

Abstract
The synaptosomal-associated protein SNAP25 is a key player in synaptic vesicle docking and fusion and has been associated with multiple psychiatric conditions, including schizophrenia, bipolar disorder, and attention-deficit/hyperactivity disorder. We recently identified a promoter variant in SNAP25, rs6039769, that is associated with early-onset bipolar disorder and a higher gene expression level in human prefrontal cortex. In the current study, we showed that this variant was associated both in males and females with schizophrenia in two independent cohorts. We then combined in vitro and in vivo approaches in humans to understand the functional impact of the at-risk allele. Thus, we showed in vitro that the rs6039769 C allele was sufficient to increase the SNAP25 transcription level. In a postmortem expression analysis of 33 individuals affected with schizophrenia and 30 unaffected control subjects, we showed that the SNAP25b/SNAP25a ratio was increased in schizophrenic patients carrying the rs6039769 at-risk allele. Last, using genetics imaging in a cohort of 71 subjects, we showed that male risk carriers had an increased amygdala–ventromedial prefrontal cortex functional connectivity and a larger amygdala than non-risk carriers. The latter association has been replicated in an independent cohort of 121 independent subjects. Altogether, results from these multilevel functional studies are bringing strong evidence for the functional consequences of this allelic variation of SNAP25 on modulating the development and plasticity of the prefrontal–limbic network, which therefore may increase the vulnerability to both early-onset bipolar disorder and schizophrenia.
SIGNIFICANCE STATEMENT Functional characterization of disease-associated variants is a key challenge in understanding neuropsychiatric disorders and will open an avenue in the development of personalized treatments. Recent studies have accumulated evidence that the SNARE complex, and more specifically the SNAP25 protein, may be involved in psychiatric disorders. Here, our multilevel functional studies are bringing strong evidence for the functional consequences of an allelic variation of SNAP25 on modulating the development and plasticity of the prefrontal–limbic network. These results demonstrate a common genetically driven functional alteration of a synaptic mechanism both in schizophrenia and early-onset bipolar disorder and confirm the shared genetic vulnerability between these two disorders.
Keywords: bipolar disorder, brain imaging, genetics, schizophrenia, SNAP25, SNARE
Introduction
Bipolar disorder (BD) and schizophrenia (SZ) share many risk factors, cognitive features, neuroanatomical characteristics, clinical symptomatology, and medication. Shared risk factors include genetic risk factors with shared heritability and some common single nucleotide polymorphisms (SNPs), increasing the risk for both conditions (Lichtenstein et al., 2009; Purcell et al., 2009). Recent genome-wide association studies (GWASs) and whole exome sequence analyses conducted in SZ and BD revealed enrichment in genes encoding molecules involved in synaptic neurotransmission (Psychiatric GWAS Consortium Bipolar Disorder Working Group, 2011; Purcell et al., 2014; Schizophrenia Working Group of the Psychiatric Genomics, 2014). One of the key steps of neurotransmission is the Ca2+-dependent assembly of the synaptosomal-associated protein SNAP25 with synaptobrevin and syntaxin to form the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Multiple SNPs located in SNAP25 have already been associated with SZ (Carroll et al., 2009; Wang et al., 2015) and other psychiatric disorders (Barr et al., 2000; Terracciano et al., 2010; Braida et al., 2015). In addition, several studies have reported alteration of the SNAP25 protein level in the brains of patients with either SZ or BD (Thompson et al., 1998, 2003a; Fatemi et al., 2001; Honer et al., 2002; Scarr et al., 2006; Gray et al., 2010). In particular, alterations at the hippocampal level have been reported in both conditions (Young et al., 1998; Fatemi et al., 2001; Thompson et al., 2003a; Scherk et al., 2008).
At a neural level, common alterations in SZ and BD are mainly frontolimbic (d'Albis and Houenou, 2015). Hippocampal volumes are generally described as decreased in both conditions (Hibar et al., 2016; van Erp et al., 2016), while findings are more heterogeneous for amygdala, with an age-dependent directionality (Hajek et al., 2009; Welch et al., 2010; Ganzola et al., 2014). Prefrontal–limbic connectivity has also repeatedly been reported to be altered in both conditions, probably leading to emotional dysregulation in both conditions (Hart et al., 2013; Cannon, 2015; d'Albis and Houenou, 2015).
We recently identified one polymorphism located in the promoter region of SNAP25 that was associated with early-onset BD (EOBD) as well as with a higher expression level in the prefrontal cortex of subjects carrying the at-risk allele (Etain et al., 2010). Among the various forms of BD, EOBD is probably a more homogeneous subtype, more severe and more frequently associated with psychotic features, genetic heritability, and neurodevelopmental abnormalities than forms with a later age of onset. Thus, EOBD may be a neurodevelopmental subtype of BD, closer to SZ than BD. Given this, we hypothesized that the rs6039769 SNAP25 at-risk variant for EOBD may be associated with an increased risk of SZ. However, one usual limitation of genetic association studies is the absence of information on the plausibility and nature of the relation between a genotype and a phenotype. Functional studies are thus necessary to bring support and shed light on such links. We thus aimed at understanding the functional impact of this DNA at-risk variant combining multiple approaches (i.e., gene expression analyses both in vitro and in brain postmortem tissues as well as in vivo using a clinical human genetics imaging approach), demonstrating that this at-risk variant is associated with common neuroanatomical features of EOBD and SZ, namely alterations in the prefrontal limbic networks.
Materials and Methods
Subjects
As false-positive results are a recurrent concern in both the genetics and neuroimaging fields (Meyer-Lindenberg et al., 2008), we used the following two independent samples: a “discovery” sample and a “replication” sample for both genetic analyses and neuroimaging genetic analyses for replication.
Genetic analyses
Discovery sample.
Four hundred sixty-one patients (67.9% males) meeting the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) criteria (American Psychiatric Association, 2000) for SZ and 315 unaffected control subjects (54.6% males) have been recruited as described previously (Dumaine et al., 2011; Schürhoff et al., 2015). All subjects were interviewed by trained investigators (psychiatrists or psychologists) in 10 Expert Centres for SZ of the Fondation FondaMental (https://www.fondation-fondamental.org), using the diagnostic interview for genetic studies (DIGS version 3), yielding DSM-IV diagnoses (Nurnberger et al., 1994).
Replication sample.
The latest Psychiatric Genomics Consortium GWAS summary statistics for rs6039769 have been downloaded from the Broad Institute web site (https://data.broadinstitute.org/mpg/ricopili/). To match with the geographical origin of our discovery sample, only data from the 49 Caucasian populations, including 34,241 patients with SZ and 45,604 control subjects, have been considered for the genetic analysis (Schizophrenia Working Group of the Psychiatric Genomics, 2014).
Neuroimaging genetics analyses
Discovery sample.
Twenty-five adult inpatients and outpatients with BD type I (by DSM-IV-R criteria) were recruited from two university-affiliated participating centers (AP-HP Henri Mondor and Lariboisière Hospitals, Paris, France; mean age, 38.9 years; SD, 12.1; 10 females); 46 control subjects were recruited from media announcements and registry offices, and had no personal or family history of Axis I mood disorder, SZ, or schizoaffective disorder (mean age, 38.7 years; SD, 11.4; age comparison with patients: t(69) = 0.07, p = 0.93; 27 females χ2 = 2.27, p = 0.13). All participants underwent clinical assessment by trained raters using the DIGS. Exclusion criteria for all participants consisted of a history of neurological disease or head trauma with loss of consciousness and contraindications for magnetic resonance imaging (MRI).
