

Abstract
Parvalbumin-expressing inhibitory neurons in the mammalian CNS are specialized for fast transmitter release at their output synapses. However, the Ca2+ sensor(s) used by identified inhibitory synapses, including the output synapses of parvalbumin-expressing inhibitory neurons, have only recently started to be addressed. Here, we investigated the roles of Syt1 and Syt2 at two types of fast-releasing inhibitory connections in the mammalian CNS: the medial nucleus of the trapezoid body to lateral superior olive glycinergic synapse, and the basket/stellate cell-Purkinje GABAergic synapse in the cerebellum. We used conditional and conventional knock-out (KO) mouse lines, with viral expression of Cre-recombinase and a light-activated ion channel for optical stimulation of the transduced fibers, to produce Syt1-Syt2 double KO synapses in vivo. Surprisingly, we found that KO of Syt2 alone had only minor effects on evoked transmitter release, despite the clear presence of the protein in inhibitory nerve terminals revealed by immunohistochemistry. We show that Syt1 is weakly coexpressed at these inhibitory synapses and must be genetically inactivated together with Syt2 to achieve a significant reduction and desynchronization of fast release. Thus, our work identifies the functionally relevant Ca2+ sensor(s) at fast-releasing inhibitory synapses and shows that two major Syt isoforms can cooperate to mediate release at a given synaptic connection.
SIGNIFICANCE STATEMENT During synaptic transmission, the influx of Ca2+ into the presynaptic nerve terminal activates a Ca2+ sensor for vesicle fusion, a crucial step in the activity-dependent release of neurotransmitter. Synaptotagmin (Syt) proteins, and especially Syt1 and Syt2, have been identified as the Ca2+ sensor at excitatory synapses, but the Ca2+ sensor(s) at inhibitory synapses in native brain tissue are not well known. We found that both Syt1 and Syt2 need to be genetically inactivated to cause a significant reduction of activity-evoked release at two types of fast inhibitory synapses in mouse brain. Thus, we identify Syt2 as a functionally important Ca2+ sensor at fast-releasing inhibitory synapses, and show that Syt1 and Syt2 can redundantly control transmitter release at specific brain synapses.
Keywords: calcium sensor, inhibitory synapse, neurotransmitter release, optogenetics, parvalbumin interneuron, synaptotagmin
Introduction
The mammalian brain contains a wide variety of inhibitory neuron types, which can act either locally or in long-range projections to inhibit their target neurons (Petilla Interneuron Nomenclature Group, 2008; Caputi et al., 2013; Kepecs and Fishell, 2014). Some inhibitory neurons, like the parvalbumin (PV)-expressing interneurons in the forebrain and cerebellum, are capable of very rapid transmitter release, helped by the tight coupling of presynaptic Ca2+ channels to the Ca2+ sensor for vesicle fusion (Bucurenciu et al., 2008; Arai and Jonas, 2014; Hu et al., 2014). To understand the mechanism of fast release at these inhibitory synapses, their Ca2+ sensor(s) should be identified. Although GABA release studied in neuronal cultures from conventional Syt1−/− mice is reduced and strongly desynchronized (Maximov and Südhof, 2005; Bacaj et al., 2013), it is only beginning to be understood which Ca2+ sensor(s) act at output synapses of identified inhibitory neurons in CNS circuits. Fast GABA release from parvalbumin (PV) interneurons in hippocampal organotypic cultures showed only minor deficits in Syt1−/− mice (Kerr et al., 2008). An alternative Ca2+ sensor was not identified, but it was observed that Syt2 mRNA was upregulated (Kerr et al., 2008).
Synaptotagmins are C2-domain-containing Ca2+-binding proteins (Pang and Südhof, 2010), and it is well established that Syt1 is the major Ca2+ sensor for transmitter release at excitatory forebrain synapses and at invertebrate synapses (Geppert et al., 1994; Yoshihara and Littleton, 2002). The Syt2 gene is found in vertebrates and drives the expression of a protein with high sequence homology to Syt1 (Geppert et al., 1991; Craxton, 2010). Syt2 is preferentially expressed in hindbrain and spinal cord (Geppert et al., 1991; Berton et al., 1997; Pang et al., 2006b) and is the main Ca2+ sensor for vesicle fusion at excitatory synapses formed by hindbrain or spinal cord neurons, like the vertebrate neuromuscular junction (Pang et al., 2006b; Wen et al., 2010), and the calyx of Held (Sun et al., 2007; Kochubey and Schneggenburger, 2011). Interestingly, gene expression studies and immunohistochemistry suggest that GABA-ergic nerve terminals of PV interneurons in the hippocampus and cortex contain Syt2, especially in older animals (Okaty et al., 2009; García-Junco-Clemente et al., 2010; Bragina et al., 2011; Sommeijer and Levelt, 2012). Recently, it was shown that a large excitatory synapse in the hindbrain, the calyx of Held, initially uses Syt1 as a Ca2+ sensor; Syt2 expression started only a few days after birth and then replaced Syt1 (Kochubey et al., 2016). However, only little is known about possible overlapping roles of Syt isoforms at identified inhibitory synapses.
Here, we studied the role of Syt2 at fast-releasing inhibitory synapses of the mouse brain, using the glycinergic inhibitory connection between the medial nucleus of the trapezoid body (MNTB) and lateral superior olive (LSO) neurons in the auditory brainstem (see Fig. 1A) (Kim and Kandler, 2003), and the basket/stellate cell-Purkinje cell GABAergic synapse in the cerebellum (Vincent et al., 1992) as model synapses. Surprisingly, we found that genetic deletion of Syt2, despite the clear presence of the protein immunohistochemically, did not lead to a significant reduction of fast release at either synapse. We then found that, at the MNTB-LSO synapse, Syt1 is weakly coexpressed with the immunohistochemically more dominant Syt2 isoform at the age studied here (P12-P15). To study redundant roles of these two major synaptotagmin isoforms at identified inhibitory synapses ex vivo, we used a conditional Syt1 knock-out (KO) mouse, combined with virus-mediated expression of Cre-recombinase and a light-sensitive ion channel to allow selective stimulation of molecularly perturbed afferent fibers. With these tools, we show that genetic inactivation of both Syt1 and Syt2 produces strong alterations in the amount and kinetics of fast inhibitory transmitter release, whereas single KO synapses had much smaller, or absent, phenotypes. This identifies the functional role of Syt2 at fast-releasing inhibitory synapses and shows that two major Syt isoforms can act redundantly at a given synaptic connection.
Figure 1.

Glycine release at the MNTB-LSO inhibitory synapse is unchanged in Syt2−/− mice. A, Scheme of the MNTB-LSO inhibitory synapse (Kim and Kandler, 2003). B, IPSCs recorded in LSO neurons of a Syt2+/+ littermate control (wild-type) mouse at P12 (left) and in a Syt2−/− mouse at P14 (right), following afferent fiber stimulation. Stimulation intensities are indicated. C, Average and individual data points for minimal and maximal IPSC amplitudes. D, Quantification of 20%–80% rise time of IPSCs (left) and IPSC decay time constants (right). These parameters were not significantly different in Syt2−/− mice (n = 10) compared with Syt2+/+ mice (n = 7; p > 0.05 for all comparisons, unpaired t test). E, Example traces for spontaneous IPSCs recorded in a Syt2+/+ mouse (left; P14) and in a Syt2−/− mouse (right; P15). F, Average and individual data points for spontaneous IPSC frequency and spontaneous IPSC amplitude. **p < 0.01. For statistical tests used, see Materials and Methods.
Materials and Methods
Animals.
All experimental procedures were approved by the veterinary office of the Canton of Vaud, Switzerland (authorizations 1880.3 and 2063.3). The Syt2+/− mouse line (RRID: MGI:3696550) was described previously (Pang et al., 2006b; Kochubey and Schneggenburger, 2011). For the fiber-stimulation experiments (see Fig. 1), Syt2−/− mice were obtained from heterozygous breeding. Because Syt2−/− mice experience a developmental aggravation of a motor phenotype and die at ∼P20 (Pang et al., 2006b), homozygous Syt2−/− animals were killed at P15 at latest to minimize animal suffering, complying with a requirement imposed by the veterinary office. A mouse line harboring a floxed Syt1 allele (Syt1tm1a(EUCOMM)Wtsi) (see Skarnes et al., 2011) was purchased from the European Mouse Mutant Archive (Monterondo, Italy; stock #EM06829, RRID: MGI:5450372), and rederived as described previously (Kochubey et al., 2016). Syt1lox/+ mice were crossed with the Syt2+/− line to generate mouse pups with the following four genotype combinations: (i) Syt1+/+, Syt2+/+; (ii) Syt1lox/lox, Syt2+/+; (iii) Syt1+/+, Syt2−/−; and (iv) Syt1lox/lox, Syt2−/−. Stereotaxic injections with a lentiviral construct driving the expression of oChIEF (Lin et al., 2009) and Cre-recombinase (see details below) in these mice produced transfected synapses with the following protein KO conditions, respectively: (1) wild-type; (2) Syt1 conditional KO (cKO) synapses; (3) Syt2 KO synapses; and (4) Syt1-Syt2 double (conditional) KO synapses (called Syt1-Syt2 cDKO synapses below). For all experiments, mice of either sex were used.
