

Abstract
Glutamate-mediated excitotoxicity induces neuronal death by altering various intracellular signaling pathways and is implicated as a common pathogenic pathway in many neurodegenerative diseases. In the case of motor neuron disease, there is significant evidence to suggest that the overactivation of AMPA receptors due to deficiencies in the expression and function of glial glutamate transporters GLT1 and GLAST plays an important role in the mechanisms of neuronal death. However, a causal role for glial glutamate transporter dysfunction in motor neuron death remains unknown. Here, we developed a new animal model of excitotoxicity by conditionally deleting astroglial glutamate transporters GLT1 and GLAST in the spinal cords of mice (GLAST+/−/GLT1-cKO). GLAST+/−/GLT1-cKO mice (both sexes) exhibited nuclear irregularity and calpain-mediated degradation of nuclear pore complexes (NPCs), which are responsible for nucleocytoplasmic transport. These abnormalities were associated with progressive motor neuron loss, severe paralysis, and shortened lifespan. The nuclear export inhibitor KPT-350 slowed but did not prevent motor neuron death, whereas long-term treatment of the AMPA receptor antagonist perampanel and the calpain inhibitor SNJ-1945 had more persistent beneficial effects. Thus, NPC degradation contributes to AMPA receptor-mediated excitotoxic motor neuronal death, and preventing NPC degradation has robust protective effects. Normalization of NPC function could be a novel therapeutic strategy for neurodegenerative disorders in which AMPA receptor-mediated excitotoxicity is a contributory factor.
SIGNIFICANCE STATEMENT Despite glial glutamate transporter dysfunction leading to excitotoxicity has been documented in many neurological diseases, it remains unclear whether its dysfunction is a primary cause or secondary outcome of neuronal death at disease state. Here we show the combined loss of glial glutamate transporters GLT1 and GLAST in spinal cord caused motor neuronal death and hindlimb paralysis. Further, our novel mutant exhibits the nuclear irregularities and calpain-mediated progressive nuclear pore complex degradation. Our study reveals that glial glutamate transporter dysfunction is sufficient to cause motor neuronal death in vivo.
Keywords: animal model, excitotoxicity, glutamate, motor neuron, transporter
Introduction
Glutamate is a major excitatory neurotransmitter that regulates virtually all activities of the CNS including motor control, learning and memory, and cognition. However, excess extracellular glutamate can result in neuronal dysfunction and death, a process known as excitotoxicity (Lucas and Newhouse, 1957; Olney et al., 1974). Excess extracellular glutamate causes neuronal death by the disruption of intracellular organelles and aberrant proteolysis via ion-sensitive protease activation that results from excessive Ca2+ influx following the overstimulation of Ca2+-permeable glutamate receptors (Hollmann et al., 1991; Verdoorn et al., 1991; Bano et al., 2005; Kwak and Weiss, 2006; Lewerenz and Maher, 2015). Accumulating evidence implicates excitotoxicity involvement in the pathogenesis of many neurodegenerative diseases including Alzheimer's disease, multiple sclerosis, Parkinson's disease, Huntington's disease, frontotemporal dementia (FTD), and amyotrophic lateral sclerosis (ALS; Kawahara et al., 2004; Kwak and Weiss, 2006; Mehta et al., 2013; Zhang et al., 2016), suggesting that these diseases may share excitotoxicity as a common pathogenic pathway. Thus, understanding the molecular mechanisms underlying chronic excitotoxic neuronal death is of critical importance for the treatment of many neurodegenerative diseases.
There may be a number of mechanisms by which excitotoxicity is initiated: increased glutamate level due to enhanced glutamate release; increased glutamate level due to glutamate transporter dysfunction; and altered expression or sensitivity of glutamate receptors. Glutamate transporter dysfunction is widely documented in many neurodegenerative diseases such as Alzheimer's disease and ALS (Fontana, 2015; Miladinovic et al., 2015). There are five subtypes of Na+-dependent glutamate transporters, GLAST (EAAT1), GLT1 (EAAT2), EAAC1 (EAAT3), EAAT4, and EAAT5. GLAST and GLT1 are mainly expressed in astrocytes, while EAAT3, EAAT4, and EAAT5 are mainly expressed in neurons (Tanaka, 2000). Astroglial glutamate transporters (GLAST and GLT1) are the glutamate transporters mainly involved in the maintenance of physiological extracellular glutamate concentrations (Tanaka, 2005). In ALS, there is significant evidence to suggest that the dysfunction of glial glutamate transporters plays an important role in the pathogenic pathways of AMPA receptor-mediated motor neuron death. ALS patients show reduced glutamate uptake in the motor cortex and spinal cord (Rothstein et al., 1990), which results from a reduction of the glial glutamate transporter GLT1 (also known as EAAT2; Rothstein et al., 1995). In addition, recent studies have reported that the spinal cord of transgenic rats expressing mutant human TDP-43 selectively in astrocytes exhibit a progressive deletion of GLAST, another glial glutamate transporter, as well as GLT1 (Tong et al., 2013). However, it remains unknown whether the dysfunction of glial glutamate transporters is a primary cause or a secondary consequence of motor neuron degeneration. To explore a causal role for glial glutamate transporter dysfunction in motor neuron death and the signaling cascades leading to glutamate-mediated neuronal death, we generated a novel mouse model of chronic glutamate excitotoxicity by deleting GLT1 and GLAST from the spinal cord, because mouse with a complete deficiency of GLT1 exhibited seizure and premature death (Tanaka et al., 1997).
Materials and Methods
Mice.
All animal procedures were performed in accordance with the animal experiment plan approved by the Tokyo Medical and Dental University Animal Care and Use Committee. Mice were housed three to five animals per cage and were maintained on a regular 12 h light/dark cycle (light period, 8:00 A.M. to 8:00 P.M.) at a constant 25°C. Food and water were available ad libitum. In particular, when animals showed complete hindlimb paralysis, moistened food and water were made easily accessible to the animals on the cage floor. In the rare presence of a moribund animal, the animal was killed. Animal experiments were performed according to the Internal Institutional Review Committee. Floxed-GLT1 mice, Hoxb8-Cre transgenic mice, and GLAST knock-out (KO) mice were previously described (Watase et al., 1998; Witschi et al., 2010; Cui et al., 2014; Aida et al., 2015). Both male and female mice were used unless otherwise noted, and the number of male and female mice is described in sample sizes by experiment. The age and number of mice used for all experiments are described in the figure legends.
Behavioral tests.
Behavioral analyses were performed by investigators blind to the condition, genotype, and drug treatment groups.
Hanging wire test.
Hanging wire tests were performed as previously described (Hanada et al., 2013). Briefly, male mice were placed on a wire netting, and the netting was turned over and held 50 cm above the tabletop for 60 s. The time until the hindlimbs disengaged from the wire netting was recorded as the latency to fall.
Hindlimb reflex score.
The posture of hindlimbs was scored as previously described (Wils et al., 2010). Briefly, male mice were tail suspended for 14 s and scored using an established definition: 0, normal; 1, failure to stretch their hindlimbs; 2, hindlimb clasping; 3, hindlimb paralysis.
Histology.
Histological analysis was performed as previously described (Cui et al., 2014; Aida et al., 2015). Mice deeply anesthetized with pentobarbital (100 mg/kg, i.p.) were fixed by perfusion with 4% paraformaldehyde (PFA) in PBS. Brains and spinal cords were dissected and further fixed with 4% PFA overnight. Tissues were transferred to 30% sucrose/PBS for cryoprotection and separated into brain, cervical, thoracic, and lumbar spinal cords in OCT compound (Sakura). Cryosections were prepared at a 20 μm thickness for spinal cords and a 50 μm thickness for brain (CM3050s, Leica) and mounted on MAS-coated slides (Matunami). Cryosections were permeabilized and blocked with 0.3% Triton X-100, 1% bovine serum albumin (BSA), and 10% normal goat serum in PBS, and were incubated with primary antibodies overnight at 4°C. For choline acetyltransferase (ChAT), sections were heated in 10 mm citrate buffer, pH 6.0, at 100°C for 20 min then were permeabilized and blocked with 0.3% Triton X-100, 1% BSA, and 10% normal horse serum in PBS. The following antibodies were used: polyclonal anti-GLT1 (1:5000; a gift from M. Watanabe, Hokkaido University, Sapporo, Japan; Yamada et al., 1998), polyclonal anti-ChAT (1:200; MAB144P, Merck Millipore), polyclonal anti-glial fibrillary acidic protein (GFAP; 1:1000; Z0334, Dako), polyclonal anti-CD68 (1:100; MCA1957, AbD Serotec), monoclonal anti-NeuN (1:500; MAB377, Millipore), monoclonal anti-Nup153 (1:100; ab24700, Abcam), rabbit polyclonal anti-TDP-43 (1:100; 10782-2AP, Proteintech), goat polyclonal anti-Lamin B1 (1:200; sc-6217, Santa Cruz Biotechnology), mouse monoclonal anti-KPNB1 (1:200; sc-137016, Santa Cruz Biotechnology), polyclonal anti-RanBP1 (1:500; NB100-79814, Novus Biologicals), and polyclonal anti-cellular apoptosis susceptibility (CAS; 1:100; ab96755, Abcam) antibodies. Sections were washed, incubated with secondary antibodies conjugated with Alexa Fluor 488, 594, or 633, and DAPI (Life Technologies) for 2 h, and mounted with Fluoromount (Diagnostic BioSystems). For ChAT and Lamin B1 staining, sections were incubated with biotinylated donkey anti-goat IgG (1:200; Vector Laboratories) for 2 h, washed, and incubated with streptavidin conjugated with Alexa Fluor 488 (1:200; Invitrogen) for 2 h. For fluorescent Nissl staining, sections were incubated with NeuroTrace 640/660 Deep-Red Fluorescent Nissl Stain (1:100; Invitrogen) for 30 min, immersed in 0.3% Triton-X in PBS for 15 min, and washed two times in PBS. For GLT1 staining, sections were incubated with biotinylated goat anti-rabbit IgG (1:1000; Vector Laboratories) for 1 h, immersed in 1% H2O2/PBS, and incubated with AB complex (1:500; Vector Laboratories) for 1 h, and then visualized with a DAB substrate kit (Vector Laboratories). Images were acquired using an LSM710 Laser Scanning Confocal Microscope (Carl Zeiss) or an SZX16 Stereoscopic Microscope for GLT1 immunostaining (Olympus).
Electron microscopy.
Mice deeply anesthetized with pentobarbital (100 mg/kg, i.p.) were fixed by perfusion with 4% PFA and 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4. Spinal cords were dissected and further fixed with same fixative overnight. After dehydration, samples were embedded in Epon 812. Ultrathin sections were stained with 2% uranyl acetate for 20 min and lead citrate solution for 5 min. Electron micrographs were taken on an H7100 electron microscope (Hitachi).
Cell counts.
Cell counts were performed using ImageJ software with the cell counter plug-in (National Institutes of Health, Bethesda, MD) by investigators who were blind to genotype and drug treatment. For motor neurons, the number of ChAT-positive cells in the bilateral ventral horns of L3–L5 in three to nine serial sections was counted. For the other neurons, NeuN-positive cells in laminae I–IV were counted. Laminae boundaries were determined according to cytoarchitectonic criteria previously described (Wootz et al., 2013). For glial cells, GFAP- and CD68-positive cells in bilateral ventral horns of L3–L5 in three serial sections were counted. Motor neurons showing nuclear irregularity were defined as motor neurons (large ventral horn neurons with a nuclear diameter of >10 μm) in which laminB1-immunolabeled nuclear contour showed discontinuous and frayed morphology, such as a ruffled edge and pedal-like shape (Freibaum et al., 2015). Nup153-negative motor neurons were defined as motor neurons (large ventral horn neurons with a nuclear diameter of >10 μm) in which more than half of Nup153-immunolabeled nuclear contours were lost. Motor neurons showing TDP-43 pathology were defined as ChAT-positive cells displaying aberrant staining patterns of TDP-43, such as mislocalization to the cytoplasm (mislocalization), nuclear reduction without cytoplasmic immunoreactivity (reduction), and absence from both the nucleus and the cytoplasm (absence). CAS-negative motor neurons were defined as ChAT-positive cells in which more than half of CAS immunoreactivity was lost from the nucleus.
Western blot analysis.