Replication sample.
An independent sample of 121 healthy control subjects (mean age, 29.7 years; SD. 10.8; 65 women) has been recruited at the Central Institute of Mental Health in Mannheim, Germany. A comparison with the discovery sample yielded a significant difference in mean age (the replication sample comprising younger subjects: t(190) = 5.46, p = 1.5 × 10−7).
Local ethics committees (CPP Ile de France IX and the ethics committee at the University of Heidelberg, Heidelberg, Germany) approved the protocols and procedures, and written informed consent was obtained from all subjects before study participation.
Genetic association
Genomic DNA has been extracted from blood lymphocyte, B-lymphoblastoid cell line, or from saliva for each subject. We performed the rs6039769 genotyping using TaqMan SNP genotyping assays (C_29497348_10) on a 7900 Real-Time PCR System (Applied Biosystems).
Functional in vitro study
The Neuro-2a (N2a) mouse neuroblastoma cell line (catalog #400394/p451_Neuro-2A, CLS; RRID:CVCL_0470) was grown at 37°C, with 5% CO2 in DMEM containing GlutaMAX (Thermo Fisher Scientific) and was supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The plasmid containing the 784 bp upstream of the SNAP25 transcription start site has been cloned in the pGL3-Basic vector (Promega) upstream of the luciferase reporter gene as described previously (Cai et al., 2008) and was provided by Professor Weihong Song (The University of British Columbia, Vancouver, Canada). The rs6039769 polymorphism was generated by in vitro mutagenesis using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). N2a cells were seeded onto 24-well plates 1 d before transfection and were transfected with 50 ng of plasmid DNA per well using 1 μl of Lipofectamine 2000 (Thermo Fisher Scientific) and 1 ng of Renilla luciferase vector to normalize the transfection efficiency. Cells were harvested at 24 h after transfection and lysed with 100 μl of passive lysis buffer (Promega) per well. Luciferase activities were measured sequentially using the Dual-Luciferase Reporter Assay System (Promega).
SNAP25 postmortem expression study
RNA, cDNA, and DNA from 33 individuals affected with SZ (25 males and 8 females) and 30 unaffected control subjects (23 males and 7 females) were donated by the Stanley Medical Research Institute (Chevy Chase, MD), as part of the Array Collection that consisted of samples from the dorsolateral prefrontal cortex (Brodmann's area 46; Torrey et al., 2000). Diagnoses were made according to the DSM-IV. The samples were coded, and genotypes and disease status were known only after expression analyses. Expression levels of the two isoforms of SNAP25 were determined using TaqMan gene expression assays as described previously (Etain et al., 2010). Normalization was performed using an endogenous housekeeping gene encoding the human β-actin (ACTB) with limited primers (Applied Biosystems). As the ratio between the two isoforms of SNAP25 has been shown to play a critical role in the synaptic maturation at puberty (Bark et al., 2004), a key step for SZ onset, we compared the ratio between SNAP25b and SNAP25a depending on the genotype.
Experimental design and statistical analysis
Statistical testing for allelic and genotypic associations was performed using the PLINK version 1.07 software (RRID:SCR_001757; http://zzz.bwh.harvard.edu/plink/). We performed statistical analyses of expression using the statistical software package R version 2.9.2 (RRID:SCR_003005; http://cran.r-project.org). Groupwise comparisons included parametric tests (Student's t test/ANOVA) and nonparametric tests (Mann–Whitney U test/Kruskal–Wallis test), according to the normality of distribution, and were tested using the Shapiro–Wilk method. A potential correlation between SNAP25a and SNAP25b expression levels and age, postmortem interval, refrigerator interval, and brain pH has been tested using Spearman's rank correlation test in case patients and control subjects.
Neuroimaging genetics study
Structural and resting-state fMRI (rs-fMRI) scans were collected at Neurospin (Commissariat à l'Énergie Atomique, Saclay, France) for the discovery sample, using a 3 T MRI (12-channel head coil; Tim Trio, Siemens) with the following acquisition parameters: 3DT1 sequence: TE = 2.98 ms; TR = 2.3 s; 160 sections; voxel size = 1 × 1 × 1.1 mm; rs-fMRI: echoplanar imaging; voxel size = 3 × 3 × 3 mm; 35 slices; TR = 2000 ms; 360 time points (12 min). For rs-fMRI, subjects were instructed to close their eyes and try not to think about anything specific without sleeping.
Structural scans were acquired at the Mannheim Central Institute for Mental Health (Mannheim, Germany) for the replication sample, using the same 3 T MRI as in Paris (12-channel head coil; Tim Trio, Siemens) with the same acquisition parameters.
Automated volumetry, as implemented in FIRST 1.2, part of FSL 4.1 software (Patenaude et al., 2011), was applied on T1 images to segment the hippocampi and amygdala. Registrations and segmentations were visually inspected as recommended by the authors of the software. The volumes of hippocampi and amygdalae were measured on segmented images. As our results with the discovery sample concerned only the amygdala, we took advantage of the larger replication sample to test for lateralized associations of the amygdala volumes with the genotype.
The rs-fMRI images were preprocessed using Data Processing Assistant for Resting-State fMRI (DPARSF) toolbox (RRID:SCR_002372; http://www.restfmri.net) with Statistical Parametric Mapping (SPM8; RRID:SCR_007037; http://www.fil.ion.ucl.ac.uk/spm/). Briefly, the 10 first volumes were discarded for the stabilization of the signal. Functional volumes were corrected for slice timing acquisition and head motion (>2 mm), then were realigned on T1 images and segmented into six tissue types (white matter, gray matter, CSF, bone, and air). We then spatially normalized these images to the Montreal Neurological Institute space using Dartel and smoothed them by a 4 mm full-width at half-maximum. We performed a linear trend using a temporal bandpass filter (0.001–0.08 Hz). Finally, we used signal in gray matter, CSF, and head motion parameters (Friston et al., 1996) as regressors of nuisance.
For the analysis of frontolimbic functional connectivity, we used a seed-based analysis approach. We defined three bilateral regions of interest (ROIs) using the Wake Forest University PickAtlas software (RRID:SCR_007378; http://www.fmri.wfubmc.edu/download.htm): amygdala, hippocampus, and the ventromedial prefrontal cortex (VMPFC), defined as the addition of Brodmann's areas 11, 25, and 47 (Ladouceur et al., 2011; Fig. 1). Using the REST toolbox (http://www.restfmri.net) for SPM8, we computed correlation coefficients between the times series for four pairs of ROIs (bilaterally amygdala/VMPFC; bilaterally hippocampus/VMPFC).
Figure 1.

Illustration of the amygdala and VMPFC on an axial view of a single subject.
To explore the effect of rs6039769, we conducted multiple linear regressions using age, sex, diagnosis, and lateralization as covariates, with hippocampal volumes (left + right/2), amygdala volumes, and correlation coefficients as dependent variables in independent analyses using R 2.14.1. The rs6039769 risk variant was modeled in a model comparing AA and AC genotype versus CC as the AA group was relatively small. For analyses of volumes, we used the total intracranial volume as an additional covariate. Total intracranial volumes were computed as the sum of gray matter, white matter, and CSF volumes obtained after segmentation of the T1 images with the “new segment” procedure implemented in SPM8 software. We checked for the normality of residuals using Q–Q plots. For significant results, we additionally checked for an interaction between genotype and gender, as such interactions have previously been described on emotional neuroimaging variables; when significant, we performed post hoc stratified analyses by gender. We also tested for an interaction between genotype and status (control/BD) and could not find any interaction.
