

Abstract
Creutzfeldt–Jakob disease (CJD) is a neurodegenerative disorder caused by prion protein (PrP) misfolding, clinically recognized by cognitive and motor deficits, electroencephalographic abnormalities, and seizures. Its neurophysiological bases are not known. To assess the potential involvement of NMDA receptor (NMDAR) dysfunction, we analyzed NMDA-dependent synaptic plasticity in hippocampal slices from Tg(CJD) mice, which model a genetic form of CJD. Because PrP depletion may result in functional upregulation of NMDARs, we also analyzed PrP knock-out (KO) mice. Long-term potentiation (LTP) at the Schaffer collateral–commissural synapses in the CA1 area of ∼100-d-old Tg(CJD) mice was comparable to that of wild-type (WT) controls, but there was an inversion of metaplasticity, with increased GluN2B phosphorylation, which is indicative of enhanced NMDAR activation. Similar but less marked changes were seen in PrP KO mice. At ∼300 d of age, the magnitude of LTP increased in Tg(CJD) mice but decreased in PrP KO mice, indicating divergent changes in hippocampal synaptic responsiveness. Tg(CJD) but not PrP KO mice were intrinsically more susceptible than WT controls to focal hippocampal seizures induced by kainic acid. IL-1β-positive astrocytes increased in the Tg(CJD) hippocampus, and blocking IL-1 receptor signaling restored normal synaptic responses and reduced seizure susceptibility. These results indicate that alterations in NMDA-dependent glutamatergic transmission in Tg(CJD) mice do not depend solely on PrP functional loss. Moreover, astrocytic IL-1β plays a role in the enhanced synaptic responsiveness and seizure susceptibility, suggesting that targeting IL-1β signaling may offer a novel symptomatic treatment for CJD.
SIGNIFICANCE STATEMENT Dementia and myoclonic jerks develop in individuals with Creutzfeldt–Jakob disease (CJD), an incurable brain disorder caused by alterations in prion protein structure. These individuals are prone to seizures and have high brain levels of the inflammatory cytokine IL-1β. Here we show that blocking IL-1β receptors with anakinra, the human recombinant form of the endogenous IL-1 receptor antagonist used to treat rheumatoid arthritis, normalizes hippocampal neurotransmission and reduces seizure susceptibility in a CJD mouse model. These results link neuroinflammation to defective neurotransmission and the enhanced susceptibility to seizures in CJD and raise the possibility that targeting IL-1β with clinically available drugs may be beneficial for symptomatic treatment of the disease.
Keywords: Creutzfeldt–Jakob disease, IL-1 receptor antagonist, prion, seizures, synaptic plasticity, transgenic mice
Introduction
Prion diseases are invariably fatal neurodegenerative disorders of humans and other mammals caused by misfolding of the cellular prion protein (PrPC), a cell surface glycoprotein of uncertain function (Chiesa, 2015). Creutzfeldt–Jakob disease (CJD) is the most common form in humans. It can arise sporadically, be dominantly inherited due to mutations in the PRNP gene encoding PrPC, or acquired by contact with exogenous PrPSc, the infectious PrP isoform (prion), which propagates by inducing misfolding of host-encoded PrPC (Colby and Prusiner, 2011; Head and Ironside, 2012).
CJD has a stereotyped clinical course. Altered mental function is the initial manifestation, including dementia, confusion, disorientation, behavior abnormalities, and depression of other higher cortical functions. Later, myoclonic jerks, rigidity, and extrapyramidal and cerebellar abnormalities become prominent.
In about two-thirds of patients, electroencephalography (EEG) detects typical periodic sharp wave complexes (PSWCs), either lateralized or generalized (Wieser et al., 2006). Epileptiform discharges and focal motor or generalized seizures may also be observed, typically in the late stage of the disease (Wieser et al., 2006). Nonconvulsive status epilepticus is sometimes a presenting symptom in CJD (Espinosa et al., 2010). This suggests that changes in neuronal network excitability occur in seizure-prone brain areas. As the patients near death, they become akinetic, unresponsive, mute, and rigid (Head and Ironside, 2012; Puoti et al., 2012).
The pathogenic mechanisms responsible for this complex symptomatology are not known. Studies in animal models have suggested several toxic mechanisms activated by abnormally folded PrP that may lead to neuronal dysfunction and death, including corruption of NMDA receptor (NMDAR) activity (Chiesa, 2015). Increased NMDAR-dependent excitation has been reported in mice inoculated with variant (v) CJD prions (Ratté et al., 2008), and when PrPSc, or the PrPSc-like PrP106–126 peptide, was exogenously presented to cultured neurons, NMDAR antagonists blocked the resulting neurotoxicity (Müller et al., 1993; Perovic et al., 1995; Brown et al., 1997; Resenberger et al., 2011; Thellung et al., 2013).
Loss of a physiological PrPC function in regulating NMDAR activity may also contribute to the pathogenic process. Genetic PrPC depletion results in increased hippocampal NMDAR-mediated excitation and glutamate exicitotoxicity (Khosravani et al., 2008). In addition, PrP knock-out (KO) mice are reported to be more susceptible to seizures induced by kainic acid (KA) than wild-type (WT) controls (Walz et al., 1999; Rangel et al., 2007), perhaps because of facilitated NMDAR-mediated excitation in the hippocampus (Maglio et al., 2004; Rangel et al., 2009); although this issue remains controversial (Striebel et al., 2013b; Carulla et al., 2015).
Deposition of misfolded/aggregated PrP and astrogliosis and microgliosis are typical neuropathological changes in CJD (Sikorska et al., 2012). In addition, CJD brains have high levels of several inflammatory cytokines, including interleukin-1β (IL-1β; Sharief et al., 1999; Shi et al., 2013; Llorens et al., 2014). However, it is not clear which cell population is responsible for the increase in IL-1β and whether this proinflammatory cytokine contributes to enhancing NMDAR-mediated glutamatergic transmission (Viviani et al., 2003; Balosso et al., 2008), lowering the threshold for seizures (Vezzani and Viviani, 2015).
We previously generated Tg(CJD) mice expressing the mouse (mo) PrP homolog of the D178N/V129 mutation linked to genetic CJD (Dossena et al., 2008). These mice synthesize a misfolded form of mutant PrP in their brains and develop clinical and neuropathological features highly reminiscent of CJD, including memory and motor deficits, abnormal EEG patterns mimicking the human PSWCs, PrP deposition and gliosis (Dossena et al., 2008).
The present study provides evidence of dysfunctional NMDA-dependent hippocampal synaptic plasticity and enhanced seizure susceptibility in Tg(CJD) mice, resulting from a combination of loss and gain of function of mutant PrP, exacerbated by neuroinflammation.
Materials and Methods
Experimental design and statistical analysis.
All animal experiments were designed in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (Kilkenny et al., 2010), with a commitment to refinement, reduction, and replacement, minimizing the numbers of mice, while using biostatistics to optimize mouse numbers (as used in our previously published peer-reviewed work: Nazzaro et al., 2012; Bouybayoune et al., 2015; Trusel et al., 2015; Iori et al., 2017). Thus, for statistical validity, we used 5–10 mice for electrophysiology, 6–10 mice for analysis of KA-induced seizures, and 4–10 mice for reverse transcription quantitative real-time PCR (qRT-PCR), biochemical analysis, and histology (the number of mice used in each experiment is reported in the figure legends). No sex-related differences in the Tg(CJD) phenotype were observed, so both male and female mice were used for qRT-PCR, biochemistry, and histology. Males were used for electrophysiology and analysis of hippocampal seizures to avoid variations due to ovarian cycles in females (Mejías-Aponte et al., 2002; Twele et al., 2016). Simple randomization was used for treatment allocation. Blinding was applied to treatment administration and data analysis.
For each variable, differences between the groups were assessed using independent-samples Student's t test, one-way ANOVA, and two-way ANOVA (genotype and/or drug treatment as independent factors) followed by Tukey's, Mann–Whitney, or Holm–Sidak post hoc analysis when appropriate (details are reported in the figure legends). Synaptic plasticity data were analyzed with one-way ANOVA for repeated measures (RM1W) or two-way ANOVA for repeated measures (RM2W) followed by Tukey's post hoc analysis; n indicates number of recordings.
Mice.
Production of Tg(CJD) mice, expressing moPrP D177N/V128 tagged with an epitope for monoclonal antibody 3F4 has already been reported (Dossena et al., 2008). We used mice of the Tg(CJD-A21+/−)/Prnp0/0 line expressing 3F4-tagged D177N/V128 PrP at ∼1× that had been backcrossed for >10 generations with an inbred colony of Zürich I Prnp0/0 mice (Büeler et al., 1992) with a pure C57BL/6J background (C57BL/6J/Prnp0/0; European Mouse Mutant Archive, Monterotondo, Rome, Italy; RRID:IMSR_EM:01723). The status of the transgene was determined by PCR (Dossena et al., 2008). PrP KO mice were nontransgenic littermates of Tg(CJD-A21+/−)/Prnp0/0 mice. Age-matched C57BL/6J (WT) mice were purchased from Envigo (http://www.envigo.com/products-services/research-models-services/models/research-models/mice/inbred/c57bl-6-inbred-mice/c57bl-6jrcchsd/). Mice were housed at constant room temperature (RT; 23°C) and relative humidity (60 ± 5%) with free access to food and water and a fixed 12 h light/dark cycle. To reduce experimental variability due to different husbandry conditions, WT mice purchased from Envigo were housed in the same animal room as Tg(CJD) and PrP KO mice for from at least 2 weeks to several months.
