

Abstract
DNA methylation has been implicated in the pathogenesis of chronic pain. However, the specific genes regulated by DNA methylation under neuropathic pain condition remain largely unknown. Here we investigated how chemokine receptor CXCR3 is regulated by DNA methylation and how it contributes to neuropathic pain induced by spinal nerve ligation (SNL) in mice. SNL increased Cxcr3 mRNA and protein expression in the neurons of the spinal cord. Meanwhile, the CpG (5′-cytosine-phosphate-guanine-3′) island in the Cxcr3 gene promoter region was demethylated, and the expression of DNA methyltransferase 3b (DNMT3b) was decreased. SNL also increased the binding of CCAAT (cytidine–cytidine–adenosine–adenosine–thymidine)/enhancer binding protein α (C/EBPα) with Cxcr3 promoter and decreased the binding of DNMT3b with Cxcr3 promoter in the spinal cord. C/EBPα expression was increased in spinal neurons after SNL, and inhibition of C/EBPα by intrathecal small interfering RNA attenuated SNL-induced pain hypersensitivity and reduced Cxcr3 expression. Furthermore, SNL-induced mechanical allodynia and heat hyperalgesia were markedly reduced in Cxcr3−/− mice. Spinal inhibition of Cxcr3 by shRNA or CXCR3 antagonist also attenuated established neuropathic pain. Moreover, CXCL10, the ligand of CXCR3, was increased in spinal neurons and astrocytes after SNL. Superfusing spinal cord slices with CXCL10 enhanced spontaneous EPSCs and potentiated NMDA-induced and AMPA-induced currents of lamina II neurons. Finally, intrathecal injection of CXCL10 induced CXCR3-dependent pain hypersensitivity in naive mice. Collectively, our results demonstrated that CXCR3, increased by DNA demethylation and the enhanced interaction with C/EBPα, can be activated by CXCL10 to facilitate excitatory synaptic transmission and contribute to the maintenance of neuropathic pain.
SIGNIFICANCE STATEMENT Peripheral nerve injury induces changes of gene expression in the spinal cord that may contribute to the pathogenesis of neuropathic pain. CXCR3 is a chemokine receptor. Whether it is involved in neuropathic pain and how it is regulated after nerve injury remain largely unknown. Our study demonstrates that spinal nerve ligation downregulates the expression of DNMT3b, which may cause demethylation of Cxcr3 gene promoter and facilitate the binding of CCAAT/enhancer binding protein α with Cxcr3 promoter and further increase CXCR3 expression in spinal neurons. The upregulated CXCR3 may contribute to neuropathic pain by facilitating central sensitization. Our study reveals an epigenetic mechanism underlying CXCR3 expression and also suggests that targeting the expression or activation of CXCR3 signaling may offer new therapeutics for neuropathic pain.
Keywords: astrocyte, CEBPα, CXCL10, CXCR3, demethylation, neuropathic pain
Introduction
Neuropathic pain resulting from nerve injury or disease is a highly debilitating chronic pain state and is often resistant to currently available treatments (Toth et al., 2009). Nerve injury induces changes in gene expression in sensory neurons of the dorsal root ganglion and spinal cord. Such changes are thought to influence the pathogenesis of neuropathic pain (Lacroix-Fralish et al., 2006; von Hehn et al., 2012; Jiang et al., 2016). Revealing the molecular mechanisms underlying the dysregulated gene expression under neuropathic pain condition is important for the discovery of new treatments.
Epigenetic regulations, including DNA methylation, histone modification, and noncoding RNA expression, play important roles in the modulation of gene expression. Functionally, these processes are involved in many physiological and pathological processes, including learning and memory (Bali et al., 2011; Zovkic et al., 2013), addiction (Tuesta and Zhang, 2014), cancer (Suzuki et al., 2012; Ma et al., 2013), neurodegenerative disease (Sanchez-Mut and Gräff, 2015), and chronic pain (Descalzi et al., 2015; Liang et al., 2015). DNA methylation is a key epigenetic mechanism controlling DNA accessibility and gene expression (Jaenisch and Bird, 2003). Evidence has shown that nerve injury changes the spinal global DNA methylation (Wang et al., 2011) and that blockade of DNA methylation affects pain behaviors implicated in chronic pain (Wang et al., 2011; Viet et al., 2014). However, the specific genes regulated by DNA methylation and its implication in neuropathic pain remain to be investigated.
Neuroinflammation, mediated by proinflammatory cytokines and chemokines, may play a pivotal role in the development and maintenance of neuropathic pain (White and Wilson, 2008; Abbadie et al., 2009; Gao and Ji, 2010). Several chemokines, such as CXCL13, CCL2, CXCL1, and CX3CL1, are upregulated in the spinal cord after nerve injury or tissue inflammation (Lindia et al., 2005; Gao et al., 2009; Zhang et al., 2013; Jiang et al., 2016). Using mouse gene-expression microarrays, we recently found that the gene for chemokine, Cxcl10, is one of the highly upregulated genes with >5-fold increase in the spinal cord after spinal nerve ligation (SNL: Jiang et al., 2016). CXCR3, as the major receptor of CXCL10, has been shown to be involved in a variety of human diseases, including chronic inflammation, immune dysfunction, cancer, metastasis, and pruritus (Groom and Luster, 2011; Qu et al., 2015; Van Raemdonck et al., 2015). A recent study has shown that CXCR3 deficiency reduced experimental autoimmune encephalomyelitis-evoked hyperalgesia, but did not affect spared nerve injury-induced mechanical allodynia (Schmitz et al., 2013). However, to date, little is known about how the expression of CXCR3 is regulated and whether CXCR3 is involved in SNL-induced neuropathic pain.
DNA methylation involves the transfer of a methyl group to cysteine residues at CpG (cytosine–phosphate–guanine) sites in the promoter regions of genes, a process that plays an important role in regulating gene expression (Bird, 1986). DNA methyltransferases (DNMTs) are important in regulating DNA methylation and can directly inhibit transcription by interfering with transcription factor binding (Poetsch and Plass, 2011; Géranton, 2012). The CCAAT (cytidine–cytidine–adenosine–adenosine–thymidine)/enhancer binding protein α (C/EBPα) is a differentiation-inducing transcription factor that belongs to a family of basic region leucine zipper transcription factors (Koschmieder et al., 2009). C/EBPα promotes the expression of certain genes through interaction with the promoter region. Whether C/EBPα regulates CXCR3 expression and further contributes to neuropathic pain has not previously been elucidated.
In the present study, we investigated how CXCR3 is regulated in the spinal cord in SNL-induced neuropathic pain and how the implied mechanisms involved in such regulation. We found that SNL induced demethylation of the Cxcr3 gene and increased the binding of C/EBPα with Cxcr3 promoter. Furthermore, inhibition of CXCR3 by different strategies markedly attenuated neuropathic pain. We also showed that CXCL10 contributed to the maintenance of neuropathic pain via enhanced CXCR3-mediated excitatory synaptic transmission.
Materials and Methods
Animals and surgery.
Adult ICR and C57BL/6 (male, 8 weeks) mice were purchased from the Experimental Animal Center of Nantong University. Cxcr3−/− mice (B6.129P2-Cxcr3tm1Dgen/J, stock #005796, RRID:IMSR_JAX:005796) were purchased from the Jackson Laboratory. Female Cxcr3tm1Dgen/J homozygous mice were mated with the C57BL/6 mice to get the F1 heterozygous offspring, and then backcrossed with the Cxcr3tm1Dgen/J homozygous mice to get F2 heterozygous or homozygous offspring. The homozygous and the littermate wild-type (WT) mice were used for experiments. Somatostatin(SST)-Tomato+ mice were generated by crossing Sst-IRES-Cre mice (Jackson Laboratory, stock #013044, RRID:IMSR_JAX:013044) with Ai9(RCL-tdT) mice (Jackson Laboratory, stock #007909, RRID:IMSR_JAX:007909). GAD2-Tomato+ mice were generated by crossing Gad2-IRES-Cremice (Jackson Laboratory, stock #019022, RRID:IMSR_JAX:019022) with Ai9(RCL-tdT) mice. The animals were maintained in specific-pathogen-free facilities on a 12:12 light/dark cycle at a room temperature of 22 ± 1°C. All animal procedures performed in this study were reviewed and approved by the Animal Care and Use Committee of Nantong University and performed in accordance with the guidelines of the International Association for the Study of Pain. SNL was produced as previously described (Gao et al., 2009). For sham operations, the L5 spinal nerve was exposed but not ligated.
Drugs and administration.
Recombinant murine CXCL10 was purchased from PeproTech. A potent and selective CXCR3 antagonist, NBI-74330, was purchased from Tocris Bioscience. Five′-cholesteryl-modified and 2′-O-methyl-modified Cebpa small interfering RNA (siRNA; 5′-GGA GTT GAC CAG TGA CAA T3′) and an additional scrambled siRNA were purchased from RiboBio. Intrathecal injection was made with a 30 G needle between the L5 and L6 intervertebral spaces to deliver the reagents to the CSF.
Real-time quantitative PCR.
The total RNA of the spinal cord or cultured cells was extracted using Trizol reagent (Invitrogen). One microgram of total RNA was reverse-transcribed using an oligo primer according to the manufacturer's protocol (Takara). Quantitative PCR (qPCR) analysis was performed in a real-time detection system (Rotor-Gene 6000, Qiagen) by SYBR green I dye detection (Takara). The detailed primer sequences for each gene (Cxcr3, Cxcl10, and Gapdh) are listed in Table 1. The PCR amplifications were performed at 95°C for 30 s, followed by 40 cycles of thermal cycling at 95°C for 5 s and 60°C for 45 s. Gapdh was used as an endogenous control to normalize differences. Melt curves were performed on completion of the cycles to ensure that nonspecific products were absent. Quantification was performed by normalizing Ct (cycle threshold) values with Gapdh Ct and analyzed with the 2−ΔΔCT method.
Table 1.
Primer sets used in qPCR
| Gene | Primer sequence | size |
|---|---|---|
| Cxcr3 | 5′-TACCTTGAGGTTAGTGAACGTCA-3′; | 100 bp |
| 5′-CGCTCTCGTTTTCCCCATAATC-3′ | ||
| Cxcl10 | 5′-TGAATCCGGAATCTAAGACCATCAA-3′; | 171 bp |
| 5′-AGGACTAGCCATCCACTGGGTAAAG-3′ | ||
| Gapdh | 5′-AAATGGTGAAGGTCGGTGTGAAC-3′; | 90 bp |
| 5′-CAACAATCTCCACTTTGCCACTG-3′ |
Bisulfite sequencing PCR.
Genomic DNA was extracted using QIAamp DNA Mini Kit (Qiagen) according to the manufacturer's protocol. Sodium bisulfite conversion of genomic DNA was performed using EpiTect Bisulfite Kit (Qiagen). PCR products of genomic DNA fragment-containing promoter amplified from bisulfite-converted genomic DNA using the primers shown in Table 2 were gel-purified with the QIAquick Gel Extraction Kit (Qiagen). The eluted DNA fragments were ligated into pGEM-T Easy Vector (Promega) for sequencing. Ten colonies for each mouse were randomly chosen for sequencing.
Table 2.