Western blot analysis was performed as previously described (Aida et al., 2015). Brains and spinal cords were dissected after perfusion with ice-cold PBS. Brain hemisphere or lumbar spinal cords were homogenized in 0.32 m sucrose, 20 mm Tris-HCl, pH 7.5, 1 mm NaVO4, 1 mm NaF, and a protease inhibitor cocktail (Roche). After the calculation of protein concentrations by BCA assay (Sigma-Aldrich), samples were mixed with an equal amount of sample loading buffer (250 mm Tris-HCl, pH 6.8, 4% SDS, 10% glycerol, and 1% β-mercaptoethanol) and denatured by heating at 95°C for 10 min. Thirty micrograms of each sample was separated by 10% SDS-PAGE, transferred to PVDF membranes (Merck Millipore), and blocked in Tris-buffered saline (TBS) with 0.5% Tween 20 (TBS-T) and 5% skim milk for 1 h at room temperature. The membranes were incubated with primary antibodies in TBS-T containing 1% skim milk at 4°C overnight, washed, and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 1 h. The following antibodies were used: polyclonal anti-GLAST (1:1000; a gift from M. Watanabe, Hokkaido University), polyclonal anti-GLT1 (1:5000; a gift from M. Watanabe, Hokkaido University), polyclonal anti-EAAC1 (1:500; sc-25658, Santa Cruz Biotechnology), monoclonal anti-Nup62 (1:500; BD Biosciences), monoclonal anti-Nup153 (1:500; Abcam), polyclonal anti-Nup88 (1:1000; ab79785, Abcam), goat polyclonal anti-Lamin B1 (1:2000; sc-6217, Santa Cruz Biotechnology), monoclonal anti-spectrin (1:5000; MAB1622, Millipore), rabbit polyclonal anti-p35 (1:250; sc-820, Santa Cruz Biotechnology), rabbit polyclonal anti-TDP-43 (1:2000; 10782-2AP, Proteintech), monoclonal anti-α-tubulin (1:5000; T7451; Sigma-Aldrich), monoclonal anti-β-actin (1:5000; sc-47778, Santa Cruz Biotechnology), and HRP-conjugated anti-rabbit or mouse IgG (1:5000; Jackson ImmunoResearch) antibodies. The membranes were washed and visualized with Luminata Forte Western HRP substrate (Millipore). Gel images were taken and quantified by measuring the ratio of the band intensities normalized to the intensity of β-actin or α-tubulin using Image Lab Software (Bio-Rad).
In vitro calpain activity analysis.
In vitro calpain activity analysis was previously described (Sato et al., 2011). The mouse brains were homogenized in 10 volumes (volume/weight) of HEPES buffer (20 mm HEPES, pH 7.5, 5 mm KCl, 2 mm MgCl2, 1 mm EGTA, and 1 mm dithiothreitol) at 4°C using a Teflon pestle in a glass homogenizer. The homogenates were centrifuged at 18,000 × g for 20 min, and the resulting supernatants were collected. The supernatant was incubated in vitro with exogenous 5 mm CaCl2 at 30°C. Proteolysis of Nup153, Nup88, Nup62, LaminB1, α-spectrin, and TDP-43 was assessed by immunoblotting, as described above.
Measurement of endogenous calpain activity.
Calpain activity was measured using a kit from BioVision following manufacturer instructions. Briefly, tissue lysates were prepared from lumbar ventral horn of each mouse in extraction buffer and the homogenates were centrifuged at 13,200 rpm for 5 min at 4°C. After collecting supernatant, protein concentration was determined by BCA assay (Sigma-Aldrich). Fifty micrograms of protein was used for the calpain assay, as described by the manufacturer. Fluorescence emitted from calpain substrates and a cleaved one was quantified using a fluorescence microplate reader equipped with a 400 nm excitation filter, a 505 nm emission filter (Spark 10M, Tecan). Calpain activity was calculated as relative fluorescence units using Spark Control Software (Tecan).
Drug treatment.
Perampanel (Hanada et al., 2011) provided by Eisai was dissolved in 0.5% (w/v) methylcellulose (Wako Pure Chemicals) and orally administered to the mice at a dose of 20 mg/kg once a day from 6 to 7 weeks of age. Memantine (Sigma-Aldrich) dissolved in saline was injected intraperitoneally into the mice at a dose of 10 mg/kg twice a day from 6 to 7 weeks of age. MK-801 (Sigma-Aldrich) dissolved in saline was injected intraperitoneally into the mice at a dose of 3 mg/kg once a day from 6 to 8 weeks of age. SNJ-1945 (Oka et al., 2006) provided by Senju Pharmaceutical Co. was dissolved in 0.5% (w/v) sodium carboxymethyl cellulose (Wako Pure Chemicals) and orally administered to the mice at a dose of 200 mg/kg once a day from 6 to 7 weeks of age. KPT-350 (Haines et al., 2015) provided by Karyopharm Therapeutics was dissolved 0.6% Pluronic F-68 (w/v) and 0.6% PVP K-29/32 (w/v) in water and orally administered to the mice at a dose of 15 mg/kg every other day from 6 to 7 weeks of age. The hanging wire test was performed 24 h after the final administration, and then the mice were fixed by perfusion with 4% PFA/PBS and subjected to quantification of motor neurons.
To examine the long-term effects of perampanel and SNJ-1945, mice were treated daily with perampanel (20 mg/kg, orally) or SNJ-1945 (200 mg/kg, orally) for 42 d starting at 6 weeks of age. Since a 15 mg/kg dose of KPT-350 is not a safe dose for long-term treatment, we orally administered KPT-350 (7.5 mg/kg) three times per week from 6 to 12 weeks of age (Haines et al., 2015). Drug-treated and vehicle-treated animals were monitored daily for survival and weekly for grip strength. In the hanging wire test, we replaced the missing values in the latency to fall with 0 s when mice died during the long-term treatment.
Mice were fixed 24 h after the final administration by perfusion with 4% PFA/PBS and subjected to the quantification of motor neurons.
Experimental design.
All experiments were performed with appropriate controls such as GLT1flox/flox mice and vehicle-treated GLAST+/−/GLT1-cKO mice. The type, number, and sexes of the animals used for each experiment are detailed in the figures as follows:
Figure 1C–F: 3 GLT1flox/flox (control, 3 females), 3 Hoxb8-Cre/GLT1flox/flox (3 females), 3 GLAST+/−/GLT1flox/flox (3 females), and 3 Hoxb8-Cre/GLT1flox/flox/GLAST+/− (GLAST+/−/GLT1-cKO, 3 females) mice.
Figure 1.

Conditional knockout of astroglial glutamate transporters in the spinal cord causes paralysis and motor neuron death. A, Schematic diagram showing the generation of GLAST+/−/GLT1-cKO mice. B, GLT1 immunohistochemistry in brain, cervival (C6), thoracic (T2 and T10), and lumbar (L5) spinal cord tissue at 3 months of age. Scale bars: top, 5 mm; bottom, 500 μm. C, D, Western blot analysis of GLT1 in the brains (C) and lumbar spinal cords (D) of control (GLT1flox/flox), Hoxb8-Cre/GLT1flox/flox, GLAST+/−/GLT1flox/flox, and Hoxb8-Cre/GLT1flox/flox /GLAST+/− (GLAST+/−/GLT1-cKO) mice at 4 weeks of age (n = 3 for each groups). GLT1 monomer band intensities were normalized to β-actin. **p < 0.01. n.s., not significant (post hoc Tukey-HSD test after one-way ANOVA). E, Western blot analysis of GLAST in the lumbar spinal cord of control, Hoxb8-Cre/GLT1flox/flox, GLAST+/−/GLT1flox/flox, and GLAST+/−/GLT1-cKO mice at 4 weeks of age (n = 3 for each groups). GLAST band intensities were normalized to β-actin. **p < 0.01 (post hoc Tukey HSD test after one-way ANOVA). F, Western blot analysis of EAAC1 in the lumbar spinal cord of control, Hoxb8-Cre/GLT1flox/flox, GLAST+/−/GLT1flox/flox, and GLAST+/−/GLT1-cKO mice at 4 weeks of age (n = 3 for each groups). EAAC1 band intensities were normalized to β-actin. n.s., not significant (one-way ANOVA). G–I, Body weight changes (G), hindlimb reflex score (H), and hanging wire test (I) of control (GLT1flox/flox, n = 15) and GLAST+/−/GLT1-cKO animals (Hoxb8-Cre/GLT1flox/flox /GLAST+/−, n = 16). ***p < 0.001 (post hoc unpaired two-tailed t test at corresponding time-point after two-way repeated-measures ANOVA). J, Percentage survival of control (n = 11) and GLAST+/−/GLT1-cKO (n = 14) animals calculated using the Kaplan–Meier method (p = 0.000016, log rank test). All data are expressed as the mean ± SEM.
Figure 1G–I: 15 control (15 males) and 16 GLAST+/−/GLT1-cKO (16 males) mice.
Figure 1J: 11 control (11 males) and 14 GLAST+/−/GLT1-cKO (14 males) mice.
Figure 2B: P5W, 4 control (4 males) and 5 GLAST+/−/GLT1-cKO (5 males) mice; P6W, 4 control (4 males) and 4 GLAST+/−/GLT1-cKO (4 males) mice; P7W, 3 control (3 males) and 4 GLAST+/−/GLT1-cKO (4 males) mice; P8W, 3 control (3 males) and 3 GLAST+/−/GLT1-cKO (3 males) mice; P9W, 4 control (4 males) and 5 GLAST+/−/GLT1-cKO (5 males) mice; and P20W, 3 control (1 male, 2 females) and 4 GLAST+/−/GLT1-cKO (1 male, 3 females) mice.
Figure 2.
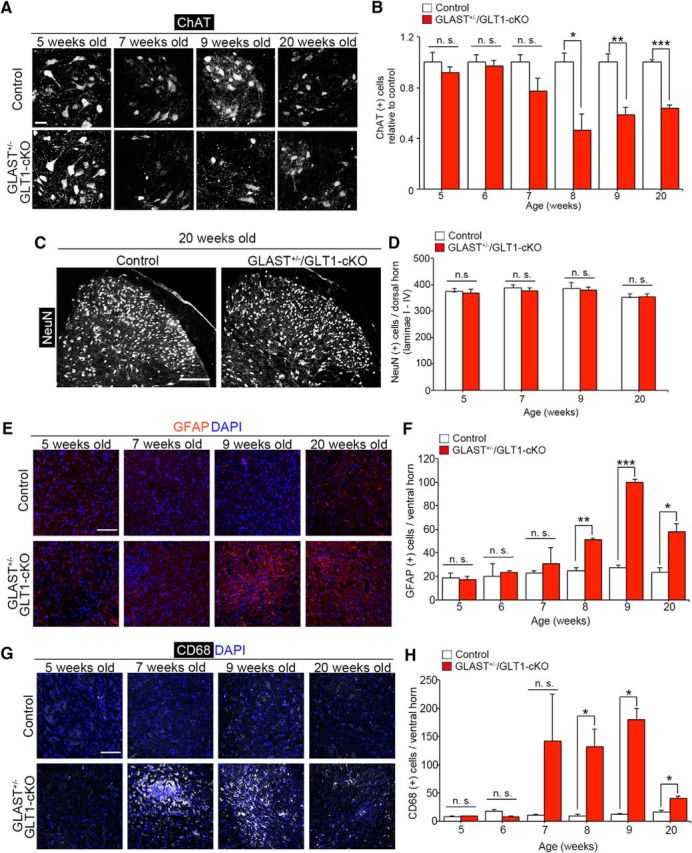
Loss of motor neurons and gliosis in GLAST+/−/GLT1-cKO mice. A, ChAT immunofluorescence of the lumbar ventral horn in control and GLAST+/−/GLT1-cKO mice at 5, 7, 9, and 20 weeks of age. Scale bar, 50 μm. B, Quantification of ChAT-positive motor neurons from 5 to 9, and 20 weeks of age of control mice (P5W, n = 4; P6W, n = 4; P7W, n = 3; P8W, n = 3; P9W, n = 4; P20W, n = 3) and GLAST+/−/GLT1-cKO mice (P5W, n = 5; P6W, n = 4; P7W, n = 4; P8W, n = 3; P9W, n = 5; P20W, n = 4). *p < 0.05, **p < 0.05, ***p < 0.001. n.s., not significant (unpaired two-tailed t test). C, NeuN immunofluorescence in the lumbar dorsal horn from control and GLAST+/−/GLT1-cKO mice at 20 weeks of age. Scale bar, 200 μm. D, Quantification of NeuN-positive neurons in the superficial dorsal horn (laminae I–IV) of the spinal cord at 5, 7, 9, and 20 weeks of control mice (P5W, n = 3; P7W, n = 4; P9W, n = 3; P20W, n = 3) and GLAST+/−/GLT1-cKO mice (P5W, n = 3; P7W, n = 4; P9W, n = 4; P20W, n = 3) mice. E, GFAP immunofluorescence in the lumbar ventral horn from control and GLAST+/−/GLT1-cKO mice at 5, 7, 9, and 20 weeks of age. Scale bar, 100 μm. F, Quantification of GFAP-positive cells in the lumbar ventral horn from 5 to 9, and 20 weeks of age of control mice (P5W, n = 3; P6W, n = 3; P7W, n = 4; P8W, n = 3; P9W, n = 3; P20W, n = 3) and GLAST+/−/GLT1-cKO mice (P5W, n = 3; P6W, n = 3; P7W, n = 4; P8W, n = 3; P9W, n = 3; P20W, n = 3) mice. *p < 0.05, **p < 0.01, ***p < 0.001. G, CD68 immunofluorescence in the lumbar ventral horns from control and GLAST+/−/GLT1-cKO mice at 5, 7, 9, and 20 weeks of age. Scale bar, 100 μm. H, Quantification of CD68-positive cells in the lumbar ventral horn from 5 to 9, and 20 weeks of age of control mice (P5W, n = 3; P6W, n = 3; P7W, n = 4; P8W, n = 3; P9W, n = 3; P20W, n = 3) and GLAST+/−/GLT1-cKO mice (P5W, n = 3; P6W, n = 3; P7W, n = 4; P8W, n = 3; P9W, n = 3; P20W, n = 3). *p < 0.05. All data are expressed as the mean ± SEM. n.s., Not significant (unpaired two-tailed t test).