Results
The SNAP25 promoter variant rs6039769 is associated with SZ
We genotyped rs6039769 in 461 patients with SZ and 315 unaffected control subjects. Both populations were in Hardy–Weinberg equilibrium (patients: χSZ2 = 1.81, 2 df, pSZ = 0.18; control subjects: χctrl2 = 0.83, 2 df, pctrl = 0.36). We showed a significant increase in the C allele frequency in patients compared with control subjects [χ2 = 5.33; 1 df; p = 0.02; odds ratio (OR) = 1.29; 95% CI = 1.04–1.60; Table 1). The genotypic association study also showed a significant difference in genotypic distribution between patients and control subjects (χ2 = 8.03, 2 df, p = 0.02). The most significant difference was observed for CC genotypes under a recessive model (χ2 = 7.76, 1 df, p = 0.006). As more males were present in the patient group than in the control subject group (χ2 = 14.11, 1 df, p = 0.0002), we checked for gender-specific difference in genotype frequencies. However, no difference was observed between males and females, either in patients (χ2 = 0.19, 1 df, p = 0.91) or in control subjects (χ2 = 0.76, 1 df, p = 0.68).
Table 1.
rs6039769 genotype and allele frequency in the population of patients with SZ and unaffected control subjects
| rs6039769 | AA | AC | CC | χ2 (p value) | A | C | χ2 (p value) | OR (95% CI) |
|---|---|---|---|---|---|---|---|---|
| SZ | 37 | 208 | 216 | 8.03 (0.02) | 282 | 640 | 5.33 (0.02) | 1.29 (1.04–1.60) |
| Unaffected control subjects | 45 | 138 | 132 | 228 | 402 |
As a replication sample, we used results from the latest Psychiatric Genomics Consortium GWAS on schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics, 2014). Similar to what we observed in the discovery sample, a significant increase in allele frequency was observed for the rs6039769 C at-risk allele in patients with SZ when compared with control subjects (χ2 = 5.03; 1 df; p = 0.02; OR = 1.03; 95% CI = 1.01–1.04).
The rs6039769 C allele is sufficient to increase the SNAP25 transcription level in vitro
We previously showed that rs6039769 was associated with an increased expression level of SNAP25 in prefrontal cortex (Etain et al., 2010). To determine whether this effect was due to this rs6039769 or another SNP in linkage disequilibrium, we cloned 784 bp of the SNAP25 promoter region upstream to the luciferase reporter gene in the pGL3-basic vector. Plasmids with either the C or the A alleles were thus transfected in mouse neuroblastoma cells. Both plasmids showed a significant increase in the expression level when compared with the empty vector (Fig. 2). The presence of the at-risk C allele resulted in a significant increase of the luciferase activity of almost 20% (Student's t test, t(40) = 4.76, p = 7.7 × 10−5).
Figure 2.

In vitro analysis of rs6039769 in N2a cells. Quantification of the SNAP25 expression level was estimated using a luciferase reporter gene under control of the human SNAP25 promoter region containing either the C or the A allele of rs6039769. Both constructs showed a higher reporter expression level than the empty vector. The C allele showed a 20% increase of the reporter expression level than the A allele. The values are reported as the mean ± SD. ***p < 0.0001.
The SNAP25b/SNAP25a ratio is increased in schizophrenic patients carrying the rs6039769 C at-risk allele
Using postmortem studies, the expression level of SNAP25 has been previously reported to be modified in patients with SZ (Thompson et al., 1998, 2003a; Fatemi et al., 2001; Honer et al., 2002; Scarr et al., 2006; Gray et al., 2010). To determine whether such modification in brains of patients was dependent on rs6039769, we analyzed the transcription level of SNAP25 using quantitative real-time PCR on postmortem prefrontal cortex from patients with SZ and control subjects. Two isoforms exist for SNAP25 (SNAP25a and SNAP25b), which differ only by 9 aa (Bark et al., 1995), but have different function and expression pattern (Sørensen et al., 2003; Bark et al., 2004; Yamamori et al., 2011). We thus analyzed both isoforms independently as well as their combination. No significant difference was observed according to the rs6039769 genotype for SNAP25a or for SNAP25b, either in patients with SZ or in control subjects (Fig. 3A–D). While no difference in the SNAP25b/SNAP25a ratio was observed according to rs6039769 genotype in control subjects (Kruskal–Wallis test, K = 0.59, p = 0.75; Fig. 3E), we showed a significant increase of this ratio when patients had the at-risk allele C (Kruskal–Wallis test, K = 6.23, p = 0.04; Fig. 3F). We checked that no significant interaction was found between the SNAP25b:SNAP25a ratio and postmortem interval, refrigerator interval, brain pH, and gender (data not shown).
Figure 3.
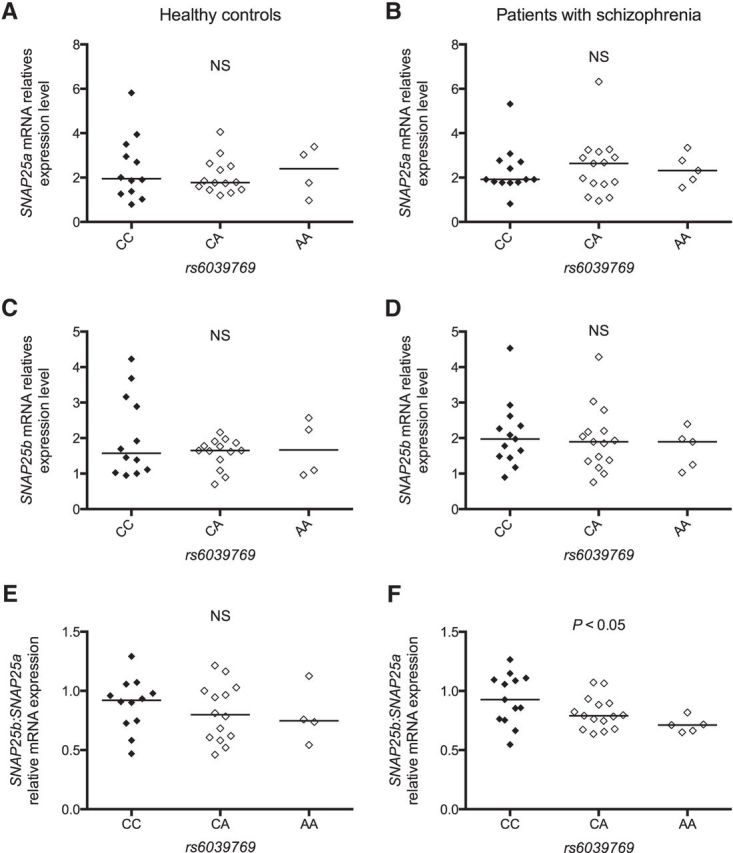
A–F, SNAP25a and SNAP25b mRNA relative expression level in postmortem prefrontal cortex of patients with schizophrenia (B, D, F) and healthy control subjects (A, C, E) according to rs6039769 genotype. Data are expressed as a mean value of relative mRNA expression level of SNAP25a (A, B), SNAP25b (C, D), and SNAP25b/SNAP25a ratio (E, F). No significant difference was observed for patients and control subjects for either SNAP25a or SNAP25b. A significant difference was observed for SNAP25b/SNAP25a ratio only for patients with schizophrenia according to the rs6039769 genotype, with an increased ratio for individuals homozygous for the C allele (Kruskal–Wallis test, K = 6.23, p = 0.04). Subjects homozygous for the rs6039769 C at-risk allele are shown with a black symbol, whereas others genotypes are shown with a clear symbol. Individual results and median values are reported. NS, Not significant.
