

Abstract
As the population ages, an increasing number of people suffer from age-related cognitive impairment. However, the mechanisms underlying this process remain unclear. Here, we found that S-nitrosoglutathione reductase (GSNOR), the key enzyme that metabolizes intracellular nitric oxide (NO) and regulates S-nitrosation, was significantly increased in the hippocampus of both aging humans and mice. Transgenic mice overexpressing GSNOR exclusively in neurons showed cognitive impairment in behavioral tests, including the Morris water maze, fear conditioning, and the Y-maze test. We also found that GSNOR transgenic mice have LTP defects and lower dendrite spine density, whereas GSNOR knock-out mice rescued the age-related cognitive impairment. Analysis of S-nitrosation showed significantly decreased hippocampal CaMKIIα S-nitrosation in naturally aged mice and GSNOR transgenic mice. Consistent with the change in CaMKIIα S-nitrosation, the accumulation of CaMKIIα in the hippocampal synaptosomal fraction, as well as its downstream signaling targets p(S831)-GLUR1, was also significantly decreased. All these effects could be rescued in the GSNOR knock-out mice. We further verified that the S-nitrosation of CaMKIIα was responsible for the CaMKIIα synaptosomal accumulation by mutating CaMKIIα S-nitrosated sites (C280/C289). Upregulation of the NO signaling pathway rescued the cognitive impairment in GSNOR transgenic mice. In summary, our research demonstrates that GSNOR impairs cognitive function in aging and it could serve as a new potential target for the treatment of age-related cognitive impairment. In contrast to the free radical theory of aging, NO signaling deficiency may be the main mediator of age-related cognitive impairment.
SIGNIFICANCE STATEMENT This study indicated that S-nitrosoglutathione reductase (GSNOR), a key protein S-nitrosation metabolic enzyme, is a new potential target in age-related cognitive impairment; and in contrast to the free radical theory of aging, NO signaling deficiency may be the main cause of this process. In addition, increased GSNOR expression during aging decreases S-nitrosation of CaMKIIα and reduces CaMKIIα synaptosomal accumulation. To our knowledge, it is for the first time to show the cellular function regulation of CaMKIIα by GSNOR-dependent S-nitrosation as a new post-translational modification after its phosphorylation was explored. These findings elucidate a novel mechanism of age-related cognitive impairment and may provide a new potential target and strategy for slowing down this process.
Keywords: age-related cognitive impairment, CaMKIIα, nitric oxide, S-nitrosation modification, S-nitrosoglutathione reductase
Introduction
As the aged population grows, brain aging and neurodegenerative diseases are increasingly common (Hung et al., 2010). The Marist poll results of 1247 adults showed that age-related cognitive impairment is the top fear of aging for 82% of people. Age-related cognitive impairment seriously reduces the quality of elderly life and the ability of elderly individuals to live independently. The condition also causes increased economic burdens of care for families and the government. Currently, no effective drugs are available to reverse age-associated cognitive decline (Mora, 2013). Thus, the development of novel and efficient cognitive enhancers is urgent, and a better understanding of the molecular and neuronal mechanisms underlying age-related cognitive impairment is crucial (Konar et al., 2016).
Recent studies have shown that a decline in cognition function with age is highly correlated with loss of synaptic plasticity rather than complete degeneration of neuron (Morrison and Baxter, 2012). Therefore, we investigated whether the dysregulation of specific molecular signaling pathways involved in synaptic plasticity and cognitive function induced age-related cognitive impairment. The process of cognitive function is dependent on the functions of multiple neurotransmitter systems, including nitric oxide (NO) signaling (Bliss and Lomo, 1973; Bliss and Collingridge, 1993; Amtul and Atta-Ur-Rahman, 2015). Following its generation at postsynaptic sites, NO acts as a retrograde messenger diffused back to the presynaptic terminal and increases cGMP levels through the activation of soluble guanylate cyclase. This process subsequently facilitates the maintenance of LTP, a form of synaptic plasticity proposed to be pivotal in memory formation (Lu et al., 1999). In addition to this classic NO-cGMP-PKC signaling pathway, NO also exerts its effects on cognition by S-nitrosation, the covalent attachment of an NO group to the thiol side chain of cysteine (Lipton et al., 1994). S-nitrosation can regulate signal modulation and stress responses by affecting protein activity, localization, stability, and protein- protein interaction (Hess et al., 2005). However, whether the protein S-nitrosation plays an active role in cognition is poorly understood. S-nitrosoglutathione reductase (GSNOR) is a key protein S-nitrosation metabolic enzyme that is widely expressed and highly conserved from bacteria to humans (Liu et al., 2001). Research has shown that GSNOR has an important role in many physiological functions, including hepatocarcinogenesis (Tang et al., 2013), liver development (Cox et al., 2014), vasculogenesis (Gomes et al., 2013), asthma (Que et al., 2009), and inflammatory response (Wu et al., 2013). However, little is known about the function of GSNOR in the nervous system. Our previous work found that Drosophila with transgenic (TG) fdh, the homolog of mammalian GSNOR, showed a deficiency in visual pattern memory (Hou et al., 2011).
CaMKII is a Ca2+-activated enzyme that is highly enriched at synapses and is a major protein of the postsynaptic density (Lisman et al., 2002). CaMKII activity is stimulated by Ca2+/CaM, but activity can be maintained beyond the initial Ca2+ stimulus by several mechanisms that generate Ca2+-independent “autonomous” CaMKII activity (Coultrap and Bayer, 2012). For instance, NO can induce autonomous activity of CaMKIIα by S-nitrosation of Cys280 and Cys289 (Coultrap and Bayer, 2014). Activation of CaMKII is necessary for memory formation and LTP by increasing the number of synaptic AMPA receptors and their conductance (Lisman et al., 2012).
Based on our previous studies and the existing literature, we sought to determine the function and mechanism of GSNOR in age-related cognitive impairment in this study. By using naturally aged mice, TG mice overexpressing GSNOR exclusively in neurons (GSNOR TG mice) and GSNOR knock-out (KO) mice as models, we demonstrated that GSNOR may be a new potential target for the treatment of age-related cognitive impairment. Meanwhile, our results showed that S-nitrosated CaMKIIα levels and CaMKIIα synaptosomal accumulation were decreased by high expression of GSNOR in aging mice. Interestingly, we found that NO signaling deficiency may be the main cause of age-related cognitive impairment rather than NO toxicity.
Materials and Methods
Antibodies and reagents.
The antibodies used were as follows: β-actin (Santa Cruz Biotechnology catalog #sc-47778, RRID:AB_626632), HA (Santa Cruz Biotechnology catalog #sc-805, RRID:AB_631618), and synaptophysin (SYP, Santa Cruz Biotechnology catalog #sc-17750, RRID:AB_628311), CaMKIIα (Santa Cruz Biotechnology catalog #sc-13141, RRID:AB_626789), GLUR1 (Millipore catalog #MAB2263, RRID:AB_11212678), and p(S831)-GLUR1 (Abcam catalog #ab109464, RRID:AB_10862154). A rabbit GSNOR antibody for Western blot was raised against His-GSNOR fusion protein and affinity purified by protein A-Sepharose (Immunogenic Biological Technology). GSNOR inhibitor C3 was purchased from ChemDiv (Sanghani et al., 2009). All chemical reagents were obtained from Sigma-Aldrich unless otherwise indicated.
Animals.
GSNOR KO mice were generated as previously described (Liu et al., 2004) and compared with age-matched littermates. GFP mice (TG (Thy1-EGFP) MJrs/J, RRID:IMSR_JAX:007788) were obtained from The Jackson Laboratory. All mice were bred in the specific pathogen-free barrier facility of the Institute of Biophysics, Chinese Academy of Science. The Animal Center approved all procedures involving animals.
Cell cultures and transfection.
HEK293 cells (ATCC catalog #CRL-3216, RRID:CVCL_0063) were cultured in DMEM (Hyclone, SH30243.01) supplemented with 10% FBS (Invitrogen, 10019-141), 100 U/ml penicillin, and 100 mg/ml streptomycin (Hyclone, SV30010). Primary hippocampal neurons were prepared as described previously (Yin et al., 2011). Briefly, hippocampus was removed from 18-d-old embryo mouse brains and dissociated with 0.25% trypsin. Hippocampal neurons were then plated on plastic dishes previously coated with poly-L-lysine (Sigma). The dissociated cells were cultured in neurobasal media (Invitrogen, 21103-049) containing B27 supplements (Invitrogen, 17504-044) and 2 mm l-glutamine (Invitrogen, 35050-061). The cells were transfected with Lipofectamine 2000 (Invitrogen) or lenti virus according to the manufacturers' instructions.
Western blot analysis.
Protein extracts form cells and tissues were separated by 10% SDS-PAGE. The separated proteins were transferred to a nitrocellulose filter membrane (Millipore). The membrane was treated with 5% (w/v) fat free milk in Tris-buffered saline containing 0.05% Tween 20 for 2 h, and incubated with the indicated antibody for 2 h, followed by an incubation of peroxidase-conjugated anti-rabbit or mouse IgG (Santa Cruz Biotechnology) for 2 h. The epitopes were visualized with an ECL Western blot detection kit (Pierce).
Quantitative real-time PCR.
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's protocol; 2 μg of total RNA was used to synthesize the first-strand cDNA by M-MLV reverse transcriptase with OligodT(18) primer. Expression of the actin gene served as an internal control.
Griess assay.
A nitrite detection kit (Beyotime Biotech) was used to test NO level in TG and wild-type (WT) mice according to the manufacturer's instructions. Fresh mouse hippocampus was lysed in RIPA cell lysis buffer. After centrifugation, 50 μl supernatant was added to the Griess reagent. After 10 min, the nitrite concentration was determined by a microplate reader at 540 nm. Each experiment was performed in triplicate. The results of 5 TG and 5 WT mice were used for statistical analysis. The NO data were normalized to WT mice.
Irreversible biotin switch assay procedure (IBP).
The IBP for detecting the S-nitrosylation was performed as previously described (Huang and Chen, 2010). Adult mouse brain or cells were lysed in HEN buffer with 1% Nonidet P-40. The supernatants were incubated with 2.5% SDS at 50°C for 30 min with frequent vortex, and excess S-methyl methanethiosulfonate was removed by ice-cold acetone precipitation followed by centrifugation at 2000 × g for 10 min. This precipitation was repeated three times. The precipitate was recovered in HEN buffer containing 2.5% SDS with 0.4 mm biotin-maleimide and 10 mm ascorbate and incubated at 37°C for 1 h or at room temperature for 2 h. The excess biotin-maleimide was removed by ice-cold acetone precipitation as previously described. The pellet was suspended in HEN buffer with 200 mm DTT and incubated for 15 min at 100°C, followed by addition neutralization buffer (250 mm HEPES, pH 7.7, 100 mm NaCl, 0.1 mm EDTA, 10 mm neocuproine) and streptavidin-agarose (50–100 μl/sample) to purify the biotinylated proteins. This material was incubated at room temperature for 2 h. The agarose was washed 3 times (800 × g for 1 min) with neutralization buffer with 0.05% SDS, and the proteins were eluted by HEN buffer containing 2.5% SDS at 100°C for 15 min. The eluted mixture was analyzed by SDS-PAGE, followed by immunoblotting with relevant antibody.
Quantitative S-nitrosation proteomic analysis.
Total SNO modification proteins were prepared according to the IBP. Protein digestion by trypsin (Invitrogen) (0.3 μg trypsin to digest 10 μg proteins) was performed after adding dissolution buffer (75 μl) dilution. After incubation at 37°C overnight, the trypsin digestion process was terminated at −20°C for 30 min. The protein digestions were dried in a centrifugal vacuum concentrator; 20 μl TEAB buffer was used to suspend the freeze-dried peptide. According to the TMT labeling kit (Pierce), we labeled the adult mice with TMT 128 and labeled the aging mice with TMT 129. After suspension of the mixed samples with spin tips C18 desalination, the peptide was analyzed by mass spectrometry detection.