Primer sequences for MSP, BSP, and ChIP experiments
| Usage | Primer name | Primer sequence | Size |
|---|---|---|---|
| MSP | Cxcr3-M-F | 5′-TAAGTGGTCGAGTTCGGATATTA-3′ | 83 bp |
| Cxcr3-M-R | 5′-CGATACTACTCACTCAACCCG-3′ | ||
| Cxcr3-U-F | 5′-GGTTAAGTGGTTGAGTTTGGATATTA-3′ | 83 bp | |
| Cxcr3-U-R | 5′-ATTCAATACTACTCACTCAACCCA-3′ | ||
| BSP | Cxcr3-F | 5′-ATTTTTTGGAAGAGGTTGTGTTG-3′ | 325 bp |
| Cxcr3-R | 5′-ACTAAATTTCCCAATCCACCAATA-3′ | ||
| ChIP | P1-F | 5′-TTCCTGGCTAGCCCCTTCTA-3′ | 169 bp |
| P1-R | 5′-TTGGCACTTGTGGTTGGGAA-3′ | ||
| P2-F | 5′-TGGCTTGGATGCCTCATCTG-3′ | 118 bp | |
| P2-R | 5′-AATGGGGGCATACCACTTCC-3′ |
Methylation-specific PCR.
The genomic DNA extraction and bisulfite treatment was performed as described in bisulfite sequencing PCR (BSP). Bisulfite-converted genomic DNA was amplified using the EpiTect Methylation-Specific PCR (MSP) PCR Kit (Qiagen) with methylation-specific or unmethylation-specific primer pairs (Table 2). The PCR products were analyzed by electrophoresis. The percentage of methylation of Cxcr3 promoter from the spinal cord of SNL and sham-operated animals was determined by densitometric analysis of MSP products (ratio of methylated products to unmethylated products).
Cxcr3 promoter reporter analysis.
Cxcr3 promoter reporter constructs, cloned into the pCpG-free basic reporter vector (InvivoGen), were either methylated or unmethylated by incubation with S-adenosyl methionine in the presence or absence of CpG methylase (M. SssI; Fisher Thermo Scientific). Methylated and unmethylated pCpG-free-Cxcr3-Lucia luciferase reporter constructs were transfected into HEK293 (CLS catalog #300192/p777_HEK293, RRID:CVCL_0045) cells using Lipofectamine 3000 (Invitrogen). The activity of secreted coelenterazine luciferase in medium was measured 24 h later with the QUANTI-Luc (InvivoGen) according to the manufacturer's instructions. In brief, 10 μl of medium sample was transferred by pipette to a 96-well white plate, then a 50-μl injector was used to add QUANTI-Luc assay solution. Measurements were performed with a luminometer (Synergy 2 Multi-Mode Reader, BioTEL).
C/EBPα expression vectors were purchased from Obio Technology. C/EBPα expression vectors and methylated or unmethylated pCpG-free-Cxcr3-Lucia promoters were transiently transfected into HEK293 cells using Lipofectamine 3000 (Invitrogen). The activity of secreted coelenterazine luciferase in medium was measured 24 h after transfection as described above.
Chromatin immunoprecipitation PCR and quantitative chromatin immunoprecipitation analyses.
Chromatin immunoprecipitation (ChIP) assays were performed using the SimpleChIP Enzymatic Chromatin IP kit (Magnetic Beads, Cell Signaling Technology) according to the manufacturer's instructions. After SNL or sham operation, the ipsilateral dorsal horn of the spinal cord was collected and exposed to 1% formaldehyde immediately to cross-link proteins to DNA. After glycine treatment and PBS washing, the tissues were homogenized, the cells were lysed, and the chromatin was harvested and fragmented using enzymatic digestion. The chromatin was then subjected to immunoprecipitation with positive control histone H3 antibody (rabbit, 1:50; Cell Signaling Technology, catalog #4620, RRID:AB_1904005), negative control (NC) normal IgG antibody (rabbit, 1:500; Cell Signaling Technology, catalog #2729, RRID:AB_2617119), C/EBPα antibody (rabbit, 1:100; Santa Cruz Biotechnology, catalog #sc-61X, RRID:AB_631233), and DNMT3b antibody (mouse, 1:250; Abcam, catalog #ab13604, RRID:AB_300494). The complex coprecipitates were captured by protein-G magnetic beads. After immunoprecipitation, the protein–DNA cross-links were reversed and the DNA was purified. The enrichment of the Cxcr3 promoter sequences were detected by ChIP-PCR and quantitative ChIP-PCR (qChIP-PCR) using the C/EBPα site-specific primer pairs in Cxcr3 promoter (Table 2). Genomic DNA fragments bound to C/EBPα were analyzed by PCR using random primers (P1 and P2) designed to respectively check C/EBPα binding site 1 and binding sites 2 and 3 in the Cxcr3 promoter region. Using P2 to check both binding sites 2 and 3 is because C/EBPα binding site 2 is positioned very close to site 3, and no suitable primers to distinguish C/EBPα binding site 2 from binding site 3. Primers for Dnmt3b enrichment of DNA sequence were identical to the ones used in C/EBPα ChIP-PCR. ChIP-PCR products were visualized by GelRed (Biotium) staining in 3.0% agarose gels. For qChIP-PCR, DNA samples and standards were analyzed using a LightCycler96 Real-Time PCR System (Roche Diagnostics) and FastStart Essential DNA Green Master for SYBR Green I-based real-time PCR (Roche Diagnostics).
ELISA.
Mouse CXCL10 ELISA kit was purchased from R&D Systems. Animals were transcardially perfused with PBS. Spinal cord tissues were homogenized in a lysis buffer containing protease and phosphatase inhibitors (Sigma-Aldrich). For each reaction in a 96-well plate, 100 μg of proteins were used, and ELISA was performed according to the manufacturer's protocol. The standard curve was included in each experiment.
Western blot.
Protein samples were prepared in the same way as for ELISA analysis. Protein samples (30 μg) were separated on SDS–PAGE gel and transferred to nitrocellulose blots. The blots were blocked with 5% milk and incubated overnight at 4°C with antibody against CXCR3 (rabbit, 1:200; Boster Biological Technology, catalog #BAO759), DNMT3b (goat, 1:600; Santa Cruz Biotechnology, catalog #sc-10236, RRID:AB_2094128), and pERK (phosphorylated extracellular signal-regulated kinase; 1:500; Cell Signaling Technology, catalog #9101, RRID:AB_331646). For loading control, the blots were incubated with GAPDH antibody (mouse, 1:20,000; Millipore, catalog #MAB374, RRID:AB_2107445). These blots were further incubated with HRP-conjugated secondary antibody, developed in ECL solution, and exposed onto Hyperfilm (Millipore) for 1–5 min. Specific bands were evaluated by apparent molecular size. The intensity of the selected bands was analyzed using ImageJ software (National Institutes of Health, RRID:SCR_003070).
Immunohistochemistry.
Animals were deeply anesthetized with isoflurane and perfused through the ascending aorta with PBS followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 m PB. After the perfusion, the L5 spinal cord segment was removed and postfixed in the same fixative overnight. Spinal cord sections (30 μm, free-floating) were cut in a cryostat and processed for immunofluorescence as we described previously (Gao et al., 2009). The sections were first blocked with 5% donkey serum for 2 h at room temperature, then incubated overnight at 4°C with the following primary antibodies: CXCR3 (rabbit, 1: 200; Boster Biological Technology, catalog #BAO759), neurokinin 1 receptor (NK1R; goat, 1:400; Santa Cruz Biotechnology, catalog #sc-14115, RRID:AB_2255736), CXCL10 (goat, 1:100; R&D Systems, catalog #AF-466-NA, RRID:AB_2292487), glial fibrillary acidic protein (GFAP; mouse, 1:5000; Millipore, catalog #MAB360, RRID:AB_2275415), NeuN (mouse, 1:1000; Millipore, catalog #MAB377, RRID:AB_2298772), IBA-1 (rabbit, 1:3000; Wako, catalog #019-19741, RRID:AB_839504), CD11b (mouse, 1:100; Serotec, catalog #MCA-257GA, RRID:AB_566455), C/EBPα (rabbit, 1:400; Boster Biological Technology, catalog #BAO669), pERK (rabbit, 1:500; Cell Signaling Technology, catalog #9101, RRID:AB_331646). The sections were then incubated for 1 h at room temperature with Cy3-conjugated or FITC-conjugated secondary antibodies (1:400; Jackson ImmunoResearch). For double immunofluorescence, sections were incubated with a mixture of different primary antibodies followed by a mixture of FITC-conjugated and Cy3-conjugated secondary antibodies. The stained sections were examined with a Leica fluorescence microscope, and images were captured with a CCD Spot camera.
In situ detection of Cxcr3 mRNA.
Digoxigenin (DIG)-labeled RNA antisense and sense probes for the Cxcr3 gene were produced using PCR and in vitro transcription with a DIG RNA Labeling Kit (SP6/T7; Roche Diagnostics). The template fragments were amplified using a set of primers (forward: 5′-AAC AGC ACC TCT CCC TAC GA-3′ and reverse: 5′-CAG CCA CTA GCT GCA GTA CA-3′) and then subcloned into pSPT18. Cellular localization of Cxcr3 was performed using a mouse Cxcr3 mRNA in situ hybridization assay kit (Boster Biological Technology) as described previously (Zhang et al., 2013). Briefly, in situ hybridizations were performed in 14 μm cryosections from the spinal cord. Sections were fixed in 4% paraformaldehyde/0.1 m PBS for 30 min followed by washes in DEPC-treated ultrapure water. Prehybridization procedures were performed under RNase-free conditions for 4 h at room temperature. Hybridization was performed with a hybridization probe specific to Cxcr3 at 42°C overnight in hybridization buffer. Sections were then incubated in blocking solution at 37°C for 30 min and in mouse anti-DIG-biotin for 60 min, washed, incubated using streptavitin–biotin complex-FITC reagent (Boster Biological Technology, SA1080) for 30 min.
To identify the cell types expressing Cxcr3 and colocalization of Cxcr3 with C/EBPα, the above sections under in situ hybridization were incubated overnight at 4°C with primary antibodies against GFAP (mouse, 1:6000; Millipore, catalog #MAB360, RRID:AB_2275415), NeuN (mouse, 1:1000; Millipore, catalog #MAB377, RRID:AB_2298772), IBA-1 (rabbit, 1:3000; Wako, catalog #019-19741, RRID:AB_839504), and C/EBPα (rabbit, 1:400; Boster Biological Technology, catalog #BAO669). On the following day, Cy3-conjugated secondary antibody was added and incubated for 2 h. The signal was detected with a Leica SP8 gated stimulated emission depletion confocal microscope (Leica Microsystems).
Lentiviral vectors production and intraspinal injection.
The shRNA targeting the sequence of mice Cxcr3 (Gene Bank Accession: NM_009910.2) was designed. An additional scrambled sequence was also designed as an NC. The recombinant lentivirus containing Cxcr3 shRNA (LV-Cxcr3 shRNA, 5′-CTG AAC TTT GAC AGA ACC T-3′) or NC shRNA (LV-NC, 5′-TTC TCC GAA CGT GTC ACG T-3′) was packaged using pGCSIL-GFP vector by Shanghai GeneChem. To test the knock-down effect, the Cxcr3-expressing plasmid and Cxcr3 shRNA plasmid were transfected to HEK293 cells. Two days after transfection, the cells were harvested and subjected to quantitative real-time PCR (RT-PCR). The coding sequences of Dnmt3b (GenBank Accession: NM_001122997.2) were synthesized by Sangon Biotech and cloned into pLV-Ubi-MCS-3FLAG to generate Dnmt3b-expressing lentiviral plasmid, which mediated transcription of Dnmt3b by ubiquitin promoter. Then the Dnmt3b-expressing plasmid and EGFP-expressing plasmid (control) were packaged into lentivirus. The intraspinal injection was performed as described previously (Zhang et al., 2013).
