

Abstract
The hormone leptin indirectly communicates metabolic information to brain neurons that control reproduction, using GABAergic circuitry. Agouti-related peptide (AgRP) neurons in the arcuate nucleus are GABAergic, express leptin receptors (LepR), and are known to influence reproduction. This study tested whether leptin actions on AgRP neurons are required and sufficient for puberty onset and subsequent fertility. First, Agrp-Cre and Lepr-flox mice were used to target deletion of LepR to AgRP neurons. AgRP-LepR knock-out female mice exhibited mild obesity and adiposity as described previously, as well as a significant delay in the pubertal onset of estrous cycles compared with control animals. No significant differences in male puberty onset or adult fecundity in either sex were observed. Next, mice with a floxed polyadenylation signal causing premature transcriptional termination of the Lepr gene were crossed with AgRP-Cre mice to generate mice with AgRP neuron-specific rescue of LepR. Lepr-null control males and females were morbidly obese and exhibited delayed puberty onset, no evidence of estrous cycles, and minimal fecundity. Remarkably, AgRP-LepR rescue partially or fully restored all of these reproductive attributes to levels similar to those of LepR-intact controls despite minimal rescue of metabolic function. These results indicate that leptin signaling in AgRP neurons is sufficient for puberty onset and normal adult fecundity in both sexes when leptin signaling is absent in all other cells and that in females, the absence of AgRP neuron leptin signaling delays puberty. These actions appear to be independent of leptin's metabolic effects.
SIGNIFICANCE STATEMENT Sexual maturation and fertility are dispensable at the individual level but critical for species survival. Conditions such as nutritional imbalance may therefore suppress puberty onset and fertility in an individual. In societies characterized by widespread obesity, the sensitivity of reproduction to metabolic imbalance has significant public health implications. Deficient leptin signaling attributable to diet-induced leptin resistance is associated with infertility in humans and rodents, and treatments for human infertility show a decreased success rate with increasing body mass index. Here we show that the transmission of metabolic information to the hypothalamo-pituitary-gonadal axis is mediated by leptin receptors on AgRP neurons. These results provide conclusive new insights into the mechanisms that cause infertility attributable to malnourishment.
Keywords: AgRP, fertility, GnRH, leptin, puberty, reproduction
Introduction
Leptin is an adipose-derived hormone that fluctuates in proportion to the nutritional status of the individual. This allows levels of oxidizable fuels to be effectively communicated to the CNS, where leptin acts to influence control of metabolic function and also fertility (Moschos et al., 2002; Quennell et al., 2009). Compromised leptin signaling caused by mutations in the leptin or leptin receptor (LepR) genes (Lepr) results in neuroendocrine dysfunction, including obesity and infertility. Leptin treatment is able to attenuate the effects of leptin deficiency, stimulating puberty onset, sexual maturation, and gonadotropin secretion (Chehab et al., 1996).
Regulation of the reproductive system is initiated at the level of the hypothalamus by a neuronal network that converges on a small population of neurosecretory cells that synthesize and secrete gonadotropin-releasing hormone (GnRH). Many of the neurons that provide important input to the reproductive axis are also involved in appetite control, and this allows the availability of metabolic fuels to be coordinated with fertility (Evans and Anderson, 2017). Leptin acts centrally via neurons to modulate the activity of the GnRH neuronal network (Quennell et al., 2009), but this occurs indirectly of GnRH neurons themselves as they do not contain LepR (Quennell et al., 2009). To identify the neurons required for leptin to communicate with GnRH neurons, Cre-LoxP technology has been used to delete LepR from specific neuronal populations, but this has mostly proved to be unfruitful (Quennell et al., 2009; Shi et al., 2010; Donato et al., 2011b). Zuure et al. (2013) used a less focused approach to show that mice with glutamate neuron-specific LepR knock-out (KO) have no reproductive deficits, whereas GABA neuron-specific LepR knock-out mice display delayed puberty and reduced fecundity. This indicates that leptin communicates with GnRH neurons through critical GABAergic neurons (Zuure et al., 2013), although the specific populations involved are yet to be identified.
Within the arcuate nucleus (ARC), LepR are colocalized with agouti-related peptide (AgRP)/neuropeptide Y (NPY) neurons (Donato et al., 2011a), among other cell types. AgRP/NPY neurons secrete two highly orexigenic peptides (Hahn et al., 1998) and are critically involved in the maintenance of energy homeostasis (Robertson et al., 2008). They are attractive candidates for metabolic control of reproduction because of their well characterized role in metabolic regulation, because they are also GABAergic (Horvath et al., 1997), and because they are known to influence the activity of GnRH neurons (Roa and Herbison, 2012; Sheffer-Babila et al., 2013). Leptin signaling inhibits the activity of AgRP/NPY neurons, so it may be that these neuropeptides cause suppression of the hypothalamo-pituitary-gonadal (HPG) axis in conditions of leptin deficiency. Consistent with this, ablation of these neurons or knock-out of the genes encoding either neuropeptide or the NPY Y4 receptor partially rescues the infertility phenotype of leptin-signaling-deficient mice (Erickson et al., 1996; Sainsbury et al., 2002; Wu et al., 2012; Sheffer-Babila et al., 2013).
The requirement or sufficiency of leptin signaling specifically in AgRP neurons for fertility has not previously been tested. Interestingly, however, van de Wall et al. (2008) reported normal fertility in mice with LepR knock-out in both AgRP and pro-opiomelanocortin (POMC) neurons but did not investigate puberty onset. Since the reproductive effects of POMC and AgRP/NPY neuropeptides may oppose each other (Roa and Herbison, 2012), deleting leptin receptors from both cell types simultaneously could potentially cancel and mask the effects of deletion from the individual populations. We therefore used Cre-LoxP technology to specifically delete LepR from AgRP neurons to assess the requirement of this signaling pathway for reproductive function. We also assessed the sufficiency of leptin signaling through AgRP neurons by crossing mice with a floxed transcription blocker sequence in the Lepr gene with AgRP-Cre mice to generate mice with AgRP neuron-specific rescue of LepR. The effects of AgRP-LepR knock-out and AgRP-LepR rescue on puberty onset and adult fecundity were evaluated in males and females to determine whether this pathway is required or is sufficient for leptin's effects on the HPG axis.
Materials and Methods
Animals.
To generate mice with deletion of LepR specifically from AgRP neurons, homozygous Lepr flox mice [Leprfl/fl; loxP sites flanking Lepr coding exon 17, a region that encodes a Janus kinase docking site required for signal transducer and activator of transcription 3 (STAT3) signaling; McMinn et al., 2004] were bred to Agrp-IRES-Cre (Jax stock number 012899; IRES-Cre inserted in exon 3 of the Agrp gene) mice (Tong et al., 2008). The resulting Leprfl/+,Agrp-IRES-Cre mice were backcrossed to Leprfl/fl mice to generate Leprfl/fl,Agrp-IRES-Cre conditional knock-out mice (referred to as AgRP-LepR KO mice). To generate mice with specific rescue of LepR only in AgRP neurons, heterozygous Cre-dependent Lepr mice (LeprloxTB/+; Jax stock number 018989; loxP flanked transcription blocker sequence between exons 16 and 17 of the Lepr gene prevents transcription of the downstream exons; Berglund et al., 2012) were bred to Agrp-IRES-Cre mice. The resulting LeprloxTB/+,Agrp-IRES-Cre mice were bred together to generate LeprloxTB/loxTB,Agrp-IRES-Cre conditional rescue mice (referred to as AgRP-LepR rescue mice). Agrp-Cre was visualized through Agrp-Cre-dependent green fluorescent protein (GFP) expression as a result of crossing Agrp-IRES-Cre mice with a Tau-GFP reporter line (Mayer et al., 2010; Wen et al., 2011) producing Agrp-GFP mice. All mouse lines were on a primarily C57BL/6J background strain. The AgRP-Cre mouse line has been previously validated (van de Wall et al., 2008). They report that 85–100% of AgRP-immunoreactive neurons express Cre recombinase and that all Cre-expressing neurons are AgRP immunoreactive. Furthermore, in AgRP-LepR KO mice, leptin treatment was unable to cause any increase in leptin signaling in AgRP neurons.
