A224V/A224V mice spent the same time at the center of the open field (Kruskal–Wallis test, p > 0.05) and presented the same locomotor activity at the center and the periphery of the open field (Kruskal–Wallis test, p > 0.05). The number of animals is indicated in the bar graph. B, Anxiety levels assessed in the elevated plus maze were similar in WT, VGLUT3A224V/+, and VGLUT3A224V/A224V mice (Kruskal–Wallis test, p > 0.05). Horizontal exploration was measured for 6 min. No difference was found in total entries or in the time spent in open arms between the WT and the VGLUT3 mutant mice (Kruskal–Wallis test, p > 0.05). C, Spontaneous locomotor activity of naive WT, VGLUT3A224V/+, and VGLUT3A224V/A224V mice. Horizontal locomotor activity was recorded for 5 h (first hour during the light cycle, followed by four hours during the dark cycle). Spontaneous locomotor activity was similar in WT and mutant animals (Kruskal–Wallis test, p > 0.05). D, Left, Time course of the locomotor effect of cocaine (10 mg/kg, i.p.) in WT (n = 7) and VGLUT3A224V/A224V (n = 6) mice. Animals were placed in the cyclotron for 240 min for habituation, given injections of saline (NaCl 0.9%), placed back in the cyclotron for 60 min, and given injections of cocaine (10 mg/kg). After cocaine injection, locomotion was recorded for 95 min. There was no significant difference in locomotor activity between WT and mutant mice that were treated with cocaine (repeated-measures ANOVA, p > 0.05). Right, Cumulative horizontal locomotor activity over 60 min for saline-treated or 95 min for cocaine-injected WT (n = 7) and VGLUT3A224V/A224V mice (n = 6). No difference was observed in cumulative locomotor activity after saline or cocaine injection between the WT and the VGLUT3A224V/A224V mice (Mann–Whitney U test, p > 0.05). E–H, Behavioral analysis of mice expressing only one copy of VGLUT3-pA224V (VGLUT3A224V/−). E, In the open field, WT and VGLUT3A224V/− mice spent the same time in the center area and crossed the periphery or the central area the same number of times (Mann–Whitney U test, p > 0.05). F, WT and VGLUT3A224V/− mice presented the same anxiety level when assessed in the elevated plus maze (Mann–Whitney U test, p > 0.05). G, Time course of the locomotor effect of cocaine (10 mg/kg, i.p.) in WT mice and VGLUT3A224V/− mice. Animals were placed in the cyclotron as described in D. No significant differences in locomotor activity were observed after cocaine injection between the WT and the mutant mice (repeated-measures ANOVA, p > 0.05). H, Cumulative horizontal locomotor activity during the first 120 min in the cyclotron or 60 min after saline injection and 95 min after cocaine injection (10 mg/kg, i.p.) in WT (n = 16) and VGLUT3A224V/− (n = 15) mice. During the first 2 h in the cyclotron, the locomotor activity of the VGLUT3A224V/− mice was higher than the locomotor activity of the WT mice (Mann–Whitney U test, p = 0.0329). After saline injection, the VGLUT3A224V/− mice were slightly hyperactive compared with the WT mice (Mann–Whitney U test, p = 0.0270). No difference was observed in cumulative locomotor activity after cocaine injection between the WT and the VGLUT3A224V/− mice (Mann–Whitney U test, p = 0.1725). Sal, Saline; Coc, cocaine.
We next investigated the behavior of VGLUT3A224V/− mice that displayed a 78% reduction of VGLUT3 expression. We first compared the anxiety behaviors of WT and VGLUT3A224V/− mice in the open field and elevated plus maze and found no differences (Fig. 7E,F). We then inspected basal and cocaine-induced locomotor activity of WT and VGLUT3A224V/− mice (Fig. 7G,H). During the first 2 h, we recorded the basal locomotor activity of mutant and WT mice and observed a significant increase in the activity in VGLUT3A224V/− mice (Fig. 7G, repeated-measures ANOVA, F(1,29) = 1.472, p < 0.05, H, cumulative analysis, +41%, Mann–Whitney U test, p = 0.0329). In contrast, the locomotor activity levels after cocaine injection were similar in both genotypes (Fig. 7H; Mann–Whitney U test, p > 0.05). Therefore, with only one copy of the VGLUT3-p.A224V isoform, and only 20% VGLUT3 expression, we detected minimal alterations in mutant mice behavior.
The p.A224V point mutation does not modify VGLUT3-dependent vesicular synergy over cortical [3H]5-HT accumulation
We then investigated whether the p.A224V mutation was able to alter the vesicular accumulation of glutamate in brain SVs of VGLUT3 mutants. VGLUT3 is a minor subtype compared with VGLUT1 and VGLUT2. It is therefore not possible to directly assess VGLUT3 activity in brain tissue. However, it has been previously established that VGLUT3 accelerates 5-HT accumulation in cortical synaptic vesicles through a molecular process called vesicular synergy (Amilhon et al., 2010; El Mestikawy et al., 2011). To estimate VGLUT3 activity in cortical synaptic vesicles, reserpine-sensitive vesicular uptake of [3H]5-HT was measured in the presence (+) or absence (−) of l-glutamate (Fig. 8). Similarly to previously reported results (Amilhon et al., 2010), l-glutamate (10 mm) increased [3H]5-HT reserpine-sensitive accumulation by 27% (Fig. 8; Mann–Whitney U test, p < 0.0001, n = 26) in the cortical synaptic vesicles of WT mice but had no effect on those of VGLUT3−/− mice. Similarly to the results for WT mice, we observed a vesicular synergy in 5-HT vesicular uptake of +27% in VGLUT3A224V/+ mice (Mann–Whitney U test, p = 0.0014, n = 7), +22% in VGLUT3A224V/A224V mice (Mann–Whitney U test, p < 0.0001, n = 19), and +22% in VGLUT3A224V/− mice (Mann–Whitney U test, p < 0.001, n = 19). The stimulatory effect of glutamate on 5-HT cortical vesicular uptake was not significantly different between the WT and the three other genotypes (one-way ANOVA, p > 0.05). We conclude from these experiments that VGLUT3 activity was probably unaltered, even in mice with only 20–30% transporter expression.