Procedures involving animals and their care were conducted in conformity with the institutional guidelines at the IRCCS (Istituto di Ricovero e Cura a Carattere Scientifico)—Mario Negri Institute for Pharmacological Research in compliance with national (D.lgs 26/2014; Authorization no. 19/2008-A issued March 6, 2008 by the Ministry of Health of Italy) and international laws and policies (European Economic Community Council Directive 2010/63/UE; the National Institutes of Health Guide for the Care and Use of Laboratory Animals, 2011 edition). They were reviewed and approved by the Mario Negri Institute Animal Care and Use Committee, which includes ad hoc members for ethical issues, and by the Italian Ministry of Health (Decreto no. 62/2012-B and 212/2016-PR). Animal facilities meet international standards and are regularly checked by a certified veterinarian who is responsible for health monitoring, animal welfare supervision, experimental protocols, and review of procedures.
Determination of NMDARs on postsynaptic density.
Subcellular fractionation of the mouse hippocampus was as described previously (Balducci et al., 2010). Tissue was homogenized in ice-cold 0.32 m sucrose containing 1 mm HEPES, 1 mm MgCl2, 1 mm NaHCO3, and 0.1 mm PMSF, at pH 7.4, with a complete set of protease inhibitors (SigmaFast, Sigma-Aldrich) and phosphatase inhibitors (PhosSTOP, Roche Life Science). The homogenized tissue was centrifuged at 1000 × g for 5 min and the supernatant was centrifuged at 13,000 × g for 15 min to obtain a crude membrane fraction. The pellet was then resuspended in 1 mm HEPES containing protease and phosphatase inhibitors and was centrifuged at 100,000 × g for 1 h. The resulting pellet was resuspended in a buffer containing 75 mm KCl, protease, and phosphatase inhibitors and 1% Triton X-100 and centrifuged at 100,000 × g for 1 h. The final pellet was homogenized in a glass-glass Potter homogenizer in 20 mm HEPES with protease and phosphatase inhibitors; this fraction, referred to as the Triton-insoluble fraction, was stored at −80°C. The protein composition of this preparation was tested for the absence of the presynaptic marker synaptophysin and enrichment in postsynaptic density (PSD) proteins. Proteins (10 μg) were resolved by SDS PAGE and electrophoretically transferred to polyvinylidene fluoride membranes. The membranes were blocked for 1 h in 5% nonfat dry milk in Tris-buffered saline 0.1 m, pH 7.4, containing 0.01% Tween 20 (TTBS), then incubated with the primary antibody diluted in blocking solution, with the exception of anti-phospho-Tyr1472 GluN2B, which was diluted in TTBS containing 5% bovine serum albumin (BSA). The antibodies were as follows: mouse monoclonal anti-GluN1 (1:1000; Synaptic Systems; RRID:AB_2113443), rabbit polyclonal anti-phospho-Tyr1472 GluN2B (1:700; Thermo Fisher Scientific; RRID:AB_325370), rabbit polyclonal anti-GluN2A (1:2000; Invitrogen; RRID:AB_2536209), anti-GluN2B (1:2000; Invitrogen; RRID:AB_2536210), mouse monoclonal anti-PSD95 (1:10,000; NeuroMab; RRID:AB_10698024), and mouse monoclonal anti-β-actin (1:20,000; Millipore; RRID:AB_2223041). After thorough rinsing in TTBS, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies, revealed using enhanced chemiluminescence (Luminata Forte, Millipore) and visualized by a BIO-RAD XRS Image Scanner. Quantity-One software (BIO-RAD) was used for quantitative densitometry of protein bands.
Histology.
Mice were deeply anesthetized by intraperitoneal injection of 100 mg/kg ketamine hydrochloride and 1 mg/kg medetomidine hydrochloride (Alcyon) and perfused through the ascending aorta with PBS (0.05 m), pH 7.4, followed by 4% paraformaldehyde in PBS. Brains were removed, postfixed, cryoprotected, and frozen at −80°C. Sections were cut throughout the septotemporal aspects of the hippocampus using a Leica cryostat and incubated for 1 h at RT with 10% normal goat serum, 1% Triton X-100 in Tris-buffered saline 0.1 m, pH 7.4, then overnight at 4°C with rabbit polyclonal anti-glial fibrillary acid protein (GFAP) antibody (1:2500; Dako; RRID:AB_10013382), rat polyclonal anti-CD11b (1:1000; Serotec; RRID:AB_321292), and goat polyclonal anti-IL-1β (1:200; Santa Cruz Biotechnology; RRID:AB_2124627), followed by visualization with the Vectastain ABC kit (Vector Laboratories), with diaminobenzidine (DAB; or nickel-intensified DAB for IL-1β) as chromogen. The TSA Cyanine 5 System (PerkinElmer) was used for immunofluorescent staining of IL-1β. Anti-rat (RRID:AB_141709) and anti-rabbit IgG Alexa Fluor 488 (1:500; Invitrogen; RRID:AB_143165) were used for immunofluorescent staining of CD11b and GFAP, respectively. Sections were reacted with 2 μg/ml Hoechst 33258 stain (Molecular Probes; RRID:AB_2651133) to stain the nuclei. Slices were matched at comparable anteroposterior and dorsoventral levels for comparison of the different experimental groups.
Reverse transcription qRT-PCR.
Total RNA from the mouse hippocampus was extracted using the SV Total RNA Isolation System (Promega) according to the manufacturer instructions; 1–2 μg of RNA was reverse transcribed with the High-Capacity cDNA Kit (Life Technologies), and cDNA was amplified by a 7900 HT Sequence Detection System (Life Technologies). Samples were always processed in triplicate. The relative gene expression was calculated by the formula 2(−ΔΔCt) using a defined group as reference (2(−ΔΔCt) = 1). The primer sequences were as follows: 5′-CTCCATGAGCTTTGTACAAGG-3′ for IL-1β forward and 5′-TGCTGATGTACCAGTTGGGG-3′ for IL-1β reverse; 5′-AGGTCGGTGTGAACGGATTTG-3′ for GAPDH forward and 5′-TGTAGACCATGTAGTAGTTGAGGTCA-3′ for GAPDH reverse (De Simoni et al., 2000).
Electrophysiology.
Male mice were anesthetized with isoflurane and decapitated, and their brains were transferred to ice-cold dissecting artificial CSF (aCSF) containing 87 mm NaCl, 75 mm sucrose, 2.5 mm KCl, 1.25 mm NaH2PO4, 7 mm MgCl2, 0.5 mm CaCl2, 25 mm NaHCO3, and 25 mm d-glucose, saturated with 95% O2 and 5% CO2 (Balducci et al., 2010). Oblique coronal sections (350 μm thick) were cut using a Vibratome 1000S Slicer (Leica), then transferred to aCSF containing 115 mm NaCl, 3.5 mm KCl, 1.2 mm NaH2PO4, 1.3 mm MgCl2, 2 mm CaCl2, 25 mm NaHCO3, and 25 mm d-glucose and aerated with 95% O2 and 5% CO2. After 15 min at 32°C, slices were kept at 22–24°C. During experiments, slices were continuously superfused with aCSF at a rate of 2 ml/min at 28°C.
Extracellular recordings of field postsynaptic potentials (fPSPs) were obtained in the CA1 stratum radiatum, using glass micropipettes filled with aCSF. Stimuli (50–160 μA, 50 μs) to excite Shaffer collaterals were delivered through a bipolar twisted tungsten electrode placed 400 μm from the recording electrode. Long-term potentiation (LTP) was induced using the following theta burst stimulation (TBS) protocol: 10 trains (4 pulses at 100 Hz) at 5 Hz, repeated twice with a 2 min interval. To induce metaplasticity, we applied a priming low-frequency stimulation (LFS) protocol (10 Hz, 10 pulses repeated twice, separated by 1 s) delivered 25 min before TBS (Costello et al., 2012). The magnitude of synaptic plasticity was evaluated by comparing the fPSP normalized slopes from the last 5 min of baseline recordings with those 18–26 min after LFS or 35–45 min after TBS planned comparison.
Mouse model of seizures.
Male mice (6–10) were surgically implanted under general gas anesthesia (1–3% isoflurane in O2) and stereotaxic guidance (Iori et al., 2013). Two nichrome-insulated bipolar depth electrodes (outer diameter, 60 μm) were implanted bilaterally into the dorsal hippocampus (from bregma: nose bar, 0 mm; anteroposterior, −1.8 mm; lateral, 1.5 and 2.0 mm below dura mater). A 23-gauge cannula was unilaterally positioned on top of the dura mater and glued to one of the depth electrodes for the intrahippocampal injection of KA (Balosso et al., 2008; Iori et al., 2013, 2017). The electrodes were connected to a multipin socket and, together with the injection cannula, were secured to the skull with acrylic dental cement. KA (Sigma-Aldrich) was injected intrahippocampally in freely moving mice, 7 d after surgery (Iori et al., 2013). KA 7 ng in 0.5 μl was dissolved in 0.1 m PBS, pH 7.4, and injected unilaterally into the dorsal hippocampus in freely moving mice using a needle protruding 2.0 mm from the bottom of the guide cannula. The needle was left in place for 1 more minute to avoid backflow through the cannula. This dose of KA induces EEG ictal episodes in the hippocampus in 100% of mice with no mortality (Iori et al., 2013). Human recombinant IL-1 receptor antagonist (anakinra; Biovitrum) was diluted in sterile saline and bilaterally injected intracerebroventricularly (0.5 μg/0.5 μl/side) in mice, 10 min before KA. For injection of saline or anakinra, mice were implanted with two additional cannulae bilaterally on top of the dura mater.