Figure 2D: P5W, 3 control (2 males, 1 female) and 3 GLAST+/−/GLT1-cKO (2 males, 1 female) mice; P7W, 4 control (4 males) and 4 GLAST+/−/GLT1-cKO (4 males) mice; P9W, 3 control (3 males) and 4 GLAST+/−/GLT1-cKO (4 males) mice; and P20W, 3 control (1 male, 2 female) and 3 GLAST+/−/GLT1-cKO (1 male, 2 female) mice.
Figure 2F,H: P5W, 3 control (2 males, 1 female) and 3 GLAST+/−/GLT1-cKO (2 males, 1 female) mice; P6W, 3 control (2 males, 1 female) and 3 GLAST+/−/GLT1-cKO (2 males, 1 female) mice; P7W, 4 control (4 males) and 4 GLAST+/−/GLT1-cKO (4 male) mice; P8W 3 control (2 males, 1 female) and 3 GLAST+/−/GLT1-cKO (3 males); P9W, 3 control (3 males) and 3 GLAST+/−/GLT1-cKO (3 males) mice; and P20W, 3 control (1 male, 2 females) and 3 GLAST+/−/GLT1-cKO (1 male, 2 females) mice.
Figure 3A: 15 vehicle-treated GLAST+/−/GLT1-cKO (15 males) and 17 perampanel-treated GLAST+/−/GLT1-cKO (17 males) mice.
Figure 3.

Overactivation of AMPA receptor but not NMDA receptor contributes to motor deficits and motor neuron loss in GLAST+/−/GLT1-cKO mice. A, Perampanel treatment delayed motor deficits in the hanging wire test (n = 15 for vehicle-treated and n = 17 for perampanel-treated GLAST+/−/GLT1-cKO mice). *p < 0.05 (post hoc unpaired two-tailed t test at corresponding time point after two-way repeated-measures ANOVA). B, Amelioration of hindlimb paralysis in GLAST+/−/GLT1-cKO mice after perampanel treatment. Arrow indicates the fully paralyzed hindlimb of vehicle-treated GLAST+/−/GLT1-cKO mice. Arrowhead indicates the hindlimb upon which the perampanel-treated GLAST+/−/GLT1-cKO mice could stand. C, ChAT immunofluorescence of the lumbar ventral horn from control, vehicle-treated, and perampanel-treated GLAST+/−/GLT1-cKO mice at 7 weeks of age. Scale bar, 100 μm. D, Quantification of ChAT-positive motor neurons at 7 weeks of age (n = 3 for control mice, n = 4 for vehicle-treated mice, and n = 5 for perampanel-treated GLAST+/−/GLT1-cKO mice). *p < 0.05, ***p < 0.001 (post hoc Tukey HSD test after one-way ANOVA). E, Memantine treatment could not ameliorate motor deficits in the hanging wire test (n = 9 for vehicle-treated and n = 7 for memantine-treated GLAST+/−/GLT1-cKO mice). n.s., Not significant (two-way repeated-measures ANOVA). F, ChAT immunofluorescence of the lumbar ventral horn from vehicle-treated and memantine-treated GLAST+/−/GLT1-cKO mice at 7 weeks of age. Scale bar, 100 μm. G, Quantification of ChAT-positive motor neurons at 7 weeks of age (n = 3 for vehicle-treated and 4 for memantine-treated GLAST+/−/GLT1-cKO mice). n.s., Not significant (unpaired two-tailed t test). H, MK-801 treatment could not ameliorate motor deficits in the hanging wire test (n = 5 for vehicle-treated and 6 for MK-801-treated GLAST+/−/GLT1-cKO mice). n.s., Not significant (two-way repeated-measures ANOVA). I ChAT immunofluorescence of the lumbar ventral horn from vehicle-treated and MK-801-treated GLAST+/−/GLT1-cKO mice at 8 weeks of age. Scale bar, 100 μm. J, Quantification of ChAT-positive motor neurons at 8 weeks of age (n = 3 for each groups). n.s., Not significant (unpaired two-tailed t test). All data are expressed as the mean ± SEM.
Figure 3D: 3 control (3 males), 4 vehicle-treated GLAST+/−/GLT1-cKO (4 males) and 5 perampanel-treated GLAST+/−/GLT1-cKO (5 males) mice.
Figure 3E: 9 vehicle-treated GLAST+/−/GLT1-cKO (5 males, 4 females) and 7 memantine-treated GLAST+/−/GLT1-cKO (4 males, 3 females) mice.
Figure 3G: 3 vehicle-treated GLAST+/−/GLT1-cKO (3 females) and 4 memantine-treated GLAST+/−/GLT1-cKO (1 male, 3 female) mice.
Figure 3H: 5 vehicle-treated GLAST+/−/GLT1-cKO (4 males, 1 females) and 6 MK-801-treated GLAST+/−/GLT1-cKO (4 males, 2 female) mice.
Figure 3J: 3 vehicle-treated GLAST+/−/GLT1-cKO (3 males) and 3 MK-801-treated GLAST+/−/GLT1-cKO (2 male, 1 female) mice.
Figure 4C: P4W, 3 control (3 females) and 3 GLAST+/−/GLT1-cKO (3 females) mice; P5W, 3 control (1 male, 2 females) and 4 GLAST+/−/GLT1-cKO (2 males, 2 females) mice; P6W, 4 control (4 males) and 6 GLAST+/−/GLT1-cKO (3 males, 3 females) mice; P7W, 5 control (5 males) and 4 GLAST+/−/GLT1-cKO (2 males, 2 females) mice; P8W, 3 control (3 females) and 3 GLAST+/−/GLT1-cKO (2 male, 1 females) mice; and P9W, 3 control (2 males, 1 female) and 3 GLAST+/−/GLT1-cKO (2 males, 1 female) mice.
Figure 4.

Nuclear contour irregularity and the loss of nuclear pore complex proteins are observed in spinal motor neurons of GLAST+/−/GLT1-cKO mice. A, Pseudocolored electron microscopic images of motor neurons in lumbar ventral horns from control and GLAST+/−/GLT1-cKO mice at 6 weeks of age. The nucleus of the motor neuron is pseudocolored in light blue. Scale bar, 2 μm. B, Lamin B1 immunofluorescence of lumbar ventral horns from control and GLAST+/−/GLT1-cKO mice at 7 weeks of age. Sections were double labeled with anti-lamin B1 (green) and DAPI (blue). Rectangles in middle panel are enlarged in the right panel. Scale bars: left, 10 μm; right, 5 μm. C, Quantitative analysis of Lamin B1-immunostained sections. The percentage of large ventral horn neurons showing nuclear contour irregularity is higher in GLAST+/−/GLT1-cKO mice from 5 weeks of age. Large ventral horn neurons showing nuclear contour irregularity were counted from control mice (P4W, n = 3; P5W, n = 3; P6W, n = 4; P7W, n = 5; P8W, n = 3; P9W, n = 3) and GLAST+/−/GLT1-cKO mice (P4W, n = 3; P5W, n = 4; P6W, n = 6; P7W, n = 4; P8W, n = 3; P9W, n = 3). *p < 0.05, **p < 0.01, ***p < 0.001. n.s., Not significant (unpaired two-tailed t test). D, Nup153 immunofluorescence of lumbar ventral horns from control and GLAST+/−/GLT1-cKO mice at 6 weeks of age. Sections were triple labeled with anti-Nup153 (magenta), DAPI (blue), and Neurotrace (fluorescent Nissl stain, white). Rectangles in the middle panel are enlarged in right panel. Scale bars: left, 10 μm; right, 5 μm. E, Quantitative analysis of Nup153-immunostained sections. The percentage of Nup153-negative large ventral horn neurons is higher in GLAST+/−/GLT1-cKO mice from 6 weeks of age. Nup153-negative large ventral horn neurons were counted from control mice (n = 3 at each age), and GLAST+/−/GLT1-cKO mice (P4W, n = 3; P5W, n = 4; P6W, n = 3; P7W, n = 4) mice. *p < 0.05, ***p < 0.001. n.s., Not significant (unpaired two-tailed t test). All data are expressed as the mean ± SEM. F, Nup153 immunofluorescence of lumbar ChAT-positive motor neurons from control and GLAST+/−/GLT1-cKO mice at 7 weeks of age. Sections were triple-labeled with anti-Nup153 (magenta), DAPI (blue), and ChAT (white). Rectangles in middle panel are enlarged in right panel. Scale bars: left, 10 μm; right, 5 μm. G, Quantitative analysis of Nup153(−)/ChAT(+) motor neurons. The percentage of Nup153 (−)/ ChAT(+) motor neurons is similar to that of Nup153(−) large ventral horn neurons in GLAST+/−/GLT1-cKO mice at 7 weeks of age (n = 4 for control and n = 5 for GLAST+/−/GLT1-cKO). **p < 0.01 (unpaired two-tailed t test). All data are expressed as the mean ± SEM.
Figure 4E: P4W, 3 control (3 females) and 3 GLAST+/−/GLT1-cKO (3 females) mice; P5W, 3 control (1 male, 2 females) and 4 GLAST+/−/GLT1-cKO (2 males, 2 females) mice; P6W, 3 control (1 male, 2 females) and 3 GLAST+/−/GLT1-cKO (1 male, 2 females) mice; and P7W, 3 control (3 males) and 4 GLAST+/−/GLT1-cKO (4 males) mice.
Figure 4G: 4 control (3 males, 1 female) and 5 GLAST+/−/GLT1-cKO (4 males, 1 female) mice.
Figure 5C: P5W, 3 control (1 male, 2 females) and 3 GLAST+/−/GLT1-cKO (1 make, 2 females) mice; and P6W, 4 control (4 females) and 3 GLAST+/−/GLT1-cKO (3 females) mice.
Figure 5.

