

Abstract
Group 1 metabotropic glutamate receptors (mGlu1/5s) are critical to synapse formation and participate in synaptic LTP and LTD in the brain. mGlu1/5 signaling alterations have been documented in cognitive impairment, neurodegenerative disorders, and psychiatric diseases, but underlying mechanisms for its modulation are not clear. Here, we report that transferrin receptor 1 (TFR1), a transmembrane protein of the clathrin complex, modulates the trafficking of mGlu1 in cerebellar Purkinje cells (PCs) from male mice. We show that conditional knock-out of TFR1 in PCs does not affect the cytoarchitecture of PCs, but reduces mGlu1 expression at synapses. This regulation by TFR1 acts in concert with that by Rab8 and Rab11, which modulate the internalization and recycling of mGlu1, respectively. TFR1 can bind to Rab proteins and facilitate their expression at synapses. PC ablation of TFR1 inhibits parallel fiber–PC LTD, whereas parallel fiber–LTP and PC intrinsic excitability are not affected. Finally, we demonstrate that PC ablation of TFR1 impairs motor coordination, but does not affect social behaviors in mice. Together, these findings underscore the importance of TFR1 in regulating mGlu1 trafficking and suggest that mGlu1- and mGlu1-dependent parallel fiber–LTD are associated with regulation of motor coordination, but not autistic behaviors.
SIGNIFICANCE STATEMENT Group 1 metabotropic glutamate receptor (mGlu1/5) signaling alterations have been documented in cognitive impairment, neurodegenerative disorders, and psychiatric diseases. Recent work suggests that altered mGlu1 signaling in Purkinje cells (PCs) may be involved in not only motor learning, but also autistic-like behaviors. We find that conditional knock-out of transferrin receptor 1 (TFR1) in PCs reduces synaptic mGlu1 by tethering Rab8 and Rab11 in the cytosol. PC ablation of TFR1 inhibits parallel fiber–PC LTD, whereas parallel fiber–PC LTP and PC intrinsic excitability are intact. Motor coordination is impaired, but social behaviors are normal in TFR1flox/flox;pCP2-cre mice. Our data reveal a new regulator for trafficking and synaptic expression of mGlu1 and suggest that mGlu1-dependent LTD is associated with motor coordination, but not autistic-like behaviors.
Keywords: autism, metabotropic glutamate receptor, motor coordination, purkinje cell, trafficking, transferrin receptor
Introduction
Metabotropic glutamate receptors are G-protein-coupled receptors enriched at excitatory synapses in the CNS. Group 1 mGlu receptors (mGlu1/5) activate phospholipase C and protein kinase C (PKC) (Niswender and Conn, 2010) and modulate signal-to-nucleus communication (Hermans and Challiss, 2001). mGlu1/5 are critical for synapse formation during development (Ichise et al., 2000; Hannan et al., 2001) and participate in LTP and LTD in the hippocampus and the cerebellum, two structures critical for declarative and procedural memory formation, respectively (Aiba et al., 1994a, 1994b; Huber et al., 2000; Ichise et al., 2000; Brasnjo and Otis, 2001). Therefore, mGlu1/5 is implicated in forms of conditioning behaviors such as associative learning (Aiba et al., 1994a) and cocaine addiction (Kenny and Markou, 2004; Bellone and Lüscher, 2006; Mameli et al., 2009).
Alterations in mGlu1/5 signaling have been documented for cognitive impairment, neurodegenerative disorders, and psychiatric diseases such as fragile X syndrome, autism, Alzheimer's disease, Parkinson's disease, Huntington's disease, and epilepsy (Ribeiro et al., 2010; for reviews, see Lüscher and Huber, 2010; D'Antoni et al., 2014). Accordingly, mGlu1/5 may form an attractive target to tackle with allosteric modulators or agonists/antagonists for treating several CNS disorders (Gregory and Conn, 2015). However, developing therapeutic strategies using mGlu1/5-related agents remains challenging. For example, applying antagonists of mGlu1/5 does not appear very efficient in treating convulsions or cognitive impairments (Löscher et al., 2006). This lack of effect may be partly ascribed to the complexity of mGlu1/5 signaling, which involves a variety of downstream cascades that remain unknown for most neurological pathologies. In particular, the mechanisms controlling the trafficking of mGlu1/5 are less understood.
In the cerebellum, mGlu1 is highly expressed in Purkinje cells (PCs) (Shigemoto et al., 1992). Inhibition or lack of mGlu1 signaling leads to PC dysfunction and motor coordination deficits (Ichise et al., 2000; Coesmans et al., 2003). Interestingly, global knock-out of Nlgn3, which is implicated in autism, causes an increased expression of mGlu1 and an inhibition of parallel fiber–LTD in the cerebellum, whereas the development of the molecular layer is affected (Baudouin et al., 2012). Therefore, a follow-up question is to what extent the alteration of mGlu1 signaling in PCs results concomitantly in specific cellular physiological abnormalities, autistic-like behaviors, and developmental morphological aberrations.
Transferrin receptor 1 (TFR1) is a transmembrane glycoprotein involved in processing of clathrin-coated vesicles. It has been found that TFR1 is present in neuronal dendrites and colocalizes with presynaptic/postsynaptic proteins (West et al., 1997; Liu et al., 2016). TFR1 is expressed abundantly in the cerebellar cortex and deep cerebellar nuclei (Moos, 1996), but its function in the cerebellum is unknown. In the present study, we created a mutant mouse line in which TFR1 is deleted specifically in PCs. Deletion of TFR1 reduces synaptic expression of mGlu1 in PCs, presumably by affecting the trafficking of mGlu1 mediated by Rab proteins. Furthermore, TFR1 turns out to be critical for the expression of parallel fiber–LTD and proper motor coordination, but not for the development of a normal cerebellar cytoarchitecture or functional social interactions. These data highlight the complexity of mGlu1 signaling and its diverse roles in cerebellar cellular and system physiology and, conversely, cerebellar development, which appear to have differential implications for cerebellar pathology when they deteriorate.
Materials and Methods
Generation of TFR1flox/flox mice and animal maintenance.
TFR1flox/flox mice were generated in the Model Animal Research Center of Nanjing University (Nanjing, China), as illustrated in Figure 1. BAC clones containing targeted gene were purchased from Invitrogen (BAC Clone #BMQ-235I21). The mouse TFR1 gene is composed of 19 exons. Targeted gene including homologous arms was retrieved from the BAC vector. The first LoxP was introduced into intron 2 and the second loxP along with a FRT-flanked PGK/neo cassette was introduced into intron 3 by homologous recombination in E. coli. The targeting vector was linearized by digesting with I-CeuI and electroporated into W4 embryonic stem (ES) cells. The recombinants were selected for their G418 and DTA resistance. The PGK/EM7-NeoR cassette served as positive selection marker and DTA was a negative selection marker at the ES targeting step. Targeted ES clones were screened by PCR and Southern blot. The positive ES cell clones were chosen for microinjection to obtain chimeras. Germline-transmitting chimeras were generated from ES lines carrying targeted TFR1 allele by the microinjection of blastocysts isolated from albino C57B6/J-Tyrc-Brd females. Chimeras were crossed to C57BL/6J to obtain TFR1fln/+ mice, which were backcrossed to C57BL/6J for >10 generations and then bred with Flper mice, which were on the C57BL/6 background, to remove the neomycin (neo) cassette so as to obtain TFR1flox/+ mice. Conditioned knock-out mice (TFR1flox/flox;pCP2-cre) were obtained by crossing TFR1flox/flox mice with pCP2-cre mice (Barski et al., 2000) and were bred with a C57BL/6 background because the genetic background for both Flper and pCP2-Cre mice was C57BL/6. The resulting offspring were genotyped using PCR of genomic DNA. Mice were kept under temperature-controlled conditions on a 12:12 h light/dark cycle with food and water ad libitum. All experiments were approved by the Animal Experimentation Ethics Committee of Zhejiang University and were done in male mice at the age of postnatal day 2 (P2) to P50 depending on experimental requirements.
Figure 1.

Gene targeting strategy for the generation of TFR1flox/flox mice. A LoxP was inserted into intron 2. Another loxP along with a FRT-flanked PGK/Neo cassette was inserted into intron 3. 5′ and 3′ probes used for Southern blot analysis of genomic DNA appear in black bars. The loxP and FRT elements are shown as filled and open triangles, respectively.
Antibodies and reagents.
Antibody against mGlu1 was purchased from BD Biosciences. Antibody to TFR1 was purchased from Novus Biologicals [coimmunoprecipitation (co-IP) and immunohistochemistry] and Life Technologies (Western blot). Antibodies to PSD95 and GAPDH were from Millipore. Anti-GluA2 antibody was a gift from Dr. Richard Huganir (Johns Hopkins University, Baltimore, MD). Anti-vesicular glutamate tranporter 1 (vGluT1) was a gift from Dr. Masahiko Watanabe (Hokkaido University, Sapporo, Japan). Antibodies against both Rab11 and Rab8 were from Cell Signaling Technology. Antibody to excitatory amino acid transporter 4 (EAAT4) was from Alpha Diagnostics. Antibodies to β-actin and rat IgG were from Santa Cruz Biotechnology. Antibodies to gephyrin and vesicular GABA transporter (vGAT) were from Synaptic Systems. Horseradish peroxidase-conjugated secondary antibodies for immunoblotting were from GE Healthcare. Nissl was from Beyotime. DMEM, DAPI, and Alexa Fluor-conjugated secondary antibodies were from Invitrogen. Protease inhibitor mixture was from Merck Chemicals. Other chemicals were from Sigma-Aldrich unless stated otherwise.
RT-PCR.
For single-cell analysis, the contents of individual PCs (P7–P21) were harvested as described in our previous work (Wu et al., 2012; Zhou et al., 2015). In brief, the tip of a conventional patch-clamp pipette was placed tightly on the soma of a selected PC. Gentle suction was applied to the pipette. After complete incorporation of the soma, the negative pressure was released and the pipette was quickly removed from the bath. The harvested contents were subjected to RT-PCR using the OneStep Kit (Qiagen). Forward (F) and reverse (R) primers used for amplification were as follows: TFR1, F: 5′-GCC AGA TCA GCA TTC TCT AAC T-3′; R: 5′-CAC TAG CCT TCA TGT TAT TGT C-3′; calbindin, F: 5′-GGC TTC ATT TCG ACG CTG AC-3′; R: 5′-ACG TGA GCC AAC TCT ACA ATT C-3′; GAPDH, F: 5′-GGT GAA GGT CGG TGT GAA CG-3′; R: 5′-CTC GCT CCT GGA AGA TGG TG-3′.
HEK293 cell cultures.
HEK293 cells were cultured in DMEM and supplemented with 10% FBS (Invitrogen), 1 mm sodium pyruvate, 100 U/ml penicillin, and 10 μg/ml streptomycin in an incubator (95% O2/5% CO2, 37°C). At 70–80% confluency, cells were transfected in OPTI-MEM (Invitrogen) with plasmids using lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. At 36 h after transfection with myc-mGlu1 alone or with myc-mGlu1 + GFP-TFR1, transfected cells were collected and extracted proteins were centrifuged at 10,000 × g at 4°C for 30 min to collect supernatant and pellet fractions, which were kept as Triton X-100-soluble and -insoluble fractions, respectively.
Synaptosome preparation and immunocytochemistry.
The purification of synaptosomes from TFR1flox/flox mouse cerebellum (P21) was modified from Ferrero et al. (2013). The cerebellar homogenate (P21) was centrifuged at 2000 × g (4°C for 2 min) and the supernatant was centrifuged again at 9500 × g for 12 min. The loosely compacted white layer containing the majority of synaptosomes was gently resuspended in 0.32 m sucrose, pH 7.4, and an aliquot of synaptosomal suspension (2 ml) was placed onto a 3 ml Percoll gradient, pH 7.4. After centrifugation at 25,000 × g (4°C for 10 min), synaptosomes were recovered from between 10% and 23% Percoll bands and then diluted in a final volume of 30 ml of HEPES-buffered medium (HBM; pH 7.4) containing the following (in mm): 140 NaCl, 5 KCl, 5 NaHCO3, 1.2 NaH2PO4, 1 MgCl2, 10 glucose, and 10 HEPES. After another centrifugation at 22,000 × g (10 min), the synaptosome pellet was resuspended in HBM (6 ml). Finally, synaptosomal suspension (0.75 mg) was diluted in HBM (2 ml) and centrifuged at 10,000 × g for 10 min. The pellets containing synaptosomes were stored on ice and remained viable for 4–6 h. For immunocytochemistry, synaptosomes (1.0 mg/ml) were added to a medium containing 0.32 m sucrose, pH 7.4, at 37°C, allowed to attach to polylysine-coated coverslips for 1 h, and fixed for 4 min in 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, at room temperature (RT). After several washes with PB, pH 7.4, synaptosomes were preincubated for 1 h in 10% normal goat serum diluted in 50 mm Tris buffer, pH 7.4, containing 0.2% Triton X-100. Subsequently, they were incubated for 24 h with primary antiserum for EAAT4 (1:1000), TFR1 (1:150), and vGluT1 (1:200). After washing in TBS, synaptosomes were incubated with secondary antibodies for 2 h. Coverslips were mounted with Prolong Antifade Kit (Invitrogen) and synaptosomes were viewed using a confocal microscope (Nikon A1R) with a 100× objective.
Immunohistochemistry.