Viral construct and stereotactic surgery.
We found that adenoviral vectors, used in previous studies of our laboratory for the ventral cochlear nucleus to MNTB calyx of Held pathway (Kochubey and Schneggenburger, 2011; Genç et al., 2014; Kochubey et al., 2016), did not efficiently transfect MNTB neurons. We therefore used a lentiviral system to express an oChIEFeYFP-IRES-Cre construct in MNTB neurons (the presynaptic neurons of the MNTB-LSO inhibitory connection). For this, DNA plasmids were constructed using standard PCR-based cloning techniques. The open reading frame of oChIEF (Lin et al., 2009) was PCR-amplified from an oChIEF-tdTomato encoding plasmid (Addgene #32846) and subcloned in-frame with the eYFP sequence (separated by an AgeI restriction site) into a pRRLsincPPT lentiviral vector (generously provided by Dr. Didier Trono, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland). Neuron-specific human synapsin1 promoter (Kügler et al., 2003) and the Kozak sequence GCCACC were cloned immediately upstream of oChIEF-eYFP. An internal ribosomal entry site (IRES) encoding sequence followed by an open reading frame of codon-optimized Cre recombinase (Gradinaru et al., 2010) was inserted downstream of the stop codon of the oChIEF-eYFP construct. A woodchuck hepatitis virus post-transcriptional regulatory element sequence was inserted at the end of the oChIEF eYFP-IRES-Cre expression cassette. Lentiviral particles were produced in 293T cells according to standard methods, aliquoted, and stored at −80°C. For injections, the viral stock was fourfold diluted in PBS.
Stereotaxic injections into the MNTB of P0 or P1 mice (genotypes i-iv; see Animals) were done under global isoflurane anesthesia and local lidocaine analgesia, using a model 900 stereotactic instrument (Kopf Instruments) and following the general procedures described previously for VCN injections at P0-P1 (Genç et al., 2014). The skull was aligned so that the midline point 3.7 mm anterior to lambda was located 0.37 mm more ventral than lambda while being in the same sagittal plane. The stereotaxic coordinates for targeting the MNTB were 0.25 mm lateral and 0.6 mm posterior from lambda. Viral suspension (0.8 μl) was continuously infused at a rate of ∼80 nl/min with a SP120 PZ syringe pump (WPI) through a 34 G stainless steel needle while retracting the needle between 5.1 and 4.8 mm depth. One injection was done on each hemisphere. The animals were used for experiments 12–15 d after surgery. For viral injections into cerebellar vermis (see Fig. 6), animals of the genotypes i-iv were used (see above) at the age of P4-P5. The skull was aligned such that the midline point 3.7 mm anterior from lambda was 2 mm more ventral, but in the same sagittal plane. The same volume of lentiviral suspension (0.8 μl, at 80 nl/min) was injected at two sites along the midline, 2.5 and 3.5 mm posterior from lambda, respectively, while retracting the needle between the depth of 2.5 and 1 mm ventral from the surface. The animals were used for experiments 9–11 d after surgery, at P12-P15.
Figure 6.
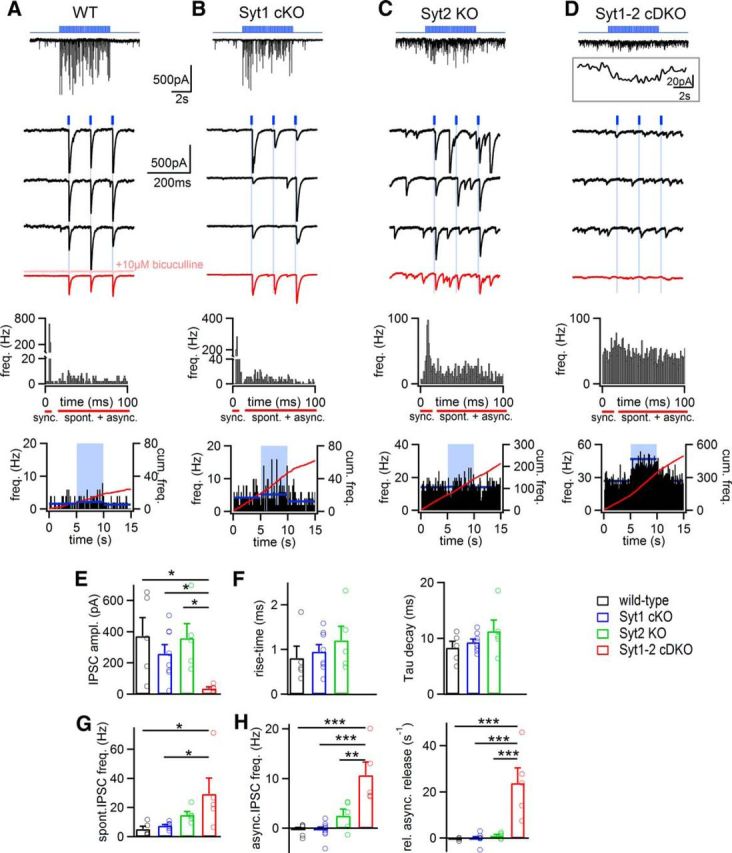
Syt1 and Syt2 redundantly support fast release at inhibitory synapses onto cerebellar Purkinje cells. A–D, Response to 10 Hz trains of optogenetic stimuli recorded in Purkinje neurons after virus-mediated expression of Cre-recombinase and oChIEF in the cerebellum. From top to bottom: blue trace represents 10 Hz light stimulus; black traces represent recorded IPSCs; black traces represent successive IPSCs in response to 10 Hz optical stimuli at higher time resolution (first three IPSCs only); red trace represents average IPSCs of 10 successive stimuli; histogram of event frequency for 100 ms following the light stimuli (average over 50 periods); histogram of spontaneous and asynchronous IPSC frequency before, during, and after the optogenetic stimulation train (red line indicates cumulative event frequency; blue line indicates average value for each 5 s interval). Light red trace in A represents the IPSCs in the presence of 10 μm bicuculline. D, Inset, Low-pass filtered (2 Hz) response to the 10 Hz optogenetic train (average of n = 10 traces). E, Individual and average values for IPSC amplitudes measured in all four genotype groups. F, The 20%–80% rise times (left) and IPSC decay time constants (right) for all four groups. In Syt2 KO synapses, the kinetic parameters could not be estimated because phasic evoked release events were absent (see D, bottom, red trace). G, Individual and average values for spontaneous IPSC frequency. H, Asynchronous IPSC frequency (left) and relative asynchronous release frequency (right). Number of recorded cells: wild-type synapses, n = 5; Syt1 KO synapses, n = 8; Syt2 KO synapses, n = 5; Syt1-Syt2 cDKO synapses, n = 5. Average data are presented as mean ± SEM. A significant difference was only observed in the comparisons involving the Syt1-Syt2 cDKO group (see asterisks), indicating that Syt1 and Syt2 act redundantly in inhibitory synapses on cerebellar Purkinje cells. *p < 0.05. **p < 0.01. ***p < 0.001.
Electrophysiology and optical stimulation.
Coronal 200 μm slices containing MNTB and LSO were cut using a VT1200s slicer (Leica Microsystems) while submerged in ice-cold preparation solution containing the following (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 25 glucose, 0.4 ascorbic acid, 3 myo-inositol, and 2 Na-pyruvate, supplemented with 0.1 CaCl2 and 3 MgCl2 (all from Sigma-Aldrich/Fluka), continuously bubbled with 95% O2, 5% CO2, pH 7.4. The slices were kept in submerged incubation chambers filled with the bicarbonate-based extracellular solution detailed above, with 2 mm CaCl2 and 1 mm MgCl2, for at least 45 min at 35°C before the recordings. For cerebellar recordings (see Fig. 6), parasagittal slices of 200 μm thickness were cut from the cerebellar vermis. Neurons were identified under transmitted light using an upright BX-51WI microscope (Olympus) equipped with an iXon ultra 897 EMCCD camera (Andor Technology) controlled with Micromanager software (Vale Laboratory, University of California San Francisco).
Whole-cell patch-clamp recordings were done using an EPC9/2 patch-clamp amplifier (HEKA Elektronik). Patch pipette solution contained the following (in mm): 140 CsCl, 10 HEPES, 20 TEA-Cl, 5 Na2-phosphocreatine, 4 MgATP, 0.3 Na2GTP, and 0.2 EGTA. Recordings were made at room temperature using the bicarbonate-based extracellular solution with 2 mm CaCl2 and 1 mm MgCl2, to which 10 μm CNQX and 50 μm APV were added (Biotrend). The holding potential was −70 mV. Series resistances (Rs) were 3–12 MOhm and were compensated by the patch-clamp amplifier by up to 70%; recordings were rejected if Rs changed by >20%. For fiber-stimulation experiments (see Fig. 1), MNTB axons were stimulated with a tungsten concentric bipolar electrode (WPI) placed on the side of the MNTB (see Fig. 1A); short pulses (0.2 ms) of increasing intensities (0–30 V) were delivered from an isolated stimulator (A-M Systems, model 2100).