Calpain overactivation contributes to motor deficits and motor neuron loss in GLAST+/−/GLT1-cKO mice. A, Representative immunoblot that shows proteolysis of Nup153, Nup88, Nup62, laminB1, and α-spectrin in mouse brain homogenate that was incubated with exogenous calcium and/or a calpain inhibitor, SNJ-1945 (100 μm). B, Representative immunoblot showing proteolysis of p35 in lumbar ventral horn from control and GLAST+/−/GLT1-cKO mice at 5 and 6 weeks of age. C, Quantification of the p25/p35 ratio from control mice (P5W, n = 3; P6W, n = 4) and GLAST+/−/GLT1-cKO mice (n = 3 at each age). Band intensities of p35 and p25 were normalized to α-tubulin. **p < 0.01. n.s., Not significant (unpaired two-tailed t test). D, Measurement of calpain activity of lumbar ventral horn form control mice (P4W, n = 5; P5W, n = 6; P6W, n = 5) and GLAST+/−/GLT1-cKO mice (P4W, n = 6; P5W, n = 5; P6W, n = 6). Relative fluorescence units (RFUs) were compared by calculating the fold difference in enzyme activity. *p < 0.05; n.s., Not significant (unpaired two-tailed t test). E, TDP-43 immunofluorescence in the lumbar ventral horns from control at 7 weeks of age (the top row of a panel) and GLAST+/−/GLT1-cKO mice at 7 (the second and third rows of a panel) and 9 (the bottom row of a panel) weeks of age. Sections were double labeled with TDP-43 (red) and ChAT (white). Nuclei were visualized with DAPI. The second row of a panel shows mislocalization of TDP-43 to the cytoplasm in motor neurons of GLAST+/−/GLT1-cKO mice. The third row of a panel shows reduction of nuclear TDP-43 without cytoplasmic TDP-43 immunoreactivity in motor neurons of GLAST+/−/GLT1-cKO mice. The bottom row of a panel shows loss of TDP-43 from both the nucleus (yellow arrowhead) and the cytoplasm in motor neurons of GLAST+/−/GLT1-cKO mice. Scale bar, 10 μm. F, The percentage of motor neurons showing aberrant staining pattern of TDP-43 from 5, 6, 7, and 9 weeks of age of control mice (P5W, n = 7; P6W, n = 6; P7W, n = 5; P9W, n = 6) and GLAST+/−/GLT1-cKO mice (P5W, n = 7; P6W, n = 5; P7W, n = 6; P9W, n = 5). *p < 0.05, **p < 0.01 (unpaired two-tailed t test). G, Representative immunoblot that shows proteolysis of TDP-43 and β-actin in mouse brain homogenate that was incubated with exogenous calcium and/or a calpain inhibitor, SNJ-1945 (100 μm). Black and white arrowheads indicate a full-length and cleaved fragment of TDP-43, respectively. H, SNJ-1945 treatment delayed motor deficits in the hanging wire test (n = 8 for each groups). *p < 0.05 (post hoc unpaired two-tailed t test at corresponding time point after two-way repeated-measures ANOVA). I, ChAT immunofluorescence of the lumbar ventral horns from vehicle-treated and SNJ-1945-treated GLAST+/−/GLT1-cKO mice at 7 weeks of age. Scale bar, 100 μm. J, Quantification of ChAT-positive motor neurons at 7 weeks of age (n = 5 for vehicle-treated and n = 6 for SNJ-1945-treated GLAST+/−/GLT1-cKO mice). **p < 0.01 (unpaired two-tailed t test). K, Nup153 immunofluorescence of lumbar ventral horns from vehicle-treated and SNJ-1945-treated GLAST+/−/GLT1-cKO mice at 7 weeks of age. Sections were triple labeled with anti-Nup153 (magenta), DAPI (blue), and Neurotrace (fluorescent Nissl stain, white). Rectangles in the middle panel are enlarged in the right panel. Scale bars: left, 10 μm; right, 5 μm. L, Quantitative analysis of Nup153-immunostained sections. The percentage of Nup153-negative large ventral horn neurons decreased in GLAST+/−/GLT1-cKO mice treated with SNJ-1945 (n = 4 for each groups). **p < 0.01 (unpaired two-tailed t test). M, Nup153 immunofluorescence of lumbar ventral horns from vehicle-treated and perampanel-treated GLAST+/−/GLT1-cKO mice at 7 weeks of age. Sections were triple labeled with anti-Nup153 (magenta), DAPI (blue), and Neurotrace (fluorescent Nissl stain, white). Rectangles in the middle panel are enlarged in the right panel. Scale bars: left, 20 μm; right, 5 μm. N, Quantitative analysis of Nup153-immunostained sections. The percentage of Nup153-negative large ventral horn neurons decreased in GLAST+/−/GLT1-cKO mice treated with perampanel (n = 4 for vehicle-treated and n = 5 for perampanel-treated GLAST+/−/GLT1-cKO mice). *p < 0.05 (unpaired two-tailed t test). All data are expressed as the mean ± SEM.
Figure 5D: P4W, 5 control (2 males, 3 females) and 6 GLAST+/−/GLT1-cKO (2 males, 4 females) mice; P5W, 6 control (3 males, 3 females) and 5 GLAST+/−/GLT1-cKO (3 males, 2 females) mice; and P6W, 5 control (3 males, 2 females) and 6 GLAST+/−/GLT1-cKO (3 males, 3 females) mice.
Figure 5F: P5W, 7 control (7 males) and 7 GLAST+/−/GLT1-cKO (7 males) mice; P6W, 6 control (6 males) and 5 GLAST+/−/GLT1-cKO (3 males, 2 females) mice; P7W, 5 control (5 males) and 6 GLAST+/−/GLT1-cKO (2 males, 4 females) mice; and P9W, 6 control (6 males) and 5 GLAST+/−/GLT1-cKO (5 males) mice.
Figure 5H: 8 vehicle-treated GLAST+/−/GLT1-cKO (6 males, 2 females) and 8 SNJ-1945-treated GLAST+/−/GLT1-cKO (7 males, 1 females) mice.
Figure 5J: 5 vehicle-treated GLAST+/−/GLT1-cKO (5 males) and 6 SNJ-1945-treated GLAST+/−/GLT1-cKO (6 males) mice.
Figure 5L: 4 vehicle-treated GLAST+/−/GLT1-cKO (4 males) and 4 SNJ-1945-treated GLAST+/−/GLT1-cKO (4 males) mice.
Figure 5N: 4 vehicle-treated GLAST+/−/GLT1-cKO (4 males) and 5 perampanel-treated GLAST+/−/GLT1-cKO (5 males) mice.
Figure 6C: 3 control (3 males) and 4 GLAST+/−/GLT1-cKO (3 males, 1 female) mice.
Figure 6.

Nuclear export inhibitor KPT-350 transiently ameliorates motor deficits, motor neuron loss in GLAST+/−/GLT1-cKO mice. A, KPNB1 immunofluorescence of motor neurons in the lumbar ventral horn from control and GLAST+/−/GLT1-cKO mice at 7 weeks of age. Sections were triple labeled with anti-KPNB1 (green), DAPI (blue), and anti-ChAT (white). Scale bar, 10 μm. B, RanBP-1 immunofluorescence of motor neurons in the lumbar ventral horn from control and GLAST+/−/GLT1-cKO mice at 7 weeks of age. Sections were triple labeled with anti-RanBP-1 (green), DAPI (blue), and anti-ChAT (white). Scale bar, 10 μm. C, CAS immunofluorescence of motor neurons in the lumbar ventral horn from control and GLAST+/−/GLT1-cKO mice at 7 weeks of age. Sections were triple labeled with anti-CAS (red), DAPI (blue), and anti-ChAT (white). Arrowheads indicate the reduction or absence of nuclear CAS immunoreactivity in GLAST+/−/GLT1-cKO mice. Scale bar, 10 μm. D, KPT-350 treatment delayed motor deficits in the hanging wire test (n = 10 for vehicle-treated and n = 9 for KPT-350-treated GLAST+/−/GLT1-cKO mice). *p < 0.05 (post hoc unpaired two-tailed t test at corresponding time point after two-way repeated-measures ANOVA). E, Amelioration of hindlimb paralysis in GLAST+/−/GLT1-cKO mice after KPT-350 treatment. Arrow indicates the fully paralyzed hindlimb of the vehicle-treated GLAST+/−/GLT1-cKO mice. Arrowhead indicates the hindlimb upon which the KPT-350-treated GLAST+/−/GLT1-cKO mice could stand. F, ChAT immunofluorescence of the lumbar ventral horn from vehicle-treated and KPT-350-treated GLAST+/−/GLT1-cKO mice at 7 weeks of age. Scale bar, 100 μm. G, Quantification of ChAT-positive motor neurons at 7 weeks of age (n = 7 for vehicle-treated and n = 5 for KPT-350-treated GLAST+/−/GLT1-cKO mice). ***p < 0.001 (unpaired two-tailed t test). H, Nup153 immunofluorescence of lumbar ventral horns from vehicle-treated and KPT-350-treated GLAST+/−/GLT1-cKO mice at 7 weeks of age. Sections were triple labeled with anti-Nup153 (magenta), DAPI (blue), and Neurotrace (fluorescent Nissl stain, white). Rectangles in the middle panel are enlarged in the right panel. Scale bars: left, 20 μm; right, 5 μm. (I) Quantitative analysis of Nup153-immunostained sections. The percentage of Nup153-negative large ventral horn neurons is not changed in GLAST+/−/GLT1-cKO mice treated with KPT-350 (n = 3 for each groups). n.s., Not significant (unpaired two-tailed t test). All data are expressed as the mean ± SEM.
Figure 6D: 10 vehicle-treated GLAST+/−/GLT1-cKO (5 males, 5 females) and 9 KPT-350-treated GLAST+/−/GLT1-cKO (3 males, 6 females) mice.
Figure 6G: 7 vehicle-treated GLAST+/−/GLT1-cKO (2 males, 5 females) and 5 KPT-350-treated GLAST+/−/GLT1-cKO (1 male, 4 females) mice.
Figure 6I: 3 vehicle-treated GLAST+/−/GLT1-cKO (1 male, 2 females) and 3 KPT-350-treated GLAST+/−/GLT1-cKO (3 females) mice.
Figure 7A,B: 20 vehicle-treated GLAST+/−/GLT1-cKO (11 males, 9 females) and 16 perampanel-treated GLAST+/−/GLT1-cKO (8 males, 8 females) mice.
Figure 7.

Effects of long-term treatment with perampanel, SNJ-1945, and KPT-350 in GLAST+/−/GLT1-cKO mice. A, Perampanel treatment for 6 weeks delayed motor deficits in the hanging wire test (n = 20 for vehicle-treated and n = 16 for perampanel-treated GLAST+/−/GLT1-cKO mice). B, Percentage survival of vehicle-treated (n = 20) and perampanel-treated (n = 16) GLAST+/−/GLT1-cKO mice calculated using the Kaplan–Meier method (p = 0.076, log rank test). n.s., Not significant. C, ChAT immunofluorescence of the lumbar ventral horn from vehicle-treated and perampanel-treated GLAST+/−/GLT1-cKO mice at 12 weeks of age. D, Quantification of ChAT-positive motor neurons at 12 weeks of age (n = 4 for vehicle-treated and n = 6 for perampanel-treated GLAST+/−/GLT1-cKO mice). E, SNJ-1945 treatment for 6 weeks delayed motor deficits in the hanging wire test (n = 18 for vehicle-treated and n = 16 for SNJ-1945-treated GLAST+/−/GLT1-cKO mice). F, Percentage survival of vehicle-treated (n = 18) and SNJ-1945-treated (n = 16) GLAST+/−/GLT1-cKO mice calculated using the Kaplan–Meier method (p = 0.026, log rank test). *p < 0.05. G, ChAT immunofluorescence of the lumbar ventral horn from vehicle-treated and perampanel-treated GLAST+/−/GLT1-cKO mice at 12 weeks of age. H, Quantification of ChAT-positive motor neurons at 12 weeks of age (n = 4 for vehicle-treated and n = 6 for SNJ-1945-treated GLAST+/−/GLT1-cKO mice). I, Amelioration of motor dysfunction by KPT-350 treatment was observed at 7 weeks of age but not at later (n = 19 for vehicle-treated and n = 14 for KPT-350-treated GLAST+/−/GLT1-cKO mice). J, Percentage survival of vehicle-treated (n = 19) and KPT-350-treated (n = 14) GLAST+/−/GLT1-cKO mice calculated using the Kaplan-Meier method (p = 0.86, logrank test). n.s., Not significant. K, ChAT immunofluorescence of the lumbar ventral horn from vehicle-treated and KPT-350-treated GLAST+/−/GLT1-cKO mice at 12 weeks of age. L, Quantification of ChAT-positive motor neurons at 12 weeks of age (n = 4 for vehicle-treated and n = 6 for KPT-350-treated GLAST+/−/GLT1-cKO mice). Scale bars: C, G, K, 100 μm. A, E, I, *p < 0.05, **p < 0.01, ***p < 0.001 (post hoc unpaired two-tailed t test at corresponding time point after two-way repeated-measures ANOVA). D, H, L, *p < 0.05. n.s., Not significant (unpaired two-tailed t test). Red broken lines represent the first day of administration. All data are expressed as the mean ± SEM.
