

Abstract
Cataplexy is a hallmark of narcolepsy characterized by the sudden uncontrollable onset of muscle weakness or paralysis during wakefulness. It can occur spontaneously, but is typically triggered by positive emotions such as laughter. Although cataplexy was identified >130 years ago, its neural mechanism remains unclear. Here, we show that a newly identified GABA circuit within the central nucleus of the amygdala (CeA) promotes cataplexy. We used behavioral, electrophysiological, immunohistochemical, and chemogenetic strategies to target and manipulate CeA activity selectively in narcoleptic (orexin−/−) mice to determine its functional role in controlling cataplexy. First, we show that chemogenetic activation of the entire CeA produces a marked increase in cataplexy attacks. Then, we show that GABA cells within the CeA are responsible for mediating this effect. To manipulate GABA cells specifically, we developed a new mouse line that enables genetic targeting of GABA cells in orexin−/− mice. We found that chemogenetic activation of GABA CeA cells triggered a 253% increase in the number of cataplexy attacks without affecting their duration, suggesting that GABA cells play a functional role in initiating but not maintaining cataplexy. We show that GABA cell activation only promotes cataplexy attacks associated with emotionally rewarding stimuli, not those occurring spontaneously. However, we found that chemogenetic inhibition of GABA CeA cells does not prevent cataplexy, suggesting these cells are not required for initiating cataplexy attacks. Our results indicate that the CeA promotes cataplexy onset and that emotionally rewarding stimuli may trigger cataplexy by activating GABA cells in the CeA.
SIGNIFICANCE STATEMENT Although cataplexy has been closely linked to positive emotions for >130 years, the neural circuitry that underlies this relationship is poorly understood. Recent work suggests that the amygdala, a brain area important for processing emotion, may be part of this circuit. This study provides the first functional evidence to implicate GABA cells in the amygdala as regulators of cataplexy triggered by positive emotions and identifies the amygdala as the brain region important more for gating the entrance into rather than the exit from cataplexy. We also generated a new mouse model for studying GABA neurons in narcoleptic mice, which could serve as a useful tool for studying the neurobiological underpinnings of narcolepsy.
Keywords: amygdala, cataplexy, chemogenetics, narcolepsy, sleep
Introduction
Narcolepsy is a sleep disorder caused by loss of hypothalamic orexin (hypocretin) cells (Peyron et al., 2000; Thannickal et al., 2000; Jones, 2008). It is typically characterized by excessive sleepiness, sleep paralysis, and hypnagogic hallucinations, but its pathognomonic symptom is cataplexy, the sudden involuntary onset of bilateral muscle paralysis or weakness during otherwise normal wakefulness (Dauvilliers et al., 2014; Fraigne et al., 2015). Cataplexy can occur spontaneously, but it is typically triggered by strong positive emotions such as laughter, joking, or elation (Overeem et al., 2011; Dauvilliers et al., 2014). Even though the link between emotions and cataplexy was identified in the late 1800s (Westphal, 1877; Gélineau, 1880), the neural pathways through which emotions trigger cataplexy remain speculative. Here, we aimed to determine whether the amygdala, a limbic structure involved in processing positive emotions (Gallagher et al., 1990; Breiter et al., 1996; Garavan et al., 2001; Paton et al., 2006; Murray, 2007; Tye and Janak, 2007), plays a functional role in triggering cataplexy in narcoleptic mice.
Evidence not only suggests that the amygdala is involved in mediating cataplexy, but also that it could be a neural substrate through which emotions trigger cataplexy. Functional imaging studies in human narcoleptics show that amygdala activity increases during cataplexy episodes associated with humorous stimuli (Hong et al., 2006; Meletti et al., 2015). Unit recordings in narcoleptic dogs show that cells in the central nucleus of the amygdala (CeA) increase their activity during cataplexy attacks triggered by rewarding stimuli such as food (Gulyani et al., 2002). Lesion studies in narcoleptic (orexin−/−) mice show that the CeA plays a functional role in promoting cataplexy because its destruction reduces cataplexy attacks associated with potentially rewarding stimuli such as running wheels (Lett et al., 2002; Burgess et al., 2013). In addition, cell-mapping studies show that the amygdala, and particularly GABA cells in the CeA, are anatomically connected to brainstem regions that are capable of triggering muscle paralysis during cataplexy (Davis, 1992; Sah et al., 2003; Burgess et al., 2013).
Here, we test the hypothesis that GABA neurons in the CeA are involved in triggering cataplexy in narcoleptic mice. We did this using behavioral, electrophysiological, immunohistochemical, and chemogenetic approaches to determine how manipulation of GABA cells in the CeA affects cataplexy in orexin−/− mice. However, to target GABA cells selectively, we first needed to develop a narcoleptic mouse suited for this purpose. We did this by creating the orexin−/−,VGAT-Cre mouse, an orexin−/− mouse (Chemelli et al., 1999) that expresses Cre recombinase only in GABA neurons (Vong et al., 2011), thereby allowing us to deliver candidate proteins (e.g., hM3Dq receptors; Armbruster et al., 2007) to GABA cells. After extensive validation, we showed that this new model experiences a typical amount of cataplexy (relative to orexin−/− mice) and that chemogenetic approaches can be used to target and manipulate GABA cells selectively. Then, we showed that switching on GABA CeA cells triggered rapid and robust increases in cataplexy, but that chemogenetic inhibition of these cells did not prevent cataplexy. Our results indicate that GABA cells in the amygdala are important in gating the entrance into, but not the exit from, cataplexy.
Materials and Methods
Animals.
We used three different lines of mice in these experiments. Male wild-type (i.e., C57BL/6; age: 19 ± 2 weeks; mass: 25 ± 2 g) and male orexin knock-out (orexin−/− on a C57BL/6 background; age: 13 ± 2 weeks; mass: 23 ± 1 g; founder line provided by Drs. Sinton and Yanagisawa) littermates were used to determine how CeA activation and inhibition affects behavior. We also used a new mouse line that we developed over the last 2 years. This line was developed so that GABA cells in the CeA could be targeted selectively using chemogenetic methods. We call these mice orexin−/−,VGAT-Cre mice (age: 13 ± 2 weeks; mass: 26 ± 2 g) because they were generated by crossing orexin−/− mice (Chemelli et al., 1999) with VGAT-Cre mice (Vong et al., 2011) on a C57BL/6 background (founder line was provided by Dr. Adamantidis).
Throughout the experiments, mice were housed individually on a 12:12 light/dark cycle (lights off at 19:00) in a temperature- and humidity-controlled environment, had food and water available ad libitum, and had access to a running wheel. All procedures and experimental protocols were approved by the University of Toronto's Local Animal Care Committee and were in accordance with the Canadian Council on Animal Care.
Chemogenetic methods.
To manipulate the activity of CeA cells, we drove expression of hM3Dq or hM4Di receptors, which, when exposed to clozapine-N-oxide (CNO), cause neuronal activation and inhibition, respectively (Armbruster et al., 2007; Rogan and Roth, 2011). We delivered hM3Dq and hM4Di receptors virally using constructs obtained from the University of North Carolina Vector Core. Constructs were packaged into adeno-associated viruses with serotype 8 (AAV8) expressed downstream of a neuron-specific promoter (hSyn) and mCherry was used as a reporter gene. For experiments involving orexin−/− mice and wild-type mice, the construct contained an excitatory hM3Dq receptor (AAV8/hSyn-hM3Dq-mCherry, 4.5 × 1012 particles/ml). For experiments involving orexin−/−,VGAT-Cre mice, the construct contained either hM3Dq (excitation experiments), no DREADD (empty vector; control experiments), or hM4Di (inhibition experiments). To ensure cellular specificity in these mice, viral constructs were in a double-floxed inverted orientation (DIO) such that the genes could be expressed only in cells containing Cre recombinase (i.e., GABA cells; excitation: AAV8/hSyn-DIO-hM3Dq-mCherry, 5.7 × 1012 particles/ml; control: AAV8/hSyn-DIO-mCherry, 8.0 × 1012 particles/ml; inhibition: AAV8/hSyn-DIO-hM4Di-mCherry, 5.3 × 1012 particles/ml).
Drug preparation.
hM3Dq and hM4Di receptors are activated exclusively by the biologically inert ligand CNO. CNO injections were prepared by dissolving CNO powder (donated by Dr. Bryan Roth) in a sterile solution of 0.9% saline and 0.05% dimethyl sulfoxide (DMSO; Sigma-Aldrich). CNO was administered intraperitoneally in doses of 1 mg/kg for wild-type mice or 2.5 mg/kg and 5 mg/kg for orexin−/− and orexin−/−,VGAT-Cre mice to activate hM3Dq receptors, and 5 mg/kg for orexin−/−,VGAT-Cre mice to activate hM4Di receptors; these doses have been shown previously to activate hM3Dq and hM4Di receptors (Farrell and Roth, 2013; Li et al., 2013; Cai et al., 2014; Weber et al., 2015). Because we found no response difference for doses in orexin−/− mice (i.e., 2.5 mg/kg and 5 mg/kg CNO all increased cataplexy by the same magnitude), we therefore combined these doses into a single CNO dataset. In all cases, we compared effects of CNO injection with vehicle injections that contained only 0.9% saline and 0.05% DMSO, but no CNO (i.e., baseline). A recent study found that 5 mg/kg CNO might be back-metabolized into clozapine (MacLaren et al., 2016) and that this could affect sleep–wake architecture (Sorge et al., 2004). However, we found that CNO did not alter sleep–wake architecture in either wild-type or orexin−/− or orexin−/−,VGAT-Cre mice compared with saline controls (see Figs. 2D, 3A, 7D, Table 1).
Figure 2.

Behavioral arrests triggered by CeA activation are cataplexy. A, B, In 5 orexin−/− mice, activation of all CeA cells produced behavioral arrests characterized by similar levels of muscle tone in the masseter and neck muscles compared with typical baseline (i.e., saline) cataplexy. C, These arrests also had similar EEG spectral profiles to baseline (i.e., saline) cataplexy (data presented as mean ± SEM). D, CeA activation did not produce any changes in the overall amount of wakefulness (W), NREM sleep, or REM sleep, nor did it alter sleep attacks (inset).
Figure 3.