For optogenetic stimulation, an LSO area containing transfected fibers was identified using the eYFP fluorescence, and a LSO principal neuron in the vicinity was recorded. Brief blue light pulses (2–5 ms) for optical stimulation were delivered from a custom-adapted high-power LED (CREE XP-E2 Royal Blue, 450–460 nm; Cree) driven by an LED controller (Doric Lenses). The LED light was coupled into the epifluorescence port of the microscope (Olympus BX-51WI; see above) through a custom-built epifluorescence condenser via a quartz glass lightguide. The light was focused onto the preparation using a 60× objective (Olympus LUM Plan FL, 0.9 NA); the measured energy at the focal plane was ∼5.8 mW/mm2. In case of cerebellar recordings (see Fig. 6), the recording site was chosen by the presence of YFP fluorescence surrounding the Purkinje cells, but the microscope objective was moved away toward the molecular layer to stimulate molecular layer interneurons.
Immunohistochemistry and confocal imaging.
An animal at a time was transcardially perfused with 4% PFA in PBS solution. Frozen brains were cut on a Hyrax S30 sliding microtome (Carl Zeiss) and processed for immunohistochemistry as described previously (Kochubey et al., 2016). Primary antibodies against Syt2 (polyclonal rabbit I735/3, 1:300; kindly provided by Dr. T. Südhof, Stanford, RRID: AB_2636925), Syt1 (monoclonal mouse clone 41.1, 1:200, Synaptic Systems, catalog #105011, RRID: AB_887832), GFP (polyclonal chicken, 1:1000, Abcam, catalog #13970; RRID: AB_300798), and VGAT (polyclonal guinea-pig, 1:500, Synaptic Systems, catalog #131004; RRID: AB_887873) were applied overnight at 4°C. The secondary fluorescently labeled antibodies (1 h incubation at room temperature, dilution 1:200) were anti-rabbit Alexa-488 (A11008, RRID: AB_143165), anti-chicken Alexa-488 (A11039, RRID: AB_2534096), anti-mouse Alexa-647 (A31571, RRID: AB_162542), and anti-guinea-pig Alexa-568 (A11075, RRID: AB_2534119, all from Thermo Fisher Scientific). Confocal images were acquired with an upright LSM 700 microscope (Carl Zeiss) equipped with 488, 563, and 633 nm laser lines and a Plan-Apochromat 40×/1.30 NA oil-immersion objective (pixel size was 90 nm).
For the side-by-side comparison of Syt1 and Syt2 expression in wild-type versus Syt2−/− mice (see Fig. 2A,B; n = 2 each), or Syt1+/+ versus Syt1lox/lox mice injected with the lenti: oChIEFeYFP-IRES-Cre virus (see Fig. 2D,E; n = 1 each), the sections were processed strictly in parallel. During confocal image acquisition, the imaging parameters were identical for the samples from the pairwise groups. Quantification of Syt1 and Syt2 expression levels at MNTB to LSO synapse (see Fig. 2) was done with custom-written procedures (IgorPro 6.3; Wavemetrics), as described previously (Kochubey et al., 2016).
Figure 2.

Syt1 is weakly coexpressed with Syt2 at inhibitory synapses on LSO neurons. A, Colabeling with antibodies against VGAT (left panel; magenta channel), Syt1 (second from left, yellow channel), Syt2 (second from right, cyan channel), and the merged image of all three channels, in an immunohistochemical experiment on the level of the LSO. A Syt2+/+ mouse at postnatal day 14 (P14) was used. B, Same immunohistochemical staining as in A, but now for a littermate Syt2−/− mouse at the same age (P14). Note the clear absence of specific signal in the Syt2 channel, whereas the Syt1 signal is unaffected. C, Quantification of pixel intensity for VGAT, Syt1, and Syt2 in VGAT-positive nerve terminals on LSO neurons. The values connected by thin lines indicate the pixel intensity averaged over all individual VGAT-positive punctae of a given section. In Syt2−/− mice, the fluorescence signal in the Syt2 channel was clearly absent (p < 0.001), whereas the Syt1 fluorescence intensity was not changed significantly (p = 0.21). D, E, Colabeling with antibodies against VGAT (left panel), Syt1 (second from left), and an anti-GFP antibody to detect oChIEFeYFP (second from right), and the overlay image (right). D, A Syt1+/+ mouse was injected with lenti:oChIEFeYFP-IRES-Cre (control). E, A littermate P15 Syt1lox/lox mouse was injected with lenti:oChIEFeYFP-IRES-Cre into the MNTB at P1 (thus producing Syt1 cKO synapses). Note the presence (D) and absence (E) of weak perisomatic Syt1 signal nerve terminals positive for VGAT and oChIEFeYFP (arrowheads). F, Histogram of the pixel intensity in the Syt1 channel of VGAT- and oChIEFeYFP-positive nerve terminals for the two conditions. Note the highly significant reduction of Syt1 immunofluorescence intensity in the Syt1 cKO synapses. Scale bar, 5 μm. ***p < 0.001.
Analysis and statistics.
Electrophysiological data were analyzed with IgorPro 6.3 (Wavemetrics). The minimal amplitude of IPSCs (see Fig. 1C) was the IPSC amplitude analyzed at the threshold for stimulation. The maximal amplitude (see Fig. 1C) is the IPSC amplitude at the maximal stimulation intensity of 30–40 V, or else the amplitude at a plateau if a plateau was observed with stimulation intensities of <30 V. IPSCs (spontaneous and evoked) were detected using a template-matching algorithm (Clements and Bekkers, 1997). Every detected IPSC was visually inspected to reject false positives. We constructed histograms of the time of occurrence of spontaneous and asynchronous IPSC events during the 100 ms intervals between adjacent optogenetic stimuli (average over 50 periods and over all repetitions). At 10–20 ms following optogenetic stimulation, we detected fewer asynchronous and spontaneous events (see, e.g., Fig. 3D1, bottom), most likely because the decay of the large evoked IPSC masked the much smaller asynchronous and spontaneous events. Therefore, the analysis of asynchronous and spontaneous release events was restricted to an interval of 20–100 ms after optogenetic stimulation. The asynchronous release frequency was estimated by subtracting the average spontaneous release frequency measured during 5 s before the train, from the frequency of all detected events at 20–100 ms.
Figure 3.

Optogenetic targeting and stimulation of MNTB-LSO synapses. A, Scheme of the lentiviral infection of MNTB neurons with a construct driving the expression of a light-activated ion channel (oChIEF) and Cre-recombinase (hSyn:oChIEFeYFP-IRES-Cre). The transfected fibers from Cre-expressing neurons are selectively amenable to optic stimulation. B, Confocal image of a brainstem slice of a P15 mouse on the level of the MNTB and LSO, 15 d after transfection. Bottom images, Higher-magnification images of the MNTB (left) and LSO (right), at the positions indicated in the top image (white squares). Scale bars: Top, 200 μm; Bottom, 50 μm. C, Optogenetically evoked IPSCs, here in response to a 10 Hz train, were blocked by bath application of 4 μm strychnine. D1–D3, Optogenetic stimulation experiment in an LSO slice of a P13 Syt1+/+, Syt2+/+ mouse, injected at P0 with the construct shown in A. D1, Three successively recorded IPSCs in response to the first optogenetic stimuli of 10 Hz trains (black traces). Red trace (here and in subsequent figures) represents the average of n = 10 successive trials. Bottom, Histogram of event frequency for 100 ms following the light stimuli (averaged over all 50 periods of the 10 Hz stimulus trains, and for n = 10 trains). Note the peak of synchronous events at the onset of stimulation. Spontaneous and asynchronous release events were detected at 20–100 ms following optical stimulation (see Materials and Methods). D2, A single IPSC train in response to a 10 Hz train. Bottom, Relative depression (normalized to the average first IPSC amplitude) for all 10 Hz trains in this recording (gray data points). The average depression time course (black data points) was fitted with an exponential function (black line). D3, Time course of spontaneous release frequency before and after the train, and of asynchronous and spontaneous release during the 10 Hz optogenetic trains. Red line indicates cumulative event frequency (see right axis). Blue lines indicate averages of the release frequencies during the three different 5 s intervals.
We furthermore calculated the “relative asynchronous release” by dividing the asynchronous release rate (calculated as explained above) by the quantal content (thus, a ratio of asynchronous relative to synchronous release rate was calculated). The quantal content was estimated by dividing the amplitude of the first evoked IPSC by the average amplitude of the spontaneous IPSCs (quantal size, see Fig. 5C).
Figure 5.

Syt1 and Syt2 act redundantly at the MNTB-LSO synapse to drive fast glycine release. Individual and average values are displayed for optogenetic stimulation experiments in wild-type synapses (black symbols), in Syt1 cKO synapses (blue), in Syt2 KO synapses (green), and in Syt1-Syt2 cDKO synapses (red). A, Individual and average values for IPSC amplitudes. B, The 20%–80% rise time (left) and IPSC decay time constants (right) for the first IPSC of the train of optical stimuli. C, Quantal size as estimated by the average spontaneous IPSC amplitude in each recording. Note the unchanged quantal size across all genotypes. D, Spontaneous release frequency. E, Asynchronous release frequency (left), and relative asynchronous frequency (right), calculated as explained in Results and Materials and Methods. F, Normalized IPSC depression curves during 10 Hz trains of optical stimuli for all genotypes, as indicated. Each gray line indicates the average relative depression curve for an individual recording. Color symbols represent the averages across all cells in each group. Note the strong reversal from depression to facilitation in Syt1-Syt2 cDKO synapses (right). Number of recorded cells were as follows: wild-type, n = 11; Syt1 cKO, n = 7; Syt2 KO, n = 11; Syt1-Syt2 cDKO, n = 8. Data are mean ± SEM. The significance was tested with ANOVA or the Kruskal–Wallis test (for IPSC amplitudes and quantal content), and is only indicated for those pairs that showed a significant difference. *p < 0.05. **p < 0.01. ***p < 0.001. A–G, For most comparisons, only the Syt1-Syt2 cDKO synapses showed significant differences to the remaining groups (see asterisks), with exception of the spontaneous release rate (D), which was also significantly different in Syt2 KO synapses.