Figure 7D: 4 vehicle-treated GLAST+/−/GLT1-cKO (3 males, 1 females) and 6 perampanel-treated GLAST+/−/GLT1-cKO (3 males, 3 females) mice.
Figure 7E,F: 18 vehicle-treated GLAST+/−/GLT1-cKO (9 males, 9 females) and 16 SNJ-1945-treated GLAST+/−/GLT1-cKO (6 males, 10 females) mice.
Figure 7H: 4 vehicle-treated GLAST+/−/GLT1-cKO (3 males, 1 female) and 6 SNJ-1945-treated GLAST+/−/GLT1-cKO (2 males, 4 females) mice.
Figure 7I,J: 19 vehicle-treated GLAST+/−/GLT1-cKO (9 males, 10 females) and 14 KPT-350-treated GLAST+/−/GLT1-cKO (7 males, 7 females) mice.
Figure 7L: 4 vehicle-treated GLAST+/−/GLT1-cKO (2 males, 2 females) and 6 KPT-350-treated GLAST+/−/GLT1-cKO (4 males, 2 females) mice.
Statistical analysis.
All data are expressed as the mean ± SEM. An unpaired two-tailed t test was used to compare differences between any two groups. One-way ANOVA with post hoc Tukey's HSD test was used to compare differences between among than three groups. For longitudinal observations, two-way repeated-measures ANOVA with post hoc unpaired two-tailed t test at the corresponding time point was performed. Significant differences were analyzed by Kaplan-Meier and log rank tests for the survival rate. All statistical analyses were performed using SPSS 24 (IBM). A p value of <0.05 was considered to be statistically significant.
Results
Generation of spinal cord-specific astroglial glutamate transporters KO mice (GLAST+/−/GLT1-cKO)
Because astroglial glutamate transporters (GLT1 and GLAST) are critical for the maintenance of extracellular glutamate and the prevention of chronic excitotoxicity (Tanaka et al., 1997; Watase et al., 1998; Matsugami et al., 2006), we generated mice that lacked GLT1 specifically in the spinal cord on a GLAST heterozygous background (Hoxb8-Cre/GLT1flox/flox/GLAST+/− mice, hereafter, denoted as “GLAST+/−/GLT1-cKO mice”; Fig. 1A; Watase et al., 1998; Witschi et al., 2010; Cui et al., 2014; Aida et al., 2015). Immunohistochemistry and Western blot analysis of GLT1 showed that the amount of GLT1 protein was gradually reduced from the spinal cord caudal to the cervical level C6 and completely lost in the lumbar spinal cord in GLAST+/−/GLT1-cKO mice, which was consistent with the pattern of Cre-recombinase expression in the Hoxb8-Cre line [Fig. 1B–D; (Fig. 1C: one-way ANOVA, F(3,8) = 0.11, p = 0.95; Fig. 1D: one-way ANOVA, F(3,8) = 17.53, p = 0.00071; post hoc Tukey HSD tests, GLT1flox/flox vs Hoxb8-Cre/GLT1flox/flox, p = 0.0039; GLT1flox/flox vs GLAST+/−/GLT1flox/flox, p = 1.00; GLT1flox/flox vs GLAST+/−/GLT1-cKO, p = 0.0044; Hoxb8-Cre/GLT1flox/flox vs GLAST+/−/GLT1flox/flox, p = 0.0036; Hoxb8-Cre/GLT1flox/flox vs GLAST+/−/GLT1-cKO, p = 1.00; GLAST+/−/GLT1flox/flox vs GLAST+/−/GLT1-cKO, p = 0.0041)]. Western blot analysis of GLAST revealed an ∼50% reduction in the spinal cords of GLAST+/−/GLT1flox/flox and GLAST+/−/GLT1-cKO mice when compared with GLT1flox/flox (hereafter denoted as “control”) and Hoxb8-Cre/GLT1flox/flox mice (Fig. 1E [one-way ANOVA, F(3,8) = 22.81, p = 0.00028; post hoc Tukey HSD tests, control vs Hoxb8-Cre/GLT1flox/flox, p = 0.95; control vs GLAST+/−/GLT1flox/flox, p = 0.0027; control vs GLAST+/−/GLT1-cKO, p = 0.0020; Hoxb8-Cre/GLT1flox/flox vs GLAST+/−/GLT1flox/flox, p = 0.0015; Hoxb8-Cre/GLT1flox/flox vs GLAST+/−/GLT1-cKO, p = 0.0012; GLAST+/−/GLT1flox/flox vs GLAST+/−/GLT1-cKO, p = 0.99]). There was no compensatory upregulation of other glutamate transporters (GLAST and EAAC1) in Hoxb8-Cre/GLT1flox/flox, GLAST+/−/GLT1flox/flox, and GLAST+/−/GLT1-cKO mice (Fig. 1E,F [Fig. 1F: one-way ANOVA, F(3,8) = 0.095, p = 0.96]). Thus, we succeeded in generating spinal cord-specific astroglial glutamate transporters knock-out mice.
Impaired motor function and motor neuron loss in GLAST+/−/GLT1-cKO mice
The most appropriate controls for GLAST+/−/GLT1-cKO mice are GLAST+/−/GLT1flox/flox mice. However, there was no difference in the expression levels of GLAST and GLT1 between GLAST+/−/GLT1flox/flox and GLAST+/− mice. In addition, GLAST+/− mice did not show apparent survival differences or any obvious abnormalities in basic motor and neurological function compared with wild-type mice (Watase et al., 1998; Karlsson et al., 2009; Aida et al., 2015). Thus, we used GLT1flox/flox mice as controls instead of GLAST+/−/GLT1flox/flox mice in this study. GLAST+/−/GLT1-cKO mice had lower body weights than did the controls starting at 7 weeks of age [Fig. 1G (two-way repeated-measures ANOVA for genotype × age interaction: F(4,116) = 41.40, p = 1.8E-21; post hoc unpaired two-tailed t tests: P5W, t(29) = 0.41, p = 0.69; P6W, t(29) = 1.10, p = 0.28; P7W, t(29) = 3.89, p = 0.00054; P8W, t(29) = 5.42, p = 7.8E-06; P9W, t(29) = 8.46, p = 2.6E-09)]. They displayed an impaired hindlimb reflex by 7 weeks of age, after which they progressively lost grip strength and became fully paralyzed in the hindlimbs by 8 weeks of age [Fig. 1H,I (Fig. 1H: two-way repeated-measures ANOVA for genotype × age interaction, F(4,116) = 62.03, p = 6.2E-28; post hoc unpaired two-tailed t tests, P5W, t(29) = 0.98, p = 0.33; P6W, t(29) = 1.14, p = 0.26; P7W, t(29) = 3.79, p = 0.0007; P8W, t(29) = 13.75, p = 3.1E-14; P9W, t(29) = 13.97, p = 2.1E-14; Fig. 1I: two-way repeated-measures ANOVA for genotype × age interaction, F(4,116) = 37.49, p = 4.2E-20; post hoc unpaired two-tailed t tests, P5W, t(29) = 1.01, p = 0.32; P6W, t(29) = 0.16, p = 0.88; P7W, t(29) = 1.89, p = 0.072; P8W, t(29) = 8.03, p = 7.4E-09; P9W, t(29) = 8.80, p = 1.1E-09)]. Thereafter, they had shortened life spans [Fig. 1J (log rank test, p = 0.000016)]. There was no significant difference in progression of motor deficits and survival rate between male and female GLAST+/−/GLT1-cKO mice (data not shown). In some GLAST+/−/GLT1-cKO mice, spasms of upper and lower limb muscles were observed, which were followed by an extremely rapid disease progression with mice dying within 3–4 d.
Because GLAST+/−/GLT1-cKO mice showed progressive motor deficits that resulted in hindlimb paralysis, we next examined whether GLT1 and GLAST deletion caused motor neuron loss and gliosis. ChAT immunolabeling showed motor neuron loss in lumbar ventral horn of GLAST+/−/GLT1-cKO mice beginning at 7 weeks of age with a peak at 8 weeks of age [Fig. 2A,B (Fig. 2B: unpaired two-tailed t tests, P5W, t(7) = 0.98, p = 0.36; P6W, t(6) = 0.38, p = 0.72; P7W, t(5) = 1.78, p = 0.14; P8W, t(4) = 3.70, p = 0.021; P9W, t(7) = 4.75, p = 0.0021; P20W, t(5) = 10.85, p = 0.00012). In contrast, there was no change in the number of NeuN-positive neurons in the superficial dorsal horn in GLAST+/−/GLT1-cKO mice [Fig. 2C,D (Fig. 2D: unpaired two-tailed t tests, P5W, t(4) = 0.26, p = 0.81; P7W, t(6) = 0.73, p = 0.49; P9W, t(5) = 0.20, p = 0.85; P20W, t(4) = 0.06, p = 0.96)]. Furthermore, enhanced immunoreactivity for GFAP, a marker of astroglia, and CD68, a marker of activated microglia, was detected in the ventral horn of the spinal cord of GLAST+/−/GLT1-cKO mice beginning at 7 weeks of age (symptomatic stage; Fig. 2E,G). Quantification indicated a significant increase of GFAP-positive and CD68-positive cells in the ventral horn of the spinal cord of GLAST+/−/GLT1-cKO mice beginning at 8 weeks of age [Fig. 2F,H [Fig. 2F: unpaired two-tailed t tests, P5W, t(4) = 0.33, p = 0.76; P6W, t(4) = 0.86, p = 0.44; P7W, t(6) = 0.55, p = 0.60; P8W, t(4) = 8.39, p = 0.0011; P9W, t(4) = 19.97, p = 3.7E-05; P20W, t(4) = 4.51, p = 0.011; Fig. 2H: P5W, t(4) = 1.09, p = 0.34; P6W, t(4) = 1.34, p = 0.25; P7W, t(6) = 1.39, p = 0.21; P8W, t(4) = 2.91, p = 0.044; P9W, t(4) = 4.10, p = 0.015; P20W, t(4) = 4.00, p = 0.016)].
Together, these results suggest that the dysfunction of astroglial glutamate transporters, GLT1 and GLAST, induced spinal motor neuron loss, resulting in severe hindlimb paralysis in vivo.
Involvement of AMPA receptors overactivation in spinal motor neuron loss of GLAST+/−/GLT1-cKO mice
Excess extracellular glutamate due to the dysfunction of astroglial glutamate transporters induces neuronal death via overactivation of ionotropic glutamate receptors. In particular, previous works have demonstrated the contribution of AMPA receptors (Couratier et al., 1993; Carriedo et al., 1996; Kwak and Weiss, 2006; Hideyama et al., 2010; Akamatsu et al., 2016; Yamashita et al., 2017b) and/or NMDA receptors to spinal motor neuron deaths (Mitchell et al., 2010; Sasabe et al., 2007, 2012). To determine the subtype of ionotropic glutamate receptor that is involved in motor neuron death in GLAST+/−/GLT1-cKO mice, we examined the effects of AMPA and NMDA receptor antagonists. We treated GLAST+/−/GLT1-cKO mice with perampanel (Hanada et al., 2011), an AMPA receptor antagonist that is used as an antiepileptic drug. Akamatsu et al. (2016) reported that perampanel (20 mg/kg) prevented the progression of the ALS phenotype in the AR2 mice, in which an RNA editing enzyme adenosine deaminase acting on RNA 2 (ADAR2) is conditionally knocked out in the motor neurons. Thus, GLAST+/−/GLT1-cKO mice were treated daily with perampanel (20 mg/kg, orally) starting at 6 weeks of age. Perampanel administration for 7 d prevented motor deficits and motor neuron loss in GLAST+/−/GLT1-cKO mice [Fig. 3A–D (Fig. 3A: two-way repeated-measures ANOVA for drug × time interaction, F(1,30) = 4.76, p = 0.037; post hoc unpaired two-tailed t test, P7W, t(30) = 2.48, p = 0.019; Fig. 3D: one-way ANOVA, F(2,9) = 18.93, p = 0.0006; post hoc Tukey's HSD tests, control vs vehicle-treated GLAST+/−/GLT1-cKO, p = 0.00048; control vs perampanel-treated GLAST+/−/GLT1-cKO, p = 0.045; vehicle-treated vs perampanel-treated GLAST+/−/GLT1-cKO, p = 0.0104)]. Next, to investigate the role of NMDA receptors in spinal motor neuron death in GLAST+/−/GLT1-cKO mice, we treated GLAST+/−/GLT1-cKO mice with memantine (Lipton, 2006), an NMDA receptor antagonist. Wang and Zhang (2005) reported that treating SOD1 (G93A) mice with memantine (10 mg/kg) significantly delayed the disease progression and prolonged survival. We administered memantine (10 mg/kg, i.p., twice per day) every day for 7 d to GLAST+/−/GLT1-cKO mice at 6 weeks of age. Memantine treatment for 7 d had no effect on motor function decline and motor neuron loss [Fig. 3E–G (Fig. 3E: two-way repeated-measures ANOVA for drug × time interaction, F(1,14) = 1.71, p = 0.21; Fig. 3G: unpaired two-tailed t test, t(5) = 1.25, p = 0.27)]. Furthermore, we examined the effect of another NMDA receptor antagonist, MK-801. Gill et al. (1987) demonstrated that systemic administration of MK-801 (3 mg/kg) completely protected ischemia-induced hippocampal damage in the gerbil. We administered MK-801 (3 mg/kg, i.p.) every day for 14 d to GLAST+/−/GLT1-cKO mice at 6 weeks of age. MK-801 treatment for 14 d could not prevent the progression of motor deficits and motor neuron loss [Fig. 3H–J (Fig. 3H: two-way repeated-measures ANOVA for drug × time interaction, F(2,18) = 0.02, p = 0.98; Fig. 3J: unpaired two-tailed t test, t(4) = 0.83, p = 0.46)].