Behavioral analysis.
Animals were habituated to the testing environment daily for ≥2 d before baseline testing. All the behavioral experiments were done by individuals blinded to the treatment or genotypes of the mice. For heat hyperalgesia, the animals were put in a plastic box placed on a glass plate, and the plantar surface was exposed to a beam of radiant heat through a transparent glass surface (IITC model 390 Analgesia Meter, Life Science). The baseline latencies were adjusted to 12–15 s with a maximum of 20 s as cutoff to prevent potential injury (Hargreaves et al., 1988). For mechanical allodynia, the animals were put in boxes on an elevated metal mesh floor and allowed 30 min for habituation before examination. The plantar surface of the hindpaw was stimulated with a series of von Frey hairs with logarithmically incrementing stiffness (0.02–2.56 g, Stoelting). The 50% paw-withdrawal threshold was determined using Dixon's up–down method (Dixon, 1980). For the tail-immersion test, the temperature of the water was set at 48, 50, or 52°C. The tail-flick latency was recorded (Ramabadran et al., 1989). For the rotarod test, the speed was set at 10 rpm for 60 s and subsequently accelerated to 80 rpm in 5 min. The time taken for mice to fall after the beginning of the acceleration was recorded (Abbadie et al., 2003).
Spinal slice preparation.
The lumbar spinal cord was carefully removed from mice (4–6 weeks) under urethane anesthesia (1.5∼2 g/kg, i.p.) and placed in preoxygenated (saturated with 95% O2 and 5% CO2) ice-cold sucrose artificial CSF (aCSF) solution. The sucrose aCSF contained the following (in mm): 234 sucrose, 3.6 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 12 glucose, and 25 NaHCO3. The pia-arachnoid membrane was gently removed from the section. The portion of the lumbar spinal cord (L4–L5) was identified by the lumbar enlargement and large dorsal roots. The spinal segment was placed in a shallow groove formed in an agar block and then glued to the button stage of a VT1000S vibratome (Leica). Transverse slices (450 μm) were cut in the ice-cold sucrose aCSF, incubated in Krebs' solution oxygenated with 95% O2 and 5% CO2 at 34○C for 30 min, and then allowed to recover 1∼2 h at room temperature before the experiment. The Krebs' solution contained the following (in mm): 117 NaCl, 3.6 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11 glucose.
Patch-clamp recordings in spinal slices.
The voltage-clamp recordings were made from neurons in outer lamina II of the dorsal horn. The slice was continuously superfused (3–5 ml/min) with Krebs' solution in room temperature, and saturated with 95% O2 and 5% CO2. Individual neurons were visualized under a stage-fixed upright infrared differential interference contrast microscope (BX51WI, Olympus) equipped with a 40× water-immersion objective. The patch pipettes were pulled using a Flaming micropipette puller (P-97, Sutter Instruments), and had initial resistance of 5∼10 MΩ when filled with the internal pipette solution contained the following (in mm): 135 potassium gluconate, 5 KCl, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, and 5 Na2ATP. Membrane voltage and current were amplified with a multiclamp 700B amplifier (Molecular Devices). Data were filtered at 2 kHz and digitized at 10 kHz using a data acquisition interface (1440A, Molecular Devices). A seal resistance (>2 GΩ) and an access resistance (<35 MΩ) were considered acceptable. The cell capacity transients were cancelled by the capacitive cancelation circuitry on the amplifier. After establishing the whole-cell configuration, the membrane potential was held at −70 mV for recording sEPSCs. NMDA-induced or AMPA-induced current was recorded by puff application of NMDA (50 μm; Sigma-Aldrich) or AMPA (10 μm, Sigma-Aldrich) at the holding potential of −45 and −70 mV, respectively. Data were stored with a personal computer using pClamp10.0 software and analyzed with Mini Analysis (Synaptosoft 6.0). Those cells that showed >10% changes from the baseline levels were regarded as responsive to the presence of drugs.
Quantification and statistics.
All data were expressed as mean ± SEM. The behavioral data were analyzed by two-way repeated-measures ANOVA followed by Bonferroni's test as the post hoc multiple-comparison analysis. For Western blot, the density of specific bands was measured with ImageJ. The levels of CXCR3, DNMT3b, C/EBPα, and pERK were normalized to loading control GAPDH. Differences between groups were compared using one-way ANOVA followed by Bonferroni's test. Student's t test was applied when only two groups needed to be compared. The criterion for statistical significance was p < 0.05.
Results
CXCR3 expression is upregulated in spinal neurons after SNL
SNL induces rapid (1 d) and persistent (>21 d) mechanical allodynia and heat hyperalgesia (Zhang et al., 2013). We first examined Cxcr3 expression in the spinal cord at days 1, 3, 10, and 21 after SNL. SNL induced persistent Cxcr3 mRNA upregulation, which started at day 3, peaked at day 10, and was still elevated at day 21 (Fig. 1A). CXCR3 protein level was also significantly increased 10 d after SNL (Fig. 1B). Immunostaining revealed basal expression of CXCR3 in the dorsal horn in naive mice (Fig. 1C). CXCR3 immunoreactivity was markedly increased in the ipsilateral dorsal horn (Fig. 1D), but not in the contralateral dorsal horn (Fig. 1E) 10 d after SNL.
Figure 1.
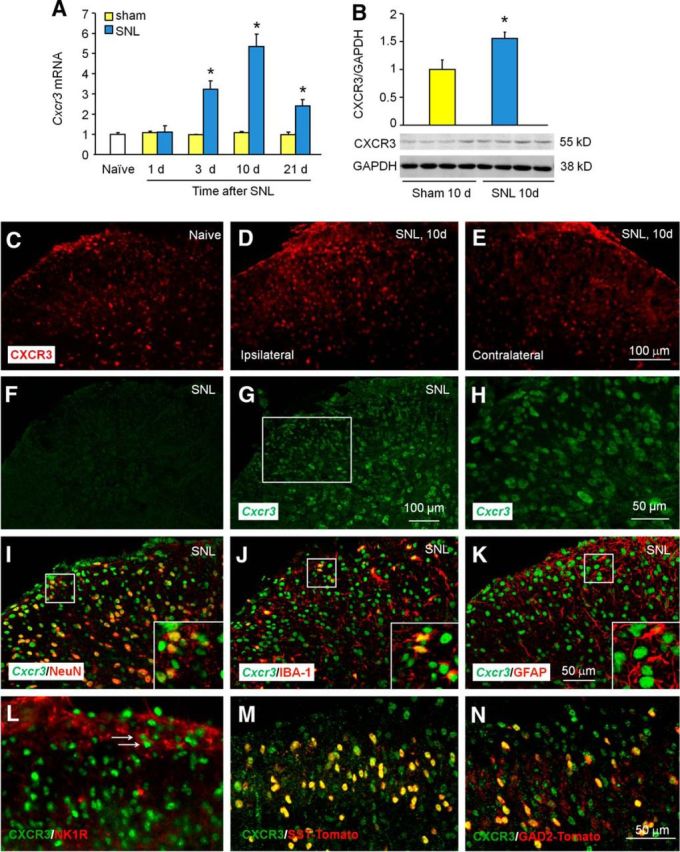
CXCR3 expression is increased mainly in spinal neurons after SNL. A, Time course of Cxcr3 mRNA expression in the ipsilateral dorsal horn in naive, sham-operated, and SNL mice. Cxcr3 expression at 3, 10, and 21 d increased more in SNL mice than in sham-operated mice. *p < 0.05, Student's t test. n = 4–5 mice/group. B, Western blotting shows the increase of CXCR3 protein in the spinal cord 10 d after SNL. *p < 0.05, Student's t test, n = 4 mice/group. C–E, Representative images of CXCR3 immunofluorescence in the spinal cord from naive and SNL mice. CXCR3 was constitutively expressed in naive mice (C), remarkably increased in the ipsilateral dorsal horn of SNL mice (D), but not in the contralateral side (E). F–H, In situ hybridization of Cxcr3 mRNA shows that no signals were found in spinal sections incubated with Cxcr3 sense probe (F), but positive signals were apparent in sections incubated with antisense probe (G, H). H, High-magnification image of G. I–K, In situ hybridization of Cxcr3 mRNA and immunofluorescence staining of NeuN (I), IBA-1 (J), and GFAP (K) shows that Cxcr3 mRNA was predominantly colocalized with neuronal marker NeuN, rarely with microglia marker IBA-1, and not at all with astrocyte marker GFAP. L, Double staining of NK1R and CXCR3 in the spinal dorsal horn. M, N, Immunofluorescence staining of CXCR3 on the spinal cord from SST-Tomato+ (M) and GAD2-Tomato+ (N) mice 10 d after SNL.
To define the cellular localization of CXCR3 in the spinal cord, we performed in situ hybridization using antisense probes for Cxcr3 on spinal sections 10 d after SNL. A Cxcr3-positive signal was not evident in sections incubated with the Cxcr3 sense probe (Fig. 1F), but was obvious in sections with the antisense probe (Fig. 1G,H). In situ hybridization combined with immunostaining showed that Cxcr3 mRNA was highly colocalized with neuronal marker NeuN (Fig. 1I), partially colocalized with microglial marker IBA-1 (Fig. 1J), but not at all colocalized with astrocytic marker GFAP (Fig. 1K), suggesting the predominant expression of Cxcr3 by spinal neurons.
We further characterized the type of CXCR3+ neurons. NK1R is expressed by ∼80% of ascending projection neurons in lamina I (Todd, 2010). Double staining of NK1R and CXCR3 showed that CXCR3 was expressed in NK1R-positive neurons in lamina I (Fig. 1L). As SST and GAD2 are respectively expressed in excitatory and inhibitory neurons (Zeilhofer et al., 2012; Duan et al., 2014), we performed CXCR3 staining on SST-Tomato+ or GAD2-Tomato+ mice. It showed that both SST-Tomato+ neurons and GAD2-Tomato+ neurons can express CXCR3, and the percentages of these neurons in CXCR3+ cells were 57 ± 2% and 32 ± 1%, respectively, in laminae I–IV (Fig. 1M,N). These data suggest that CXCR3 is widely distributed in all three populations of neurons.
Demethylation of the CpG sites of the Cxcr3 gene promoter region after SNL
DNA methylation is an important epigenetic mechanism controlling gene expression (Jaenisch and Bird, 2003). To determine whether DNA methylation contributes to Cxcr3 upregulation, we examined the methylation status of the Cxcr3 promoter region by MSP and BSP assays. The genomic structure of the Cxcr3 gene contains one CpG dinucleotide region with nine CpG sites around the transcriptional starting site (TSS; Fig. 2A). The MSP assay showed that the methylation degree of Cxcr3 promoter in the spinal cord of SNL mice was lower than that in sham-operated mice (Fig. 2B). To further confirm the methylation status of the nine CpG sites within Cxcr3 promoter, DNA sequencing was performed on PCR products of the 325 bp fragment obtained after the treatment of genomic DNA samples with sodium bisulfite. As shown in Figure 2C, all spinal cord samples were successfully sequenced. Consistent with the MSP assay, DNA methylation in CpG dinucleotide regions of the Cxcr3 gene was less in SNL mice compared with sham-operated mice (Fig. 2D).