Error bars indicate SEM, and statistical significance was assessed either by the unpaired Student's t test (see Fig. 1) or by one-way ANOVA with post hoc Bonferroni test for multiple comparisons in case of more than two groups. Alternatively, a nonparametric Kruskal–Wallis test was performed, followed by the Dunn's multiple comparison test, as indicated. Statistical significance of distributions of fluorescence values (see Fig. 2) was tested by the Kolmogorov–Smirnov test. Statistical tests were performed using Prism (GraphPad, RRID: SCR_002798).
Results
Fast glycine release at the MNTB-LSO synapse is unchanged in Syt2 KO mice
Previous immunohistochemical studies showed that inhibitory nerve terminals on LSO neurons, which represent putative nerve terminals of the MNTB-LSO glycinergic synapse, express Syt2 from ∼P5 onwards (Cooper and Gillespie, 2011). Thus, we reasoned that this inhibitory projection in the auditory brainstem, at which glycine release occurs in a highly synchronous and multiquantal fashion (Kim and Kandler, 2010), would be a suitable model to study the function of Syt2 at fast-releasing inhibitory synapses. We used conventional Syt2−/− mice, which initially do not show phenotypes, but then develop progressive motor phenotypes and muscle weakness, and finally die at ∼2–3 weeks of age (Pang et al., 2006b). This limited our study to an age of P12-P15 (see Materials and Methods).
We recorded fiber-stimulation evoked IPSCs in LSO neurons of Syt2−/− mice and their wild-type littermates at P12-P15. To our surprise, we observed evoked IPSCs with large amplitudes and fast rise and decay times in Syt2−/− mice (Fig. 1B). When varying the stimulation strength, the IPSC amplitudes showed discrete steps, suggesting the recruitment of presynaptic fibers each mediating a well-resolvable unitary IPSC (Fig. 1B) (Kim and Kandler, 2003; Michalski et al., 2013). The IPSC amplitudes upon minimal and maximal stimulation were unchanged between wild-type and Syt2−/− mice (Fig. 1C), which suggests that neither the amplitudes nor the number of unitary IPSCs was significantly changed. The rise times of the IPSCs were unchanged; the decay times showed a tendency to be faster in Syt2−/− mice, but the difference did not reach statistical significance (Fig. 1D; p > 0.05 for both comparisons). The spontaneous IPSC frequency measured in LSO neurons was increased from ∼3 Hz to ∼15 Hz in Syt2−/− mice, whereas the amplitude of sIPSCs was unchanged (Fig. 1E,F). The increased spontaneous IPSC frequency likely indicates clamping of spontaneous release by Syt2 at the MNTB-LSO synapse. Nevertheless, Syt2−/− mice did not show a measurable deficit in the amount or in the kinetics of action potential (AP)-evoked fast glycine release at the MNTB-LSO synapse.
Syt1 is weakly coexpressed with Syt2 at the inhibitory MNTB-LSO synapse
A previous immunohistochemical study showed that Syt1 is expressed early on at inhibitory synaptic terminals that form onto LSO neurons in the rat (Cooper and Gillespie, 2011). However, the earlier study suggested that Syt1 has a function in early glutamate release, known to occur at P3-P6 at this connection (Gillespie et al., 2005; Noh et al., 2010). Because in Syt2−/− mice we did not observe a reduction of AP-evoked glycine release (see above), we next verified whether Syt1 might be coexpressed with Syt2 at P12-P15, the age of mice used in our study.
Using immunohistochemistry on the level of the LSO (Fig. 2A), we observed a clear Syt2-signal in VGAT-positive nerve terminals on LSO neurons (Fig. 2A, cyan channel) (Cooper and Gillespie, 2011). In a P14 Syt2−/− mouse (a littermate of the Syt2+/+ mouse shown in Fig. 2A), the Syt2 signal was absent (Fig. 2B,C; p < 0.001; Kolmogorov–Smirnov test), confirming the specificity of the Syt2 antibody, and the effective removal of Syt2 protein in the Syt2−/− mice (Pang et al., 2006b). Using a monoclonal antibody against Syt1, we observed a weak, but clearly detectable, signal in VGAT-positive nerve terminals adjacent to LSO neuron somata in wild-type mice (Fig. 2A). Neighboring nerve terminals were strongly Syt1-positive (Fig. 2A,B,D,E, yellow channels), suggesting different Syt1 protein levels in inhibitory nerve terminals, versus nonidentified nerve terminals in the LSO neuropil. In the Syt2−/− mice, the Syt1 immunofluorescence signal was qualitatively similar as in the Syt2−/− littermate mouse (compare Fig. 2A,B, yellow). Quantitative analysis showed a slight increase in the Syt1 immunofluorescence intensity in some but not in other sections (Fig. 2C; each thin line indicates the averaged data from a single section); this difference did not reach statistical significance (p = 0.21; Kolmogorov–Smirnov test; n = 7 and 7 sections from n = 2 Syt2+/+ and n = 2 Syt2−/− mice, respectively). Therefore, if there is a compensatory increase in Syt1 expression in Syt2 −/− mice, it is small (maximally ∼1.4-fold).
We next investigated whether the weak Syt1 immunofluorescence signal in inhibitory nerve terminals was specific, by genetically inactivating the Syt1 protein. For this purpose, we used a floxed allele of Syt1 (Syt1lox) (Zhou et al., 2015; Kochubey et al., 2016), and injected a lentivirus driving the expression of Cre-recombinase and oChIEFeYFP (for details on the construct, see below) in the MNTB of a Syt1lox/lox mouse, and a littermate Syt1+/+ (control) mouse at P1. Immunohistochemistry of the LSO was done at P15, with antibodies against VGAT (to identify inhibitory nerve terminals), GFP (to identify oChIEFeYFP and thereby, virally transduced nerve terminals), and Syt1 (Fig. 2D,E). This showed that Cre expression in MNTB neurons of Syt1lox/lox mice caused the ablation of the weak, perisomatic Syt1 signals, whereas the control mice showed weak perisomatic Syt1 signals (Fig. 2D,E; yellow channels, white arrowheads). Quantitative analysis showed a highly significant reduction of the perisomatic Syt1 immunofluorescence signal (Fig. 2F; p < 0.001; Kolmogorov–Smirnov test; n = 567 and 781 VGAT- and eYFP-positive terminals analyzed in n = 3 and n = 4 sections from 1 Syt1+/+ mouse and 1 Syt1lox/lox mouse, respectively). This experiment validates the specificity and the sensitivity of the anti-Syt1 antibody. It also shows that the perisomatic Syt1-positive inhibitory synapses in the LSO originate from MNTB neurons.
Combined Cre expression and optogenetic stimulation of presynaptic fibers
To investigate whether the weakly coexpressed Syt1 can functionally replace Syt2 in KO experiments and thereby explain the absence of a release phenotype in Syt2−/− mice (Fig. 1), it is necessary to genetically delete both proteins. To manipulate Syt1 expression, we used Syt1lox/lox mice (see above, and Materials and Methods). We attempted to produce Syt1-Syt2 cDKO mice but found that crossing Syt1+/lox mice with a standard Cre driver mouse for the auditory brainstem (Krox20Cre) (Voiculescu et al., 2000; Han et al., 2011), in the background of Syt2+/−, did not give rise to viable cDKO mice. Therefore, we used an alternative approach, in which we expressed Cre-recombinase virally in a spatially more restricted manner, with the aim to produce Syt1-Syt2 cDKO synapses specifically at the MNTB-LSO inhibitory connection. This was done by coexpressing Cre-recombinase, and a light-activated ion channel in MNTB neurons in vivo, using a bicystronic lentiviral vector (lenti hSyn: oChIEFeYFP-IRES-Cre; for oChIEF, see Lin et al., 2009). This approach made sure that optogenetic activation of the transduced afferent fibers leads to selective stimulation of those nerve terminals that have also undergone molecular perturbation (Fig. 3A). As shown above, Cre expression in MNTB neurons of Syt1lox/lox mice leads to the ablation of Syt1 protein at the MNTB-LSO inhibitory synapses (Fig. 2D–F).