Sagittal sections (20 μm) from mice at P2–P21 were prepared and placed in blocking solution for 1 h at RT. After washing with PBS, sections were incubated with primary antibodies overnight at 4°C and incubated with secondary antibodies for 1 h at RT. Primary antibody dilutions used for immunohistochemistry were EAAT4 (1:200), vGAT (1:500), gephyrin (1:500), TFR1 (1:150), calbindin (1:1000), and secondary antibodies (1:1000). All antibodies were diluted in PBS containing 1% BSA and 1% normal goat serum.
Morphometric analysis.
Sagittal cerebellar slices (10 μm) were immuonostained with antibodies against calbindin and EAAT4. For dendritic branching analysis, images of PCs (P21) were captured by a confocal microscope (FV1000; Olympus) and dendritic tracing was performed using MetaMorph software (Olympus). Spine analysis was based on previous studies (Lee et al., 2004; Zhou et al., 2015). In brief, images of distal dendrites of PCs were captured by a SIM super resolution confocal microscope (N-SIM; Nikon). Spine density was evaluated as the relative spine number over a 10 μm dendritic fragment. The measurements of the area and lengths of the dendrites were performed in the bank area of lobule V. Initially, the dendritic area and length of single PCs were acquired and then pooled for the averages. Imaging data analysts were blinded to experimental conditions during the measurements.
Postsynaptic density fraction isolation.
Cerebellar tissues (P21) were homogenized in SHEEP buffer containing the following (in mm): 380 sucrose, 4 HEPES, pH 7.5, 0.1 EDTA, and 0.1 EGTA supplemented with protease inhibitors (Merck) and centrifuged at 800 × g (4°C for 15 min). The supernatant was centrifugated at 9200 × g (4°C for 15 min). The pellet was the crude synaptosomal fraction (P2). The P2 fraction was resuspended in buffer D (20 mm Tris-HCl-pH 8.0, 1 mm EDTA, 100 mm NaCl, and 1% Triton X-100) and was centrifuged at 100,000 × g for 1 h to produce a supernatant and a pellet. The pellet represented the PSD fraction.
Co-IP.
Co-IP experiments were performed according to previous work (Wang et al., 2015). Mouse cortices were lysed in RIPA buffer plus the protease inhibitor. The protein concentration was measured using the BCA assay after the centrifugation at 16,000 × g at 4°C for 10 min. Decimus supernatant was used for input and the remainder was for IP. Precleared solubilized preparations were incubated with rat anti-TFR1 antibody, which was precoupled to protein A-Sepharose beads (GE Healthcare) at 2–4 μg of antibody/1 ml of beads for 2 h in 50 mm Tris-HCl. Proteins on the beads were extracted with 2× SDS sample buffer and boiled for 5 min before Western blot analysis.
Western blot.
Cerebellar tissues or cultures were rinsed with PBS and diluted in 1% SDS containing protease inhibitors. After determining protein concentration with BCA protein assay (Bio-Rad), equal quantities of proteins were loaded and fractionated by SDS-PAGE and transferred to PVDF membrane (Immobilon P; Millipore), immunoblotted with antibodies, and visualized by enhanced chemiluminescence (Pierce Biotechnology). Primary antibody dilutions used were as follows: mGlu1 (1:5000), TFR1 (1:1000), GluA2 (1:2000), PSD95 (1:10,000), calbindin (1:10,000), Rab11 (1:1000), Rab 8 (1:1000), GAPDH (1:10,000), and secondary antibodies (1:10,000). Film signals were digitally scanned and quantitated using ImageJ version 1.42q.
Recombinant virus and in vivo injection.
To express mGlu1WT in PCs, recombinant Semliki Forest virus (SFV) was constructed in combination with enhanced GFP (eGFP) as described previously (Jia et al., 2017). Under deep anesthesia with an intraperitoneal injection of 4% chloral hydrate (10 μl/g), a viral solution (300 nl) containing recombinant SFV (2.3E6 FFU/ml) was injected into lobules V–VI of the cerebellar vermis of TFR1flox/flox;pCP2-cre mice (P21) at a rate of 30 nl/min using a glass pipette (30 μm in diameter) and a microinjector (World Precision Instruments). Eighteen hours later, the injected animals were decapitated and eGFP signals were observed in sporadic PCs in affected lobules.
Slice preparation.
Sagittal slices of cerebellar vermis (250 μm) were prepared from anesthetic mice (P21) using a vibrating tissue slicer (VT1000S; Leica) and ice-cold standard artificial CSF (aCSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 26 NaHCO3, and 25 D-glucose bubbled with 95% O2/5% CO2. After recovery for 30 min at 37°C, slices were placed in a submerged chamber that was perfused at 2 ml/min with aCSF. Patch-clamp electrodes (3–5 MΩ) were filled with an intracellular solution composed of one of the following (in mm): (1) 135 Cs-methanesulfonate, 10 CsCl, 10 HEPES, 0.2 EGTA, 4 Na2ATP, and 0.4 Na3GTP, pH 7.3, osmolality 90, for voltage-clamp recordings; (2) 134 K-gluconate, 6 KCl, 4 NaCl, 10 HEPES, 0.2 EGTA, 4 Na2ATP, 0.3 Na3GTP, and 14 Na2 phosphocreatine, pH 7.3, osmolality 290, for current-clamp recordings; or (3) 150 Cs-methanesulfonate, 5 KCl, 0.1 EGTA, 5 HEPES, 3 MgATP, and 0.4 Na3GTP, pH 7.3, osmolality 290, for IPSC recording (Ikeda et al., 2013; Peter et al., 2016).
Whole-cell recording.
Purkinje cells were visualized under an upright microscope (BX51; Olympus) with a 40× water-immersion objective and equipped with infrared differential interference contrast enhancement. Whole-cell recordings were obtained with an Axon MultiClamp 700B amplifier (Molecular Devices). Currents were digitized at 10 kHz and filtered at 3 kHz. To obtain parallel fiber–EPSCs, standard patch pipettes were filled with aCSF and placed in the middle third of the molecular layer. mGlu1-EPSCs were induced by burst parallel fiber stimulations (10 stimuli at 100 Hz) delivered in the presence of NBQX (5 μm). Gabazine (10 μm) was supplemented into aCSF when EPSCs and mGlu1-EPSCs were recorded. To record evoked IPSCs (eIPSCs), stimulation electrodes were placed in the inner fourth of the molecular layer to activate axons of interneurons while PCs were held at 0 mV in the presence of NBQX (5 μm) (Ikeda et al., 2013). For climbing fiber stimulation, constant current steps (20–200 mA/100 ms) were applied to a patch pipette that was positioned in the granule cell layer close to the vicinity of recorded neuron. The recording was performed at a holding potential of −10 mV while stimulus intensity and electrode position were adjusted so that an all-or-none response was elicited (Su and Shen, 2009; Baudouin et al., 2012). Synaptic responses were evoked every 20 s (0.05 Hz) using ∼10 μA pulses (100 μs). Recordings were excluded from analysis if the series or input resistance varied by >15% over the course of an experiment. In voltage-clamp mode, holding potential at −70 mV was chosen to prevent spontaneous spike activity that might escape clamp.
LTP, LTD, and intrinsic plasticity protocols were recorded from lobules V–VI or IX–X neurons. The lobules were identified by their locations with a 10× objective and then zoomed in with a 40× objective for recording. LTD of parallel fiber–EPSCs was induced by a conjunction of five parallel fiber pulses at 100 Hz and a 100-ms-long depolarization of PC to 0 mV repeated 30 times with an interval of 2 s (Sun et al., 2011; Zhou et al., 2015). LTP of parallel fiber–EPSCs was obtained when parallel fibers were stimulated at 1 Hz for 5 min in current-clamp mode (Wang et al., 2014a). Intrinsic plasticity was induced using LTP tetanus without bias currents when action potentials (APs) were evoked by 400 pA current injections (500 ms) at 0.05 (Peter et al., 2016). Miniature EPSCs (mEPSCs) and miniature IPSCs (mIPSCs) were recorded in whole-cell configuration in the presence of 0.5 μm tetrodotoxin (TTX) plus gabazine (10 μm) or NBQX (5 μm), respectively (Sun et al., 2011; Peter et al., 2016). The offline analysis of mEPSCs and mIPSCs was conducted using a sliding template algorithm (ClampFit 10; Molecular Devices) (Sun et al., 2011). The criteria for inclusion were the amplitude larger than 6 pA and a rise time (10–90%) longer than 1 ms. Overlapping events were rejected.
Cell-attached recordings.
Cell-attached recordings were made in lobule I–III or IX–X neurons. The recording pipette contained the following (in mm): 125 NaCl, 10 HEPES, 3 KCl, and 2 CaCl2. Recordings were performed in voltage-clamp mode (0 injections) using a MultiClamp 700B amplifier. Membrane voltage/current was filtered at 3 kHz and digitized at 8 kHz. Gabazine (20 μm), NBQX (10 μm), and D-AP5 (50 μm) were added to perfusing aCSF to inhibit GABAA and ionotropic glutamate receptor currents. To induce the plasticity of spontaneous spikes, burst stimuli (15 pulses at 100 Hz, repeated at 1 Hz for 5 min) were delivered to parallel fibers (Belmeguenai et al., 2010).
AP parameters.
In the majority of recordings, a hyperpolarizing bias current was applied to prevent spontaneous spike activity. Cells were discarded when the bias current was >400 pA. The membrane voltage of clamped cells was ∼−70 mV. A series of current steps (500 ms) was delivered to PCs with an interval of 10 s. The upstroke of APs was measured from the threshold to the peak. The AP amplitude from the peak to the afterhyperpolarization (AHP) and AP widths were measured at half amplitude. The AHP size was measured from AP threshold to the negative peak of the AHP. The voltage threshold was measured in the first derivative of AP (dV/dt) considering the point where the velocity was close to 50 mV/ms.
mGlu1 trafficking assay.
This assay was consistent with previous work (Hong et al., 2009; Pandey et al., 2014; Zhou et al., 2015). In brief, acute cerebellar slices (250 μm) were incubated in aCSF containing 3,5-dihydroxyphenylglycine (DHPG; 100 μm) for 10 min at RT. Subsequently, slices were chased at two time points (30 min or 180 min) in plain aCSF without DHPG at RT. Then, slices for different conditions were homogenized to determine protein concentrations in the total and PSD fractions.
Motor coordination test.
The elevated beam balancing and rotarod tests were conducted as described previously (Hartmann et al., 2014; Zhou et al., 2015). After habituation to the rotarod, target mice (P45–P50) were tested twice a day at a time interval of ∼8 h for 4 consecutive days. In each session, the velocity of the rotation increased with a constant acceleration of 9 rpm/min starting from 5 rpm. Behavioral experimenters were blinded to the mouse genotype until the data were integrated.
Open-field test.
Mice (P45–P50) were placed in a novel, brightly lit rectangular (46 cm × 33 cm × 28 cm, length × width × height) Plexiglas chamber for 15 min. The activity of mice was recorded and analyzed by an infrared tracking software (ViewPoint Life Sciences). During analysis, the arena was subdivided into three concentric zones: the inner (23 cm × 16.5 cm, length × width), middle (35 cm × 25 cm, length × width), and outer zones. The percentage of time in each zone was calculated.
Three-chamber test.
The three-chamber social test was conducted in mice of (P45–P50) as described previously (Neul et al., 2010; Peter et al., 2016; Zhang et al., 2016). The apparatus consisted of a rectangular Plexiglas box (60 cm × 35 cm × 10 cm, length × width × height) evenly divided into three chambers. Age- and gender-matched WT target subjects (strangers 1 and 2) were habituated for 5 consecutive days before the test by being placed inside metal-wired cages. On the test day, test mice were placed in the central chamber for a 10 min habituation. Stranger 1 was introduced into a wire cage in one chamber and the empty chamber served as an inanimate object with no social valence. The dividers were then raised to allow test mice to explore all three chambers freely over a 5 min session. The time spent in each chamber was recorded and the ratio of (S1 − E) to (S1 + E) was calculated as the preference index (S1 − E). Subsequently, Stranger 2 was introduced to the other chamber. Again, test mice spent another 10 min exploring the entire apparatus. The total time spent in each chamber was recorded and the ratio of (S2 − S1) to (S2 + S1) was measured as the preference index (S2 − S1).
Statistics.
Data analyses were performed using Excel 2003 (Microsoft), Clampfit 10, and Igor Pro 6.0 (Wavemetrics) software. Statistical differences were determined using the unpaired two-sided Student's t test for two group comparisons or a one-way ANOVA test followed by Tukey's post hoc test for multiple comparisons. The accepted level of significance was p < 0.05. Error bars in the text and figures are presented as mean ± SEM. n represents the number of cells, cultures, or animals tested.
Results
TFR1 deficiency in PCs does not affect cytoarchitecture in the cerebellum
We first observed the expression of TFR1 in PCs in the developing cerebellum. Imunostaining showed that TFR1 was increasingly expressed in mouse PCs at young ages (P2, P7, and P14; Fig. 2A), which was confirmed by TFR1 bands after RT-PCR of mRNA extracted from PCs at P7 and P14 (Fig. 2B). We next examined the presence of TFR1 at parallel fiber–PC synapses using a synaptosome assay from TFR1flox/flox mice (P21). Our results demonstrated that the signal of vGluT1, a putative marker of parallel fiber terminal, overlapped with TFR1 (Fig. 2C). EAAT4, which is recognized as the marker of PC spines (Dehnes et al., 1998), also colocalized with TFR1 (Fig. 2D). These data indicate that TFR1 is expressed at both presynaptic and postsynaptic sites of parallel fiber–PC synapses.