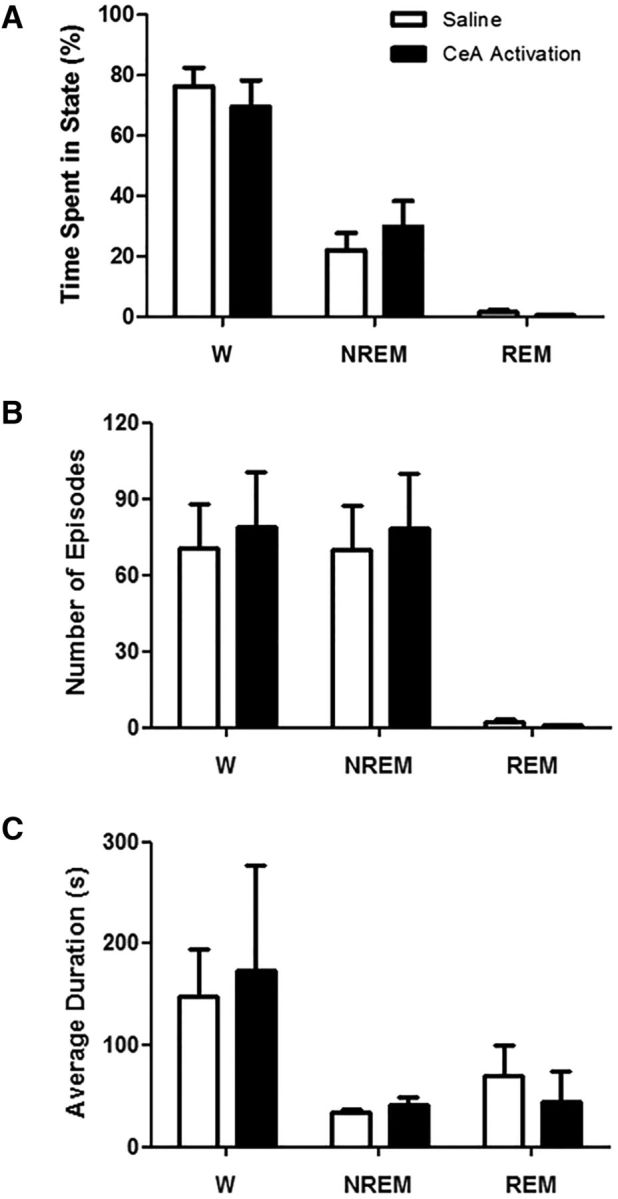
CeA activation does not alter sleep–wake architecture in wild-type mice. A, Bilateral activation of all CeA cells in wild-type mice (n = 4) did not influence the amounts of time they spent in wakefulness (W), NREM sleep, or REM sleep. CeA activation produced no differences in the frequency (B) or duration (C) of W, NREM, or REM, nor was there evidence of overt behaviors resembling cataplexy during the 3 h recording period.
Figure 7.

Activating GABA CeA cells produces cataplexy but does not influence sleep-wake behavior. A, B, In 13 orexin−/−,VGAT-Cre mice, activation of GABA CeA cells produced behavioral arrests characterized by similar levels of muscle tone in the masseter and neck muscles compared with typical baseline (i.e., saline) cataplexy. C, These arrests also had similar EEG spectral profiles to baseline (i.e., saline) cataplexy (data presented as mean ± SEM), suggesting that they were indeed cataplexy. D, GABA CeA activation did not produce any changes in the overall amounts of NREM sleep or REM sleep, nor did it alter sleep attacks (inset), but it did decrease the amount of wakefulness (W) mice experienced. E, However, this decrease in wakefulness was an artifact resulting from mice spending significantly more of their waking time in cataplexy and was nullified when time awake and time in cataplexy were combined. **p < 0.01.
Table 1.
Behavioral state architecture in orexin−/− and orexin−/−,VGAT-Cre mice
| orexin−/− | orexin−/−,VGAT-Cre | p-value | |
|---|---|---|---|
| Wake | |||
| Percentage of time | 83.65 ± 5.04 | 84.60 ± 2.98 | 0.8693 |
| Number of episodes | 31.20 ± 5.67 | 33.54 ± 6.72 | 0.8419 |
| Average duration of episodes (s) | 342.7 ± 74.49 | 483.6 ± 106.1 | 0.4446 |
| Total time spent (min) | 150.57 ± 9.08 | 152.29 ± 5.36 | 0.8695 |
| NREM sleep | |||
| Percentage of time | 10.77 ± 3.74 | 10.24 ± 2.52 | 0.9117 |
| Number of episodes | 25.20 ± 5.86 | 28.62 ± 6.87 | 0.7761 |
| Average duration of episodes (s) | 43.07 ± 5.41 | 39.04 ± 2.86 | 0.4888 |
| Total time spent (min) | 19.38 ± 6.73 | 18.43 ± 4.53 | 0.9117 |
| REM sleep | |||
| Percentage of time | 2.74 ± 1.39 | 2.36 ± 0.62 | 0.7766 |
| Number of episodes | 3.40 ± 1.54 | 3.92 ± 1.19 | 0.8119 |
| Average duration of episodes (s) | 69.04 ± 24.39 | 65.09 ± 11.39 | 0.8690 |
| Total time spent (min) | 4.93 ± 2.50 | 4.26 ± 1.12 | 0.7798 |
| Sleep attacks | |||
| Percentage of time | 0.57 ± 0.35 | 1.06 ± 0.29 | 0.2498 |
| Number of episodes | 1.60 ± 0.68 | 2.46 ± 0.47 | 0.3405 |
| Average duration of episodes (s) | 23.75 ± 8.62 | 47.67 ± 9.44 | 0.1037 |
| Total time spent (min) | 1.02 ± 0.63 | 1.91 ± 0.39 | 0.2468 |
Viral injection surgery.
Mice were anesthetized using isoflurane (2–5%) and secured in a stereotaxic frame (model 902; David Kopf Instruments). Then, 200 nl of virus was slowly (50 nl/min) infused into the left and right CeA (1.35 mm posterior to bregma, ±2.75 mm lateral, 4.50 mm ventral). This was performed through a 28-gauge cannula connected to a digital microinjection syringe pump (Pump 11 Elite; Harvard Apparatus). Mice were administered ketoprofen (5 mg/kg, s.c.) up to 48 h after surgery and allowed to recover for at least 14 d.
EEG and EMG instrumentation.
At the end of this 14 d recovery period, mice underwent a second surgery during which electroencephalogram (EEG) and electromyogram (EMG) electrodes were implanted to identify sleep states and cataplexy. Electrodes were constructed from multistranded stainless steel wire (AS 632; Cooner Wire). For EEG recordings, we implanted stainless steel screws (P0090CE125; J.I. Morris) bilaterally into the frontal and parietal bones (1.5 mm anterior and ±1.5 mm lateral to bregma; 2 mm posterior and ±2.75 mm lateral to bregma). For EMG recordings, we sutured two electrodes into the neck extensor muscles and two electrodes into the right masseter muscle. All EEG and EMG electrodes were soldered to a microstrip connector (CLP-105-02-L-D; Electrosonic) that we affixed to the skull using dental cement (Ketac-cem and C&B Metabond Cement System; K-dental). Mice were administered ketoprofen (5 mg/kg, s.c.) up to 48 h after surgery and allowed to recover for at least 14 d before experiments began.
Experimental protocols.
After the second surgery, mice were transferred into a plexiglass recording chamber containing a running wheel (Bio-Serv). Because mice require 10–12 d to maximize their wheel running (España et al., 2007), this provided sufficient time for them to habituate to the wheel before the experiments, which began 14 d after being placed into the recording chamber. Orexin−/− mice experience more cataplexy when they have access to running wheels, presumably because running wheels represent a rewarding stimulus (Lett et al., 2002; España et al., 2007; Burgess et al., 2013). Therefore, we provided mice with running wheels to study how rewarding conditions affect cataplexy.
After being transferred to the recording chamber, we gently handled mice at the start of the dark phase (19:00) on three separate occasions to habituate them to human contact. One week after being transferred to the recording chamber, we fastened a lightweight recording cable (CW7117; Cooner Wire) to the microstrip connector and gave mice an additional week to habituate to this cable. During this habituation period, mice were given 3 intraperitoneal injections of 0.3 ml of lactated Ringer's solution (B. Braun Medical) to habituate them to CNO injections.
At the end of this week, we recorded mice up to four consecutive nights for 12 h each. No intervention was performed on the first night of recording so that we could observe the natural behavior of the mice. On the subsequent nights, we randomly gave mice intraperitoneal injections of saline or CNO at the start of the dark phase (19:00). EEG, EMG, and video were recorded each night to observe changes in cataplexy and sleep–wake behaviors.
Data acquisition.
EEG and EMG signals were passed through a Super-Z Head-Stage and a BMA-400 Bioamplifier (CWE), which amplified signals by a factor of 500. All signals were digitized at 1000 Hz (Spike2 Software, 1401 Interface; Cambridge Electronic Design), had a DC offset applied with a time constant of 0.4 s, and were digitally filtered (EEG, 1–100 Hz; EMG, 30–1000 Hz). For signals in which electrical noise was detected, a 60 Hz Notch filter was applied in all recordings for that mouse. EMG signals were rectified. We simultaneously captured and synchronized videos to electrophysiological recordings to assist in identifying behaviors of interest.
Data analysis.
We analyzed the first 6 h of recording for orexin−/− mice to determine the time course of any CNO-induced effects on cataplexy and behavior (see Fig. 1E). Because we found that CNO effects persisted for only 3 h after injection, all subsequent analyses were performed only on the first 3 h of the recording period. We scored behavioral states in 5 s epochs based on EEG, EMG, and video using a custom-made script for Spike2 (sleepscore version 1.01). We identified wakefulness, non-rapid eye movement (NREM) sleep, and rapid eye movement (REM) sleep using standard criteria described previously (Burgess et al., 2010; Brooks and Peever, 2012; Burgess and Peever, 2013). However, we did not score transitions between different states (i.e., NREM to REM sleep or REM sleep to wakefulness) as separate events. Instead, we scored each epoch based on the predominating state within it. For example, if a 5 s epoch contains more that 60% of a given state (e.g., REM sleep), then it is scored as REM sleep even though 40% of that epoch could contain either wakefulness or NREM sleep. Therefore, “bouts” of wakefulness, NREM sleep, or REM sleep could be as short as 5 s; however, this is exceedingly rare.
Figure 1.

CeA activation exacerbates cataplexy in narcoleptic mice. A, Stereotaxic map displaying the location of expression of the Cre-independent hM3Dq construct in the CeA of 5 orexin−/− mice. B, Extent of expression was determined on the basis of mCherry fluorescence. A representative sample of mCherry-positive neurons is shown, as is a single neuron (inset). C, D, Hypnograms displaying behavioral states of orexin−/− mice during baseline conditions (saline) or CeA activation (CNO). E, Compared with saline levels of cataplexy, CNO produced increases in cataplexy that persisted for 3 h after injection before returning to control levels, so this time frame was examined in detail. F, After global CeA activation, a large increase in the total time spent in cataplexy was observed. G, This increase in cataplexy arose from an increase in the number of cataplexy episodes mice experienced per 3 h recording. H, Average duration of cataplexy episodes did not change. W, Wakefulness; SA, sleep attacks; CAT, cataplexy. *p < 0.05 compared with saline.