SNAP25 rs6039769 C at-risk allele and the prefrontal–limbic network in humans
We combined genetic and brain imaging data in two independent cohorts to determine the in vivo functional consequences of the SNAP25 at-risk allele. No difference in genotype frequencies was observed between the discovery and replication cohorts (χ2 = 1.39, 2 df, pdisc = 0.50) and both were in Hardy–Weinberg equilibrium (discovery cohort: χdisc2 = 1.96, 2 df, pdisc = 0.16; replication cohort: χrep2 = 0.03, 2 df, prep = 0.86; Table 2). Our analyses revealed a gender-dependent significant correlation between rs6039769 genotype and the amygdala volume (multiple linear regression genotype × gender interaction, t = −2.8; p = 0.008; Fig. 4A). Both in patients with BD and healthy control subjects, males with an at-risk genotype (CC) had a larger amygdala than non-risk carriers (AA or AC; Student's t test, t(31) = 2.73, p = 0.01), but this difference was not observed in females (Student's t test, t(33) = 1.31, p = 0.20). We did not find any effect on the hippocampal volume. In the “replication” sample, we also detected a significant association between the risk genotype and a larger amygdala volume, localized on the left side (ANCOVA, F(1,117) = 8.05; p = 0.005).
Table 2.
rs6039769 genotype and allele frequency in the two populations used for imaging genetic studies
| rs6039769 | AA | AC | CC | χ2 (p value) | A | C | χ2 (p value) |
|---|---|---|---|---|---|---|---|
| Discovery cohort | 9 | 25 | 37 | 1.39 (0.50) | 43 | 99 | 0.41 (0.52) |
| Replication cohort | 14 | 53 | 54 | 81 | 161 |
Figure 4.

Structural and functional prefrontal limbic network characterization in regard to SNAP25 genotype. A, Amygdala volume in patients with schizophrenia and unaffected control subjects. Males homozygous for the SNAP25 rs6039769 C at-risk allele have a larger amygdala than non-risk carriers (AA or AC) or than females regardless of their genotype. B, Amygdala–VMPFC connectivity in patients with schizophrenia and unaffected control subjects. Again, males homozygous for the SNAP25 rs6039769 C at-risk allele have a higher connectivity than non-risk carriers (AA or AC) or than females regardless of their genotype. Squares show males, and circles show females. Black symbols show individuals with CC genotype and clear symbols show those with AA or AC genotype. Individual results are reported with mean values ± SD.
For prefrontal–limbic functional connectivity, we identified a gender-dependent significant relationship between rs6039769 genotype and functional connectivity measures between the amygdala and the VMPFC (multiple linear regression genotype × gender interaction, t = −2.3, p = 0.03; Fig. 4B). In both patients with BD and healthy control subjects, male risk carriers had a greater correlation coefficient than non-risk carriers or than females regardless of their genotype.
Discussion
SNAP25 genetic variants are associated with an increased risk of multiple psychiatric disorders
In this study, we showed that the rs6039769 SNAP25 at-risk allele for EOBD was also associated with SZ. We then explored the functional impact of this variant both in vitro and in vivo using postmortem studies and brain imaging. We showed that this variation was associated with an increased transcription level, which was consistent with the increased SNAP25b/SNAP25a ratio observed in patients with SZ. This was also consistent with a larger amygdala volume and an increased amygdala–VMPFC connectivity reported in men carrying the CC at-risk genotype.
The association between rs6039769 and SZ is consistent with the abundant literature identifying SNAP25 genetic variations as risk factors for many psychiatric conditions, such as SZ (Wang et al., 2015), BD (Etain et al., 2010) or attention-deficit/hyperactivity disorder (ADHD; Liu et al., 2017), and strongly supports the hypothesis of a shared genetic vulnerability that has been suggested for these mental disorders (Lichtenstein et al., 2009; Lee et al., 2013). The association that we previously reported between rs6039769 and BD was specific to early-onset forms of the disorder, which is highly comorbid with ADHD (West et al., 1995; Wozniak et al., 1995). This common vulnerability allele may thus be associated with a specific comorbid condition shared among several psychiatric disorders, such as psychotic symptoms that are more frequently observed in early-onset forms of BD (Leboyer et al., 2005).
Patients with psychiatric disorders exhibit alterations in the expression level of SNAP25
The functional consequences of this promoter variation help us in understanding how it may contribute to a higher liability for SZ and EOBD. The increased transcription level we observed is consistent with the increased SNAP25 levels reported in CSF of patients with SZ (Thompson et al., 1999, 2003b) and with altered SNAP25 levels in postmortem brain samples from patients with SZ or BD. These reports have highlighted a decreased level of SNAP25 in the hippocampus (Thompson et al., 2003a) but increased densities in prefrontal cortex area 9 in patients with SZ or BD (Thompson et al., 1998; Scarr et al., 2006). Animal models of SZ, such as mutant human DISC1 mice, also exhibit altered SNAP25 expression (Pletnikov et al., 2008), and mutant mice for SNAP25 exhibit several phenotypic traits similar to those present in psychosis, such as impaired sensorimotor gating (Jeans et al., 2007).
The SNARE complex, including SNAP25, is crucial for vesicular membrane fusion and neurotransmitter release in synapses (Graham et al., 2002). SNAP25 is also necessary for axonal growth (Osen-Sand et al., 1993; Shimojo et al., 2015), dendrite formation (Kimura et al., 2003), and calcium homeostasis (Verderio et al., 2004; Pozzi et al., 2008). Therefore, an altered expression level of SNAP25 may impact the brain neurotransmission. More specifically, we observed an altered balance in the SNAP25b/SNAP25a isoforms ratio, which probably results from the SNAP25 expression increase in subjects with rs6039769 CC genotype. The two isoforms are differentially expressed during the development, with the SNAP25a being associated with development and synaptogenesis, while SNAP25b is the major form in mature brains (Prescott and Chamberlain, 2011; Yamamori et al., 2011). Note that during postnatal development both isoforms have a similar expression level and SNAP25b is dramatically upregulated in early adulthood (Bark et al., 1995), a critical period for SZ and BD onset. A modified SNAP25b/SNAP25a balance may alter the brain maturation or the balance among neurite outgrowth, synaptogenesis, and neurotransmitter release (Yamamori et al., 2011). This increase of SNAP25b compared with SNAP25a may modify the stability of vesicles in the releasable vesicle pool (Sørensen et al., 2003) and thus increase neurotransmitter exocytosis.