Figure 8.

The p.A224V mutation does not influence VGLUT3-dependent vesicular synergy. [3H]5-HT reserpine-sensitive uptake in cortical synaptic vesicles was measured in the presence (+) or absence (−) of l-glutamate (Glut). [3H]5-HT reserpine-sensitive vesicular accumulation was augmented by l-glutamate (10 mm) in the cortical synaptic vesicles of WT (+27%), VGLUT3A224V/+ (+27%), VGLUT3A224V/A224V (+22%), and VGLUT3A224V/− (+22%) mice but not in VGLUT3−/− mice because of the absence of VGLUT3 from synaptic vesicles. ** p < 0.01; *** p < 0.001.
High-resolution fluorescence microscopy suggests that the p.A224V mutation alters the number of VGLUT3-positive vesicles
The aforementioned results strongly suggested that the p.A224V mutation markedly reduces the number of copies of VGLUT3 in synaptic boutons. This decrease in copy number may result in (1) a reduction in the number of copies of the transporter on each vesicle or/and (2) a reduction in the number of VGLUT3-positive vesicles in each terminal. In an attempt to gain further insight in this issue, we used STED super-resolution imaging of VGLUT3 in the striatum of mutant mice (Fig. 9).
Figure 9.

High-resolution fluorescence imaging by STED microscopy revealed a decrease in VGLUT3-positive vesicles in mutant mice. A–E, Codetection by STED microscopy of VGLUT3 (red) and synaptophysin (green) in axonal varicosities of the striatum of WT (A), VGLUT3A224V/+ (B), VGLUT3A224V/A224V (C), VGLUT3A224V/− (D), and VGLUT3−/− (E) mice. VGLUT3 and synaptophysin immunofluorescence events were observed as round-shaped elements within striatal varicosities of WT, VGLUT3A224V/+, VGLUT3A224V/A224V, and VGLUT3A224V/− mice. In VGLUT3−/− mice, only synaptophysin was detected. Note the decrease in the number of VGLUT3-immunopositive puncta in VGLUT3A224V/+, VGLUT3A224V/A224V, and VGLUT3A224V/− mice compared with WT mice. Scale bar, 500 nm. F, Quantification of VGLUT3 and synaptophysin immunofluorescent events per varicosities. The numbers of VGLUT3-immunopositive puncta were quantified in striatal axonal varicosities of WT, VGLUT3A224V/+, VGLUT3A224V/A224V, VGLUT3A224V/−, and VGLUT3−/− mice after double immunofluorescence (80, 76, 76, 90, and 105 varicosities per animal were quantified in 6 WT, 6 VGLUT3A224V/+, 6 VGLUT3A224V/A224V, 6 VGLUT3A224V/−, and 5 VGLUT3−/− mice, respectively). Note that the number of VGLUT3 puncta decreased with the genotype. G, Correlation between number of VGLUT3-positive puncta determined by STED microscopy and VGLUT3 expression determined by immunoautoradiography. The correlation was statistically significant (linear regression, r2 = 0.9528; p = 0.0044). H, Effect of laser power on the number of events of VGLUT3-fluorescence detection. The blue arrowheads indicate the laser power that was selected for the images shown in A–E. The number of puncta per varicosity did not differ significantly when the laser power increased (Kruskal–Wallis test, p > 0.05).
STED microscopic observations after immunodetection showed spotty labeling of VGLUT3 and synaptophysin (Fig. 9A–E). The number of VGLUT3-positive puncta was highest in WT mice, was virtually nonexistent in VGLUT3−/− mice, and was decreased in VGLUT3A224V/+, VGLUT3A224V/A224V, and VGLUT3A224V/− mice. The quantification of VGLUT3-immunopositive puncta revealed decreases of 45% (Kruskal–Wallis test, p = 0.0029), 75% (Kruskal–Wallis test, p = 0.0022), and 84% (Kruskal–Wallis test, p = 0.0022) in VGLUT3A224V/+, VGLUT3A224V/A224V, and VGLUT3A224V/− mice, respectively (Fig. 9F). These decreases were significantly correlated with the expression of VGLUT3 detected by immunoautoradiography (Fig. 9G; linear regression, r2 = 0.9528, p = 0.0044). In contrast, the expression of synaptophysin was found to be similar in WT and in VGLUT3 mutants (Fig. 9F). Interestingly, increasing laser power neither increased the number of detected puncta (Fig. 9H; Kruskal–Wallis test, p > 0.05) nor allowed the detection of a population of puncta with low-intensity labeling.
Therefore, although it was not possible to determine whether the number of VGLUT3 copy per vesicle was reduced, STED imaging suggested that diminishing the expression of VGLUT3 reduced the number of VGLUT3-positive vesicles.