Figure 2.
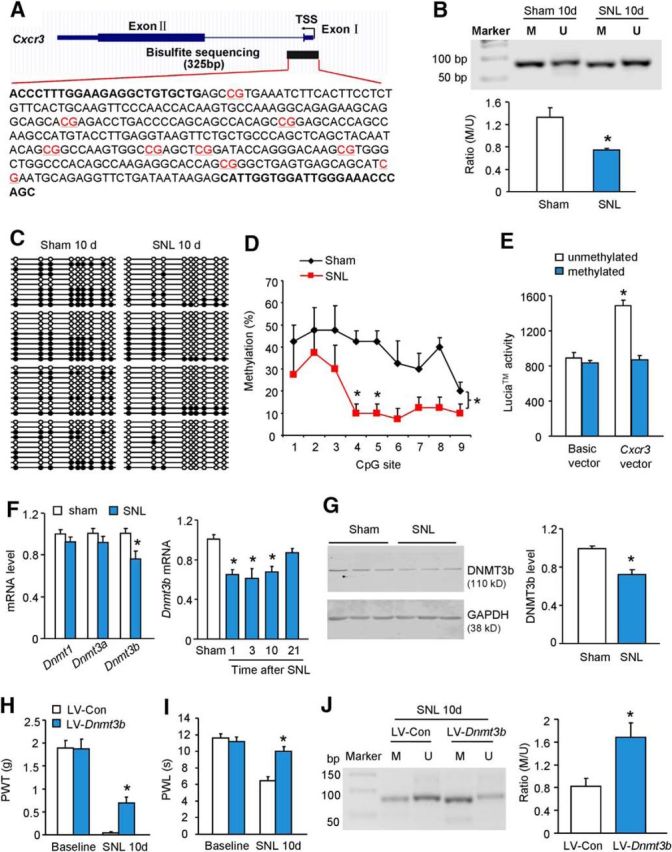
Demethylation of Cxcr3 gene promoter region after SNL. A, Schematic of a CpG island showing the locations of the nine CpG sites (in red) in the Cxcr3 gene promoter area. B, Representative PCR results of the Cxcr3 promoter region using primers for methylation-specific or unmethylation-specific amplifications of genomic DNA from the spinal cord of sham-operated or SNL-operated mice. SNL reduced the ratio of methylated (M) to unmethylated (U) products of CpG sites. *p < 0.05, SNL versus sham, Student's t test, n = 3 mice/group. C, Bisulfite sequencing data of Cxcr3 promoter of spinal dorsal horn from SNL (n = 4) and sham-operated (n = 4) mice. Ten clones were randomly collected from each mouse. Filled circles, Methylated CpG sites; unfilled circles, unmethylated CpG sites. D, Overall methylation of Cxcr3 promoter was decreased in SNL mice. *p < 0.05, SNL versus sham, two-way ANOVA, n = 4 mice/group. E, Luciferases assay using coelenterazine shows that the luciferase activity was increased when HEK293 cells were transfected with unmethylated pCpG-free-Cxcr3-promoter-Lucia vector compared with that transfected with methylated pCpG-free-Cxcr3-promoter-Lucia vector. *p < 0.05, unmethylated versus methylated, n = 3/group. F, Dnmt1 mRNA and Dnmt3a mRNA were not changed 10 d after SNL. However, Dnmt3b mRNA level decreased at days 1, 3, and 10 after SNL. *p < 0.05, one-way ANOVA, n = 5–6 mice/group. G, DNMT3b protein was decreased in the spinal cord 10 d after SNL. *p < 0.05, Student's t test, n = 3 mice/group. H, I, Intraspinal injection of LV-Dnmt3b, 7 d before SNL, attenuated SNL-induced mechanical allodynia (H) and heat hyperalgesia (I). *p < 0.05, LV-Dnmt3b versus LV-Con, two-way repeated-measures ANOVA followed by Bonferroni's tests, n = 7 mice/group. J, Pretreatment with LV-Dnmt3b increased the methylation of Cxcr3 promoter in the spinal cord 10 d after SNL. *p < 0.05, LV-Dnmt3b vs LV-Con, Student's t test, n = 4–6 mice/group.
The effect of DNA methylation on Cxcr3 promoter activity was further analyzed in an in vitro cell culture system using a luciferase assay employing coelenterazine. The luciferase activity was not different between transfection with methylated pCpG-free and unmethylated pCpG-free basic reporter vector in HEK293 cells (Fig. 2E). However, luciferase activity for cells transfected with unmethylated pCpG-free-Cxcr3-promoter-Lucia vector was more than that of cells transfected with methylated pCpG-free-Cxcr3-promoter-Lucia vector (Fig. 2E), indicating that the promoter activity of the mouse Cxcr3 gene is increased by DNA demethylation.
Knowing that DNMTs are important in directly regulating DNA methylation (Jurkowska et al., 2011), we then examined the expression of three Dnmts: Dnmt1, Dnmt3a, and Dnmt3b in the spinal cord 10 d after SNL. As shown in Figure 2F, Dnmt3b, but not Dnmt1 or Dnmt3a, decreased in the spinal cord after SNL. The time course of Dnmt3b mRNA expression showed that the Dnmt3b level was reduced at days 1, 3, and 10 (Fig. 2F). DNMT3b protein was also remarkably decreased in the spinal cord after SNL (Fig. 2G). These data suggest that downregulated DNMT3b may be responsible for the demethylation of Cxcr3 promoter in neuropathic pain condition.
To check whether DNMT3b could directly regulate Cxcr3 methylation, we intraspinally injected Dnmt3b-expressing lentivirus (LV-Dnmt3b) and control lentivirus (LV-Con). Seven days after injection, LV-Dnmt3b effectively increased Dnmt3b mRNA expression by 10.7 ± 2.2-fold (p < 0.05, vs LV-Con, n = 7 mice per group) in the spinal cord. In addition, injection of LV-Dnmt3b 7 d before SNL significantly alleviated SNL-induced mechanical allodynia (Fig. 2H) and heat hyperalgesia (Fig. 2I), besides increasing the methylation of Cxcr3 promoter in the spinal cord 10 d after SNL (Fig. 2J). These data confirmed the regulation of DNMT3b on Cxcr3 methylation and the involvement of DNMT3b in the pathogenesis of neuropathic pain.
Increased binding of C/EBPα with Cxcr3 gene promoter is associated with decreased binding of DNMT3b with Cxcr3 gene promoter after SNL
DNA methylation can regulate transcription by interfering with transcription factor binding (Poetsch and Plass, 2011). To reveal the transcriptional factor that may regulate Cxcr3 expression, the sequence from −1500 to +500 of Cxcr3 promoter was analyzed. Three C/EBPα binding sites (site 1: at 4–14 from TSS; site 2: at 256–267 from TSS; site 3: at 337–347 from TSS; Fig. 3A) were predicted within the Cxcr3 promoter region based on Core vertebrata Jaspar matrix models with a defined 80% profile score threshold (http://jaspar.genereg.net/). A conservation analysis using the University of California Santa Cruz genome browser showed that sites 1 and 2, but not site 3, are well conserved among mice, rats, and humans (data not shown).
Figure 3.
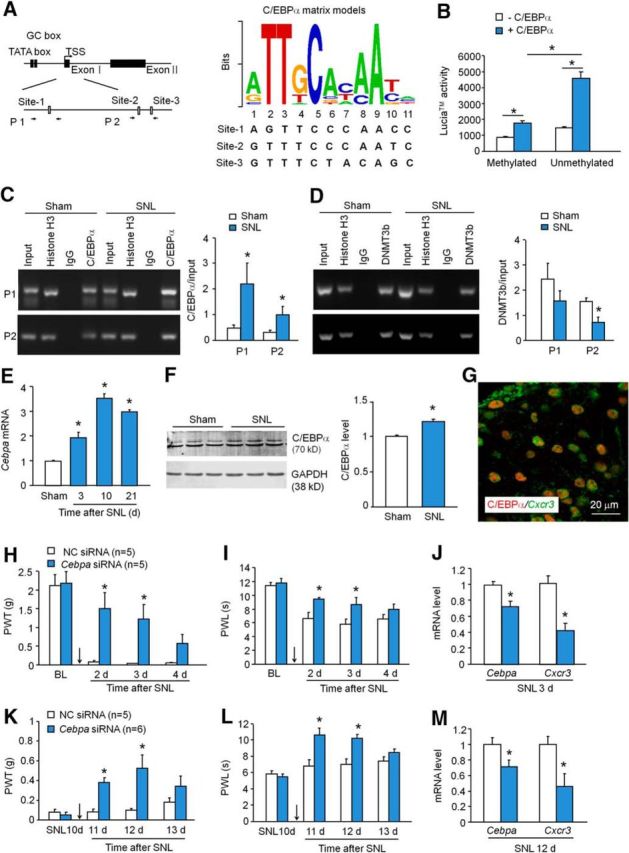
C/EBPα increases CXCR3 expression in the spinal cord after SNL. A, Schematic of Cxcr3 gene promoter showing locations of the three binding sites for C/EBPα with Cxcr3 within the CpG islands (left). P1 and P2 were ChIP-PCR primer pairs. Right, The logos of the standard C/EBPα motif and three predicted C/EBPα binding sites in Cxcr3 gene promoter. B, Cotransfection of C/EBPα-expressing vector with unmethylated pCpG-free-Cxcr3-promoter-Lucia vector dramatically increased the luciferase activity. *p < 0.05, two-way ANOVA. C, ChIP assay shows the increased binding of C/EBPα with the binding sites 1 and 2 at Cxcr3 promoter after SNL. *p < 0.05, Student's t test, n = 5–6 mice/group. D, The binding of DMNT3b with Cxcr3 promoter on the binding site 2 was decreased after SNL. *p < 0.05, Student's t test, n = 4 mice/group. E, Cebpa mRNA expression was increased at days 3, 10, and 21 after SNL. p < 0.05, one-way ANOVA, n = 3 mice/group. F, C/EBPα protein was increased 3 d in the spinal cord after SNL. p < 0.05, Student's t test. G, Immunofluorescence staining of C/EBPα and in situ hybridization of Cxcr3 mRNA shows that C/EBPα was highly colocalized with Cxcr3 in the spinal cord dorsal horn 10 d after SNL. H, I, Intrathecal injection of C/EBPα siRNA 1 d before SNL attenuated SNL-induced mechanical allodynia (H) and heat hyperalgesia (I) for 2 d. *p < 0.05, two-way repeated-measures ANOVA followed by Bonferroni's tests. J, Pretreatment with Cebpa siRNA reduced the expression of Cebpa mRNA and Cxcr3 mRNA in the spinal cord 2 d after SNL. *p < 0.05, Student's t test, n = 5 mice/group. K–M, Intrathecal injection of C/EBPα siRNA 10 d after SNL attenuated mechanical allodynia (K) and heat hyperalgesia (L), and also reduced mRNA expression of Cebpa and Cxcr3 in the spinal cord (M). *p < 0.05, n = 5–6 mice/group.