Synaptic transmission is impaired in SZ and BD and is also the target of their common medication (i.e., antipsychotic agents). Haloperidol has been shown to increase SNAP25 levels in the striatum (Barakauskas et al., 2010). The mouse mutant coloboma, which bears a mutation on chromosome 2 including SNAP25, displays a decreased glutamate level in the neocortex and a low dopamine level in the ventral striatum (Raber et al., 1997). In another mouse model, carrying a single amino acid change in SNAP25, Kataoka et al. (2011) reported abnormal serotonin and dopamine release in the amygdala, along with anxiety-related behavior. This rodent model exemplifies the potential impact of SNAP25 on serotonin, amygdala, and, in turn, behaviors. Though we did not have human behavioral dimensional measures in this study, a previous study has reported a link between a SNAP25 genotype and emotional regulation in a population of children between the ages of 6 and 20 months (Sheese et al., 2009). Another group identified an association between neuroticism and SNAP25 in a GWAS that included 3972 individuals (Terracciano et al., 2010).
SNAP25 genetic variants are associated with structural and functional brain alterations
Consistent with what was observed in animal models, we found, through our imaging genetics approach in humans, that the rs6039769 CC genotype was associated with a larger amygdala volume and an increased functional connectivity between VMPFC and amygdala. The amygdala is a core component of emotion production and emotional processing, mostly implicated in automatic emotional regulation (Phillips et al., 2008). Alterations in amygdala volume, function, and/or connectivity have been repeatedly reported in individuals with BD and SZ (Hajek et al., 2009; van Erp et al., 2016). Amygdala volume has been found to be enlarged in at-risk offspring of patients with BD (Bauer et al., 2014) and in adolescents at risk for SZ (Welch et al., 2010; Ganzola et al., 2014). Intriguingly, a meta-analysis (Hajek et al., 2009) identified a decreased volume in children and adolescents with BD compared with control subjects and a trend toward a larger left amygdala volume in adult patients with BD. In our study, we only studied adult subjects in both of our samples (discovery and replication) and identified an increase in the amygdala volume associated with the risk variant. In our replication sample, our effect was also left sided. One possible interpretation is that both BD and the SNAP25 risk variant are associated with an abnormal neurodevelopment of the amygdala, leading to a smaller amygdala in children and adolescents and a larger volume in adults. This is speculative and should be tested in children and adolescents or through longitudinal studies. A higher SNAP25 expression in patients with psychiatric disorders may lead to a chronic hyperactivity, which has been proposed as a putative mechanism for an enlargement of amygdala volumes, resulting in emotional and social dysregulation and increasing the risk for BD, SZ, and ADHD (Rosenfeld et al., 2011). Prefrontal limbic connectivity is also a key network implicated in emotion regulation, allowing a prefrontal modulation over the amygdala activation (Phillips et al., 2008). An increased functional connectivity between VMPFC and amygdala is thus a crucial feature of BD (d'Albis and Houenou, 2015). This altered connectivity is also present in healthy relatives of patients with BD, suggesting it as a possible endophenotype, a marker of a genetic liability to BD. This altered prefrontal–limbic connectivity may explain the inability of patients with BD to efficiently regulate emotional states, leading to emotional hyperreactivity (Henry et al., 2012). Altered prefrontal–amygdala functional connectivity has also been reported to be altered in patients with SZ (Vai et al., 2015). Structural connectivity of the uncinate fasciculus that connects VMPFC and amygdala has consistently been found altered in first-episode patients with SZ (Wheeler and Voineskos, 2014) and in healthy relatives (Muñoz Maniega et al., 2008). Prefrontal–limbic dysconnectivity is assumed to be one of the neural networks abnormalities common to SZ and BD (d'Albis and Houenou, 2015). Overall, our results suggest that the increased liability to SZ and EOBD associated with the rs6039769 risk allele may be mediated by its action on limbic networks (amygdala volume and prefrontal–limbic connectivity). Our associations between the SNP variation and neuroimaging variables were only present in male, suggesting a gender-dependent impact of SNAP25 on limbic networks. Gender differences in emotion processing are largely described in the literature (Nolen-Hoeksema, 2012; Lungu et al., 2015). In parallel, the regional SNAP25 density varies according to gender with a higher density in men than in women in frontal regions, with an inverse relationship in temporal areas (Downes et al., 2008). In addition, hormones have been found to regulate the expression of SNAP25 in animal models (Jacobsson et al., 1998). One study (Cagliani et al., 2012) also identified a gender-specific correlation between a SNAP25 allelic variation and verbal performance. All these studies, along with the fact that SNAP25 has been associated with several male-predominant psychiatric conditions (ADHD, SZ, and autism spectrum disorder) shed light on our gender-specific finding. However, we cannot fully explain why this result was gender specific while the other ones were not. Note that all of our samples were enriched in males and may thus not be fully appropriate to observe significant differences between genders.
We selected different populations for the samples of the imaging genetics study: patients and control subjects for the discovery sample, and control subjects only for the replication group. Selecting the better population for imaging genetics is highly debated in the literature (Bigos and Weinberger, 2010). Further, the replication sample was significantly younger than the discovery sample. However, we found similar results in those two different groups. These points strengthen the validity and generalizability of our results but may also explain why we did not identify gender effects in the replication sample.
Despite bringing evidence from multiple levels, we recognize some limitations in our study. First, the sample size for the MRI study is relatively modest, and these results should be taken with caution. We thus did not correct our analyses for multiple comparisons. However, we were able to replicate the association between the risk genotype and a larger amygdala volume in an independent sample. Independent samples with a larger sample size would also allow conducting whole-brain structural and connectivity analyses. Second, the sample size of the genetic study was also modest, but the same genetic effect was observed in the Psychiatric Genomics Consortium cohorts. Third, the limited number of available brain samples prevents any replication of our data and more specifically of the importance of the SNAP25b/SNAP25a ratio. Because of the crucial importance of the amygdala and hippocampus in SZ and BD, studying SNAP25 expression levels in these regions depending on the genotype would be of high interest. However, alteration of the SNAP25 level and its binding partners of the SNARE complex have been consistently reported in humans with psychiatric disorders as well as in mouse models, demonstrating the importance of the regulation of this complex. Further, study of animal models modulating the ratio between the two SNAP25 isoforms and its interaction with environmental factors, such as pre-perinatal stress, should help in understanding the physiological impact of such deregulation and its impact on behaviors.
In conclusion, through our multimodal approach, from in vitro construct to imaging genetics and postmortem brain expression, we brought strong evidence for the functional consequences of this allelic variation of SNAP25. Our findings suggest that rs6039769, as a functional polymorphism located in the promoter region of SNAP25, may modulate the development and plasticity of the prefrontal–limbic network and therefore increase vulnerability to EOBD and SZ.
Footnotes
This work was supported by the Investissements d'Avenir program managed by the Agence Nationale pour la Recherche (ANR) under reference ANR-11-IDEX-0004-02 (Labex BioPsy). This work also received financial support from the ANR (Grant ANR-08-MNPS-041), the Institut National Pour La Sante et la Recherche Medicale, the Réseau Thématique de Recherche et de Soins en Santé Mentale (Fondation FondaMental), and the Institut Thématique Multi Organismes Neurosciences, Sciences Cognitives, Neurologie et Psychiatrie. The Jamain group is affiliated with the Paris School of Neuroscience and the Bio-Psy Laboratory of Excellence. We thank the patients and control subjects for their participation. We also thank Pr. W. Song for SNAP25 promoter constructs. In addition, we thank E. Abadie, the Cochin Hospital Cell Library (Professor J. Chelly), the Clinical Investigation Centre (Dr. P. Lecorvoisier). and the Biological Resources Platform (Professor B. Ghaleh) of Mondor Hospital for technical assistance. Finally, We thank the personnel of the UNIACT, NeuroSpin Platform for their help in the MRI scanning of patients and control subjects.