Discussion
In two human families, a point mutation in the gene encoding VGLUT3 exchanges the amino acid alanine 211 for a valine and segregates with an early-onset form of presbycusis (named DFNA25; Ruel et al., 2008). The VGLUT3-p.A211V (VGLUT3-p.A224V in rodent) point mutation is the first identified mutation of a VGLUT that is responsible for a human pathology. The impact of this mutation on the auditory system of the rodent is currently under investigation. Our preliminary results show that the p.A224V mutation recapitulates the human auditory pathology. The aim of the present study was to determine whether and how the p.A211V mutation influences CNS activity. Consequently, functions of mutant VGLUT3 were investigated in cell cultures, as well as in a genetic mouse model.
Interestingly, alanine 211 is part of a peptide sequence (KWAPPLER) that is conserved among VGLUTs and is even present in the more distant transporter named sialin. According to a theoretical model (Fig. 3A–H), the KWAPPLER motif is part of a small cytoplasmic loop that faces the pore of the transporter. Both the conservation and the localization of alanine 211 argue for its functional importance. Structurally, our 3D model predicted that exchanging alanine for valine would have little influence on the structure of VGLUT3. In line with this conclusion, glutamate vesicular accumulation in BON cells and evoked EPSCs observed in isolated neuronal cultures expressing the VGLUT3-p.A211V isoform were not different from that observed with the WT isoform. Furthermore, indirect estimation of VGLUT3 activity in cortical synaptic vesicles provided evidence that the ability of VGLUT3 to load glutamate in vesicles was not different in the brains of WT or mutant mice.
However, in both cell cultures and in a mutant mouse model, the mutation had a dramatic effect on the expression of the transporter. The average decrease across investigated brain areas of the mutant transporter in VGLUT3A224V/A224V mice was 70 ± 2.7%. As expected, mice with a single copy of the mutated allele (VGLUT3A224V/−) express ≈15–20% of transporter whereas those with two copies (VGLUT3A224V/A224V) express ≈20–30% of transporter (Table 2). These decreases could impact more severely synapses expressing low levels of VGLUT3. It thus cannot be ruled out that the VGLUT3-p.A221V mutation could have stronger effects in some terminals present in noninspected brain regions.
In the mutant mouse brain, the impact of the mutation on the expression of VGLUT3 appears to be constant between P10 and 12 months of age. This observation suggests that the reduction of VGLUT3 triggered by the mutation is independent from aging. Interestingly when the equivalent alanine in VGLUT1 (alanine 198) was mutated into a valine residue, an even stronger decrease of the transporter was observed (−90%; Fig. 2F). Therefore, alanine in the KWAPPLER motif could be pivotal for the expression levels (or stability) of all VGLUTs.
In VGLUT3A224V/A224V mice, the amount of the mutant transporter was dramatically reduced in hippocampal, striatal, and cortical terminals. However, this point mutation had no effect on the mRNA or on protein levels in the soma and proximal dendrites of VGLUT3-positive neurons. This result implies that the mutation does not alter the transcription or translation of VGLUT3. Furthermore, the mobility of VGLUT3-p.A211V was not significantly modified in the axons. These results suggested that the mutant transporter could be less stable or more rapidly degraded in the terminals of neurons. However, it should be kept in mind that the expression of mutant VGLUT3 was as low in BON as in primary neurons or in the mouse brain. Further investigation of the half-life of the mutated isoform will be needed to clarify this issue.
We estimated the activity of the mutated isoform directly by measuring glutamate uptake in vesicles of BON cells and indirectly by measuring synaptic transmission in single neurons expressing the mutant transporter or by measuring vesicular synergy in cortical vesicles. In all cases, we found very minimal or no modifications of VGLUT3 activity.
A previous study reported that in Caenorhabditis elegans, four different point mutations of VAChT profoundly altered transporter activity (mostly the Km) without decreasing its expression or altering its targeting (Zhu et al., 2001). This illustrates the complex and diverse effects of point mutations on transporters activity.
Previous reports established that mice completely lacking VGLUT3 demonstrate increased anxiety and sensitivity to cocaine (Gras et al., 2008; Amilhon et al., 2010; Sakae et al., 2015). Here, none of these phenotypes were observed with VGLUT3A224V/A224V mice lacking >70% of VGLUT3, and only moderate effects were observed on locomotor activity in a mutant lacking up to 80% of VGLUT3 (VGLUT3A224V/−). Our results showed that despite a substantial reduction in VGLUT3 expression, its biochemical and many integrated functions were unchanged. However, we cannot rule out that VGLUT3A224V/A224V or VGLUT3A224V/+ mice may display some phenotypes that were not investigated in the present study. For example, mice lacking VGLUT3 demonstrate abnormal interictal discharges (Seal et al., 2008). These cortical generalized synchronous discharges are not accompanied by convulsive electrographic seizures. Interestingly, knock-out mice heterozygous for VGLUT3 (VGLUT3+/−) also show sharp interictal spike discharges. We therefore cannot exclude the possibility that VGLUT3A224V/A224V or VGLUT3A224V/+ mice present the abnormal interictal discharges reported in VGLUT3+/− mice (Seal et al., 2008).