Tandem liquid chromatography-mass spectrometry (MS) analysis.
All nano-tandem liquid chromatography-mass spectrometry experiments were performed on a Q Exactive (Thermo Scientific) equipped with an Easy n-LC 1000 HPLC system (Thermo Scientific). The labeled peptides were loaded onto a 100 μm id × 2 cm fused silica trap column packed in-house with reversed-phase silica (Reprosil-Pur C18 AQ, 5 μm, Dr. Maisch) and then separated on an a 75 μm id × 20 cm C18 column packed with reversed phase silica (Reprosil-Pur C18 AQ, 3 μm, Dr. Maisch). The peptides bounded on the column were eluted with a 78 min linear gradient. Solvent A consisted of 0.1% formic acid in water solution, and Solvent B consisted of 0.1% formic acid in acetonitrile solution. The segmented gradient was 5%–8% B, 8 min; 8%–22% B, 50 min; 22%–32% B, 12 min; 32%–95% B, 1 min; 95% B, 7 min at a flow rate of 280 nl/ min.
The MS analysis was performed with Q Exactive mass spectrometer (Thermo Scientific). With the data-dependent acquisition mode, the MS data were acquired at a high resolution 70,000 (m/z 200) across the mass range of 300–1600 m/z. The target value was 3.00E+06 with a maximum injection time of 60 ms. The top 20 precursor ions were selected from each MS full scan with isolation width of 2 m/z for fragmentation in the HCD collision cell with normalized collision energy of 27%. Subsequently, MS/MS spectra were acquired at resolution 17,500 at m/z 200. The target value was 5.00E+04 with a maximum injection time of 80 ms; the dynamic exclusion time was 40 s. For nano electrospray ion source setting, the spray voltage was 2.0 kV; no sheath gas flow; the heated capillary temperature was 320°C.
Protein identification and quantification analysis.
The raw data from Q Exactive were analyzed with Proteome Discovery version 1.4 using Sequest HT search engine for protein identification and Percolator for false discovery rate analysis. The Uniprot mice protein database (updated on 06-2013) was individually used for searching the data from the mice sample. Some important searching parameters were set as follows: trypsin was selected as enzyme, and two missed cleavages were allowed for searching; the mass tolerance of precursor was set as 10 ppm, and the product ion tolerance was 0.02 Da.; the cysteine carbamidomethylation was selected as a fixed modification, and methionine oxidation and TMT-labeled lysine and N terminus of peptides were specified as variable modifications. False discovery rate analysis was performed with Percolator, and false discovery rate <1% was set for protein identification. The peptide confidence was set as high for peptide filter.
Protein quantification was also performed on Proteome Discovery version 1.4 using the ratio of the intensity of reporter ions from the MS/MS spectra. Only unique peptides of proteins or protein groups were selected for protein relative quantification. The total SNO proteins of adult mouse hippocampus tissue from two groups labeled with TMT 128 were considered as control reference for calculating the ratios of TMT 129/TMT 128, in which total SNO proteins of aging mouse hippocampus tissue were labeled with TMT 129. The normalization to the protein median of each sample was used to corrected experimental bias, and the number of minimum protein count must be >20.
Synaptosomal fraction extraction.
Adult mouse brain or cells were lysed in TEVP buffer ice (10 mm Tris-HCl, pH 7.4, 5 mm NaF, 1 mm Na3VO4, 1 mm EDTA, 1 mm EGTA, and 320 mm sucrose). The detailed assay for extracting synaptosomal fraction was performed as described previously (Yin et al., 2011).
Immunofluorescence staining.
Cells were fixed by 4% formaldehyde in PBS for 30 min at room temperature and subsequently treated with 0.4% Triton X-100 in PBS for 10 min at room temperature. Cells were blocked with 10% donkey serum in PBS for 1 h at room temperature and then incubated with primary antibody at 4°C overnight. Cells were washed in PBS and incubated with the corresponding secondary antibody for 1 h at room temperature. DNA was stained with Hoechst 33342 (Invitrogen). Microscopy was performed using a TCS SP5 II laser scanning confocal imaging system (HC PL FLUOTAR 10×/0.30 and HC PL APO 20×/0.70 CS objective lens; Leica) in conjunction with LAS AF 2.2 software (Leica).
Electrophysiology.
Experiments were performed in accordance with the National Institutes of Health Guide for the care and use of laboratory animals and were approved by the local animal care committee. GSNOR-overexpressed mice (2–5 months, both sexes) were anesthetized with pentobarbital sodium. After decapitation, whole brains were quickly removed and placed in ice-chilled sucrose slicing solution, which contained the following (in mm): 213 sucrose, 3 KCl, 1 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, and 10 glucose, pH 7.4. Hippocampal slices (400 μm) were cut with a vibratome (VT1200, Leica) and recovered in ACSF for at least 1 h. The ACSF contained the following (in mm): 125 NaCl, 5 KCl, 1.2 NaH2PO4, 26 NaHCO3, 1.3 CaCl2, 1.3 MgCl2, and 10 glucose, pH 7.4, equilibrated with 95% O2 and 5% CO2. Before recording, the slices were transferred to a recording chamber and continuously superfused with ACSF throughout the experiments.
We used normal procedures to record field EPSPs (fEPSP) with a glass microelectrode (3–5 mΩ, filled with ASCF) in the stratum radiatum of the hippocampal CA1 area as described previously. Evoked fEPSPs were elicited by stimulation of the Schaffer collateral fibers with a bipolar stimulating electrode (FHC) via a 2100 isolated pulse stimulator (A-M Systems). The slopes of fEPSP were calculated. Test stimulation was applied at 0.067 Hz. To measure the input–output relationship, the stimulation intensity was increased from 10 μA to 45 μA (in 5 μA increments) and then adjusted to evoke 30%–50% of maximal fEPSP. The effects of paired pulses at different (20, 40, 60, 80, 100, 200, and 300 ms) interstimulus intervals were also checked. Before LTP induction, at least 20 min of stable fEPSP was recorded for a baseline. LTP was induced by five episodes of theta burst stimulation (TBS) delivered at 0.1 Hz. Each episode contained 10 stimulus trains (5 pulses at 100 Hz) delivered at 5 Hz. Responses were recorded for 60 min after TBS induction. For better display, eight slopes of successive fEPSP were averaged, and the data were presented as the percentages of mean fEPSP slopes recorded during the baseline (before TBS) period. Electrophysiological experiments were performed using a Patch Clamp EPC 10 (HEKA). The data were sampled at 10 kHz, filtered at 2 kHz, and analyzed using Igor Pro (Wavemetrics). Electrophysiological experiments of GSNOR KO aging mice were performed in Capital Medical University of China. We used the Med64 system to record fEPSP in the hippocampal slices as described previously (Shimono et al., 2002) with specially modified ACSF for aging mice (Ting et al., 2014). Statistical analyses were performed by Student's t tests. Data are mean ± SEM, and n indicates the number of mice.
Spine density image analysis.
To quantify dendritic spine density, we mated GSNOR TG mice with Thy 1:GFP mice to make the dendritic spine visible under a fluorescent microscope. Spines were measured by using a Leica DM LB microscope TCS SP5 II laser scanning confocal imaging system (HC PL FLUOTAR 10×/0.30 and HC PL APO 20×/0.70 CS objective lens; Leica) in conjunction with LAS AF 2.2 software (Leica). Microscopic analysis was performed by the image analysis system ImageJ software. The average spine density (number of spines per μm of dendrite length) was calculated for the complete dendrite (Poeggel et al., 2003) from Thy 1:GFP-GSNOR TG mice and WT littermates as controls (n = 6, 10 brain slices per mice).
Generation of TG mice and analysis of transgene expression.
To generate TG mice overexpressing GSNOR, the GSNOR gene was amplified by PCR using a plasmid encoding WT GSNOR fused with the Kozak and HA-tag sequences at the N terminus. The resultant PCR fragment was ligated into the murine Thy-1.2 expression cassette (gifts of Dr. Pico Caroni), which drives constitutive transgene expression in postnatal and adult neurons (Caroni, 1997). The linearized GSNOR expression cassette driven by Thy-1.2 promoter was digested, and TG mice were generated by injecting the purified insert into the pronuclei of CD-1 zygotes. The GSNOR founder was crossed with CD-1 mice. Genotyping was performed by PCR analysis. Distribution of TG GSNOR in fresh frozen sections was analyzed by in situ hybridization as previously described. Protein expression was assessed by Western blots of tissue homogenates using anti-HA and anti-actin antibodies.
Tissue slice preparation.
For immunohistochemical studies, mice were anesthetized with pentobarbital and perfused via the ascending aorta with PBS, pH 7.4, until the outflow became clear, followed by 0.1 m phosphate buffer, pH 7.4, containing 4% PFA. The brain was postfixed in the same solution at 4°C and then sliced using a vibratome (Leica). Coronal sections and sagittal sections were prepared for immunohistochemistry.
Behavioral tests.
For all behavioral tasks, 2- to 3-month-old GSNOR TG mice and WT littermates were used. All the behavioral tests were conducted at approximately the same time of day (9:00 A.M. to 5:00 P.M.). Mice were extensively handled for 3 d before behavioral tests. Data are presented as mean ± SEM.
Open field.
Spontaneous locomotor activity was measured with an automated Open Field system (Lin et al., 2008). Mice were individually placed in a clear chamber (41 × 41 × 30 cm), and their activities were monitored for 20 min. Total distances traveled in the open field were analyzed.
Morris water maze.
The procedure was performed essentially as described previously (Vorhees and Williams, 2006). For the visible platform task, the platform was marked with a visible cue (colored flag). Mice were given four trials (60 s) per day using a fixed platform position and with changes of the starting position in each new trial. Then, the mice were given four trials every day (60 s) with a hidden platform, and the starting position varied between trials. If the mice failed to find the platform within 60 s, it was picked up and placed on the platform for 15 s. Probe trials were given the day after the last training. During probe trials, the platform was removed, and the mice were allowed to search for it for 60 s. Latencies and swim speed in the hidden and visible tasks were analyzed. Time spent in the target quadrant (%) compared with other quadrants was analyzed in the probe trials.
Y-maze.
Y-maze was performed as described previously (Arendash et al., 2001). The Y-maze apparatus, used to measure spontaneous alternation behavior, consisted of three equal-size arms oriented at 60° angles from each other. The mice were placed at the end of one fixed arm and allowed to move freely through the maze during an 8 min session. The sequence of arm entries was recorded, and three consecutive choices were defined as one succeeded alternation. The percentage of alternations was calculated as (actual alternations/maximum alternations) × 100. The total number of arms entered during the session was also determined.
Contextual fear-conditioning test.
The contextual fear-conditioning test was performed as described previously (Yin et al., 2011). Briefly, on the training day, mice were transported to the testing area at least 2 h before fear conditioning. In the conditioning session, mice were placed in the conditioning chamber and allowed to explore for 120 s. An electric foot shock (0.5 mA, 100 V, 2 s) was delivered, and a 30 s/2 s explore/shock paradigm was repeated for a total of five shocks. After the last shock, the mice were allowed to explore the chamber for an additional 1 min before returning to the home cage. For contextual fear memory tests, freezing behavior was measured 24 h after training by placing the mice back in the conditioning chamber to explore for 5 min. Freezing was defined as immobility of mice lasting >0.5 s.
Data analysis.
