

Abstract
Repulsive guidance molecule member a (RGMa) is a membrane-associated or released guidance molecule that is involved in axon guidance, cell patterning, and cell survival. In our previous work, we showed that RGMa is significantly upregulated in the substantia nigra of patients with Parkinson's disease. Here we demonstrate the expression of RGMa in midbrain human dopaminergic (DA) neurons. To investigate whether RGMa might model aspects of the neuropathology of Parkinson's disease in mouse, we targeted RGMa to adult midbrain dopaminergic neurons using adeno-associated viral vectors. Overexpression of RGMa resulted in a progressive movement disorder, including motor coordination and imbalance, which is typical for a loss of DA release in the striatum. In line with this, RGMa induced selective degeneration of dopaminergic neurons in the substantia nigra (SN) and affected the integrity of the nigrostriatal system. The degeneration of dopaminergic neurons was accompanied by a strong microglia and astrocyte activation. The behavioral, molecular, and anatomical changes induced by RGMa in mice are remarkably similar to the clinical and neuropathological hallmarks of Parkinson's disease. Our data indicate that dysregulation of RGMa plays an important role in the pathology of Parkinson's disease, and antibody-mediated functional interference with RGMa may be a disease modifying treatment option.
SIGNIFICANCE STATEMENT Parkinson's disease (PD) is a neurodegenerative disease characterized by severe motor dysfunction due to progressive degeneration of mesencephalic dopaminergic (DA) neurons in the substantia nigra. To date, there is no regenerative treatment available. We previously showed that repulsive guidance molecule member a (RGMa) is upregulated in the substantia nigra of PD patients. Adeno-associated virus-mediated targeting of RGMa to mouse DA neurons showed that overexpression of this repulsive axon guidance and cell patterning cue models the behavioral and neuropathological characteristics of PD in a remarkable way. These findings have implications for therapy development as interfering with the function of this specific axon guidance cue may be beneficial to the survival of DA neurons.
Keywords: AAV-mediated overexpression, dopamine neuron degeneration, Parkinson's disease, RGMa
Introduction
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease. Patients suffer from motor (Dauer and Przedborski, 2003; Jankovic, 2008) and cognitive dysfunction (Jankovic, 2008; Olanow et al., 2009). The motor symptoms are mainly attributed to the loss of mesencephalic dopaminergic neurons in the substantia nigra (SN). The etiology of PD is multifactorial (Kalia and Lang, 2015), including both genetic (Kumaran and Cookson, 2015; Hernandez et al., 2016) and environmental components (Gorell et al., 2004). Transcriptional profiling (for review, see Cooper-Knock et al., 2012) and pathway analysis (Lesnick et al., 2007; Lin et al., 2009; Srinivasan et al., 2009; Sutherland et al., 2009; Edwards et al., 2011) have revealed dysregulation of genes involved in processes previously implicated in PD (the ubiquitin/proteasome system, heat shock regulation, iron and vesicular transport, neurotransmission, oxidative stress), yet also implicated novel pathways in PD, including axon guidance, extracellular matrix, polyamine signaling, and microRNA modulation of mitochondrial function (Miñones-Moyano et al., 2011).
Repulsive guidance molecule a (RGMa) is upregulated in DA neurons of PD patients (Bossers et al., 2009; Barrett et al., 2013; GEO accession GSE7307), suggesting a link between RGMa and PD. RGMa has also been implicated in other neurodegenerative diseases. Its increased expression contributes to the repulsive environment of the neural scar (Schwab et al., 2005a, b; Hata et al., 2006; Mueller et al., 2006; Yamashita et al., 2007) and a chromosome microdeletion in the RGMa gene has been linked to Angelman syndrome (Capelli et al., 2012). RGMa is also implicated in multiple sclerosis (MS) (Nohra et al., 2010; Muramatsu et al., 2011; Kubo et al., 2012; Tanabe and Yamashita, 2014; Demicheva et al., 2015) where RGMa antibodies increased axonal outgrowth, ameliorated remyelination, and improved function in a rodent MS model (Muramatsu et al., 2011; Kubo et al., 2012; Tanabe and Yamashita, 2014).