Nonetheless, for phenotypes such as locomotor activity, sensitivity to cocaine, or anxiety VGLUT3A224V/A224V or VGLUT3A224V/+ mice demonstrated no behavioral alterations. As depicted in the model in Figure 10A, there may be a substantial safety factor in the level of VGLUT3 required to sustain glutamate uptake such that only a small copy number is required to maintain these physiological functions. The threshold where a decrease of the VGLUT3 level will start to impact its function is situated within a narrow range between 20 and 0% of the wild-type level (Fig. 10A). Therefore, unlike those of the monoamine and acetylcholine vesicular transporters, VMAT2 and VAChT (Fon et al., 1997; Prado et al., 2006), the safety factor for VGLUT3 may be higher, with normal function being maintained, even with protein levels that are below 70% of WT levels.
Figure 10.

Putative models depicting the reduction in VGLUT3 at the synapses of different mouse genotypes. In this study, we investigated mouse models with variable levels of VGLUT3 expression: WT mice (2 copies of WT VGLUT3 isoform; 100%), heterozygous mice (1 copy of WT VGLUT3 isoform and 1 copy of VGLUT3-p.A224V allele; 67%), homozygous mice (2 copies of the mutated VGLUT3-p.A224V allele; 28%), VGLUT3A224V/− mice (expressing only 1 copy of the mutated VGLUT3-p.A224V allele; 21%), and VGLUT3 knock-out mice (no copy of VGLUT3; 0%). A, The black curve shows the relationship between VGLUT3 expression (detected by immunoautoradiography) and the activity of VGLUT3 (indirectly assessed by measuring vesicular synergy). VGLUT3 activity did not decline in proportion to the amount of VGLUT3 in the expression range between 100% and 21% (black curve). The absence of a correlation between these two sets of measurements explains the virtual lack of a VGLUT3-dependent phenotype in our panel of mutants. This model predicts that these phenotypes will be observed in the gray zone of the curve (red curve). B, Three putative models that may account for the reduction in VGLUT3 at the synapses are compared with our experimental results. In WT mice, numerous copies of VGLUT3 are uniformly distributed between synaptic vesicles, and these vesicles are “normally” loaded (100% gray level) with glutamate. In Model 1 (molecular model), VGLUT3 copies are uniformly decreased in all vesicles of VGLUT3A224V/A224V mice. In this model, the vesicular content of glutamate is minimally decreased in all vesicles. This decrease in glutamatergic quantal size cannot be observed with bulk methods, such as vesicular uptake or behavioral measurements, but can be detected by more sensitive electrophysiological techniques. Model 1 would be compatible with (1) an absence of change in vesicular uptake (observed in BON cells) and vesicular synergy (VS; observed in cortical vesicles) and (2) a decrease in the amplitudes of the mEPSCs that were observed in recordings of isolated neurons. Model 2 (vesicular model) is based on the STED high-resolution inspection of VGLUT3-positive terminals in our panel of mutants. In this model, in a small proportion of vesicles, the number of VGLUT3-p.A224V copies per vesicles is similar (or minimally decreased) to that found in WT neurons. According to the electrophysiological recordings of autapses, these vesicles may contain normal levels of glutamate and be preferentially docked. The remaining vesicles (80%) will contain neither VGLUT3 nor glutamate. This model is consistent with the decreased frequency of the mEPSCs. A mixed model (Model 3) of these two models may better explain all of our experimental results. In this model, we found vesicles without VGLUT3, a small proportion of vesicles expressing the correct number of VGLUT3-p.A224V copies, and a third population of vesicles in which the number of VGLUT3 copies were uniformly decreased in all vesicles of the synapses from VGLUT3A224V/A224V mice.
A key question is whether these observations are also valid for VGLUT1 and VGLUT2. Whether vesicular accumulation of glutamate catalyzed by VGLUT1 or VGLUT2 is proportional to the amount of transporters has been a matter of debate (for review, see Fremeau et al., 2004; Schuske and Jorgensen, 2004; Wojcik et al., 2004; Wilson et al., 2005). Wojcik et al. (2004) reported decreases in mEPSC amplitude and frequency in neurons lacking VGLUT1 and, conversely, increased quantal size in neurons overexpressing VGLUT1. Furthermore, Wilson et al. (2005) reported that in hippocampal neuronal culture as well as during postnatal development, increasing the density of VGLUT1 results in enhanced glutamate release into the synaptic cleft. These observations support a linear relationship between VGLUT1 amount and VGLUT1 activity (i.e., glutamate vesicular packaging). However, if glutamate vesicular loading and transmission were proportional to the copy number of VGLUTs, then heterozygous knock-out mice (with 50% VGLUT expression) should display a 50% decrease in glutamatergic signaling, and this in turn should impact related behaviors. Fremeau et al. (2004) showed that VGLUT1+/− heterozygous mice display normal excitatory transmission. In contrast, impaired mood regulation and memory have been reported in VGLUT1+/− heterozygous mice (Tordera et al., 2007; Balschun et al., 2010). VGLUT2+/− mice express only 50% of the WT level but demonstrate only discrete phenotypes in taste aversion, nociceptive responses, and clonic seizures (Moechars et al., 2006; Leo et al., 2009; Schallier et al., 2009). However, these phenotypes are often modest.
Takamori et al. (2006) showed that, on average, synaptic vesicles contain 10 copies of VGLUT1 or 14 copies of VGLUT2 protein. Hence, there are multiple copies of VGLUT1–2 inserted into vesicular membranes. The exact number of copies of VGLUT3 that are present on individual synaptic vesicles has not yet been determined. However, it can be reasonably assumed that, as with VGLUT1 and VGLUT2, multiple copies of VGLUT3 are inserted in the membrane of synaptic vesicles.