Transgenic mice were identified by PCR analysis of genomic DNA using the following primer sets, at an annealing temperature of 59°C (Lepr flox), 61°C (LeprloxTB), or 60°C (Agrp-Cre): for Lepr flox identification, AAT GAA AAA GTT GTT TTG GGA CGA and CAG GCT TGA GAA CAT GAA CAC AAC AAC and CTG ATT TGA TAG ATG GTC TTG AG (200 bp product indicates the wild-type gene, 250 bp indicates the floxed gene); for LeprloxTB identification, TGG CTT TTA AGC TCT GCA GTC and TAG GGC CAA ACC CAC ATT TA and CCC AAG GCC ATA CAA GTG TT (522 bp product indicates the wild-type gene, 360 bp indicates the floxed gene); for Agrp-Cre identification, GCT TCT TCA ATG CCT TTT GC and GTG TGT GGT TCC AGC ATG AC and GG AAC TGC TTC CTT CAC GA (199 bp product indicates the wild-type gene, 280 bp indicates the Cre gene). Animals were group housed or paired with an animal of the opposite sex (for the fecundity experiments), except during food intake assessments. Mice were housed under conditions of controlled lighting (lights on from 6:00 A.M. to 6:00 P.M.) and temperature (22 ± 1°C). They had ad libitum access from the date of weaning to standard rodent chow, except during overnight fasting as described. All mice were weighed every 2 weeks except for female mice when they were paired with male mice for the fecundity experiments. The University of Otago Animal Ethics Committee approved all animal experimental protocols.
Tissue collection and immunohistochemistry to identify leptin-responsive cells.
At the end of the fecundity studies when mice were 5 months old, they were assessed for food intake over a 24 h period in individual cages. The animals were then fasted overnight to reduce the concentration of endogenous circulating leptin and given injections of recombinant leptin (1 mg/kg, s.c.; National Hormone and Peptide Program). Two hours after injection, they were anesthetized with sodium pentobarbital (240 mg/kg, i.p.) and transcardially perfused with 4% paraformaldehyde in 0.1 m PBS, pH 7.4. Total abdominal fat mass was measured at this time. Coronal (30-μm-thick) sections were cut throughout the ARC for each brain on a sliding microtome to be used for immunohistochemical staining. To visualize leptin-responsive cells in the ARC of AgRP-LepR KO and AgRP-LepR rescue mice and their respective control groups, immunohistochemical labeling of phosphorylated STAT3 (pSTAT3) was performed. Antigen retrieval was performed by incubating for 15 min in 1 mm EDTA, pH 8.0, at 90°C. To quench endogenous peroxidase activity, the tissues were incubated in 1% H2O2 for 30 min. Sections were incubated for 24 h in the primary antibody, monoclonal rabbit anti-pSTAT3 (Tyr705, D3A7 XP; 1:1000 dilution, Cell Signaling Technology). Tissue was then incubated for 1 h in the secondary antibody, biotinylated goat anti-rabbit Ig (1:1000 dilution, Vector Laboratories). The signal was amplified by incubating in Vector Elite avidin–biotin peroxidase (Vector Laboratories) and was stained in diaminobenzidine solution to visualize pSTAT3 immunoreactivity. Omission of the primary antibody resulted in a complete absence of staining. Stained cells in the ventromedial ARC (vmARC) and ventromedial hypothalamic nucleus (VMH; at least three sections per area from each animal) were counted.
Experiment 1: Are leptin actions on AgRP neurons required for normal puberty onset and fertility.
AgRP-LepR KO and Lepfl/fl littermate controls were used to evaluate the requirement of leptin signaling in AgRP neurons for puberty onset and subsequent fertility. In female mice, puberty onset was measured by assessing the age of vaginal opening and first estrus. From 21 d of age, all mice were checked daily for vaginal opening. Once this had occurred, vaginal cytology was used to detect occurrence of first estrus. Estrous cyclicity of adult females was then assessed for 14 consecutive days, starting at least 10 d after first estrus. Experimental and control female animals were paired with adult wild-type C57BL/6J males between 60 and 140 d of age to assess their fecundity (body weight measurements were not obtained from females over this time because of pregnancies). Male puberty progression was assessed visually based on the date of separation of the prepuce from the glans penis. Once this had occurred, male mice were paired with adult wild-type C57BL/6J females. The date of first successful mating was used as another measure of pubertal progression, calculated by subtracting the gestation period (21 d) from the date when their first litter was born. Assessment of male fecundity was performed over a 100 d period. Cages were checked daily during male and female fecundity assessments for the presence of pups, and the date and size of the litter were recorded before pups were removed and culled.
Experiment 2: Are leptin actions on AgRP neurons sufficient for normal puberty onset and fertility.
To test the sufficiency of leptin signaling in AgRP neurons for puberty onset and subsequent fertility, we generated mice with specific rescue of LepR only in AgRP neurons (AgRP-LepR rescue mice). LeprloxTB/loxTB and Leprwt/wt littermates were used for the two control groups (referred to as Lepr-null and Lepr-intact controls, respectively). The Lepr-null control group has previously been shown to be essentially infertile (Cravo et al., 2013) and was included as a baseline reference against which any improvement of fertility could be compared in the AgRP-LepR rescue group. In male and female AgRP-LepR rescue, Lepr-null, and Lepr-intact control mice, puberty onset, estrous cycles, and adult fecundity were assessed as described for Experiment 1. At the completion of these studies, when the mice were 5 months old, we also assessed NPY fiber immunoreactivity in the paraventricular nucleus of the hypothalamus (PVN), since it is known that leptin deficiency causes profound disruptions in the development of AgRP feeding regulatory circuits to this region (Bouret et al., 2004). Hypothalamic tissue sections from Lepr-null, Lepr-intact control, and AgRP-LepR rescue mice were quenched as described for pSTAT3 immunohistochemistry and incubated for 48 h in polyclonal rabbit anti-neuropeptide Y (T-4070; 1:2000, Bachem), followed by biotinylated goat anti-rabbit Ig, avidin–biotin peroxidase, and diaminobenzidine solutions as described for pSTAT3 immunohistochemistry. Omission of the primary antibody or overnight preabsorption of the primary antibody with 20 μg/ml human NPY (H6375; Bachem) resulted in a complete absence of staining. Fiber density within the PVN was quantified using ImageJ software after first thresholding the grayscale image to binary values.
The infertility anticipated in LepR-null (and possibly AgRP-LepR rescue) mice in Experiment 2 could reflect impaired estrogenic feedback on the hypothalamo-pituitary-gonadal axis. Therefore, negative feedback assessment was performed as described by Zuure et al. (2013). Briefly, female AgRP-LepR rescue, Lepr-null, and Lepr-intact control mice were blood sampled (4 μl) from the tail tip (day 0; ovary intact). Ovariectomies (OVX) were performed under isoflurane anesthesia, and 8 d later another blood sample was taken. Animals were subsequently implanted with a chronic slow-release 17β-estradiol subcutaneous implant (50 μg/ kg; 10–30 mm long depending on body weight); 8 d later, another blood sample was taken (OVX plus estradiol implant). A sandwich ELISA (Evans et al., 2014) was used to measure luteinizing hormone (LH) concentrations in whole blood samples. The sensitivity of the assay was 0.2 ng/ml after correction for sample dilution, the intra-assay coefficient of variation was 6%, and the interassay variation was 15%. All samples were analyzed in duplicate.