The authors declare no competing financial interests.
References
- American Psychiatric Association (2000) Diagnostic criteria from DSM-IV-TR. Washington, DC: American Psychiatric Association. [Google Scholar]
- Barakauskas VE, Beasley CL, Barr AM, Ypsilanti AR, Li HY, Thornton AE, Wong H, Rosokilja G, Mann JJ, Mancevski B, Jakovski Z, Davceva N, Ilievski B, Dwork AJ, Falkai P, Honer WG (2010) A novel mechanism and treatment target for presynaptic abnormalities in specific striatal regions in schizophrenia. Neuropsychopharmacology 35:1226–1238. 10.1038/npp.2009.228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark C, Bellinger FP, Kaushal A, Mathews JR, Partridge LD, Wilson MC (2004) Developmentally regulated switch in alternatively spliced SNAP-25 isoforms alters facilitation of synaptic transmission. J Neurosci 24:8796–8805. 10.1523/JNEUROSCI.1940-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark IC, Hahn KM, Ryabinin AE, Wilson MC (1995) Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci U S A 92:1510–1514. 10.1073/pnas.92.5.1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CL, Feng Y, Wigg K, Bloom S, Roberts W, Malone M, Schachar R, Tannock R, Kennedy JL (2000) Identification of DNA variants in the SNAP-25 gene and linkage study of these polymorphisms and attention-deficit hyperactivity disorder. Mol Psychiatry 5:405–409. 10.1038/sj.mp.4000733 [DOI] [PubMed] [Google Scholar]
- Bauer IE, Sanches M, Suchting R, Green CE, El Fangary NM, Zunta-Soares GB, Soares JC (2014) Amygdala enlargement in unaffected offspring of bipolar parents. J Psychiatr Res 59:200–205. 10.1016/j.jpsychires.2014.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigos KL, Weinberger DR (2010) Imaging genetics—days of future past. Neuroimage 53:804–809. 10.1016/j.neuroimage.2010.01.035 [DOI] [PubMed] [Google Scholar]
- Braida D, Guerini FR, Ponzoni L, Corradini I, De Astis S, Pattini L, Bolognesi E, Benfante R, Fornasari D, Chiappedi M, Ghezzo A, Clerici M, Matteoli M, Sala M (2015) Association between SNAP-25 gene polymorphisms and cognition in autism: functional consequences and potential therapeutic strategies. Transl Psychiatry 5:e500. 10.1038/tp.2014.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagliani R, Riva S, Marino C, Fumagalli M, D'Angelo MG, Riva V, Comi GP, Pozzoli U, Forni D, Cáceres M, Bresolin N, Clerici M, Sironi M (2012) Variants in SNAP25 are targets of natural selection and influence verbal performances in women. Cell Mol Life Sci 69:1705–1715. 10.1007/s00018-011-0896-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai F, Chen B, Zhou W, Zis O, Liu S, Holt RA, Honer WG, Song W (2008) SP1 regulates a human SNAP-25 gene expression. J Neurochem 105:512–523. 10.1111/j.1471-4159.2007.05167.x [DOI] [PubMed] [Google Scholar]
- Cannon TD. (2015) How schizophrenia develops: cognitive and brain mechanisms underlying onset of psychosis. Trends Cogn Sci 19:744–756. 10.1016/j.tics.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll LS, Kendall K, O'Donovan MC, Owen MJ, Williams NM (2009) Evidence that putative ADHD low risk alleles at SNAP25 may increase the risk of schizophrenia. Am J Med Genet B Neuropsychiatr Genet 150B:893–899. 10.1002/ajmg.b.30915 [DOI] [PubMed] [Google Scholar]
- d'Albis MA, Houenou J (2015) The kraepelinian dichotomy viewed by neuroimaging. Schizophr Bull 41:330–335. 10.1093/schbul/sbu174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downes EC, Robson J, Grailly E, Abdel-All Z, Xuereb J, Brayne C, Holland A, Honer WG, Mukaetova-Ladinska EB (2008) Loss of synaptophysin and synaptosomal-associated protein 25-kDa (SNAP-25) in elderly Down syndrome individuals. Neuropathol Appl Neurobiol 34:12–22. 10.1111/j.1365-2990.2007.00899.x [DOI] [PubMed] [Google Scholar]
- Dumaine A, Maucuer A, Barbet A, Manceau V, Deshommes J, Méary A, Szöke A, Schürhoff F, Llorca PM, Lancon C, Leboyer M, Jamain S (2011) Genetic and molecular exploration of UHMK1 in schizophrenic patients. Psychiatr Genet 21:315–318. 10.1097/YPG.0b013e3283458a37 [DOI] [PubMed] [Google Scholar]
- Etain B, Dumaine A, Mathieu F, Chevalier F, Henry C, Kahn JP, Deshommes J, Bellivier F, Leboyer M, Jamain S (2010) A SNAP25 promoter variant is associated with early-onset bipolar disorder and a high expression level in brain. Mol Psychiatry 15:748–755. 10.1038/mp.2008.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Earle JA, Stary JM, Lee S, Sedgewick J (2001) Altered levels of the synaptosomal associated protein SNAP-25 in hippocampus of subjects with mood disorders and schizophrenia. Neuroreport 12:3257–3262. 10.1097/00001756-200110290-00023 [DOI] [PubMed] [Google Scholar]
- Friston KJ, Williams S, Howard R, Frackowiak RS, Turner R (1996) Movement-related effects in fMRI time-series. Magn Reson Med 35:346–355. 10.1002/mrm.1910350312 [DOI] [PubMed] [Google Scholar]
- Ganzola R, Maziade M, Duchesne S (2014) Hippocampus and amygdala volumes in children and young adults at high-risk of schizophrenia: research synthesis. Schizophr Res 156:76–86. 10.1016/j.schres.2014.03.030 [DOI] [PubMed] [Google Scholar]
- Graham ME, Washbourne P, Wilson MC, Burgoyne RD (2002) Molecular analysis of SNAP-25 function in exocytosis. Ann N Y Acad Sci 971:210–221. 10.1111/j.1749-6632.2002.tb04465.x [DOI] [PubMed] [Google Scholar]
- Gray LJ, Dean B, Kronsbein HC, Robinson PJ, Scarr E (2010) Region and diagnosis-specific changes in synaptic proteins in schizophrenia and bipolar I disorder. Psychiatry Res 178:374–380. 10.1016/j.psychres.2008.07.012 [DOI] [PubMed] [Google Scholar]
- Hajek T, Kopecek M, Kozeny J, Gunde E, Alda M, Höschl C (2009) Amygdala volumes in mood disorders–meta-analysis of magnetic resonance volumetry studies. J Affect Disord 115:395–410. 10.1016/j.jad.2008.10.007 [DOI] [PubMed] [Google Scholar]
- Hart SJ, Bizzell J, McMahon MA, Gu H, Perkins DO, Belger A (2013) Altered fronto-limbic activity in children and adolescents with familial high risk for schizophrenia. Psychiatry Res 212:19–27. 10.1016/j.pscychresns.2012.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry C, Phillips M, Leibenluft E, M'Bailara K, Houenou J, Leboyer M (2012) Emotional dysfunction as a marker of bipolar disorders. Front Biosci (Elite Ed) 4:2622–2630. 10.2741/578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar DP, Westlye LT, van Erp TG, Rasmussen J, Leonardo CD, Faskowitz J, Haukvik UK, Hartberg CB, Doan NT, Agartz I, Dale AM, Gruber O, Krämer B, Trost S, Liberg B, Abé C, Ekman CJ, Ingvar M, Landén M, Fears SC, et al. (2016) Subcortical volumetric abnormalities in bipolar disorder. Mol Psychiatry 21:1710–1716. 10.1038/mp.2015.227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honer WG, Falkai P, Bayer TA, Xie J, Hu L, Li HY, Arango V, Mann JJ, Dwork AJ, Trimble WS (2002) Abnormalities of SNARE mechanism proteins in anterior frontal cortex in severe mental illness. Cereb Cortex 12:349–356. 10.1093/cercor/12.4.349 [DOI] [PubMed] [Google Scholar]