The redundancy of vesicular glutamate copies per vesicles could provide a safety factor in case of loss of transporters. For example, in Drosophila, reduced levels of DVGLUTs result in a reduction of mEPSC frequency, with no change in quantal size (Daniels et al., 2006). Additional studies support the notion that one copy of a VGLUT is sufficient to fill synaptic vesicles and to maintain a normal quantal size (Wojcik et al., 2004; Daniels et al., 2006; Schenck et al., 2009; Preobraschenski et al., 2014), as depicted in Model 1 (Fig. 10A). Model 1 predicts that the decreased expression of VGLUT3-p.A224V could be attributable to a homogeneous reduction of the number of copies of transporter per vesicles. According to this model, as long as there is at least 20% residual VGLUT3, the filling of vesicles with glutamate should be normal. The exact number of copies of VGLUT3 per vesicle corresponding to this percentage remains to be determined.
In the present study, we observed normal loading of [3H]l-glutamate into VGLUT3-p.A211V-positive BON vesicles. However, a small significant decrease in mEPSC amplitude was observed in isolated neurons expressing the mutant isoform. It should be noted that electrophysiological recordings in autapses were obtained with increased amounts of mutant isoform with the objective of normalizing levels with those of the WT isoform. This represents a limitation of the present study, as studying the effects of the mutation on synaptic activity without artificially increasing the level of the mutant transporter may have better represented the situation occurring in mutation carriers. Discrepancies between vesicular uptake and electrophysiological measurements can easily be explained by a difference in the sensitivity of both methods. In line with this explanation, a small but nonsignificant decrease was observed in vesicles of BON cells expressing VGLUT3-p.A211V. However, Model 1 does not explain why a significant decrease of mEPSC frequency was observed in isolated neurons expressing the mutant isoform. Furthermore, this model is not readily compatible with our observations obtained by STED microscopy. Surprisingly, STED microscopy showed a decrease of VGLUT3-positive fluorescent puncta per terminal. This suggests that the reduced expression of the mutated isoform in terminals is attributable to a reduction of the number of VGLUT3-positive vesicles as described by Model 2.
This second model is well in line with the decreased frequency of mEPSCs observed in hippocampal autapses expressing VGLUT3-p.A211V. Indeed, an increased number of “empty” vesicles could result in an increased number of silent events and, therefore, in a decreased frequency of mEPSCs. However, Model 2 is not compatible with the fact that vesicular uptake (in BON cells and in brain vesicles) and release probability (in hippocampal autapses) were virtually unchanged.
The discrepancy between the predictions of Models 1 and 2 could be resolved if there is a nonhomogeneous distribution of the mutant isoform between different pools of vesicles as shown with Model 3. In this third putative model, we propose the existence of at least three populations of synaptic vesicles in VGLUT3A224V/A224V mice. Synaptic vesicles could contain either high-level (or normal) or low level VGLUT3-p.A224V copies. Synaptic vesicles with a low copy number of mutant VGLUT3 would not be detected by STED microscopy. They could contain a slightly decreased amount of glutamate, therefore explaining the small decrease in the amplitude of mEPSCs in hippocampal autapses. The vesicular populations with “high and low” VGLUT3-p.A211V content could allow a normal or only slightly altered quantum of glutamate. They also could explain why VGLUT3 biochemical, electrophysiological, and behavioral function are preserved in VGLUT3A224V/A224V mice. In addition, a small fraction of synaptic vesicles are present with no copy of VGLUT3-p.A221V. These vesicles account for the decreased frequency of mEPSC depicted in Figure 3, P and Q. If this model is correct, it implies that the KWAPPLER motif plays a central role in the vesicular targeting of VGLUT3. In particular, the trafficking of VGLUT3-p.A211V between different vesicular pools appears to be profoundly altered. Interestingly, synaptophysin labeling remained constant in the various VGLUT3 mutant mice analyzed in this study. This observation suggests that the number of synaptic vesicles is unaltered in the mutant mice.
The validation or invalidation of Model 3 will necessitate additional experiments such as the use of super-resolution microscopic approaches [STochastic Optical Reconstruction Microscopy (STORM) or Photo-Activated Localization Microscopy (PALM)]. These methods that allow detection of single molecules may help to quantify the number of VGLUT3 per vesicle.
Rare variants of VGLUTs have begun to be identified in human pathologies (Ruel et al., 2008; Shen et al., 2010; Sakae et al., 2015). It is a key challenge to understand how these mutations can affect VGLUT functions and glutamatergic transmission. As shown here, the A224V mutation that causes deafness in humans profoundly alters the protein levels of VGLUT3 but minimally alters its functions. Our study reveals an unexpected redistribution of VGLUT3 in the synaptic vesicles of VGLUT3A224V/A224V mice brain. Furthermore, we suggest the existence of a large safety factor in the number of VGLUT3 molecules required to sustain normal physiological functions. Additional experiments will be required to validate or invalidate the three models proposed in Figure 10. Clarifying this question will be important for gaining a complete understanding of the pathologies that involve VGLUTs.