RGMa acts as a repulsive axon guidance molecule in the developing amphibian, bird, and mammalian brain (Monnier et al., 2002; Niederkofler et al., 2004; Samad et al., 2004; Matsunaga et al., 2006; Mueller et al., 2006; Yamashita et al., 2007). RGMa is processed by extracellular proteases to generate membrane-bound and soluble forms, which function as short- and a long-range guidance cues (Tassew et al., 2012) by interacting with its receptor neogenin (Yamashita et al., 2007). Many embryonic neurons are sensitive to RGMa neogenin repulsive signaling (Monnier et al., 2002; Conrad et al., 2007; Metzger et al., 2007; Kubo et al., 2008; Yoshida et al., 2008; Tassew et al., 2012). Neogenin is expressed by developing DA neurons (van den Heuvel et al., 2013) and continues to be expressed by adult neurons in the brain (Rodriguez et al., 2007).
RGMa neogenin signaling also plays a role in neuronal survival, proliferation, and differentiation (Matsunaga and Chédotal, 2004; Matsunaga et al., 2004, 2006; Cole et al., 2007; Metzger et al., 2007; Lah and Key, 2012). Administration of RGMa promotes retinal ganglion cell survival after an optic nerve lesion (Koeberle et al., 2010) and improves neuronal survival following stroke (Paxinos, 2001). The prosurvival signaling of RGMa is dependent on the interaction of neogenin with lipid rafts (Shabanzadeh et al., 2015).
Because RGMa is upregulated in the SN of human PD brains (Bossers et al., 2009; RRID: GSE7307 Barrett et al., 2013), has deleterious effects in other disorders such as MS, and has a well-documented role in axon repulsion (Monnier et al., 2002; Niederkofler et al., 2004; Samad et al., 2004; Matsunaga et al., 2006; Mueller et al., 2006; Yamashita et al., 2007), we hypothesized that overexpression of RGMa in the mouse SN would negatively impact the midbrain dopaminergic system. We show that neuronal overexpression of RGMa induced a movement disorder typical for loss of striatal DA, degeneration of DA neurons in the SN, loss of DA nigrostriatal axonal projections, and microglial and astrocyte activation. These data indicate that RGMa is a negative regulator of DA neuron survival and may play an important role in PD pathology.
Materials and Methods
Human brain tissue samples.
Brain tissue used in these studies was from the same controls and PD patients as described by Bossers et al. (2009). In brief, formalin-fixed, paraffin-embedded postmortem human SN tissue from 7 PD patients and 9 controls was obtained from the Netherlands Brain Bank (NBB, Amsterdam, The Netherlands). Table 1 summarizes their clinicopathological data.
Table 1.
Clinicopathological data of human postmortem tissue samples used for in situ hybridization and immunohistochemistrya
| Subject no. | Diagnosis | Sex | Age (yr) | PMI (h) | pH | BW (g) | RIN | Cause of death |
|---|---|---|---|---|---|---|---|---|
| 00–115 | PD/DEM | Male | 70 | 9:05 | 6.33 | 1258 | 6.2 | Pneumonia, septic shock |
| 04–045 | PD/DEM | Male | 71 | 6:58 | 6.55 | 1358 | 8.4* | Pneumonia |
| 00–139 | PD/DEM | Male | 72 | 7:15 | 6.55 | 1546 | 6.7 | Uremia |
| 02–003 | PD | Female | 75 | 5:00 | 6.52 | 1218 | 9.6* | Euthanasia |
| 02–011 | PD | Female | 79 | 5:45 | 6.37 | 1203 | 8.7* | Myocardial infraction |
| 00–034 | PD | Male | 86 | 8:30 | 6.52 | 1178 | 9.2* | Unknown |
| 02–064 | PD | Male | 87 | 7:20 | 6.37 | 1166 | 7.4 | Respiratory insufficiency |
| 98–126 | CTRL | Male | 71 | 6:00 | 6.54 | 1385 | 8.8 | Respiratory insufficiency |