To determine the effect of C/EBPα on Cxcr3 expression, we conducted a luciferase activity assay in HEK293 cells. C/EBPα-expressing vector and pCpG-free-Cxcr3-promoter-Lucia vector (methylated or unmethylated) were cotransfected. As shown in Figure 3B, in HEK293 cells transfected with methylated pCpG-free-Cxcr3-promoter-Lucia vector, cotransfection of C/EBPα-expressing vectors slightly increased the promoter luciferase activity, compared with absence of C/EBPα-expressing vector. However, in HEK293 cells transfected with unmethylated pCpG-free-Cxcr3-promoter-Lucia vector, cotransfection of C/EBPα-expressing vector dramatically increased the luciferase activity, compared with absence of C/EBPα-expressing vector. In addition, C/EBPα induced more luciferase activity increase in cells transfected with unmethylated pCpG-free-Cxcr3-promoter-Lucia vector than in cells transfected with methylated pCpG-free-Cxcr3-promoter-Lucia vector (Fig. 3B). These results suggest that C/EBPα can increase the transcription of the Cxcr3 gene and that this increase is further enhanced when Cxcr3 promoter is demethylated.
We then examined the binding of C/EBPα with Cxcr3 promoter in the spinal cord after SNL by ChIP-PCR. After C/EBPα antibody immunoprecipitation, the DNA fragments amplified by primer pairs either P1 (primers for site 1) or P2 (primers for sites 2 and 3) were increased in the spinal cord of SNL mice compared with sham-operated mice (Fig. 3C), indicating that the binding of C/EBPα with Cxcr3 promoter in the spinal cord is specific and also enhanced by SNL.
To further check whether the increased binding of C/EBPα with Cxcr3 promoter is associated with the decreased binding of DNMT3b with Cxcr3 promoter, we examined the binding of Dnmt3b with Cxcr3 in the same binding regions of C/EBPα with Cxcr3 promoter. ChIP-PCR using primers P2, but not P1, revealed that the binding of DNMT3b to Cxcr3 promoter regions was decreased in SNL mice compared with sham-operated mice (Fig. 3D). These data suggest that the decreased binding of DNMT3b with Cxcr3 promoter at binding sites 2 and 3 demethylates Cxcr3 promoter and further facilitates the binding of C/EBPα with Cxcr3 promoter.
Cebpa siRNA attenuates SNL-induced pain hypersensitivity and decreases Cxcr3 expression in the spinal cord
We then examined C/EBPα expression in the spinal cord after SNL. RT-PCR showed that Cebpa mRNA was significantly increased at days 3, 10, and 21 after SNL (Fig. 3E). Western blot further confirmed the increase of C/EBPα protein 10 d after SNL (Fig. 3F). Immunofluorescence staining combined with in situ hybridization showed that C/EBPα was highly colocalized with Cxcr3 mRNA 10 d after SNL (Fig. 3G), supporting the regulation of C/EBPα on CXCR3 in spinal neurons.
To determine whether C/EBPα plays a role in the development of SNL-induced neuropathic pain, we intrathecally injected Cebpa siRNA 1 d before SNL. Behavioral data showed that Cebpa siRNA dramatically increased the paw-withdrawal threshold, compared with NC-injected mice, 2 and 3 d after SNL, but this effect was insignificant at day 4 (Fig. 3H). Meanwhile, Cebpa siRNA also attenuated SNL-induced heat hyperalgesia at days 2 and 3 (Fig. 3I). To confirm the knock-down effect of Cebpa siRNA, we checked mRNA level in another sets of animals 3 d after SNL. As shown in Figure 3J, Cebpa siRNA significantly reduced Cebpa mRNA expression and also reduced Cxcr3 mRNA expression in the spinal cord.
To check whether CEBP/α is involved in the maintenance of neuropathic pain, we intrathecally injected Cebpa siRNA 10 d after SNL. As shown in Figure 3K,L, Cebpa siRNA attenuated both mechanical allodynia (Fig. 3K) and heat hyperalgesia (Fig. 3L) at 1 and 2 d postinjection. The same treatment also reduced the expression of Cebpa mRNA and Cxcr3 mRNA (Fig. 3M). These results suggest that C/EBPα regulates CXCR3 expression and is involved in SNL-induced neuropathic pain.
Deletion or inhibition of CXCR3 persistently attenuates SNL-induced pain hypersensitivity
To determine the role of CXCR3 in pain sensation, we first examined whether acute pain or neuropathic pain are changed in Cxcr3 knock-out (KO; Cxcr3−/−) mice. Acute thermal sensitivity tested by hot-water immersion (Fig. 4A) and radiant heat (Fig. 4B) were indistinguishable in WT and Cxcr3−/− mice. Acute mechanical sensitivity was not different between the two genotypes either (Fig. 4C). The rotarod test revealed a similar falling latency in WT and Cxcr3−/− mice (Fig. 4D). These data indicate that Cxcr3 deletion did not cause deficits in acute pain sensation and motor function. Consistently, Cxcr3−/− mice show normal distribution patterns of the neurochemical markers NeuN and PKCγ, astrocyte marker GFAP, and microglia marker IBA-1. The mice also show normal innervations of the primary afferents, labeled with CGRP and IB4 in the spinal cord dorsal horn (Fig. 4E–P).
Figure 4.
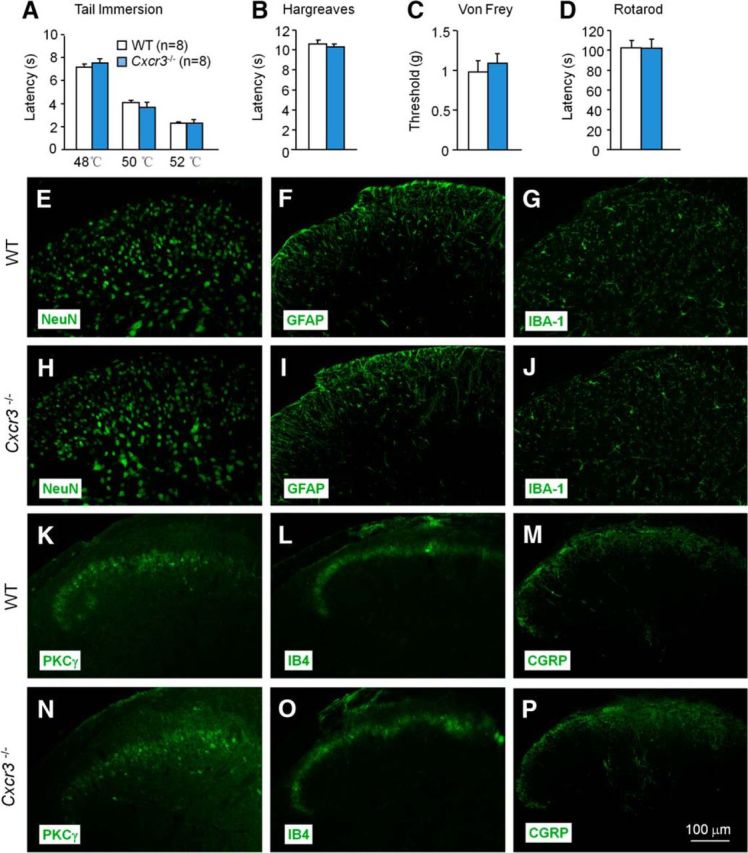
Cxcr3 KO mice are normal in basal pain and in the expression of cellular markers and neurochemical markers. A–D, Cxcr3 KO mice show normal acute pain threshold and motor function. Acute pain thresholds tested by tail immersion (A), Hargreaves test (B), and von Frey test (C). D, Motor function assessed by recording the falling latency on a rotarod. E–P, The distribution of the cellular marker NeuN (E, H), GFAP (F, I), and IBA-1(G, J), and of neurochemical markers PKCγ (K, N), IB4 (L, O), and CGRP (M, P) in the spinal dorsal horn of WT and Cxcr3 KO mice.
Next, we tested pain behaviors after SNL. Consistent with previous reports (Jiang et al., 2016), SNL induced persistent mechanical allodynia (Fig. 5A) and heat hyperalgesia (Fig. 5B) in WT mice. However, SNL-induced mechanical allodynia was substantially reduced in Cxcr3−/− mice from 3 to 28 d (Fig. 5A). In addition, heat hyperalgesia was also markedly reduced in Cxcr3−/− mice at 28 d (Fig. 5B).
Figure 5.
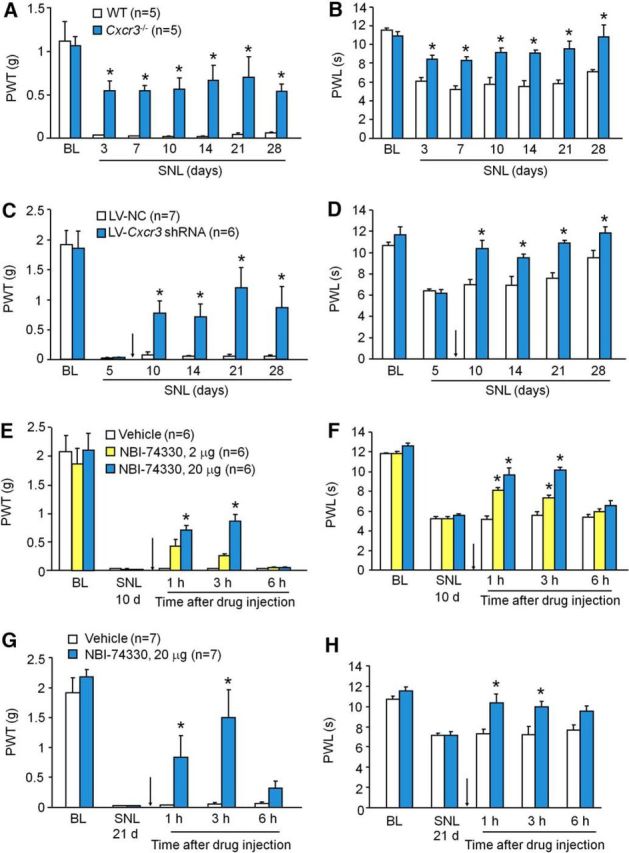
Deletion or inhibition of CXCR3 alleviates SNL-induced neuropathic pain. A, B, SNL-induced mechanical allodynia (A) and heat hyperalgesia (B) were markedly attenuated in Cxcr3−/− mice. p < 0.05, Cxcr3−/− versus WT. Two-way repeated-measures ANOVA followed by Bonferroni's tests. C, D, Intraspinal infusion of LV-Cxcr3 shRNA lentivirus in the spinal cord 5 d after SNL persistently alleviated SNL-induced mechanical allodynia (C) and reversed heat hyperalgesia (D). *p < 0.05, LV-Cxcr3 shRNA versus LV-NC. Two-way repeated-measures ANOVA followed by Bonferroni's tests. E, F, Intrathecal injection of CXCR3 antagonist NBI-774330 10 d after SNL transiently and dose-dependently attenuated SNL-induced mechanical allodynia (E) and heat hyperalgesia (F). G, H, Intrathecal injection of NBI-774330 21 d after SNL also attenuated SNL-induced mechanical allodynia (G) and heat hyperalgesia (H). *p < 0.05, NBI-74330 versus vehicle. Two-way repeated-measures ANOVA followed by Bonferroni's tests.