EEG activity was recorded using the Twin EEG Recording System (version 4.5.3.23) connected with a Comet AS-40 32/8 Amplifier (sampling rate, 400 Hz; high-pass filter, 0.3 Hz; low-pass filter, 70 Hz; sensitivity, 2000 mV/cm; Grass-Telefactor). Digitized EEG data were processed using the Twin record and review software. EEG was recorded for 30 min before KA injection to assess baseline activity, and for 180 min after KA. At least one 30 min recording similar to baseline was required before ending the experiment.
Ictal episodes are characterized by high-frequency (7–10 Hz) and/or multispike complexes and/or high-voltage (700 μV to 1.0 mV) synchronized spikes occurring simultaneously in the injected and contralateral hippocampi. Seizure activity was quantified by measuring the number and total duration of seizures (summing up the duration of each ictal episode during the EEG recording). Seizures occurred with an average latency of ∼10 min from KA injection, then recurred for ∼120 min, and were associated with motor arrest of the mice.
Results
Tg(CJD) mice show an inversion of synaptic metaplasticity associated with alterations in NMDAR composition
The Tg(CJD) mice we used express mutant PrP at a level similar to that of endogenous PrP in WT mice, on a PrP KO genetic background (C57BL/6J/Prnp0/0); therefore, they express transgenic but not endogenous WT PrP (Dossena et al., 2008). They develop deficits in spatial working memory between 200 and 300 d of age; progressive motor dysfunction, first detectable in the accelerating Rotarod test between 300 and 350 d; overt clinical signs, such as ataxia, kyphosis, and foot clasp reflex, by ∼450 d; and die prematurely at ∼700 d (Dossena et al., 2008; Senatore et al., 2012).
We examined synaptic NMDA-dependent long-term plasticity at Schaffer collateral–commissural synapses in the CA1 area on ex vivo brain slices from presymptomatic ∼100-d-old Tg(CJD) mice, nontransgenic PrP KO littermates, and age-matched C57BL/6J (WT) controls. We first examined the ability of CA3-CA1 synapses to undergo LTP. TBS resulted in robust LTP in all groups of mice (WT mice, 134 ± 6% of baseline; PrP KO mice, 143 ± 8% of baseline; Tg(CJD) mice mice, 144 ± 10% of baseline; n = 7–9, p < 0.05; Fig. 1A).
Figure 1.

Young adult Tg(CJD) mice have normal hippocampal plasticity but abnormal metaplasticity. A, TBS of Schaffer collaterals induces significant LTP of fPSP responses in CA1 of ∼100-d-old WT mice (white circles; RM1W, F(7,9) = 30, p = 0.0003; Tukey's test, p < 0.05). LTP was similar in age-matched PrP KO mice (gray circles; RM1W, F(8,9) = 26, p = 0.0004; Tukey's test, p < 0.05) and Tg(CJD) mice (green circles; RM1W, F(6,9) = 18, p = 0.005; Tukey's test, p < 0.05; RM2W, F(2,21) = 0.7, p = 0.7; WT vs CJD mice, p > 0.05, Tukey's post hoc test). Insets, Superimposed averaged records (5 traces) from WT (left) PrP KO (center), and Tg(CJD) mice (right) before (gray line) and 35–45 min after the TBS stimulation (red line). B, Preceding the TBS with a priming stimulation (2 trains of 10 pulses at 10 Hz, spaced 1 s apart) prevented the LTP in WT mice (white circles; RM1W, F(6,9) = 0.4, p = 0.6) but not in PrP KO mice (gray circles; RM1W, F(9,9) = 13, p = 0.003; Tukey's test, p < 0.05) or Tg(CJD) mice (green circles; RM1W, F(8,9) = 22, p = 0.001; Tukey's test, p < 0.05; RM2W, F(2,23) = 0.9, p = 0.5; WT vs KO mice, p > 0.05; WT vs CJD mice, p < 0.001; KO vs CJD mice, p < 0.5, Tukey's post hoc test). The priming stimulus did not significantly alter the fPSP slope in Tg(CJD) mice compared with controls (WT mice: 100 ± 4% of baseline, 7 mice, RM1W, F(6,8) = 0.5, p = 0.5; PrP KO mice: 99 ± 2% of baseline, 10 mice, RM1W, F(9,8) = 0.2, p = 0.7; Tg(CJD) mice: 111 ± 4% of baseline, 9 mice, RM1W, F(8,9) = 7, p = 0.02; Tukey's test, p > 0.05; RM2W, F(2,23) = 1.0, p = 0.5; WT vs PrP KO mice, p > 0.05; WT vs Tg(CJD) mice, p > 0.05, Tukey's post hoc test). Insets, Superimposed averaged records (5 traces) from WT (left) PrP KO (center), and Tg(CJD) mice (right) before the priming stimulus (gray line), 18–26 min after the low-frequency priming stimulus (LFS; blue line) and 35–45 min after the TBS (red line). A, B, Averaged time courses (mean ± SEM) of normalized fPSP slopes. TBS or LFS (in the metaplasticity experiments) was performed at the red and the black arrows, respectively. Dashed horizontal lines define baseline responses.
Preactivation of various intracellular signaling pathways before the delivery of a plasticity induction protocol can prime hippocampal synapses by modifying NMDAR activity (Blitzer et al., 1998; Lu et al., 1998; Huang et al., 2001). This priming inhibits the subsequent induction of LTP (MacDonald et al., 2006), a process conceptualized as synaptic metaplasticity (Hulme et al., 2013). When we investigated metaplasticity at CA3–CA1 synapses of WT mice, we found that a low-frequency (10 Hz) priming stimulus (LFS) did not change basal neurotransmission (p > 0.05) but prevented the LTP after TBS (100 ± 3% of baseline; n = 7; p > 0.05; Fig. 1B), which is consistent with previous findings (Balducci et al., 2010). As in WT mice, LFS priming did not significantly affect basal synaptic responsiveness in Tg(CJD) and PrP KO animals (p > 0.05) but failed to inhibit LTP induction in Tg(CJD) mice (152 ± 11%; n = 9; p < 0.05), and to a lesser extent in PrP KO mice (125 ± 6%; n = 10; p < 0.05; Fig. 1B).
Hippocampal metaplasticity depends on changes in the localization and composition of postsynaptic NMDARs triggered by the priming stimulation (Christie et al., 1995; Gisabella et al., 2003). The inversion of metaplasticity in Tg(CJD) and PrP KO mice raises the possibility that the composition of NMDARs may be constitutively altered in these mice, impairing further changes after priming. To test this, we analyzed NMDAR subunit composition in hippocampal PSD-enriched fractions by immunoblotting. We found no changes in the total levels of GluN2A, GluN2B, and GluN1, but found a significant increase in phospho-Tyr1472 GluN2B in Tg(CJD) mice (Fig. 2), which is the phosphorylated isoform that potentiates NMDA-induced Ca2+ influx. The level of phospho-Tyr1472 GluN2B in PrP KO mice was intermediate between those of WT and Tg(CJD) mice (Fig. 2C).
Figure 2.

Increased phospho-Tyr GluN2B in hippocampal postsynaptic fractions of Tg(CJD) and PrP KO mice. A, Triton X-100-insoluble fractions representing the postsynaptic density were isolated from the hippocampi of WT, KO, and Tg(CJD) mice. Samples corresponding to 10 μg of protein were analyzed by Western blot with the antibodies indicated. B, The protein levels were quantified by densitometric analysis of Western blots, normalized for the level of actin and expressed as percentages of the level in WT mice. C, Tyrosine residue 1472-phosphorylated GluN2B was normalized on total GluN2B and expressed as percentages of the level in WT mice. Data are the mean ± SEM of 8–10 animals at 65–80 d of age; F(2,86) = 6.541, p = 0.0023 by one-way ANOVA; **p < 0.01 vs WT by Tukey's post hoc test.
These results indicate abnormal hippocampal NMDAR trafficking in the PSD fractions, with an increase in GluN2B synaptic expression in Tg(CJD) mice and, to a lesser extent, in PrP KO mice. This correlates well with the impairment in synaptic metaplasticity, which is also more evident in Tg(CJD) mice.
Tg(CJD) mice show age-dependent increases in IL-1β levels in the hippocampus
There is evidence that IL-1β promotes Src family kinase (SFK)-mediated Tyr1472 phosphorylation of GluN2B (Viviani et al., 2003), and its brain levels are high in CJD (Sharief et al., 1999; Shi et al., 2013; Llorens et al., 2014). We investigated IL-1β expression in the hippocampus of Tg(CJD) mice at presymptomatic (60 and 120 d), early (240 d), and advanced (> 400 d) symptomatic stages of their illness (Dossena et al., 2008; Senatore et al., 2012). Immunohistochemistry with an anti-GFAP antibody confirmed the proliferation and hypertrophy of astrocytes previously documented in Tg(CJD) mice (Dossena et al., 2008). Astrocytosis was already observed in the hippocampi of 60-d-old mice and increased with age (Fig. 3A). Staining with the microglial marker CD11b showed marked microglial activation already at 60 d of age (Fig. 3B).