Statistical analysis.
Values are presented as mean ± SEM. Differences were considered significant at p < 0.05. In Experiment 1, unpaired Student's t tests were used to identify significant differences between control and AgRP-LepR KO animals when group sizes were greater than n = 10. For smaller sample sizes, the nonparametric Mann–Whitney U test was used. In Experiment 2, one-way ANOVA followed by the post hoc Holm–Sidak test was used to identify significant differences between the Lepr-null, AgRP-LepR rescue, and Lepr-intact groups when group sizes were greater than n = 10. For smaller sample sizes, the nonparametric Kruskal–Wallis H test was used, followed by Dunn's post hoc test. Body weight data were analyzed using a repeated-measures two-way ANOVA followed by the Holm–Sidak test.
Results
Experiment 1: Are leptin actions on AgRP neurons required for normal puberty onset and fertility?
GFP expression indicative of cell bodies in the Agrp-GFP reporter mice was localized to the ventromedial portion of the ARC (Fig. 1D), with virtually no soma seen elsewhere in the hypothalamus. This pattern of expression closely matches that previously reported for Agrp mRNA (Broberger et al., 1998) and AgRP immunohistochemistry using colchicine-treated rodents (Kloukina et al., 2012). This indicates that these animals were appropriate to use in experiments assessing the necessity and sufficiency of leptin signaling through AgRP neurons.
Figure 1.

Leptin-induced pSTAT3 signaling in the vmARC and VMH. A, B, Representative examples of pSTAT3 immunoreactivity in the ARC and VMH of control (A) and AgRP-LepR KO (B) animals. dlARC, Dorsolateral ARC. C, No difference in leptin-induced pSTAT3 immunoreactivity between the control and KO groups was observed in the VMH, but a significant decrease (p = 0.004) was seen in the vmARC of AgRP-LepR KO animals compared with controls. D, Representative coronal section showing GFP immunofluorescence in AgRP Cre-positive neurons of the vmARC. Controls, n = 7–8; AgRP-LepR KO, n = 6–8. **p < 0.01. 3V, Third ventricle. Scale bars, 200 μm.
Phosphorylated STAT3 immunohistochemistry was used to detect the presence of any leptin-induced STAT3 signaling, which is a functional indicator of leptin-responsive cells, in all mice. Counting of pSTAT3-stained cells was restricted to the vmARC as this corresponded to the location of AgRP neurons in the reporter mice, and pSTAT3-responsive cells in the VMH were counted in an internal control. Shown in Figure 1, A and B, are representative images of the vmARC and VMH of control and AgRP-LepR KO mice used in Experiment 1. Consistent with a previous report based on these AgRP-LepR KO mice (van de Wall et al., 2008), a significant decrease in pSTAT3 cell numbers was observed in the vmARC of AgRP-LepR KO mice compared with controls (U(12) = 0, p < 0.01), but no difference was seen for the VMH (Fig. 1C) or for the dorsolateral ARC (where few, if any, AgRP neurons are located; data not shown). Normal pSTAT3 staining was also evident in other hypothalamic regions in all mice, suggesting that the widespread “ectopic” Cre expression previously reported in up to 5% of AgRP-Cre mice (Dietrich et al., 2015) did not occur in this experiment. (It should be noted that LepR excision-specific primers were not used in this study to check for excision outside the ARC). The loss of leptin responsiveness was previously shown to be specific to AgRP cells (van de Wall et al., 2008).
As reported previously (van de Wall et al., 2008), the body weight of both male and female AgRP-LepR KO mice was slightly but significantly greater than control littermates (males: F(1,16) = 6.04, p < 0.05; females: F(1,16) = 15.79, p < 0.05), and this was confirmed by post hoc tests at a number of time points as shown in Figure 2, A and B. This effect was particularly apparent for female mice. Female AgRP-LepR KO mice also had significantly heavier abdominal fat pads compared with the control group (U(10) = 4, p < 0.05), as shown in Figure 2C. Adiposity of male mice was not significantly different between the two groups, suggesting that lack of leptin signaling in AgRP neurons may have a greater impact on metabolism in female animals than in males. These observations align with the well characterized role of AgRP in metabolism and validate the animal model used. Figure 2D illustrates the amount of food consumed by control and AgRP-LepR KO animals over a 24 h period. Consistent with a previous report (van de Wall et al., 2008), no significant difference was seen between the groups for either male or female mice, suggesting that the slight obese phenotype of the KO mice was not caused by increased food intake.
Figure 2.

Effects of AgRP-LepR KO on body weight, adiposity, and food intake in male and female mice. A, AgRP-LepR KO male mice were significantly heavier than control males at the marked time points (n = 9). B, AgRP-LepR KO female mice were significantly heavier than control females from 49 d of age (controls, n = 8; AgRP-LepR KO, n = 10). C, There was no significant difference in adiposity of male AgRP-LepR KO and control animals (n = 9), but AgRP-LepR KO females had significantly increased (p = 0.030) abdominal adiposity compared with control females (controls, n = 5; AgRP-LepR KO, n = 7). BW, Body weight. D, Daily food intake for male or female AgRP-LepR KO animals was not significantly different from male or female control mice (males, n = 9; female controls, n = 8; female AgRP-LepR KO, n = 10). *p < 0.05; **p < 0.01; ***p < 0.001.
Although no significant difference in age at vaginal opening was observed between control and AgRP-LepR KO females, the onset of first estrus was significantly delayed in AgRP-LepR KO mice by 3.4 ± 1.0 d compared with control littermates (Fig. 3B; U(16) = 16, p < 0.05), indicating that AgRP leptin signaling is required for normally timed female puberty. No significant difference was seen in the age of onset of male puberty (preputial separation or first fertile mating) between the control and AgRP-LepR KO groups (Fig. 3A).
Figure 3.
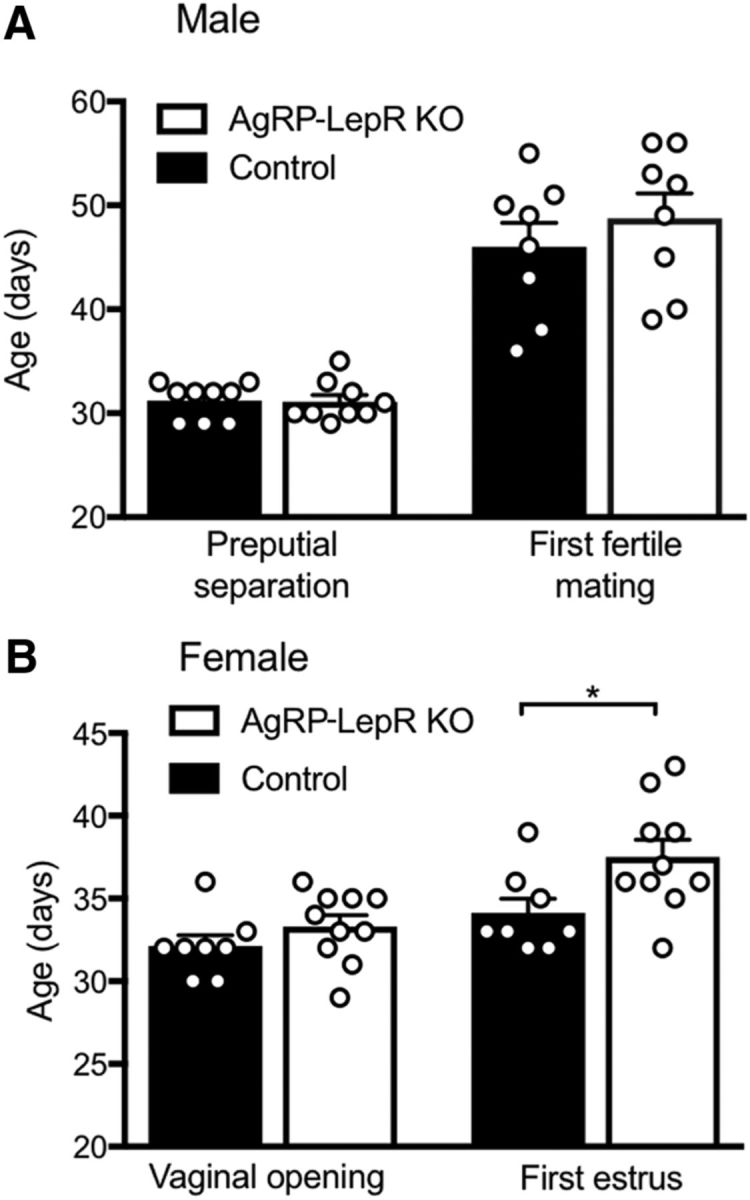
Age at puberty onset in male and female AgRP-LepR KO mice compared with control animals. A, No significant difference in puberty onset was observed between male AgRP-LepR KO and control animals (n = 9). B, No significant difference in vaginal opening was observed between female AgRP-LepR KO and control animals, but a significant delay (p = 0.030) in age at first estrus was observed in the AgRP-LepR KO group (controls, n = 8; AgRP-LepR KO, n = 10). *p < 0.05.