We first established the optogenetic stimulation approach in wild-type synapses, by injecting the lentiviral construct into the MNTB of Syt1+/+, Syt2+/+ mice at P0-P1. We made recordings of LSO neurons in brainstem slices 12–14 d after infection (thus, at P12-P15) and stimulated the transduced afferent fibers optogenetically (Fig. 3A,B). We used 10 Hz trains of brief (1–5 ms) pulses of blue light (470 nm) applied to the surrounding tissue, to measure phasic release, synaptic depression, and asynchronous release. Optogenetic stimulation caused phasic IPSCs with large, but quite variable, amplitudes between recordings (2.1 ± 0.9 nA; n = 11 cells) and fast rise and decay times (0.48 ± 0.05 and 12.1 ± 2.4 ms, respectively) (Fig. 5 reports all quantifications of the optogenetic experiments made at the MNTB-LSO synapse with the different genotypes). Repetitive optogenetic stimulation at 10 Hz evoked synaptic depression (Fig. 3C); the optogenetically evoked currents were blocked by both TTX (1 μm; see Fig. 4D, bottom) and by strychnine (4 μm; Fig. 3C), identifying them as postsynaptic currents mediated by glycine receptors and thus, as IPSCs.
Figure 4.
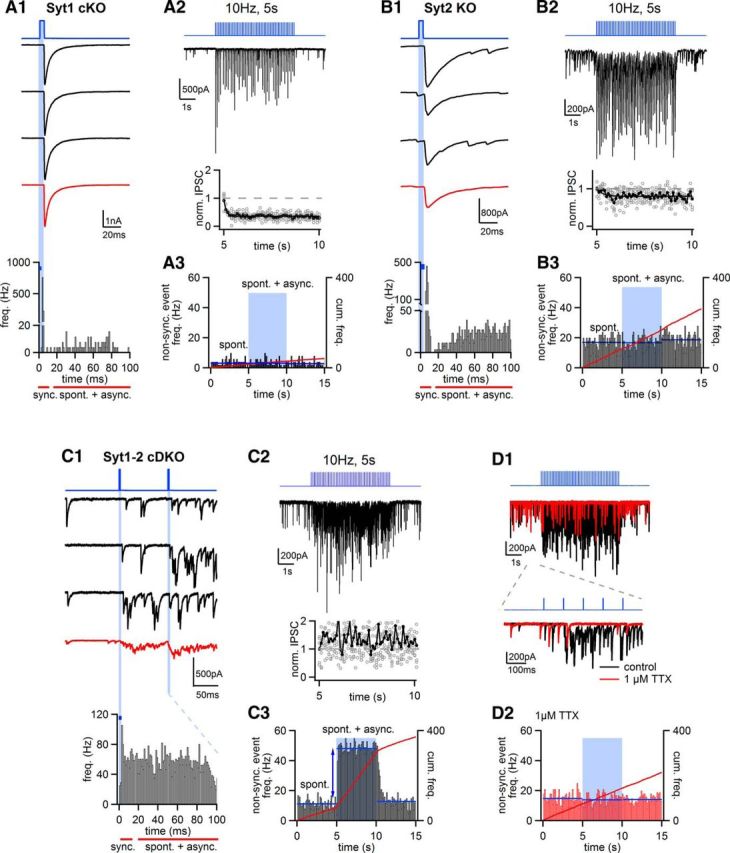
Disrupted fast inhibitory transmission and increased asynchronous release are limited to Syt1-Syt2 cDKO synapses. A, Optogenetically evoked IPSCs in Syt1 cKO synapses at P14, produced by injecting the lenti: hSyn: oChIEFeYFP-IRES-Cre virus (Fig. 3A) into the MNTB of a Syt1lox/lox, Syt2+/+ mouse at P0. The arrangement of traces follows that shown for the optogenetic experiment in wild-type synapses (Fig. 3D1–D3). A1, Three consecutive IPSCs (black traces) and the average of n = 10 successive IPSCs (red), and the histogram of event frequency for all light stimuli at 10 Hz (bottom). A2, A single example IPSC trace in response to a 10 Hz train of optical stimuli, as well as average and single data points for relative depression (bottom). A3, Histogram of spontaneous and asynchronous IPSC frequency before, during, and after the optogenetic stimulation train. Blue line indicates average spontaneous release frequency during three 5 s intervals. Red line indicates the cumulative release frequency (right axis). Note the clear phasic release and the absence of an increase in spontaneous and asynchronous release in Syt1 cKO synapses. A1–A3, All traces are from the same recording. B, Example of an optogenetic recording in a P12 Syt2−/− mouse, injected at P0 with the lentivirus (thus, Syt2 KO synapses were studied). B1–B3, The arrangement is the same as in A. Note the increased spontaneous release in the Syt2 KO synapses, but no additional asynchronous release during the optogenetic stimulation train. C, Optogenetic stimulation of IPSCs in a Syt1lox/lox, Syt2−/− mouse at P15 (thus, Syt1-Syt2 cDKO synapses were studied after expression of Cre-recombinase and oChIEF). Note the absence of time-locked IPSCs (C1) and the strongly increased asynchronous release frequency (C1, bottom; C3). Upon 10 Hz trains, the remaining IPSC (see C1, red average IPSC trace) does not show depression but rather facilitation (C2, bottom). D, Application of 1 μm TTX in the same recording as shown in C leads to suppression of evoked asynchronous release (D1), but spontaneous release persists as expected. The histogram of spontaneous, and spontaneous plus asynchronous release events (D2), demonstrates that events sampled during the optogenetic stimulation train (D2, time indicated by blue shaded area) correctly quantify the spontaneous release rate.
We next analyzed asynchronous release in between optogenetic stimuli, at a time window of 20–100 ms after each light stimulus (Fig. 3D1; see Materials and Methods). To calculate the stimulus-evoked asynchronous release, the spontaneous release measured before optogenetic stimulation (Fig. 3D3) was subtracted (see Materials and Methods). In wild-type synapses, asynchronous release did not rise above the spontaneous release frequency of ∼2–3 Hz measured before the stimulus (Fig. 3D3). Thus, optogenetic stimulation of the MNTB-LSO synapse in wild-type mice produces large and fast-decaying synchronous IPSCs, whereas asynchronous release in response to 10 Hz trains is small or absent.
Syt1 and Syt2 compensate for each other in single KO experiments
We next used the virus-mediated Cre expression and optogenetic stimulation method to measure transmitter release in single Syt1 or Syt2 KO synapses (Fig. 4A,B). This was done by injecting the same viral construct as used above (lenti hSyn: oChIEFeYFP-IRES-Cre), but now using either Syt1lox/lox mice or Syt2−/− mice. All mice in the four genotype comparison done here, giving rise to wild-type synapses (Syt1+/+, Syt2+/+), Syt1 cKO synapses (Syt1lox/lox, Syt2+/+), Syt2 KO synapses (Syt1+/+, Syt2−/−), and Syt1–2 cDKO synapses (Syt1lox/lox, Syt2−/−), were obtained from the same breeding pairs (see Materials and Methods).
In Syt1 cKO synapses, optogenetic stimulation caused phasic IPSCs with amplitudes of 0.50 ± 0.31 nA (Fig. 4A; n = 7). These amplitudes were smaller than the ones observed in wild-type synapses (see above), but the difference did not reach statistical significance (p > 0.5; Fig. 5A). We cannot rule out the possibility that this sample of cells (Syt1 cKO; n = 7) represented a subset of connections in which Syt1 played a more dominant role. However, it is difficult to compare absolute IPSC amplitudes because the number of virally transfected fibers is most likely variable across recordings. The kinetics of IPSCs in Syt1 cKO synapses showed fast rise and decay times indistinguishable from the wild-type synapses (Fig. 4A; see Fig. 5B for summary data), and synaptic depression was unchanged compared with control synapses (Figs. 4A2, 5F). Similarly, both the spontaneous as well as the asynchronous release frequencies were small (Fig. 4A3) and indistinguishable from the control data (Fig. 5D,E). We conclude, therefore, that transmitter release at the MNTB-LSO synapse is normal when the Syt1 protein alone is genetically inactivated.
In Syt2−/− mice, we observed clear phasic IPSCs (Fig. 4B; 0.55 ± 0.16 nA; n = 11; see also Fig. 5A). The IPSC rise times were slightly slowed compared with control synapses, but this tendency did not reach statistical significance (p > 0.05; Fig. 5B); the IPSC decay times were unchanged (Fig. 5B). The spontaneous release was strongly increased (Fig. 4B), as expected from the fiber stimulation experiments (Fig. 1). Interestingly, the asynchronous release rate was the same as that of spontaneous release before and after the stimulus (Fig. 4B3), an unexpected finding given that genetic inactivation of Syt2 in a Syt2-dominated connection should lead to a strong increase in asynchronous release on the expense of synchronous release (Sun et al., 2007). Across all cells, asynchronous release was slightly elevated in Syt2 KO synapses (to 2.9 ± 2.1 Hz), but this rate was small compared with the spontaneous release in Syt2 KO synapses (∼15 Hz; Fig. 5D), and the change was not statistically significant with respect to wild-type or Syt1 cKO synapses (for quantification, see Fig. 5E).
As mentioned above, a possible concern in the comparison of absolute measures of synchronous and asynchronous release rates is a possible cell-to-cell variability in the number of virally transduced afferent fibers. At the MNTB-LSO synapse, single afferent fibers can generate large IPSCs (E.G. et al., manuscript in preparation), and thus small variations in the number of transfected fibers are expected to cause a large variability in the absolute IPSC amplitudes. We therefore also analyzed the ratio of asynchronous release over synchronous release (the latter was measured as quantal content; see Materials and Methods). This “relative asynchronous release” was not significantly different between single Syt1 and Syt2 KO synapses versus wild-type synapses (p > 0.05; see Fig. 5E, right). Taken together, in Syt1- or Syt2 single KO synapses at the MNTB-LSO connection, clear phasic release was present, and asynchronous release was not significantly increased, despite an increased spontaneous release in Syt2 KO synapses. This suggests that Syt1 and Syt2 compensate for each other in triggering fast release in the single KO synapses.