Figure 3.

Tg(CJD) mice have proliferation of astrocytes and microglia in the hippocampus. A, Brain sections from Tg(CJD) mice at the ages indicated were stained with anti-GFAP antibody. Immunostaining revealed marked astrocytosis in the hippocampus. B, Immunostaining with anti-CD11b shows marked microgliosis in the hippocampus of a Tg(CJD) mouse at 60 d of age compared with an age-matched WT control. Scale bars, 100 μm. CA1, Field CA1 of hippocampus; h, hilus of the dentate gyrus. Insets, High-magnification images of resting (WT) and activated (CJD) microglia.
IL-1β-immunopositive cells were detected in the hippocampi of Tg(CJD) mice, but not in WT and PrP KO mice (Fig. 4A). There were gradually more of these cells in older Tg(CJD) mice (Fig. 4B). Coimmunofluorescent staining of IL-1β with GFAP, CD11b, or the neuronal marker NeuN (data not shown), showed that IL-1β was exclusively expressed by astrocytes (Fig. 4C). Quantitative RT-PCR showed a significant increase in IL-1β mRNA in the hippocampus of Tg(CJD) mice already at 60 d of age (Fig. 4D).
Figure 4.

Tg(CJD) mice have an age-related increase in IL-1β-positive astrocytes in the hippocampus. A, Brain sections from the Tg(CJD), WT, and PrP KO mice of the ages indicated were immunostained with anti-IL-1β antibody and nickel-intensified diaminobenzidine. Scale bar, 100 μm. Inset, High-magnification image of IL-1β-positive cells. B, The IL-1β-immunopositive cells in the hippocampus of mice of different genotypes and ages were counted and expressed as the fold change over the numbers in WT mice. Data are the mean ± SEM of 4–6 animals (2–3 brain sections each); F(4,18) = 22.20, p < 0.0001 by one-way ANOVA; ***p < 0.001; ****p < 0.0001 vs WT and KO by Tukey's post hoc test. C, Immunofluorescence staining of IL-1β (red) and GFAP or CD11b (green) in the dentate gyrus of the hippocampus of a Tg(CJD) mouse at 72 d of age. Sections were reacted with Hoechst 33258 to stain the cell nuclei (blue). The merged images show colocalization of IL-1β with GFAP, but not with CD11b. Scale bar, 10 μm. D, Total RNA was extracted from the hippocampi of 60- to 66-d-old WT, PrP KO, and Tg(CJD) mice, and was analyzed by qRT-PCR for IL-1β and GAPDH. mRNAs were quantified by the ΔΔCt method and expressed as the fold increase over WT. Data are the mean ± SEM of 4–5 animals per group; F(2,17) = 4.506, p = 0.0269 by one-way ANOVA; *p < 0.05 vs WT and KO by Holm–Sidak post hoc test.
These results indicate proinflammatory changes in the hippocampi of Tg(CJD) mice starting from a presymptomatic stage. This proinflammatory milieu, and particularly IL-1β released from activated astrocytes, may promote neuronal GluN2B phosphorylation over the level induced by the loss of PrP function in PrP KO mice. This may contribute to enhancing NMDAR activity in the mutant mice (Viviani et al., 2003).
The IL-1 receptor antagonist rescues the defect in hippocampal metaplasticity and the age-dependent increase in LTP in Tg(CJD) mice
IL-1β signaling participates in the regulation of synaptic plasticity in physiological and pathological conditions (Ross et al., 2003; Costello et al., 2011; Vezzani and Viviani, 2015). To investigate whether the age-dependent elevation of IL-1β in Tg(CJD) mice was associated with changes in hippocampal synaptic plasticity, we analyzed LTP and metaplasticity in ∼300-d-old mice. TBS resulted in efficient LTP in WT mice (129 ± 5% of baseline; 10 mice; p < 0.01; Fig. 5A). LTP was significantly reduced in slices from PrP KO mice (106 ± 5%; 13 mice; p > 0.05; Fig. 5A), which is consistent with the age-dependent impairment in LTP previously documented in these mice (Curtis et al., 2003). In contrast, TBS triggered a greater LTP in Tg(CJD) animals than in WT controls (171 ± 12%; 5 mice; p < 0.05; Fig. 5A), indicating enhanced synaptic responsiveness.
Figure 5.
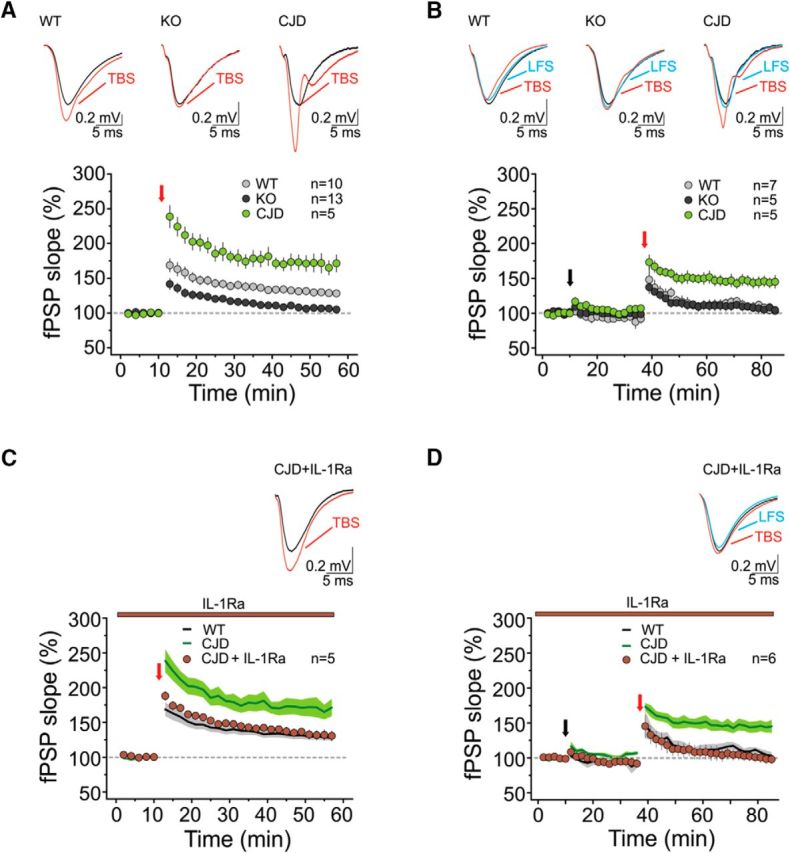
Aged Tg(CJD) mice have altered hippocampal plasticity and metaplasticity, which are normalized by IL-1 receptor antagonism. A, TBS induces LTP at the CA3–CA1 synapses of ∼300-d-old WT mice (gray circles; RM1W, F(9,9) = 36, p < 0.0001; Tukey's test, p < 0.01). The same protocol fails to induce LTP in brain slices of age-matched PrP KO mice (dark gray circles; RM1W, F(12,9) = 1.6, p = 0.2). In slices from Tg(CJD) mice, TBS results in LTP (light green circles; RM1W, F(4,9) = 31, p = 0.003; Tukey's test, p < 0.05) larger than in controls (RM2W, F(2,25) = 0.9, p = 0.6; WT vs KO, p < 0.05; WT vs CJD, p < 0.001; KO vs CJD, p < 0.001; Tukey's post hoc test). Insets, Superimposed averaged records (5 traces) from WT (left), PrP KO (center), and Tg(CJD) mice (right) before TBS (gray line) and 35–45 min after the TBS (red line). B, Upon LFS priming, LTP was absent in WT mice (gray circles; RM1W, F(6,9) = 3, p = 0.1) and PrP KO mice (dark gray circles; RM1W, F(4,9) = 1, p = 0.4), but not in Tg(CJD) mice (light green circles; RM1W, F(4,9) = 28, p = 0.0024; Tukey's test, p < 0.01; RM2W, F(2,14) = 0.6, p = 0.7; WT vs KO, p > 0.05; WT vs CJD, p < 0.001; KO vs CJD, p < 0.001; Tukey's post hoc test). The priming stimulus did not significantly alter fPSP responses in Tg(CJD) mice compared with controls (WT mice: 92 ± 5% of baseline, 7 mice, RM1W, F(6,8) = 1.3, p = 0.3; PrP KO mice: 98 ± 5% of baseline, 5 mice, RM1W, F(4,8) = 0.4, p = 0.6; Tg(CJD) mice: 104 ± 3% of baseline, 5 mice, RM1W, F(4,8) = 16, p = 0.001; Tukey's test, p > 0.05; RM2W, F(2,14) = 1.6, p = 0.1; WT vs KO, p > 0.05; WT vs CJD, p > 0.05; Tukey's post hoc test). Insets, Superimposed averaged records (5 traces) from WT (left), PrP KO (center), and Tg(CJD) mice (right) before the priming stimulus (gray line), 15–25 min after the LFS (blue line), and 35–45 min after the TBS (red line). A, B, Averaged time courses (mean ± SEM) of normalized fPSP slopes. TBS or LFS (in the metaplasticity experiments) was performed at the red and the black arrows, respectively. Dashed horizontal lines define baseline responses. C, Bath application of IL1-Ra reduced the magnitude of LTP in Tg(CJD) mice (brown circles; RM1W, F(4,9) = 22, p = 0.0055; Tukey's test, p < 0.05; the green line from A is reported here for comparison) to levels comparable to those of WT mice (black line, reported from A for comparison; RM2W, F(2,17) = 0.9, p = 0.5; CJD + IL-1Ra vs CJD, p < 0.05; CJD + IL-1Ra vs WT, p > 0.05; Tukey's post hoc test). Inset, Superimposed averaged records (5 traces) from CJD + IL-1Ra before the TBS (gray line) and 35–45 min after the TBS (red line). D, Upon bath application of IL-1Ra, the LFS priming protocol prevented LTP in Tg(CJD) mice (brown circles; RM1W, F(5,9) = 0.2, p = 0.7; the green line from B is reported here for comparison), similar to WT controls (black line, reported from B for comparison; RM2W, F(2,15) = 1.5, p = 0.2; CJD + IL-1Ra vs CJD, p < 0.05; CJD + IL-1Ra vs WT, p > 0.05; Tukey's post hoc test). The priming stimulus did not significantly alter the fPSP responses in CJD mice (94 ± 6% of baseline, 6 mice, RM1W, F(5,8) = 1.2, p = 0.3) compared with controls (RM2W, F(2,15) = 0.9, p = 0.6; CJD + IL-1Ra vs CJD, p > 0.05; CJD + IL-1Ra vs WT, p > 0.05; Tukey's post hoc test). Inset, Superimposed averaged records (5 traces) from CJD + IL-1Ra before the priming stimulus (gray line), 18–26 min after the LFS (blue line), and 35–45 min after the TBS (red line). C, D, Averaged time courses (mean ± SEM) of normalized fPSP amplitudes. TBS or LFS (in the metaplasticity experiments) was performed at the red and the black arrows, respectively. Dashed horizontal lines define baseline responses.