Expression data of GSNOR in the frontal cortex were retrieved from Brain Cloud (http://braincloud.jhmi.edu/, Gene Expression Omnibus accession no. GSE30272), which provides temporal dynamics of mRNA transcription in 269 human prefrontal cortex samples across the lifespan (Colantuoni et al., 2011). Hippocampal expression data from the Human Brain Transcriptome project, including 82 hippocampus aged from infancy to 80 s were used to confirm the trend of GSNOR expression alteration in hippocampus. The expression of GSNOR (probe 2779095), measured in signal intensity, was retrieved from the processed matrix generated by the Human Brain Transcriptome project (Kang et al., 2011; Pletikos et al., 2014) (Gene Expression Omnibus accession no. GSE25219). The relationship of the gene expression level with age was measured by linear regression analysis, with age as the independent factor and gene expression level as the dependent factor. Two-tailed Student's t tests were used for two-group comparisons. ANOVA and appropriate post hoc analyses were used for comparisons of more than two groups. p < 0.05 was considered statistically significant.
Results
GSNOR expression increases in the hippocampus of aged humans and mice
To explore the function of GSNOR in age-related cognitive impairment, we first sought to determine whether there is a change in the transcription and expression levels of GSNOR in the aging process. First, we retrieved publicly available microarray expression profiles of the human frontal cortex and hippocampus from the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). Using the human brain database Brain Cloud (http://braincloud.jhmi.edu/) (Colantuoni et al., 2011), we found that GSNOR expression levels increased with age in the human prefrontal cortex (n = 231 human; correlation analysis, p = 0.0008, r = 0.22; Fig. 1A). Moreover, using the Human Brain Transcriptome database (http://hbatlas.org/) (Kang et al., 2011; Pletikos et al., 2014), the expression level of GSNOR in the human hippocampus remarkably increased with age (n = 26 human; correlation analysis, p = 0.0091, r = 0.501; Fig. 1B). These findings informed our choice of naturally aged mice as our aging model. According to the growth pattern and the average life expectancy of these mice, we selected 22-month-old mice as the aged group and 7-month-old mice as the control adult group. We assessed GSNOR transcription and expression levels in the hippocampus of aged mice and controls. The results showed that GSNOR transcription (n = 6 mice; two-tailed Student's t test, p = 0.019; Fig. 1C) and expression levels (adult mice, n = 4; old mice, n = 5; two-tailed Student's t test, p = 0.0008; Fig. 1D) were both significantly increased in the aged mouse hippocampus. In addition, the expression level of GSNOR in aged mice was approximately twice that of adult mice (Fig. 1D).
Figure 1.

GSNOR expression increased in aging brain of human and mice. A, GSNOR gene expression over life span in human prefrontal cortex (n = 231 human from GSE30272 (Colantuoni et al., 2011); correlation analysis, p = 0.0008, r = 0.22). B, GSNOR gene expression over life span in human hippocampus (n = 26 human from GSE25219 (Kang et al., 2011; Pletikos et al., 2014); correlation analysis, p = 0.0091, r = 0.501). C, GSNOR mRNA level in adult (7-month-old) and old (22-month-old) mouse hippocampus (n = 6 mice; two-tailed Student's t test, p = 0.019). D, GSNOR protein expression in adult (7-month-old) and old (22-month-old) mouse hippocampus (adult mice, n = 4; old mice, n = 5; two-tailed Student's t test, p = 0.0008). Quantitation of GSNOR protein levels in right. Data are mean ± SEM. *p < 0.05, **p < 0.01.
Overexpressing GSNOR exclusively in neurons impairs cognitive function
To explore whether the elevated expression of GSNOR would induce age-related cognitive impairment, we generated TG GSNOR mice that was affixed to an HA epitope tag at the N terminus. The postnatal, neuron-specific transcription of GSNOR was under the control of the Thy1.2 promoter (Vidal et al., 1990) (Fig. 2A). After the F0 generation of mice was obtained by microinjection, we used primers for the Thy-1.2 control element and HA-GSNOR to identify TG mice (data not shown). We tested the HA expression in different tissues, including the brain, liver, spleen, lung, kidney, heart, and muscle by Western blot, and found that HA-GSNOR was only expressed in the brain and not in other tissues (Fig. 2B). Western blot analysis using an HA antibody showed that GSNOR was widely expressed in the brain, including the cerebral cortex, hippocampus, cerebellum, olfactory bulb, and medulla oblongata (Fig. 2C). In situ immunofluorescence hybridization on sections from GSNOR TG mice revealed a distribution of HA-GSNOR throughout the forebrain and with expression in the hippocampal CA1 pyramidal cell layer (Fig. 2D). We simultaneously tested the expression of endogenous and TG GSNOR in GSNOR TG and WT mouse hippocampus by Western blot using a GSNOR antibody (Fig. 2E). We found that the overexpression of TG GSNOR was twice that of WT mice, in which the GSNOR expression level was similar to the aged mice (WT, n = 3; TG, n = 3; two-tailed Student's t test, p = 0.0037; Fig. 2F). The NO level in primary hippocampal neurons from WT and TG mice treated with glutamate (GLU, 100 or 500 μm) was tested using a DAF-FM-DA fluorescent probe for 10 min. The results showed that the NO levels in TG mouse hippocampal neurons were lower than those in WT mice, regardless of GLU treatment (n = 4 experiments, two-tailed Student's t test, p < 0.01; Fig. 2G). S-nitrosoglutathione (GSNO), the main substrate of GSNOR, is an intermediate in either production or metabolism of SNO derived from NO. Although SNO proteins and nitric oxide are not the directly substrates of GSNOR, it may govern NO level and protein S-nitrosylation by influencing the cellular equilibrium between GSNO/NO and SNO proteins (as shown below). Overexpression of GSNOR in the TG mice may decrease NO level by increasing the transform of NO to GSNO and GSNO metabolism in cultured neurons induced by glutamate (Foster et al., 2009; Lima et al., 2010). We also found that the mRNA level (WT, n = 4; TG, n = 4; two-tailed Student's t test, p = 0.006; Fig. 2H) and enzyme activity (WT, n = 4 mice; TG, n = 4 mice; two-tailed Student's t test, p = 0.045; Fig. 2I) increased in GSNOR TG mice compared with WT mice. Furthermore, overexpression of GSNOR did not affect the reproduction, weight, and tissue morphology or brain anatomy of these mice (Fig. 2J,K).
Figure 2.

Generation and characterization of TG mice overexpressing GSNOR exclusively in neurons (GNSOR TG mice). A, Schematic representation of TG mice overexpressing GSNOR exclusively in neuron constructs. B, Mouse tissue lysates were analyzed by immunoblot using anti-HA antibody. Br, Brain; Li, liver; Sp, spleen; Lu, lung; Ki, kidney; He, heart; Mu, muscle. C, Mouse brain tissue lysates were analyzed by immunoblot using anti-HA antibody. CT, Cerebral cortex; HI, hippocampal; CB, cerebellum; OB, olfactory bulb; MO, medulla oblongata. D, Immunofluorescence detection of TG GSNOR by anti-HA antibody on coronal sections of adult mouse brain. E, GSNOR protein expression in GSNOR TG mouse hippocampus using GSNOR antibody (WT, n = 3; TG, n = 3; two-tailed Student's t test, p = 0.0037). F, Quantitation of GSNOR protein levels in E. G, NO level is tested by DAF-FM-DA fluorescence probe. Primary hippocampal neuron derived from WT and TG mice was treated by GLU (100 or 500 μm) for 10 min (n = 4 experiments, one-way ANOVA, p < 0.01). H, Real-time PCR demonstrates relative GSNOR mRNA level in mouse hippocampus (WT, n = 4; TG, n = 4; two-tailed Student's t test, p = 0.0061). I, Relative GSNOR enzyme activity in GSNOR WT and TG mouse hippocampus (WT, n = 4 mice; TG, n = 4 mice; two-tailed Student's t test, p = 0.045). J, Number of birth of GSNOR TG mice (WT, n = 5 mice; TG, n = 5 mice). K, Measurements of body weight of TG mice and age-/sex-matched WT-mice at 21 d and 8 weeks (male: TG, n = 15–21 mice, WT, n = 15–19 mice; female: TG, n = 11–21 mice, WT, n = 8–18 mice). Data are mean ± SEM. *p < 0.05, **p < 0.01.
To further confirm whether upregulation of GSNOR in adult mouse neurons impaired cognitive function in GSNOR TG mice, we compared GSNOR TG mice with their WT littermates in the open-field test (Lin et al., 2008). The GSNOR TG mice showed the same amount of exploratory and locomotor activity by measuring the total distance traveled within 20 min, as their WT littermates in the open field (Fig. 3A,B). Then, GSNOR TG mice and their WT littermates were subjected to three cognitive functional tasks, including the Morris water maze (Vorhees and Williams, 2006), the Y-maze (Arendash et al., 2001), and the fear conditioning test (Yin et al., 2011).
Figure 3.