Syt1-Syt2 cDKO inhibitory synapses show drastically reduced phasic release and increased asynchronous release
We next investigated Syt1-Syt2 cDKO synapses. For this purpose, we delivered the lentiviral construct to the MNTB of Syt1lox/lox, Syt2−/− mice, thereby producing Syt1 cKO synapses in the background of the conventional Syt2−/− mice. Strikingly, optogenetic stimulation of the resulting Syt1-Syt2 cDKO synapses was unable to cause phasic release (Fig. 4C1). Instead, there was a large increase in asynchronous release, which occurred immediately after the optogenetic stimulus and lasted for at least 100 ms, with rates of ∼35 Hz in the example recording of Figure 4C1, C3. The asynchronous release rate was strongly elevated above the spontaneous release rate; the latter was ∼15 Hz in the recording of Figure 4C. Indeed, across all cells, the spontaneous release was not significantly higher in Syt1-Syt2 cDKO synapses compared with Syt2 KO synapses (Fig. 5D), whereas the asynchronous release frequency was significantly increased in the Syt1-Syt2 cDKO synapses compared with all other groups (p < 0.001 for all comparisons; see Fig. 5E). Similarly, the “relative asynchronous release” was strongly enhanced in Syt1-Syt2 cDKO synapses compared with all other groups (Fig. 5E, right; p < 0.001). Thus, both Syt1 and Syt2 must be deleted to cause a significant decrease in synchronous release (Fig. 5A) and to lead to the appearance of significant asynchronous release at the glycinergic MNTB-LSO synapse.
We applied TTX (1 μm) in some of the recordings in Syt1-Syt2 cDKO synapses (Fig. 4D1), to validate that the evoked asynchronous release of Syt1-Syt2 cDKO synapses depended on presynaptic APs. TTX reduced the frequency of release events during the optogenetic train to exactly the frequency of spontaneous release events before and after the optogenetic train (Fig. 4D1,D2). This shows that the asynchronous release evoked by optogenetic stimulation depends on AP activity in the presynaptic afferent fibers (compare Fig. 4C3 with Fig. 4D2; both histograms are from the same recording).
Finally, we analyzed the degree of synaptic depression across all four genotype conditions (Fig. 5F,G). Because synaptic depression depends on vesicle use and is correlated with the initial release probability (Scheuss et al., 2002), molecular perturbations of synapses, which reduce the release probability, often lead to a decreased depression (Reim et al., 2001; Cornelisse et al., 2012). We found that the amount of depression was slightly reduced in Syt1 cKO synapses, but this effect did not reach statistical significance; depression in Syt2 KO synapses was unchanged compared with wild-type synapses (Fig. 5F,G). In contrast, in Syt1-Syt2 cDKO synapses, depression upon 10 Hz stimulation was converted to facilitation (p < 0.001 for the comparisons with all other groups; Fig. 5F,G, red symbols). Thus, analyzing synaptic depression further corroborates our findings that the phasic release component is affected at most partially in single KO synapses, whereas the double Syt1 and Syt2 KO synapses have a strongly reduced synaptic depression, suggesting a reduced release probability.
Syt1 and Syt2 function redundantly also at cerebellar inhibitory synapses
We next wished to investigate whether other fast-releasing inhibitory synapses might also show a redundant function of Syt1 and Syt2. We studied the synapse between basket/stellate cells and Purkinje neurons (Vincent et al., 1992; Arai and Jonas, 2014), again using the optogenetic stimulation approach after virus-mediated expression of Cre-recombinase and oChIEFeYFP. A Purkinje cell surrounded by eYFP-positive fibers was recorded, and optogenetic stimulation was then centered onto the molecular layer.
In wild-type cerebellar inhibitory synapses, optogenetically evoked IPSCs were clearly resolvable, with amplitudes of ∼400 pA across cells and rapid rise and decay times (Fig. 6A; for all quantifications, see Fig. 6E–H). These currents were blocked by bath application of 10 μm bicuculline, identifying them as GABAA-receptor mediated IPSCs (Fig. 6A, pink trace). The 10 Hz optogenetic stimulus train did not notably increase the asynchronous release frequency above the spontaneous IPSC frequency (Fig. 6A, bottom; H, black symbols), similarly as we observed at the MNTB-LSO synapse. Conditional deletion of Syt1 (by using Syt1lox/lox mice) showed essentially unchanged spontaneous, asynchronous and fast release (for an example cell, see Fig. 6B; for quantification, see Fig. 6E–H). The recordings in Syt2−/− mice showed an increased spontaneous IPSC frequency of 14.8 ± 2.4 Hz (Fig. 6C,G; p < 0.05). In addition, the asynchronous release rate was somewhat higher than in wild-type and Syt1 cKO synapses, but this comparison did not reach statistical significance (p > 0.05; Fig. 6H, left), and normalization by the synchronous release made the difference with respect to wild-type synapses smaller (Fig. 6H, right, green data symbols). In the Syt1 and Syt2 single KO synapses, the IPSC amplitude was not significantly different from the one measured in wild-type synapses (Fig. 6E; p > 0.05), and a slight increase in the IPSC rise and decay times did not reach statistical significance (Fig. 6F; p > 0.05). Thus, the single Syt1 and Syt2 KO synapses did not show significantly desynchronized or reduced fast release compared with wild-type synapses.
In Syt1-Syt2 cDKO cerebellar synapses, on the other hand, there were marked deficits in the fast release component (Fig. 6D). Instead of a train of phasic IPSCs, we observed an increase in asynchronous release events during optogenetic stimulation (Fig. 6D). In many recordings, the increased asynchronous release was difficult to detect against an already high spontaneous release rate. However, evoked release responses were evident by averaging and low-pass filtering (n = 10 traces), which revealed a tonic response without apparent phasic release (Fig. 6D, inset).
Across all recordings, evoked IPSC amplitudes were drastically smaller in Syt1-Syt2 cDKO synapses (Fig. 6E). Furthermore, both the asynchronous release frequency and the relative asynchronous release frequency were significantly higher in Syt1-Syt2 cDKO synapses than the corresponding values in wild-type and single KO synapses (Fig. 6H; asterisks indicate statistical significance of each comparison). Thus, similar to the MNTB-LSO glycinergic synapse, the full phenotype of genetic removal of presynaptic Ca2+ sensor proteins is achieved only when both Syt1 and Syt2 are removed at the cerebellar inhibitory synapses.
Finally, we investigated the expression of both synaptotagmin isoforms in cerebellar inhibitory synapses with immunohistochemical experiments, using both parasagittal (data not shown) and coronal sections of P14 wild-type mice (Fig. 7). In both types of sections, we observed weak Syt1 staining in VGAT- and Syt2-positive perisomatic punctae close to Purkinje cells (Fig. 7B, white arrows). In addition, especially in coronal sections, we observed VGAT-positive inhibitory synapses on the proximal dendrite of the Purkinje neuron that clearly coexpress Syt1 and Syt2 (Fig. 7B, white arrowheads). These Syt1-positive inhibitory synapses were less apparent in parasagittal section because here, the Syt1 staining from parallel fibers seen in the molecular layer (see also Fig. 7A, asterisk) was very strong and dense, making the detection of occasional Syt1-positive GABAergic synapses more difficult (data not shown). These results do not confirm a recent study, which reported that Syt1 is absent from Syt2-expressing inhibitory synapses near cerebellar Purkinje cells (Chen et al., 2017) (see Discussion). However, they corroborate our functional findings that Syt1 and Syt2 redundantly cause fast GABA release at the stellate/basket cell to Purkinje cell synapses.
Figure 7.

Immunohistochemical evidence for coexpression of Syt1 and Syt2 in inhibitory synapses onto cerebellar Purkinje cells. A, Coronal sections of a P14 wild-type mouse were stained with anti-VGAT antibody (left), anti-Syt1 antibody (middle), and anti-Syt2 antibody (right); the overlay image is shown in the rightmost panel. Note the strongly Syt1-positive parallel fibers in the molecular layer (second panel, *). B, Higher-magnification images of the area highlighted by a white box in A. Note three VGAT-, Syt1-, and Syt2-positive punctae on the proximal dendrite of the Purkinje cell (white arrowheads), and more weakly Syt1-expressing inhibitory terminals close to the soma (arrows). Scale bars: A, 20 μm; B, 5 μm.
Discussion
We have manipulated the expression of two major Syt isoforms, Syt1 and Syt2, in two types of fast-releasing inhibitory synapses of the hindbrain, with the aim to identify the Ca2+ sensor used by specific inhibitory connections in native brain tissue. In both synapses, we found that genetic deletion of only one of the proteins did not significantly reduce fast transmitter release. Only the simultaneous deletion of both Syt1 and Syt2 led to a significantly reduced fast release component, as would be expected for the complete removal of the fast Ca2+ sensor for vesicle fusion in a synapse (Geppert et al., 1994; Kochubey et al., 2016). These experiments demonstrate the functional significance of Syt2 at the fast-releasing output synapses of PV-expressing inhibitory neurons, and furthermore show that at these synapses, Syt1 can act redundantly with Syt2.