| 00–049 | CTRL | Male | 78 | 6:55 | 6.42 | 1332 | 9.2* | Cardiac failure |
| 97–144 | CTRL | Male | 78 | 4:00 | 6.43 | 1160 | 9* | Pulmonary carcinoma |
| 00–142 | CTRL | Female | 82 | 5:30 | 6.60 | 1280 | 9.2* | Myocardial infarct |
| 00–022 | CTRL | Female | 83 | 7:45 | 6.52 | 1102 | 9.2* | Acute myocardial infraction |
| 98–062 | CTRL | Male | 85 | 4:35 | 6.95 | 1332 | 7.5 | Respiratory insufficiency |
| 99–046 | CTRL | Female | 89 | 5:10 | 6.62 | 1168 | 9.5 | Cardiac arrest |
| 01–029 | CTRL | Female | 90 | 5:25 | 6.58 | 1066 | 7.6 | Myocardial infraction |
| 00–050 | CTRL | Female | 52 | 6:50 | 7.16 | 1258 | — | Leiomyosarcoma with metastasis |
aAll brain tissue was collected from donors from whose written informed consent for a brain autopsy and the use of the material and clinical information for research purposes had been obtained by the NBB. For further diagnosis, an extensive neuropathological investigation was performed on all PD and control tissue. Control tissue did not present any Braak pathology score for neurofibrillary tangles >2 (Braak et al., 2003), and neither control nor PD subject had a known history of neurological or psychiatric disease other than PD or PD-related dementia. All PD patients received dopamine replacement therapy during the course of the disease. CTRL, Control; PD/DEM, Parkinson's disease with dementia; PMI, postmortem interval; BW, brain weight; RIN, RNA integrity number.
*Sample used in the microarray study. All samples, except Subject 00–050, were used for qPCR analysis as described previously (Bossers et al., 2009). Subject 00–050 was only used for in situ hybridization.
In situ hybridization on human brain tissue samples.
For semiquantitative analysis of mRNA expression, one SN section from every subject was used (Table 1). The sections were deparaffinized, rehydrated, and heated in a microwave in 0.1 m citrate buffer, pH 6.0, for 20 min. Sections were washed 2 times in PBS for 5 min, deproteinated for 20 min in 0.2 N HCl, washed twice for 5 min in PBS, and treated with proteinase K (10 μg/ml, Invitrogen) in proteinase K buffer (2 mm CaCl2, 10 mm Tris-HCl, pH 7.5) for 15 min at 37°C. The proteinase K digestion was stopped by a 30 min incubation in glycine buffer (27 mm glycine in PBS) followed by two 5 min washes in PBS. Finally, sections were delipidated for 10 min in PBS, 0.1% Triton X-100 (Sigma-Aldrich) and washed twice for 5 min in PBS.
An RGMA-specific LNA-2′O-methyl-RNA modified oligonucleotide probe 5′-FAM-TugAccAcuTccTcuGgcA-3′, recognizing nucleotides 1169–1187 of the human RGMa mRNA (NM_020211.2), was obtained from RiboTask Aps. Sections were prehybridized overnight at room temperature (RT) in 200 μl LNA hybridization buffer (50% formamide, 600 mm NaCl, 10 mm HEPES buffer, pH 7.5, 5× Denhardt's, 1 mm EDTA, 200 μg/ml denatured herring sperm DNA). Next, the probe was diluted in LNA hybridization buffer to a concentration of 25 nm, denatured at 95°C for 5 min, and cooled on ice for 5 min. Sections were hybridized in this buffer at 55°C for 90 min, followed by a series of wash steps: 5 min in 5× SSC at 55°C, 5 min in 2× SSC at 55°C, 5 min in 0.2× SSC at 55°C, and 5 min in PBS at RT. To detect the probes, sections were preincubated with 1% milk-TBS, pH 7.6, for 1 h, followed by 3 h incubation with sheep IgG, anti-fluorescein-AP-Fab fragments (Roche) diluted 1:3000 in 1% milk-Super Mix all at RT.