Figure 2.

TFR1 is expressed at parallel fiber–PC synapses. A, Immunohistochemical staining for calbindin (calb, red) and TFR1 (green) in PCs from P2, P7, and P14 control mice. Scale bars, 20 μm. B, Electrophoresis of TFR1, calbindin, and GAPDH amplicons in individual PCs from control mice at P7 (n = 5) and P14 (n = 6). C, Synaptosomes were stained with antibodies against vGluT1 and TFR1 (white arrows). Scale bar, 5 μm. D, Synaptosomes were stained with antibodies against EAAT4 and TFR1 (white arrows). Scale bar, 5 μm.
To assess the roles of TFR1 in PCs, we generated conditional knock-out mice (TFR1flox/flox;pCP2-cre) that ablates TFR1 specifically in PCs. The efficiency of deletion was confirmed by lack of TFR1 mRNA (Fig. 3A) and TFR1 immunostaining in TFR1flox/flox;pCP2-cre PCs (Fig. 3B). The body weight and size of the cerebellum of TFR1flox/flox;pCP2-cre mice appeared normal at P21 (Fig. 3C). In terms of cerebellar cyto-architecture, PC-specific TFR1 deletion did not interfere with the formation or thickness of lobules (Fig. 3D). The average lobule V–VI thickness was 715 ± 55 μm in TFR1flox/flox mice (n = 7) and 747 ± 67 μm in TFR1flox/flox;pCP2-cre mice (n = 7; p = 0.35). In addition, dendritic branching and spine density of PCs were kept intact in TFR1flox/flox;pCP2-cre mice (Fig. 3E), as shown by the immunostaining for calbindin and EAAT4. Average dendritic tree area was 5.3 ± 0.3 × 103 μm2 (TFR1flox/flox; n = 14) and 5.2 ± 0.2 × 103 μm2 (TFR1flox/flox;pCP2-cre; n = 14; p = 0.54). Total dendritic length was 664 ± 96 μm (TFR1flox/flox; n = 14) or 679 ± 101 μm (TFR1flox/flox;pCP2-cre; n = 14; p = 0.28). The number of spines over 10 μm dendritic fragment was 19 ± 1.5 (TFR1flox/flox; n = 12) or 19 ± 1.3 (TFR1flox/flox;pCP2-cre; n = 12; p = 0.72). Unlike the result from TFR1flox/flox;nestin-cre mice (Liu et al., 2016), the present findings indicate that PC deletion of TFR1 does not affect the morphology of neuronal dendrites.
Figure 3.

PC morphogenesis is normal in TFR1flox/flox;pCP2-cre mice. A, Electrophoresis of TFR1 (154 bp), calbindin (184 bp), and GAPDH (220 bp) amplicons from individual TFR1flox/flox (control, n = 10) and TFR1flox/flox;pCP2-cre (cKO, n = 10) PCs. B, Immunohistochemical staining for calbindin (red) and TFR1 (green) in the cerebellum from control and cKO mice. Arrowheads show TFR1 signal was absent in PCs of cKO mice. ML, Molecular layer; PCL, Purkinje cell layer; GCL, granule cell layer. Scale bars, 20 μm. C, cKO mice (P21) displayed normal body weight and brain size. Average body weights were 17.3 ± 1.7 g (control) and 16.5 ± 1.5 g (cKO; n = 14 pairs; p = 0.38). n.s., Not significant. D, Nissl staining in the cerebellum from control and cKO mice (P21). Scale bars, 100 μm. E, Immunostaining for calbindin (calb, red) and EAAT4 (green) showing that dendrites and spine formation were normal in cKO mice. Scale bars, 10 μm.
mGlu1 expression is reduced at PC synapses in TFR1flox/flox;pCP2-cre mice
Synaptic expression of ionotropic glutamate receptor subunits is downregulated in hippocampal CA1 neurons of TFR1flox/flox;nestin-cre mice (Liu et al., 2016). We therefore examined the expression of the AMPAR subunit GluA2 in the cerebellum in relation to TFR1 and calbindin. TFR1 expression was significantly reduced in both total and PSD fractions of TFR1flox/flox;pCP2-cre mice compared with TFR1flox/flox mice (P21; Fig. 4A). The remaining TFR1 in the molecular layer was probably from granule cells, which also expressed TFR1 (Fig. 3B). The amount of calbindin in TFR1flox/flox;pCP2-cre mice was similar to that in TFR1flox/flox mice (p = 0.69; Fig. 4A), again indicating that PC development is normal in TFR1flox/flox;pCP2-cre mice. GluA2 expression at synapses was unaltered in TFR1flox/flox;pCP2-cre mice (p = 0.37; Fig. 4A). Likewise, the frequency and amplitude of AMPA mEPSCs were unchanged (frequency: p = 0.56; amplitude: p = 0.67; Fig. 4B). Consistent with these observations, evoked climbing fiber transmissions were unaltered in TFR1flox/flox;pCP2-cre mice (p = 0.25; Fig. 4C). Climbing fiber–EPSCs were elicited in an all-or-none fashion in the vast majority of PCs in both TFR1flox/flox and TFR1flox/flox;pCP2-cre mice (Fig. 4C), suggesting that the elimination of multiple climbing fiber innervation develops normally.
Figure 4.
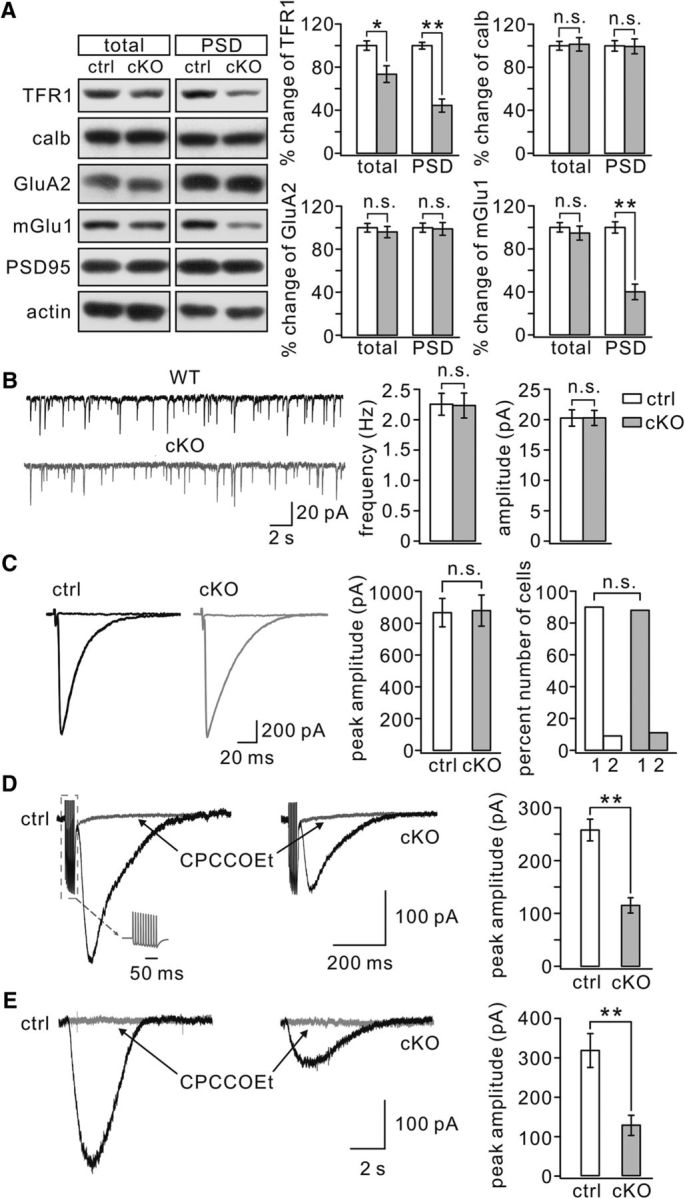
Synaptic mGlu1 is reduced in TFR1flox/flox;pCP2-cre mice. A, Cerebellar (total) and PSD fractions from control and cKO mice were probed with antibodies to TFR1, calbindin (calb), GluA2, and mGlu1. GAPDH and PSD95 were internal controls for total and PSD, respectively. Histograms show percentage changes of proteins in cKO mice relative to control (n = 4 pairs). TFR1: 73 ± 7% (total; p = 0.014) and 44 ± 6% (PSD; p = 0.0067). calb: 101 ± 6% (total; p = 0.69) and 99 ± 7% (PSD; p = 0.45). GluA2: 96 ± 5% (total; p = 0.44) and 99 ± 6% (PSD; p = 0.37). mGlu1: 90 ± 6% (total; p = 0.31) and 40 ± 7% (PSD; p = 0.0076). B, mEPSCs recorded from control (n = 10) and cKO (n = 10) PCs. Averages of frequency were 2.3 ± 0.2 Hz (control) and 2.2 ± 0.2 Hz (cKO; p = 0.56). Averages of amplitude were 21 ± 1.3 pA (control) and 21 ± 1.2 pA (cKO; p = 0.67). C, Sample recordings of climbing fiber–EPSCs from control and cKO PCs. Three traces evoked by different intensities are superimposed. The peak averages of climbing fiber–EPSCs were 867 ± 89 pA (control, n = 23) and 880 ± 98 pA (cKO, n = 25; p = 0.25). Right, Percentages of discrete steps (1 or 2) of climbing fiber–EPSCs. For control: 90% (1) and 9% (2), n = 23. For cKO: 88% (1) and 11% (2), n = 25. D, Example mGlu1-EPSCs in response to burst stimulations (gray inset) in control and cKO cells. mGlu1-EPSCs were blocked by its antagonist CPCCOEt (100 μm). E, Slow currents were evoked by a pulse (10 psi, 20 ms) of aCSF containing DHPG (100 μm) and blocked by mGlu1 antagonist CPCCOEt (100 μm). *p < 0.05. **p < 0.01. n.s., Not significant.
Interestingly, we found that synaptic mGlu1 was reduced significantly in TFR1flox/flox;pCP2-cre mice (40 ± 7% of TFR1flox/flox), whereas its total expression was not affected (90 ± 6% of TFR1flox/flox; Fig. 4A). Because mGlu1 is normally highly expressed in PCs (Ichise et al., 2000), this result suggests that PC ablation of TFR1 may reduce mGlu1 expression at PC synapses. Therefore, we recorded mGlu1 currents in PCs using ex vivo whole-cell recordings. After burst stimulation (100 Hz) of parallel fibers under a holding voltage of −70 mV in aCSF in the presence of 5 μm NBQX, PCs displayed AMPAR-mediated EPSCs and a slow mGlu1-EPSC (Fig. 4D; Kim et al., 2008; Zhou et al., 2015). We found that the peak amplitude of mGlu1-EPSC was significantly reduced in TFR1flox/flox;pCP2-cre mice (115 ± 14 pA; n = 21) compared with the current recorded in TFR1flox/flox mice (257 ± 21 pA; n = 20; p = 0.0071; Fig. 4D). Furthermore, mGlu1 currents were evoked in PCs by locally applying a brief pulse of aCSF containing its agonist, DHPG (100 μm) (Kim et al., 2008; Zhou et al., 2015). Similar to the results obtained with electric stimulation, DHPG-induced currents were markedly reduced in TFR1flox/flox;pCP2-cre mice (127 ± 25 pA; n = 15) compared with the current recorded in TFR1flox/flox mice (315 ± 43 pA; n = 15; p = 0.0083; Fig. 4E). Together, these results indicate that PC ablation of TFR1 affects mainly the expression and function of mGlu1 at PC synapses.
mGlu1 activation triggers an endocannabinoid production in PCs (Maejima et al., 2001; Varma et al., 2001), which suppresses synaptic transmission (Maejima et al., 2001; Crepel and Daniel, 2007). Therefore, we investigated whether endocannabinoid-mediated suppression of synaptic transmission is affected by TFR1 ablation. In TFR1flox/flox mice (P8–P12), applying DHPG (100 μm) to the bath for 5 min induced a significant decrease in the mean amplitude of parallel fiber–EPSCs to 48 ± 6% (n = 10) of basal (p = 0.0045; Fig. 5A), which was accompanied by an increase in paired-pulse facilitation (PPF) from 1.69 ± 0.05 to 2.25 ± 0.07 at the peak of DHPG effect (n = 10; p = 0.0079; Fig. 5A). Similar results were obtained in TFR1flox/flox;pCP2-cre mice (P8–P12), in which DHPG also induced a decrease (49 ± 4%; n = 10; p = 0.0037) compared with basal in the amplitude and an increase from 1.68 ± 0.06 to 2.17 ± 0.07 (n = 10; p = 0.0037) in PPF (Fig. 5A). Furthermore, the application of DHPG (100 μm) caused a depression of climbing fiber–EPSCs and an increase in PPD in both TFR1flox/flox and TFR1flox/flox;pCP2-cre mice (P8–P12; Fig. 5B). These results suggest that encocannabinoid-dependent suppression of PC neurotransmission is intact in TFR1flox/flox;pCP2-cre mice.
Figure 5.