We started our study at the MNTB-LSO synapse. MNTB neurons, which receive the large excitatory calyx of Held synapse, in turn make fast-releasing glycinergic inhibitory synapses onto neurons in a neighboring nucleus, the LSO (Kim and Kandler, 2003, 2010) (Fig. 1A). We chose this connection because Syt2 is clearly expressed at this inhibitory synapse (Cooper and Gillespie, 2011), and we expected that genetic deletion would reveal a functional role of Syt2. However, we found in fiber-stimulation experiments (Fig. 1) as well as with optogenetic stimulation (Figs. 4, 5), that the fast release component was essentially unaffected in Syt2−/− mice, despite the clear presence of Syt2 at inhibitory nerve terminals surrounding LSO neurons in wild-type mice (Fig. 2) (Cooper and Gillespie, 2011). This strongly suggested that another Ca2+ sensor is expressed and acts redundantly with Syt2. We investigated Syt1 because it was shown recently that this isoform acts as an early Ca2+ sensor at the excitatory calyx of Held synapse before being developmentally replaced by Syt2, which showed that overlapping roles between Syt1 and Syt2 are possible in the same nerve terminal (Kochubey et al., 2016). In quantitative immunohistochemistry experiments, we detected a weak, but specific, Syt1 signal as verified by conditional ablation of the floxed Syt1 allele (Fig. 2D–F). Thus, immunohistochemistry and conditional genetic elimination clearly establish a weak coexpression of Syt1 at a Syt2-positive inhibitory nerve terminal.
To address the functionally redundant roles of the two major Syt isoforms 1 and 2 in inhibitory synapses of native mouse brain, we devised an approach that used virus-mediated expression of both Cre-recombinase and a light-activated ion channel (see also Jackman et al., 2016; Acuna et al., 2016). This allowed us to create Syt1-Syt2 cDKO synapses while avoiding perinatal lethality which we observed in Syt1-Syt2 cDKO mice upon the more widespread expression of Cre from the Krox20 locus. At the same time, the optogenetic approach ensured that only the molecularly perturbed fibers were stimulated. With this approach, we show at the MNTB-LSO connection that the effects on fast glycine release was minor in single KO synapses, whereas deletion of both Syts caused a strong reduction of fast AP-driven release (Figs. 4, 5). This directly shows that Syt1 and Syt2 act redundantly to trigger fast release at an inhibitory synapse.
Previous work has demonstrated quite complex developmental changes in the neurotransmitter used at the MNTB-LSO synapse. In the first postnatal week, both GABA and glutamate are released; and consistently, there is an early expression of VGluT3 (Gillespie et al., 2005). With further development, GABA and glutamate release becomes eliminated, and only glycine persists at ∼2 weeks of age (Kotak et al., 1998; Gillespie et al., 2005). We showed that Syt1 triggers fast transmitter release redundantly with Syt2 at a developmental time point when the transmitter switching toward glycine is complete (see also Fig. 3C). Based on the immunohistochemical finding of early Syt1 expression, Cooper and Gillespie (2011) suggested that Syt1 plays a functional role for early glutamate release at the MNTB-LSO synapse; note, however, that this possibility has not yet been addressed experimentally. Thus, it could be argued that the primary function of Syt1 at the MNTB-LSO synapse is to drive early glutamate release from VGluT3-positive vesicles. We do not think that this is a compelling explanation for our results because Syt1 must be targeted to glycine-containing vesicles to explain its critical role in fast glycine release. Thus, in addition to a possible earlier role in triggering glutamate release from immature synapses, which should be addressed in future work, Syt1 acts redundantly with Syt2 in fast glycine release up to at least postnatal day 15.
To study whether a redundant action of Syt1 and Syt2 is also found at other fast-releasing inhibitory synapses, we investigated inhibitory synapses in the cerebellum. The cerebellar stellate/basket cell to Purkinje neuron synapse is a more typical inhibitory interneuron synapse in a cortical structure. Using the optogenetic stimulation approach, we found very similar results as at the MNTB-LSO synapse: KO of Syt1 or of Syt2 alone caused only minor phenotypes in the fast release response, whereas the combined deletion of both isoforms caused a drastic reduction of fast, AP-evoked GABA release (Fig. 6). Thus, Syt1 and Syt2 act redundantly not only at the MNTB-LSO inhibitory synapse, but also in PV-expressing inhibitory synapses of the cerebellum.
A recent study reported that the synchronous component of GABA release at the basket cell-Purkinje cell synapse of Syt2−/− mice was reduced to 16% of control (Chen et al., 2017); the previous study did not manipulate Syt1 expression. It is at present unclear what might be the reason for the difference between the present findings and the results of the previous study. Differences in the exact age of mice (P14-P16 in their experiments; P12-P15 used here because of the lethality of Syt2−/− mice; see Materials and Methods), and in the recording-and stimulation techniques could explain part of the discrepancies. Also, Chen et al. (2017) observed a clear residual fast release component in Syt2−/− mice, which was further increased by repetitive stimulation. This remaining synchronous release in Syt2−/− mice of the previous study was most likely mediated by weak, but functionally relevant, coexpression of Syt1 because we show that genetic elimination of both Syt2 and Syt1 completely abolishes fast release (Fig. 6D) and because Syt1 is coexpressed in many Syt2-expressing inhibitory nerve terminals on Purkinje cells (Fig. 7). Taken together, the present and the previous study agree in their conclusion that Syt2 is an important Ca2+ sensor at fast-releasing inhibitory synapses. In addition, the present study highlights the redundancy caused by weak coexpression of Syt1 at two types of fast-releasing inhibitory synapses in the juvenile mouse brain.
One notable exception from the functional Syt1-Syt2 redundancy at both inhibitory synapses studied here was a strong increase in spontaneous IPSC frequency that was observed in the single Syt2 KO synapses, but not in the Syt1 cKO synapses (Figs. 1, 4, 6) (Chen et al., 2017). Previous work at the calyx of Held found that the elevated spontaneous release frequency in Syt2−/− mice is suppressed by the fast-acting Ca2+ buffer BAPTA but not by EGTA, but was unaffected by removal of extracellular Ca2+ (Kochubey and Schneggenburger, 2011; see also Pang et al., 2006a; Xu et al., 2009). Thus, spontaneous release in the absence of Syt1 or Syt2 is likely caused by short-lived intracellular Ca2+ transients acting on a secondary (slow) Ca2+ sensor which, at fast-releasing synapses of PV-expressing inhibitory neurons, can be clamped by Syt2 but not by Syt1. It is possible that its relatively weak expression level did not enable Syt1 to engage in spontaneous release clamping at the inhibitory synapses studied here, although Syt1 is able to clamp spontaneous release in synapses in which release is primarily driven by Syt1 (Geppert et al., 1994; Xu et al., 2009).
It was recently shown that the excitatory calyx of Held terminals undergo a Syt1 to Syt2 expression change, which starts at ∼P3 and is completed at P12-P14 (Kochubey et al., 2016). Thus, it seems possible that the redundant action of Syt1 and Syt2, which we observed here at the output synapses of PV-expressing inhibitory neurons, reflects a similar but temporally delayed expression switch. This possibility could be addressed in future studies using conditional KO or shRNA mediated knockdown of Syt2, to circumvent the lethality of the conventional Syt2−/− mice. Such approaches will also be relevant to study the roles of Syt1 and Syt2 at the output synapses of cortical PV interneurons, which express Syt2, especially at later stages of development (García-Junco-Clemente et al., 2010; Bragina et al., 2011; Sommeijer and Levelt, 2012). The transcriptional mechanisms of Syt2 expression (Pang et al., 2010; Lucas et al., 2014), and of a possible Syt1-Syt2 expression switch in PV interneurons, should be investigated in future studies.
Footnotes
This work was supported by Swiss National Science Foundation Grant 310030B_156934/1 and National Competence Center for Research “Synapsy” and by the German Research Foundation (DFG Priority Program 1608 “Ultrafast and temporally precise information processing: Normal and dysfunctional hearing”, SCHN 451/5-2). Confocal image acquisition was performed at the Bioimaging and optics platform of École Polytechnique Fédérale de Lausanne. We thank Ayah Khubieh and Dr. David Perkel for help with initial experiments; and Heather Murray and Jessica Dupasquier for expert technical assistance and genotyping.
The authors declare no competing financial interests.