Cataplexy was scored according to the consensus definition (Scammell et al., 2009) and both the electrophysiological signals and video recording were used to determine the activity the mouse that was engaged in immediately before the attack began. Almost all cataplexy attacks were triggered by wheel running, grooming, exploring, eating, or drinking. We considered cataplexy episodes without a clear preceding behavior to be spontaneous attacks and had the trigger classified as “other.” We identified sleep attacks on the basis of a gradual loss of neck muscle tone and the appearance of EEG slow waves in addition to automatic behaviors such as chewing that were evident in the masseter EMG (Burgess et al., 2010). Sleep attacks progressed into wakefulness, NREM sleep, or REM sleep and the subsequent state was scored as such.
We quantified the number and length of cataplexy attacks during both CNO and saline (i.e., baseline) treatments. We calculated percentage time in cataplexy by summing the total time spent in cataplexy and dividing this value by the length of the recording period. The number of cataplexy episodes recorded was used to divide the total time spent in cataplexy to determine the average duration of episodes. We quantified muscle tone during cataplexy in each mouse by normalizing the average integrated EMG activity of the neck and masseter muscles during cataplexy to that muscle's activity during NREM sleep during the saline treatment. EMG activity was quantified for each muscle using a custom-made Spike2 script (5 s epoch analysis). We generated EEG power spectra during cataplexy by performing a fast Fourier transform of the EEG signal during cataplexy using Spike2 software. Relative power was achieved by summing the absolute power of 1–20 Hz and dividing the absolute power of each intermediate frequency by that sum.
Histology, immunohistochemistry, and fluorescence in situ hybridization.
After experiments, mice were deeply anesthetized with Avertin (250 mg/kg, i.p.) produced from 2,2,2-tribromoethanol and 2-methyl-2-butanol (Sigma-Aldrich) and isoflurane. Upon loss of the foot withdrawal and righting reflexes, mice were immobilized and transcardially perfused with 0.1 m PBS and 4% paraformaldehyde (PFA). The brain was isolated and immersed in 4% PFA for 24 h, then transferred to a 30% sucrose solution for 48 h for cryoprotection. Cryoprotected brains were frozen in Tissue-Tek optimal cutting temperature compound (Electron Microscopy Sciences) and coronally sectioned at 40 μm using a cryostat (CM3050 S; Leica). Enhanced yellow fluorescent protein (eYFP) signal was amplified by incubating tissue in monoclonal mouse antibody anti-GFP (1:1000, MAB3580; Millipore) followed by Alexa Fluor 488-conjugated goat anti-mouse antibodies (1:500, 115-547-003; Jackson ImmunoResearch). Slices were subsequently mounted on glass slides with Permafluor and expression of mCherry or eYFP was confirmed visually with fluorescence microscopy. Only mice in which expression encompassed the CeA bilaterally were analyzed.
For c-Fos/mCherry immunohistochemistry, mice were given either saline or 5 mg/kg CNO injection intraperitoneally 2 h before being euthanized, which allowed for sufficient activation of hM3Dq receptors and expression of c-Fos. After perfusion, cryoprotection, and sectioning of brain tissue, sections were washed in 0.1 m PBS, pH 7.4 (2 changes) and then incubated in the primary antiserum for 72 h at 4°C. For c-Fos, we used a rabbit polyclonal antiserum (1:5000, 26209; Immunostar) against residues 4–17 from human c-Fos. For mCherry, we used a polyclonal mouse antiserum (1:5000, T513; Signalway Antibody). Sections were then washed in PBS and incubated in biotinylated secondary antiserum against rabbit (1:800, BA-1000; Vector Laboratories) for 1 h, washed in PBS, and incubated in avidin-biotin-horseradish peroxidase conjugate (ABC solution; Vector Laboratories) for 1 h. Sections were then washed again and incubated in a 0.06% solution of 3,3-diaminobenzidine tetrahydrochloride containing nickel (DAB, purple/black staining; Vector Laboratories) plus 0.02% H2O2. After DAB staining, sections were washed and then incubated in biotinylated secondary antiserum against mouse (1:800, BA-9200; Vector Laboratories) for 1 h, washed in PBS, and incubated again in ABC solution for 1 h. Sections were washed again and then incubated in NovaRed (red staining; Vector Laboratories). Tissue was mounted on glass slides with Permount and c-Fos/mCherry expression was examined with light microscopy. mCherry-expressing CeA cells (i.e., mCherry-positive with or without c-fos) were counted (Cell Counter plug-in, ImageJ) in 3 slices for each mouse separated by 160 μm.
We used fluorescence in situ hybridization to verify that chemogenetic constructs were targeted selectively to GABA CeA cells. After mice were anesthetized, brains were immediately frozen in −30°C isopentane and stored at −80°C. Brains were coronally sectioned at 16 μm and mounted on glass slides, which were stored at −80°C. Sections containing the CeA were immersed in 4% PFA for 20 min, then incubated in 0.1 m PBS containing 0.3% H2O2 for 10 min at room temperature. Slices were acetylated for 10 min using 0.1 m TEA buffer containing 0.25% acetic anhydride, dehydrated with ethanol, then transferred to a humid chamber saturated with formamide and incubated in a hybridization buffer (40% formamide, 10 mm Tris-HCl, pH 8.0, 200 μg/ml yeast tRNA, 10% dextran sulfate, 1× Denhardt's solution, 600 mm NaCl, 1 mm EDTA, pH 8.0) for 2 h at 56°C. Sections were next transferred to a hybridization buffer containing the antisense GAD67 riboprobe (1:1000), incubated overnight at 56°C, then washed with serial SSC buffers under gentle agitation. Sections were next incubated in a blocking solution containing 4% goat serum and 0.5% blocking reagent (Roche) for 1 h at room temperature and then incubated in a polyclonal mouse antibody to mCherry (1:1000, T513; Signalway Antibody) for 36 h at room temperature. At the end of this period, sections were incubated in sheep anti-DIG-POD (1:500, 11207733910; Roche) overnight at 4°C, washed 5 times for 5 min washes in 0.1 m PBS containing 0.1% Triton-X (PBS-T), and transferred to a solution containing the TSA Plus Fluorescein System (1:100, NEL741001KT; PerkinElmer) for 10 min. Slices were next incubated in Cy3-conjugated goat anti-mouse antibodies (1:200, CLCC35010; Cedarlane) for 3 h at room temperature and then stained with DAPI (1:1000) for 5 min. Slices were left to dry overnight, coverslipped with Permafluor, and examined with a fluorescent microscope. mCherry-expressing CeA cells (i.e., mCherry-positive with or without GAD67) were counted (Cell Counter plug-in, ImageJ) in three slices for each mouse separated by 160 μm.
Circuit-mapping methods.
In addition to functional studies, we also wanted to determine how GABA CeA neurons communicate with the downstream circuits that mediate muscle tone. We hypothesized that GABA CeA cells produce muscle atonia during cataplexy by projecting to and inhibiting GABA cells in the ventrolateral periaqueductal gray (vlPAG) and lateral pontine tegmentum (LPT). GABA cells in the vlPAG and LPT normally function to promote waking muscle tone by inhibiting atonia-generating cells in the subcoeruleus nucleus (SubC; Lu et al., 2006). However, when GABA CeA cells switch on during cataplexy, they inhibit GABA cells in the vlPAG/LPT, which in turn causes muscle atonia through disinhibition of SubC cells (Burgess et al., 2013).
To determine whether GABA CeA cells project to GABA cells in the vlPAG, an AAV containing the sequence for a YFP (rAAV5/EF1a-DIO-ChETA-eYFP, 200 nl, 3.9 × 1012 particles/ml) was injected bilaterally into the CeA of orexin−/−,VGAT-Cre mice. Simultaneously, another AAV containing the sequence for a red fluorescent protein (rAAV8/hSyn-DIO-mCherry, 200 nl, 3.7 × 1012 particles/ml) was injected into the vlPAG (5 mm posterior to bregma, ±0.75 mm lateral, 2.75 mm ventral). In a separate experimental group of orexin−/−,VGAT-Cre mice, projections from GABA CeA neurons to GABA neurons in the LPT were also examined. An AAV vector containing the sequence for a yellow fluorescent protein (rAAV5/EF1a-DIO-ChETA-eYFP, 200 nl, 3.9 × 1012 particles/ml) was injected into the CeA to visualize its axonal projections. Simultaneously, an AAV vector containing the sequence for a red fluorescent protein (rAAV8/hSyn-DIO-mCherry, 200 nl, 3.7 × 1012 particles/ml) was injected into the LPT (4.84 mm posterior to bregma, ±1.25 mm lateral, 3.50 mm ventral). To determine whether GABA vlPAG/LPT neurons project to cells within the SubC region, a viral vector containing the sequence for a red fluorescent protein (rAAV8/hSyn-DIO-mCherry, 200 nl, 3.7 × 1012 particles/ml) was injected bilaterally into the vlPAG and LPT of orexin−/−,VGAT-Cre mice. Confocal microscopy was used to examine the projection patterns and locations of candidate GABA cells (i.e., CeA→vlPAG/LPT and vlPAG/LPT→SubC). Confocal microscopy was used to assess the proximity of CeA axonal terminals on GABA cells in both the vlPAG and LPT.
Statistical analyses.
We performed between-group comparisons using unpaired two-tailed t tests and within-group comparisons using paired two-tailed t tests. For data that were not normally distributed, we instead used Mann–Whitney U or Wilcoxon signed-rank tests. We compared EEG power spectra using two-way repeated-measures ANOVA within groups or two-way ANOVA without repeated measures between groups. All statistical analyses were performed using GraphPad Prism software and had a critical α value of p < 0.05 applied. All data are presented as mean + SEM unless otherwise indicated.
Results
CeA activation exacerbates cataplexy in narcoleptic mice
The CeA may have a pivotal role in triggering cataplexy. For example, CeA cells increase and decrease their discharge activity at the beginning and end of cataplexy in narcoleptic dogs (Gulyani et al., 2002) and CeA lesions suppress the amount of cataplexy in orexin−/−mice (Burgess et al., 2013). Therefore, our first goal was to determine whether activation of CeA neurons would promote cataplexy. The CeA is composed of at least two distinct cell populations. A minority of CeA cells are glutamatergic and have relatively sparse projections beyond the amygdala (Boissard et al., 2003; Xi et al., 2011), whereas the majority of CeA cells contain GABA and densely innervate multiple other brain regions, including those that control muscle tone (Swanson and Petrovich, 1998; Sah et al., 2003; Burgess et al., 2013). Instead of targeting either of these cell groups selectively, we decided to first use a Cre-independent strategy to activate all CeA cells so that we could establish a functional role for the CeA in mediating cataplexy. We did this by using AAVs to drive bilateral hM3Dq receptor expression in CeA neurons of orexin−/− mice and then determined how CNO-induced activation of these receptors influenced cataplexy, sleep attacks, and general sleep–wake behavior.