These results suggest that overstimulation of AMPA receptors, but not NMDA receptors, induces motor neuron loss and severe motor deficits in this new in vivo model of chronic spinal cord excitotoxicity.
Calpain-dependent degradation of nuclear pore complex proteins in GLAST+/−/GLT1-cKO mice
We next examined the mechanisms by which the overactivation of AMPA receptors results in motor neuron death. An electron microscopic analysis revealed nuclear irregularities in spinal motor neurons of GLAST+/−/GLT1-cKO mice (Fig. 4A), which resembles the pathologies of ALS and Parkinson's disease patients (Kinoshita et al., 2009; Liu et al., 2012). The nuclear irregularity seen by electron microscopy was accentuated by lamin B1 immunostaining. In the large ventral horn neurons of control mice with a nuclear diameter of >10 μm, lamin B1 immunoreactivity was distributed predominantly as smooth nuclear contours (Fig. 4B). In contrast, the percentage of large ventral horn neurons exhibiting nuclear contour irregularity such as flayed and discontinuous morphology is significantly higher in GLAST+/−/GLT1-cKO mice as early as 5 weeks of age (presymptomatic stage; Fig. 4B,C). The large ventral horn neurons with an irregular nuclear contour became the most common type of large ventral horn neurons in GLAST+/−/GLT1-cKO mice from 7 week of age to 9 weeks of age [symptomatic stage; Fig. 4C (unpaired two-tailed t tests, P4W, t(4) = 0.33, p = 0.76; P5W, t(5) = 3.72, p = 0.014; P6W, t(8) = 4.38, p = 0.0024; P7W, t(7) = 7.39, p = 0.00015; P8W, t(4) = 5.16, p = 0.0067; P9W, t(4) = 6.60, p = 0.0027)]. These observations prompted an examination of nuclear pore complexes (NPCs) that were embedded in the nuclear envelope in the large ventral horn neurons of GLAST+/−/GLT1-cKO mice. We examined the immunohistochemical distribution of the major protein component of NPCs, Nup153 (Fahrenkrog and Aebi, 2003; Kinoshita et al., 2009; Freibaum et al., 2015). Whereas Nup153 immunoreactivity was distributed predominantly along smooth nuclear contours in controls, it was reduced in large ventral horn neurons of GLAST+/−/GLT1-cKO mice at 6 weeks of age [Fig. 4D,E (Fig. 4E: unpaired two-tailed t tests, P4W, t(4) = 1.48, p = 0.21; P5W, t(5) = 1.30, p = 0.25; P6W, t(4) = 3.27, p = 0.031; P7W, t(5) = 10.68, p = 0.00012)]. Double immunostaining of Nup153 and ChAT revealed that the percentage of Nup 153-negative motor neurons is significantly increased to the same extent as Nup 153-negative large ventral horn neurons in GLAST+/−/GLT1-cKO mice at 7 weeks of age [Fig. 4F,G (Fig. 4G: unpaired two-tailed t test, t(7) = 5.12, p = 0.0014)]. These results indicate the reduced NPC expression was occurred in the spinal motor neurons of GLAST+/−/GLT1-cKO mice.
The loss of NPC protein immunolabeling on the nuclear envelope suggests NPC protein degradation. Recent studies showed that nucleoporins were cleaved by Ca2+-activated protease calpain (Bano et al., 2010; Yamashita et al., 2017a). Consistent with this, an in vitro assay (Sato et al., 2011) using mouse brain homogenates displayed that the amounts of detectable full-length Nup62, Nup88, and Nup153 were decreased in a Ca2+-dependent manner, whereas SNJ-1945 (Oka et al., 2006), a calpain inhibitor, reversed the loss of these proteins (Fig. 5A). However, laminB1 was not a calpain substrate (Fig. 5A). In addition, we observed that the cleavage of p35, which is well known as a calpain substrate (Lee et al., 2000), was significantly enhanced in the spinal cords of GLAST+/−/GLT1-cKO mice at 6 weeks of age compared with the control mice [Fig. 5B,C (Fig. 5C: unpaired two-tailed t tests, P5W, t(4) = 0.97, p = 0.39; P6W, t(5) = 5.10, p = 0.0038)]. These results suggest that calpain is hyperactivated in the spinal cord of GLAST+/−/GLT1-cKO mice. To confirm this, we measured calpain activity of the spinal cords. An increase in calpain activity in lumbar ventral horn was observed in GLAST+/−/GLT1-cKO mice as early as 5 weeks of age [Fig. 5D (unpaired two-tailed t tests, P4W, t(9) = 0.28, p = 0.79; P5W, t(9) = 3.05, p = 0.014; P6W, t(9) = 2.90, p = 0.018)]. Yamashita et al. (2012, 2017b) and Yamashita and Kwak (2014) have reported that TAR DNA binding protein (TDP-43), one of the disease-related proteins of ALS and FTD, was also cleaved by calpain (Akamatsu et al., 2016). Thus, we assessed the immunohistochemical analysis for TDP-43. Immunofluorescence for TDP-43 demonstrated that TDP-43 localized exclusively to the nuclei in all of the spinal motor neurons in control mice, whereas some spinal motor neurons in GLAST+/−/GLT1-cKO mice exhibited aberrant staining patterns of TDP-43, such as mislocalization to the cytoplasm (mislocalization), nuclear reduction without cytoplasmic immunoreactivity (reduction), and the absence from both the nucleus and the cytoplasm (absence; Fig. 5E). Although the number of motor neurons showing aberrant staining patterns of TDP-43 was very low, we detected such motor neurons in GLAST+/−/GLT1-cKO mice as early as 5 weeks of age [Fig. 5F (unpaired two-tailed t tests, P5W, t(12) = 3.32, p = 0.0061; P6W, t(9) = 2.84, p = 0.019; P7W, t(9) = 3.94, p = 0.0034; P9W, t(9) = 2.53, p = 0.032)]. The percentage of motor neurons devoid of TDP-43 increased between 7 and 9 weeks of age when motor neuron loss was prominent (Fig. 5F). Consistent with previous reports (Yamashita et al., 2012, 2017b; Akamatsu et al., 2016), TDP-43 was cleaved by calpain (Fig. 5G), suggesting that various immunostaining patterns of TDP-43 in motor neurons of GLAST+/−/GLT1-cKO mice may result from cleavage by calpain.
To verify whether calpain-mediated NPC degradation was linked to motor deficits and neuronal death of GLAST+/−/GLT1-cKO mice, we examined the effects of a blood–brain barrier-permeable calpain inhibitor, SNJ-1945, in GLAST+/−/GLT1-cKO mice. Imai et al. (2010) reported that SNJ-1945 (200 mg/kg) significantly prevented light-induced photoreceptor degeneration in the mice. Thus, we treated mice daily with SNJ-1945 (200 mg/kg, orally) starting at 6 weeks of age. One week of SNJ-1945 treatment prevented the loss of grip strength of the hindlimbs in GLAST+/−/GLT1-cKO mice [Fig. 5H (two-way repeated-measures ANOVA for drug × time interaction, F(1,14) = 8.59, p = 0.011; post hoc unpaired two-tailed t test, P7W, t(14) = 2.93, p = 0.011)]. Furthermore, SNJ-1945 delayed the loss of spinal motor neurons and Nup153 immunoreactivity [Fig. 5I–L (Fig. 5J: unpaired two-tailed t test, t(9) = 3.30, p = 0.0093; Fig. 5L: unpaired two-tailed t test, t(6) = 4.86, p = 0.0028)]. In addition, we also investigated a role for AMPA receptors in the degradation of Nup153 in GLAST+/−/GLT1-cKO mice. One week of perampanel treatment (20 mg/kg, orally) prevented the degradation of Nup153 [Fig. 5M,N (Fig. 5N: unpaired two tailed t test, t(7) = 3.34, p = 0.012)] as well as the death of spinal motor neurons (Fig. 3C,D) in GLAST+/−/GLT1-cKO mice.
Together, these findings suggest that the hyperactivation of calpain due to the overactivation of AMPA receptors is involved in the degradation of the major protein components of NPCs, which is associated with excitotoxic spinal motor neuron death in GLAST+/−/GLT1-cKO mice.
Effects of short-term treatment with the nuclear export inhibitor KPT-350 in GLAST+/−/GLT1-cKO mice
Nucleocytoplasmic transport has a crucial role in cell viability, and the loss of nucleoporins has been reported to impair nuclear import and export (Roth et al., 2003; Pemberton and Paschal, 2005; Bano et al., 2010; Nishimura et al., 2010; Makise et al., 2012; Yamashita et al., 2017a). Because GLAST+/−/GLT1-cKO mice showed the reduction of Nup153 in motor neurons, we examined the changes in the subcellular localization of proteins involved in nucleocytoplasmic trafficking. The spinal motor neurons in GLAST+/−/GLT1-cKO mice exhibited normal nuclear immunoreactivity for KPNB1, which is the member of karyopherin protein family responsible for protein import, and normal cytoplasmic immunoreactivity for RanBP1, which regulates the assembly and disassembly of karyopherin-cargo complex (Fig. 6A,B). In contrast, subcellular localization of CAS protein, known as exportin-2, was different between controls and GLAST+/−/GLT1-cKO mice. Immunoreactivity for CAS was predominant in the nucleus of motor neurons in control mice, whereas some spinal motor neurons in GLAST+/−/GLT1-cKO mice showed reduced levels of CAS in the nucleus at 7 weeks of age (early symptomatic stage, Fig. 6C). Although the number of CAS-negative motor neurons was low, the percentage of CAS-negative motor neurons was increased in GLAST+/−/GLT1-cKO mice at 7 weeks of age [CAS (−)/ChAT (+) cells (%): control, 2.3 ± 1.3%; GLAST+/−/GLT1-cKO, 12.7 ± 2.6%; unpaired two-tailed t test, t(5) = 3.21, p = 0.024]. These results indicate that nucleocytoplasmic transport was slightly impaired in some motor neurons in GLAST+/−/GLT1-cKO mice.
To examine whether nucleocytoplasmic transport deficits are involved in motor dysfunction and motor neuron loss in GLAST+/−/GLT1-cKO mice, we analyzed the effect of KPT-350, a nuclear export inhibitor (Haines et al., 2015), because the in vivo toxicity of all the other modifiers of nucleocytoplasmic transport precluded their use. GLAST+/−/GLT1-cKO mice were treated every other day with KPT-350 (15 mg/kg, orally) starting at 6 weeks of age. GLAST+/−/GLT1-cKO mice treated with KPT-350 delayed motor deficits in the hanging wire test, whereas GLAST+/−/GLT1-cKO mice treated with vehicle showed complete hindlimb paralysis [Fig. 6D,E (Fig. 6D: two-way repeated-measures ANOVA for drug × time interaction, F(1,17) = 7.98, p = 0.012; post hoc unpaired two-tailed t test, P7W, t(17) = 2.82, p = 0.012)]. KPT-350 treatment also delayed motor neuron loss in GLAST+/−/GLT1-cKO mice [Fig. 6F,G (Fig. 6G: unpaired two-tailed t test, t(10) = 4.83, p = 0.00069)]. However, the effects of KPT-350 were transient, and long-term treatment of KPT-350 did not prevent motor neuron loss and motor deficits in GLAST+/−/GLT1-cKO mice (Fig. 7I,K,L). Although KPT-350 delayed motor neuron death and motor deficits in GLAST+/−/GLT1-cKO mice, the degradation of Nup153 was not prevented [Fig. 6H,I (Fig. 6I: unpaired two-tailed t test, t(4) = 0.13, p = 0.90)].