To further investigate the role of spinal CXCR3 in the maintenance of neuropathic pain, we intraspinally injected Cxcr3 shRNA lentivirus (LV-Cxcr3 shRNA) and LV-NC 5 d after SNL in WT mice. An in vitro study in HEK293 cells showed that LV-Cxcr3 shRNA reduced Cxcr3 expression by 87.0 ± 0.5% compared with LV-NC (p < 0.05, n = 4). Intraspinal injection of LV-Cxcr3 shRNA attenuated SNL-induced mechanical allodynia from 10 to 28 d after SNL (Fig. 5C). LV-Cxcr3 shRNA also reversed SNL-induced heat hyperalgesia (Fig. 5D). qPCR data showed that spinal injection of LV-Cxcr3 shRNA reduced Cxcr3 mRNA expression in lumbar segments by 82.9 ± 12.7% compared with LV-NC 21 d after SNL (p < 0.05, n = 3), confirming the knock-down effect of LV-Cxcr3 shRNA.
To further check whether a single bolus injection of CXCR3 antagonist could attenuate established neuropathic pain, NBI-74330, a potent and selective CXCR3 antagonist, was intrathecally injected 10 d after SNL. NBI-74330 at the dose of 20 μg significantly attenuated the threshold at 1 and 3 h (Fig. 5E). For the thermal test, NBI-74330 at the dose of 2 μg alleviated SNL-induced heat hyperalgesia at 1 and 3 h. The high dose (20 μg) reversed SNL-induced heat hyperalgesia (Fig. 5F). Furthermore, intrathecal injection of NBI-74330 (20 μg) 21 d after SNL also markedly relieved SNL-induced mechanical allodynia (Fig. 5G) and heat hyperalgesia (Fig. 5H). The same effect was observed when NBI-74330 was injected 28 d after SNL (data not shown). Collectively, the behavioral results obtained by different strategies suggest that CXCR3 play a pivotal role in the maintenance of neuropathic pain.
SNL increases CXCL10 expression in spinal neurons and astrocytes
CXCL10 is the major ligand of CXCR3 (Ransohoff et al., 2007). We examined the expression of CXCL10 in the spinal cord after SNL or sham operation. As shown in Figure 6A, Cxcl10 mRNA was significantly increased at day 3, peaked at day 10, and was still elevated at day 21 in SNL mice compared with sham-operated mice. The mRNA level did not significantly differ between naive and sham-operated mice at any time point (Fig. 6A). ELISA results further confirmed the increased expression of CXCL10 protein in the spinal cord 10 d after SNL (Fig. 6B).
Figure 6.

CXCL10 is increased in spinal neurons and astrocytes after SNL. A, The time course of Cxcl10 mRNA expression in the spinal cord from naive, sham-operated, and SNL-operated mice. SNL increased Cxcl10 expression at 3, 10, and 21 d. *p < 0.05, SNL versus sham, Student's t test, n = 5 mice/group. B, ELISA shows increased CXCL10 protein levels in the spinal cord 10 d after SNL. *p < 0.05, SNL vs sham, Student's t test, n = 3 mice/group. C–E, Representative images of CXCL10 immunofluorescence in the L5 dorsal horn. CXCL10 immunoreactivity was constitutively expressed in naive mice (C), with similar expression in sham-operated mice (D) and increased expression in SNL mice (E). F–H, Immunofluorescence double staining shows that CXCL10 was highly colocalized with NeuN (F), slightly colocalized with GFAP (G), and not at all colocalized with CD11b (H) in naive mice. I–K, CXCL10 was colocalized with NeuN (I) and GFAP (J), but not with CD11b (K) 10 d after SNL.
We then examined the expression and distribution of CXCL10 in the spinal cord by immunofluorescence staining. CXCL10 was expressed in the dorsal horn of naive mice (Fig. 6C) and sham-operated mice (Fig. 6D). At SNL day 10, CXCL10 immunoreactivity was markedly increased in the ipsilateral dorsal horn (Fig. 6E). As the cellular distribution of CXCL10 in naive and SNL mice seems different, we further did double staining. In naive mice, CXCL10 was highly colocalized with NeuN (Fig. 6F), sparely colocalized with GFAP (Fig. 6G), but not colocalized at all with CD11b (Fig. 6H). In SNL mice, in addition to the colocalization with NeuN (Fig. 6I), CXCL10 was highly colocalized with GFAP in superficial lamina (Fig. 6J), and not colocalized with CD11b (Fig. 6K). These results suggest that CXCL10 expression was constitutively expressed in spinal neurons and was increased in both neurons and astrocytes after SNL.
CXCL10 enhances excitatory synaptic transmission in spinal cord neurons via CXCR3
Since CXCR3 is mainly expressed in spinal neurons, we asked whether CXCL10 regulates neuronal function via CXCR3. We prepared spinal cord slices and performed patch-clamp recordings in lamina II neurons in which nociceptive information is modulated and conveyed to projection neurons (Kawasaki et al., 2008). We first recorded sEPSCs from WT mice. As shown in Figure 7A,B, superfusion of CXCL10 (100 ng/ml) significantly increased the amplitude of sEPSCs in six (40.0%) neurons (1.26 ± 0.04-fold) of 15 neurons recorded from naive mice. However, the frequency was not significantly changed (1.17 ± 0.15-fold). Given that the expression of CXCR3 was increased after SNL (Fig. 1D), we further examined sEPSCs from mice 10 d after SNL. The same concentration of CXCL10 induced a similar increase of the amplitude of sEPSCs in 53.8% of neurons (1.22 ± 0.04-fold). In addition, the frequency of sEPSCs was increased in neurons from SNL mice (1.55 ± 0.16-fold; Fig. 7A,B). As the amplitude and frequency of the sEPSCs respectively reflects postsynaptic glutamate receptor function and presynaptic glutamate release (Kohno et al., 2005), these data suggest that CXCL10 contributes to synaptic transmission by postsynaptic mechanisms in naive mice and by both postsynaptic and presynaptic mechanisms in mice with SNL.
Figure 7.

CXCL10 enhances excitatory synaptic transmission in lamina II neurons. A, Patch-clamp recording of sEPSCs shows an increase in the amplitude of sEPSCs from WT naive animals and in both amplitude and frequency from WT SNL animals after perfusion of CXCL10 (100 ng/ml, 2 min). However, neither amplitude nor frequency was changed after perfusion of CXCL10 in slices from KO mice. a1 and a2 are enlarged recordings before and after CXCL10 treatment, respectively. B, The ratio of amplitude and frequency of sEPSCs after CXCL10 treatment in WT mice. *p < 0.05 versus pretreatment baseline, Student's t test, n = 4–5 mice/group. C–F, Patch-clamp recording demonstrates increase in NMDA-induced (C, D) and AMPA-induced currents (E, F) from WT mice after CXCL10 treatment (100 ng/ml). CXCL10 did not change the amplitude of NMDA-induced or AMPA-induced currents in KO mice (C, E). The amplitude of NMDA-induced and AMPA-induced currents from WT mice is shown in F and H, respectively. *p < 0.05 versus pretreatment baseline, Student's t test, n = 4–6 mice/group. Inside each column, the number of total recorded neurons and number of responding neurons are indicated (B, D, F).
We then recorded sEPSCs from Cxcr3 KO mice. Of 11 neurons recorded from mice, the sEPSC amplitude was not affected by CXCL10 (100 ng/ml) in nine. Thus, the amplitude was increased only in two (18.2%) neurons. In slices from SNL mice, none of the 11 neurons responded to CXCL10 (Fig. 7A). These data suggest that CXCR3 is important in CXCL10-mediated excitatory transmission of lamina II neurons.
As excitatory synaptic transmission is mainly mediated by NMDA and AMPA receptors, we further examined the effects of the CXCL10 on inward currents induced by NMDA (50 μm, 30 s) or AMPA (10 μm, 30 s) when holding the voltage at −45 and −70 mV, respectively. In WT mice, CXCL10 (100 ng/ml) significantly enhanced the inward currents elicited by NMDA in 44.4% (four of nine) neurons from naive (1.18 ± 0.03-fold) and 77.8% (seven of nine) neurons from SNL (1.33 ± 0.09-fold, Fig. 7C,D). However, in Cxcr3 KO mice, most of the neurons were unaffected by CXCL10 (Fig. 6C). CXCL10 enhanced NMDA-induced currents in only 27.2% (3 of 11) neurons from naive mice and 18.2% (2 of 11) neurons from SNL mice. For AMPA-induced inward currents, CXCL10 (100 ng/ml) enhanced the currents in 33.3% (three of nine) neurons from naïve WT mice (1.25 ± 0.05-fold, p < 0.05) and 70.0% (7 of 10) neurons from SNL WT mice (1.33 ± 0.05-fold, Fig. 6E,F). In KO mice, CXCL10 did not change AMPA-induced currents in most of the cells (Fig. 7E). The amplitude in only 18.2% (2 of 11) neurons from naive mice and 20% (2 of 10) from SNL mice was increased by CXCL10. These data suggest that (1) SNL increases the percentage of CXCL10-responding neurons, and (2) CXCL10 enhances NMDA-induced or AMPA-induced current via CXCR3. In addition, as CXCL10 increased sEPSCs and NMDA-induced or AMPA-induced currents in ∼20% of the neurons in Cxcr3 KO mice, researchers need to investigate whether CXCL10 has other receptors in the spinal cord.
Spinal injection of CXCL10 induces CXCR3-dependent pain hypersensitivity and ERK activation in the spinal cord
To investigate whether CXCL10 is sufficient to induce pain, we intrathecally injected CXCL10 in WT and Cxcr3 KO mice. CXCL10, but not PBS induced mechanical allodynia in WT mice for >6 h (Fig. 8A), whereas CXCL10 did not significantly reduce the paw-withdrawal threshold in Cxcr3 KO mice. Intrathecal CXCL10 also induced heat hyperalgesia in WT mice, but this hyperalgesia was abrogated in KO mice (Fig. 8B). These data indicate that CXCL10 is sufficient to induce pain hypersensitivity via its receptor CXCR3.
Figure 8.

CXCL10 induces CXCR3-dependent pain hypersensitivity and ERK activation in spinal neurons. A, B, Intrathecal injection of CXCL10 (100 ng) induced mechanical allodynia (A) and heat hyperalgesia (B) in WT mice, but not in KO mice. *p < 0.05, two-way repeated-measures ANOVA followed by Bonferroni's tests. C, Intrathecal injection of CXCL10 increased pERK expression at 1 h in WT mice, but not in Cxcr3 KO mice. p < 0.05, Student's t test, n = 3 mice/group. D, E, pERK expression in the spinal cord after incubation of the isolated spinal cord slices with CXCL10 (100 ng/ml, 5 min). CXCL10 induces marked ERK activation in the superficial dorsal horn. E–G, Majority of pERK-immunoreactive cells were colocalized with NeuN.