Encocannabinoid-dependent suppression of PC synaptic transmission is unchanged by TFR1 deletion. A, Superimposed parallel fiber–EPSCs by two successive parallel fiber stimulations in the absence (basal) and presence of DHPG in control and cKO mice. The bar graph shows the percentage changes of first EPSC amplitudes relative to basal values. The scatterd points show changes in PPD. B, Examples of climbing fiber–EPSCs in response to paired stimuli in control and cKO mice. Records obtained before (basal) and during the presence of DHPG are superimposed. The bar graph shows percentage changes of first EPSC amplitudes relative to basal values. Control: 58 ± 8% (n = 10). cKO: 60 ± 5% (n = 10). p = 0.34. The right panel shows changes in PPD. Control: 0.57 ± 0.04 (basal) and 0.76 ± 0.07 (DHPG), n = 10. cKO: 0.59 ± 0.05 (basal) and 0.80 ± 0.08 (DHPG), n = 10. p = 0.52. n.s., Not significant.
The GABAergic transmission from interneurons to PCs was measured in TFR1flox/flox and TFR1flox/flox;pCP2-cre mice (P21). We found that neither the frequency nor the amplitude of mIPSCs was changed (Fig. 6A). eIPSCs were also recorded from PCs according to previous literature (Ikeda et al., 2013). As we increased the stimulation intensity, eIPSC amplitudes recorded from TFR1flox/flox and TFR1flox/flox;pCP2-cre mice increased almost linearly and reached a similar plateau level (Fig. 6B). We found that mean eIPSC amplitudes at a series of intensities were not different between TFR1flox/flox and TFR1flox/flox;pCP2-cre mice (Fig. 6B). These recordings suggest that GABAergic transmission onto PCs is not affected by TFR1 deletion. This conclusion was corroborated by the immunohistochemical analysis using the presynaptic and postsynaptic markers vGAT and gephyrin, respectively. Our results indicated that the distribution and intensity of GABAergic synapses are quite similar among TFR1flox/flox and TFR1flox/flox;pCP2-cre mice (Fig. 6C).
Figure 6.
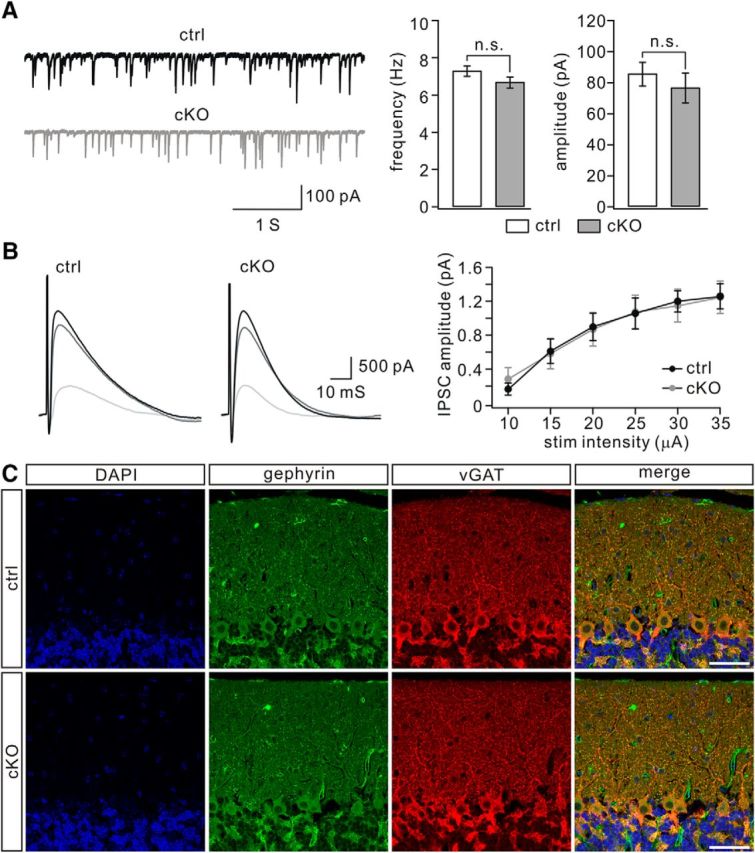
GABAergic synaptic transmission onto PCs is not changed by TFR1 deletion. A, mIPSCs recorded from control (n = 9) and cKO (n = 9) PCs. Averages of frequency were 7.3 ± 0.3 Hz (control) and 6.7 ± 0.3 Hz (cKO; p = 0. 26). Averages of amplitude were 85 ± 8 pA (control) and 76 ± 10 pA (cKO; p = 0.18). n.s., Not significant. B, Superimposed eIPSCs were recorded from a single PC of control and cKO mice in response to stimulations at 15, 25, and 35 μA. Each trace is derived from averaging IPSCs of three successive traces recorded every 20 s. The right panel shows the relationship between eIPSC amplitude and a series of stimulation intensity for control (n = 12) and cKO (n = 11) mice. C, Immunofluorescence using antibodies to gephyrin and vGAT in control and cKO mice (P21). Nuclei were stained with DAPI (blue). Both gephyrin and vGAT showed a dotted appearance in the molecular layer and were mostly overlapped, suggesting the putative GABAergic synapses. The density of double-labeled puncta was 5.4 ± 0.2/100 μm2 in TFR1flox/flox mice (n = 6) and 5.2 ± 0.2/100 μm2 in TFR1flox/flox;pCP2-cre mice (n = 6). Scale bars, 50 μm.
TFR1 affects the trafficking of mGlu1
mGlu1 is internalized and recycled back to the cell membrane upon ligand exposure (Dhami and Ferguson, 2006; Francesconi et al., 2009; Choi et al., 2011). Therefore, the intracellular trafficking of mGlu1 was examined to find out how synaptic mGlu1 can be reduced in mutant mice. DHPG (100 μm) was applied to cerebellar slices for 10 min and the level of synaptic mGlu1 was measured 30–40 min (for endocytosis) or 180–190 min (for recycling) later (Pandey et al., 2014; Zhou et al., 2015). In control slices, synaptic mGlu1 decreased to 44 ± 4% of basal level at t = 40 min (n = 4) and returned to 80 ± 6% at t = 190 min (n = 4; Fig. 7A), consistent with previous work (Zhou et al., 2015). In TFR1flox/flox;pCP2-cre slices, synaptic mGlu1 decreased to 34 ± 5% of basal level at t = 40 min (n = 4) and returned to 36 ± 7% at t = 190 min (n = 4; Fig. 7A), suggesting that TFR1 deletion exaggerates the internalization and inhibits the recycling of mGlu1. To further investigate the effect of TFR1 on mGlu1 expression, myc-mGlu1α was transfected to HEK293 cells alone or with GFP-TFR1 (Fig. 7B). Both myc-mGlu1 and GFP-TFR1 were detected in the Triton X-100-insoluble and -soluble fractions (Fig. 7C). The action of TFR1 on the membrane expression of mGlu1α was examined by quantifying the mGlu1 ratio of insoluble/soluble fractions (Ohtsuka et al., 2002). Coexpression of TFR1 significantly increased mGlu1α expression in the Triton X-100-insoluble fraction compared with that in absence of TFR1 (p = 0.0073; Fig. 7C). These results support the idea that TFR1 promotes the presence of mGlu1α on the membrane.
Figure 7.

mGlu1 trafficking is inhibited in TFR1flox/flox;pCP2-cre mice. A, mGlu1 in total and PSD fractions was immunoblotted 30 min or 180 min after DHPG challenge. β-actin was the control. B, HEK293 cells were transfected with myc-mGlu1α and GFP-TFR1. mGlu1α and TFR1 were visualized by fluorescent antibody and GFP signals, respectively. C, Histogram showing the ratios of the Triton X-100-insoluble (I) and Triton X-100-soluble (S) fraction in HEK293 cells. TFR1, 35 ± 11% (mGlu1) and 115 ± 23% (mGlu1+TFR1), p = 0.0023. mGlu1, 134 ± 17% (mGlu1) and 273 ± 26% (mGlu1+TFR1), p = 0.0073. Rab8, 7 ± 3% (mGlu1) and 31 ± 10% (mGlu1+TFR1), p = 0.0047. Rab11, 5 ± 3% (mGlu1) and 15 ± 4% (mGlu1+TFR1), p = 0.0051. The experiment was repeated for four times. D, Precleared brain lysates from WT mice were immunoprecipitated with rat anti-TFR1 antibody and immunoprecipitates were probed with antibodies to Rab11, Rab8, mGlu1, and TFR1. The experiment was performed three times. Rat IgG was used as the negative control. E, Expression of Rab proteins in PSD fraction was decreased in cKO mice. β-actin and PSD-95 were used as loading controls of total and PSD fractions, respectively. Percentage changes of Rab proteins in PSD fraction from cKO mice were 47 ± 6% (Rab8; p = 0.0075) and 35 ± 5% (Rab11; p = 0.0068). n = 4 pairs (P21). F, Working model showing that TFR1 facilitates the trafficking of mGlu1 from the intracellular reserve pool to the membrane by binding to Rab11 and decreases the endocytosis of mGlu1 by recruiting Rab8. *p < 0.05, **p < 0.01. n.s., Not significant.
In vitro studies have shown that Rab8 antagonizes mGlu1 internalization (Esseltine et al., 2012) and that Rab11 participates in mGlu1 recycling (Pandey et al., 2014). To determine whether TFR1 interacts with Rab proteins, we set up some co-IP experiments, which showed that both Rab8 and Rab11 were robustly precipitated by TFR1 in vivo (Fig. 7D), indicating the ability of TFR1 to interact with Rab8 and Rab11. Unexpectedly, TFR1 did not bind directly to mGlu1 (Fig. 7D). Rab8 and Rab11 require secure association with the membrane to execute their functions (Li et al., 2009; Esseltine et al., 2012). Accordingly, the levels of Rab8 and Rab11 at PC synapses were measured in TFR1flox/flox and TFR1flox/flox;pCP2-cre mice. We found that both Rab8 and Rab11 were significantly attenuated in PSD fraction of TFR1flox/flox;pCP2-cre mice compared with that in TFR1flox/flox mice (Rab8: 47 ± 6%, p = 0.0075; Rab11: 35 ± 5%, p = 0.0068), whereas their total expressions were not altered (Rab8, p = 0.87; Rab11, p = 0.54; Fig. 7E), indicating that Rab8 and Rab11 are tethered in the cytoplasm due to TFR1 deficiency. Interestingly, we also found that TFR1 increased the expression of Rab8 and Rab11 significantly in the Triton X-100-insoluble fraction in HEK293 cells (Fig. 7C). Together, these results suggest that TFR1 contributes to the regulation of mGlu1 trafficking at least in part by regulating the actions of Rab8 and Rab11. The presence of TFR1 enhances the synaptic expression of Rab proteins and thereby facilitates surface expression of mGlu1; instead, the absence of TFR1 may keep Rab8 and Rab11 tethered in the cytoplasm of the dendrites, thus increasing the internalization of mGlu1 and impeding the recycling of endosomes (Fig. 7F).
Deletion of TFR1 impairs parallel fiber–LTD, but not parallel fiber–LTP
mGlu1 is essential for the induction of parallel fiber–LTD (Ichise et al., 2000; Brasnjo and Otis, 2001). Given that mGlu1 at PC synapses was reduced in TFR1flox/flox;pCP2-cre mice, LTD was examined at parallel fiber–PC synapses in voltage-clamp mode (−70 mV) at RT. As shown by example responses to parallel fiber stimulation, TFR1flox/flox PCs showed robust LTD at the parallel fiber to PC synapse (t = 38 min: 59 ± 6% of baseline; n = 15; Fig. 8A,B) in response to repetitive parallel fiber stimulation at 100 Hz for 100 ms paired with PC depolarization. In contrast, LTD induction was strongly inhibited in TFR1flox/flox;pCP2-cre PCs (t = 38 min: 94 ± 4% of baseline; n = 14; p = 0.0045; Fig. 8A,B). The ratio of PPF, as measured with an interstimulus interval of 80 ms, was not changed in the TFR1flox/flox;pCP2-cre mice (p > 0.94 at t = 38 min; Fig. 8C). We also examined LTP at parallel fiber synapses in TFR1flox/flox and TFR1flox/flox;pCP2-cre mice using a 1 Hz tetanus protocol (Wang et al., 2014a; Zhou et al., 2015). After acquiring stable EPSCs in voltage-clamp mode (−70 mV), a tetanus stimulation (1 Hz for 5 min) was delivered to parallel fibers (Fig. 8D). The potentiation of EPSCs was 130 ± 5% of baseline in TFR1flox/flox mice (t = 38 min; n = 11) and 129 ± 6% (t = 38 min; n = 12) in TFR1flox/flox;pCP2-cre mice (Fig. 8E), suggesting that TFR1 is not associated with parallel fiber–LTP (p = 0.87 at t = 38 min). Here too, the PPF ratio was unaffected (p = 0.90 at t = 38 min; Fig. 8F).
Figure 8.