The hybridization signal was developed as follows. Sections were washed twice in Buffer 1 (100 mm Tris, 150 mm NaCl, pH 7.5) and once in Buffer 2 (100 mm Tris-HCl pH 9.0, 100 mm NaCl, 5 mm MgCl2) for 5 min and further incubated in 10 ml Buffer 2 containing 3.4 mg nitro-blue tetrazolium chloride (Roche), 1.75 mg 5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt (Roche), and 2.4 mg levamisole (Sigma-Aldrich) for 25 min. Reaction was stopped in water, and slides were treated with 100% methanol for 5 min and coverslipped with aquamount (Merck). An adjacent section was incubated with sense probe to test the specificity of the observed in situ hybridization signal.
Immunohistochemistry on human postmortem brain tissue.
For protein localization, one SN section was used from each PD and control sample. The sections were deparaffinized in xylene, rehydrated in a graded series of ethanol, and washed twice for 2 min in distilled water. Antigen retrieval was performed by microwave heating (2× 5 min at 700 W) in 50 mm Tris-HCl, pH 9.0, and sections were washed in TBS twice for 5 min. Sections were blocked with TBS-0.1% milk for 1 h at RT. Sections were then incubated with anti-RGMa antibody (SC-46482, Santa Cruz Biotechnology, 1:25, antibody raised against a 15–25 amino acid peptide mapping within the region of amino acids 300–350 of C-terminal human RGMa) and either anti-TH antibody (Jacques Boy, 1:1000) or anti-neogenin antibodies (SC-15337, Santa Cruz Biotechnology, 1:25) diluted in Super Mix-0.1% milk solution at pH 7.6 (1× TBS, 0.25% gelatin, Merck; and 0.5% Triton X-100, Sigma-Aldrich) for 1 h at RT followed by overnight incubation at 4°C. After the primary antibody incubation, sections were washed 3 times with TBS. To enhance the staining for RGMa, we first amplified the signal by applying a biotin-labeled secondary antibody (1:400; Vector Laboratories) diluted in SuperMix for 1 h at RT to the sections. Sections were subsequently incubated in ABC solution (1:800 in TBS, Vector Laboratories) for 1 h at RT followed by a second round of signal amplification with a biotinylated tyramine incubation for 10 min in a TBS-0.01% H2O2 solution (1:750). Finally, the sections were incubated for 2 h at RT with a streptavidin-conjugated Alexa-594 antibody (1:800 in TBS, Invitrogen) and for TH and neogenin detection, an anti-rabbit Alexa-488-conjugated antibody (1:800 in TBS, Invitrogen). To quench autofluorescence, sections were treated with 0.5% filtered Sudan Black solution (BDH) for 7 min, briefly washed in 70% ethanol and TBS. Sections were embedded in Mowiol (0.1 m Tris, pH 8.5, 25% glycerol, 10% w/v Mowiol 4–88, Sigma-Aldrich) containing Hoechst 33258 (Bio-Rad; 1:10,000). Images were acquired on the confocal laser scanning microscope (Zeiss).
Adeno-associated virus (AAV) constructs and viral vector production.
Plasmids pAAV2Sna-SW and pTRUF20B-SEW (generous gifts from Prof. Deniz Kirik, Lund University, Lund, Sweden) formed the basis for the production of the AAV vectors used in this study. Each plasmid contained two inverted terminal repeats of AAV2 flanking a human synapsin 1 (SYN) promoter driving expression of either human α-synuclein (pAAV2Sna-SW plasmid) or GFP (pTRUF20B-SEW plasmid), followed by a woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) and a polyadenylation signal. For the construction of the empty vector (pAAV2-SYN), the pAAV2Sna-SW plasmid was cut with BamHI to remove the α-synuclein sequence and religated. For the construction of the vector containing mouse RGMa (pAAV2-SYN-RGMa), plasmid pcDNA4/HisB-RGMaFL was cut with Dra1 and Xho1 to isolate the full-length mouse RGMa sequence (NM_177740), and this fragment was ligated into the pAAV2Sna-SW cut with EcoRV and Xho1.