We successfully targeted hM3Dq receptors to cells in the left and right CeA in five orexin−/− mice (Fig. 1A,B). CNO-induced activation of CeA cells had no observable impact on typical waking behaviors such as eating, drinking, exploring, grooming, or wheel running; however, it induced an intense increase in the number of cataplexy attacks (Fig. 1C,D). Amounts of cataplexy were elevated for 3 h after CNO administration and returned to baseline levels thereafter, suggesting that the stimulatory effects of CNO on hM3Dq receptors last ∼3 h (Fig. 1E).
Analysis of group data (n = 5) showed that CeA activation triggered up to 3.5 times more cataplexy during the 3 h period (paired t test, t(4) = 3.232, p = 0.0319; Fig. 1F). CeA activation exacerbated cataplexy by increasing the number of episodes (paired t test, t(4) = 2.811, p = 0.0483; Fig. 1G), but had no impact on the length of cataplexy events (paired t test, t(4) = 0.2547, p = 0.8115; Fig. 1H). This observation suggests that cells in the CeA play a functional role in initiating, but not maintaining, cataplexy.
From a behavioral perspective, attacks were indistinguishable from typical baseline cataplexy in orexin−/− mice. They featured the standard hallmarks of cataplexy in that they were characterized by an abrupt postural collapse and a sudden loss of EMG tone and the appearance of theta-rich EEG activity, both of which punctuated normal waking behavior (see Fig. 4A,B). Like typical baseline cataplexy, they lasted ∼50 s and ended with an abrupt and rapid return to active wakefulness (Fig. 1H).
Figure 4.

Orexin−/− mice that express Cre in GABA neurons (orexin−/−,VGAT-Cre mice) exhibit typical cataplexy. A, In orexin−/− mice, cataplexy is characterized by the abrupt loss of skeletal muscle tone during wakefulness and a theta-rich EEG. B, Magnification of boxed area in A. Orexin−/−,VGAT-Cre mice express cataplexy electrophysiologically indistinguishable from that seen in conventional orexin−/− mice as measured by masseter EMG (C), neck EMG (D), and EEG (E). F-H, There were no significant differences in the time spent in, number of episodes of, or average duration of cataplexy attacks in the first 3 h of behavioral recording of orexin−/−,VGAT-Cre mice compared with orexin−/− mice. A.U., Arbitrary units.
Although CeA activation produced attacks that were behaviorally indistinguishable from typical cataplexy, we nonetheless wanted to verify that these events were indeed cataplexy and not some other form of behavioral arrest such as sleep attacks. To do this, we compared EEG power spectral profiles and levels of EMG tone during baseline cataplexy with that elicited during CeA activation. We quantified these two particular variables because murine cataplexy is characterized by both muscle paralysis (Burgess and Peever, 2013) and a wake-like, theta-rich cortical EEG (Vassalli et al., 2013). We found that CeA activation produced cataplexy episodes with levels of muscle tone that were comparable to those during typical baseline cataplexy (masseter tone: paired t test, t(4) = 0.9472, p = 0.3972; neck tone: paired t test, t(4) = 0.1637, p = 0.8779; Fig. 2A,B). We also found that EEG activity peaked in the theta band during baseline and CeA activation-induced cataplexy and overall power spectral profiles were identical in both conditions (2-way RM ANOVA, F = 3.458 × 10−4, p = 0.9852; Fig. 2C). Together, these data suggest that the cataplexy produced by CeA activation is physiologically and behaviorally identical to cataplexy attacks that occur during baseline conditions in orexin−/− mice.
Because the amygdala has been implicated in sleep regulation (Smith and Miskiman, 1975; Calvo et al., 1996; Sanford et al., 2006), we wanted to determine whether CeA activation would affect either sleep–wake behavior or sleep attacks, which are common in orexin−/− mice. Although CNO-induced CeA activation triggered marked increases in cataplexy, this same intervention had no measureable effect on the amount of wakefulness (paired t test, t(4) = 2.315, p = 0.0816), NREM sleep (paired t test, t(4) = 1.1319, p = 0.2576), or REM sleep (paired t test, t(4) = 0.6870, p = 0.5299), nor did it affect sleep attacks (paired t test, t(4) = 2.497, p = 0.0670; Fig. 2D). These findings indicate that CeA activation does not have broad effects on behavior; rather, its activation targets the mechanisms mediating cataplexy selectively.
CeA activation does not cause cataplexy in wild-type mice
Next, we wanted to verify that cataplexy was only triggered by CeA activation in orexin−/− mice and that it could not be triggered by CeA stimulation it in wild-type mice with an intact orexin system. Therefore, we used a Cre-independent strategy to drive hM3Dq expression in CeA cells of wild-type mice (n = 4) and then examined how CNO-induced activation of these cells affected behavior. Using EEG, EMG, and videography, we found no evidence indicating that CeA activation causes cataplexy in wild-type mice. Unlike orexin−/− mice, which experienced as many as 12 cataplexy attacks during the 3 h recording period (Fig. 1G), we saw no evidence of postural collapses or intermittent losses of EMG tone during waking behaviors in wild-type mice after CeA stimulation. We also found no evidence that CeA activation influenced the overall amount or architecture of sleep–wake behavior (Fig. 3A–C). These findings suggest that the orexin system protects against cataplexy and that CeA activation only promotes cataplexy in narcoleptic mice that already lack an intact orexin signaling system (see Fig. 10). Although previous studies showed that the amygdala can promote REM sleep (Calvo et al., 1996; Sanford et al., 2006), our results indicate that CeA activation does not affect sleep–wake behavior.
Figure 10.

Circuits controlling cataplexy. A, Positive emotions activate cortical areas such as the mPFC, which in turn activate the CeA. GABA cells in the CeA then inhibit midbrain regions, including the LPT and vlPAG, which normally function to inhibit the atonia-generating network in the brainstem (i.e., SubC). Inhibition of these midbrain regions disinhibits the muscle paralysis circuit during cataplexy. B, Mapping of the cataplexy circuit using virally tractable tracers. rAAV5/EF1a-DIO-ChETA-eYFP and rAAV8/hSyn-DIO-mCherry were injected into the CeA and vlPAG/LPT, respectively, of orexin−/−,VGAT-Cre mice. C, GABA CeA neurons expressed eYFP. D, GABA vlPAG/LPT neurons expressed mCherry. E, GABA vlPAG/LPT neurons expressing mCherry send projections to the SubC region. F, GABA CeA fibers expressing eYFP are located in the vicinity of a GABA vlPAG cell body expressing mCherry. G, GABA CeA fibers expressing eYFP are located in the vicinity of a GABA LPT cell body expressing mCherry. H, GABA vlPAG/LPT projections are located within the SubC region. Aq, Aqueduct; Mo5, trigeminal motor nucleus; ic, internal capsule; mPFC, medial prefrontal cortex; LH, lateral hypothalamus; Orx, orexin; Glut, glutamate.
Orexin−/− mice that express Cre in GABA neurons exhibit typical cataplexy
Having shown that general CeA stimulation promotes cataplexy, our next step was to identify which type of CeA cell is responsible for this effect. Here, we test the role for GABA neurons because they are the primary extrinsic pathway from the CeA and they are anatomically connected to nuclei that regulate skeletal muscle tone (Swanson and Petrovich, 1998; Sah et al., 2003; Burgess et al., 2013; see Fig. 10). However, to understand how GABA cells in the CeA regulate cataplexy, we needed to develop a new narcoleptic mouse line in which we could target and manipulate these cells selectively. To do this, we crossed orexin−/− mice with VGAT-Cre mice (Chemelli et al., 1999; Vong et al., 2011) to produce so-called orexin−/−,VGAT-Cre mice so that Cre-dependent AAVs could be used to deliver either hM3Dq or hM4Di receptors directly to GABA cells in the CeA. However, before using these new mice, we needed to determine whether they behave like typical orexin−/− mice. We did this by comparing the amounts of cataplexy, sleep attacks, NREM/REM sleep, and wakefulness in orexin−/− (n = 5) and orexin−/−,VGAT-Cre (n = 13) mice.
First, we showed that general sleep–wake behavior is unaffected in orexin−/−,VGAT-Cre mice. Specifically, we found that orexin−/− and orexin−/−,VGAT-Cre mice spent the same amount of time in wakefulness, NREM sleep, and REM sleep, and that the length and number of each behavioral state was comparable (Table 1). We also found that the number and length of sleep attacks was similar between genotypes (Table 1). Together, these results suggest that orexin−/−,VGAT-Cre mice are phenotypically indistinguishable from orexin−/−mice.
Next, we showed that cataplexy was behaviorally and electrophysiologically identical in orexin−/−,VGAT-Cre and orexin−/− mice. In both mouse lines, we found that cataplexy occurred during active wakefulness when mice were engaged in purposeful activities such as wheel running, grooming, exploring, eating, and drinking. Attacks were characterized by abrupt postural collapse and rapid loss of EMG tone (Fig. 4A,B). Mice remained immobile throughout attacks, which were terminated by a swift return of skeletal muscle tone and resumption of prior waking activities (Fig. 4A,B). Levels of EMG tone and cortical EEG activity during cataplexy were similar in both orexin−/−,VGAT-Cre and orexin−/− mice (masseter EMG: t test, t(11) = 1.525, p = 0.1554; neck EMG: t test, t(13) = 0.4438; p = 0.6645; EEG: 2-way ANOVA, F = 2.756 × 10−3, p = 0.9582; Fig. 4C–E).
Both mouse lines also spent comparable amounts of time in cataplexy. During the 3 h recording period, orexin−/− and orexin−/−,VGAT-Cre mice spent 2.3 ± 0.5% and 1.7 ± 0.5% of their time in cataplexy (Mann–Whitney U test, U(16) = 22.50, p = 0.3488; Fig. 4F). They also had the same number of attacks that lasted for the same amount of time. Orexin−/− mice experienced 5 ± 1 attacks that lasted for 53 ± 8 s and orexin−/−, VGAT-Cre mice experienced 3 ± 1 attacks that lasted for 49 ± 7 s (number: Mann–Whitney U test, U(16) = 16.50, p = 0.1248; duration: t test, t(16) = 0.3527, p = 0.7289; Fig. 4G,H). Together, these data indicate that orexin−/−,VGAT-Cre mice exhibit cataplexy that is behaviorally and electrophysiologically indistinguishable from that of orexin−/− mice, making this new mouse line useful for determining how GABA cell manipulation affects cataplexy.
hM3Dq receptors are expressed in and activate GABA cells of the CeA
Our next step was to determine whether Cre-dependent AAV delivery targeted GABA cells in the CeA. In 13 orexin−/−,VGAT-Cre mice, we successfully targeted and bilaterally delivered hM3Dq receptors to GABA cells in the left and right CeA (Fig. 5A). To verify the specificity of hM3Dq expression, we used fluorescence in situ hybridization to examine the distribution of GAD67 (a specific marker of GABA cells) in the CeA. We found that, not only was the CeA densely populated by GAD67-positive neurons, but that hM3Dq receptors also colocalized extensively with these cells (Fig. 5B,C), suggesting that our subsequent CNO manipulations would primarily target GABA cells in the CeA. We found that 96 ± 2% of GAD67-positive neurons expressed mCherry and that 94 ± 1% of mCherry-positive cells expressed GAD67 (Fig. 5C).