Knowing that activation (phosphorylation) of ERK in dorsal horn neurons could serve as a marker for central sensitization (Ji et al., 1999; Gao and Ji, 2009; Gao et al., 2009), we then checked pERK expression in the spinal cord after intrathecal CXCL10. CXCL10 (100 ng) induced pERK expression in the spinal dorsal horn in WT mice but not in Cxcr3 KO mice 1 h after injection (Fig. 8C). Further, incubation of the spinal slices with CXCL10 (100 ng/ml, 5 min) induced a marked ERK activation in the superficial spinal cord, predominantly in the laminae I–III (Fig. 8D). The number of pERK-immunoreactive cells per spinal cord section increased from 5.6 ± 0.4 in control sections (n = 8) to 22.5 ± 2.1 in sections treated with CXCL10 (n = 10). Double staining showed that pERK was colocalized with NeuN (Fig. 8E–G), indicating that CXCL10 can activate spinal neurons.
Discussion
Accumulating evidence has shown that several chemokines and chemokine receptors are upregulated in the spinal cord in neuropathic pain (Verge et al., 2004; Zhuang et al., 2007; Gao et al., 2009; Zhang et al., 2013; Jiang et al., 2016). However, the mechanisms underlying the upregulation of these genes are not well investigated. Our study provides the first evidence that DNA demethylation of Cxcr3 promoter in spinal neurons is involved in CXCR3 upregulation after SNL. Our results further demonstrate that Cxcr3 demethylation, which may be caused by the downregulation of DNMT3b and decreased binding of DNMT3b with Cxcr3, increases the binding of transcription factor C/EBPα with Cxcr3 and thus facilitates CXCR3 expression (Fig. 9A). In addition, inhibition of CXCR3 effectively attenuates neuropathic pain in mice. Finally, we found that CXCL10, a major ligand of CXCR3, is induced in spinal astrocytes and further acts on neuronal CXCR3 to enhance the excitatory synaptic transmission (Fig. 9B). Thus, our results reveal an epigenetic mechanism underlying CXCR3 expression and the important role of CXCR3 in mediating neuropathic pain.
Figure 9.

Schematic shows the regulation of CXCR3 expression and the involvement of CXCL10/CXCR3 in neuropathic pain. A, SNL decreases the expression of DNMT3b and increased the expression of C/EBPα in the neurons of the spinal dorsal horn. The decreased DNMT3b causes DNA demethylation of Cxcr3 promoter, which increases the binding of C/EBPα to Cxcr3 promoter and further increases the transcription of Cxcr3 mRNA and the expression of CXCR3 protein on cytoplasm and membrane. B, SNL increases CXCL10 expression in spinal astrocytes and neurons. The released CXCL10 acts on neuronal CXCR3, which activates ERK. Activation of ERK may phosphorylate NR2B to enhance NMDA receptor activity (Hu et al., 2015) and further enhance AMPA receptor activity (Lu et al., 2001), thus increasing excitatory synaptic transmission and contributing to the pathogenesis of neuropathic pain.
A growing body of evidence shows that epigenetic mechanisms, including DNA methylation, histone modification, and noncoding RNA, contribute to the regulation of gene expression (Denk and McMahon, 2012). DNA methylation is the most stable epigenetic modification, which occurs at cytosine residues, predominantly in the context of CpG dinucleotides in mammals (Jeltsch, 2002). DNA methylation is often associated with gene silencing (Kass et al., 1997). Recent studies have shown that DNA methylation is involved in chronic pain induced by nerve injury (Massart et al., 2016), inflammation (Qi et al., 2013), or diabetes (Zhang et al., 2015). We for the first time show that SNL persistently increased Cxcr3 mRNA expression in spinal neurons, which was associated with decreased DNA methylation levels at the CpG sites within Cxcr3 promoter region. In vitro experiments also confirmed that methylation of the Cxcr3 promoter region decreased Cxcr3 promoter activity. These data indicate that DNA methylation contributes to SNL-induced Cxcr3 upregulation in the spinal cord.
DNA methylation can be achieved by the action of three enzymes: DNMT1, DNMT3a, and DNMT3b. DNMT1 is the key maintenance methyltransferase (Howell et al., 2001), whereas DNMT3a and DNMT3b are responsible for de novo methylation (Okano et al., 1999). The expression patterns of these DMNTs in the spinal cord after peripheral nerve injury, inflammation, or paw incision (Tochiki et al., 2012; Sun et al., 2015; Wang et al., 2016) are quite inconsistent, which may be due to different animal models, different species, or different time points. In the present study, neither Dnmt1 nor Dnmt3a was changed, but both mRNA and protein level of DNMT3b were decreased 10 d after SNL in mice. Overexpression of DNMT3b by lentivirus increased methylation of Cxcr3 promoter and attenuated neuropathic pain, suggesting that the methylation status of Cxcr3 can be regulated by the expression level of DNMT3b.
C/EBPα belongs to the C/EBP family of transcription factors, which consists of six isoforms: C/EBPα, C/EBPβ, C/EBPγ, C/EBPδ, C/EBPε, and C/EBPζ (Tsukada et al., 2011). C/EBPs are widely expressed and involved in regulation of a variety of functions, including inflammation, and innate and adaptive immunity (Miller et al., 2003; Tsukada et al., 2011). C/EBPβ was recently reported to be upregulated in dorsal root ganglion neurons after SNL and may be involved in neuropathic pain via downstream signaling through TNF-α (Sasaki et al., 2014). Here our results demonstrate a pivotal role of C/EBPα in neuropathic pain via regulation of CXCR3 expression. First, the ChIP-PCR data showed that C/EBPα positively regulated Cxcr3 transcription via direct binding to the promoter region of Cxcr3. Second, luciferase reporter experiments showed that binding ability of C/EBPα to Cxcr3 promoter was enhanced when Cxcr3 promoter was demethylated. Third, SNL increased C/EBPα mRNA and protein expression in the spinal cord, and knockdown of C/EBPα with siRNAs reduced CXCR3 expression and attenuated nerve injury-induced pain hypersensitivity. Moreover, as C/EBPα is an important transcription factor in microglia and involved in proinflammatory transcriptional activation and expression (Ponomarev et al., 2011), C/EBPα may indirectly regulate CXCR3 expression and contribute to neuropathic pain via increasing neuroinflammation (White et al., 2005; Gao and Ji, 2010).
Previous studies showed that CXCR3 was expressed in neurons of the hippocampus (Vlkolinský et al., 2004) and microglia in the entorhinal cortex (Rappert et al., 2004). In the present study, CXCR3 was found to be constitutively expressed in spinal neurons and increased after SNL. SNL also induced the expression of CXCR3 in a few microglia in the superficial dorsal horn. Moreover, CXCR3 was not only expressed in projection neurons, also in SST+-excitatory neurons and GAD2+-inhibitory neurons. Recent studies have revealed that the microcircuit formed by excitatory neurons (e.g., SST+ or VGLUT3+) and inhibitory neurons [e.g., GAD2+ or Dynorphin (Dyn)+] play an important role in mechanical allodynia (Duan et al., 2014; Peirs et al., 2015). Especially, ablation of SST+ neurons causes loss of mechanical pain, whereas ablation of Dyn+ neurons (>80% in GAD67+ neurons) induces spontaneous mechanical allodynia (Duan et al., 2014), suggesting the different role of spinal excitatory neurons and inhibitory neurons in pain modulation. Although CXCR3 is expressed in projection, excitatory, and inhibitory neurons, behavioral data showed that SNL-induced pain hypersensitivity was remarkably reduced in Cxcr3-deficient mice, and inhibition of CXCR3 by siRNA or antagonist alleviated established neuropathic pain, suggesting that CXCR3 in projection neurons and excitatory neurons may play a major role in mediating the maintenance of neuropathic pain. In addition, electrophysiological data (Fig. 7) showed that CXCL10 failed to increase synaptic transmission in some neurons, even after SNL. Whether these neurons are inhibitory neurons and how CXCR3 in different populations regulates pain still need to be clarified.
CXCR3 can be activated by three ligands: CXCL9, CXCL10, and CXCL11. These ligands have different temporal and spatial patterns of expression, are regulated by different stimuli, and are expressed by distinct cell types during the course of immune response (Groom and Luster, 2011; Van Raemdonck et al., 2015). As CXCL10 is a major ligand of CXCR3, and gene-expression microarray data showed that Cxcl10 was remarkably increased after SNL (Jiang et al., 2016), we focused on CXCL10 in this study. The qPCR and ELISA data further confirmed the upregulation of CXCL10 in the spinal cord after SNL. In addition, CXCL10 was increased from 3 d after SNL, whereas SNL-induced pain hypersensitivity was shown 1 d after SNL, suggesting that CXCL10 may be induced by other mediators. Our previous study showed that CXCL10 was induced by TNF-α in cultured astrocytes (Gao et al., 2009). Consistently, immunostaining showed that CXCL10 was markedly upregulated in spinal astrocytes in vivo. In addition, TNF-α was rapidly (1 d) upregulated in the spinal cord after SNL (Zhang et al., 2013). Therefore, the delayed increase of CXCL10 in the spinal cord may be induced by TNF-α.
Our behavioral data showed that intrathecal injection of CXCL10 induced rapid and CXCR3-dependent pain hypersensitivity, suggesting that activation of CXCL10/CXCR3 is sufficient to induce pain. Previous studies have shown that chemokines, such as CCL2 and CXCL1, with the expression of their receptors in spinal neurons, contribute to central sensitization via increased excitatory synaptic transmission (Gao et al., 2009; Zhang et al., 2013). We also found that CXCL10 increased the amplitude of sEPSCs of lamina II neurons and enhanced NMDA-induced and AMPA-induced currents via CXCR3, supporting the role of CXCL10/CXCR3 in mediating excitatory synaptic transmission. In addition, CXCL10 also increased the frequency of sEPSCs of lamina II neurons from slices after SNL, suggesting that CXCR3 in the dorsal root ganglion may also contribute to the pathogenesis of neuropathic pain.
ERK activation in dorsal horn neurons contributes importantly to the induction of central sensitization (Ji et al., 1999; Gao et al., 2009). We showed that CXCL10 induced CXCR3-dependent ERK activation in spinal neurons. As ERK is involved in the synaptic transmission mediated by GluN2B subunit-containing NMDA receptors (Hu et al., 2015), and pERK translocates to the nucleus and activates the transcription factor CREB to initiate gene transcription and maintain central sensitization and chronic pain (Ji and Strichartz, 2004), we speculate that CXCL10/CXCR3 may contribute to neuropathic pain via regulation of synaptic transmission and gene transcription.
In summary, our results for the first time demonstrate the regulation of Cxcr3 expression in the spinal cord by DNA methylation after SNL-induced neuropathic pain. Our data support the hypothesis that the decreased expression of DNMT3b may cause the demethylation of Cxcr3 promoter and further increase the binding of C/EBPα with Cxcr3 to promote its transcription. Furthermore, CXCL10, expressed by spinal neurons and astrocytes, increases the excitatory synaptic transmission via CXCR3 and contributes to the maintenance of neuropathic pain. Collectively, our results suggest that targeting the expression and activation of CXCR3 may offer new therapeutics for neuropathic pain.
Footnotes
This work was supported by grants from the National Natural Science Foundation of China (NSFC 31371121, 81300954, 81400915, and 31671091), the National Science Foundation for Young Scientists of Jiangsu Province (BK20140427), the Natural Science Research Program of Jiangsu Province (13KJB180017), the Innovation Fund for Graduate Students in Nantong University (YKC16021), and the Priority Academic Program Development of Jiangsu Higher Education Institutions. We thank Prof. Gary R. Strichartz for critical reading and language editing of the manuscript. We also thank Prof. Ru-Rong Ji for providing somatostatin (Sst)-Tomato+ and Gad2-Tomato+ mice.