Together, these findings suggest that nucleocytoplasmic transport deficits may partially contribute to excitotoxic spinal motor neuron degeneration in GLAST+/−/GLT1-cKO mice, but may be not a major cause of motor neuron loss in GLAST+/−/GLT1-cKO mice.
Effects of long-term treatment with perampanel, SNJ-1945 and KPT-350 in GLAST+/−/GLT1-cKO mice
We next evaluated the efficacy of long-term treatment with perampanel, SNJ-1945, and KPT-350 on the phenotypes of GLAST+/−/GLT1-cKO mice. GLAST+/−/GLT1-cKO mice were treated daily with perampanel (20 mg/kg, orally) or SNJ-1945 (200 mg/kg, orally) for 42 d starting at 6 weeks of age. Since the treatment of control mice with a 15 mg/kg dose of KPT-350 three times per week for 3 weeks causes serious weight loss, we orally administered KPT-350 (7.5 mg/kg) three times per week from 6 to 12 weeks of age. Drug-treated and vehicle-treated animals were monitored daily for survival and weekly for grip strength. The results showed that motor function decline was delayed by the administration of perampanel or SNJ-1945 [Fig. 7A,E (Fig. 7A: two-way repeated-measures ANOVA for drug × time interaction, F(6,204) = 5.59, p = 2.2E-05; post hoc unpaired two-tailed t tests, P7W, t(34) = 2.44, p = 0.020; P8W, t(34) = 3.18, p = 0.0031; P9W, t(34) = 2.70, p = 0.011; P10W, t(34) = 4.28, p = 0.00014; P11W, t(34) = 4.64, p = 5.0E-05; P12W, t(34) = 5.37, p = 5.8E-06; Fig. 7E: two-way repeated-measures ANOVA for drug × time interaction, F(6,192) = 7.16, p = 6.6E-07; post hoc unpaired two-tailed t tests, P7W, t(32) = 3.79, p = 0.00062; P8W, t(32) = 2.63, p = 0.013; P9W, t(32) = 2.86, p = 0.0074; P10W, t(32) = 3.64, p = 0.00095; P11W, t(32) = 3.88, p = 0.000495; P12W, t(32) = 4.12, p = 0.00025)]. In contrast, KPT-350 treatment delayed motor deficits at 7 weeks of age but not later in GLAST+/−/GLT1-cKO mice [Fig. 7I (two-way repeated-measures ANOVA for drug × time interaction, F(6,186) = 8.85, p = 1.7E-08; post hoc unpaired two-tailed t test, P7W, t(31) = 3.03, p = 0.0049)]. SNJ-1945 treatment prolonged survival in GLAST+/−/GLT1-cKO mice [Fig. 7F (log rank test, p = 0.026)], whereas KPT-350 treatment had no effect on survival [Fig. 7J (log rank test, p = 0.86)]. Although there is a tendency for survival rate to be higher in perampanel-treated GLAST+/−/GLT1-cKO mice, there is no significant difference between perampanel-treated and vehicle-treated GLAST+/−/GLT1-cKO mice (Fig. 7B (log rank test, p = 0.076)]. Consistent with these results, 6 weeks of treatment with perampanel or SNJ-1945 prevented motor neuron loss in GLAST+/−/GLT1-cKO mice at 12 weeks of age [Fig. 7C,D,G,H (unpaired two-tailed t test: Fig. 7D: t(8) = 2.93, p = 0.019; Fig. 7H: t(8) = 3.21, p = 0.013)]. On the other hand, the neuroprotective effect of KPT-350 was observed at 7 weeks of age but not at 12 weeks of age [Figs. 6F,G, 7K,L (Fig. 7L: unpaired two-tailed t test, t(8) = 0.01, p = 0.98)]. These results indicate that both perampanel and SNJ-1945 have continued robust protective effects, while the protective effects of KPT-350 in GLAST+/−/GLT1-cKO mice are transient. Thus, preventing NPC degradation by peramapanel or SNJ-1945 has robust protective effects on motor neurons.
Discussion
In this study, we generated a new animal model of excitotoxicity: a spinal cord-specific astroglial glutamate transporter (GLT1 and GLAST) double-knock-out mouse. We further demonstrated that GLAST+/−/GLT1-cKO mice showed selective motor neuron loss and severe hindlimb paralysis, which recapitulated the symptom of motor neuron diseases such as ALS. These findings suggest that combined loss of astroglial glutamate transporters GLT1 and GLAST is required to induce motor neuron death leading to paralysis in vivo.
Our results suggest a possible mechanism for spinal motor neuron death that is induced by excitotoxicity (Fig. 8), which includes the following: (1) dysfunction or reduction of astroglial glutamate transporters, which increases extracellular glutamate; (2) an increase in glutamate-induced AMPA receptor activation, not NMDA receptor activation, thereby leading to an increase in intracellular Ca2+ levels; (3) calpain hyperactivation by Ca2+ overload; (4) the degradation of NPCs by calpain; (5) the loss of nuclear integrity or other unknown mechanisms; and (6) the induction of motor neuron death (Fig. 8).
Figure 8.
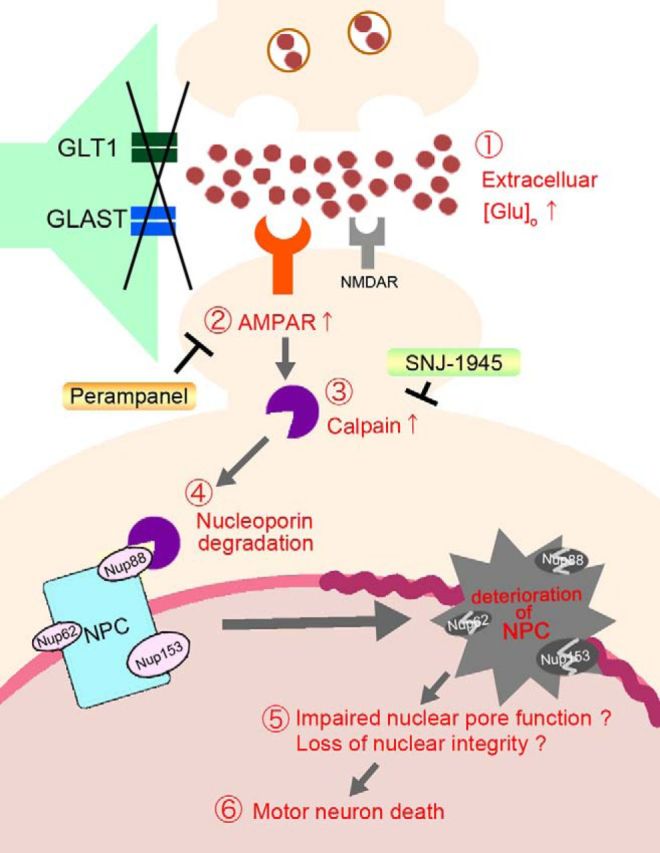
Model of AMPA receptor-induced spinal motor neuron death. In spinal cord-specific GLT1 and GLAST double-knock-out mice, the reduction of astroglial glutamate transporters (GLAST and GLT1) induces spinal motor neuron death as follows: (1) an increase in extracellular glutamate concentration; (2) an increase in glutamate-induced AMPA receptor activation, thereby leading to an increase in intracellular Ca2+ levels; (3) the Ca2+ overload-induced activation of calpain; (4) hyperactivated calpain degradation of NPCs; (5) impairment of nuclear pore function may progress to nuclear “leakiness”; and (6) induces motor neuron death. Glutamate-induced spinal motor neuron death can be reversed following long-term treatment with the antiepileptic AMPA receptor antagonist perampanel and the calpain inhibitor SNJ-1945. AMPAR, AMPA receptor; NMDAR, NMDA receptor; Nup62, Nucleoporin 62; Nup88, Nucleoporin 88; Nup153, Nucleoporin 153.
GLAST+/−/GLT1-cKO mice exhibited nuclear membrane abnormalities and calpain-mediated degradation of NPCs. An increase in the percentage of motor neurons exhibiting nuclear pathology preceded motor neuron death in GLAST+/−/GLT1-cKO mice, suggesting that nuclear pathology is an early pathological event following calpain activation in GLAST+/−/GLT1-cKO mice. In addition, long-term treatment of perampanel and SNJ-1945 has continued beneficial effects in motor neuron loss and the degradation of Nup153. Consistent with this, a recent study has demonstrated that activated calpain degraded NPCs via upregulation of Ca2+-permeable AMPA receptors in motor neurons of ADAR2 conditional knock-out (AR2) mice (Yamashita et al., 2017a). Together, these results indicate that nuclear membrane abnormalities and degradation of NPCs may be involved in motor neuron death that is driven by AMPA receptor-mediated excitotoxicity.
How do nuclear membrane abnormalities and degradation of NPCs actually lead to motor neuron death? The nucleocytoplasmic transport through NPCs plays a crucial role in maintaining cell viability and cellular functions (Pemberton and Paschal, 2005). Furthermore, previous studies have shown that nucleocytoplasmic transport deficits due to calpain-dependent degradation of the NPCs were observed in glutamate-treated cultured neurons and AR2 mice (Bano et al., 2010; Yamashita et al., 2017a). In this study, we showed that a selective inhibitor of nuclear export KPT-350 delayed but did not prevent motor neuron death in GLAST+/−/GLT1-cKO mice. In addition, only a small number of motor neurons showed the change in the subcellular localization of CAS in GLAST+/−/GLT1-cKO mice. Together, the nucleocytoplasmic transport deficits may transiently contribute to motor neuron death, but may not be a major downstream pathway to cause motor neuron death following NPCs degradation in GLAST+/−/GLT1-cKO mice. Thus, further studies are required to reveal the contribution of nucleocytoplasmic transport defects to motor neuron death and the downstream mechanisms of the NPCs degradation in GLAST+/−/GLT1-cKO mice.
Although our study suggests that the combined loss of glial glutamate transporters is required to recapitulate the ALS-like symptoms such as motor neuron death leading to paralysis, there was no report showing a reduction of GLAST expression in ALS patients. Thus, additional studies are required to evaluate the clinical relevance of GLAST+/−/GLT1-cKO mice to human ALS.
In this study, we demonstrated that motor neuron death was partially recovered by the pharmacological blockade of AMPA receptor and calpain activation, which could inhibit the degradation of Nup153. Therefore, our findings suggest that perampanel and SNJ-1945 could be a novel therapeutic strategy for many acute and chronic neurological diseases that involve AMPA receptor-mediated excitotoxicity, such as ischemic or traumatic injury and ALS.
Footnotes
This work was supported by the Strategic Research Program for Brain Sciences from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to K.T.). Additional support was provided by a grant from Medical Research Institute (MRI), Tokyo Medical and Dental University (TMDU; to T.A.), a Grant-in-Aid for Japan Society for the Promotion of Science Fellows (to K.S.), and Nanken-Kyoten, TMDU (to K.T.). We thank Dr. Masahiko Watanabe (Hokkaido University) for providing antibodies; Kei Watase (Center for Brain Integration Research, TMDU) for valuable suggestions; and Shizuko Ichinose, Yuriko Sakamaki (Research Center for Medical and Dental Science, TMDU), Harumi Ishikubo, and Masako Hidaka (MRI, TMDU) for their technical support.
The authors declare no competing financial interests.