Figure 5.

hM3Dq receptors are expressed in and activate GABA CeA cells. A, Stereotaxic map displaying the location of the hM3Dq construct in the CeA of 13 orexin−/−,VGAT-Cre mice. B, Extent of expression was determined on the basis of mCherry fluorescence (red; single mCherry-positive neuron showed in inset), which encompassed the CeA where GABA cells predominate (green; single GAD67-positive neuron shown in inset). Most cells expressing mCherry also expressed GAD67 (orange, overlay). C, Quantification of the number of GAD67-positive cells that also expressed mCherry and number of mCherry-positive cells that also expressed GAD67. D, Expression of c-Fos (black), a neural marker of activity, in the region of the CeA. GABA cells (red) of the CeA are activated by CNO, as seen by the greater number of GABAergic cells expressing c-Fos compared with saline controls. E, Quantification of the number of mCherry-positive neurons expressing c-Fos under saline and CNO administration. ic, Internal capsule; BLA, basolateral nucleus of the amygdala. ***p < 0.001.
Next, we wanted to verify that CNO activates GABA CeA cells expressing hM3Dq receptors. We did this by immunostaining for c-Fos, a marker of cell activity, in mice expressing hM3Dq that received either a saline or CNO injection. We found that, after saline injection, <5% of mCherry-positive cells were activated (i.e., expressed c-Fos); however, after CNO injection, 83 ± 0.4% of mCherry-positive cells were activated (t test, t(6) = 106.8, p < 0.001; Fig. 5D,E), suggesting that CNO activates hM3Dq receptors expressed in GABA cells of the CeA. This observation is important because it indicates that changes in cataplexy following chemogenetic manipulation are due to changes in GABA cell activity per se.
GABA cells in the CeA promote cataplexy
Next, we aimed to determine whether GABA cells in the CeA are responsible for promoting cataplexy. In 13 orexin−/−,VGAT-Cre mice, we successfully delivered hM3Dq receptors to GABA cells in both the left and right CeA (Fig. 5A,B) and found that CNO-induced activation of these cells triggered a robust increase in cataplexy that was identical in effect to that produced by activating the entire CeA cell population. Cataplexy occupied 3.7 times more of the 3 h recording period after GABA cell activation than it did before (paired t test, t(12) = 4.573; p = 0.0006; Fig. 6A–C). Increases in cataplexy were due to an increase in the number of attacks rather than the duration of attacks. Before CeA activation, mice experienced 5 ± 1 episodes in 3 h, but during GABA cell stimulation, they experienced 16 ± 3 arrests during the same time period (Wilcoxon signed-rank test, W(12) = −89; p = 0.0021; Fig. 6D). In contrast, CeA stimulation had no effect on the length of cataplexy episodes (baseline vs CNO: 43 ± 6 s and 42 ± 4 s; paired t test, t(12) = 0.1341, p = 0.8956; Fig. 6E), suggesting that GABA CeA cells gate the entrance into, but not the exit from, cataplexy.
Figure 6.

GABA cells in the CeA promote cataplexy. A, B, Hypnograms displaying behavioral states of one orexin−/−,VGAT-Cre mouse during baseline (i.e., saline) conditions and activation of GABA CeA cells. C, During activation of GABA CeA cells (denoted as “hM3Dq”), a large increase in the total time spent in cataplexy was observed. This same increase was not apparent in a group of 6 hM3Dq-negative orexin−/−,VGAT-Cre mice (denoted as “mCherry”). D, This cataplexy change arose from an increase in the number of cataplexy episodes (per 3 h recording) that hM3Dq-expressing mice experienced. Mice lacking the hM3Dq receptor experienced similar numbers of cataplexy episodes after CNO injection. E, Average duration of cataplexy episodes did not change after CNO injection in hM3dq-positive and hM3Dq-negative mice. W, Wakefulness; SA, sleep attacks; CAT, cataplexy. ***p < 0.001; **p < 0.01.
Once again, we wanted to confirm that the behavioral arrests produced by specifically activating GABA CeA cells were in fact cataplexy. Although these events were behaviorally identical to typical baseline cataplexy, we again performed a rigorous comparison of the electrophysiological features that characterized these attacks. We found that activation of GABA CeA cells produced cataplexy attacks with levels of muscle tone (masseter EMG: paired t test, t(9) = 1.056 × 10−7, p = 1.0000; neck EMG: paired t test t(11) = 0.2364, p = 0.8175; Fig. 7A,B) and EEG activity (2-way repeated-measures ANOVA, F = 0.001451, p = 0.9697; Fig. 7C) indistinguishable from baseline cataplexy. These observations suggest that activating GABA CeA cells is capable of evoking cataplexy attacks.
Next, we wanted to verify that activation of GABA CeA cells did not affect sleep–wake behavior. We found that CNO-induced CeA stimulation had no effect on the overall amount of NREM sleep (paired t test, t(12) = 1.526; p = 0.1529), REM sleep (Wilcoxon signed-rank test, W(12) = 24; p = 0.3804), or sleep attacks (paired t test, t(12) = 0.03691; p = 0.9712). However, GABA cells activation did decrease the amount of wakefulness by ∼7% (paired t test, t(12) = 3.437; p = 0.0049; Fig. 7D). However, this change was actually due to the substantial increase in the amount of time spent in cataplexy and this apparent decrease disappeared when time awake and time in cataplexy were combined (paired t test, t(12) = 0.8996; p = 0.3861; Fig. 7E). Together, these results indicate that activation of GABA CeA neurons promotes cataplexy, but does not influence sleep–wake activity.
Last, we needed to demonstrate that increases in cataplexy were in fact mediated by CNO-induced activation of hM3Dq receptors on CeA cells. To do this, we drove bilateral expression of the physiologically inert mCherry protein in GABA CeA cells in orexin−/−,VGAT-Cre mice (n = 6). We found that CNO application had no effect on the amount of cataplexy in these “hM3Dq-null” mice (paired t test, t(5) = 0.8153, p = 0.4520; Fig. 6C). CNO injection in hM3Dq-null mice had no impact on either the number (paired t test, t(5) = 1.360, p = 0.2321) or duration (paired t test, t(5) = 1.530, p = 0.1867) of cataplexy attacks (Fig. 6D,E). These findings demonstrate that CNO administration triggers cataplexy by activating GABA CeA cells through an hM3Dq-dependent mechanism. They also suggest that neither CNO application nor mCherry expression within the CeA affects cataplexy.
GABA cells in the CeA mediate cataplexy associated with rewarding stimuli
In narcoleptic mice, cataplexy can occur spontaneously, but it is typically associated with positive or rewarding stimuli such as wheel running, grooming, and palatable food (Chemelli et al., 1999; España et al., 2007; Clark et al., 2009; Burgess et al., 2013). However, the neural mechanism by which such stimuli elicit cataplexy remains unidentified. Because the CeA is involved in processing positive emotions and rewarding stimuli and is anatomically connected to structures that mediate muscle tone (Murray, 2007; Burgess et al., 2013), we hypothesized that GABA CeA cells could be a neural substrate through which these stimuli trigger cataplexy. To test this hypothesis, we documented the types of behaviors that triggered cataplexy under baseline conditions and how they changed during GABA CeA activation in orexin−/−,VGAT-Cre mice.
We found that virtually all cataplexy attacks occurred when mice were engaged in purposeful behaviors such as wheel running, grooming, exploring, eating, or drinking and rarely occurred during nonpurposeful behaviors such as sitting stationary. During baseline recordings, 77% of cataplexy events occurred when mice were either wheel running or grooming and only 23% of events occurred spontaneously or when mice were exploring, eating, or drinking (data not shown). Remarkably, CeA activation almost exclusively triggered cataplexy attacks associated with either wheel running or grooming, increasing cataplexy triggered during wheel running by 385% (Wilcoxon signed-rank test, W(12) = −78; p = 0.0025) and during grooming by 182% (paired t test, t(12) = 2.878; p = 0.0139), with proportionally more wheel-running-triggered cataplexy (Fig. 8A). Cataplexy associated with exploring (paired t test, t(12) = 0, p = 1.0000), eating (Wilcoxon signed-rank test, W(12) = −34, p = 0.0843), drinking (Wilcoxon signed-rank test, W(12) = −20, p = 0.1725), and other behaviors (Wilcoxon signed-rank test, (12) = −1, p = 1.0000) did not change during CeA activation (Fig. 8A), suggesting that GABA CeA cells facilitate cataplexy associated with rewarding stimuli such as wheel running.
Figure 8.

GABA cells in the CeA mediate cataplexy associated with rewarding stimuli. A, GABA CeA activation significantly increased the number of cataplexy episodes triggered by wheel running and grooming without influencing cataplexy triggered by other behaviors. B, The increase in running-triggered cataplexy episodes was not due to more wheel running because mice spent the same fraction of their time awake engaged in this activity. C, Number of wheel running bouts did not change. D, GABA CeA activation caused mice to experience cataplexy after significantly less wheel running compared with baseline (i.e., saline) conditions, indicating a reduction in the stimulus threshold required to trigger cataplexy. *p < 0.05; **p < 0.01.
Because CeA stimulation produced the largest increase in running-triggered cataplexy attacks, we wanted to determine whether this intervention affected cataplexy expression by changing the levels of wheel-running activity. CeA activation did not change the overall amounts of wheel running (paired t test, t(12) = 0.5476, p = 0.5940) or the number of running bouts that mice engaged in (Wilcoxon signed-rank test, W(12) = −54, p = 0.0639; Fig. 8B,C). However, we found that running bouts were shortened because they were punctuated by more frequent cataplexy attacks during CeA activation. During baseline conditions, cataplexy occurred every 43 ± 13 min of wheel running, but during CeA activation, it occurred every 10 ± 3 min of wheel running, indicating a 77% increase in the probability of running-induced cataplexy (paired t test, t(9) = 3.138; p = 0.0120; Fig. 8D). We therefore suggest that activation of GABA CeA neurons lowers the threshold for triggering cataplexy associated rewarding stimuli such as wheel running.