Production of AAV2/7-SYN-Empty, AAV2/7-SYN-RGMa, and AAV2/7-SYN-GFP viral vectors was performed using capsid and helper plasmids provided by J.M. Wilson (Gao et al., 2002). For each viral vector stock, eight 15 cm Petri dishes containing 1 × 107 human embryonic kidney 293T (HEK293T) cells were transfected using polyethylenimine (MW 25000; Polysciences). Cells were grown in DMEM containing 10% FCS and 1% penicillin/streptomycin (Invitrogen). pAAV2-SYN, pTRUF20B-SEW, and pAAV2-SYN-RGMa plasmids were cotransfected with packaging plasmids in a 1:2:2 ratio (AAV-gene plasmid: helper plasmid pAdΔF6: AAV2/7 capsid plasmid) with a total amount of 62.5 μg of DNA per plate. Two days after transfection, cells were harvested in Dulbecco's PBS (D-PBS containing Ca2+ and Mg; Invitrogen) containing 10 μg/ml DNaseI (Roche Diagnostics) and incubated for 1 h at 37°C. Cells were lysed by three freeze-thaw cycles, centrifuged for 30 min at 4000 rpm, and the crude lysate was collected. The virus was purified by iodixanol gradient ultracentrifugation (Hermens et al., 1999; Zolotukhin et al., 1999) diluted in D-PBS (with Ca2+ and Mg; Invitrogen) containing 5% sucrose and concentrated using an Amicon, 100 kDA MWCO Ultra-15 device (Millipore). Viral vector stocks were aliquoted and stored at −80°C until use. Titers were determined by quantitative PCR on DNase-treated viral particles using WPRE-directed primers (forward, CAGGTGTATTGCCACAAGACAAA; and reverse, TGCACAGGTGAAGACCAAGCAA). AAV-Empty viral vector gave a titer of 8.6 × 1012 genomic copies per milliliter (gc/ml), AAV-GFP viral vector gave a titer of 3.0 × 1012 gc/ml, and AAV-RGMa virus gave a titer of 9.7 × 1012 gc/ml. Virus was used in two experiments using either a low- or a high-titer dosage: 3.0 × 1012 gc/ml and 9.0 × 1012 gc/ml, respectively (Table 2). For the low-titer experiment, the AAV-Empty and AAV-RGMa viruses were diluted in D-PBS/5% sucrose accordingly to titer-match the AAV-GFP virus.
Table 2.
Description of the experimental animal groups, the viral vectors used, and the viral vector dose injected in the two overexpression experiments
| Virus | Low-dose RGMa overexpression |
High-dose RGMa overexpression |
|---|---|---|
| AAV2/7-RGMa, AAV2/7-Empty, AAV2/7-GFP, saline | AAV2/7-RGMa unilateral, AAV2/7-RGMa bilateral, AAV2/7-Empty | |
| Titer | 3.0 × 1012 | 9.0 × 1012 |
| No. of animals per group | N = 7–8, for saline N = 6 | N = 6 |
| Survival time (wk) | 12 | 18.5 |
AAV vector validation by Western blot analysis.
N2A cells were plated in 24-well plates and transfected with pTRUF20B-SEW and pAAV2-SYN-RGMa plasmids 1 d after plating using polyethylenimine. On day 6, media and cells were harvested. Cells were lysed on ice for 10 min in 70 μl RIPA buffer (25 mm Tris-HCl, pH 7.4, Sigma; 150 mm NaCl, Sigma; 1% NP40, AppliChemicals; 1% sodium deoxycholate, Sigma; 0.1% SDS and Complete Protease Inhibitor, Roche). The cell lysate was sonicated and its protein concentration determined using the bicinchoninic acid protein assay kit (Pierce, Thermo Scientific). For Western blot analysis, each sample was heated in 5× loading buffer containing 10% SDS (MP Biomedicals) and 5% β-mercaptoethanol (Sigma) at 95°C for 5 min and separated on an 10% polyacrylamine-SDS gel. Proteins were transferred to nitrocellulose membranes and treated with block mix (5% milk in 1× TBS/0.5% Triton X-100) for 1 h at RT. Blots were incubated with goat anti-mouse RGMa antibody (1:100, R&D Systems, AF2458, an antibody raised against amino acids 48–421 of human RGMa) and β-actin (1:1000, Sigma-Aldrich, A5316) at 4°C overnight in block mix. The primary antibodies were detected with anti-goat-Cy5 (1:800, Jackson ImmunoResearch Laboratories) and anti-mouse IR-dye 800-conjugated antibodies (1:2000, Thermo Scientific). Blots were scanned using the Odyssey infrared imager and Odyssey 2.1 scanning software (LI-COR Biosciences).