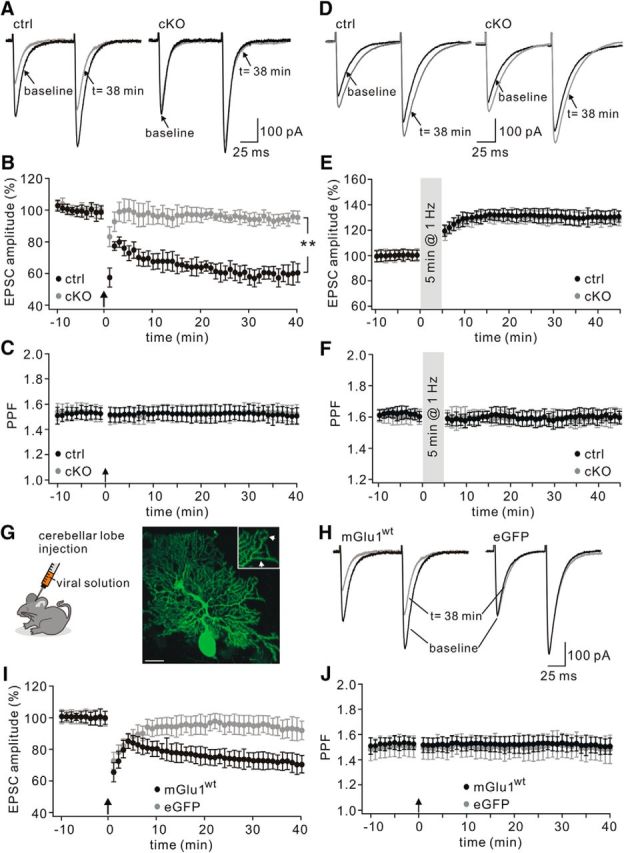
LTD is deficient whereas LTP is normal in TFR1flox/flox;pCP2-cre mice. A, Example parallel fiber–EPSCs before (baseline) and after (t = 38 min) LTD stimulation in control and cKO PCs. B, Time course of percentage changes of EPSC1 amplitudes in control and cKO animals. Each data point represents the average of three successive EPSCs evoked at 0.05 Hz. The upward arrow shows LTD tetnus. **p < 0.01. C, Time course of PPF (EPSC2/EPSC1) from the cells shown in B. D, Example EPSCs before (baseline) and after (t = 38 min) LTP stimulation. E, Time course of percentage changes of EPSC amplitude in control and cKO mice. Each data point represents the average of three successive EPSCs evoked at 0.05 Hz. F, Time courses of PPF ratios from the subset of cells shown in E. G, Schema showing SFV injection into the vermis cerebellum. Eighteen hours after injection, eGFP signals were observed in scattered PCs (right). The amplification shows the affected spines (arrowheads) in distal dendrites. Scale bar, 20 μm. H, Representative traces of EPSCs recorded from cKO PCs expressing eGFP or eGFP+mGlu1 (mGlu1) before (baseline) and after (t = 38 min) LTD induction. I, Time course of percentage changes of EPSC amplitudes in cKO PCs transduced by eGFP or mGlu1. J, Time course of PPFs shown in I.
We continued to investigate whether parallel fiber–LTD could be restored in TFR1flox/flox;pCP2-cre PCs when they were compensated for a lack of functional mGlu1. TFR1flox/flox;pCP2-cre PCs in lobule V were transfected with SFV-packed eGFP-mGlu1 or eGFP (Fig. 8G) and subsequently subjected to LTD induction. We found that parallel fiber–LTD could be induced successfully in PCs expressing mGlu1-eGFP (t = 38 min: 71 ± 6% of baseline; n = 14), but not in those cells only expressing eGFP (t = 38 min: 93 ± 7% of baseline; n = 14; Fig. 8H,I). PPF ratios were not changed in either condition (Fig. 8J). Therefore, we conclude that the abrogation of parallel fiber–LTD is due to the reduction of mGlu1 in TFR1flox/flox;pCP2-cre PCs.
Deletion of TFR1 has no effect on mGlu1 plasticity
mGlu1-EPSCs exhibit a persistent depression (mGlu1-LTD) after climbing fiber stimulation-like depolarization (Kim et al., 2008; Zhou et al., 2015). To address whether TFR1 might be involved in mGlu1-LTD, we recorded mGlu1-EPSCs for a stable period and applied a 5 s step depolarization from −70 to 0 mV to clamped PCs (Fig. 9A). In TFR1flox/flox PCs, mGlu1-EPSCs decreased immediately and showed a strong depression 30 min later (25 ± 6% of baseline; n = 12; Fig. 9B). Likewise, the amplitude of mGlu1-EPSC was reduced after depolarization in TFR1flox/flox;pCP2-cre PCs (t = 30 min: 30 ± 6% of baseline; n = 12; Fig. 9B), showing no difference between controls and mutants (p = 0.83). These data suggest that TFR1 is not involved in mGlu1-LTD. We further investigated whether TFR1 is involved in a transient upregulation of mGlu1 currents (mGlu1-STP), which can be induced by brief depolarizing currents on PCs (Batchelor and Garthwaite, 1997; Kim et al., 2008; Zhou et al., 2015). After recording a test mGlu1-EPSC, somatic depolarization to 0 mV was delivered for 100 ms to the recorded PC and a second mGlu1-EPSC was evoked at different intervals (Fig. 9C). The conditioning depolarization produced a potentiation of mGlu1 current, but had no effect on AMPAR-EPSCs (Fig. 9C). We found that mGlu1-STP was robustly induced in both control and TFR1-cKO PCs at similar degrees across a range of intervals (p > 0.05; Fig. 9C), indicating that TFR1 is not involved in mGlu1-STP. These results suggest that stimulation protocols for mGlu1-STP and mGlu1-LTD do not require dynamic transport of mGlu1 and that mGlu1 plasticity may be triggered by altered activity of mGlu1-associated proteins.
Figure 9.

LTD and STP of mGlu1 are unaltered in TFR1flox/flox;pCP2-cre mice. A, Example mGlu1-EPSCs before and after mGlu1-LTD induction in control and cKO cells. Current amplitudes were measured at the peak of slow EPSCs. B, Time course of percentage changes in mGlu1-EPSC amplitudes from control (n = 12) and cKO (n = 12) cells induced by 5 s depolarization at the time indicated by the arrow. C, Representative mGlu1 traces showing four different trials before (pre) and 10, 30, 60, and 180 s after a transient depolarization from one cell. Right, Time course of percentage changes of mGlu1-EPSCs in control (n = 10) and cKO (n = 9) cells.
TFR1 ablation does not change intrinsic excitability in PCs
The intrinsic properties of PCs govern the functions of cerebellar circuits and have a major impact on its simple spike output (Zhou et al., 2014). We therefore examined the intrinsic excitability of PCs by measuring their simple spike (here referred to as APs) evoked by depolarizing current steps with increasing amplitude (Fig. 10A). Evoked APs showed comparable thresholds (TFR1flox/flox: 52 ± 3 mV; TFR1flox/flox;pCP2-cre: 51 ± 3 mV), amplitudes (TFR1flox/flox: 41 ± 6 mV; TFR1flox/flox;pCP2-cre: 40 ± 6 mV), half-widths (TFR1flox/flox: 0.3 ± 0.03 ms; TFR1flox/flox;pCP2-cre: 0.3 ± 0.02 ms), and AHP amplitudes (TFR1flox/flox: 8 ± 2 mV; TFR1flox/flox;pCP2-cre: 7 ± 2 mV) in TFR1flox/flox PCs (n = 22) and TFR1flox/flox;pCP2-cre PCs (n = 23; Fig. 10B–E). Moreover, PC intrinsic excitability was normal, as shown by PC responses to 400 pA current injection in TFR1flox/flox and TFR1flox/flox;pCP2-cre mice (Fig. 10F). Current step injections of increasing amplitude resulted in a linear current-to-firing frequency relationship with similar (p = 0.48) slopes between TFR1flox/flox (16 ± 1; n = 11) and TFR1flox/flox;pCP2-cre (18 ± 1; n = 11) mice (Fig. 10G). Together, these findings indicate that PC intrinsic excitability remains intact in TFR1flox/flox;pCP2-cre mice.
Figure 10.

Normal intrinsic plasticity in TFR1flox/flox;pCP2-cre mice. A, Recording configuration for whole-cell recording. Inset, Example AP. B, AP threshold. C, AP amplitude. D, AP half-width. E, AP AHP. F, Example traces of intrinsic PC excitability as apparent from AP firing evoked by 400 pA current injections. G, No difference in evoked firing frequency relative to various levels of current injections. Inset barplot, Average slope of firing rate per current step. H, Example of traces for intrinsic plasticity with current injections of 400 pA. I, LTP induction protocol induced enhanced spike output in both control and cKO PCs. n.s., Not significant.
Because LTP was reported to facilitate adaptation of intrinsic properties and to drive spike activity (Schonewille et al., 2010; Peter et al., 2016), we next examined PC intrinsic plasticity before and after parallel fiber–LTP induction (Fig. 10H). Our results indicated that both TFR1flox/flox and TFR1flox/flox;pCP2-cre mice readily demonstrated a potentiation of intrinsic excitability (TFR1flox/flox: 130 ± 5%, n = 11; TFR1flox/flox;pCP2-cre: 131 ± 6%, n = 12; p = 0.62) 40 min after induction (Fig. 10I). These results suggest that TFR1 is not a modulator for intrinsic plasticity in PCs.
PC spontaneous spikes are unaltered in TFR1flox/flox;pCP2-cre mice
Although whole-cell patch-clamp recordings from PCs may disrupt physiological PC responses, cell-attached recordings provide a less invasive technique for measuring PC excitability (Perkins, 2006; Belmeguenai et al., 2010). Here, we recorded spontaneous spikes of PCs in cerebellar slices from TFR1flox/flox and TFR1flox/flox;pCP2-cre mice with cell-attached recordings to determine PC excitability when TFR1 is deleted. The recordings of spontaneous spikes were made in anterior (I–III) and posterior (IX–X) lobules because they are dominated by different expression patterns of proteins involved in intrinsic excitability (Zhou et al., 2014) and because abnormalities in different lobules may have different clinical implications (Mosconi et al., 2015; Peter et al., 2016). In both the anterior and posterior lobules, the firing frequency of TFR1flox/flox;pCP2-cre PCs was not significantly different from that of controls. The averages of firing frequency were 72 ± 15 (TFR1flox/flox, n = 15) and 80 ± 20 (TFR1flox/flox;pCP2-cre, n = 18; p = 0.35) in anterior lobules (Fig. 11A) and 52 ± 14 (TFR1flox/flox, n = 14) and 58 ± 14 (TFR1flox/flox;pCP2-cre, n = 20; p = 0.27) in posterior lobules (Fig. 11B).
Figure 11.

Normal spontaneous spike firing in TFR1flox/flox;pCP2-cre mice. A, Example spontaneous spikes of PCs recorded from anterior lobules (I–III) in control and cKO mice. B, Example spontaneous spikes of PCs recorded from posterior lobules (IX–X) in control and cKO mice. C, A 100 Hz parallel fiber tetanization caused increased spontaneous spike firing in cell attached recordings in control and cKO PCs. For these recordings, only cells that showed regular simple spike firing were used. Arrows indicate the time point of tetanization. n.s., Not significant.
The protocol for parallel fiber–LTP can trigger intrinsic plasticity of PCs (Belmeguenai et al., 2010; Peter et al., 2016). To further characterize the intrinsic plasticity of TFR1flox/flox;pCP2-cre PCs, we delivered a 100 Hz burst protocol to parallel fibers in cerebellar slices and recorded spontaneous spikes of PCs in lobules IX–X before and after the tetanus (Fig. 11C). The application of parallel fiber bursts resulted in comparable increases in the spontaneous spike rate of both TFR1flox/flox PCs (123 ± 5% of baseline at t = 40 min; n = 7) and TFR1flox/flox;pCP2-cre mice (124 ± 5% at t = 40 min; n = 8; p = 0.52; Fig. 11D). Together, these data indicate that ablation of TFR1 does not affect the excitability of PCs.
TFR1 ablation in PCs impairs motor coordination, but not social behaviors
We have demonstrated previously that downregulation of mGlu1 and lack of parallel fiber–LTD by protein Numb deletion can be associated with impairment in motor coordination (Zhou et al., 2015). Because synaptic mGlu1 was also reduced in TFR1flox/flox;pCP2-cre mice, we predicted that TFR1 deficiency in PCs might impair motor coordination. Although TFR1flox/flox;pCP2-cre mice did not show overt ataxia in standard cages, they indeed performed poorly when walking on a narrow elevated beam, with a remarkably higher number of hindpaw slips (Fig. 12A). They also exhibited impaired motor learning in that they showed limited improvement on the accelerating rotarod compared with controls after six sessions (Fig. 12B). These results indicate that TFR1 deficiency in PCs impairs motor coordination.
Figure 12.