Analysis of vaginal cytology revealed no significant difference in the time spent in each phase of the estrous cycle between control and AgRP-LepR KO animals (Fig. 4A). The average length of the estrous cycle was not significantly different between the two groups. The similarity between estrous cyclicity profiles is demonstrated in Figure 4B. Fecundity of adult mice was assessed by litter frequency, number of pups per litter, and interlitter interval over 80 d (females) or 100 d (males). None of these measures differed between the groups in both males and females (Fig. 4, C and D). Reproductive function was also assessed by measuring the weight of reproductive organs. No significant difference was observed in the weight of the testes and seminal vesicles in males or of the uteri in females (data not shown).
Figure 4.

Estrous cyclicity and fecundity of AgRP-LepR KO male and female mice compared with control animals. A, Frequency of occurrence of cycle stages in females. The stage of the estrous cycle was determined by the predominant presence of leukocytes (proestrus), cornified epithelial cells (estrus), or nucleated epithelial cells (metestrus or diestrus). There were no significant differences between AgRP-LepR KO and control animals. B, Representative examples of the cyclicity of AgRP-LepR KO and control female mice. C, There were no significant differences in the number of litters produced over 100 d between male or female AgRP-LepR KO and control animals. D, There were no significant differences in the average number of pups produced per litter between male or female AgRP-LepR KO and control animals. Male controls, n = 9; male AgRP-LepR KO, n = 8; female controls, n = 8; female AgRP-LepR KO, n = 10. P, Proestrus; E, estrus; M/D, metestrus/diestrus.
Experiment 2: Are leptin actions on AgRP neurons sufficient for normal puberty onset and fertility?
Consistent with the vmARC-specific loss of leptin-responsive cells in AgRP LepR KO mice in Experiment 1, AgRP-LepR rescue restored responsiveness to leptin specifically in the vmARC. Figure 5A–C shows representative images from Lepr-intact control, AgRP-LepR rescue, and Lepr-null control animals used in Experiment 2. In all Lepr-null mice, no pSTAT3-positive cells were visible in any hypothalamic region. In all AgRP-LepR rescue animals, a group of leptin-responsive cells in the vmARC was visible, but no staining was seen in any other hypothalamic region (females only examined). The number of cells counted in the vmARC of AgRP-LepR rescue mice was not statistically different from Lepr-intact control animals but was significantly greater than Lepr-null animals (Fig. 5D; F(2,20) = 4.80, p < 0.001), consistent with AgRP neuron-specific LepR rescue. Staining in the VMH of Lepr-intact control animals was significantly greater than both the Lepr-null and AgRP-LepR rescue groups, in which pSTAT3 was essentially undetectable in this region (Fig. 5; F(2,20) = 9.22, p < 0.001).
Figure 5.

Leptin-induced pSTAT3 signaling in the ARC and VMH of Lepr-null control, AgRP-LepR, rescue and Lepr-intact control animals. A, Representative Lepr-intact control section showed staining throughout both the vmARC and VMH. B, Representative AgRP-LepR rescue section showing staining in the vmARC. C, No pSTAT3 staining was observed in any region in Lepr-null animals. D, Quantification of leptin-induced pSTAT3 immunoreactivity, showing that leptin responsiveness was rescued in the vmARC of AgRP-LepR rescue mice (p = 0.0003 vs Lepr-null mice). In the VMH, the response to leptin remained undetectable in both AgRP-LepR rescue and Lepr-null animals (Lepr-intact controls, n = 7; AgRP-LepR rescue and Lepr-null, n = 8). ***p < 0.001. Scale bar, 200 μm.
As expected, Lepr-null male and female mice were significantly heavier than Lepr-intact control animals (males: F(2,28) = 126.2, p < 0.0001; females: F(2,28) = 62.33, p < 0.0001), and post hoc tests showed this occurred from 34 d of age (Fig. 6A,B). AgRP-LepR rescue mice were also metabolically compromised by lack of intact leptin signaling. This was demonstrated by their obesity compared with Lepr-intact control animals from 34 d of age. Post hoc testing also showed that AgRP-LepR rescue mice of both sexes were significantly lighter than Lepr-null animals from 49 d of age (males, p < 0.001; females, p < 0.01). Abdominal adiposity of male and female Lepr-null and female AgRP-LepR rescue animals was also significantly increased compared with Lepr-intact controls (Fig. 6C; males: F(2,22) = 3.48, p < 0.05; females: H(2,23) = 14.98, p < 0.05). Food intake results for male animals showed that both Lepr-null and AgRP-LepR rescue animals consumed significantly more food than Lepr-intact controls over a 24 h period (Fig. 6E; F(2,28) = 12.23; p < 0.001 and p < 0.01, respectively). There was no significant difference in food intake between Lepr-null and AgRP-LepR rescue mice. Food intake values for female animals are not shown because of the confounding effects of the different levels of parity across the treatment groups. Consistent with the minimal rescue from obesity by AgRP-LepR rescue, this group and the Lepr-null mice both exhibited impaired NPY fiber density in the PVN compared with Lepr-intact control animals (Fig. 6G; F(2,16) = 4.70, p < 0.05), which has been previously shown to be a function of leptin-dependent outgrowth from ARC NPY/AgRP neurons during neonatal development (Bouret et al., 2004).
Figure 6.