Experimental animals and surgical procedures.
Male C57BL/6 mice weighing 20–25 g (Harlan) were socially housed with food and water ad libitum, in 12 h light and dark cycles. The experimental procedures and postoperative care were performed in accordance with the Institutional Animal Care and Use Committee of the Royal Netherlands Academy of Arts and Sciences.
The viral vector injections were performed using glass capillaries (1.0 mm external diameter) with an 80 μm tip diameter connected via Portex polyethylene tubing to a Hamilton syringe fixed in a micro-infusion pump (PHD2000, Harvard Apparatus). Two experiments were performed with animals injected unilaterally (into the right SN) with a low-titer (3.0 × 1012 gc.ml) and unilaterally or bilaterally with a high-titer (8.7–9.0 × 1012 gc.ml) virus (Table 2).
Mice were intraperitoneally injected with a mix of Hypnorm (0.1 mg/kg fentanyl citrate/ 3.3 mg/kg fluanisone HCl, Janssen Pharmaceuticals) and Dormicum (8.3 mg/kg midazolam, Roche) and placed into a stereotactic device (David Kopf Instruments). The skull was leveled using the heights of bregma, lambda, and two lateral measurements 2.0 mm from bregma. The injection coordinates from bregma were −2.8 mm anteroposterior and ±1.3 mm lateral and −4.3 mm ventrodorsal from the dura. Subsequently, the needle was lowered into the brain 0.1 mm below the ventrodorsal coordinate and retracted back up to the correct level. We infused 1 μl of volume at a speed of 0.2 μl/min. After the infusion, the needle was left in place for 3 min before retraction. Animals recovered from the anesthesia in a heated incubator set to 37°C and were monitored until fully recovered. Experimenters were blinded regarding the viral vector genotype.
Behavioral testing.
The behavior of animals was assessed with the following tests: narrow beam test, grid test, cylinder test, swing test, and tremor assessment. The week before surgery, animals received at least three pretraining sessions on the narrow beam test. A baseline measurement was obtained 2 d before surgery, and the first measurement was performed 1 week after the surgery. During the first 3 weeks after surgery, all tests were performed twice a week and subsequently once a week until the termination of the experiment. The two investigators scoring the behavior tests were blinded for the treatment groups.
Grid test.
The grid test was used to study forepaw use, in particular the use of distal musculature and digit manipulation, which is sensitive to dopaminergic input from the striatum (Tillerson and Miller, 2003). Mice were suspended upside down on a metal grid and allowed to move freely across the grid. A successful trial occurred when the animal held on to the grid for a minimal of 10 s and took at least 10 steps. The maximum trial length was 30 s. The total number of successful and unsuccessful steps (overshoot, misplacement, loss of grip) with either forepaw was counted by two blinded observers independently. Each animal performed three trials, and the average ratio between the total forepaw faults/total forepaw steps over these trials was calculated (Meredith and Kang, 2006).
Cylinder test.
The cylinder test was performed to assess preference of front paw use during rearing behavior (Liu et al., 1999; Ulusoy et al., 2009). Animals were allowed to move freely in a glass cylinder for 5 min or until they performed 20 full rearing movements. During a successful rearing movement (i.e., the mouse reached at least 5 cm from the base of the cylinder), the placement of the right, left, or both forepaws was recorded. A new rearing movement was considered only once the mouse had returned its forepaws to the base of the cylinder. Additionally, paw placements were not recorded if the animal's torso was rotated during a rearing movement. The percentage of right paw use was calculated over the total number of the rearing score.