When we examined synaptic metaplasticity, we found that the priming stimulus efficiently suppressed LTP in slices from WT animals (107 ± 5% of baseline; 7 mice; p > 0.05) but failed to abolish it in Tg(CJD) mice (145 ± 8%; 5 mice; p < 0.01; Fig. 5B), similar to what was seen at ∼100 d. In ∼300-d-old PrP KO mice, we could not detect any metaplastic phenomena (107 ± 6%; 5 mice; p > 0.05), due to impaired LTP even in the absence of LFS (Fig. 5B). As in ∼100-d-old animals, the priming protocol did not modify the basal synaptic responses per se (Fig. 5B).
Next, we tested whether blocking IL-1β signaling could correct the abnormal plasticity in Tg(CJD) mice. Bath application of anakinra, the human recombinant IL-1 receptor type 1 antagonist (IL-1Ra), on brain slices from Tg(CJD) mice reduced the LTP to a level comparable to that of WT controls (133 ± 6% of baseline; 5 mice; p < 0.05; Fig. 5C). IL-1Ra also normalized metaplasticity in the mutant mice (100 ± 7%; 6 mice; p > 0.05), restoring the effectiveness of the priming protocol in preventing the induction of LTP (Fig. 5D).
These results indicate that IL-1β contributes to the metaplastic abnormalities and the age-dependent increase in hippocampal synaptic responsiveness in Tg(CJD) mice.
Tg(CJD) mice have enhanced susceptibility to kainate-induced seizures, which depends on IL-1β signaling
Since IL-1β, by promoting SFK activation and GluN2B phosphorylation (Viviani et al., 2003), has proconvulsive effects (Balosso et al., 2008; Galic et al., 2008), we tested whether the Tg(CJD) mice were more prone to seizures. Focal-onset acute seizures were induced by unilateral intrahippocampal injection of KA in Tg(CJD), PrP KO, and WT mice between the ages of 210 and 310 d. EEG recordings showed there were more seizures and longer times in ictal activity in Tg(CJD) mice than in PrP KO and WT mice (Fig. 6A–C). To test the contribution of IL-1β signaling in this seizure susceptibility, we injected Tg(CJD) mice with IL-1Ra (0.5 μg in 0.5 μl, i.c.v.) 10 min before intrahippocampal KA. The number of seizures and time in ictal activity were significantly reduced in Tg(CJD) mice but not in WT mice (Fig. 6D,E).
Figure 6.

Tg(CJD) mice have increased susceptibility to kainate-induced seizures, which depends on IL-1β signaling. A, Representative EEG tracings depicting baseline recordings (top) and ictal activity after intrahippocampal KA injection (bottom) in the left (LHP) and right (RHP) hippocampus in freely moving mice. B, C, Number of seizures and time spent in seizures in WT (n = 8), PrP KO (n = 9), and Tg(CJD) mice (n = 6) injected with KA. Data are the mean ± SEM; **p < 0.01 vs WT and KO mice by one-way ANOVA (F(2,20) = 19.41 in B and F(2,20) = 17.25 in C, p < 0.0001) followed by Tukey's test. D, E, Number of seizures and time spent in seizures in WT and Tg(CJD) mice injected with IL-1Ra (anakinra) or saline (intracerebroventricularly and bilaterally; 0.5 μg in 0.5 μl/side), 10 min before KA. Data are the mean ± SEM (9–10 each group); **p < 0.01 vs WT; and #p < 0.05 vs Tg(CJD) + saline by Mann–Whitney test (D; p = 0.0102; E, p = 0.0133; Tg(CJD)+IL-1Ra vs Tg(CJD)).
To exclude that differences in rearing conditions between commercial WT mice and Tg(CJD) and PrP KO mice raised in-house could affect the results, we compared the susceptibility to kainate-induced seizures of C57BL/6J adult male mice purchased from our external provider and exposed to seizures 2 weeks after arrival in our animal facility to that of mice of the same strain, age, and sex that were born and raised in-house (7 mice/group). We found no significant differences in time to seizure onset, number of seizures, and time in seizure between the two groups (mean ± SD; WT raised in-house: 10.0 ± 4.0 min, 7.9 ± 2.3, 6.7 ± 2.3 min; WT from Envigo: 8.0 ± 2.6 min, 6.9 ± 3.7, 5.0 ± 1.8 min).
Discussion
The present study found that Tg(CJD) mice, which express a misfolded mutant PrP, had alterations in hippocampal metaplasticity and an age-related increase in the magnitude of LTP. They also had high levels of phosphorylated GluN2B, indicative of enhanced NMDAR activity, and were intrinsically more susceptible to hippocampal seizures. Subtle alterations in synaptic metaplasticity and a modest increase in GluN2B phosphorylation were also seen in PrP KO mice. However, these mice had an age-dependent decrease in LTP, and no alterations in hippocampal seizure susceptibility. Tg(CJD) mice had proinflammatory changes, with progressive astrogliosis and microgliosis, and more IL-1β-positive astrocytes in the hippocampus. Blocking IL-1β signaling rescued the abnormalities in metaplasticity and the age-related increase in LTP, and significantly reduced susceptibility to seizures.
These findings indicate an enhancement of NMDA-dependent synaptic signaling in the Tg(CJD) hippocampus, which may result from a combination of the loss and gain of function of mutant PrP and involves neuroinflammation. They suggest that anti-inflammatory drugs that block IL-1β signaling, perhaps in combination with NMDAR antagonists, may help to normalize hippocampal synaptic activity, with beneficial effects on the symptomatic expression of disease.
Tg(CJD) and PrP KO mice develop similar and divergent abnormalities in hippocampal synaptic plasticity
In hippocampal slices from ∼100-d-old Tg(CJD) mice, LTP was normal at the CA3–CA1 synapses, but there was an inversion of metaplasticity, with a significant increase in phosphorylation of the NMDAR GluN2B subunit at tyrosine residue 1472. Phosphorylation at this C-terminal regulatory site prevents endocytosis of GluN2B, enhancing its cell surface expression and NMDAR activity (Salter and Kalia, 2004). In the hippocampus, low-frequency (10 Hz) presynaptic stimulation triggers NMDA-dependent metaplastic changes that inhibit the induction of LTP (Zhang et al., 2005). This homeostatic control may serve to prevent LTP from occurring too readily in response to weak stimuli or to restrain LTP induction after strong stimuli, thus helping to maintain synaptic efficacy within a dynamic range permissive for a learning-ready state and memory retention (Abraham, 2008). The GluN2B-type NMDARs mediate maximal increases in intracellular Ca2+ in response to low-frequency stimulation, while GluN2A-type receptors activate maximally in response to high-frequency synaptic inputs (Erreger et al., 2005). The hyperphosphorylation of GluN2B in Tg(CJD) mice may change the biophysical properties of NMDAR, with consequent alterations in postsynaptic Ca2+ dynamic and downstream Ca2+-dependent signaling after the 10 Hz priming stimulus. This may lead to the inversion of metaplasticity without affecting LTP and potentially contribute to the memory impairment that develops in these mice (Dossena et al., 2008).