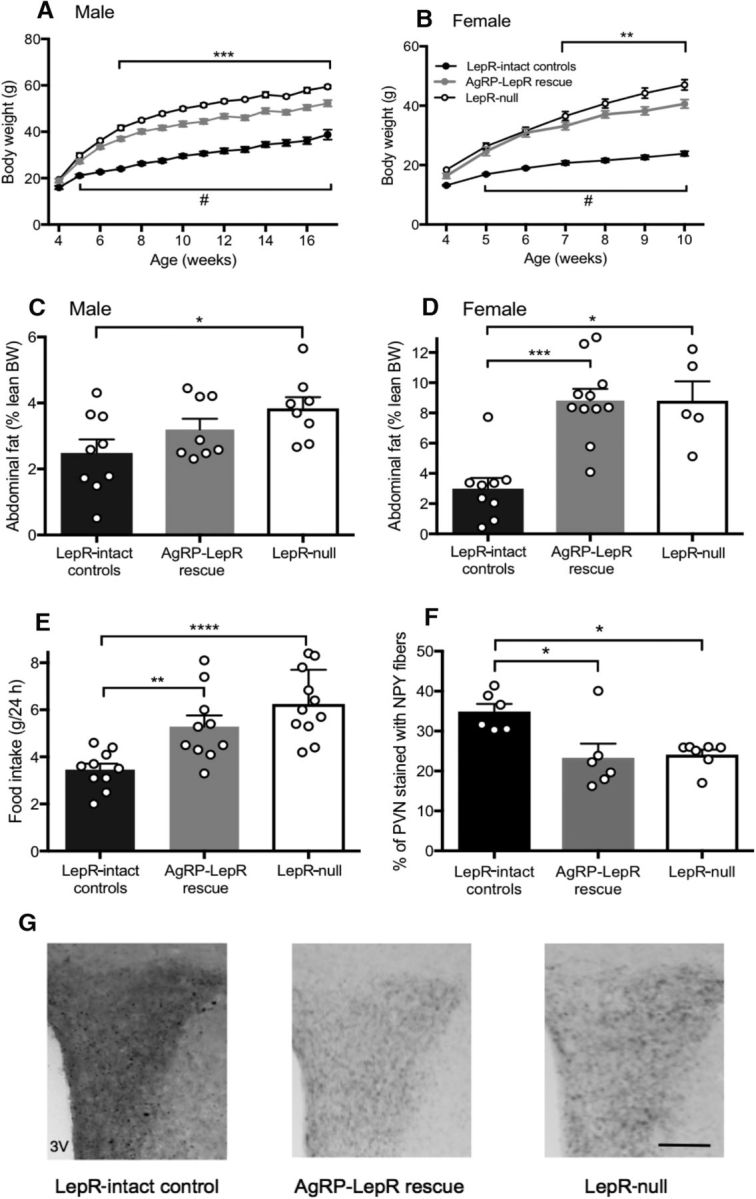
The effect of AgRP-LepR rescue on body weight, adiposity, food intake, and NPY fiber density. A, B, AgRP-LepR rescue and Lepr-null male (A) and female (B) mice were significantly heavier than Lepr-intact control animals from 5 weeks of age (#), whereas AgRP-LepR rescue animals were significantly lighter than Lepr-null animals (**, ***) from 6–7 weeks of age (n = 10–11). C, D, Lepr-null male (C) and female (D) animals had significantly increased abdominal fat mass compared with Lepr-intact controls, and AgRP-LepR rescue females also had significantly increased (p = 0.0003) adiposity compared with Lepr-intact control females (male Lepr-intact controls, n = 9; male AgRP-LepR rescue and Lepr-null, n = 8; female Lepr-intact controls, n = 9; female AgRP-LepR rescue, n = 11; Lepr-null, n = 5). E, daily food intake of both Lepr-null (p = 0.0001) and AgRP-LepR rescue (p = 0.008) males was significantly increased compared with Lepr-intact controls (Lepr-intact controls and AgRP-LepR rescue, n = 10; Lepr-null, n = 11). F, NPY fiber density in the paraventricular nucleus was significantly reduced in both Lepr-null (p = 0.046) and AgRP-LepR rescue (p = 0.012) animals compared with Lepr-intact controls (Lepr-intact controls and AgRP-LepR rescue, n = 6; Lepr-null, n = 7). G, Representative examples. *p < 0.05; **p < 0.01; ***p < 0.001. 3V, Third ventricle. Scale bar, 100 μm.
As expected, a significant delay in preputial separation of male mice was observed for Lepr-null mice when compared with Lepr-intact controls (Fig. 7A; F(2,28) = 17.56, p < 0.001). Whereas AgRP-LepR rescue mice were also significantly delayed compared with control animals (p < 0.001), preputial separation in this group happened significantly earlier than the Lepr-null group (p < 0.05), indicating that the presence of leptin signaling in AgRP neurons reduced the delay in this aspect of puberty onset experienced by Lepr-null mice. Age at first successful mating was also significantly delayed for Lepr-null mice compared with Lepr-intact control mice (Fig. 7B; F(2,23) = 4.77, p < 0.05), but remarkably no such delay occurred for AgRP-LepR rescue animals (Fig. 7B). This further indicates that the presence of LepR in AgRP neurons was sufficient to normalize puberty onset in male mice. For female mice, as expected, vaginal opening in Lepr-null mice was significantly delayed when compared with Lepr-intact controls (Fig. 7C; H(2,23) = 12.1, p < 0.01). As was the case with mating onset in males, AgRP-LepR rescue was sufficient to completely normalize this aspect of female puberty compared with the Lepr-intact control animals (Fig. 7C; p < 0.05 vs Lepr-null mice). Lepr-null animals did not undergo first estrus during the monitoring period whereas, in marked contrast, all AgRP-LepR rescue animals did, albeit with a significant delay compared with Lepr-intact controls (U(18) = 0, p < 0.001; Fig. 7D). Collectively, these data suggest that the presence of leptin signaling in AgRP neurons is essentially sufficient to normalize puberty onset in these mice. In the month after puberty onset in Lepr-intact controls and AgRP-LepR rescue animals, Lepr-null mice showed no evidence of reproductive cycling (Fig. 8A,B); rather, their vaginal cytological smears remained in a constant diestrus-like state. In marked contrast, AgRP-LepR rescue mice exhibited cycling patterns that were not significantly different from control animals in terms of cycle length or frequency of cycle stages (Fig. 8A,B).
Figure 7.

Puberty onset in male and female AgRP-LepR rescue compared with Lepr-null and Lepr-intact control animals. A, B, Age at preputial separation (A) and the first fertile mating (B) was significantly delayed in Lepr-null compared with Lepr-intact control males, and this was partially overcome by AgRP-LepR rescue (preputial separation, p = 0.046 vs Lepr-null mice; first fertile mating, not significant vs either Lepr-intact or Lepr-null mice). Note that only 6 of 11 of Lepr-null males were able to sire a litter. Lepr-intact controls and AgRP-LepR rescue, n = 10; Lepr-null, n = 11. C, Age at vaginal opening in Lepr-null females was significantly delayed compared with both Lepr-intact control (p = 0.002) and AgRP-LepR rescue (p = 0.012) females. D, First estrus did not occur in any Lepr-null animals during the monitoring time whereas first estrus occurred in all AgRP-LepR rescue females, albeit delayed (p = 0.0001), compared with Lepr-intact control animals (Lepr-intact controls and AgRP-LepR rescue, n = 10; Lepr-null, n = 5). *p < 0.05; ** p < 0.01; ***p < 0.001.
Figure 8.

Estrous cyclicity and fecundity of AgRP-LepR rescue males and females compared with Lepr-null and Lepr-intact control animals and plasma LH concentration in female mice. A, Frequency of occurrence of estrous cycle stages. Lepr-null mice showed lack of cycling, remaining in a constant diestrus-like state, whereas no statistically significant differences between AgRP-LepR rescue and control females. B, Representative examples of the cycling pattern seen in AgRP-LepR rescue and Lepr-intact control animals and the lack of cycling in Lepr-null animals. C, A significant reduction in litter frequency was observed when Lepr-null male and female animals were compared with AgRP-LepR rescue mice (males, p = 0.0001; females, p = 0.008) and Lepr-intact controls (males, p = 0.0001; females, p = 0.0003), whereas no significant difference was observed between LepR rescue mice and Lepr-intact controls. Male Lepr-intact controls and male AgRP-LepR rescue, n = 10; male Lepr-null, n = 11; female Lepr-intact controls, n = 14; female AgRP-LepR rescue, n = 11; female Lepr-null, n = 9. D, There was no significant difference in litter size between any of the male groups, but AgRP-LepR rescue females had smaller litters (p = 0.024) when compared with Lepr-intact control females. Not enough litters were born to Lepr-null females to enable comparison. E, Plasma LH concentration in female mice in the intact state, OVX state, and OVX plus estradiol implanted state. A significant increase in LH levels in response to ovariectomy and a subsequent decrease after estradiol replacement was seen in both Lepr-intact control (p = 0.001) and AgRP-LepR rescue (p = 0.0001) mice, but not Lepr-null mice (Lepr-intact controls, n = 11; female AgRP-LepR rescue, n = 6; Lepr-null, n = 5). *p < 0.05; **p < 0.01; ***p < 0.001. P, Proestrus; E, estrus; M/D, metestrus/diestrus.