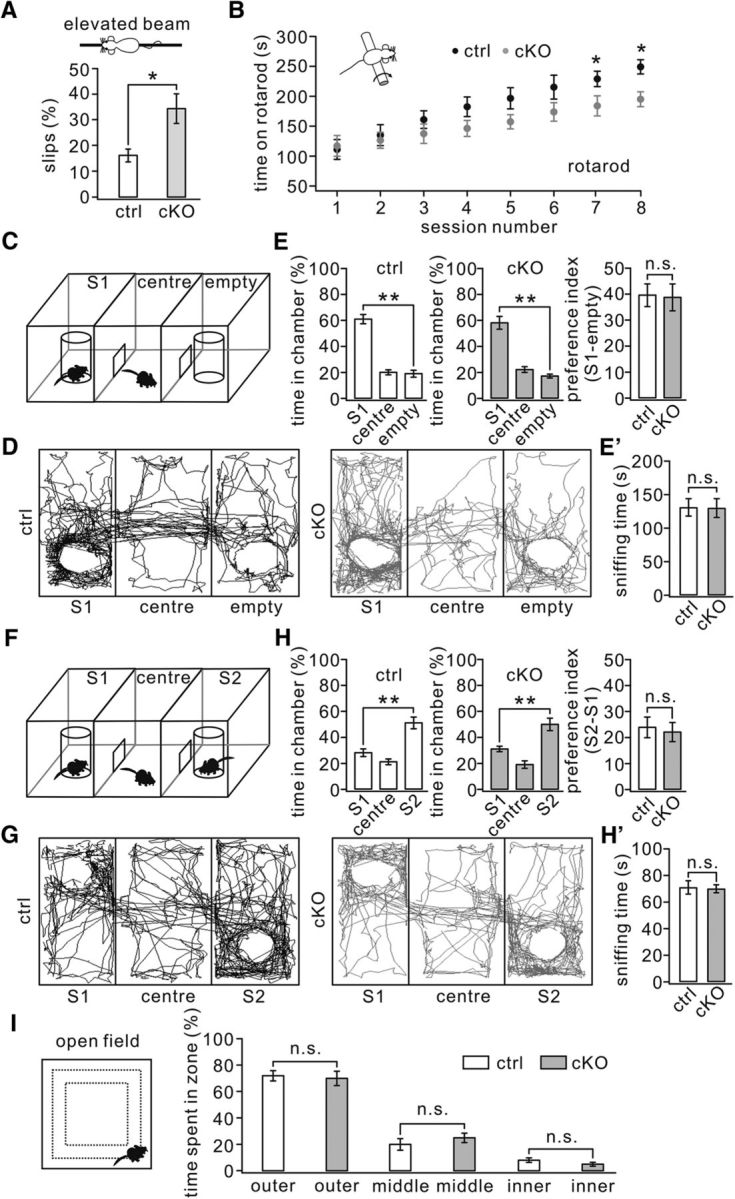
Impaired motor learning but normal social interactions were seen in TFR1flox/flox;pCP2-cre mice. A, Percentage of steps with hindpaw slips during runs on an elevated horizontal beam. TFR1flox/flox: 16 ± 2%. TFR1flox/flox;pCP2-cre: 34 ± 6%. n = 10 pairs; p = 0.023. B, Time spent on the accelerating rotarod for control and cKO mice. TFR1flox/flox: 229 ± 13 s; TFR1flox/flox;pCP2-cre: 184 ± 17 s for session 7 (p = 0.019); TFR1flox/flox: 249 ± 12 s; TFR1flox/flox;pCP2-cre: 195 ± 12 s for session 8. n = 10 pairs. p = 0.014. C, Configuration of a three-chamber social interaction evaluated by relative time spent in each chamber. D, Example movement traces of a control mouse and a cKO mouse. E, Summary of spent time in S1, center, and empty chambers of control (n = 12) and cKO (n = 11) mice showing that both genotypes preferred to spend time in the room with S1 compared with the empty room. E′, Summary of sniffing time onto S1 of control and cKO mice. F, Configuration of a three-chamber after the introduction of S2. G, Example movement traces of a control mouse and a cKO mouse mice in the three-chamber with S1 and S2. H, Summary of spent time in S1, center, and S2 chambers of control (n = 12) and cKO (n = 11) mice showing that both genotypes preferred to spend time in the room with S2 compared with S1. H′, Summary of sniffing time onto S2 of control and cKO mice. I, In the open-field test, control (n = 17) and cKO mice (n = 15) showed no differences in time spent in the inner (control: 8 ± 4%; cKO: 5 ± 1%), middle (control: 20 ± 4%; cKO: 25 ± 3%), or outer (control: 72 ± %; cKO: 70 ± 5%) zones. *p < 0.05, **p < 0.01. n.s., Not significant.
Increasing evidence has highlighted the importance of PC synaptic function for autism beyond classical motor-related behavior. Global or PC-specific deletion of syndromic or nonsyndromic autism-related genes such as Fmr1 (Koekkoek et al., 2005), Nlgn3 (Baudouin et al., 2012; Zhang et al., 2015), TSC1 (Tsai et al., 2012), and Shank2 (Peter et al., 2016) results in autistic-like behaviors. In all of these genetic mouse models, some deficits in PC physiology, such as parallel fiber–LTP, mGlu1-dependent LTD, and intrinsic excitability, have been found to possibly explain the formation of autistic-like behaviors (Koekkoek et al., 2005; Baudouin et al., 2012; Tsai et al., 2012; Piochon et al., 2014; Peter et al., 2016). Given that mGlu1 was downregulated (Fig. 4) and that induction of parallel fiber–LTD was affected (Fig. 8) in TFR1flox/flox;pCP2-cre mice, we investigated autism-related behaviors of these mutant mice using the three-chamber social interaction task, a widely used social interaction paradigm to evaluate autistic-like behavior in mouse models of autism (Schmeisser et al., 2012; Won et al., 2012). TFR1flox/flox mice exhibited a normal preference for the chamber in which a stranger mouse (S1) was present compared with the empty chamber (Fig. 12C,D). Similarly, TFR1flox/flox;pCP2-cre mice also displayed a preference for S1 (Fig. 12C,D). The time of sniffing onto the S1 cage was similar among TFR1flox/flox (131 ± 13 s; n = 13) and TFR1flox/flox;pCP2-cre (130 ± 14 s; n = 10) groups (Fig. 12E′; p = 0.22). Comparing the preference index (stranger empty) revealed no difference (p = 0.47) for the stranger mouse between TFR1flox/flox mice (40 ± 4; n = 12) and TFR1flox/flox;pCP2-cre mice (39 ± 5; n = 11; Fig. 12E). With the introduction of a second stranger in the previously empty chamber, both the TFR1flox/flox and TFR1flox/flox;pCP2-cre mice demonstrated an increased preference for the novel stranger (S2) compared with the familiar mouse (S1; Fig. 12F,G). The time of sniffing onto the S2 cage was similar among TFR1flox/flox (71 ± 5 s; n = 13) and TFR1flox/flox;pCP2-cre (70 ± 3 s; n = 10) groups (Fig. 12H′; p = 0.34). Again, no difference (p = 0.53) was found between TFR1flox/flox (24 ± 4; n = 12) and TFR1flox/flox;pCP2-cre (22 ± 3; n = 11) mice comparing the preference index (S2 − S1; Fig. 12H). Finally, we looked at the anxiety of TFR1flox/flox;pCP2-cre mice that could have potentially biased the social behavior assessments (Fig. 12I). The open-field test showed no differences in time spent in the inner, middle, or outer zones between TFR1flox/flox (n = 17) and TFR1flox/flox;pCP2-cre mice (n = 15; Fig. 12I). Together, these results indicate that TFR1flox/flox;pCP2-cre mice do not exhibit deficits in social interaction.
Discussion
In the present study, we investigated the regulation of mGlu1 expression as well as motor coordination and autism-related behaviors in TFR1flox/flox;pCP2-cre mice. We show that: (1) TFR1 deficiency does not overtly influence PC development; (2) TFR1 deficiency decreases constitutive expression of synaptic mGlu1 in PCs and inhibits the induction of parallel fiber–LTD, but not parallel fiber–LTP and intrinsic plasticity of PCs; (3) TFR1 modulates the trafficking of mGlu1 through Rab8 and Rab11; and (4) PC-specific TFR1 deletion impairs motor coordination, but not autistic-like behaviors. Together, these findings underscore the importance of TFR1 in regulating the trafficking and constitutive expression of mGlu1 and suggest that mGlu1-dependent parallel fiber–LTD can be associated with motor coordination, but not autistic-like behaviors.
As an iron transporter, TFR1 is critical for iron uptake and homeostasis in erythrocyte and immune systems (Tacchini et al., 2002; Ng et al., 2006). However, it appears that, in neurons, the role of TFR1 is more prominent in non-iron transport than in iron homeostasis (Liu et al., 2016). Accordingly, TFR1 is distributed in a polarized fashion in that it is prominently present in neuronal dendrites and that it colocalizes with presynaptic/postsynaptic proteins (West et al., 1997; Liu et al., 2016), implicating a role in synaptic function. Indeed, Liu et al. (2016) found that both the postsynaptic density and the level of presynaptic/postsynaptic proteins such as synaptophysin and PSD95 are reduced in mutant mice lacking TFR1 in their neurons. However, our experiments showed that ablation of TFR1 selectively in PCs does not affect the gross morphology of cerebellar lobules or the development of spine and dendrites in PCs. These differences can possibly be attributed to the knock-out strategy in that a nestin promoter-driven knock-out will suffer not only from postsynaptic, but also presynaptic deficits. This hypothesis is supported by electrophysiological recordings showing that presynaptic release is reduced in TFR1flox/flox;nestin-cre mice (Liu et al., 2016), but not in the TFR1flox/flox;pCP2-cre mice, as indicated by the unaltered frequency of PC-mEPSCs and unchanged parallel fiber–EPSCs and climbing fiber–EPSCs (Fig. 4). The presynaptic defect may explain increased synaptic expression of GluA2 in hippocampal CA1 neurons of TFR1flox/flox;nestin-cre mice (Liu et al., 2016) because the expression of postsynaptic glutamate receptors is closely related to synaptogenesis (Garner et al., 2002). Therefore, TFR1 may play a role in regulating synapse development and neurotransmission efficacy at the presynaptic site, but how it is involved in the modulation of presynaptic proteins and transmitter-containing vesicles needs to be further investigated using specific presynaptic promoter-driven TFR1 knock-out mice.
mGlu1/5, which is critical for synapse formation and synaptic plasticity, is implicated in various neurological diseases such as ataxia, memory deficits, mental retardation, and epilepsy (Aiba et al., 1994a; Aiba et al., 1994b; Huber et al., 2000; Ichise et al., 2000; Brasnjo and Otis, 2001; Hannan et al., 2001; Coesmans et al., 2003; Koekkoek et al., 2005; Lüscher and Huber, 2010; Ribeiro et al., 2010; D'Antoni et al., 2014). However, the mechanisms controlling trafficking of mGlu1/5 remains largely unkown. Our present work suggests that TFR1 regulates endocytosis and recycling of mGlu1 and thereby affects constitutive expression of mGlu1 at PC synapses. The molecular mechanism underlying the regulation of TFR1 on mGlu1 remains elusive because in vivo co-IP showed no direct interaction between TFR1 and mGlu1 (Fig. 7D). However, TFR1 bound to Rab8 and Rab11 (Fig. 7D) and loss of TFR1 reduced the level of these proteins at synapses (Fig. 7E). These findings are consistent with previous work. The endocytosis of G-protein-coupled receptors mainly requires clathrin-coated pits and machinery proteins including arrestin and dynamin (Mundell et al., 2001), PKC (Mundell et al., 2003), and Rab8 (Esseltine et al., 2012). Moreover, the recycling of mGlu1 depends largely on protein phosphatase 2A and Rab11 (Pandey et al., 2014). Therefore, we hypothesize that TFR1 might control mGlu1 trafficking at PC synapses by facilitating the recruitment of Rab8 and Rab11 into the membrane area, thus strengthening constitutive expression of mGlu1. However, other possibilities cannot be excluded. For example, TFR1 may regulate other mGlu1-trafficking proteins and may interact with the clathrin complex through links to adaptor protein adaptin-2 (Ohno et al., 1995). It is possible that TFR1 works as a specified protein to select cargos recruited by clathrin-mediated vesicles. To address this issue, correlations between TFR1 and recycling and endocytic vesicles need to be investigated.
Given that mGlu1 deletion impairs both parallel fiber–LTD and synapse formation during development (Ichise et al., 2000), mGlu1-knock-out mice are not ideal for studying the specific roles of mGlu1 in PC function in isolation. Certainly, the ideal strategy might be PC deletion of mGlu1 in adult animals in an inducible fashion (e.g., tamoxifen-mediated Cre recombination; Yang et al., 2014). However, ablation of TFR1 specifically in PCs allowed us to examine how dys-regulated mGlu1 acts on cerebellar plasticity and motor behaviors because PC morphology and synapse formation were not affected by TFR1 deletion. We found that parallel fiber–LTD and motor coordination were impaired, whereas parallel fiber–LTP and intrinsic plasticity were unaffected by reduced synaptic mGlu1, which is consistent with the historic view that parallel fiber–LTD may be one of the mechanisms for cerebellar learning (Linden and Connor, 1995; Ito, 2002; Yamaguchi et al., 2016). Similar conclusions were drawn after application of auto-antibodies against mGlu1 to cerebellar tissue in vitro and in vivo (Coesmans et al., 2003); here too, a combination of deficits in LTD induction and motor coordination was found. However, an increasing body of evidence is suggesting an even more prominent role of parallel fiber–LTP in motor coordination (Andreescu et al., 2007; Schonewille et al., 2010; Schonewille et al., 2011; Hesslow et al., 2013; Peter et al., 2016; Gutierrez-Castellanos et al., 2017). Possibly, the dominant roles of the various forms of plasticity is to a large extent determined by the intrinsic properties of the PCs of the various types of olivocerebellar modules involved (De Zeeuw and Ten Brinke, 2015). Indeed, the zebrin-positive zones, the PCs of which fire at relatively low firing frequencies (e.g., those controlling adaptation of vestibulo-ocular reflex), may benefit in particular from potentiation mechanisms that increase the firing rate, whereas the predominant zebrin-negative zones, the PCs of which fire at relatively high firing frequencies (e.g., those controlling locomotion and eyeblink conditioning), may benefit in particular from suppression mechanisms that decrease the firing rate (De Zeeuw and Ten Brinke, 2015; Ten Brinke et al., 2015). Therefore, synaptic and intrinsic plasticity processes at different levels, including those regulating excitatory and inhibitory transmissions and PC intrinsic excitability, might underlie particular aspects of motor behaviors (Wulff et al., 2007; Schonewille et al., 2010; Galliano et al., 2013). To what extent a deficit in mGlu1-dependent LTD contributes to the phenotype on the accelerating rotarod remains to be determined.