There was a modest increase in phospho-Tyr1472 GluN2B in PrP KO mice. There is evidence that PrP participates in a signaling cascade that activates SFK Fyn and promotes GluN2B phosphorylation (Um et al., 2012), and the loss of PrP may result in constitutive activation of this signaling. We have also found that PrP interacts physically with GluN2B (unpublished observation), possibly promoting its intracellular transport, or acting as a scaffold protein to target the receptor to specific microdomains of the synaptic membrane (Senatore et al., 2013). GluN2B may be hyperphosphorylated in PrP KO mice as an adaptive response to permit interaction with other proteins that favor its cell surface expression (Maier et al., 2013). The modest increase of phospho-Tyr1472 GluN2B in the hippocampus of PrP KO mice, however, does not appear to have important functional consequences, since metaplasticity is only slightly altered, and mice are not impaired in hippocampus-dependent learning and memory (Dossena et al., 2008; Bouybayoune et al., 2015; unpublished data). Other mechanisms should therefore presumably be operative in Tg(CJD) mice that raise GluN2B phosphorylation over the level induced by the loss of PrP, leading to hippocampal dysfunction.
We previously reported that PrP deposition in the brains of Tg(CJD) mice was associated with prominent hypertrophy and proliferation of astrocytes in the hippocampus (Dossena et al., 2008). We have now shown that microglia are also activated, and that both microgliosis and astrogliosis prefigure the appearance of neurological deficits and increase further during the symptomatic phase of the disease, most likely reflecting the progressive accumulation of misfolded PrP (Bouybayoune et al., 2015). The novel information is that hippocampal IL-1β is specifically raised in astrocytes, in accordance with previous evidence of high whole-tissue levels in variant and sporadic CJD and in CJD-infected mice (Kordek et al., 1996; Sharief et al., 1999; Shi et al., 2013; Llorens et al., 2014).
The mechanism by which misfolded mutant PrP triggers IL-1β biosynthesis in astroglial cells in vivo is not known. The application of the neurotoxic PrP106–126 peptide to cultured astrocytes and microglial cells stimulates their proliferation and IL-1β release through the activation of nuclear factor-κB (Forloni et al., 1994; Hafiz and Brown, 2000; Lu et al., 2012), suggesting that the misfolded PrP in the brains of Tg(CJD) mice may have a similar effect.
Electrophysiological analysis detected a dramatic increase in hippocampal LTP magnitude in ∼300-d-old Tg(CJD) mice and weakened LTP in PrP KO littermates, clearly differentiating the effects of mutant PrP from those of PrP deletion. At this age, we saw a marked increase of IL-1β-positive astrocytes in the Tg(CJD) hippocampus. IL-1β dose-dependently induced GluN2B phosphorylation in cultured hippocampal neurons through the activation of SFK, and this effect was abolished by the IL-1 receptor antagonist (Viviani et al., 2003). Consistent with a role of IL-1β in altering NMDAR responses in Tg(CJD) mice, IL-1Ra restored normal LTP and rescued the metaplastic defect in brain slices.
Hypersynchronous hippocampal bursting caused by enhanced NMDAR-mediated excitation has been reported in mice infected with vCJD prions (Ratté et al., 2008). In the light of the high IL-1β levels in vCJD patients (Sharief et al., 1999), it may be worth measuring this cytokine in the brain of vCJD-infected mice and test whether anakinra prevents these hypersynchronous hippocampal bursting.
LTP was significantly impaired in ∼300-d-old PrP KO mice. An age-related alteration in hippocampal LTP was previously reported in Zürich I PrP KO mice (the strain also used in our study) and in an independently generated line of coisogenic 129/Ola mice in which the Prnp locus was disrupted using a different strategy (Curtis et al., 2003). However, analysis of hippocampal synaptic plasticity in PrP KO mice has produced conflicting results (Collinge et al., 1994; Manson et al., 1995; Lledo et al., 1996; Maglio et al., 2004, 2006). Differences in the induction protocol, animal model, and/or age of the mice may account for these. Zürich I mice, as well as mice in which PrP was postnatally ablated only in neurons, also had a significant attenuation of afterhyperpolarization potentials in CA1 pyramidal neurons (Colling et al., 1996; Mallucci et al., 2002), supporting a direct role for PrP in hippocampal excitability.
Tg(CJD) but not PrP KO mice show enhanced susceptibility to hippocampal seizures, which depends on IL-1β signaling
Tg(CJD) mice were intrinsically more susceptible to hippocampal seizures, suggesting a low seizure threshold. The competitive IL-1R type 1 antagonist IL-1Ra significantly blocked this effect, indicating that it was mediated by the endogenous IL-1β, which was increased in Tg(CJD) mouse hippocampus. In full accordance with this, there is evidence that intrahippocampally injected IL-1β increases kainate seizures—and other types of seizures—in rodents (Vezzani et al., 1999, 2000; Maroso et al., 2011; Iori et al., 2017), and IL-1β proictogenic effects were blocked by IL-1Ra (Vezzani et al., 1999, 2000). Gliosis was attenuated, disease onset was delayed, and survival was significantly prolonged in prion-infected mice lacking IL-1R type 1 (Schultz et al., 2004; Tamgüney et al., 2008). The effects of IL-1β on seizures are mediated by Src family kinase phosphorylation of GluN2B, which promotes neuronal Ca2+ influx (Viviani et al., 2003; Balosso et al., 2008). Since IL-1β is increased in the brain and CSF of CJD patients, this may contribute to the synaptic alterations and seizures in patients in advanced stages of the disease (Wieser et al., 2006). These data support the idea that anakinra, or other anti-IL-1β drugs, might have therapeutic effects in prion diseases. Notably, two proof-of-concept clinical studies have recently shown that anakinra is effective in treating drug-resistant seizures in epilepsy patients (Jyonouchi and Geng, 2016; Kenney-Jung et al., 2016).
At variance with previous observations (Walz et al., 1999; Rangel et al., 2007), we found no difference in susceptibility to seizures between PrP KO and WT mice; this might be due to focal rather than systemic administration of KA, the latter also involving extrahippocampal brain regions contributing to seizures. In addition, we used mice with a homogeneous C57BL/6J background, whereas other studies used PrP KO mice with mixed genetic backgrounds (typically 129Sv × C57BL/6J), and genetic heterogeneity may profoundly affect seizure susceptibility in mice (Schauwecker, 2011; Striebel et al., 2013a,b).
Animal husbandry and rearing conditions can also affect susceptibility to seizures (Leussis and Heinrichs, 2006, 2009), and our C57BL/6J controls were purchased from an external provider whereas the Tg(CJD) and PrP KO mice were raised in-house. However, this difference is unlikely to have affected our results since we found no differences in seizure susceptibility between C57BL/6J mice purchased from Envigo and those that were born and raised in-house. Moreover, all mice from the external vendor were housed for at least 2 weeks in our animal facilities together with Tg(CJD) and PrP KO mice before being used for the experiments.
In summary, we provide evidence of dysfunctional NMDA-dependent hippocampal synaptic plasticity and enhanced seizure susceptibility in Tg(CJD) mice. These effects appear to result from altered NMDAR activity and neuroinflammation. Targeting NMDAR and IL-1β signaling with clinically available drugs may be beneficial in the symptomatic treatment of the disease.
Footnotes
This work was supported by grants from Telethon-Italy (GGP12115), Fondazione Cariplo (2012-0560), the Italian Ministry of Health (RF-2010-2314035), the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 602102 (EPITARGET), and the Fondazione Istituto Italiano di Tecnologia.
The authors declare no competing financial interests.