Overexpressing GSNOR exclusively in neurons impaired cognitive function. TG female mice, n = 18 mice; WT female, n = 18 mice; age, 8–10 weeks. TG male mice, n = 12 mice; WT male, n = 10 mice; age, 8–10 weeks. TG 5 line mice, n = 11 mice; WT 5 line, n = 10 mice; female; age, 8–10 weeks. A, B, Measurement of spontaneous locomotion in the open field between female (A) or male (B) TG mice and sex-matched WT littermate mice. C, D, Learning curves of female (C) or male (D) TG mice and sex-matched WT littermate mice in Morris water maze tasks for 4 d visible platform training. Escape latencies were averages of four trials for each day, 60 s per trial. E, H, Average velocity (cm/s) traveled to the platform in the Morris water maze during the training of female (E) or male (H) TG mice and sex-matched WT littermate mice. F, Learning curves of Morris water maze tasks for 8 d spatial reference memory training of female TG mice and sex-matched WT littermate mice. Escape latencies were averages of four trials for each day, 60 s per trial: multiple t test, p = 0.04 (4 d), p = 0.005 (5 d), p = 0.014 (6 d), p = 0.02 (7 d), p = 0.001 (8 d). G, Comparison of quadrant time in the probe test after the last-platform training. PL, Platform quadrant; TQ, target quadrant; AL, adjacent left; AR, adjacent right; OP, opposite. Multiple t test, p = 0.04 (platform), p = 0.02 (target). I, Learning curves of Morris water maze tasks for 8 d spatial reference memory training of male TG mice and sex-matched WT littermate mice. Escape latencies were averages of four trials for each day, 60 s per trial (multiple t test, p = 0.002, 8 d). J, Comparison of quadrant time in the probe test after the last-platform training. Multiple t test, p = 0.03 (target), p = 0.02 (opposite). K, Percentage time of mice spent freezing at exploration, immediately acquired after the foot shock and 24 h after context training of female TG mice in a fear-conditioning task (multiple t test, p = 0.04, 24 h retention). L, The freezing time of every minute in the 24 h retention test was shown: multiple t test, p = 0.02 (2 min), 0.017 (3 min). M, Number of arm entries that female TG mice explored the Y-maze during 8 min. N, Percentage spontaneous alternations among arms by TG female and WT mice during exploration of Y-maze (two-tailed Student's t test, p = 0.012). O, Percentage time of mice spent freezing at exploration, immediately acquired after the foot shock and 24 h after context training of male TG mice in a fear-conditioning task: multiple t test, p = 0.04 (24 h retention). P, The freezing time of every minute in the 24 h retention test was shown: multiple t test, p = 0.016 (4 min). Q, Number of arm entries that male TG mice explored the Y-maze during 8 min. R, Percentage spontaneous alternations among arms by TG male and WT mice during exploration of Y-maze (two-tailed Student's t test, p = 0.0035). S, Percentage time of mice spent freezing at exploration, immediately acquired after the foot shock and 24 h after context training of line 5 TG mice in a fear-conditioning task: multiple t test, p = 0.03 (24 h retention). T, The freezing time of every minute in the 24 h retention test was shown: multiple t test, p = 0.024 (2 min), p = 0.026 (3 min). U, Number of arm entries that line 5 TG mice explored the Y-maze during 8 min. V, Percentage spontaneous alternations among arms by line 5 TG male and WT mice during exploration of Y-maze (two-tailed Student's t test, p = 0.001). Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
In the visible version of the Morris water maze, we found that both groups learned to swim to the marked platform equally well, with the average latencies decreasing from ∼40 s on the first day to ∼15 s on the third to fourth day (Fig. 3C,D). In this task, the similarities in latencies were accompanied by similar swimming speeds (Fig. 3E,H). We next trained these mice to swim to a hidden platform located in a fixed location of the pool. As expected, GSNOR TG mice required more time to arrive at the hidden platform than WT mice in 4–8 training day: TG female, n = 18 mice; WT female, n = 18 mice; multiple t test, p = 0.04 (4 d), p = 0.005 (5 d), p = 0.014 (6 d), p = 0.02 (7 d), p = 0.001 (8 d) (Fig. 3F) or in 8 training day: (TG male, n = 12 mice; WT male, n = 10 mice; multiple t test, p = 0.002 (8 d); Fig. 3I). After the hidden-platform training, the platform was removed, and spatial memory retention was assessed in a probe trial. The GSNOR TG mice spent less time in the target quadrant: multiple t test, p = 0.04 (plat), p = 0.02 (target) (Fig 3G); multiple t test, p = 0.03 (target), p = 0.02 (OP) (Fig 3J). This result indicated that GSNOR TG mice have cognitive impairment in the Morris water maze. Thereafter, we tested GSNOR TG and WT mice in the contextual fear memory test, which is dependent on the hippocampus, by assessing freezing behavior in the same environmental context (Yin et al., 2011). There is no difference in freezing behavior between GSNOR TG mice and WT mice indicated by the exploration time or immediately acquire freezing percentage after the foot shock (Fig. 3K,O). However, GSNOR TG mice showed less freezing than WT mice after 24 h retention: multiple t test, p = 0.04 (24 h retention) (Fig. 3K); multiple t test, p = 0.04 (24 h retention) (Fig. 3O). We further analyzed the freezing percentage in every minute with the line chart. The results showed that the freezing of GSNOR TG mice at 2–3 or 4 min was significantly lower than that of WT mice: multiple t test, p = 0.02 (2 min), 0.017 (3 min) (Fig. 3L); multiple t test, p = 0.016 (4 min) (Fig. 3P). The spontaneous alteration Y-maze task is another hippocampus-dependent spatial task commonly used in rodents (Arendash et al., 2001; Conrad et al., 2003). GSNOR TG mice showed fewer alternations than controls: two-tailed Student's t test, p = 0.012 (Fig. 3N); two-tailed Student's t test, p = 0.0035 (Fig. 3R), with similar times of entry to the arms (Fig. 3M,Q), indicating an impaired working memory (Dellu et al., 2000; Jung et al., 2008). Cognitive function tests showed that overexpression of GSNOR induced cognitive impairment in the three behavior tests in female (Fig. 3E–G,K–N) or male mice (Fig. 3H–J,O–R).
GSNOR TG mice (line 5) are another strain of GSNOR TG mice that were used to rule out false-positive results induced by the overexpression gene fragment inserted into other endogenous genes. To confirm the cognitive impairment effect in GSNOR TG mice, we compared another strain of GSNOR TG mice with WT littermate mice (line 5) apart from line 6, which we usually used in experiments. There is no difference in freezing between GSNOR TG mice (line 5) and WT mice, indicated by exploration time or immediately acquire freezing percentage after the foot shock (Fig. 3S). GSNOR TG mice (line 5) showed less freezing than WT mice after 24 h retention (Fig. 3S). We further analyzed the freezing percentage in every minute with the line chart. The results showed that the freezing of GSNOR TG mice (line 5) at 2–3 min was significantly lower than that of WT mice (Fig. 3T). GSNOR TG mice (line 5) showed fewer alternations than controls (Fig. 3V), with similar times of entry to the arms (Fig. 3U). The Y-maze and fear conditioning test results showed that line 5 GSNOR TG mice also had cognitive impairment. In conclusion, overexpression of GSNOR impairs cognitive function, and these effects are independent of TG strains.
GSNOR overexpression impaired hippocampal synaptic plasticity
Considering that synaptic transmission and plasticity are widely regarded as the basic cellular mechanisms of cognitive function (Amtul and Atta-Ur-Rahman, 2015), we used hippocampal slices prepared from GSNOR TG and WT littermates to measure the effect of GSNOR overexpression on synaptic properties in the Schaffer collateral-CA1 pathway.
Basal synaptic transmission was first examined in the GSNOR TG and WT mice. When the stimulus intensity was increased from 10 to 45 μA, the input–output curves for the slope of extracellular fEPSP revealed no differences between slices from TG and WT mice (Fig. 4A). We also investigated paired-pulse facilitation (PPF), which indicates presynaptic function, to exclude the possibility that the overexpression of GSNOR influenced the probability of neurotransmitter release. There was also no difference in PPF between slices from TG and WT mice at interstimulus intervals ranging from 20 to 300 ms (WT, n = 10 mice; TG, n = 10 mice; Fig. 4B). These results indicated that both basal synaptic transmission and presynaptic function are normal in GSNOR TG mice (Tsien et al., 1996). LTP is known as a model of processes associated with cognitive function (Lynch, 2004; Kandel et al., 2014). We next examined whether LTP was affected by the overexpression of GSNOR. In WT hippocampal slices, LTP was readily induced by TBS stimulation and lasted for at least 60 min (Fig. 4C). Compared with the mean values at baseline, the average slope of fEPSP in the last 10 min was 146.1 ± 12.0% (Fig. 4D). However, TBS stimulation failed to induce LTP in GSNOR TG hippocampus slices (TG, n = 10 mice; WT, n = 10 mice; two-way ANOVA, p < 0.0001; Fig. 4C). The average slope of fEPSP in the last 10 min was 97.3 ± 15.9%, and it was significantly different in WT hippocampus slices (TG, n = 10 mice; WT, n = 10 mice; two-tailed Student's t test, p = 0.033; Fig. 4D). These results suggest that overexpression of GSNOR can block LTP in the hippocampus, and it is likely the cellular mechanism underlying the cognitive impairment of GSNOR TG mice showed in behavior tests.
Figure 4.

GSNOR overexpression impaired hippocampal synaptic plasticity. A, Input–output curve. Data from WT and TG slices at stimulus intensity from 10 to 45 μA. B, PPF. Data from WT and TG slices from recordings at different interval (WT, n = 10 mice; TG, n = 10 mice). C, LTP. TBS (arrow) induces LTP in WT mice but not in TG mice, indicating that GSNOR overexpression impairs hippocampus LTP in TG mice. Top, Representative traces before (gray) and after (black) TBS. Data are percentage of mean fEPSP slopes recorded during the baseline (before TBS) (TG, n = 10 mice; WT, n = 10 mice; two-way ANOVA, p < 0.0001). D, Mean ± SEM of LTP levels recorded 50–60 min after TBS. WT: 146.1 ± 12.0%; TG: 97.3 ± 15.9% (TG, n = 10 mice; WT, n = 10 mice; two-tailed Student's t test, p = 0.033). E, Representative images of hippocampal neuronal dendrite spine from Thy1:GFP-GSNOR TG mice and littermate control. F, Quantitation of dendrite spine density in hippocampal neuron from Thy1:GFP-GSNOR TG mice and littermate control (WT, n = 183 neurons form 6 mice; TG, n = 200 neurons form 6 mice; two-tailed Student's t test, p = 0.0003). Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
The density, morphology, and function of dendritic spines are closely related to synaptic plasticity, which is a key mechanism for cognitive function (Bosch and Hayashi, 2012; Amtul and Atta-Ur-Rahman, 2015). To examine the effect of GSNOR overexpression on synaptic plasticity, we mated GSNOR TG mice with Thy 1:GFP mice to make the morphology of neuronal dendritic spines visible under a fluorescent microscope. The density of dendritic spines as an indicator of synaptic plasticity was analyzed by laser confocal microscopy in brain tissue frozen sections (Fig. 4E). The results showed that the hippocampal neuronal dendritic spine density of GSNOR TG mice was significantly lower than that of their WT littermates (WT, n = 183 neurons form 6 mice; TG, n = 200 neurons form 6 mice; two-tailed Student's t test, p = 0.0003; Fig. 4F).
GSNOR deficiency rescues age-related cognitive impairment
If the high expression of GSNOR in the hippocampus is responsible for age-related cognitive impairment, then downregulation of its expression in aged mice should ameliorate the memory deficiency. Indeed, we tested cognitive function of GSNOR KO aged mice and their littermates (22-month-old) in the Y-maze test and context fear conditioning test.
In the Y-maze test, the number of arm entries among the three groups of aged mice (+/+, +/−, −/−) was not significantly different (Fig. 5A). The alternation percentage of GSNOR KO aged mice (−/−) was significantly higher than that of the other two groups of aged mice (+/+, +/−), suggesting that the working memory of GSNOR KO aged mice is stronger than that of the control mice (+/+, n = 8 mice; +/−, n = 9 mice; −/−, n = 9 mice; one-way ANOVA/Turkey's post hoc test, p = 0.014, +/+ vs −/−; p = 0.047, +/− vs −/−; Fig 5B).
Figure 5.
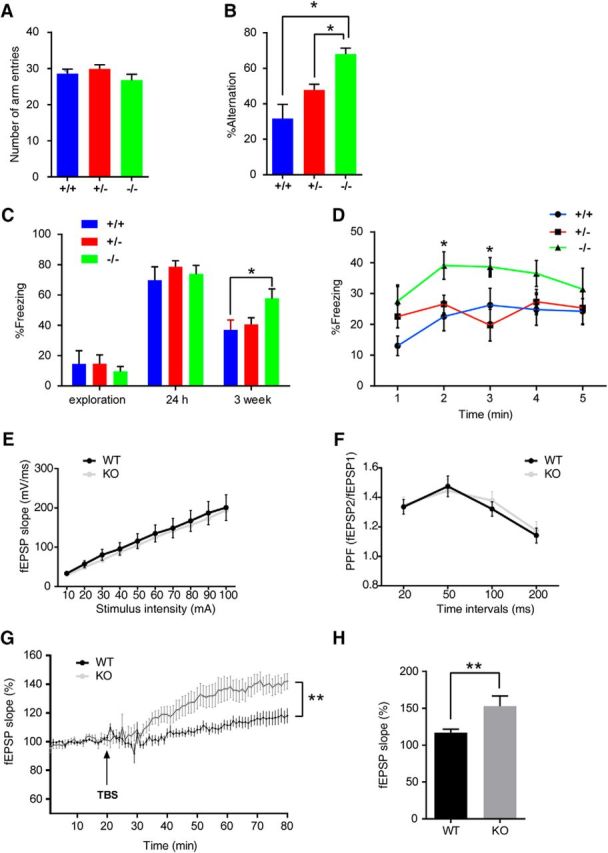
GSNOR KO mice rescued age-related cognitive impairment (GSNOR KO mice, 22-month-old, male, +/+, n = 8 mice; +/−, n = 9 mice; −/−, n = 9 mice). A, Number of arm entries that mice explored the Y-maze during 8 min. B, Percentage alternations among Y-maze arms that mice showed during 8 min exploration: one-way ANOVA, p = 0.014 (+/+ vs −/−); p = 0.047 (+/− vs −/−). C, Percentage time of mice spent freezing at exploration, 24 h, 3 weeks after context training in a fear-conditioning task: one-way ANOVA, p = 0.028 (+/+ vs −/−). D, The freezing time of every minute in the 3 weeks retention test: one-way ANOVA, p = 0.019 (+/+ vs −/−, 2 min); p = 0.027 (+/+ vs −/−, 3 min). E, Input–output curve. Data from WT and KO slices at stimulus intensity from 10 to 100 μA. F, PPF. Data from WT and KO slices from recordings at different intervals (WT, n = 12 slices form 4 mice; KO, n = 11 slices from 4 mice). G, LTP. Data are the percentage of mean fEPSP slopes recorded during the baseline (before TBS) period (WT, n = 12 slices form 4 mice; KO, n = 11 slices from 4 mice; two-way ANOVA, p < 0.0001). H, Mean ± SEM of LTP levels recorded 50–60 min after TBS (two-tailed Student's t test, p = 0.018). Data are mean ± SEM. *p < 0.05, **p < 0.01.