Footnotes
This work was supported by the Fondation pour la Recherche Médicale (Équipe FRM DEQ20130326486), the Agence Nationale pour la Recherche, the Fédération pour la Recherche sur le Cerveau, Labex (Bio-Psy Laboratory of Excellence), Institut National de la Santé et de la Recherche Médicale (Inserm), CNRS, and Université Pierre et Marie Curie (UPMC). L.R. received a PhD fellowship from the Ministère de l'enseignement supérieur et de la recherche and from the Fondation pour la Recherche Médicale (FDT20140930909). We thank Géraldine Toutirais from the Service de Microscopie Electronique of the Institut de Biologie Paris-Seine (UPMC, Paris, France). We thank Valérie Nicolas from the Plateforme d'imagerie cellulaire of the Institut Paris Saclay d'Innovation Thérapeutique (Unité Mixte de Service [UMS] Institut Paris Saclay d'Innovation Thérapeutique [IPSIT] Université Paris-Sud–US 31 Inserm–UMS 3679 CNRS, Châtenay-Malabry, France) for assistance with STED microscopy. We thank Christoph Biesemann for the lentivirus constructs and Stéphanie Pons, Martine Soudant, and l'École des Neurosciences de Paris (“Network for Viral Transfer”) for producing the lentiviruses.
The authors declare no competing financial interests.
References
- Almqvist J, Huang Y, Laaksonen A, Wang DN, Hovmöller S (2007) Docking and homology modeling explain inhibition of the human vesicular glutamate transporters. Protein Sci 16:1819–1829. 10.1110/ps.072944707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amilhon B, Lepicard E, Renoir T, Mongeau R, Popa D, Poirel O, Miot S, Gras C, Gardier AM, Gallego J, Hamon M, Lanfumey L, Gasnier B, Giros B, El Mestikawy S (2010) VGLUT3 (vesicular glutamate transporter type 3) contribution to the regulation of serotonergic transmission and anxiety. J Neurosci 30:2198–2210. 10.1523/JNEUROSCI.5196-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balschun D, Moechars D, Callaerts-Vegh Z, Vermaercke B, Van Acker N, Andries L, D'Hooge R (2010) Vesicular glutamate transporter VGLUT1 has a role in hippocampal long-term potentiation and spatial reversal learning. Cereb Cortex 20:684–693. 10.1093/cercor/bhp133 [DOI] [PubMed] [Google Scholar]
- Bellocchio EE, Reimer RJ, Fremeau RT Jr, Edwards RH (2000) Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289:957–960. 10.1126/science.289.5481.957 [DOI] [PubMed] [Google Scholar]
- Bernard V, Levey AI, Bloch B (1999) Regulation of the subcellular distribution of m4 muscarinic acetylcholine receptors in striatal neurons in vivo by the cholinergic environment: evidence for regulation of cell surface receptors by endogenous and exogenous stimulation. J Neurosci 19:10237–10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A (2004) Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci 24:10466–10474. 10.1523/JNEUROSCI.3001-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Chen K, Gelfand MV, Featherstone DE, DiAntonio A (2006) A single vesicular glutamate transporter is sufficient to fill a synaptic vesicle. Neuron 49:11–16. 10.1016/j.neuron.2005.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Miller BR, DiAntonio A (2011) Increased vesicular glutamate transporter expression causes excitotoxic neurodegeneration. Neurobiol Dis 41:415–420. 10.1016/j.nbd.2010.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gois S, Slama P, Pietrancosta N, Erdozain AM, Louis F, Bouvrais-Veret C, Daviet L, Giros B (2015) Ctr9, a protein in the transcription complex Paf1, regulates dopamine transporter activity at the plasma membrane. J Biol Chem 290:17848–17862. 10.1074/jbc.M115.646315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Mestikawy S, Wallén-Mackenzie A, Fortin GM, Descarries L, Trudeau LE (2011) From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nat Rev Neurosci 12:204–216. 10.1038/nrn2969 [DOI] [PubMed] [Google Scholar]
- Fasano C, Thibault D, Trudeau LE (2008) Culture of postnatal mesencephalic dopamine neurons on an astrocyte monolayer. Curr Protoc Neurosci Chap 3:Unit 3.21. 10.1002/0471142301.ns0321s44 [DOI] [PubMed] [Google Scholar]
- Feyfant E, Sali A, Fiser A (2007) Modeling mutations in protein structures. Protein Sci 16:2030–2041. 10.1110/ps.072855507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fon EA, Pothos EN, Sun BC, Killeen N, Sulzer D, Edwards RH (1997) Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron 19:1271–1283. 10.1016/S0896-6273(00)80418-3 [DOI] [PubMed] [Google Scholar]
- Fremeau RT Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH (2001) The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31:247–260. 10.1016/S0896-6273(01)00344-0 [DOI] [PubMed] [Google Scholar]
- Fremeau RT Jr, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH (2002) The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci U S A 99:14488–14493. 10.1073/pnas.222546799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH (2004) Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 304:1815–1819. 10.1126/science.1097468 [DOI] [PubMed] [Google Scholar]
- Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S (2002) A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci 22:5442–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras C, Amilhon B, Lepicard EM, Poirel O, Vinatier J, Herbin M, Dumas S, Tzavara ET, Wade MR, Nomikos GG, Hanoun N, Saurini F, Kemel ML, Gasnier B, Giros B, El Mestikawy S (2008) The vesicular glutamate transporter VGLUT3 synergizes striatal acetylcholine tone. Nat Neurosci 11:292–300. 10.1038/nn2052 [DOI] [PubMed] [Google Scholar]
- Herman MA, Ackermann F, Trimbuch T, Rosenmund C (2014) Vesicular glutamate transporter expression level affects synaptic vesicle release probability at hippocampal synapses in culture. J Neurosci 34:11781–11791. 10.1523/JNEUROSCI.1444-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B, El Mestikawy S (2001) The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci 21:RC181(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog E, Gilchrist J, Gras C, Muzerelle A, Ravassard P, Giros B, Gaspar P, El Mestikawy S (2004) Localization of VGLUT3, the vesicular glutamate transporter type 3, in the rat brain. Neuroscience 123:983–1002. 10.1016/j.neuroscience.2003.10.039 [DOI] [PubMed] [Google Scholar]
- Herzog E, Nadrigny F, Silm K, Biesemann C, Helling I, Bersot T, Steffens H, Schwartzmann R, Nägerl UV, El Mestikawy S, Rhee J, Kirchhoff F, Brose N (2011) In vivo imaging of intersynaptic vesicle exchange using VGLUT1 Venus knock-in mice. J Neurosci 31:15544–15559. 10.1523/JNEUROSCI.2073-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Gittis AH, Oldenburg IA, Balthasar N, Seal RP, Edwards RH, Lowell BB, Kreitzer AC, Sabatini BL (2011) Cholinergic interneurons mediate fast VGluT3-dependent glutamatergic transmission in the striatum. PLoS One 6:e19155. 10.1371/journal.pone.0019155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- Leo S, Moechars D, Callaerts-Vegh Z, D'Hooge R, Meert T (2009) Impairment of VGLUT2 but not VGLUT1 signaling reduces neuropathy-induced hypersensitivity. Eur J Pain 13:1008–1017. 10.1016/j.ejpain.2008.12.001 [DOI] [PubMed] [Google Scholar]
- McAnaney TB, Zeng W, Doe CF, Bhanji N, Wakelin S, Pearson DS, Abbyad P, Shi X, Boxer SG, Bagshaw CR (2005) Protonation, photobleaching, and photoactivation of yellow fluorescent protein (YFP 10C): a unifying mechanism. Biochemistry 44:5510–5524. 10.1021/bi047581f [DOI] [PubMed] [Google Scholar]
- Moechars D, Weston MC, Leo S, Callaerts-Vegh Z, Goris I, Daneels G, Buist A, Cik M, van der Spek P, Kass S, Meert T, D'Hooge R, Rosenmund C, Hampson RM (2006) Vesicular glutamate transporter VGLUT2 expression levels control quantal size and neuropathic pain. J Neurosci 26:12055–12066. 10.1523/JNEUROSCI.2556-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Bussert TG, Kreitzer AC, Seal RP (2014) Striatal cholinergic neurotransmission requires VGLUT3. J Neurosci 34:8772–8777. 10.1523/JNEUROSCI.0901-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirs C, Williams SP, Zhao X, Walsh CE, Gedeon JY, Cagle NE, Goldring AC, Hioki H, Liu Z, Marell PS, Seal RP (2015) Dorsal horn circuits for persistent mechanical pain. Neuron 87:797–812. 10.1016/j.neuron.2015.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado VF, Martins-Silva C, de Castro BM, Lima RF, Barros DM, Amaral E, Ramsey AJ, Sotnikova TD, Ramirez MR, Kim HG, Rossato JI, Koenen J, Quan H, Cota VR, Moraes MF, Gomez MV, Guatimosim C, Wetsel WC, Kushmerick C, Pereira GS, et al. (2006) Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron 51:601–612. 10.1016/j.neuron.2006.08.005 [DOI] [PubMed] [Google Scholar]
- Preobraschenski J, Zander JF, Suzuki T, Ahnert-Hilger G, Jahn R (2014) Vesicular glutamate transporters use flexible anion and cation binding sites for efficient accumulation of neurotransmitter. Neuron 84:1287–1301. 10.1016/j.neuron.2014.11.008 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. 10.1016/S0896-6273(00)80146-4 [DOI] [PubMed] [Google Scholar]
- Ruel J, Emery S, Nouvian R, Bersot T, Amilhon B, Van Rybroek JM, Rebillard G, Lenoir M, Eybalin M, Delprat B, Sivakumaran TA, Giros B, El Mestikawy S, Moser T, Smith RJ, Lesperance MM, Puel JL (2008) Impairment of SLC17A8 encoding vesicular glutamate transporter-3, VGLUT3, underlies nonsyndromic deafness DFNA25 and inner hair cell dysfunction in null mice. Am J Hum Genet 83:278–292. 10.1016/j.ajhg.2008.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakae DY, Marti F, Lecca S, Vorspan F, Martín-García E, Morel LJ, Henrion A, Gutiérrez-Cuesta J, Besnard A, Heck N, Herzog E, Bolte S, Prado VF, Prado MA, Bellivier F, Eap CB, Crettol S, Vanhoutte P, Caboche J, Gratton A, et al. (2015) The absence of VGLUT3 predisposes to cocaine abuse by increasing dopamine and glutamate signaling in the nucleus accumbens. Mol Psychiatry 20:1448–1459. 10.1038/mp.2015.104 [DOI] [PubMed] [Google Scholar]
- Schäfer MK, Varoqui H, Defamie N, Weihe E, Erickson JD (2002) Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J Biol Chem 277:50734–50748. 10.1074/jbc.M206738200 [DOI] [PubMed] [Google Scholar]