References
- Abraham WC. (2008) Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci 9:387. 10.1038/nrn2356 [DOI] [PubMed] [Google Scholar]
- Balducci C, Tonini R, Zianni E, Nazzaro C, Fiordaliso F, Salio M, Vismara L, Gardoni F, Di Luca M, Carli M, Forloni G (2010) Cognitive deficits associated with alteration of synaptic metaplasticity precede plaque deposition in AbetaPP23 transgenic mice. J Alzheimers Dis 21:1367–1381. 10.3233/JAD-2010-100675 [DOI] [PubMed] [Google Scholar]
- Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, Vezzani A (2008) A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain 131:3256–3265. 10.1093/brain/awn271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM (1998) Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 280:1940–1942. 10.1126/science.280.5371.1940 [DOI] [PubMed] [Google Scholar]
- Bouybayoune I, Mantovani S, Del Gallo F, Bertani I, Restelli E, Comerio L, Tapella L, Baracchi F, Fernández-Borges N, Mangieri M, Bisighini C, Beznoussenko GV, Paladini A, Balducci C, Micotti E, Forloni G, Castilla J, Fiordaliso F, Tagliavini F, Imeri L, et al. (2015) Transgenic fatal familial insomnia mice indicate prion infectivity-independent mechanisms of pathogenesis and phenotypic expression of disease. PLoS Pathog 11:e1004796. 10.1371/journal.ppat.1004796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Herms JW, Schmidt B, Kretzschmar HA (1997) Prp and beta-amyloid fragments activate different neurotoxic mechanisms in cultured mouse cells. Eur J Neurosci 9:1162–1169. 10.1111/j.1460-9568.1997.tb01470.x [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C (1992) Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356:577–582. 10.1038/356577a0 [DOI] [PubMed] [Google Scholar]
- Carulla P, Llorens F, Matamoros-Angles A, Aguilar-Calvo P, Espinosa JC, Gavín R, Ferrer I, Legname G, Torres JM, del Río JA (2015) Involvement of PrP(C) in kainate-induced excitotoxicity in several mouse strains. Sci Rep 5:11971. 10.1038/srep11971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R. (2015) The elusive role of the prion protein and the mechanism of toxicity in prion disease. PLoS Pathog 11:e1004745. 10.1371/journal.ppat.1004745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie BR, Stellwagen D, Abraham WC (1995) Reduction of the threshold for long-term potentiation by prior theta-frequency synaptic activity. Hippocampus 5:52–59. 10.1002/hipo.450050107 [DOI] [PubMed] [Google Scholar]
- Colby DW, Prusiner SB (2011) Prions. Cold Spring Harb Perspect Biol 3:a006833. 10.1101/cshperspect.a006833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colling SB, Collinge J, Jefferys JG (1996) Hippocampal slices from prion protein null mice: disrupted Ca(2+)-activated K+ currents. Neurosci Lett 209:49–52. 10.1016/0304-3940(96)12596-9 [DOI] [PubMed] [Google Scholar]
- Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG (1994) Prion protein is necessary for normal synaptic function. Nature 370:295–297. 10.1038/370295a0 [DOI] [PubMed] [Google Scholar]
- Costello DA, Watson MB, Cowley TR, Murphy N, Murphy Royal C, Garlanda C, Lynch MA (2011) Interleukin-1α and HMGB1 mediate hippocampal dysfunction in SIGIRR-deficient mice. J Neurosci 31:3871–3879. 10.1523/JNEUROSCI.6676-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello DA, Claret M, Al-Qassab H, Plattner F, Irvine EE, Choudhury AI, Giese KP, Withers DJ, Pedarzani P (2012) Brain deletion of insulin receptor substrate 2 disrupts hippocampal synaptic plasticity and metaplasticity. PLoS One 7:e31124. 10.1371/journal.pone.0031124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis J, Errington M, Bliss T, Voss K, MacLeod N (2003) Age-dependent loss of PTP and LTP in the hippocampus of PrP-null mice. Neurobiol Dis 13:55–62. 10.1016/S0969-9961(03)00017-2 [DOI] [PubMed] [Google Scholar]
- De Simoni MG, Perego C, Ravizza T, Moneta D, Conti M, Marchesi F, De Luigi A, Garattini S, Vezzani A (2000) Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur J Neurosci 12:2623–2633. 10.1046/j.1460-9568.2000.00140.x [DOI] [PubMed] [Google Scholar]
- Dossena S, Imeri L, Mangieri M, Garofoli A, Ferrari L, Senatore A, Restelli E, Balducci C, Fiordaliso F, Salio M, Bianchi S, Fioriti L, Morbin M, Pincherle A, Marcon G, Villani F, Carli M, Tagliavini F, Forloni G, Chiesa R (2008) Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron 60:598–609. 10.1016/j.neuron.2008.09.008 [DOI] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ, Traynelis SF (2005) Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol 563:345–358. 10.1113/jphysiol.2004.080028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa PS, Bensalem-Owen MK, Fee DB (2010) Sporadic Creutzfeldt-Jakob disease presenting as nonconvulsive status epilepticus case report and review of the literature. Clin Neurol Neurosurg 112:537–540. 10.1016/j.clineuro.2010.03.025 [DOI] [PubMed] [Google Scholar]
- Forloni G, Del Bo R, Angeretti N, Chiesa R, Smiroldo S, Doni R, Ghibaudi E, Salmona M, Porro M, Verga L (1994) A neurotoxic prion protein fragment induces rat astroglial proliferation and hypertrophy. Eur J Neurosci 6:1415–1422. 10.1111/j.1460-9568.1994.tb01003.x [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ (2008) Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 28:6904–6913. 10.1523/JNEUROSCI.1901-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisabella B, Rowan MJ, Anwyl R (2003) Mechanisms underlying the inhibition of long-term potentiation by preconditioning stimulation in the hippocampus in vitro. Neuroscience 121:297–305. 10.1016/S0306-4522(03)00440-8 [DOI] [PubMed] [Google Scholar]
- Hafiz FB, Brown DR (2000) A model for the mechanism of astrogliosis in prion disease. Mol Cell Neurosci 16:221–232. 10.1006/mcne.2000.0868 [DOI] [PubMed] [Google Scholar]
- Head MW, Ironside JW (2012) Review: Creutzfeldt-Jakob disease: prion protein type, disease phenotype and agent strain. Neuropathol Appl Neurobiol 38:296–310. 10.1111/j.1365-2990.2012.01265.x [DOI] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF (2001) CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 29:485–496. 10.1016/S0896-6273(01)00220-3 [DOI] [PubMed] [Google Scholar]
- Hulme SR, Jones OD, Abraham WC (2013) Emerging roles of metaplasticity in behaviour and disease. Trends Neurosci 36:353–362. 10.1016/j.tins.2013.03.007 [DOI] [PubMed] [Google Scholar]
- Iori V, Maroso M, Rizzi M, Iyer AM, Vertemara R, Carli M, Agresti A, Antonelli A, Bianchi ME, Aronica E, Ravizza T, Vezzani A (2013) Receptor for Advanced Glycation Endproducts is upregulated in temporal lobe epilepsy and contributes to experimental seizures. Neurobiol Dis 58:102–114. 10.1016/j.nbd.2013.03.006 [DOI] [PubMed] [Google Scholar]
- Iori V, Iyer AM, Ravizza T, Beltrame L, Paracchini L, Marchini S, Cerovic M, Hill C, Ferrari M, Zucchetti M, Molteni M, Rossetti C, Brambilla R, Steve White H, D'Incalci M, Aronica E, Vezzani A (2017) Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis 99:12–23. 10.1016/j.nbd.2016.12.007 [DOI] [PubMed] [Google Scholar]
- Jyonouchi H, Geng L (2016) Intractable epilepsy (IE) and responses to anakinra, a human recombinant IL-1 receptor agonist (IL-1ra): case reports. J Clin Cell Immunol 7:5 10.4172/2155-9899.1000456 [DOI] [Google Scholar]
- Kenney-Jung DL, Vezzani A, Kahoud RJ, LaFrance-Corey RG, Ho ML, Muskardin TW, Wirrell EC, Howe CL, Payne ET (2016) Febrile infection-related epilepsy syndrome treated with anakinra. Ann Neurol 80:939–945. 10.1002/ana.24806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, Zamponi GW (2008) Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol 181:551–565. 10.1083/jcb.200711002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8:e1000412. 10.1371/journal.pbio.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordek R, Nerurkar VR, Liberski PP, Isaacson S, Yanagihara R, Gajdusek DC (1996) Heightened expression of tumor necrosis factor alpha, interleukin 1 alpha, and glial fibrillary acidic protein in experimental Creutzfeldt-Jakob disease in mice. Proc Natl Acad Sci U S A 93:9754–9758. 10.1073/pnas.93.18.9754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leussis MP, Heinrichs SC (2006) Routine tail suspension husbandry facilitates onset of seizure susceptibility in EL mice. Epilepsia 47:801–804. 10.1111/j.1528-1167.2006.00525.x [DOI] [PubMed] [Google Scholar]
- Leussis MP, Heinrichs SC (2009) Quality of rearing guides expression of behavioral and neural seizure phenotypes in EL mice. Brain Res 1260:84–93. 10.1016/j.brainres.2009.01.007 [DOI] [PubMed] [Google Scholar]
- Lledo PM, Tremblay P, DeArmond SJ, Prusiner SB, Nicoll RA (1996) Mice deficient for prion protein exhibit normal neuronal excitability and synaptic transmission in the hippocampus. Proc Natl Acad Sci U S A 93:2403–2407. 10.1073/pnas.93.6.2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens F, López-González I, Thüne K, Carmona M, Zafar S, Andréoletti O, Zerr I, Ferrer I (2014) Subtype and regional-specific neuroinflammation in sporadic Creutzfeldt-Jakob disease. Front Aging Neurosci 6:198. 10.3389/fnagi.2014.00198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Roder JC, Davidow J, Salter MW (1998) Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science 279:1363–1367. 10.1126/science.279.5355.1363 [DOI] [PubMed] [Google Scholar]