In the fear conditioning test, the freezing percentage of the three groups of aged mice (+/+, +/−, −/−) in the exploration stage showed no significant differences among the groups (Fig. 5C). After 5 consecutive electric shocks, the percentage of freezing in the three groups of aged mice was very high, up to 90%, with no significant differences (data not shown). After 24 h, there was still no significant difference in the freezing percentage among the three groups of mice. We concluded that the C57 aged mice were perhaps too sensitive to the electrical stimulation, such that our relatively strong electric shock (previous experimental condition in CD1 WT mice or TG mice) could have masked the difference. Based on such judgments, we opined that the freezing memory could likely be regulated over a longer period of time. Hence, we detected the freezing percentage again 3 weeks later. Results showed that the freezing percentage of GSNOR KO aged mice (−/−) was significantly higher than the other two groups of aged mice (+/+, +/−) in 3 weeks retention (+/+, n = 8 mice; +/−, n = 9 mice; −/−, n = 9 mice; one-way ANOVA/Turkey's post hoc test, p = 0.028, +/+ vs −/−; Fig. 5C). Then the freezing percentages in every minute were analyzed with the line chart. The results showed that the freezing of GSNOR KO aged mice (−/−) at 2 and 3 min was significantly higher than that of the other two groups of aged mice (+/+, n = 8 mice; +/−, n = 9 mice; −/−, n = 9 mice; one-way ANOVA/Turkey's post hoc test, p = 0.019, +/+ vs −/−, 2 min; p = 0.027, +/+ vs −/−, 3 min; Fig 5D). This is an indication that the fear-related cognitive function of GSNOR KO mice was significantly stronger than the control mice. In summary, we found that the cognitive function of GSNOR KO aged mice is stronger than that of WT or heterozygous control mice.
We examined whether LTP was affected in GSNOR KO aging mice (22-month-old). We first detected the input–output curves and PPF in GSNOR KO aging mice compared with WT aging mice. When the stimulus intensity was increased from 10 to 100 μA, the input–output curves for the slope of extracellular fEPSP revealed no differences between slices from KO and WT mice (Fig. 5E). There was also no difference in PPF between slices from KO and WT aging mice at interstimulus intervals ranging from 20 to 200 ms (WT, n = 12 slices; KO, n = 11 slices; Fig. 5F). In WT and GSNOR KO aging hippocampal slices, LTP was induced by TBS stimulation and lasted for at least 60 min. TBS stimulation induced higher LTP in GSNOR KO aging mice than WT (WT, n = 12 slices; KO, n = 11 slices; two-way ANOVA, p < 0.001; Fig. 5G). Compared with the mean values at baseline, the average slope of fEPSP in the last 10 min of GSNOR KO mice was significantly higher in WT hippocampus slices (WT, n = 12 slices; KO, n = 11 slices; two-tailed Student's t test, p = 0.018; Fig. 5H). These results suggest that knock-out GSNOR can increase LTP in the hippocampus of aging mice.
GSNOR decreases CaMKIIα S-nitrosation modification in the aged hippocampus
GSNOR is considered to be a key metabolic enzyme for S-nitrosation modification homeostasis (Liu et al., 2001, 2004; Benhar et al., 2009). Thus, we screened the S-nitrosation protein targets in the aged mouse hippocampus by S-nitrosation modification quantitative proteomics. We obtained 627 S-nitrosation protein targets, 103 of which were significantly decreased (20%). Then, we did the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis and tried to find related targets. Because LTP is known as a key mechanism of cognitive function, we first focused on the protein target listed in Table 1 involved in LTP process of KEGG analysis. Among them, we found an important target, CaMKIIα, which is directly related to cognitive function and synaptic plasticity (Lisman et al., 2012).
Table 1.
S-Nitrosation modification quantitative proteomics in aging mice hippocampusa
| Accession no. | Name | Scoreb | Aging S-nitrosation/adult S-nitrosationc |
|---|---|---|---|
| P11798 | CaMKIIα | 40.15 | 0.706 |
| P63328 | Serine/threonine-protein phosphatase 2B catalytic subunit α | 39.40 | 1.007 |
| E0CZ78 | Serine/threonine-protein phosphatase | 24.67 | 1.150 |
| Q5SVI9 | CaMKIIβ | 23.55 | 0.519 |
| P21279 | Guanine nucleotide-binding protein G(q) subunit α | 13.90 | 1.191 |
| P31938 | Dual-specificity mitogen-activated protein kinase 1 | 9.99 | 0.833 |
| P63085 | Mitogen-activated protein kinase 1 | 9.35 | 0.707 |
| Q2NKI4 | PKC | 6.23 | 1.169 |
| P63087 | Serine/threonine-protein phosphatase PP1-γ catalytic subunit | 2.48 | 1.174 |
| B1AUP0 | Serine/threonine-protein kinase A | 2.15 | 0.886 |
aTargets are from LTP classification in KEGG analysis of S-nitrosation modification quantitative proteomics in aging mice compared with adult mice hippocampus. S-nitrosation modification protein targets are labeled by TMT 129 (aging mice hippocampus) or TMT 128 (adult mice hippocampus) in quantitative proteomics.
bProteome Discovery version 1.4 software indicates the sum of Xcorr (the score of the cross correlation with the theoretical spectrum) of all identified peptide of this protein. The higher the score, the higher the confidence level of identified protein.
cTMT 129/TMT 128.
First, consistent with S-nitrosation modification quantitative proteomics, we verified the downregulation of SNO-CaMKIIα in aged mouse hippocampus by the IBP method (7-month-old, n = 5 mice; 22-month-old, n = 5 mice; two-tailed Student's t test, p = 0.0047; Fig. 6A,B). To study the effect of GSNOR on SNO-CaMKIIα, we detected SNO-CaMKIIα level in GSNOR TG mice and GSNOR KO aged mouse hippocampus by the IBP method. Consistent with previous results, SNO-CaMKIIα levels in the hippocampus were indeed downregulated in GSNOR TG mice (n = 6 experiments; two-tailed Student's t test, p = 0.0031; Fig. 6C,D) but were upregulated in the GSNOR KO aged mouse (WT, n = 4 mice; KO, n = 4 mice; two-tailed Student's t test, p = 0.0026, Fig. 6E,F) hippocampus.
Figure 6.

GSNOR decreased CaMKIIα S-nitrosation modification in aging hippocampus. A, Detection of CaMKIIα S-nitrosation modification level in 7-month-old (adult) and 22-month-old (old) mouse hippocampus. B, Quantitation of SNO-CaMKIIα level in in 7-month-old (adult) and 22-month-old (old) mouse hippocampus (7-month-old, n = 5 mice; 22-month-old, n = 5 mice; two-tailed Student's t test, p = 0.0047). C, Detection of CaMKIIα S-nitrosation modification level in GSNOR TG mouse hippocampus. D, Quantitation of SNO-CaMKIIα level in GSNOR TG mice hippocampal (n = 6 experiments; two-tailed Student's t test, p = 0.0031). E, Detection of CaMKIIα S-nitrosation modification level in GSNOR KO aging mice (22-month-old) hippocampus. F, Quantitation of SNO-CaMKIIα level in GSNOR KO aging mice (22-month-old) hippocampus (WT, n = 4 mice; KO, n = 4 mice; two-tailed Student's t test, p = 0.0026). Data are mean ± SEM. **p < 0.01.
GSNOR downregulates CaMKIIα signaling in the aged hippocampus by decreasing S-nitrosation of CaMKIIα
Our previous results have demonstrated that GSNOR downregulates SNO-CaMKIIα levels in the aged mouse hippocampus (Fig. 7A,B). However, whether CaMKIIα function is regulated by GSNOR in the aging process is unknown. Synaptosomal accumulation of CaMKIIα is essential for its function in cognition (Lisman et al., 2012). Therefore, we detected CaMKIIα synaptosomal accumulation in old mouse hippocampus compared with adult mice. The results showed that CaMKIIα synaptosomal accumulation was downregulated in old mice (7-month-old, n = 6 mice; 22-month-old, n = 6 mice; two-tailed Student's t test, p = 0.042; Fig. 7A). We further verified the effect of GSNOR on CaMKIIα accumulation in mature hippocampal neurons. Overexpression of GSNOR decreased CaMKIIα accumulation in hippocampal neuronal synaptosome but not in the GSNOR mutant R115D (mutation of arginine 115 to aspartic acid, enzyme activity mutant) (Hedberg et al., 2003; Wu et al., 2014) (n = 3 experiments; one-way ANOVA/Turkey's post hoc test, p = 0.009, ctrl vs GSNOR; p = 0.008, GSNOR vs R115D; Fig 7B). Interestingly, the downregulation of both GSNOR by the shGSNOR plasmid (n = 3 experiments; two-tailed Student's t test, p = 0.0071; Fig. 7C) and the GSNOR inhibitor C3 (33 μm for 3 h; n = 3 experiments; two-tailed Student's t test, p = 0.036; Fig. 7D) increased CaMKIIα accumulation in hippocampal neuronal synaptosome. Verification of the results in GSNOR TG mice and GSNOR KO aged mice revealed that CaMKIIα synaptosomal accumulation level was downregulated in GSNOR TG mice (n = 4 experiments; two-tailed Student's t test, p = 0.0062; Fig. 7E) and upregulated in GSNOR KO aged mouse (WT, n = 4 mice; KO, n = 4 mice; two-tailed Student's t test, p = 0.0093; Fig. 7F) hippocampus.
Figure 7.

GSNOR decreased CaMKIIα accumulation in aging hippocampal synaptosomal fraction. A, Left, CaMKIIα accumulation level in adult (7 months) and old (22 months) mice hippocampal synaptosomal fraction. Right, Quantitation of CaMKIIα accumulation level normalized by synaptosomal marker SYP (7-month-old, n = 6 mice; 22-month-old, n = 6 mice; two-tailed Student's t test, p = 0.042). B, Top, CaMKIIα accumulation level in primary hippocampal neuron synaptosomal fraction by overexpression of GSNOR. Neuron was transfected by Ctrl, GSNOR, or R115D (GSNOR enzyme activity mutant) lentivirus plasmids. Bottom, Quantitation of CaMKIIα accumulation level normalized by synaptosomal marker SYP: n = 3 experiments; one-way ANOVA, p = 0.009 (ctrl vs GSNOR); p = 0.008 (GSNOR vs R115D). C, Top, CaMKIIα accumulation level in primary hippocampal neuron synaptosomal fraction by downregulation of GSNOR. Neuron was transfected by shGSNOR to downregulated GSNOR protein level. Bottom, Quantitation of CaMKIIα accumulation level normalized by synaptosomal marker SYP (n = 3 experiments; two-tailed Student's t test, p = 0.0071). D, Top, CaMKIIα accumulation level in primary hippocampal neuron synaptosomal fraction by inhibition of GSNOR activity. Neuron was treated by GSNOR inhibitor C3 (33 μm) for 3 h. Bottom, Quantitation of CaMKIIα accumulation level normalized by synaptosomal marker SYP (n = 3 experiments; two-tailed Student's t test, p = 0.036). E, Top, CaMKIIα accumulation level in GSNOR TG mice hippocampal synaptosomal fraction. Bottom, Quantitation of CaMKIIα accumulation level normalized by synaptosomal marker SYP (n = 4 experiments; two-tailed Student's t test, p = 0.0062). F, Top, CaMKIIα accumulation level in GSNOR KO aging mice hippocampal synaptosomal fraction. Bottom, Quantitation of CaMKIIα accumulation level normalized by synaptosomal marker SYP (WT, n = 4 mice; KO, n = 4 mice; two-tailed Student's t test, p = 0.0093). Data are mean ± SEM. *p < 0.05, **p < 0.01.