Fecundity of adult mice was measured by assessing litter frequency and average number of pups. In this experiment, the interlitter interval was not a valid measurement for assessing fecundity as the majority of Lepr-null animals, and some of the AgRP-LepR rescue animals, had only one litter. Males were left in breeding pairs for 100 d, whereas females were paired for 80 d. This was attributable to welfare considerations; Lepr-null and AgRP-LepR rescue females were prone to dystocia complications because of their obesity. Lepr-null male mice sired very few litters (6 of 11 males sired one to three litters each before becoming infertile) compared with both the AgRP-LepR rescue and Lepr-intact control groups (F(2,28) = 27.21, p < 0.001). AgRP-LepR rescue males had a litter frequency similar to that of Lepr-intact control males (Fig. 8C). There were no significant differences in the average number of pups per litter between any of the male groups (Fig. 8D). Leptin- or LepR-deficient female mice are usually infertile (Chehab et al., 1996; Quennell et al., 2009). Consistent with this, Lepr-null female mice produced almost no litters compared with both the AgRP-LepR rescue and Lepr-intact control groups (F(2,32) = 16.2; p < 0.05 and p < 0.01, respectively; Fig. 8C). Surprisingly, given their apparent absence of reproductive cycles immediately after vaginal opening, two of the nine Lepr-null female mice produced a single litter. It is possible that pairing with a male provided an additional stimulus for reproductive cyclicity. These two females were not included in the analysis of litter size. Lepr-intact control animals had significantly larger litters compared with AgRP-LepR rescue mice (t(22) = 2.41, p < 0.05; Fig. 8D).
To test whether the efficacy of estrogenic negative feedback is reduced in infertile LepR-null mice and whether AgRP-LepR rescue overcomes this, measurements of LH concentration in whole blood samples were used to assess the effect of ovariectomy and subsequent estradiol replacement. As expected for female Lepr-intact control mice, there was a significant elevation of blood LH concentration in the ovariectomized state relative to both the ovary intact and ovariectomized plus estradiol implant states (F(1.6,16) = 7.1, p < 0.01), indicating their HPG axis response to estrogenic negative feedback. In contrast in Lepr-null mice, there was no statistically significant ovariectomy-induced rise in blood LH levels (ovary intact, p = 0.13; ovariectomized plus estradiol implant, p = 0.13 vs ovariectomized state). In the AgRP-LepR rescue groups, the LH increase in response to ovariectomy was restored (F(1.2,5.9) = 30.13, p = 0.001 when compared with the intact and ovariectomy plus estradiol implant states; Fig. 8E). Indeed, when compared with the other two groups, AgRP-LepR rescue mice had significantly increased LH levels in the ovariectomized state (F(2,18) = 3.76, p < 0.05).
Discussion
Under conditions of undernutrition, decreased circulating leptin levels are thought to lead to reproductive suppression, since exogenous leptin treatment is able to overcome this situation in female mice (Ahima et al., 1996) and women (Welt et al., 2004). Humans and mice with a congenital leptin or LepR deficiency are infertile despite being energy replete, and leptin treatment is sufficient to restore reproductive function in leptin-deficient individuals (Chehab et al., 1996; Mounzih et al., 1997; Farooqi et al., 1999). Mice exhibiting forebrain neuron-specific deletion of LepR are also infertile, highlighting the importance of leptin's central actions in regulating reproductive activity (Quennell et al., 2009). Recent experiments have narrowed down the pool of candidate neuronal populations that are required for control of reproduction by leptin, so that we now know that these neurons are likely to coexpress GABA rather than glutamate (Zuure et al., 2013). In this study, the importance of LepR signaling through GABAergic AgRP neurons for the functioning of the hypothalamo-pituitary-gonadal axis was assessed to determine whether this pathway is required and/or sufficient for fertility. Although the effects of deletion of LepR from AgRP neurons were limited to delayed female puberty, rescuing LepR expression solely in AgRP neurons revealed that leptin signaling through this population is almost entirely sufficient for normal puberty onset and fecundity in both sexes.
To assess the specificity of Cre recombinase-induced LepR knock-out or rescue in AgRP neurons, leptin-induced pSTAT3 signaling was evaluated as a functional indicator of leptin-responsive cells. In Experiment 1 when pSTAT3-stained cells in the vmARC were counted, significantly less pSTAT3 staining was observed in AgRP-LepR KO animals compared with Lepr-intact controls. This indicates that the animal model used was successful in preventing leptin signaling in this region, and presumably specifically in the AgRP neuronal population. In Experiment 2, Lepr-null animals were completely devoid of leptin signaling whereas AgRP-LepR rescue animals only expressed leptin signaling in the vmARC, presumably in the AgRP neurons.
Leptin signaling in AgRP neurons exerts minor body weight and food intake effects
AgRP acts as an antagonist to melanocortin receptors to promote feeding, whereas overexpression of NPY is associated with hyperphagia and obesity (Sheffer-Babila et al., 2013). Leptin inhibits the activity of AgRP neurons, so deficient leptin signaling within these cells should lead to hyperphagia and increased body weight (van de Wall et al., 2008). In Experiment 1, both male and female mice that lacked leptin signaling in AgRP neurons showed significantly increased body weight compared with control littermates, although this difference was relatively minor and no difference in food intake was observed compared with controls. The body weight increase was greatest in female mice, in which a significant increase in abdominal adiposity also occurred. This mild and sex-specific metabolic phenotype and lack of difference in caloric intake of AgRP-LepR KO mice compared with controls has been confirmed by other researchers using this model (van de Wall et al., 2008). Consistent with the moderate body weight effect of AgRP-LepR KO, in Experiment 2, rescue of leptin signaling only in AgRP neurons led to a slight reduction in body weight in both sexes compared with the profoundly obese Lepr-null animals from 34 d of age. Abdominal fat mass and food intake were also unaffected or only minimally rescued in mice with restored leptin signaling in AgRP neurons, and the density of AgRP/NPY fibers in one of their primary target nuclei, the PVN, remained as defective as in the Lepr-null mice compared with Lepr-intact controls. The latter results may indicate that leptin's trophic actions on AgRP neuronal wiring (Bouret et al., 2004) occurs indirectly to these cells and are consistent with the idea that direct leptin signaling in AgRP neurons is not the key regulator of food intake circuitry. Previously, it has been demonstrated by deletion of LepR from either AgRP or POMC neurons, or both, that the actions of leptin on these cell types are additive in regard to body weight and adiposity, but even collectively they do not account for the full extent of leptin's metabolic effects, particularly in regard to hyperphagia (van de Wall et al., 2008).