The authors declare no competing financial interests.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbadie C, Bhangoo S, De Koninck Y, Malcangio M, Melik-Parsadaniantz S, White FA. Chemokines and pain mechanisms. Brain Res Rev. 2009;60:125–134. doi: 10.1016/j.brainresrev.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bali P, Im HI, Kenny PJ. Methylation, memory and addiction. Epigenetics. 2011;6:671–674. doi: 10.4161/epi.6.6.15905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- Denk F, McMahon SB. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron. 2012;73:435–444. doi: 10.1016/j.neuron.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descalzi G, Ikegami D, Ushijima T, Nestler EJ, Zachariou V, Narita M. Epigenetic mechanisms of chronic pain. Trends Neurosci. 2015;38:237–246. doi: 10.1016/j.tins.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren X, Ross SE, Lowell BB, Wang Y, Goulding M, Ma Q. Identification of spinal circuits transmitting and gating mechanical pain. Cell. 2014;159:1417–1432. doi: 10.1016/j.cell.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J. 2009;2:11–17. doi: 10.2174/1876386300902010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126:56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géranton SM. Targeting epigenetic mechanisms for pain relief. Curr Opin Pharmacol. 2012;12:35–41. doi: 10.1016/j.coph.2011.10.012. [DOI] [PubMed] [Google Scholar]
- Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89:207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, Chaillet JR. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104:829–838. doi: 10.1016/S0092-8674(01)00280-X. [DOI] [PubMed] [Google Scholar]
- Hu XD, Liu YN, Zhang ZY, Ma ZA, Suo ZW, Yang X. Spinophilin-targeted protein phosphatase-1 alleviated inflammatory pain by negative control of MEK/ERK signaling in spinal cord dorsal horn of rats. J Neurosci. 2015;35:13989–14001. doi: 10.1523/JNEUROSCI.2293-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jeltsch A. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem. 2002;3:274–293. doi: 10.1002/1439-7633(20020402)3:4<274::AID-CBIC274>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Jiang BC, Cao DL, Zhang X, Zhang ZJ, He LN, Li CH, Zhang WW, Wu XB, Berta T, Ji RR, Gao YJ. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest. 2016;126:745–761. doi: 10.1172/JCI81950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12:206–222. doi: 10.1002/cbic.201000195. [DOI] [PubMed] [Google Scholar]
- Kass SU, Pruss D, Wolffe AP. How does DNA methylation repress transcription? Trends Genet. 1997;13:444–449. doi: 10.1016/S0168-9525(97)01268-7. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Ji RR, Ito N, Allchorne AJ, Befort K, Karchewski LA, Woolf CJ. Peripheral axonal injury results in reduced mu opioid receptor pre- and post-synaptic action in the spinal cord. Pain. 2005;117:77–87. doi: 10.1016/j.pain.2005.05.035. [DOI] [PubMed] [Google Scholar]
- Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol. 2009;27:619–628. doi: 10.1200/JCO.2008.17.9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix-Fralish ML, Tawfik VL, Tanga FY, Spratt KF, DeLeo JA. Differential spinal cord gene expression in rodent models of radicular and neuropathic pain. Anesthesiology. 2006;104:1283–1292. doi: 10.1097/00000542-200606000-00025. [DOI] [PubMed] [Google Scholar]
- Liang L, Lutz BM, Bekker A, Tao YX. Epigenetic regulation of chronic pain. Epigenomics. 2015;7:235–245. doi: 10.2217/epi.14.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindia JA, McGowan E, Jochnowitz N, Abbadie C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J Pain. 2005;6:434–438. doi: 10.1016/j.jpain.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/S0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Ma X, Wang YW, Zhang MQ, Gazdar AF. DNA methylation data analysis and its application to cancer research. Epigenomics. 2013;5:301–316. doi: 10.2217/epi.13.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massart R, Dymov S, Millecamps M, Suderman M, Gregoire S, Koenigs K, Alvarado S, Tajerian M, Stone LS, Szyf M. Overlapping signatures of chronic pain in the DNA methylation landscape of prefrontal cortex and peripheral T cells. Sci Rep. 2016;6:19615. doi: 10.1038/srep19615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Shuman JD, Sebastian T, Dauter Z, Johnson PF. Structural basis for DNA recognition by the basic region leucine zipper transcription factor CCAAT/enhancer-binding protein alpha. J Biol Chem. 2003;278:15178–15184. doi: 10.1074/jbc.M300417200. [DOI] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Peirs C, Williams SP, Zhao X, Walsh CE, Gedeon JY, Cagle NE, Goldring AC, Hioki H, Liu Z, Marell PS, Seal RP. Dorsal Horn Circuits for Persistent Mechanical Pain. Neuron. 2015;87:797–812. doi: 10.1016/j.neuron.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetsch AR, Plass C. Transcriptional regulation by DNA methylation. Cancer Treat Rev. 2011;37(Suppl 1):S8–S12. doi: 10.1016/j.ctrv.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med. 2011;17:64–70. doi: 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F, Zhou Y, Xiao Y, Tao J, Gu J, Jiang X, Xu GY. Promoter demethylation of cystathionine-beta-synthetase gene contributes to inflammatory pain in rats. Pain. 2013;154:34–45. doi: 10.1016/j.pain.2012.07.031. [DOI] [PubMed] [Google Scholar]
- Qu L, Fu K, Yang J, Shimada SG, LaMotte RH. CXCR3 chemokine receptor signaling mediates itch in experimental allergic contact dermatitis. Pain. 2015;156:1737–1746. doi: 10.1097/j.pain.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramabadran K, Bansinath M, Turndorf H, Puig MM. Tail immersion test for the evaluation of a nociceptive reaction in mice. Methodological considerations. J Pharmacol Methods. 1989;21:21–31. doi: 10.1016/0160-5402(89)90019-3. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Liu L, Cardona AE. Chemokines and chemokine receptors: multipurpose players in neuroinflammation. Int Rev Neurobiol. 2007;82:187–204. doi: 10.1016/S0074-7742(07)82010-1. [DOI] [PubMed] [Google Scholar]
- Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C, Boddeke HW, Nitsch R, Kettenmann H. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci. 2004;24:8500–8509. doi: 10.1523/JNEUROSCI.2451-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Mut JV, Gräff J. Epigenetic alterations in Alzheimer's disease. Front Behav Neurosci. 2015;9:347. doi: 10.3389/fnbeh.2015.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Hashimoto S, Sawa T, Amaya F. Tumor necrosis factor-alpha induces expression of C/EBP-beta in primary afferent neurons following nerve injury. Neuroscience. 2014;279:1–9. doi: 10.1016/j.neuroscience.2014.08.032. [DOI] [PubMed] [Google Scholar]
- Schmitz K, Pickert G, Wijnvoord N, Häussler A, Tegeder I. Dichotomy of CCL21 and CXCR3 in nerve injury-evoked and autoimmunity-evoked hyperalgesia. Brain Behav Immun. 2013;32:186–200. doi: 10.1016/j.bbi.2013.04.011. [DOI] [PubMed] [Google Scholar]
- Sun Y, Sahbaie P, Liang D, Li W, Shi X, Kingery P, Clark JD. DNA methylation modulates nociceptive sensitization after incision. PloS One. 2015;10:e0142046. doi: 10.1371/journal.pone.0142046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Maruyama R, Yamamoto E, Kai M. DNA methylation and microRNA dysregulation in cancer. Mol Oncol. 2012;6:567–578. doi: 10.1016/j.molonc.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochiki KK, Cunningham J, Hunt SP, Géranton SM. The expression of spinal methyl-CpG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Mol Pain. 2012;8:14. doi: 10.1186/1744-8069-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11:823–836. doi: 10.1038/nrn2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth C, Lander J, Wiebe S. The prevalence and impact of chronic pain with neuropathic pain symptoms in the general population. Pain Med. 2009;10:918–929. doi: 10.1111/j.1526-4637.2009.00655.x. [DOI] [PubMed] [Google Scholar]
- Tsukada J, Yoshida Y, Kominato Y, Auron PE. The CCAAT/enhancer (C/EBP) family of basic-leucine zipper (bZIP) transcription factors is a multifaceted highly-regulated system for gene regulation. Cytokine. 2011;54:6–19. doi: 10.1016/j.cyto.2010.12.019. [DOI] [PubMed] [Google Scholar]
- Tuesta LM, Zhang Y. Mechanisms of epigenetic memory and addiction. EMBO J. 2014;33:1091–1103. doi: 10.1002/embj.201488106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev. 2015;26:311–327. doi: 10.1016/j.cytogfr.2014.11.009. [DOI] [PubMed] [Google Scholar]
- Verge GM, Milligan ED, Maier SF, Watkins LR, Naeve GS, Foster AC. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci. 2004;20:1150–1160. doi: 10.1111/j.1460-9568.2004.03593.x. [DOI] [PubMed] [Google Scholar]
- Viet CT, Dang D, Ye Y, Ono K, Campbell RR, Schmidt BL. Demethylating drugs as novel analgesics for cancer pain. Clin Cancer Res. 2014;20:4882–4893. doi: 10.1158/1078-0432.ccr-14-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlkolinský R, Siggins GR, Campbell IL, Krucker T. Acute exposure to CXC chemokine ligand 10, but not its chronic astroglial production, alters synaptic plasticity in mouse hippocampal slices. J Neuroimmunol. 2004;150:37–47. doi: 10.1016/j.jneuroim.2004.01.011. [DOI] [PubMed] [Google Scholar]
- von Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron. 2012;73:638–652. doi: 10.1016/j.neuron.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu C, Guo QL, Yan JQ, Zhu XY, Huang CS, Zou WY. Intrathecal 5-azacytidine inhibits global DNA methylation and methyl-CpG-binding protein 2 expression and alleviates neuropathic pain in rats following chronic constriction injury. Brain Res. 2011;1418:64–69. doi: 10.1016/j.brainres.2011.08.040. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lin ZP, Zheng HZ, Zhang S, Zhang ZL, Chen Y, You YS, Yang MH. Abnormal DNA methylation in the lumbar spinal cord following chronic constriction injury in rats. Neurosci Lett. 2016;610:1–5. doi: 10.1016/j.neulet.2015.10.048. [DOI] [PubMed] [Google Scholar]
- White FA, Wilson NM. Chemokines as pain mediators and modulators. Curr Opin Anaesthesiol. 2008;21:580–585. doi: 10.1097/ACO.0b013e32830eb69d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Bhangoo SK, Miller RJ. Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov. 2005;4:834–844. doi: 10.1038/nrd1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Wildner H, Yévenes GE. Fast synaptic inhibition in spinal sensory processing and pain control. Physiol Rev. 2012;92:193–235. doi: 10.1152/physrev.00043.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HH, Hu J, Zhou YL, Qin X, Song ZY, Yang PP, Hu S, Jiang X, Xu GY. Promoted interaction of nuclear factor-kappaB with demethylated purinergic P2X3 receptor gene contributes to neuropathic pain in rats with diabetes. Diabetes. 2015;64:4272–4284. doi: 10.2337/db15-0138. [DOI] [PubMed] [Google Scholar]
- Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. 2013;154:2185–2197. doi: 10.1016/j.pain.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21:642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learn Mem. 2013;20:61–74. doi: 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]