Inactivation of GABA CeA cells does not prevent cataplexy
Our last aim was to determine whether GABA CeA cells are required for initiating cataplexy. To do this, we inactivated these cells chemogenetically and examined cataplexy over a 3 h period. In 5 orexin−/−,VGAT-Cre mice, we successfully delivered hM4Di receptors to GABA cells in both the left and right CeA (Fig. 9A,B) and found that CNO-induced inactivation of GABA CeA cells did not affect the amounts of cataplexy. First, we investigated whether inactivation of GABA CeA cells influenced sleep–wake behavior (Fig. 9C) and found that CNO-induced inhibition did not affect overall amounts of wakefulness (paired t test, t(4) = 1.407; p = 0.2322), NREM sleep (paired t test, t(4)=1.429; p = 0.2263), REM sleep (paired t test, t(4) = 1.085; p = 0.3388), or sleep attacks (paired t test, t(4) = 1.320; p = 0.2573). Then, we found that CeA inactivation had no effect on the total time spent in cataplexy across the 3 h recording period (paired t test, t(4) = 1.177; p = 0.3046; Fig. 9D). Neither the number of attacks (paired t test, t(4) = 1.488; p = 0.2109; Fig. 9E) nor the duration of these attacks (paired t test, t(4) = 1.470; p = 0.2155; Fig. 9F) was affected by CNO-induced inhibition of GABA CeA cells, suggesting that these cells are not required for initiating cataplexy in narcoleptic mice.
Figure 9.

GABA CeA cell inhibition does not affect cataplexy or sleep–wake architecture in orexin−/−,VGAT-Cre mice. A, Stereotaxic map displaying the location of expression of the Cre-dependent hM4Di construct in the CeA of five orexin−/−,VGAT-Cre mice. B, Extent of expression was determined on the basis of mCherry fluorescence. Representative sample of mCherry-positive neurons is shown, as is a single neuron (inset). C, Inhibition of GABA CeA cells (i.e., CNO) did not influence the amounts of time mice spent in wakefulness (W), NREM sleep, REM sleep, or sleep attacks. D, Compared with baseline levels of cataplexy, inhibition of GABA CeA cells did not affect the total time spent in cataplexy. E, F, Neither the number of cataplexy episodes (E) nor the average duration of cataplexy episodes (F) changed during the 3 h recording period. ic, Internal capsule; BLA, basolateral nucleus of the amygdala.
GABA CeA cells are anatomically positioned to regulate cataplexy
GABA cells in the vlPAG and LPT are important for preventing muscle atonia during wakefulness because lesions of these cells can cause cataplexy-like attacks of muscle atonia during wakefulness (Lu et al., 2006; Kaur et al., 2009). We hypothesized that, during cataplexy, GABA CeA cells inhibit GABA cells in the vlPAG/LPT, leading to disinhibition of SubC neurons and consequently the generation of muscle atonia (Fig. 10A,B; Dauvilliers et al., 2014). Previously, we showed that GABA CeA cells innervate the vlPAG/LPT (Burgess et al., 2013), which function to facilitate waking muscle tone by silencing atonia-generating neurons in the SubC. However, it remains unclear whether GABA CeA cells innervate bona fide GABA cells in the vlPAG/LPT and if GABA vlPAG/LPT cells innervate neurons in the SubC region.
Here, we show that GABA CeA neurons do indeed project to GABA cells in the vlPAG and LPT. We found clear expression of eYFP (i.e., GABA CeA terminals; n = 3 mice; Fig. 10C) within the vicinity of GABA cell bodies and dendrites in both the vlPAG and LPT (Fig. 10D–G), which expressed mCherry. We also showed that GABA neurons in the vlPAG and LPT project to neurons in the SubC region (n = 3 mice; Fig. 10H). Together, these data indicate that GABA CeA cells are anatomically positioned to regulate muscle atonia during cataplexy (Fig. 10A).
Discussion
Our results indicate that positive stimuli may trigger cataplexy in orexin−/− mice by recruiting GABA CeA neurons. We found that activation of these cells increased the frequency of cataplexy attacks without influencing their length, demonstrating that GABA cells play a role in triggering, but not maintaining, cataplexy. We also found that GABA cell activation only promotes cataplexy associated with rewarding conditions, suggesting that positive stimuli trigger cataplexy by activating these cells. However, GABA CeA cells are not required for triggering cataplexy because their inhibition did not prevent cataplexy, suggesting that additional pathways are involved in controlling this behavior.
The amygdala promotes cataplexy
Although the link between positive emotions and cataplexy was identified >130 years ago, the pathways by which emotions trigger cataplexy remains speculative. A longstanding hypothesis is that the amygdala plays a causal role in mediating cataplexy (Luppi et al., 2012; Dauvilliers et al., 2014; Fraigne et al., 2015). This idea stems from the fact that the amygdala plays a role in processing emotional stimuli (Murray, 2007; Bonnet et al., 2015; Janak and Tye, 2015) and that cataplexy is typically elicited by positive stimuli (Nishino and Mignot, 1997; España et al., 2007; Clark et al., 2009; Overeem et al., 2011).
Evidence indicates that the amygdala is capable of mediating cataplexy, especially in response to positive stimuli. Imaging studies indicate that amygdala activity increases during cataplexy associated with emotional stimuli in humans (Hong et al., 2006; Meletti et al., 2015) and that the intensity of the amygdala response to positive stimuli is abnormally high in narcoleptics (Schwartz et al., 2008; Ponz et al., 2010), whereas amygdala discharge increases during cataplexy in narcoleptic dogs (Gulyani et al., 2002). These observations suggest that amygdala activity in response to positive or rewarding stimuli may trigger cataplexy, but it is unclear whether changes in amygdala activity are a cause or consequence of cataplexy. Our current results indicate that the amygdala, and particularly the CeA, plays a role in mediating cataplexy because they show that CeA activation increases cataplexy in response to rewarding stimuli such as wheel running. These findings also complement work demonstrating that amygdala lesions reduce cataplexy associated with rewarding stimuli in orexin−/− mice (Burgess et al., 2013).
The amygdala also influences generalized arousal and motivated behaviors (Davis and Whalen, 2001; Sah et al., 2003; Tye and Janak, 2007; Robinson et al., 2014; Janak and Tye, 2015). Therefore, it is conceivable that CeA activation enhanced cataplexy by enhancing these phenomena. However, we found that CeA stimulation had no effect on sleep–wake activity or the time that mice spent running on wheels (an index of motivated behaviors), suggesting that changes in cataplexy are primarily associated with augmented amygdala activity rather than secondary effects arising from behavioral changes associated with CeA activation.
GABA cells in the CeA mediate cataplexy
Despite evidence suggesting that the amygdala is the neural substrate through which emotions trigger cataplexy, the specific cellular mechanism by which the amygdala promotes cataplexy had until now remained unidentified. Our data suggest that GABA cells in the CeA play a permissive role in controlling cataplexy. We show that targeted stimulation of GABA CeA cells triggered a 3.5-fold increase in cataplexy events, most of which were associated with rewarding conditions such as wheel running. We therefore suggest that GABA CeA cells represent a cellular mechanism through which positive stimuli trigger cataplexy.
How do GABA cells in the CeA function to promote cataplexy? We hypothesize that they act as a “relay center” between the cortical structures that interpret emotional stimuli and the brainstem circuits that generate motor paralysis during cataplexy. This hypothesis is based on data showing that rewarding conditions activate the medial prefrontal cortex (mPFC), which innervates circuits within the amygdala (Vertes, 2004; Etkin et al., 2011; Oishi et al., 2013). Connections between the mPFC and CeA are integral in promoting cataplexy because removing either of them suppresses cataplexy in orexin−/− mice (Burgess et al., 2013; Oishi et al., 2013). GABA cells, which form the primary extrinsic pathway from the CeA (Nitecka and Ben-Ari, 1987; Pitkänen and Amaral, 1994; Sah et al., 2003), innervate the LPT, vlPAG, and locus ceruleus (LC), which collectively function to facilitate waking muscle tone by silencing atonia-generating regions in the pons (e.g., SubC). We hypothesize that positive emotions elicit cataplexy by activating the mPFC, which switches on GABA CeA cells that inhibit the LC, LPT, and vlPAG and thereby generate muscle paralysis (Fig. 10A).
Why does amygdala activation not cause cataplexy in individuals with an intact orexin system? We found that CeA activation did not trigger cataplexy in wild-type mice; its activation only promoted cataplexy in animals that lack orexin. We hypothesize that the orexin system normally protects against cataplexy so that GABA inhibition from the CeA is offset by excitatory signaling from the orexin neurons, which are active during rewarding conditions (e.g., grooming; Lee et al., 2005; Mileykovskiy et al., 2005; Siegel and Boehmer, 2006; Blouin et al., 2013; Giardino and de Lecea, 2014) and innervate the same structures as GABA CeA cells (e.g., vlPAG; Peyron et al., 1998; Kiyashchenko et al., 2001; Mieda and Sakurai, 2012; Hasegawa et al., 2014). However, loss of orexin signaling in narcolepsy upsets this balance, so that GABA CeA cells are unopposed in inhibiting the LC, LPT, and vlPAG, thus creating an environment conducive to cataplexy. Therefore, one of the major outcomes of orexin loss in narcolepsy is destabilization of the circuits that control how positive stimuli affect normal motor regulation.
GABA cells in the CeA initiate cataplexy
Our results also indicate that the CeA plays a role in triggering but not maintaining cataplexy because we show that CeA activation increases the frequency but not the duration of cataplexy attacks. If CeA activity contributes to the length of cataplexy, its activation should have produced status cataplecticus (prolonged periods of cataplexy; Overeem et al., 2001; Dauvilliers et al., 2007; Dauvilliers et al., 2014). Because we found no change in the length of cataplexy episodes, our results suggest that the CeA acts as a cataplexy “on switch” by gating the entrance into, but not the exit from, cataplexy. The circuit mechanisms that function to switch off cataplexy remain unidentified.
The CeA may increase the incidence of cataplexy by lowering the stimulus threshold for attack induction. To test this hypothesis, we examined wheel-running behavior in our orexin−/− mice as an index of this threshold. Although CeA activation did not change overall amounts of wheel running in our mice, it significantly shortened the length of individual running bouts because they were punctuated by more frequent cataplexy attacks. Furthermore, CeA stimulation caused mice to experience cataplexy attacks after 77% less wheel-running compared with control conditions, suggesting that CeA activation lowered the threshold stimulus required to trigger cataplexy. These findings are consistent with the idea that the amygdala acts as a cataplexy “on switch,” particularly in light of observations that the amygdala response to positive emotions is heightened in human narcolepsy (Schwartz et al., 2008; Ponz et al., 2010) and may thereby predispose affected individuals to experience cataplexy.