- Jacobsson G, Razani H, Ogren SO, Meister B (1998) Estrogen down-regulates mRNA encoding the exocytotic protein SNAP-25 in the rat pituitary gland. J Neuroendocrinol 10:157–163. 10.1046/j.1365-2826.1998.00178.x [DOI] [PubMed] [Google Scholar]
- Jeans AF, Oliver PL, Johnson R, Capogna M, Vikman J, Molnár Z, Babbs A, Partridge CJ, Salehi A, Bengtsson M, Eliasson L, Rorsman P, Davies KE (2007) A dominant mutation in Snap25 causes impaired vesicle trafficking, sensorimotor gating, and ataxia in the blind-drunk mouse. Proc Natl Acad Sci U S A 104:2431–2436. 10.1073/pnas.0610222104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka M, Yamamori S, Suzuki E, Watanabe S, Sato T, Miyaoka H, Azuma S, Ikegami S, Kuwahara R, Suzuki-Migishima R, Nakahara Y, Nihonmatsu I, Inokuchi K, Katoh-Fukui Y, Yokoyama M, Takahashi M (2011) A single amino acid mutation in SNAP-25 induces anxiety-related behavior in mouse. PLoS One 6:e25158. 10.1371/journal.pone.0025158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Mizoguchi A, Ide C (2003) Regulation of growth cone extension by SNARE proteins. J Histochem Cytochem 51:429–433. 10.1177/002215540305100404 [DOI] [PubMed] [Google Scholar]
- Ladouceur CD, Farchione T, Diwadkar V, Pruitt P, Radwan J, Axelson DA, Birmaher B, Phillips ML (2011) Differential patterns of abnormal activity and connectivity in the amygdala-prefrontal circuitry in bipolar-I and bipolar-NOS youth. J Am Acad Child Adolesc Psychiatry 50:1275–1289.e2. 10.1016/j.jaac.2011.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leboyer M, Henry C, Paillere-Martinot ML, Bellivier F (2005) Age at onset in bipolar affective disorders: a review. Bipolar Disord 7:111–118. 10.1111/j.1399-5618.2005.00181.x [DOI] [PubMed] [Google Scholar]
- Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, Mowry BJ, Thapar A, Goddard ME, Witte JS, Absher D, Agartz I, Akil H, Amin F, Andreassen OA, Anjorin A, Anney R, Anttila V, Arking DE, Asherson P, et al. (2013) Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet 45:984–994. 10.1038/ng.2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P, Yip BH, Björk C, Pawitan Y, Cannon TD, Sullivan PF, Hultman CM (2009) Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 373:234–239. 10.1016/S0140-6736(09)60072-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YS, Dai X, Wu W, Yuan FF, Gu X, Chen JG, Zhu LQ, Wu J (2017) The association of SNAP25 gene polymorphisms in attention deficit/hyperactivity disorder: a systematic review and meta-analysis. Mol Neurobiol 54:2189–2200. 10.1007/s12035-016-9810-9 [DOI] [PubMed] [Google Scholar]
- Lungu O, Potvin S, Tikàsz A, Mendrek A (2015) Sex differences in effective fronto-limbic connectivity during negative emotion processing. Psychoneuroendocrinology 62:180–188. 10.1016/j.psyneuen.2015.08.012 [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Nicodemus KK, Egan MF, Callicott JH, Mattay V, Weinberger DR (2008) False positives in imaging genetics. Neuroimage 40:655–661. 10.1016/j.neuroimage.2007.11.058 [DOI] [PubMed] [Google Scholar]
- Muñoz Maniega S, Lymer GK, Bastin ME, Marjoram D, Job DE, Moorhead TW, Owens DG, Johnstone EC, McIntosh AM, Lawrie SM (2008) A diffusion tensor MRI study of white matter integrity in subjects at high genetic risk of schizophrenia. Schizophr Res 106:132–139. 10.1016/j.schres.2008.09.016 [DOI] [PubMed] [Google Scholar]
- Nolen-Hoeksema S. (2012) Emotion regulation and psychopathology: the role of gender. Annu Rev Clin Psychol 8:161–187. 10.1146/annurev-clinpsy-032511-143109 [DOI] [PubMed] [Google Scholar]
- Nurnberger JI Jr, Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J, Severe JB, Malaspina D, Reich T (1994) Diagnostic interview for genetic studies. Rationale, unique features, and training. NIMH Genetics Initiative. Arch Gen Psychiatry 51:849–859. [DOI] [PubMed] [Google Scholar]
- Osen-Sand A, Catsicas M, Staple JK, Jones KA, Ayala G, Knowles J, Grenningloh G, Catsicas S (1993) Inhibition of axonal growth by SNAP-25 antisense oligonucleotides in vitro and in vivo. Nature 364:445–448. 10.1038/364445a0 [DOI] [PubMed] [Google Scholar]
- Patenaude B, Smith SM, Kennedy DN, Jenkinson M (2011) A Bayesian model of shape and appearance for subcortical brain segmentation. Neuroimage 56:907–922. 10.1016/j.neuroimage.2011.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips ML, Ladouceur CD, Drevets WC (2008) A neural model of voluntary and automatic emotion regulation: implications for understanding the pathophysiology and neurodevelopment of bipolar disorder. Mol Psychiatry 13:829, 833–857. 10.1038/mp.2008.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnikov MV, Ayhan Y, Nikolskaia O, Xu Y, Ovanesov MV, Huang H, Mori S, Moran TH, Ross CA (2008) Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry 13:173–186, 115. 10.1038/sj.mp.4002079 [DOI] [PubMed] [Google Scholar]
- Pozzi D, Condliffe S, Bozzi Y, Chikhladze M, Grumelli C, Proux-Gillardeaux V, Takahashi M, Franceschetti S, Verderio C, Matteoli M (2008) Activity-dependent phosphorylation of Ser187 is required for SNAP-25-negative modulation of neuronal voltage-gated calcium channels. Proc Natl Acad Sci U S A 105:323–328. 10.1073/pnas.0706211105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott GR, Chamberlain LH (2011) Regional and developmental brain expression patterns of SNAP25 splice variants. BMC Neurosci 12:35. 10.1186/1471-2202-12-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychiatric GWAS Consortium Bipolar Disorder Working Group (2011) Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 43:977–983. 10.1038/ng.943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kähler A, Duncan L, Stahl E, Genovese G, Fernández E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, et al. (2014) A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506:185–190. 10.1038/nature12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, Sklar P (2009) Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460:748–752. 10.1038/nature08185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J, Mehta PP, Kreifeldt M, Parsons LH, Weiss F, Bloom FE, Wilson MC (1997) Coloboma hyperactive mutant mice exhibit regional and transmitter-specific deficits in neurotransmission. J Neurochem 68:176–186. [DOI] [PubMed] [Google Scholar]