References
- Aida T, Yoshida J, Nomura M, Tanimura A, Iino Y, Soma M, Bai N, Ito Y, Cui W, Aizawa H, Yanagisawa M, Nagai T, Takata N, Tanaka KF, Takayanagi R, Kano M, Götz M, Hirase H, Tanaka K (2015) Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsycopharmacology 40:1569–1579. 10.1038/npp.2015.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akamatsu M, Yamashita T, Hirose N, Teramoto S, Kwak S (2016) The AMPA receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (ALS) pathology in sporadic ALS model mice. Sci Rep 6:28649. 10.1038/srep28649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P (2005) Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120:275–285. 10.1016/j.cell.2004.11.049 [DOI] [PubMed] [Google Scholar]
- Bano D, Dinsdale D, Cabrera-Socorro A, Maida S, Lambacher N, McColl B, Ferrando-May E, Hengartner MO, Nicotera P (2010) Alteration of the nuclear pore complex in Ca(2+)-mediated cell death. Cell Death Differ 17:119–133. 10.1038/cdd.2009.112 [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Weiss JH (1996) Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci 16:4069–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couratier P, Hugon J, Sindou P, Vallat JM, Dumas M (1993) Cell culture evidence for neuronal degeneration in amyotrophic lateral sclerosis being linked to glutamate AMPA/kainate receptors. Lancet 341:265–268. 10.1016/0140-6736(93)92615-Z [DOI] [PubMed] [Google Scholar]
- Cui W, Mizukami H, Yanagisawa M, Aida T, Nomura M, Isomura Y, Takayanagi R, Ozawa K, Tanaka K, Aizawa H (2014) Glial dysfunction in the mouse habenula causes depressive-like behaviors and sleep disturbance. J Neurosci 34:16273–16285. 10.1523/JNEUROSCI.1465-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenkrog B, Aebi U (2003) The nuclear pore complex: nucleocytoplasmic transport and beyond. Nat Rev Mol Cell Biol 4:757–766. 10.1038/nrm1230 [DOI] [PubMed] [Google Scholar]
- Fontana AC. (2015) Current approaches to enhance glutamate transporter function and expression. J Neurochem 134:982–1007. 10.1111/jnc.13200 [DOI] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133. 10.1038/nature14974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R, Foster AC, Woodruff GN (1987) Systemic administration of MK-801 protects against ischemia-induced hippocampal neurodegeneration in the gerbil. J Neurosci 7:3343–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines JD, Herbin O, de la Hera B, Vidaurre OG, Moy GA, Sun Q, Fung HY, Albrecht S, Alexandropoulos K, McCauley D, Chook YM, Kuhlmann T, Kidd GJ, Shacham S, Casaccia P (2015) Nuclear export inhibitors avert progression in preclinical models of inflammatory demyelination. Nat Neurosci 18:511–520. 10.1038/nn.3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Hashizume Y, Tokuhara N, Takenaka O, Kohmura N, Ogasawara A, Hatakeyama S, Ohgoh M, Ueno M, Nishizawa Y (2011) Perampanel: a novel, orally active, noncompetitive AMPA-receptor antagonist that reduces seizure activity in rodent models of epilepsy. Epilepsia 52:1331–1340. 10.1111/j.1528-1167.2011.03109.x [DOI] [PubMed] [Google Scholar]
- Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, Hanada R, Orthofer M, Cronin SJ, Komnenovic V, Minis A, Sato F, Mimata H, Yoshimura A, Tamir I, Rainer J, Kofler R, Yaron A, Eggan KC, Woolf CJ, Glatzel M, Herbst R, Martinez J, Penninger JM (2013) CLP1 links tRNA metabolism to progressive motor-neuon loss. Nature 495:474–480. 10.1038/nature11923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideyama T, Yamashita T, Suzuki T, Tsuji S, Higuchi M, Seeburg PH, Takahashi R, Misawa H, Kwak S (2010) Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J Neurosci 30:11917–11925. 10.1523/JNEUROSCI.2021-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S (1991) Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science 252:851–853. 10.1126/science.1709304 [DOI] [PubMed] [Google Scholar]
- Imai S, Shimazawa M, Nakanishi T, Tsuruma K, Hara H (2010) Calpain inhibitor protects cells against light-induced retinal degeneration. J Pharmacol Exp Ther 335:645–652. 10.1124/jpet.110.171298 [DOI] [PubMed] [Google Scholar]
- Karlsson RM, Tanaka K, Saksida LM, Bussey TJ, Heilig M, Holmes A (2009) Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology 34:1578–1589. 10.1038/npp.2008.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S (2004) Glutamate receptors: RNA editing and death of motor neurons. Nature 427:801. 10.1038/427801a [DOI] [PubMed] [Google Scholar]
- Kinoshita Y, Ito H, Hirano A, Fujita K, Wate R, Nakamura M, Kaneko S, Nakano S, Kusaka H (2009) Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 68:1184–1192. 10.1097/NEN.0b013e3181bc3bec [DOI] [PubMed] [Google Scholar]
- Kwak S, Weiss JH (2006) Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr Opin Neurobiol 16:281–287. 10.1016/j.conb.2006.05.004 [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405:360–364. 10.1038/35012636 [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Maher P (2015) Chronic glutamate toxicity in neurodegenerative diseases—what is the evidence? Front Neurosci 9:469. 10.3389/fnins.2015.00469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. (2006) Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov 5:160–170. 10.1038/nrd1958 [DOI] [PubMed] [Google Scholar]
- Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, Yi F, Xu X, Ruiz S, Zhang W, Wagner U, Kim A, Ren B, Li Y, Goebl A, Kim J, Soligalla RD, Dubova I, Thompson J, Yates J 3rd, Esteban CR, Sancho-Martinez I, Izpisua Belmonte JC (2012) Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 491:603–607. 10.1038/nature11557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas DR, Newhouse JP (1957) The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol 58:193–201. 10.1001/archopht.1957.00940010205006 [DOI] [PubMed] [Google Scholar]
- Makise M, Mackay DR, Elgort S, Shankaran SS, Adam SA, Ullman KS (2012) The Nup153-Nup50 protein interface and its role in nuclear import. J Biol Chem 287:38515–38522. 10.1074/jbc.M112.378893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsugami TR, Tanemura K, Mieda M, Nakatomi R, Yamada K, Kondo T, Ogawa M, Obata K, Watanabe M, Hashikawa T, Tanaka K (2006) Indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc Natl Acad Sci U S A 103:12161–12166. 10.1073/pnas.0509144103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL (2013) Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur J Pharmacol 698:6–18. 10.1016/j.ejphar.2012.10.032 [DOI] [PubMed] [Google Scholar]
- Miladinovic T, Nashed MG, Singh G (2015) Overview of glutamatergic dysregulation in central pathologies. Biomolecules 5:3112–3141. 10.3390/biom5043112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J, Paul P, Chen HJ, Morris A, Payling M, Falchi M, Habgood J, Panoutsou S, Winkler S, Tisato V, Hajitou A, Smith B, Vance C, Shaw C, Mazarakis ND, de Belleroche J (2010) Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc Natl Acad Sci U S A 107:7556–7561. 10.1073/pnas.0914128107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, Gallo JM, Hortobágyi T, Shaw CE, Rogelj B (2010) Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 133:1763–1771. 10.1093/brain/awq111 [DOI] [PubMed] [Google Scholar]
- Oka T, Walkup RD, Tamada Y, Nakajima E, Tochigi A, Shearer TR, Azuma M (2006) Amelioration of retinal degeneration and proteolysis in acute ocular hypertensive rats by calpain inhibitor ((1S)-1-((((1S)-1-benzyl-3-cyclopropylamino-2,3-di-oxopropyl)amino)carbonyl)-3-methylbutyl)carbamic acid 5-methoxy-3-oxapentyl ester. Neuroscience 141:2139–2145. 10.1016/j.neuroscience.2006.05.060 [DOI] [PubMed] [Google Scholar]
- Olney JW, Rhee V, Ho OL (1974) Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res 77:507–512. 10.1016/0006-8993(74)90640-4 [DOI] [PubMed] [Google Scholar]
- Pemberton LF, Paschal BM (2005) Mechanisms of receptor-mediated nuclear import and nuclear export. Traffic 6:187–198. 10.1111/j.1600-0854.2005.00270.x [DOI] [PubMed] [Google Scholar]
- Roth P, Xylourgidis N, Sabri N, Uv A, Fornerod M, Samakovlis C (2003) The Drosophila nucleoporin DNup88 localizes DNup214 and CRM1 on the nuclear envelope and attenuates NES-mediated nuclear export. J Cell Biol 163:701–706. 10.1083/jcb.200304046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT (1990) Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol 28:18–25. 10.1002/ana.410280106 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84. 10.1002/ana.410380114 [DOI] [PubMed] [Google Scholar]
- Sasabe J, Chiba T, Yamada M, Okamoto K, Nishimoto I, Matsuoka M, Aiso S (2007) D-serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J 26:4149–4159. 10.1038/sj.emboj.7601840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasabe J, Miyoshi Y, Suzuki M, Mita M, Konno R, Matsuoka M, Hamase K, Aiso S (2012) D-amino acid oxidase controls motoneuron degeneration through D-serine. Proc Natl Acad Sci U S A 109:627–632. 10.1073/pnas.1114639109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Minegishi S, Takano J, Plattner F, Saito T, Asada A, Kawahara H, Iwata N, Saido TC, Hisanaga S (2011) Calpastatin, an endogenous calpain-inhibitor protein, regulates the cleavage of the Cdk5 activator p35 to p25. J Neurochem 117:504–515. 10.1111/j.1471-4159.2011.07222.x [DOI] [PubMed] [Google Scholar]
- Tanaka K. (2000) Functions of glutamate transporters in the brain. Neurosci Res 37:15–19. 10.1016/S0168-0102(00)00104-8 [DOI] [PubMed] [Google Scholar]
- Tanaka K. (2005) Antibiotics rescue neurons from glutamate attack. Trends Mol Med 11:259–262. 10.1016/j.molmed.2005.04.002 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699–1702. 10.1126/science.276.5319.1699 [DOI] [PubMed] [Google Scholar]
- Tong J, Huang C, Bi F, Wu Q, Huang B, Liu X, Li F, Zhou H, Xia XG (2013) Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J 32:1917–1926. 10.1038/emboj.2013.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B (1991) Structural determinants of ion flow through recombinant glutamate receptor channels. Science 252:1715–1718. 10.1126/science.1710829 [DOI] [PubMed] [Google Scholar]
- Wang R, Zhang D (2005) Memantine prolongs survival in an amyotrophic lateral sclerosis mouse model. Eur J Neurosci 22:2376–2380. 10.1111/j.1460-9568.2005.04431.x [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K (1998) Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci 10:976–988. 10.1046/j.1460-9568.1998.00108.x [DOI] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S (2010) TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristics of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A 107:3858–3863. 10.1073/pnas.0912417107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witschi R, Johansson T, Morscher G, Scheurer L, Deschamps J, Zeilhofer HU (2010) Hoxb8-Cre mice: a tool for brain-sparing conditional gene deletion. Genesis 48:596–602. 10.1002/dvg.20656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootz H, Fitzsimons-Kantamneni E, Larhammar M, Rotterman TM, Enjin A, Patra K, André E, Van Zundert B, Kullander K, Alvarez FJ (2013) Alterations in the motor neuron-Renshaw cell circuit in the Sod1G93A mouse model. J Comp Neurol 521:1449–1469. 10.1002/cne.23266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Watanabe M, Shibata T, Nagashima M, Tanaka K, Inoue Y (1998) Glutamate transporter GLT-1 is transiently localized on growing axons of the mouse spinal cord before establishing astrocytic expression. J Neurosci 18:5706–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Kwak S (2014) The molecular link between inefficient GluA2 Q/R site-RNA editing and TDP-43 pathology in motor neurons of sporadic amyotrophic lateral sclerosis patients. Brain Res 1584:28–38. 10.1016/j.brainres.2013.12.011 [DOI] [PubMed] [Google Scholar]
- Yamashita T, Hideyama T, Hachiga K, Teramoto S, Takano J, Iwata N, Saido TC, Kwak S (2012) A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat Commun 3:1307. 10.1038/ncomms2303 [DOI] [PubMed] [Google Scholar]
- Yamashita T, Aizawa H, Teramoto S, Akamatsu M, Kwak S (2017a) Calpain-dependent disruption of nucleo-cytoplasmic transport in ALS motor neurons. Sci Rep 7:39994. 10.1038/srep39994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Akamatsu M, Kwak S (2017b) Altered intracellular milieu of ADAR2-deficient motor neurons in amyotrophic lateral sclerosis. Genes (Basel) 8:60 10.3390/genes8020060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhang L, Liang B, Schroeder D, Zhang ZW, Cox GA, Li Y, Lin DT (2016) Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat Neurosci 19:557–559. 10.1038/nn.4257 [DOI] [PMC free article] [PubMed] [Google Scholar]