References
- Acuna C, Liu X, Südhof TC (2016) How to make an active zone: unexpected universal functional redundancy between RIMs and RIM-BPs. Neuron 91:792–807. 10.1016/j.neuron.2016.07.042 [DOI] [PubMed] [Google Scholar]
- Arai I, Jonas P (2014) Nanodomain coupling explains Ca2+ independence of transmitter release time course at a fast central synapse. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Südhof TC (2013) Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80:947–959. 10.1016/j.neuron.2013.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton F, Iborra C, Boudier JA, Seagar MJ, Marquèze B (1997) Developmental regulation of synaptotagmin I, II, III, and IV mRNAs in the rat CNS. J Neurosci 17:1206–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragina L, Fattorini G, Giovedí S, Melone M, Bosco F, Benfenati F, Conti F (2011) Analysis of synaptotagmin, SV2, and Rab3 expression in cortical glutamatergic and GABAergic axon terminals. Front Cell Neurosci 5:32. 10.3389/fncel.2011.00032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucurenciu I, Kulik A, Schwaller B, Frotscher M, Jonas P (2008) Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron 57:536–545. 10.1016/j.neuron.2007.12.026 [DOI] [PubMed] [Google Scholar]
- Caputi A, Melzer S, Michael M, Monyer H (2013) The long and short of GABAergic neurons. Curr Opin Neurobiol 23:179–186. 10.1016/j.conb.2013.01.021 [DOI] [PubMed] [Google Scholar]
- Chen C, Arai I, Satterfield R, Young SM Jr, Jonas P (2017) Synaptotagmin 2 is the fast Ca2+ sensor at a central inhibitory synapse. Cell Rep 18:723–736. 10.1016/j.celrep.2016.12.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM (1997) Detection of spontaneous synaptic events with an optimally scaled template. Biophys J 73:220–229. 10.1016/S0006-3495(97)78062-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AP, Gillespie DC (2011) Synaptotagmins I and II in the developing rat auditory brainstem: synaptotagmin I is transiently expressed in glutamate-releasing immature inhibitory terminals. J Comp Neurol 519:2417–2433. 10.1002/cne.22634 [DOI] [PubMed] [Google Scholar]
- Cornelisse LN, Tsivtsivadze E, Meijer M, Dijkstra TM, Heskes T, Verhage M (2012) Molecular machines in the synapse: overlapping protein sets control distinct steps in neurosecretion. PLoS Comput Biol 8:e1002450. 10.1371/journal.pcbi.1002450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craxton M. (2010) A manual collection of Syt, Esyt, Rph3a, Rph3al, Doc2, and Dblc2 genes from 46 metazoan genomes: an open access resource for neuroscience and evolutionary biology. BMC Genomics 11:37. 10.1186/1471-2164-11-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Junco-Clemente P, Cantero G, Gómez-Sánchez L, Linares-Clemente P, Martínez-López JA, Luján R, Fernández-Chacón R (2010) Cysteine string protein-alpha prevents activity-dependent degeneration in GABAergic synapses. J Neurosci 30:7377–7391. 10.1523/JNEUROSCI.0924-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genç Ö, Kochubey O, Toonen RF, Verhage M, Schneggenburger R (2014) Munc18–1 is a dynamically regulated PKC target during short-term enhancement of transmitter release. eLife 3:e01715. 10.7554/eLife.01715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Archer BT 3rd, Südhof TC (1991) Synaptotagmin II: a novel differentially distributed form of synaptotagmin. J Biol Chem 266:13548–13552. [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79:717–727. 10.1016/0092-8674(94)90556-8 [DOI] [PubMed] [Google Scholar]
- Gillespie DC, Kim G, Kandler K (2005) Inhibitory synapses in the developing auditory system are glutamatergic. Nat Neurosci 8:332–338. 10.1038/nn1397 [DOI] [PubMed] [Google Scholar]
- Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K (2010) Molecular and cellular approaches for diversifying and extending optogenetics. Cell 141:154–165. 10.1016/j.cell.2010.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Kaeser PS, Südhof TC, Schneggenburger R (2011) RIM determines Ca2+ channel density and vesicle docking at the presynaptic active zone. Neuron 69:304–316. 10.1016/j.neuron.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Gan J, Jonas P (2014) Interneurons: fast-spiking, parvalbumin(+) GABAergic interneurons: from cellular design to microcircuit function. Science 345:1255263. 10.1126/science.1255263 [DOI] [PubMed] [Google Scholar]
- Jackman SL, Turecek J, Belinsky JE, Regehr WG (2016) The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 529:88–91. 10.1038/nature16507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepecs A, Fishell G (2014) Interneuron cell types are fit to function. Nature 505:318–326. 10.1038/nature12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr AM, Reisinger E, Jonas P (2008) Differential dependence of phasic transmitter release on synaptotagmin 1 at GABAergic and glutamatergic hippocampal synapses. Proc Natl Acad Sci U S A 105:15581–15586. 10.1073/pnas.0800621105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim G, Kandler K (2003) Elimination and strengthening of glycinergic/GABAergic connections during tonotopic map formation. Nat Neurosci 6:282–290. 10.1038/nn1015 [DOI] [PubMed] [Google Scholar]
- Kim G, Kandler K (2010) Synaptic changes underlying the strengthening of GABA/glycinergic connections in the developing lateral superior olive. Neuroscience 171:924–933. 10.1016/j.neuroscience.2010.09.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochubey O, Schneggenburger R (2011) Synaptotagmin increases the dynamic range of synapses by driving Ca2+-evoked release and by clamping a near-linear remaining Ca2+ sensor. Neuron 69:736–748. 10.1016/j.neuron.2011.01.013 [DOI] [PubMed] [Google Scholar]
- Kochubey O, Babai N, Schneggenburger R (2016) A synaptotagmin isoform switch during the development of an identified CNS synapse. Neuron 90:984–999. 10.1016/j.neuron.2016.04.038 [DOI] [PubMed] [Google Scholar]
- Kotak VC, Korada S, Schwartz IR, Sanes DH (1998) A developmental shift from GABAergic to glycinergic transmission in the central auditory system. J Neurosci 18:4646–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kügler S, Kilic E, Bähr M (2003) Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther 10:337–347. 10.1038/sj.gt.3301905 [DOI] [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P, Tsien RY (2009) Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys J 96:1803–1814. 10.1016/j.bpj.2008.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Dougherty SE, McMeekin LJ, Reid CS, Dobrunz LE, West AB, Hablitz JJ, Cowell RM (2014) PGC-1α provides a transcriptional framework for synchronous neurotransmitter release from parvalbumin-positive interneurons. J Neurosci 34:14375–14387. 10.1523/JNEUROSCI.1222-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Südhof TC (2005) Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron 48:547–554. 10.1016/j.neuron.2005.09.006 [DOI] [PubMed] [Google Scholar]
- Michalski N, Babai N, Renier N, Perkel DJ, Chédotal A, Schneggenburger R (2013) Robo3-driven axon midline crossing conditions functional maturation of a large commissural synapse. Neuron 78:855–868. 10.1016/j.neuron.2013.04.006 [DOI] [PubMed] [Google Scholar]
- Noh J, Seal RP, Garver JA, Edwards RH, Kandler K (2010) Glutamate co-release at GABA/glycinergic synapses is crucial for the refinement of an inhibitory map. Nat Neurosci 13:232–238. 10.1038/nn.2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaty BW, Miller MN, Sugino K, Hempel CM, Nelson SB (2009) Transcriptional and electrophysiological maturation of neocortical fast-spiking GABAergic interneurons. J Neurosci 29:7040–7052. 10.1523/JNEUROSCI.0105-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Südhof TC (2010) Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol 22:496–505. 10.1016/j.ceb.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Sun J, Rizo J, Maximov A, Südhof TC (2006a) Genetic analysis of synaptotagmin 2 in spontaneous and Ca2+-triggered neurotransmitter release. EMBO J 25:2039–2050. 10.1038/sj.emboj.7601103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Südhof TC (2006b) Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci 26:13493–13504. 10.1523/JNEUROSCI.3519-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Xu W, Cao P, Südhof TC (2010) Calmodulin suppresses synaptotagmin-2 transcription in cortical neurons. J Biol Chem 285:33930–33939. 10.1074/jbc.M110.150151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petilla Interneuron Nomenclature Group (2008) Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci 9:557–568. 10.1038/nrn2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C (2001) Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell 104:71–81. 10.1016/S0092-8674(01)00192-1 [DOI] [PubMed] [Google Scholar]
- Scheuss V, Schneggenburger R, Neher E (2002) Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPCSs at the calyx of Held synapse. J Neurosci 22:728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474:337–342. 10.1038/nature10163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommeijer JP, Levelt CN (2012) Synaptotagmin-2 is a reliable marker for parvalbumin positive inhibitory boutons in the mouse visual cortex. PLoS One 7:e35323. 10.1371/journal.pone.0035323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC (2007) A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450:676–682. 10.1038/nature06308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent P, Armstrong CM, Marty A (1992) Inhibitory synaptic currents in rat cerebellar Purkinje cells: modulation by postsynaptic depolarization. J Physiol 456:453–471. 10.1113/jphysiol.1992.sp019346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voiculescu O, Charnay P, Schneider-Maunoury S (2000) Expression pattern of a Krox-20/Cre knock-in allele in the developing hindbrain, bones, and peripheral nervous system. Genesis 26:123–126. [DOI] [PubMed] [Google Scholar]
- Wen H, Linhoff MW, McGinley MJ, Li GL, Corson GM, Mandel G, Brehm P (2010) Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc Natl Acad Sci U S A 107:13906–13911. 10.1073/pnas.1008598107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC (2009) Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci 12:759–766. 10.1038/nn.2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT (2002) Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron 36:897–908. 10.1016/S0896-6273(02)01065-6 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, Soltis SM, Alonso-Mori R, Chollet M, Lemke HT, Pfuetzner RA, Choi UB, Weis WI, Diao J, Südhof TC, Brunger AT (2015) Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525:62–67. 10.1038/nature14975 [DOI] [PMC free article] [PubMed] [Google Scholar]