Postsynaptic AMPAR activity plays a key role in LTP (Collingridge et al., 2004; Matsuzaki et al., 2004; Kessels and Malinow, 2009). According to current evidence, the increase in AMPAR conductance that is observed during the LTP is due to S831 phosphorylation of GLUR1, which is phosphorylated by CaMKIIα (Lisman et al., 2002, 2012; Halt et al., 2012). Thus, we tried to evaluate the impact of reduced SNO-CaMKIIα by GSNOR on S831 phosphorylation. We found that p(S831)-GLUR1 was significantly downregulated in aged mouse hippocampal synaptosome (adult, n = 3 mice; old, n = 3 mice; two-tailed Student's t test, p = 0.0043; Fig. 8A). Overexpression of GSNOR decreased p(S831)-GLUR1 in hippocampal neuronal synaptosome (n = 3 experiments; two-tailed Student's t test, p = 0.0061; Fig. 8B). On the contrary, p(S831)-GLUR1 was upregulated in GSNOR KO mice compared with WT aged hippocampal synaptosome (WT, n = 4 mice; KO, n = 4 mice; two-tailed Student's t test, p = 0.033; Fig. 8C). It is reported that NO induced autonomous activity of CaMKIIα by S-nitrosation of Cys280 and Cys289 and contributes to its pathological functions (Coultrap and Bayer, 2014). However, whether there is a link between SNO-CaMKIIα and synaptosomal CaMKIIα accumulation remains unknown. To answer this question, we constructed CaMKIIα S-nitrosation site mutant C280/C289V lentivirus plasmid. WT GFP-CaMKIIα and mutant CaMKIIα C280/C289V were transfected into HEK293 cells treated with 500 μm or 1 mm NO donor SNOC. The results showed that SNOC induced nitrosation modification in WT but not in mutant CaMKIIα-transfected cells (Fig. 9A), indicating that C280/C289 are the key nitrosation sites of CaMKIIα. Synaptosomal accumulation in DIV14 hippocampal neurons was further detected to investigate the effect of SNO-CaMKIIα on function by transfecting hippocampal neurons with WT and mutant CaMKIIα. The synaptosomal accumulation of the C280/C289V mutant CaMKIIα was lower than WT CaMKIIα (WT, n = 4; C280/C289V, n = 4; two-tailed Student's t test, p = 0.0027; Fig. 9B). The GSNOR inhibitor C3 increased synaptosomal accumulation in WT CaMKIIα but not in the C280/C289V mutant (n = 4 experiments; two-tailed Student's t test, p = 0.007, WT vs IH; p = 0.028, WT vs C2V; Fig. 9C). These results demonstrated that GSNOR inhibits CaMKIIα synaptosomal accumulation by downregulating its nitrosation modification.
Figure 8.

GSNOR decreased p(S831)-GLUR1 in aging hippocampus. A, p-GLUR1 (S831) level in adult (7 months) and old (22 months) mouse hippocampal synaptosomal fraction. Quantitation of p-S831 level normalized by synaptosomal marker SYP at right (adult, n = 3 mice; old, n = 3 mice; two-tailed Student's t test, p = 0.0043). B, p-GLUR1 (S831) level in primary hippocampal neuron synaptosomal fraction. Neuron was transfected by ctrl or GSNOR lentivirus plasmid. Quantitation of p-S831 level normalized by synaptosomal marker SYP at right (n = 3 experiments; two-tailed Student's t test, p = 0.0061). C, p-GLUR1 (S831) level in GSNOR KO aging mice (22 months) hippocampal synaptosomal fraction. Quantitation of p-S831 level normalized by synaptosomal marker SYP at right (WT, n = 4 mice; KO, n = 4 mice; two-tailed Student's t test, p = 0.033). Data are mean ± SEM. *p < 0.05, **p < 0.01.
Figure 9.

S-nitrosation site mutants inhibit CaMKIIα accumulation in synaptosome. A, CaMKIIα S-nitrosation modification level in HEK293 cells transfected by GFP-CaMKIIα or GFP-CaMKIIα (C280/C289V) mutants and then treated by 200 μm SNOC for 15 min. B, CaMKIIα accumulation level in primary hippocampal neuron synaptosomal fraction. Neuron was transfected by GFP-CaMKIIα or GFP-CaMKIIα (C280/C289V) mutants (WT, n = 4; C280/C289V, n = 4; two-tailed Student's t test, p = 0.0027). C, CaMKIIα accumulation level in primary hippocampal neuron synaptosomal fraction. Neuron was transfected by GFP-CaMKII or GFP-CaMKIIα (C280/C289V) mutants and treated with GSNOR inhibitor C3 (33 μm for 3 h). Quantitation of CaMKIIα accumulation level normalized by total GFP-CaMKIIα at right (n = 4 experiments; two-tailed Student's t test, p = 0.007, WT vs IH; p = 0.028, WT vs C2V). Data are mean ± SEM. **p < 0.01.
Increasing NO signaling rescues GSNOR TG mouse cognitive impairment
It has been reported that supplementing l-Arg by intraperitoneal injection can increase NO levels in mice (Rezayof et al., 2006). GSNOR TG and WT mice were injected with intraperitoneal l-Arg. As shown in Figure 10A (WT, n = 5; TG, n = 5), l-Arg administered 30 min before the test rescued the attenuation of NO levels in the GSNOR TG mouse hippocampus. We went further to determine whether this treatment could also rescue the GSNOR TG mouse cognitive impairment in the Y-maze and fear conditioning test. Like the performance described above, the GSNOR TG mice showed fewer alternations than WT, with similar numbers of entry to the arms. l-Arg treatment significantly increased GSNOR TG mice alternations in the Y-maze test, whereas the same dose of l-Arg did not show any significant differences in the number of arm entries (WT, n = 20; TG, n = 23; one-way ANOVA/Turkey's post hoc test, p = 0.038, TG vehicle vs WT vehicle; p = 0.047, TG vehicle vs TG l-Arg; Fig 10B,C), indicating that l-Arg treatment could rescue the impaired working memory of GSNOR TG mice. In the contextual fear memory test, GSNOR TG mice showed less freezing than WT mice, and l-Arg treatment significantly increased the freezing percentage in the fear conditioning test in GSNOR TG mice after 24 h retention. No differences were observed in the freezing or exploration times between GSNOR TG mice and WT mice after injection of l-Arg or immediately after the foot shock (TG, n = 13; WT, n = 14; two-tailed Student's t test, p = 0.034; Fig. 10D). Upon analyzing the freezing percentage at every minute, l-Arg treatment significantly increased the freezing percentage in the fear conditioning test of GSNOR TG mice in the line chart (TG, n = 13; WT, n = 14; one-way ANOVA/Turkey's post hoc test, p = 0.0083, TG vs TG l-Arg, 5 min; Fig. 10E). Consistent with the effect on the NO level and working memory in the Y-maze test, l-Arg treatment rescued the contextual fear memory impairment of GSNOR TG mice in the fear conditioning test.
Figure 10.

l-Arg rescued GSNOR TG mouse cognitive impairment in behavior tests. A, NO level is tested by the Griess method in mouse hippocampus. l-Arg (500 mg/kg) was intraperitoneally injected 30 min earlier (WT, n = 5; TG, n = 5). B, Number of arm entries that mice explored the Y-maze. l-Arg (500 mg/kg) was intraperitoneally injected 30 min earlier. C, Percentage alternations among Y-maze arms in exploration. l-Arg (500 mg/kg) was intraperitoneally injected 30 min earlier (WT, n = 20; TG, n = 23; one-way ANOVA, p = 0.038, TG vehicle vs WT vehicle, p = 0.047, TG vehicle vs TG l-Arg). D, Percentage time of mice spent freezing at exploration and 24 h after context training in a fear-conditioning task (TG, n = 13; WT, n = 14; two-tailed Student's t test, p = 0.034). E, The freezing time of every minute in the 24 h retention test was shown. l-Arg (500 mg/kg) was intraperitoneally injected 30 min earlier (TG, n = 13; WT, n = 14; one-way ANOVA, p = 0.0083, TG vs TG l-Arg, 5 min). Data are mean ± SEM. *p < 0.05. **p < 0.01.
Discussion
In this study, our research demonstrated that GSNOR expression increased significantly in hippocampal tissue samples with the aging process. TG mice overexpressing GSNOR exclusively in neurons (GNSOR TG mice) showed cognitive defects, whereas GSNOR KO mice could rescue from the effects of age-related cognitive impairment. Further study demonstrated that overexpression of GSNOR decreased S-nitrosated CaMKIIα, CaMKIIα synaptosomal accumulation, and GLUR1 phosphorylation (S831). Downregulation of S-nitrosated CaMKIIα by GSNOR was responsible for the decreased CaMKIIα synaptosomal accumulation. Upregulation of either the NO signaling pathway could rescue the cognitive impairment in GSNOR TG mice (Fig. 11).
Figure 11.

Summarized schematic model. GSNOR expression increased in aging mouse hippocampus. TG mice overexpressing GSNOR exclusively in neurons (GNSOR TG mice) had cognitive impairment, LTP impairment, and lower dendrite spine. GSNOR KO mice rescued age-related cognitive impairment. GSNOR induced cognitive impairment by downregulated S-nitrosated CaMKIIα, inhibited CaMKIIα synaptosomal accumulation, and downregulated p(S831)-GLUR1. Supplement of the NO signaling pathway rescued GSNOR TG mouse cognitive impairment.
GSNOR is a new potential target of age-related cognitive impairment
Scientists are searching for key driver molecules in this process as specific targets to decrease age-related cognitive impairment (Peleg et al., 2010; Ménard et al., 2013; Pavlopoulos et al., 2013; Johnson et al., 2015; Sharma et al., 2015; Smith et al., 2015; Chen et al., 2016). However, at present, there is no effective clinical treatment for age-related cognitive impairment, and the molecular basis, especially SNO signaling pathway underlying this process, is still poorly understood. Based on our experimental results, we provide a new specific target, GSNOR, for age-related cognitive impairment. In other words, age-related cognitive impairment could be slowed down by using GSNOR as a direct target and screening small-molecule inhibitors of GSNOR as effective treatments. Moreover, our study demonstrated a new function of GSNOR in the brain. The functions of this enzyme in the brain have always remained unclear since the past three decades when questions relating to these enzymes were first raised (Beisswenger et al., 1985). The results of this study provide the first substantive answers to these speculations by revealing that the important physiological function of GSNOR in synaptic plasticity and cognitive function is via modulating NO homeostasis.