Leptin signaling in AgRP neurons is sufficient for puberty onset
In our study, female AgRP-LepR KO mice displayed a 3 d delay in onset of first estrus compared with control females. This indicates that lack of leptin signaling through AgRP neurons is a barrier for puberty onset in female mice but that this can eventually be overcome so that reproductive function in adults is normal. It may be that redundant pathways such as ventral premammillary nucleus glutamatergic neurons (Donato et al., 2011b) or preoptic nitric oxide neurons (Bellefontaine et al., 2014) eventually compensate for lack of leptin signaling in AgRP neurons. In Experiment 2, the rescue of LepR in AgRP neurons was almost completely sufficient to allow normal puberty onset to occur, although first estrus in AgRP LepR recue mice was delayed by 11 d compared with the Lepr-intact control group. Despite the delay, it is remarkable that the presence of leptin signaling solely in AgRP neurons is sufficient to restore onset of estrus cycles in these animals. This clearly indicates, for the first time, that leptin signaling in AgRP neurons is sufficient for puberty onset in mice. Presumably, AgRP neurons are not the only leptin target population sufficient for puberty onset, since LepR re-expression in the ventral premammillary nucleus also rescued puberty onset (Donato et al., 2011b).
Leptin signaling in AgRP neurons is sufficient, but not required, for adult fertility
While no requirement of AgRP leptin actions for estrous cyclicity was evident in Experiment 1, results of Experiment 2 clearly showed that leptin signaling through AgRP neurons is completely sufficient for normal reproductive cycles and to maintain male and female fecundity in the absence of all other leptin signaling pathways. This role is consistent both with previous reports that ablation of these neurons or knock-out of the genes encoding AgRP, NPY, or the NPY Y4 receptor partially rescues the infertility phenotype of leptin-signaling-deficient mice (Erickson et al., 1996; Sainsbury et al., 2002; Wu et al., 2012; Sheffer-Babila et al., 2013) and the idea that leptin's inhibition of AgRP neurons reduces their suppression of GnRH neuronal activity.
Surprisingly, a few Lepr-null male and female mice were able to sire or give birth to litters. This suggests that the dogma of leptin requirement for fertility is not absolute in all cases. In fact, the degree of infertility in leptin-signaling-deficient mice has been reported to be dependent of the genetic background and sex of the mice (Ewart-Toland et al., 1999).
In female mice, removal of the ovaries disrupts negative feedback resulting in a gradual increase in circulating LH levels on the days after ovariectomy. The expected increase in LH levels after OVX was observed in Lepr-intact control mice, but in Lepr-null animals, this effect appeared to be blunted, suggesting mild hypogonadotropic hypogonadism in the absence of gonadal steroids. In contrast, in AgRP-LepR rescue mice, a marked increase in circulating LH levels was seen after ovariectomy. As we reported previously for GABA-specific LepR knock-out females (Zuure et al., 2013), the negative feedback actions of estradiol remained intact in both Lepr-null and AgRP-LepR rescue mice, suggesting that impairments in this neuroendocrine action are not to blame for the infertility of leptin-signaling-deficient females.
The transgenic models used in this study relied on the removal of LepR early in development. This may permit other types of leptin-responsive neurons to develop sufficient roles to compensate where previously they may have been only minor players. The reciprocal approaches of Experiments 1 and 2 helps to reveal roles that might otherwise be masked by a network of compensatory mechanisms. A role that was compensated for in a knock-out experiment would be expected to be apparent or even exacerbated in a “rescue” experiment, where all other leptin–GnRH pathways are absent. Although it is possible obesity may contribute to reduced fertility, it has been shown that fertility can be maintained in morbidly obese mice (Bates and Myers, 2003; Singireddy et al., 2013). The AgRP-LepR rescue mice in Experiment 2 provide another example of a mouse model that is essentially fully fertile while being morbidly obese and suggest that it is lack of leptin signaling rather than obesity per se that is primarily responsible for infertility in leptin-signaling-deficient mouse lines.
These results demonstrate that AgRP neurons are involved in the transmission of information from leptin receptors to the hypothalamo-pituitary-gonadal axis. It is likely this action occurs via modulation of GnRH neuronal activity. AgRP and NPY inhibit pulsatile LH release (Catzeflis et al., 1993; Vulliémoz et al., 2005), which directly reflects GnRH release. It seems that one mechanism by which leptin signaling deficiency leads to infertility is through overexpression of AgRP and NPY, which in turn leads to the suppression of GnRH release. Consistent with AgRP neurons exerting direct actions on GnRH neurons (Roa and Herbison, 2012), AgRP is also a potent antagonist of the stimulatory effects of αMSH on the melanocortin-4 receptor (MC4R) (Butler and Cone, 2002). Approximately half of murine GnRH neurons express Mc4r, and MC4R activation can increase c-Fos coexpression and firing rate in GnRH neurons (Israel et al., 2012). AgRP may also influence GnRH neurons indirectly since AgRP deficiency upregulates Tac2 (coexpressed by a subpopulation of arcuate kisspeptin neurons) gene expression in LepR-deficient mice (Sheffer-Babila et al., 2013). It is likely that a complex network exists between AgRP neurons and other neurons of the GnRH neuronal network to coordinate reproduction.
In summary, we have demonstrated here that leptin signaling in arcuate AgRP neurons is sufficient to permit all aspects of puberty onset and fertility in male and female mice, and this action appears to be independent of leptin's metabolic affects. The requirement of leptin actions in these neurons for fertility is relatively minimal, however. These findings are consistent with the existence of multiple redundant leptin-responsive inputs to the GnRH neurons that govern the reproductive axis.
Footnotes
The New York Obesity Research Center supported the development of the floxed leptin receptor mouse (Grant 1PO1DK26687 to S. Chua). We thank S. Chua for supplying this mouse.
The authors declare no competing financial interests.
References
- Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS (1996) Role of leptin in the neuroendocrine response to fasting. Nature 382:250–252. 10.1038/382250a0 [DOI] [PubMed] [Google Scholar]
- Bates SH, Myers MG Jr (2003) The role of leptin receptor signaling in feeding and neuroendocrine function. Trends Endocrinol Metab 14:447–452. 10.1016/j.tem.2003.10.003 [DOI] [PubMed] [Google Scholar]
- Bellefontaine N, Chachlaki K, Parkash J, Vanacker C, Colledge W, De Tassigny XDA, Garthwaite J, Bouret SG, Prevot V (2014) Leptin-dependent neuronal NO signaling in the preoptic hypothalamus facilitates reproduction. The J of clinical investigation 124:2550–2559. 10.1172/JCI65928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund ED, Vianna CR, Donato J, Kim MH, Chuang J-C, Lee CE, Lauzon DA, Lin P, Brule LJ, Scott MM (2012) Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Invest 122:1000–1009. 10.1172/JCI59816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB (2004) Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 304:108–110. 10.1126/science.1095004 [DOI] [PubMed] [Google Scholar]
- Broberger C, Johansen J, Johansson C, Schalling M, Hökfelt T (1998) The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci U S A 95:15043–15048. 10.1073/pnas.95.25.15043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, Cone RD (2002) The melanocortin receptors: lessons from knockout models. Neuropeptides 36:77–84. 10.1054/npep.2002.0890 [DOI] [PubMed] [Google Scholar]