Neuroimaging studies from patients and animals have revealed that the limbic system and cortico-striatothalamo-cortical circuit are major pathological loci for autism spectrum disorders (Park et al., 2016). Nevertheless, recent evidence shows the importance of cerebellar dysfunction in autistic behaviors. Many genes disrupted in autism are expressed in the cerebellum (Fatemi et al., 2012; Wang et al., 2014b). Moreover, PC-specific mutations of several syndromic or nonsyndromic autism genes result in autistic-like behaviors (Tsai et al., 2012; Zhang et al., 2015; Cupolillo et al., 2016; Peter et al., 2016). Therefore, an attractive question is how these mutations cause PC dysfunction and autistic behaviors. Evidence obtained in both rodents and patients shows a close relationship between mGlu1/5 and autism. In Shank3 mutant mice, mGlu5 is increased selectively at synapses of striatal neurons (Wang et al., 2016). mGlu5 antagonists revert neuronal phenotypes in the limbic system and attenuate autistic behaviors in Fmr1 knock-out and Shank3 mutant mice (Koekkoek et al., 2005; Lüscher and Huber, 2010; Vinueza Veloz et al., 2012; Gantois et al., 2013; Wang et al., 2016). The alteration in mGlu1/5 signaling is therefore regarded as one of the main causes of the autistic features because it disrupts synaptogenesis and synaptic function (Zoghbi and Bear, 2012). In the cerebellum, knock-out of Nlgn3 causes an increase in mGlu1 expression in PCs and inhibits mGlu1-dependent LTD, implying that altered mGlu1-dependent LTD in PCs is involved in autism (Baudouin et al., 2012). Our present work showed that the reduction in synaptic mGlu1 was sufficient to impair parallel fiber–LTD and motor learning, but it did not affect autism-related social behaviors. These findings indicate that a deficit in mGlu1-dependent LTD by itself is not sufficient to cause autism.
So what are the critical factors for the generation of autism? One of the main factors that might determine that autistic features may be linked to mutations of, for example, Tsc1 or Nlgn3 is the fact that, in contrast to the current genetic deletion of TFR1, these latter mutations introduce structural aberrations of the cyto-architecture of the cerebellar cortex during early development (Baudouin et al., 2012; Tsai et al., 2012; Zhang et al., 2015). Such early deterioration in cerebellar function might affect coherent firing in the cortico-striatothalamo-cortical circuits from early on and prevent for example expectation of reward processing (Wagner et al., 2017), generating an increase in repetitive behavior and abnormal social interactions. Therefore, the contributions of autism-related genes in PC dysfunction may reside in various mechanisms that might be involved differentially in motor coordination and social-related behaviors. Future molecular and functional studies must elucidate the exact mechanisms by which syndromic and nonsyndromic autism-related genes regulate PC function and influence autistic-like behaviors. The current findings on TFR1 and its role in mGlu1/5 trafficking through Rab8 and Rab11 in PCs may help to design future strategies to treat related motor disorders in adults, but it does not appear to be the first target of choice for intervening in autism-like disorders early in development.
Footnotes
This work was supported by the National Natural Science Foundation of China (Grants 81625006, 31471024, and 31571051), Natural Science Foundation of Zhejiang Province (Grants Z15C090001 and LY15C090001), Chinese Ministry of Education (Project 111 Program Grant B13026 to Y.S.), Zhejiang Province Medical and Health Research Foundation (Grant 2017200463), the Dutch Organization for Medical Sciences and Life Sciences (C.I.D.Z.), and the ERC-adv and ERC-POC of the European Union (C.I.D.Z.). We thank Dr. Jia-Dong Chen for reading the manuscript and the Core Facility of Zhejiang University Institute of Neuroscience for technical assistance.
The authors declare no competing financial interests.
References
- Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S (1994a) Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell 79:365–375. 10.1016/0092-8674(94)90204-6 [DOI] [PubMed] [Google Scholar]
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S (1994b) Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 79:377–388. 10.1016/0092-8674(94)90205-4 [DOI] [PubMed] [Google Scholar]
- Andreescu CE, Milojkovic BA, Haasdijk ED, Kramer P, De Jong FH, Krust A, De Zeeuw CI, De Jeu MT (2007) Estrodiol improves cerebellar memory formation by activating estrogen receptor beta. J Neurosci 27:10832–10839. 10.1523/JNEUROSCI.2588-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski JJ, Dethleffsen K, Meyer M (2000) Cre recombinase expression in cerebellar Purkinje cells. Genesis 28:93–98. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J (1997) Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature 385:74–77. [DOI] [PubMed] [Google Scholar]
- Baudouin SJ, Gaudias J, Gerharz S, Hatstatt L, Zhou K, Punnakkal P, Tanaka KF, Spooren W, Hen R, De Zeeuw CI, Vogt K, Scheiffele P (2012) Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science 338:128–132. 10.1126/science.1224159 [DOI] [PubMed] [Google Scholar]
- Bellone C, Lüscher C (2006) Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci 9:636–641. 10.1038/nn1682 [DOI] [PubMed] [Google Scholar]
- Belmeguenai A, Hosy E, Bengtsson F, Pedroarena CM, Piochon C, Teuling E, He Q, Ohtsuki G, De Jeu MT, Elgersma Y, De Zeeuw CI, Jörntell H, Hansel C (2010) Intrinsic plasticity complements long-term potentiation in parallel fiber input gain control in cerebellar Purkinje cells. J Neurosci 30:13630–13643. 10.1523/JNEUROSCI.3226-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasnjo G, Otis TS (2001) Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron 31:607–616. 10.1016/S0896-6273(01)00377-4 [DOI] [PubMed] [Google Scholar]
- Choi KY, Chung S, Roche KW (2011) Differential binding of calmodulin to group I metabotropic glutamate receptors regulates receptor trafficking and signalling. J Neurosci 31:5921–5930. 10.1523/JNEUROSCI.6253-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coesmans M, Smitt PA, Linden DJ, Shigemoto R, Hirano T, Yamakawa Y, van Alphen AM, Luo C, van der Geest JN, Kros JM, Gaillard CA, Frens MA, de Zeeuw CI (2003) Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann Neurol 53:325–336. 10.1002/ana.10451 [DOI] [PubMed] [Google Scholar]
- Crepel F, Daniel H (2007) Developmental changes in agonist-induced retrograde signaling at parallel fiber-Purkinje cell synapses: role of calcium-induced calcium release. J Neurophysiol 98:2550–2565. 10.1152/jn.00376.2007 [DOI] [PubMed] [Google Scholar]
- Cupolillo D, Hoxha E, Faralli A, De Luca A, Rossi F, Tempia F, Carulli D (2016) Autistic-like traits and cerebellar dysfunction in Purkinje cell PTEN knock-out mice. Neuropsychopharmacology 41:1457–1466. 10.1038/npp.2015.339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antoni S, Spatuzza M, Bonaccorso CM, Musumeci SA, Ciranna L, Nicoletti F, Huber KM, Catania MV (2014) Dysregulation of group-I metabotropic glutamate (mGlu) receptor mediated signalling in disorders associated with intellectual disability and autism. Neurosci Biobehav Rev 46:228–241. 10.1016/j.neubiorev.2014.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehnes Y, Chaudhry FA, Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1998) The glutamate transporter EAAT4 in rat cerebellar Purkinje cells: a glutamate-gated chloride channel concentrated near the synapse in parts of the dendritic membrane facing astroglia. J Neurosci 18:3606–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Zeeuw CI, Ten Brinke MM (2015) Motor learning and the cerebellum. Cold Spring Harb Perspect Biol 7:a021683. 10.1101/cshperspect.a021683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami GK, Ferguson SS (2006) Regulation of metabotropic glutamate receptor signalling, desensitization and endocytosis. Pharmacol Ther 111:260–271. 10.1016/j.pharmthera.2005.01.008 [DOI] [PubMed] [Google Scholar]
- Esseltine JL, Ribeiro FM, Ferguson SS (2012) Rab8 modulates metabotropic glutamate receptor subtype 1 intracellular trafficking and signaling in a protein kinase C-dependent manner. J Neurosci 32:16933–16942. 10.1523/JNEUROSCI.0625-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ, Chauhan A, Chauhan V, Dager SR, Dickson PE, Estes AM, Goldowitz D, Heck DH, Kemper TL, King BH, Martin LA, Millen KJ, Mittleman G, Mosconi MW, Persico AM, Sweeney JA, Webb SJ, Welsh JP (2012) Consensus paper: pathological role of the cerebellum in autism. Cerebellum 11:777–807. 10.1007/s12311-012-0355-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero JJ, Alvarez AM, Ramírez-Franco J, Godino MC, Bartolomé-Martín D, Aguado C, Torres M, Luján R, Ciruela F, Sánchez-Prieto J (2013) β-Adrenergic receptors activate exchange protein directly activated by cAMP (Epac), translocate Munc13-1, and enhance the Rab3A-RIM1 interaction to potentiate glutamate release at cerebrocortical nerve terminals. J Biol Chem 288:31370–31385. 10.1074/jbc.M113.463877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francesconi A, Kumari R, Zukin RS (2009) Regulation of group I metabotropic glutamate receptor trafficking and signalling by the caveolar/lipid raft pathway. J Neurosci 29:3590–3602. 10.1523/JNEUROSCI.5824-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliano E, Gao Z, Schonewille M, Todorov B, Simons E, Pop AS, D'Angelo E, van den Maagdenberg AM, Hoebeek FE, De Zeeuw CI (2013) Silencing the majority of cerebellar granule cells uncovers their essential role in motor learning and consolidation. Cell Rep 3:1239–1251. 10.1016/j.celrep.2013.03.023 [DOI] [PubMed] [Google Scholar]
- Gantois I, Pop AS, de Esch CE, Buijsen RA, Pooters T, Gomez-Mancilla B, Gasparini F, Oostra BA, D'Hooge R, Willemsen R (2013) Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav Brain Res 239:72–79. 10.1016/j.bbr.2012.10.059 [DOI] [PubMed] [Google Scholar]
- Garner CC, Zhai RG, Gundelfinger ED, Ziv NE (2002) Molecular mechanisms of CNS synaptogenesis. Trends Neurosci 25:243–251. 10.1016/S0166-2236(02)02152-5 [DOI] [PubMed] [Google Scholar]
- Gregory KJ, Conn PJ (2015) Molecular insights into metabotropic glutamate receptor allosteric modulation. Mol Pharmacol 88:188–202. 10.1124/mol.114.097220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Castellanos N, Da Silva-Matos CM, Zhou K, Canto CB, Renner MC, Koene LM, Ozyildirim O, Sprengel R, Kessels HW, De Zeeuw CI (2017) Motor learning requires Purkinje cell synaptic potentiation through activation of AMPA-receptor subunit GluA3. Neuron 93:409–424. 10.1016/j.neuron.2016.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M, Roder J, Kim D, Shin HS, Kind PC (2001) PLC-β1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat Neurosci 4:282–288. 10.1038/85132 [DOI] [PubMed] [Google Scholar]
- Hartmann J, Karl RM, Alexander RP, Adelsberger H, Brill MS, Rühlmann C, Ansel A, Sakimura K, Baba Y, Kurosaki T, Misgeld T, Konnerth A (2014) STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 82:635–644. 10.1016/j.neuron.2014.03.027 [DOI] [PubMed] [Google Scholar]
- Hermans E, Challiss RA (2001) Structural, signaling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. Biochem J 359:465–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesslow G, Jirenhed DA, Rasmussen A, Johansson F (2013) Classical conditioning of motor responses: what is the learning mechanism? Neural Netw 47:81–87. 10.1016/j.neunet.2013.03.013 [DOI] [PubMed] [Google Scholar]
- Hong YH, Kim JY, Lee JH, Chae HG, Jang SS, Jeon JH, Kim CH, Kim J, Kim SJ (2009) Agonist-induced internalization of mGluR1a is mediated by caveolin. J Neurochem 111:61–71. 10.1111/j.1471-4159.2009.06289.x [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288:1254–1257. 10.1126/science.288.5469.1254 [DOI] [PubMed] [Google Scholar]
- Ichise T, Kano M, Hashimoto K, Yanagihara D, Nakao K, Shigemoto R, Katsuki M, Aiba A (2000) mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science 288:1832–1835. 10.1126/science.288.5472.1832 [DOI] [PubMed] [Google Scholar]
- Ikeda K, Satake S, Onaka T, Sugimoto H, Takeda N, Imoto K, Kawakami K (2013) Enhanced inhibitory neurotransmission in the cerebellar cortex of Atp1a3-deficient heterozygous mice. J Physiol 591:3433–3449. 10.1113/jphysiol.2012.247817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. (2002) Historical review of the significance of the cerebellum and the role of Purkinje cells in motor learning. Ann N Y Acad Sci 978:273–288. 10.1111/j.1749-6632.2002.tb07574.x [DOI] [PubMed] [Google Scholar]
- Jia F, Miao H, Zhu X, Xu F (2017) Pseudo-typed Semliki Forest virus delivers EGFP into neurons. J Neurovirol 23:205–215. 10.1007/s13365-016-0486-8 [DOI] [PubMed] [Google Scholar]
- Kenny PJ, Markou A (2004) The ups and downs of addiction: role of metabotropic glutamate receptors. Trends Pharmacol Sci 25:265–272. 10.1016/j.tips.2004.03.009 [DOI] [PubMed] [Google Scholar]