NO signaling pathway deficiency could be an important cause of age-related cognitive impairment
Cognitive dysfunction is most prevalent in patients with brain neurodegenerative diseases, including AD, PD, and HD. Earlier research work on NO and brain diseases mainly focused on neuronal damage and impaired cognition function possibly resulting from excessive NO. However, the links between NO and cognitive dysfunction diseases were not limited to NO-mediated neurotoxicity, and the potential neuroprotective function of NO signaling should be considered (Law et al., 2001). Our findings established that GSNOR expression increased (Fig. 1B), nNOS expression decreased, and iNOS expression was not induced in the aged mouse hippocampus (data not shown). In the aged mice, hippocampal NO bioactivity was lower than in adult mice and NO level in GSNOR TG mouse hippocampal neurons was lower than WT, regardless of GLU treatment. Next, we proved that intraperitoneal injection of the NOS substrate l-arginine could rescue GSNOR TG mouse cognitive impairment. These results suggest that the lack of NO signaling is likely the key reason for age-related cognitive impairment. Many studies support our results. For example, it was found that glutamate-induced NO concentration dynamics decrease in the hippocampus, striatum, and cerebral cortex through aging (Ledo et al., 2015) and that an enriched environment prevents age-related cognitive impairment by upregulating NO production (Arnaiz et al., 2004). Similarly, curcumin could rescue age-related cognitive impairment by increasing the nNOS expression level (Yu et al., 2013). The present results show that the lack of NO signaling mediated by GSNOR impairs cognitive function. In addition, increased GSNOR levels have been found in a Huntington's disease (HD) mouse model and in Down syndrome patient brains with dementia (Lockstone et al., 2007; Becanovic et al., 2010). In an AD pathogenesis study, NO signaling was found to be downregulated by Aβ when modulating hippocampal synaptic plasticity (Virarkar et al., 2013). The NO donor DEA/NO could rescue LTP impairment in hippocampal slices perfused with Aβ (Law et al., 2001). Moreover, NO signaling attenuation was also detected in Pb-induced neuronal damage and cognitive disruption (Nava-Ruiz et al., 2012). This result implied that GSNOR-mediated NO signaling attenuation may not be its only role in age-related cognitive impairment; rather, it also has relationships with other cognitive dysfunction diseases. Based on these experimental results, we may reconsider the potential neuroprotective function of NO signaling in cognitive dysfunction disease and slow down the age-related cognitive impairment by increasing NO signaling.
The physiological function and regulation of S-nitrosation CaMKIIα by GSNOR are shown for the first time
In 2014, it was reported that NO could activate the autonomous activity of CaMKIIα by nitrosation modifications at the cys280 and cys289 sites (Coultrap and Bayer, 2014). However, whether CaMKIIα nitrosation modification exists in vivo as well as the function of SNO-CaMKIIα were still unclear. In our research, we first found that the SNO-CaMKIIα level was downregulated in the aged mouse hippocampus. At the same time, synaptosomal accumulation of CaMKIIα was also downregulated in aged mouse hippocampus. More importantly, mutating the CaMKIIα S-nitrosation sites C280/C289 could directly inhibit its synaptosomal accumulation. However, the role of CaMKIIα S-nitrosation in LTP and cognitive function remains unclear. CaMKIIα activity is a sufficient and necessary condition for LTP formation, which plays a vital role in cognitive function (Lisman et al., 2012). CaMKII autonomous activity regulated by autophosphorylation indicates that this kinase could serve as a molecular switch that is capable of long-term memory storage (Lisman et al., 2002). In recent years, scientists have found that, in addition to phosphorylation modification, some other post-translational modifications of CaMKII, including oxidation modification and glycosylation modification, can also induce autonomous activity of CaMKII and affect its function (Erickson et al., 2008, 2013). Except for post-translational modifications, binding to the NMDAR subunit GluN2B is another way to induce autonomous activity of CaMKII (Bayer et al., 2001; Halt et al., 2012). The different mechanism for generating autonomy activity seems to play different function in LTP as well as cognitive function (Coultrap and Bayer, 2012). T286 autophosphorylation of CaMKII play an important role in mediating LTP induction and memory formation but not in LTP maintenance and memory storage (Giese et al., 1998; Buard et al., 2010; Coultrap and Bayer, 2012). Meanwhile, the activity-driven interaction of CaMKII with the NMDAR is important for the storage of synaptic information and memory consolidation (Coultrap and Bayer, 2012; Halt et al., 2012; Barcomb et al., 2016). Our work revealed that SNO-CaMKIIα was responsible for synaptosomal accumulation of CaMKIIα (Fig 9) similar to the function of interaction of CaMKII with the NMDAR. However, our experiments are under the basal condition, but activity-driven postsynaptic accumulation of CaMKII was impaired in GluN2B KI mice (Halt et al., 2012). To further study the physiological function of SNO-CaMKIIα, we are constructing the CaMKIIα mutant mice of C280/C289V nitrosation modification sites. The function of SNO-CaMKIIα in contributing to LTP or cognitive function will be further elucidated in the future.
GSNOR is the key regulator of protein SNO modification levels in vivo. Because there may be a risk of neurotoxicity or other tissue NO toxicity by direct use of an NO donor or nNOS activation agent, effective and specific nitrosation proteins may be better targets for clinical treatment (Nakamura and Lipton, 2016). Our experiments showed that SNO-CaMKIIα is likely a new target of GSNOR in age-related cognitive impairment. However, the possibility cannot be excluded that GSNOR overexpression impaired cognitive function in aging and reduced surface expression of AMPA receptors by other SNO target pathways. Nevertheless, we provided a new strategy to slow down age-related cognitive impairment by screening small specific molecule activators of SNO protein targets.
In conclusion, our research demonstrates that GSNOR is a new potential target for the treatment of age-related cognitive impairment. An NO signaling deficiency may be the main cause of age-related cognitive impairment. In addition, we first proved the regulatory mechanisms of S-nitrosation CaMKIIα by GSNOR. These results provide a new potential target and strategy for slowing age-related cognitive impairment.
Footnotes
Author contributions: Y.Z., W.S., Y.-G.Y., and C.C. designed research; Y.Z., K.W., W.S., P.W., X.Q., Q.Y., and Z.Y. performed research; C.Z. and L.L. contributed unpublished reagents/analytic tools; Y.Z., K.W., W.S., D.-F.Z., P.W., X.Q., Z.Y., Y.-G.Y., G.L., and C.Z. analyzed data; Y.Z., D.-F.Z., and C.C. wrote the paper.
This work was supported by National Key R&D Program of China Grants 2017YFA0504000 and 2016YFC0903100 to C.C., 2015CB964800 to G.L., National Natural Sciences Foundation of China Grants 31570857 and 31225012 to C.C., Personalized Medicines-Molecular Signature-based Drug Discovery and Development, and Chinese Academy of Sciences Strategic Priority Research Program Grant XDA12020316 to C.C. We thank Dr. Pico Caroni for providing the Thy1.2 promoter for GSNOR TG mice; Jifeng Wang and Peng Xue (Institute of Biophysics Core Facility) for assistance with the Q Exactive mass spectrometer analyses; and Professor Zhixin Wang and Professor Jiawei Wu (Tsinghua University) for valuable discussion about CaMKIIα activity regulation.
The authors declare no competing financial interests.
References
- Amtul Z, Atta-Ur-Rahman (2015) Neural plasticity and memory: molecular mechanism. Rev Neurosci 26:253–268. 10.1515/revneuro-2014-0075 [DOI] [PubMed] [Google Scholar]
- Arendash GW, Gordon MN, Diamond DM, Austin LA, Hatcher JM, Jantzen P, DiCarlo G, Wilcock D, Morgan D (2001) Behavioral assessment of Alzheimer's transgenic mice following long-term Abeta vaccination: task specificity and correlations between Abeta deposition and spatial memory. DNA Cell Biol 20:737–744. 10.1089/10445490152717604 [DOI] [PubMed] [Google Scholar]
- Arnaiz SL, D'Amico G, Paglia N, Arismendi M, Basso N, del Rosario Lores Arnaiz M (2004) Enriched environment, nitric oxide production and synaptic plasticity prevent the aging-dependent impairment of spatial cognition. Mol Aspects Med 25:91–101. 10.1016/j.mam.2004.02.011 [DOI] [PubMed] [Google Scholar]
- Barcomb K, Hell JW, Benke TA, Bayer KU (2016) The CaMKII/GluN2B protein interaction maintains synaptic strength. J Biol Chem 291:16082–16089. 10.1074/jbc.M116.734822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411:801–805. 10.1038/35081080 [DOI] [PubMed] [Google Scholar]
- Becanovic K, Pouladi MA, Lim RS, Kuhn A, Pavlidis P, Luthi-Carter R, Hayden MR, Leavitt BR (2010) Transcriptional changes in Huntington disease identified using genome-wide expression profiling and cross-platform analysis. Hum Mol Genet 19:1438–1452. 10.1093/hmg/ddq018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisswenger TB, Holmquist B, Vallee BL (1985) Chi-Adh is the sole alcohol-dehydrogenase isozyme of mammalian brains: implications and inferences. Proc Natl Acad Sci U S A 82:8369–8373. 10.1073/pnas.82.24.8369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhar M, Forrester MT, Stamler JS (2009) Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol 10:721–732. 10.1038/nrm2764 [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39. 10.1038/361031a0 [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T (1973) Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 232:331–356. 10.1113/jphysiol.1973.sp010273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M, Hayashi Y (2012) Structural plasticity of dendritic spines. Curr Opin Neurobiol 22:383–388. 10.1016/j.conb.2011.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buard I, Coultrap SJ, Freund RK, Lee YS, Dell'Acqua ML, Silva AJ, Bayer KU (2010) CaMKII “autonomy” is required for initiating but not for maintaining neuronal long-term information storage. J Neurosci 30:8214–8220. 10.1523/JNEUROSCI.1469-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P. (1997) Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods 71:3–9. 10.1016/S0165-0270(96)00121-5 [DOI] [PubMed] [Google Scholar]
- Chen C, Meng SQ, Xue YX, Han Y, Sun CY, Deng JH, Chen N, Bao YP, Zhang FL, Cao LL, Zhu WG, Shi J, Song WH, Lu L (2016) Epigenetic modification of PKMzeta rescues aging-related cognitive impairment. Sci Rep 6:22096. 10.1038/srep22096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, Kleinman JE (2011) Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478:519–523. 10.1038/nature10524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT (2004) Receptor trafficking and synaptic plasticity. Nat Rev Neurosci 5:952–962. 10.1038/nrn1556 [DOI] [PubMed] [Google Scholar]
- Conrad CD, Grote KA, Hobbs RJ, Ferayorni A (2003) Sex differences in spatial and non-spatial Y-maze performance after chronic stress. Neurobiol Learn Mem 79:32–40. 10.1016/S1074-7427(02)00018-7 [DOI] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU (2012) CaMKII regulation in information processing and storage. Trends Neurosci 35:607–618. 10.1016/j.tins.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coultrap SJ, Bayer KU (2014) Nitric oxide induces Ca2+-independent activity of the Ca2+/calmodulin-dependent protein kinase II (CaMKII). J Biol Chem 289:19458–19465. 10.1074/jbc.M114.558254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AG, Saunders DC, Kelsey PB Jr, Conway AA, Tesmenitsky Y, Marchini JF, Brown KK, Stamler JS, Colagiovanni DB, Rosenthal GJ, Croce KJ, North TE, Goessling W (2014) S-Nitrosothiol signaling regulates liver development and improves outcome following toxic liver injury. Cell Rep 6:56–69. 10.1016/j.celrep.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellu F, Contarino A, Simon H, Koob GF, Gold LH (2000) Genetic differences in response to novelty and spatial memory using a two-trial recognition task in mice. Neurobiol Learn Mem 73:31–48. 10.1006/nlme.1999.3919 [DOI] [PubMed] [Google Scholar]