- Lu Y, Liu A, Zhou X, Kouadir M, Zhao W, Zhang S, Yin X, Yang L, Zhao D (2012) Prion peptide PrP106–126 induces inducible nitric oxide synthase and proinflammatory cytokine gene expression through the activation of NF-kappaB in macrophage cells. DNA Cell Biol 31:833–838. 10.1089/dna.2011.1362 [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Jackson MF, Beazely MA (2006) Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors. Crit Rev Neurobiol 18:71–84. 10.1615/CritRevNeurobiol.v18.i1-2.80 [DOI] [PubMed] [Google Scholar]
- Maglio LE, Perez MF, Martins VR, Brentani RR, Ramirez OA (2004) Hippocampal synaptic plasticity in mice devoid of cellular prion protein. Brain Res Mol Brain Res 131:58–64. 10.1016/j.molbrainres.2004.08.004 [DOI] [PubMed] [Google Scholar]
- Maglio LE, Martins VR, Izquierdo I, Ramirez OA (2006) Role of cellular prion protein on LTP expression in aged mice. Brain Res 1097:11–18. 10.1016/j.brainres.2006.04.056 [DOI] [PubMed] [Google Scholar]
- Maier W, Bednorz M, Meister S, Roebroek A, Weggen S, Schmitt U, Pietrzik CU (2013) LRP1 is critical for the surface distribution and internalization of the NR2B NMDA receptor subtype. Mol Neurodegener 8:25. 10.1186/1750-1326-8-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci GR, Ratté S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J (2002) Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J 21:202–210. 10.1093/emboj/21.3.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson JC, Hope J, Clarke AR, Johnston A, Black C, MacLeod N (1995) PrP gene dosage and long term potentiation. Neurodegeneration 4:113–114. 10.1006/neur.1995.0014 [DOI] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Iori V, Wright CI, French J, Vezzani A (2011) Interleukin-1beta biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics 8:304–315. 10.1007/s13311-011-0039-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejías-Aponte CA, Jiménez-Rivera CA, Segarra AC (2002) Sex differences in models of temporal lobe epilepsy: role of testosterone. Brain Res 944:210–218. 10.1016/S0006-8993(02)02691-4 [DOI] [PubMed] [Google Scholar]
- Müller WE, Ushijima H, Schröder HC, Forrest JM, Schatton WF, Rytik PG, Heffner-Lauc M (1993) Cytoprotective effect of NMDA receptor antagonists on prion protein (PrionSc)-induced toxicity in rat cortical cell cultures. Eur J Pharmacol 246:261–267. 10.1016/0922-4106(93)90040-G [DOI] [PubMed] [Google Scholar]
- Nazzaro C, Greco B, Cerovic M, Baxter P, Rubino T, Trusel M, Parolaro D, Tkatch T, Benfenati F, Pedarzani P, Tonini R (2012) SK channel modulation rescues striatal plasticity and control over habit in cannabinoid tolerance. Nat Neurosci 15:284–293. 10.1038/nn.3022 [DOI] [PubMed] [Google Scholar]
- Perovic S, Pergande G, Ushijima H, Kelve M, Forrest J, Müller WE (1995) Flupirtine partially prevents neuronal injury induced by prion protein fragment and lead acetate. Neurodegeneration 4:369–374. 10.1006/neur.1995.0044 [DOI] [PubMed] [Google Scholar]
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P (2012) Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol 11:618–628. 10.1016/S1474-4422(12)70063-7 [DOI] [PubMed] [Google Scholar]
- Rangel A, Burgaya F, Gavín R, Soriano E, Aguzzi A, Del Río JA (2007) Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: role of AMPA/kainate receptors. J Neurosci Res 85:2741–2755. 10.1002/jnr.21215 [DOI] [PubMed] [Google Scholar]
- Rangel A, Madroñal N, Gruart i Massó A, Gavín R, Llorens F, Sumoy L, Torres JM, Delgado-García JM, Del Río JA (2009) Regulation of GABA(A) and glutamate receptor expression, synaptic facilitation and long-term potentiation in the hippocampus of prion mutant mice. PLoS One 4:e7592. 10.1371/journal.pone.0007592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratté S, Prescott SA, Collinge J, Jefferys JG (2008) Hippocampal bursts caused by changes in NMDA receptor-dependent excitation in a mouse model of variant CJD. Neurobiol Dis 32:96–104. 10.1016/j.nbd.2008.06.007 [DOI] [PubMed] [Google Scholar]
- Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, Multhaup G, Winklhofer KF, Tatzelt J (2011) The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J 30:2057–2070. 10.1038/emboj.2011.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FM, Allan SM, Rothwell NJ, Verkhratsky A (2003) A dual role for interleukin-1 in LTP in mouse hippocampal slices. J Neuroimmunol 144:61–67. 10.1016/j.jneuroim.2003.08.030 [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5:317–328. 10.1038/nrn1368 [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. (2011) The relevance of individual genetic background and its role in animal models of epilepsy. Epilepsy Res 97:1–11. 10.1016/j.eplepsyres.2011.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Schwarz A, Neidhold S, Burwinkel M, Riemer C, Simon D, Kopf M, Otto M, Baier M (2004) Role of interleukin-1 in prion disease-associated astrocyte activation. Am J Pathol 165:671–678. 10.1016/S0002-9440(10)63331-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatore A, Colleoni S, Verderio C, Restelli E, Morini R, Condliffe SB, Bertani I, Mantovani S, Canovi M, Micotti E, Forloni G, Dolphin AC, Matteoli M, Gobbi M, Chiesa R (2012) Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC alpha(2)delta-1 subunit. Neuron 74:300–313. 10.1016/j.neuron.2012.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senatore A, Restelli E, Chiesa R (2013) Synaptic dysfunction in prion diseases: a trafficking problem? Int J Cell Biol 2013:543803. 10.1155/2013/543803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharief MK, Green A, Dick JP, Gawler J, Thompson EJ (1999) Heightened intrathecal release of proinflammatory cytokines in Creutzfeldt-Jakob disease. Neurology 52:1289–1291. 10.1212/WNL.52.6.1289 [DOI] [PubMed] [Google Scholar]
- Shi Q, Xie WL, Zhang B, Chen LN, Xu Y, Wang K, Ren K, Zhang XM, Chen C, Zhang J, Dong XP (2013) Brain microglia were activated in sporadic CJD but almost unchanged in fatal familial insomnia and G114V genetic CJD. Virol J 10:216. 10.1186/1743-422X-10-216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorska B, Knight R, Ironside JW, Liberski PP (2012) Creutzfeldt-Jakob disease. Adv Exp Med Biol 724:76–90. 10.1007/978-1-4614-0653-2_6 [DOI] [PubMed] [Google Scholar]
- Striebel JF, Race B, Chesebro B (2013a) Prion protein and susceptibility to kainate-induced seizures: genetic pitfalls in the use of PrP knockout mice. Prion 7:280–285. 10.4161/pri.25738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striebel JF, Race B, Pathmajeyan M, Rangel A, Chesebro B (2013b) Lack of influence of prion protein gene expression on kainate-induced seizures in mice: studies using congenic, coisogenic and transgenic strains. Neuroscience 238:11–18. 10.1016/j.neuroscience.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamgüney G, Giles K, Glidden DV, Lessard P, Wille H, Tremblay P, Groth DF, Yehiely F, Korth C, Moore RC, Tatzelt J, Rubinstein E, Boucheix C, Yang X, Stanley P, Lisanti MP, Dwek RA, Rudd PM, Moskovitz J, Epstein CJ, et al. (2008) Genes contributing to prion pathogenesis. J Gen Virol 89:1777–1788. 10.1099/vir.0.2008/001255-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thellung S, Gatta E, Pellistri F, Corsaro A, Villa V, Vassalli M, Robello M, Florio T (2013) Excitotoxicity through NMDA receptors mediates cerebellar granule neuron apoptosis induced by prion protein 90–231 fragment. Neurotox Res 23:301–314. 10.1007/s12640-012-9340-9 [DOI] [PubMed] [Google Scholar]
- Trusel M, Cavaccini A, Gritti M, Greco B, Saintot PP, Nazzaro C, Cerovic M, Morella I, Brambilla R, Tonini R (2015) Coordinated regulation of synaptic plasticity at striatopallidal and striatonigral neurons orchestrates motor control. Cell Rep 13:1353–1365. 10.1016/j.celrep.2015.10.009 [DOI] [PubMed] [Google Scholar]
- Twele F, Töllner K, Brandt C, Löscher W (2016) Significant effects of sex, strain, and anesthesia in the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy. Epilepsy Behav 55:47–56. 10.1016/j.yebeh.2015.11.027 [DOI] [PubMed] [Google Scholar]
- Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM (2012) Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15:1227–1235. 10.1038/nn.3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Viviani B (2015) Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 96:70–82. 10.1016/j.neuropharm.2014.10.027 [DOI] [PubMed] [Google Scholar]
- Vezzani A, Conti M, De Luigi A, Ravizza T, Moneta D, Marchesi F, De Simoni MG (1999) Interleukin-1β immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci 19:5054–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Moneta D, Conti M, Richichi C, Ravizza T, De Luigi A, De Simoni MG, Sperk G, Andell-Jonsson S, Lundkvist J, Iverfeldt K, Bartfai T (2000) Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc Natl Acad Sci U S A 97:11534–11539. 10.1073/pnas.190206797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M (2003) Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 23:8692–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz R, Amaral OB, Rockenbach IC, Roesler R, Izquierdo I, Cavalheiro EA, Martins VR, Brentani RR (1999) Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia 40:1679–1682. 10.1111/j.1528-1157.1999.tb01583.x [DOI] [PubMed] [Google Scholar]
- Wieser HG, Schindler K, Zumsteg D (2006) EEG in Creutzfeldt-Jakob disease. Clin Neurophysiol 117:935–951. 10.1016/j.clinph.2005.12.007 [DOI] [PubMed] [Google Scholar]
- Zhang L, Kirschstein T, Sommersberg B, Merkens M, Manahan-Vaughan D, Elgersma Y, Beck H (2005) Hippocampal synaptic metaplasticity requires inhibitory autophosphorylation of Ca2+/calmodulin-dependent kinase II. J Neurosci 25:7697–7707. 10.1523/JNEUROSCI.2086-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]