- Kim SJ, Jin Y, Kim J, Shin JH, Worley PF, Linden DJ (2008) Transient upregulation of postsynaptic IP3-gated Ca release underlies short-term potentiation of metabotropic glutamate receptor 1 signaling in cerebellar Purkinje cells. J Neurosci 28:4350–4355. 10.1523/JNEUROSCI.0284-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koekkoek SK, Yamaguchi K, Milojkovic BA, Dortland BR, Ruigrok TJ, Maex R, De Graaf W, Smit AE, VanderWerf F, Bakker CE, Willemsen R, Ikeda T, Kakizawa S, Onodera K, Nelson DL, Mientjes E, Joosten M, De Schutter E, Oostra BA, Ito M, De Zeeuw CI (2005) Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47:339–352. 10.1016/j.neuron.2005.07.005 [DOI] [PubMed] [Google Scholar]
- Lee KJ, Kim H, Kim TS, Park SH, Rhyu IJ (2004) Morphological analysis of spine shapes of Purkinje cell dendrites in the rat cerebellum using high-voltage electron microscopy. Neurosci Lett 359:21–24. 10.1016/j.neulet.2004.01.071 [DOI] [PubMed] [Google Scholar]
- Li X, Sapp E, Chase K, Comer-Tierney LA, Masso N, Alexander J, Reeves P, Kegel KB, Valencia A, Esteves M, Aronin N, Difiglia M (2009) Disruption of Rab11 activity in a knock-in mouse model of Huntington's disease. Neurobiol Dis 36:374–383. 10.1016/j.nbd.2009.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ, Connor JA (1995) Long-term synaptic depression. Annu Rev Neurosci 18:319–357. 10.1146/annurev.ne.18.030195.001535 [DOI] [PubMed] [Google Scholar]
- Liu K, Lei R, Li Q, Wang XX, Wu Q, An P, Zhang J, Zhu M, Xu Z, Hong Y, Wang F, Shen Y, Li H, Li H (2016) Transferrin receptor controls AMPA receptor trafficking efficiency and synaptic plasticity. Sci Rep 6:21019. 10.1038/srep21019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Dekundy A, Nagel J, Danysz W, Parsons CG, Potschka H (2006) mGlu1 and mGlu5 receptor antagonists lack anticonvulsant efficacy in rodent models of difficult-to-treat partial epilepsy. Neuropharmacology 50:1006–1015. 10.1016/j.neuropharm.2006.02.001 [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM (2010) Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron 65:445–459. 10.1016/j.neuron.2010.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31:463–475. 10.1016/S0896-6273(01)00375-0 [DOI] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Lüscher C (2009) Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci 12:1036–1041. 10.1038/nn.2367 [DOI] [PubMed] [Google Scholar]
- Moos T. (1996) Immunohistochemical localization of intraneuronal transferrin receptor immunoreactivity in the adult mouse central nervous system. J Comp Neurol 375:675–692. [DOI] [PubMed] [Google Scholar]
- Mosconi MW, Wang Z, Schmitt LM, Tsai P, Sweeney JA (2015) The role of cerebellar circuitry alterations in the pathophysiology of autism spectrum disorders. Front Neurosci 9:296. 10.3389/fnins.2015.00296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundell SJ, Matharu AL, Pula G, Roberts PJ, Kelly E (2001) Agonist-induced internalization of the metabotropic glutamate receptor 1a is arrestin- and dynamindependent. J Neurochem 78:546–551. 10.1046/j.1471-4159.2001.00421.x [DOI] [PubMed] [Google Scholar]
- Mundell SJ, Pula G, Carswell K, Roberts PJ, Kelly E (2003) Agonist-induced internalization of metabotropic glutamate receptor 1A: Structural determinants for protein kinase C- and G-protein-coupled receptor kinase-mediated internalization. J Neurochem 84:294–304. 10.1046/j.1471-4159.2003.01515.x [DOI] [PubMed] [Google Scholar]
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, Leonard H, Bailey ME, Schanen NC, Zappella M, Renieri A, Huppke P, Percy AK; RettSearch Consortium (2010) Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 68:944–950. 10.1002/ana.22124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PP, Helguera G, Daniels TR, Lomas SZ, Rodriguez JA, Schiller G, Bonavida B, Morrison SL, Penichet ML (2006) Molecular events contributing to cell death in malignant human hematopoietic cells elicited by an IgG3-avidin fusion protein targeting the transferrin receptor. Blood 108:2745–2754. 10.1182/blood-2006-04-020263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322. 10.1146/annurev.pharmtox.011008.145533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, Miyatake S, Saito T, Gallusser A, Kirchhausen T, Bonifacino JS (1995) Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 269:1872–1875. 10.1126/science.7569928 [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Takao-Rikitsu E, Inoue E, Inoue M, Takeuchi M, Matsubara K, Deguchi-Tawarada M, Satoh K, Morimoto K, Nakanishi H, Takai Y (2002) Cast: a novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and munc13-1. J Cell Biol 158:577–590. 10.1083/jcb.200202083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Mahato PK, Bhattacharyya S (2014) Metabotropic glutamate receptor 1 recycles to the cell surface in protein phosphatase 2A-dependent manner in non-neuronal and neuronal cell lines. J Neurochem 131:602–614. 10.1111/jnc.12930 [DOI] [PubMed] [Google Scholar]
- Park HR, Lee JM, Moon HE, Lee DS, Kim BN, Kim J, Kim DG, Paek SH (2016) A short review on the current understanding of autism spectrum disorders. Exp Neurobiol 25:1–13. 10.5607/en.2016.25.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins KL. (2006) Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods 154:1–18. 10.1016/j.jneumeth.2006.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter S, Ten Brinke MM, Stedehouder J, Reinelt CM, Wu B, Zhou H, Zhou K, Boele HJ, Kushner SA, Lee MG, Schmeisser MJ, Boeckers TM, Schonewille M, Hoebeek FE, De Zeeuw CI (2016) Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat Commun 7:12627. 10.1038/ncomms12627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Kloth AD, Grasselli G, Titley HK, Nakayama H, Hashimoto K, Wan V, Simmons DH, Eissa T, Nakatani J, Cherskov A, Miyazaki T, Watanabe M, Takumi T, Kano M, Wang SS, Hansel C (2014) Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nat Commun 5:5586. 10.1038/ncomms6586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro FM, Paquet M, Cregan SP, Ferguson SS (2010) Group I metabotropic glutamate receptor signalling and its implication in neurological disease. CNS Neurol Disord Drug Targets 9:574–595. 10.2174/187152710793361612 [DOI] [PubMed] [Google Scholar]
- Schmeisser MJ, et al. (2012) Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486:256–260. [DOI] [PubMed] [Google Scholar]
- Schonewille M, Belmeguenai A, Koekkoek SK, Houtman SH, Boele HJ, van Beugen BJ, Gao Z, Badura A, Ohtsuki G, Amerika WE, Hosy E, Hoebeek FE, Elgersma Y, Hansel C, De Zeeuw CI (2010) Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron 67:618–628. 10.1016/j.neuron.2010.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonewille M, Gao Z, Boele HJ, Veloz MF, Amerika WE, Simek AA, De Jeu MT, Steinberg JP, Takamiya K, Hoebeek FE, Linden DJ, Huganir RL, De Zeeuw CI (2011) Reevaluating the role of LTD in cerebellar motor learning. Neuron 70:43–50. 10.1016/j.neuron.2011.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R, Nakanishi S, Mizuno N (1992) Distribution of the mRNA for a metabotropic glutamate receptor (mGluR1) in the central nervous system: an in situ hybridization study in adult and developing rat. J Comp Neurol 322:121–135. 10.1002/cne.903220110 [DOI] [PubMed] [Google Scholar]
- Su LD, Shen Y (2009) Blockade of glutamate transporters facilitates cerebellar synaptic long-term depression. Neuroreport 20:502–507. 10.1097/WNR.0b013e328328f397 [DOI] [PubMed] [Google Scholar]
- Sun CL, Su LD, Li Q, Wang XX, Shen Y (2011) Cerebellar long-term depression is deficient in Niemann-Pick type C disease mice. Cerebellum 10:88–95. 10.1007/s12311-010-0233-2 [DOI] [PubMed] [Google Scholar]
- Tacchini L, Fusar Poli D, Bernelli-Zazzera A, Cairo G (2002) Transferrin receptor gene expression and transferrin-bound iron uptake are increased during postischemic rat liver reperfusion. Hepatology 36:103–111. [DOI] [PubMed] [Google Scholar]
- Ten Brinke MM, Boele HJ, Spanke JK, Potters JW, Kornysheva K, Wulff P, IJpelaar AC, Koekkoek SK, De Zeeuw CI (2015) Evolving models of pavlovian conditioning: cerebellar cortical dynamics in awake behaving mice. Cell Rep 13:1977–1988. 10.1016/j.celrep.2015.10.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, Sahin M (2012) Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488:647–651. 10.1038/nature11310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE (2001) Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21:RC188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinueza Veloz MF, Buijsen RA, Willemsen R, Cupido A, Bosman LW, Koekkoek SK, Potters JW, Oostra BA, De Zeeuw CI (2012) The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav 11:325–331. 10.1111/j.1601-183X.2011.00763.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner MJ, Kim TH, Savall J, Schnitzer MJ, Luo L (2017) Cerebellar granule cells encode the expectation of reward. Nature 544:96–100. 10.1038/nature21726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DJ, Su LD, Wang YN, Yang D, Sun CL, Zhou L, Wang XX, Shen Y (2014a) Long-term potentiation at cerebellar parallel fiber-Purkinje cell synapses requires presynaptic and postsynaptic signaling cascades. J Neurosci 34:2355–2364. 10.1523/JNEUROSCI.4064-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Kloth AD, Badura A (2014b) The cerebellum, sensitive periods, and autism. Neuron 83:518–532. 10.1016/j.neuron.2014.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bey AL, Katz BM, Badea A, Kim N, David LK, Duffney LJ, Kumar S, Mague SD, Hulbert SW, et al. (2016) Altered mGluR5-Homer scaffolds and corticostriatal connectivity in a Shank3 complete knockout model of autism. Nat Commun 7:11459. 10.1038/ncomms11459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YN, Zhou L, Li YH, Wang Z, Li YC, Zhang YW, Wang Y, Liu G, Shen Y (2015) Protein interacting with C-kinase 1 deficiency impairs glutathione synthesis and increases oxidative stress via reduction of surface excitatory amino acid carrier 1. J Neurosci 35:6429–6443. 10.1523/JNEUROSCI.3966-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Neve RL, Buckley KM (1997) Identification of a somatodendritic targeting signal in the cytoplasmic domain of the transferrin receptor. J Neurosci 17:6038–6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, Park SG, Lee JS, Lee K, Kim D, Bae YC, Kaang BK, Lee MG, Kim E (2012) Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486:261–265. 10.1038/nature11208 [DOI] [PubMed] [Google Scholar]
- Wu ZY, Zhu LJ, Zou N, Bombek LK, Shao CY, Wang N, Wang XX, Liang L, Xia J, Rupnik M, Shen Y (2012) AMPA receptors regulate exocytosis and insulin release in pancreatic beta cells. Traffic 13:1124–1139. 10.1111/j.1600-0854.2012.01373.x [DOI] [PubMed] [Google Scholar]
- Wulff P, Goetz T, Leppä E, Linden AM, Renzi M, Swinny JD, Vekovischeva OY, Sieghart W, Somogyi P, Korpi ER, Farrant M, Wisden W (2007) From synapse to behavior: rapid modulation of defined neuronal types with engineered GABAA receptors. Nat Neurosci 10:923–929. 10.1038/nn1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K, Itohara S, Ito M (2016) Reassessment of long-term depression in cerebellar Purkinje cells in mice carrying mutated GluA2 C terminus. Proc Natl Acad Sci U S A 113:10192–10197. 10.1073/pnas.1609957113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Huang S, Gaertig MA, Li XJ, Li S (2014) Age-dependent decrease in chaperone activity impairs MANF expression, leading to Purkinje cell degeneration in inducible SCA17 mice. Neuron 81:349–365. 10.1016/j.neuron.2013.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chen LY, Liu X, Maxeiner S, Lee SJ, Gokce O, Südhof TC (2015) Neuroligins sculpt cerebellar Purkinje-cell circuits by differential control of distinct classes of synapses. Neuron 87:781–796. 10.1016/j.neuron.2015.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Cao SX, Sun P, He HY, Yang CH, Chen XJ, Shen CJ, Wang XD, Chen Z, Berg DK, Duan S, Li XM (2016) Loss of MeCP2 in cholinergic neurons causes part of RTT-like phenotypes via α7 receptor in hippocampus. Cell Res 26:728–742. 10.1038/cr.2016.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Lin Z, Voges K, Ju C, Gao Z, Bosman LWJ, Ruigrok TJ, Hoebeek FE, De Zeeuw CI, Schonewille M (2014) Cerebellar modules operate at different frequencies. eLife 3:e02536. 10.7554/eLife.02536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Yang D, Wang DJ, Xie YJ, Zhou JH, Zhou L, Huang H, Han S, Shao CY, Li HS, Zhu JJ, Qiu MS, De Zeeuw CI, Shen Y (2015) Numb deficiency in cerebellar Purkinje cells impairs synaptic expression of metabotropic glutamate receptor and motor coordination. Proc Natl Acad Sci U S A 112:15474–15479. 10.1073/pnas.1512915112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Bear MF (2012) Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4: pii: a009886. 10.1101/cshperspect.a009886 [DOI] [PMC free article] [PubMed] [Google Scholar]