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE, Aykin-Burns N, Zimmerman MC, Zimmerman K, Ham AJ, Weiss RM, Spitz DR, Shea MA, Colbran RJ, Mohler PJ, Anderson ME (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133:462–474. 10.1016/j.cell.2008.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM (2013) Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502:372–376. 10.1038/nature12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MW, Liu L, Zeng M, Hess DT, Stamler JS (2009) A genetic analysis of nitrosative stress. Biochemistry 48:792–799. 10.1021/bi801813n [DOI] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ (1998) Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279:870–873. 10.1126/science.279.5352.870 [DOI] [PubMed] [Google Scholar]
- Gomes SA, Rangel EB, Premer C, Dulce RA, Cao Y, Florea V, Balkan W, Rodrigues CO, Schally AV, Hare JM (2013) S-nitrosoglutathione reductase (GSNOR) enhances vasculogenesis by mesenchymal stem cells. Proc Natl Acad Sci U S A 110:2834–2839. 10.1073/pnas.1220185110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW (2012) CaMKII binding to GluN2B is critical during memory consolidation. EMBO J 31:1203–1216. 10.1038/emboj.2011.482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedberg JJ, Griffiths WJ, Nilsson SJ, Höög JO (2003) Reduction of S-nitrosoglutathione by human alcohol dehydrogenase 3 is an irreversible reaction as analysed by electrospray mass spectrometry. Eur J Biochem 270:1249–1256. 10.1046/j.1432-1033.2003.03486.x [DOI] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS (2005) Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6:150–166. 10.1038/nrm1569 [DOI] [PubMed] [Google Scholar]
- Hou Q, Jiang H, Zhang X, Guo C, Huang B, Wang P, Wang T, Wu K, Li J, Gong Z, Du L, Liu Y, Liu L, Chen C (2011) Nitric oxide metabolism controlled by formaldehyde dehydrogenase (fdh, homolog of mammalian GSNOR) plays a crucial role in visual pattern memory in Drosophila. Nitric Oxide 24:17–24. 10.1016/j.niox.2010.09.007 [DOI] [PubMed] [Google Scholar]
- Huang B, Chen C (2010) Detection of protein S-nitrosation using irreversible biotinylation procedures (IBP). Free Radic Biol Med 49:447–456. 10.1016/j.freeradbiomed.2010.05.001 [DOI] [PubMed] [Google Scholar]
- Hung CW, Chen YC, Hsieh WL, Chiou SH, Kao CL (2010) Ageing and neurodegenerative diseases. Ageing Res Rev 9:S36–S46. 10.1016/j.arr.2010.08.006 [DOI] [PubMed] [Google Scholar]
- Johnson JL, Huang W, Roman G, Costa-Mattioli M (2015) TORC2: a novel target for treating age-associated memory impairment. Sci Rep 5:15193. 10.1038/srep15193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung WR, Kim HG, Kim KL (2008) Ganglioside GQ1b improves spatial learning and memory of rats as measured by the Y-maze and the Morris water maze tests. Neurosci Lett 439:220–225. 10.1016/j.neulet.2008.05.020 [DOI] [PubMed] [Google Scholar]
- Kandel ER, Dudai Y, Mayford MR (2014) The molecular and systems biology of memory. Cell 157:163–186. 10.1016/j.cell.2014.03.001 [DOI] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G, Guennel T, Shin Y, Johnson MB, Krsnik Z, Mayer S, Fertuzinhos S, Umlauf S, Lisgo SN, Vortmeyer A, Weinberger DR, et al. (2011) Spatio-temporal transcriptome of the human brain. Nature 478:483–489. 10.1038/nature10523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61:340–350. 10.1016/j.neuron.2009.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konar A, Singh P, Thakur MK (2016) Age-associated cognitive decline: insights into molecular switches and recovery avenues. Aging Dis 7:121–129. 10.14336/AD.2015.1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law A, Gauthier S, Quirion R (2001) Say NO to Alzheimer's disease: the putative links between nitric oxide and dementia of the Alzheimer's type. Brain Res Rev 35:73–96. 10.1016/S0165-0173(00)00051-5 [DOI] [PubMed] [Google Scholar]
- Ledo A, Lourenço CF, Caetano M, Barbosa RM, Laranjinha J (2015) Age-associated changes of nitric oxide concentration dynamics in the central nervous system of Fisher 344 rats. Cell Mol Neurobiol 35:33–44. 10.1007/s10571-014-0115-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima B, Forrester MT, Hess DT, Stamler JS (2010) S-Nitrosylation in cardiovascular signaling. Circ Res 106:633–646. 10.1161/CIRCRESAHA.109.207381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Koizumi A, Tanaka N, Panda S, Masland RH (2008) Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc Natl Acad Sci U S A 105:16009–16014. 10.1073/pnas.0806114105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Singel DJ, Stamler JS (1994) Nitric oxide in the central nervous system. Prog Brain Res 103:359–364. 10.1016/S0079-6123(08)61149-8 [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3:175–190. 10.1038/nrn753 [DOI] [PubMed] [Google Scholar]
- Lisman J, Yasuda R, Raghavachari S (2012) Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci 13:169–182. 10.1038/nrn3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS (2001) A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 410:490–494. 10.1038/35068596 [DOI] [PubMed] [Google Scholar]
- Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS (2004) Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116:617–628. 10.1016/S0092-8674(04)00131-X [DOI] [PubMed] [Google Scholar]
- Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S (2007) Gene expression profiling in the adult Down syndrome brain. Genomics 90:647–660. 10.1016/j.ygeno.2007.08.005 [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD (1999) Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci 19:10250–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA. (2004) Long-term potentiation and memory. Physiol Rev 84:87–136. 10.1152/physrev.00014.2003 [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. 10.1038/nature02617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ménard C, Tse YC, Cavanagh C, Chabot JG, Herzog H, Schwarzer C, Wong TP, Quirion R (2013) Knockdown of prodynorphin gene prevents cognitive decline, reduces anxiety, and rescues loss of group 1 metabotropic glutamate receptor function in aging. J Neurosci 33:12792–12804. 10.1523/JNEUROSCI.0290-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora F. (2013) Successful brain aging: plasticity, environmental enrichment, and lifestyle. Dialogues Clin Neurosci 15:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Baxter MG (2012) The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci 13:240–250. 10.1038/nrn3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA (2016) Protein S-nitrosylation as a therapeutic target for neurodegenerative diseases. Trends Pharmacol Sci 37:73–84. 10.1016/j.tips.2015.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava-Ruiz C, Méndez-Armenta M, Ríos C (2012) Lead neurotoxicity: effects on brain nitric oxide synthase. J Mol Histol 43:553–563. 10.1007/s10735-012-9414-2 [DOI] [PubMed] [Google Scholar]
- Pavlopoulos E, Jones S, Kosmidis S, Close M, Kim C, Kovalerchik O, Small SA, Kandel ER (2013) Molecular mechanism for age-related memory loss: the histone-binding protein RbAp48. Sci Transl Med 5:200ra115. 10.1126/scitranslmed.3006373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S, Agis-Balboa RC, Cota P, Wittnam JL, Gogol-Doering A, Opitz L, Salinas-Riester G, Dettenhofer M, Kang H, Farinelli L, Chen W, Fischer A (2010) Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328:753–756. 10.1126/science.1186088 [DOI] [PubMed] [Google Scholar]
- Pletikos M, Sousa AM, Sedmak G, Meyer KA, Zhu Y, Cheng F, Li M, Kawasawa YI, Sestan N (2014) Temporal specification and bilaterality of human neocortical topographic gene expression. Neuron 81:321–332. 10.1016/j.neuron.2013.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeggel G, Helmeke C, Abraham A, Schwabe T, Friedrich P, Braun K (2003) Juvenile emotional experience alters synaptic composition in the rodent cortex, hippocampus, and lateral amygdala. Proc Natl Acad Sci U S A 100:16137–16142. 10.1073/pnas.2434663100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que LG, Yang Z, Stamler JS, Lugogo NL, Kraft M (2009) S-Nitrosoglutathione reductase: an important regulator in human asthma. Am J Respir Crit Care Med 180:226–231. 10.1164/rccm.200901-0158OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezayof A, Amini R, Rassouli Y, Zarrindast MR (2006) Influence of nitric oxide on morphine-induced amnesia and interactions with dopaminergic receptor agents. Physiol Behav 88:124–131. 10.1016/j.physbeh.2006.03.017 [DOI] [PubMed] [Google Scholar]
- Sanghani PC, Davis WI, Fears SL, Green SL, Zhai L, Tang Y, Martin E, Bryan NS, Sanghani SP (2009) Kinetic and cellular characterization of novel inhibitors of S-nitrosoglutathione reductase. J Biol Chem 284:24354–24362. 10.1074/jbc.M109.019919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Shetty MS, Arumugam TV, Sajikumar S (2015) Histone deacetylase 3 inhibition re-establishes synaptic tagging and capture in aging through the activation of nuclear factor kappa B. Sci Rep 5:16616. 10.1038/srep16616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimono K, Baudry M, Ho L, Taketani M, Lynch G (2002) Long-term recording of LTP in cultured hippocampal slices. Neural Plast 9:249–254. 10.1155/NP.2002.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LK, He Y, Park JS, Bieri G, Snethlage CE, Lin K, Gontier G, Wabl R, Plambeck KE, Udeochu J, Wheatley EG, Bouchard J, Eggel A, Narasimha R, Grant JL, Luo J, Wyss-Coray T, Villeda SA (2015) β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat Med 21:932–937. 10.1038/nm.3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CH, Wei W, Hanes MA, Liu L (2013) Hepatocarcinogenesis driven by GSNOR deficiency is prevented by iNOS inhibition. Cancer Res 73:2897–2904. 10.1158/0008-5472.CAN-12-3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G (2014) Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol 1183:221–242. 10.1007/978-1-4939-1096-0_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S (1996) The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87:1327–1338. 10.1016/S0092-8674(00)81827-9 [DOI] [PubMed] [Google Scholar]
- Vidal M, Morris R, Grosveld F, Spanopoulou E (1990) Tissue-specific control elements of the Thy-1 gene. EMBO J 9:833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virarkar M, Alappat L, Bradford PG, Awad AB (2013) l-Arginine and nitric oxide in CNS function and neurodegenerative diseases. Crit Rev Food Sci Nutr 53:1157–1167. 10.1080/10408398.2011.573885 [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858. 10.1038/nprot.2006.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KY, Zhang YY, Su WT, Chen C (2013) GSNOR: a novel regulator of inflammation. Prog Biochem Biophys 40:731–738. [Google Scholar]
- Wu K, Ren R, Su W, Wen B, Zhang Y, Yi F, Qiao X, Yuan T, Wang J, Liu L, Izpisua Belmonte JC, Liu GH, Chen C (2014) A novel suppressive effect of alcohol dehydrogenase 5 in neuronal differentiation. J Biol Chem 289:20193–20199. 10.1074/jbc.C114.561860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Takei Y, Kido MA, Hirokawa N (2011) Molecular motor KIF17 is fundamental for memory and learning via differential support of synaptic NR2A/2B levels. Neuron 70:310–325. 10.1016/j.neuron.2011.02.049 [DOI] [PubMed] [Google Scholar]
- Yu SY, Zhang M, Luo J, Zhang L, Shao Y, Li G (2013) Curcumin ameliorates memory deficits via neuronal nitric oxide synthase in aged mice. Prog Neuropsychopharmacol Biol Psychiatry 45:47–53. 10.1016/j.pnpbp.2013.05.001 [DOI] [PubMed] [Google Scholar]