- Schallier A, Massie A, Loyens E, Moechars D, Drinkenburg W, Michotte Y, Smolders I (2009) vGLUT2 heterozygous mice show more susceptibility to clonic seizures induced by pentylenetetrazol. Neurochem Int 55:41–44. 10.1016/j.neuint.2008.12.019 [DOI] [PubMed] [Google Scholar]
- Schenck S, Wojcik SM, Brose N, Takamori S (2009) A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat Neurosci 12:156–162. 10.1038/nn.2248 [DOI] [PubMed] [Google Scholar]
- Schuske K, Jorgensen EM (2004) Neuroscience. Vesicular glutamate transporter–shooting blanks. Science 304:1750–1752. 10.1126/science.1100475 [DOI] [PubMed] [Google Scholar]
- Seal RP, Akil O, Yi E, Weber CM, Grant L, Yoo J, Clause A, Kandler K, Noebels JL, Glowatzki E, Lustig LR, Edwards RH (2008) Sensorineural deafness and seizures in mice lacking vesicular glutamate transporter 3. Neuron 57:263–275. 10.1016/j.neuron.2007.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal RP, Wang X, Guan Y, Raja SN, Woodbury CJ, Basbaum AI, Edwards RH (2009) Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature 462:651–655. 10.1038/nature08505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YC, Liao DL, Lu CL, Chen JY, Liou YJ, Chen TT, Chen CH (2010) Resequencing of the vesicular glutamate transporter 2 gene (VGLUT2) reveals some rare genetic variants that may increase the genetic burden in schizophrenia. Schizophr Res 121:179–186. 10.1016/j.schres.2010.05.015 [DOI] [PubMed] [Google Scholar]
- Siksou L, Silm K, Biesemann C, Nehring RB, Wojcik SM, Triller A, El Mestikawy S, Marty S, Herzog E (2013) A role for vesicular glutamate transporter 1 in synaptic vesicle clustering and mobility. Eur J Neurosci 37:1631–1642. 10.1111/ejn.12199 [DOI] [PubMed] [Google Scholar]
- Somogyi J, Baude A, Omori Y, Shimizu H, El Mestikawy S, Fukaya M, Shigemoto R, Watanabe M, Somogyi P (2004) GABAergic basket cells expressing cholecystokinin contain vesicular glutamate transporter type 3 (VGLUT3) in their synaptic terminals in hippocampus and isocortex of the rat. Eur J Neurosci 19:552–569. 10.1111/j.0953-816X.2003.03091.x [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407:189–194. 10.1038/35025070 [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R (2001) Identification of differentiation-associated brain-specific phosphate transporter as a second vesicular glutamate transporter (VGLUT2). J Neurosci 21:RC182 (1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Malherbe P, Broger C, Jahn R (2002) Molecular cloning and functional characterization of human vesicular glutamate transporter 3. EMBO Rep 3:798–803. 10.1093/embo-reports/kvf159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, et al. (2006) Molecular anatomy of a trafficking organelle. Cell 127:831–846. 10.1016/j.cell.2006.10.030 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordera RM, Totterdell S, Wojcik SM, Brose N, Elizalde N, Lasheras B, Del Rio J (2007) Enhanced anxiety, depressive-like behaviour and impaired recognition memory in mice with reduced expression of the vesicular glutamate transporter 1 (VGLUT1). Eur J Neurosci 25:281–290. 10.1111/j.1460-9568.2006.05259.x [DOI] [PubMed] [Google Scholar]
- Tusnády GE, Simon I (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17:849–850. 10.1093/bioinformatics/17.9.849 [DOI] [PubMed] [Google Scholar]
- Varga V, Losonczy A, Zemelman BV, Borhegyi Z, Nyiri G, Domonkos A, Hangya B, Holderith N, Magee JC, Freund TF (2009) Fast synaptic subcortical control of hippocampal circuits. Science 326:449–453. 10.1126/science.1178307 [DOI] [PubMed] [Google Scholar]
- Varoqui H, Schäfer MK, Zhu H, Weihe E, Erickson JD (2002) Identification of the differentiation-associated Na+/PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci 22:142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneault É, Poirel O, Riad M, Prud'homme J, Dumas S, Turecki G, Fasano C, Mechawar N, El Mestikawy S (2015) Distribution of vesicular glutamate transporters in the human brain. Front Neuroanat 9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MC, Nehring RB, Wojcik SM, Rosenmund C (2011) Interplay between VGLUT isoforms and endophilin A1 regulates neurotransmitter release and short-term plasticity. Neuron 69:1147–1159. 10.1016/j.neuron.2011.02.002 [DOI] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G (2005) Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci 25:6221–6234. 10.1523/JNEUROSCI.3003-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C (2004) An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci U S A 101:7158–7163. 10.1073/pnas.0401764101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander JF, Münster-Wandowski A, Brunk I, Pahner I, Gómez-Lira G, Heinemann U, Gutierrez R, Laube G, Ahnert-Hilger G (2010) Synaptic and vesicular coexistence of VGLUT and VGAT in selected excitatory and inhibitory synapses. J Neurosci 30:7634–7645. 10.1523/JNEUROSCI.0141-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Duerr JS, Varoqui H, McManus JR, Rand JB, Erickson JD (2001) Analysis of point mutants in the Caenorhabditis elegans vesicular acetylcholine transporter reveals domains involved in substrate translocation. J Biol Chem 276:41580–41587. 10.1074/jbc.M103550200 [DOI] [PubMed] [Google Scholar]