- Rosenfeld AJ, Lieberman JA, Jarskog LF (2011) Oxytocin, dopamine, and the amygdala: a neurofunctional model of social cognitive deficits in schizophrenia. Schizophr Bull 37:1077–1087. 10.1093/schbul/sbq015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarr E, Gray L, Keriakous D, Robinson PJ, Dean B (2006) Increased levels of SNAP-25 and synaptophysin in the dorsolateral prefrontal cortex in bipolar I disorder. Bipolar Disord 8:133–143. 10.1111/j.1399-5618.2006.00300.x [DOI] [PubMed] [Google Scholar]
- Scherk H, Backens M, Schneider-Axmann T, Kemmer C, Usher J, Reith W, Falkai P, Gruber O (2008) Neurochemical pathology in hippocampus in euthymic patients with bipolar I disorder. Acta Psychiatr Scand 117:283–288. 10.1111/j.1600-0447.2007.01142.x [DOI] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics C (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature 511:421–427. 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürhoff F, Fond G, Berna F, Bulzacka E, Vilain J, Capdevielle D, Misdrahi D, Leboyer M, Llorca PM, FondaMental Academic Centers of Expertise for Schizophrenia c (2015) A National network of schizophrenia expert centres: an innovative tool to bridge the research-practice gap. Eur Psychiatry 30:728–735. 10.1016/j.eurpsy.2015.05.004 [DOI] [PubMed] [Google Scholar]
- Sheese BE, Voelker P, Posner MI, Rothbart MK (2009) Genetic variation influences on the early development of reactive emotions and their regulation by attention. Cogn Neuropsychiatry 14:332–355. 10.1080/13546800902844064 [DOI] [PubMed] [Google Scholar]
- Shimojo M, Courchet J, Pieraut S, Torabi-Rander N, Sando R 3rd, Polleux F, Maximov A (2015) SNAREs controlling vesicular release of BDNF and development of callosal axons. Cell Rep 11:1054–1066. 10.1016/j.celrep.2015.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003) Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114:75–86. 10.1016/S0092-8674(03)00477-X [DOI] [PubMed] [Google Scholar]
- Terracciano A, Sanna S, Uda M, Deiana B, Usala G, Busonero F, Maschio A, Scally M, Patriciu N, Chen WM, Distel MA, Slagboom EP, Boomsma DI, Villafuerte S, Sliwerska E, Burmeister M, Amin N, Janssens AC, van Duijn CM, Schlessinger D, et al. (2010) Genome-wide association scan for five major dimensions of personality. Mol Psychiatry 15:647–656. 10.1038/mp.2008.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PM, Sower AC, Perrone-Bizzozero NI (1998) Altered levels of the synaptosomal associated protein SNAP-25 in schizophrenia. Biol Psychiatry 43:239–243. 10.1016/S0006-3223(97)00204-7 [DOI] [PubMed] [Google Scholar]
- Thompson PM, Rosenberger C, Qualls C (1999) CSF SNAP-25 in schizophrenia and bipolar illness. A pilot study. Neuropsychopharmacology 21:717–722. 10.1016/S0893-133X(99)00068-8 [DOI] [PubMed] [Google Scholar]
- Thompson PM, Egbufoama S, Vawter MP (2003a) SNAP-25 reduction in the hippocampus of patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 27:411–417. 10.1016/S0278-5846(03)00027-7 [DOI] [PubMed] [Google Scholar]
- Thompson PM, Kelley M, Yao J, Tsai G, van Kammen DP (2003b) Elevated cerebrospinal fluid SNAP-25 in schizophrenia. Biol Psychiatry 53:1132–1137. 10.1016/S0006-3223(02)01599-8 [DOI] [PubMed] [Google Scholar]
- Torrey EF, Webster M, Knable M, Johnston N, Yolken RH (2000) The stanley foundation brain collection and neuropathology consortium. Schizophr Res 44:151–155. 10.1016/S0920-9964(99)00192-9 [DOI] [PubMed] [Google Scholar]
- Vai B, Sferrazza Papa G, Poletti S, Radaelli D, Donnici E, Bollettini I, Falini A, Cavallaro R, Smeraldi E, Benedetti F (2015) Abnormal cortico-limbic connectivity during emotional processing correlates with symptom severity in schizophrenia. Eur Psychiatry 30:590–597. 10.1016/j.eurpsy.2015.01.002 [DOI] [PubMed] [Google Scholar]
- van Erp TG, Hibar DP, Rasmussen JM, Glahn DC, Pearlson GD, Andreassen OA, Agartz I, Westlye LT, Haukvik UK, Dale AM, Melle I, Hartberg CB, Gruber O, Kraemer B, Zilles D, Donohoe G, Kelly S, McDonald C, Morris DW, Cannon DM, et al. (2016) Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry 21:547–553. 10.1038/mp.2015.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Pozzi D, Pravettoni E, Inverardi F, Schenk U, Coco S, Proux-Gillardeaux V, Galli T, Rossetto O, Frassoni C, Matteoli M (2004) SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron 41:599–610. 10.1016/S0896-6273(04)00077-7 [DOI] [PubMed] [Google Scholar]
- Wang Q, Wang Y, Ji W, Zhou G, He K, Li Z, Chen J, Li W, Wen Z, Shen J, Qiang Y, Ji J, Wang Y, Shi Y, Yi Q, Wang Y (2015) SNAP25 is associated with schizophrenia and major depressive disorder in the Han Chinese population. J Clin Psychiatry 76:e76–e82. 10.4088/JCP.13m08962 [DOI] [PubMed] [Google Scholar]
- Welch KA, Stanfield AC, Moorhead TW, Haga K, Owens DC, Lawrie SM, Johnstone EC (2010) Amygdala volume in a population with special educational needs at high risk of schizophrenia. Psychol Med 40:945–954. 10.1017/S0033291709990870 [DOI] [PubMed] [Google Scholar]
- West SA, McElroy SL, Strakowski SM, Keck PE Jr, McConville BJ (1995) Attention deficit hyperactivity disorder in adolescent mania. Am J Psychiatry 152:271–273. 10.1176/ajp.152.2.271 [DOI] [PubMed] [Google Scholar]
- Wheeler AL, Voineskos AN (2014) A review of structural neuroimaging in schizophrenia: from connectivity to connectomics. Front Hum Neurosci 8:653. 10.3389/fnhum.2014.00653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak J, Biederman J, Kiely K, Ablon JS, Faraone SV, Mundy E, Mennin D (1995) Mania-like symptoms suggestive of childhood-onset bipolar disorder in clinically referred children. J Am Acad Child Adolesc Psychiatry 34:867–876. 10.1097/00004583-199507000-00010 [DOI] [PubMed] [Google Scholar]
- Yamamori S, Itakura M, Sugaya D, Katsumata O, Sakagami H, Takahashi M (2011) Differential expression of SNAP-25 family proteins in the mouse brain. J Comp Neurol 519:916–932. 10.1002/cne.22558 [DOI] [PubMed] [Google Scholar]
- Young CE, Arima K, Xie J, Hu L, Beach TG, Falkai P, Honer WG (1998) SNAP-25 deficit and hippocampal connectivity in schizophrenia. Cereb Cortex 8:261–268. 10.1093/cercor/8.3.261 [DOI] [PubMed] [Google Scholar]