Additional circuits involved in cataplexy
We found that chemogenetic inhibition of GABA CeA cells did not prevent cataplexy, suggesting they might not be required for triggering cataplexy. However, it is possible that CeA inhibition did not reduce cataplexy because of a “floor effect.” This idea is supported by Mahoney et al.'s (2017) finding that CeA inactivation only decreased cataplexy during stimulating conditions such as palatable foods. This observation suggests that mutliple brain circuits are involved in controlling cataplexy.
Cells in the zona incerta, laterodorsal tegmentum, and dorsal pons have been shown to suppress cataplexy (Burlet et al., 2002; Liu et al., 2011; Blanco-Centurion et al., 2013; Oishi et al., 2013). Drugs that affect cholinergic tone (e.g., cholinesterase inhibitors) can worsen cataplexy, suggesting that cholinergic circuits may also be involved (Reid et al., 1994; Kalogiannis et al., 2011; Kohlmeier et al., 2013). The noradrenergic, serotonergic, and dopaminergic systems also play roles in modulating cataplexy (Mignot et al., 1988; Mieda et al., 2004; Liu et al., 2008; Burgess et al., 2010; Burgess and Peever, 2013; Hasegawa et al., 2014).
Importantly, the mPFC is a critical site through which positive emotions trigger cataplexy. Neurons in the mPFC not only innervate the basolateral nucleus of the amygdala (BLA), but also melanin-concentrating hormone (MCH) cells, both of which are active during cataplexy and innervate regions that regulate motor tone (Fig. 10; Oishi et al., 2013). Therefore, it is possible that cataplexy is unaffected by CeA inhibition because the mPFC still has control over BLA and MCH neurons, which themselves initiate cataplexy.
A new resource for studying narcolepsy mechanisms
To identify the role of GABA CeA cells in regulating cataplexy, we developed a new orexin−/− mouse line that enabled selective targeting and manipulation of GABA cells. Detailed analysis showed that orexin−/−,VGAT-Cre mice were phenotypically indistinguishable from orexin−/− mice (Table 1). They experienced the same quality and quantity of cataplexy, as well as comparable amounts of sleep, wakefulness, and sleep attacks as their founder line. We showed that hM3Dq receptors could be delivered to GABA cells reliably and that systemic CNO administration increased the activity of hM3Dq-expressing GABA cells. Because mass activation of GABA CeA cells produced robust and consistent increases in cataplexy, we suggest that orexin−/−,VGAT-Cre mice could be a valuable resource for dissecting the roles that other GABA circuits (e.g., vlPAG cells) play in controlling cataplexy and other features of narcolepsy such as sleepiness. They will also be useful for determining how GABA mechanisms influence other behaviors that are orexin-dependent (e.g., addiction).
Conclusions
Current data show that positive stimuli may trigger cataplexy by recruiting GABA CeA cells in orexin−/− mice. We propose that GABA CeA cells act as a “relay center” between the cortical structures that interpret emotional stimuli and the brainstem circuits that trigger motor paralysis during cataplexy. However, additional circuitry is involved in mediating cataplexy because chemogenetic inactivation of CeA cells was unable to prevent cataplexy. Although multiple brain circuits are clearly involved in controlling cataplexy, it remains unclear how they communicate with and influence one another. Understanding how these systems function together represents a major challenge in identifying circuit mechanisms of cataplexy.
Footnotes
This work was supported by the Canadian Institutes of Health Research (CIHR), the Natural Sciences and Engineering Research Council of Canada (NSERC), and Sleep and Biological Rhythms Toronto, a CIHR-funded research and training program. We thank Carolina Gutierrez, Antoine Adamantidis, Ashley Bruce, and Daniel Li for assistance with fluorescence in situ hybridization; and Wendy Xie, Shirin Mollayeva, Hinal Patel, Amanda Stojcevski, and Dorsa Derakhshan for technical support.
The authors declare no competing financial interests.
References
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL (2007) Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 104:5163–5168. 10.1073/pnas.0700293104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Centurion C, Liu M, Konadhode R, Pelluru D, Shiromani PJ (2013) Effects of orexin gene transfer in the dorsolateral pons in orexin knockout mice. Sleep 36:31–40. 10.5665/sleep.2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin AM, Fried I, Wilson CL, Staba RJ, Behnke EJ, Lam HA, Maidment NT, Karlsson KÆ, Lapierre JL, Siegel JM (2013) Human hypocretin and melanin-concentrating hormone levels are linked to emotion and social interaction. Nat Commun 4:1547. 10.1038/ncomms2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissard R, Fort P, Gervasoni D, Barbagli B, Luppi PH (2003) Localization of the GABAergic and non-GABAergic neurons projecting to the sublaterodorsal nucleus and potentially gating paradoxical sleep onset. Eur J Neurosci 18:1627–1639. 10.1046/j.1460-9568.2003.02861.x [DOI] [PubMed] [Google Scholar]
- Bonnet L, Comte A, Tatu L, Millot JL, Moulin T, Medeiros de Bustos E (2015) The role of the amygdala in the perception of positive emotions: an “intensity detector”. Front Behav Neurosci 9:178. 10.3389/fnbeh.2015.00178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiter HC, Etcoff NL, Whalen PJ, Kennedy WA, Rauch SL, Buckner RL, Strauss MM, Hyman SE, Rosen BR (1996) Response and habituation of the human amygdala during visual processing of facial expression. Neuron 17:875–887. 10.1016/S0896-6273(00)80219-6 [DOI] [PubMed] [Google Scholar]
- Brooks PL, Peever JH (2012) Identification of the transmitter and receptor mechanisms responsible for REM sleep paralysis. J Neurosci 32:9785–9795. 10.1523/JNEUROSCI.0482-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess CR, Peever JH (2013) A noradrenergic mechanism functions to couple motor behavior with arousal state. Curr Biol 23:1719–1725. 10.1016/j.cub.2013.07.014 [DOI] [PubMed] [Google Scholar]
- Burgess CR, Tse G, Gillis L, Peever JH (2010) Dopaminergic regulation of sleep and cataplexy in a murine model of narcolepsy. Sleep 33:1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess CR, Oishi Y, Mochizuki T, Peever JH, Scammell TE (2013) Amygdala lesions reduce cataplexy in orexin knock-out mice. J Neurosci 33:9734–9742. 10.1523/JNEUROSCI.5632-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlet S, Tyler CJ, Leonard CS (2002) Direct and indirect excitation of laterodorsal tegmental neurons by Hypocretin/Orexin peptides: implications for wakefulness and narcolepsy. J Neurosci 22:2862–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Haubensak W, Anthony TE, Anderson DJ (2014) Central amygdala PKC-delta(+) neurons mediate the influence of multiple anorexigenic signals. Nat Neurosci 17:1240–1248. 10.1038/nn.3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo JM, Simón-Arceo K, Fernández-Mas R (1996) Prolonged enhancement of REM sleep produced by carbachol microinjection into the amygdala. Neuroreport 7:577–580. 10.1097/00001756-199601310-00048 [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M (1999) Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98:437–451. 10.1016/S0092-8674(00)81973-X [DOI] [PubMed] [Google Scholar]
- Clark EL, Baumann CR, Cano G, Scammell TE, Mochizuki T (2009) Feeding-elicited cataplexy in orexin knockout mice. Neuroscience 161:970–977. 10.1016/j.neuroscience.2009.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauvilliers Y, Arnulf I, Mignot E (2007) Narcolepsy with cataplexy. Lancet 369:499–511. 10.1016/S0140-6736(07)60237-2 [DOI] [PubMed] [Google Scholar]
- Dauvilliers Y, Siegel JM, Lopez R, Torontali ZA, Peever JH (2014) Cataplexy: clinical aspects, pathophysiology and management strategy. Nat Rev Neurol 10:386–395. 10.1038/nrneurol.2014.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. (1992) The role of the amygdala in fear and anxiety. Annu Rev Neurosci 15:353–375. [DOI] [PubMed] [Google Scholar]
- Davis M, Whalen PJ (2001) The amygdala: vigilance and emotion. Mol Psychiatry 6:13–34. 10.1038/sj.mp.4000812 [DOI] [PubMed] [Google Scholar]
- España RA, McCormack SL, Mochizuki T, Scammell TE (2007) Running promotes wakefulness and increases cataplexy in orexin knockout mice. Sleep 30:1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Egner T, Kalisch R (2011) Emotional processing in anterior cingulate and medial prefrontal cortex. Trends Cogn Sci 15:85–93. 10.1016/j.tics.2010.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell MS, Roth BL (2013) Pharmacosynthetics: reimagining the pharmacogenetic approach. Brain Res 1511:6–20. 10.1016/j.brainres.2012.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraigne JJ, Torontali ZA, Snow MB, Peever JH (2015) REM sleep at its core: circuits, neurotransmitters, and pathophysiology. Front Neurol 6:123. 10.3389/fneur.2015.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher M, Graham PW, Holland PC (1990) The amygdala central nucleus and appetitive Pavlovian conditioning: lesions impair one class of conditioned behavior. J Neurosci 10:1906–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavan H, Pendergrass JC, Ross TJ, Stein EA, Risinger RC (2001) Amygdala response to both positively and negatively valenced stimuli. Neuroreport 12:2779–2783. 10.1097/00001756-200108280-00036 [DOI] [PubMed] [Google Scholar]
- Gélineau J. (1880) De la narcolepsie. Gazette des Hôpitaux 53:626–628. [Google Scholar]
- Giardino WJ, de Lecea L (2014) Hypocretin (orexin) neuromodulation of stress and reward pathways. Curr Opin Neurobiol 29:103–108. 10.1016/j.conb.2014.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyani S, Wu MF, Nienhuis R, John J, Siegel JM (2002) Cataplexy-related neurons in the amygdala of the narcoleptic dog. Neuroscience 112:355–365. 10.1016/S0306-4522(02)00089-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa E, Yanagisawa M, Sakurai T, Mieda M (2014) Orexin neurons suppress narcolepsy via 2 distinct efferent pathways. J Clin Invest 124:604–616. 10.1172/JCI71017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SB, Tae WS, Joo EY (2006) Cerebral perfusion changes during cataplexy in narcolepsy patients. Neurology 66:1747–1749. 10.1212/01.wnl.0000218205.72668.ab [DOI] [PubMed] [Google Scholar]
- Janak PH, Tye KM (2015) From circuits to behaviour in the amygdala. Nature 517:284–292. 10.1038/nature14188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE. (2008) Modulation of cortical activation and behavioral arousal by cholinergic and orexinergic systems. Ann N Y Acad Sci 1129:26–34. 10.1196/annals.1417.026 [DOI] [PubMed] [Google Scholar]