- Catzeflis C, Pierroz DD, Rohner-Jeanrenaud F, Rivier JE, Sizonenko PC, Aubert ML (1993) Neuropeptide Y administered chronically into the lateral ventricle profoundly inhibits both the gonadotropic and the somatotropic axis in intact adult female rats. Endocrinology 132:224–234. 10.1210/endo.132.1.8380374 [DOI] [PubMed] [Google Scholar]
- Chehab FF, Lim ME, Lu R (1996) Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet 12:318–320. 10.1038/ng0396-318 [DOI] [PubMed] [Google Scholar]
- Cravo RM, Frazao R, Perello M, Osborne-Lawrence S, Williams KW, Zigman JM, Vianna C, Elias CF (2013) Leptin signaling in Kiss1 neurons arises after pubertal development. PloS One 8:e58698. 10.1371/journal.pone.0058698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich MO, Zimmer MR, Bober J, Horvath TL (2015) Hypothalamic Agrp neurons drive stereotypic behaviors beyond feeding. Cell 160:1222–1232. 10.1016/j.cell.2015.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato J Jr, Cravo RM, Frazão R, Elias CF (2011a) Hypothalamic sites of leptin action linking metabolism and reproduction. Neuroendocrinology 93:9–18. 10.1159/000322472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato J Jr, Cravo RM, Frazão R, Gautron L, Scott MM, Lachey J, Castro IA, Margatho LO, Lee S, Lee C, Richardson JA, Friedman J, Chua S Jr, Coppari R, Zigman JM, Elmquist JK, Elias CF (2011b) Leptin's effect on puberty in mice is relayed by the ventral premammillary nucleus and does not require signaling in Kiss1 neurons. J Clin Invest 121:355–368. 10.1172/JCI45106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JC, Hollopeter G, Palmiter RD (1996) Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science 274:1704–1707. 10.1126/science.274.5293.1704 [DOI] [PubMed] [Google Scholar]
- Evans MC, Anderson GM (2017) Neuroendocrine integration of nutritional signals on reproduction. J Mol Endocrinol 58:R107–R128. 10.1530/JME-16-0212 [DOI] [PubMed] [Google Scholar]
- Evans MC, Rizwan MZ, Anderson GM (2014) Insulin action on GABA neurons is a critical regulator of energy balance but not fertility in mice. Endocrinology 155:4368–4379. 10.1210/en.2014-1412 [DOI] [PubMed] [Google Scholar]
- Ewart-Toland A, Mounzih K, Qiu J, Chehab FF (1999) Effect of the genetic background on the reproduction of leptin-deficient obese mice*. Endocrinology 140:732–738. 10.1210/endo.140.2.6470 [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O'Rahilly S (1999) Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med 341:879–884. 10.1056/NEJM199909163411204 [DOI] [PubMed] [Google Scholar]
- Hahn TM, Breininger JF, Baskin DG, Schwartz MW (1998) Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci 1:271–272. 10.1038/1082 [DOI] [PubMed] [Google Scholar]
- Horvath TL, Bechmann I, Naftolin F, Kalra SP, Leranth C (1997) Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res 756:283–286. 10.1016/S0006-8993(97)00184-4 [DOI] [PubMed] [Google Scholar]
- Israel DD, Sheffer-Babila S, de Luca C, Jo YH, Liu SM, Xia Q, Spergel DJ, Dun SL, Dun NJ, Chua SC Jr (2012) Effects of leptin and melanocortin signaling interactions on pubertal development and reproduction. Endocrinology 153:2408–2419. 10.1210/en.2011-1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloukina V, Herzer S, Karlsson N, Perez M, Daraio T, Meister B (2012) G-protein-gated inwardly rectifying K+ channel 4 (GIRK4) immunoreactivity in chemically defined neurons of the hypothalamic arcuate nucleus that control body weight. J Chem Neuroanat 44:14–23. 10.1016/j.jchemneu.2012.03.003 [DOI] [PubMed] [Google Scholar]
- Mayer C, Acosta-Martinez M, Dubois SL, Wolfe A, Radovick S, Boehm U, Levine JE (2010) Timing and completion of puberty in female mice depend on estrogen receptor α-signaling in kisspeptin neurons. Proc Natl Acad Sci U S A 107:22693–22698. 10.1073/pnas.1012406108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMinn JE, Liu SM, Dragatsis I, Dietrich P, Ludwig T, Eiden S, Chua SC Jr (2004) An allelic series for the leptin receptor gene generated by CRE and FLP recombinase. Mamm Genome 15:677–685. 10.1007/s00335-004-2340-1 [DOI] [PubMed] [Google Scholar]
- Moschos S, Chan JL, Mantzoros CS (2002) Leptin and reproduction: a review. Fertil Steril 77:433–444. 10.1016/S0015-0282(01)03010-2 [DOI] [PubMed] [Google Scholar]
- Mounzih K, Lu R, Chehab FF (1997) Leptin treatment rescues the sterility of genetically obese ob/ob males. Endocrinology 138:1190–1193. 10.1210/endo.138.3.5024 [DOI] [PubMed] [Google Scholar]
- Quennell JH, Mulligan AC, Tups A, Liu X, Phipps SJ, Kemp CJ, Herbison AE, Grattan DR, Anderson GM (2009) Leptin indirectly regulates gonadotropin-releasing hormone neuronal function. Endocrinology 150:2805–2812. 10.1210/en.2008-1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roa J, Herbison AE (2012) Direct regulation of GnRH neuron excitability by arcuate nucleus POMC and NPY neuron neuropeptides in female mice. Endocrinology 153:5587–5599. 10.1210/en.2012-1470 [DOI] [PubMed] [Google Scholar]
- Robertson SA, Leinninger GM, Myers MG Jr (2008) Molecular and neural mediators of leptin action. Physiol Behav 94:637–642. 10.1016/j.physbeh.2008.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainsbury A, Schwarzer C, Couzens M, Jenkins A, Oakes SR, Ormandy CJ, Herzog H (2002) Y4 receptor knockout rescues fertility in ob/ob mice. Genes Dev 16:1077–1088. 10.1101/gad.979102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffer-Babila S, Sun Y, Israel DD, Liu S-M, Neal-Perry G, Chua SC (2013) Agouti-related peptide plays a critical role in leptin's effects on female puberty and reproduction. Am J Physiol Endocrinol Metab 305:E1512–E1520. 10.1152/ajpendo.00241.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Sorrell JE, Clegg DJ, Woods SC, Seeley RJ (2010) The roles of leptin receptors on POMC neurons in the regulation of sex-specific energy homeostasis. Physiol Behav 100:165–172. 10.1016/j.physbeh.2010.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singireddy AV, Inglis MA, Zuure WA, Kim JS, Anderson GM (2013) Neither signal transducer and activator of transcription 3 (STAT3) or STAT5 signaling pathways are required for leptin's effects on fertility in mice. Endocrinology 154:2434–2445. 10.1210/en.2013-1109 [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB (2008) Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11:998–1000. 10.1038/nn.2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van De Wall E, Leshan R, Xu AW, Balthasar N, Coppari R, Liu SM, Jo YH, MacKenzie RG, Allison DB, Dun NJ, Elmquist J, Lowell BB, Barsh GS, de Luca C, Myers MG Jr, Schwartz GJ, Chua SC Jr (2008) Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 149:1773–1785. 10.1210/en.2007-1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulliémoz NR, Xiao E, Xia-Zhang L, Wardlaw SL, Ferin M (2005) Central infusion of agouti-related peptide suppresses pulsatile luteinizing hormone release in the ovariectomized rhesus monkey. Endocrinology 146:784–789. 10.1210/en.2004-1093 [DOI] [PubMed] [Google Scholar]
- Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, Karalis A, Mantzoros CS (2004) Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med 351:987–997. 10.1056/NEJMoa040388 [DOI] [PubMed] [Google Scholar]
- Wen S, Go Tze IN, Mai O, Schauer C, Leinders-Zufall T, Boehm U (2011) Genetic identification of GnRH receptor neurons: a new model for studying neural circuits underlying reproductive physiology in the mouse brain. Endocrinology 152:1515–1526. 10.1210/en.2010-1208 [DOI] [PubMed] [Google Scholar]
- Wu Q, Whiddon BB, Palmiter RD (2012) Ablation of neurons expressing agouti-related protein, but not melanin concentrating hormone, in leptin-deficient mice restores metabolic functions and fertility. Proc Natl Acad Sci U S A 109:3155–3160. 10.1073/pnas.1120501109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuure WA, Roberts AL, Quennell JH, Anderson GM (2013) Leptin signaling in GABA neurons, but not glutamate neurons, is required for reproductive function. J Neurosci 33:17874–17883. 10.1523/JNEUROSCI.2278-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]