- Kalogiannis M, Hsu E, Willie JT, Chemelli RM, Kisanuki YY, Yanagisawa M, Leonard CS (2011) Cholinergic modulation of narcoleptic attacks in double orexin receptor knockout mice. PLoS One 6:e18697. 10.1371/journal.pone.0018697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Thankachan S, Begum S, Liu M, Blanco-Centurion C, Shiromani PJ (2009) Hypocretin-2 saporin lesions of the ventrolateral periaquaductal gray (vlPAG) increase REM sleep in hypocretin knockout mice. PLoS One 4:e6346. 10.1371/journal.pone.0006346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyashchenko LI, Mileykovskiy BY, Lai YY, Siegel JM (2001) Increased and decreased muscle tone with orexin (hypocretin) microinjections in the locus coeruleus and pontine inhibitory area. J Neurophysiol 85:2008–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier KA, Tyler CJ, Kalogiannis M, Ishibashi M, Kristensen MP, Gumenchuk I, Chemelli RM, Kisanuki YY, Yanagisawa M, Leonard CS (2013) Differential actions of orexin receptors in brainstem cholinergic and monoaminergic neurons revealed by receptor knockouts: implications for orexinergic signaling in arousal and narcolepsy. Front Neurosci 7:246. 10.3389/fnins.2013.00246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Jones BE (2005) Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci 25:6716–6720. 10.1523/JNEUROSCI.1887-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lett BT, Grant VL, Koh MT, Flynn G (2002) Prior experience with wheel running produces cross-tolerance to the rewarding effect of morphine. Pharmacol Biochem Behav 72:101–105. 10.1016/S0091-3057(01)00722-5 [DOI] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, Li B (2013) Experience-dependent modification of a central amygdala fear circuit. Nat Neurosci 16:332–339. 10.1038/nn.3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Thankachan S, Kaur S, Begum S, Blanco-Centurion C, Sakurai T, Yanagisawa M, Neve R, Shiromani PJ (2008) Orexin (hypocretin) gene transfer diminishes narcoleptic sleep behavior in mice. Eur J Neurosci 28:1382–1393. 10.1111/j.1460-9568.2008.06446.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Blanco-Centurion C, Konadhode R, Begum S, Pelluru D, Gerashchenko D, Sakurai T, Yanagisawa M, van den Pol AN, Shiromani PJ (2011) Orexin gene transfer into zona incerta neurons suppresses muscle paralysis in narcoleptic mice. J Neurosci 31:6028–6040. 10.1523/JNEUROSCI.6069-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB (2006) A putative flip-flop switch for control of REM sleep. Nature 441:589–594. 10.1038/nature04767 [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clement O, Sapin E, Peyron C, Gervasoni D, Léger L, Fort P (2012) Brainstem mechanisms of paradoxical (REM) sleep generation. Pflugers Arch 463:43–52. 10.1007/s00424-011-1054-y [DOI] [PubMed] [Google Scholar]
- MacLaren DA, Browne RW, Shaw JK, Krishnan Radhakrishnan S, Khare P, España RA, Clark SD (2016) Clozapine N-oxide administration produces behavioral effects in Long-Evans rats: implications for designing DREADD experiments. eNeuro 3: pii: ENEURO.0219–16.2016. 10.1523/ENEURO.0219-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney CE, Agostinelli LJ, Brooks JNK, Lowell BB, Scammell TE (2017) GABAergic neurons of the central amygdala promote cataplexy. J Neurosci 37:3995–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meletti S, Vaudano AE, Pizza F, Ruggieri A, Vandi S, Teggi A, Franceschini C, Benuzzi F, Nichelli PF, Plazzi G (2015) The brain correlates of laugh and cataplexy in childhood narcolepsy. J Neurosci 35:11583–11594. 10.1523/JNEUROSCI.0840-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieda M, Sakurai T (2012) Overview of orexin/hypocretin system. Prog Brain Res 198:5–14. 10.1016/B978-0-444-59489-1.00002-1 [DOI] [PubMed] [Google Scholar]
- Mieda M, Willie JT, Hara J, Sinton CM, Sakurai T, Yanagisawa M (2004) Orexin peptides prevent cataplexy and improve wakefulness in an orexin neuron-ablated model of narcolepsy in mice. Proc Natl Acad Sci U S A 101:4649–4654. 10.1073/pnas.0400590101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot E, Guilleminault C, Bowersox S, Rappaport A, Dement WC (1988) Role of central alpha-1 adrenoceptors in canine narcolepsy. J Clin Invest 82:885–894. 10.1172/JCI113694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM (2005) Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron 46:787–798. 10.1016/j.neuron.2005.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray EA. (2007) The amygdala, reward and emotion. Trends Cogn Sci 11:489–497. 10.1016/j.tics.2007.08.013 [DOI] [PubMed] [Google Scholar]
- Nishino S, Mignot E (1997) Pharmacological aspects of human and canine narcolepsy. Prog Neurobiol 52:27–78. 10.1016/S0301-0082(96)00070-6 [DOI] [PubMed] [Google Scholar]
- Nitecka L, Ben-Ari Y (1987) Distribution of GABA-like immunoreactivity in the rat amygdaloid complex. J Comp Neurol 266:45–55. 10.1002/cne.902660105 [DOI] [PubMed] [Google Scholar]
- Oishi Y, Williams RH, Agostinelli L, Arrigoni E, Fuller PM, Mochizuki T, Saper CB, Scammell TE (2013) Role of the medial prefrontal cortex in cataplexy. J Neurosci 33:9743–9751. 10.1523/JNEUROSCI.0499-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overeem S, Mignot E, van Dijk JG, Lammers GJ (2001) Narcolepsy: clinical features, new pathophysiologic insights, and future perspectives. J Clin Neurophysiol 18:78–105. 10.1097/00004691-200103000-00002 [DOI] [PubMed] [Google Scholar]
- Overeem S, van Nues SJ, van der Zande WL, Donjacour CE, van Mierlo P, Lammers GJ (2011) The clinical features of cataplexy: a questionnaire study in narcolepsy patients with and without hypocretin-1 deficiency. Sleep Med 12:12–18. 10.1016/j.sleep.2010.05.010 [DOI] [PubMed] [Google Scholar]
- Paton JJ, Belova MA, Morrison SE, Salzman CD (2006) The primate amygdala represents the positive and negative value of visual stimuli during learning. Nature 439:865–870. 10.1038/nature04490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron C, et al. (2000) A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med 6:991–997. 10.1038/79690 [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS (1998) Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18:9996–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A, Amaral DG (1994) The distribution of GABAergic cells, fibers, and terminals in the monkey amygdaloid complex: an immunohistochemical and in situ hybridization study. J Neurosci 14:2200–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponz A, Khatami R, Poryazova R, Werth E, Boesiger P, Bassetti CL, Schwartz S (2010) Abnormal activity in reward brain circuits in human narcolepsy with cataplexy. Ann Neurol 67:190–200. 10.1002/ana.21825 [DOI] [PubMed] [Google Scholar]
- Reid MS, Siegel JM, Dement WC, Mignot E (1994) Cholinergic mechanisms in canine narcolepsy–II. Acetylcholine release in the pontine reticular formation is enhanced during cataplexy. Neuroscience 59:523–530. 10.1016/0306-4522(94)90174-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MJ, Warlow SM, Berridge KC (2014) Optogenetic excitation of central amygdala amplifies and narrows incentive motivation to pursue one reward above another. J Neurosci 34:16567–16580. 10.1523/JNEUROSCI.2013-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan SC, Roth BL (2011) Remote control of neuronal signaling. Pharmacol Rev 63:291–315. 10.1124/pr.110.003020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J (2003) The amygdaloid complex: anatomy and physiology. Physiol Rev 83:803–834. 10.1152/physrev.00002.2003 [DOI] [PubMed] [Google Scholar]
- Sanford LD, Yang L, Liu X, Tang X (2006) Effects of tetrodotoxin (TTX) inactivation of the central nucleus of the amygdala (CNA) on dark period sleep and activity. Brain Res 1084:80–88. 10.1016/j.brainres.2006.02.020 [DOI] [PubMed] [Google Scholar]
- Scammell TE, Willie JT, Guilleminault C, Siegel JM; International Working Group on Rodent Models of Narcolepsy (2009) A consensus definition of cataplexy in mouse models of narcolepsy. Sleep 32:111–116. [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Ponz A, Poryazova R, Werth E, Boesiger P, Khatami R, Bassetti CL (2008) Abnormal activity in hypothalamus and amygdala during humour processing in human narcolepsy with cataplexy. Brain 131:514–522. 10.1093/brain/awm292 [DOI] [PubMed] [Google Scholar]
- Siegel JM, Boehmer LN (2006) Narcolepsy and the hypocretin system–where motion meets emotion. Nat Clin Pract Neurol 2:548–556. 10.1038/ncpneuro0300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CT, Miskiman DE (1975) Increases in paradoxical sleep as a result of amygdaloid stimulation. Physiol Behav 15:17–19. 10.1016/0031-9384(75)90272-3 [DOI] [PubMed] [Google Scholar]
- Sorge S, Pollmächer T, Lancel M (2004) Clozapine alters sleep-wake behavior in rats. Neuropsychopharmacology 29:1462–1469. 10.1038/sj.npp.1300445 [DOI] [PubMed] [Google Scholar]
- Swanson LW, Petrovich GD (1998) What is the amygdala? Trends Neurosci 21:323–331. 10.1016/S0166-2236(98)01265-X [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM (2000) Reduced number of hypocretin neurons in human narcolepsy. Neuron 27:469–474. 10.1016/S0896-6273(00)00058-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Janak PH (2007) Amygdala neurons differentially encode motivation and reinforcement. J Neurosci 27:3937–3945. 10.1523/JNEUROSCI.5281-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassalli A, Dellepiane JM, Emmenegger Y, Jimenez S, Vandi S, Plazzi G, Franken P, Tafti M (2013) Electroencephalogram paroxysmal theta characterizes cataplexy in mice and children. Brain 136:1592–1608. 10.1093/brain/awt069 [DOI] [PubMed] [Google Scholar]
- Vertes RP. (2004) Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse 51:32–58. 10.1002/syn.10279 [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S Jr, Lowell BB (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71:142–154. 10.1016/j.neuron.2011.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Chung S, Beier KT, Xu M, Luo L, Dan Y (2015) Control of REM sleep by ventral medulla GABAergic neurons. Nature 526:435–438. 10.1038/nature14979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal C. (1877) Eigentümliche mit Einschlafen verbundene Anfälle. Archiv für Psychiatrie und Nervenkrankheiten 7:631–635. [Google Scholar]
- Xi M, Fung SJ, Sampogna S, Chase MH (2011) Excitatory projections from the amygdala to neurons in the nucleus pontis oralis in the rat: an intracellular study. Neuroscience 197:181–190. 10.1016/j.neuroscience.2011.09.029 [DOI] [PubMed] [Google Scholar]