Narrow beam test.
The hindlimb placement was tested using the narrow beam test adapted from Fleming et al. (2004) and Drucker-Colín and García-Hernández (1991). Mice crossed an 8-mm-wide and 100-cm-long beam, elevated 15 cm above a table. The total number of correct hindlimb steps and hindlimb slips were counted and averaged over three complete runs.
Tremor.
A semisubjective tremor assessment was performed during the narrow beam test when animals were stationary on the platforms. A positive tremor score required the animal to shake while stationary, show a shaky tail when stretched, and unstable and shaky front paw placement when exploring the environment.
Swing test.
The swing test was adapted from Roghani et al. (2002). The direction of body rotation was scored by suspending mice 5 cm above the bottom of a cage while holding them at the base of their tail for 30 s. During that time, the direction of each swing above a 30° angle was scored.
Tissue processing.
Animals were killed by an intraperitoneal overdose with pentobarbital (50 mg/μl) and transcardially perfused with 0.9% saline followed by 4% PFA (Sigma-Aldrich) in sodium phosphate buffer (PBS, Sigma) pH 7.4. The brains were postfixed overnight, cryoprotected in 30% sucrose/PBS, and 6 series of 30 μm thick coronal sections containing the SN and striatum were cut on a cryostat. Sections were stored free floating at 4°C in 1% PFA in PBS, pH 7.6.
Fluorescent immunohistochemistry.
All immunohistochemical stainings were performed on free-floating sections. Before the staining, sections were blocked in blocking buffer (TBS with 2.5% FCS, DAKO; and 0.2% Triton X-100, Sigma) for 1 h at RT. Sections were incubated with anti-TH antibody (either rabbit polyclonal, 1:500; Pel-Freez Biologicals; or mouse monoclonal, clone LNC1, 1:1000; Millipore MAB318). The staining was combined with anti-RGMa (1:100; D-16 sc-46482, Santa Cruz Biotechnology), goat polyclonal antibody, anti-GFAP-Cy3-conjugated mouse monoclonal antibody (1:1500;G-A-5, Sigma), anti-Iba1 rabbit polyclonal antibody (1:2000; Wako), or anti-Akt or phosphorylated Akt (pAkt) antibodies (1:25; 9272 and 4060; Cell Signaling Technology). Primary antibody incubations were performed in blocking buffer for 1 h at RT followed by overnight incubation at 4°C. Alexa-488-, Cy3-, and DyLight 680-coupled secondary antibodies (1:800; Invitrogen) were used for detection of the primary antibodies by incubation for 1 h at RT in blocking buffer followed by 20 min incubation in PBS containing Hoechst 33258 (1:10,000; Bio-Rad). Sections were mounted on gelatin-coated glass slides and embedded in Mowiol (0.1 m Tris, pH 8.5, 25% glycerol, 10% w/v Mowiol 4–88, Sigma).
TH immunohistochemistry with cresyl violet counterstaining.
Sections were blocked as described above and subsequently incubated with anti-TH rabbit polyclonal antibody (1:500; Pel-Freez Biologicals) blocking buffer for 1 h at RT followed by overnight incubation at 4°C. Next, sections were washed 3 times in TBS and incubated with biotin-labeled donkey anti-rabbit antibody (1:400; Vector Laboratories) for 1 h at RT followed by incubation with ABC Vectastain complex (1:800, Vector Laboratories) in TBS for 1 h. Finally, sections were washed 3 times in TBS and incubated with DAB solution for 10 min, washed in water, mounted onto gelatin-coated glass slides, and dried overnight at 37°C. Sections were counterstained with 0.1% cresyl violet for 30 s (Aldrich Chemical) and subsequently dehydrated through a series of ethanol washes (70%, 80%, 90%, 96%, 2 × 100%; 5 min each) followed by two xylene washes (10 min each). Slides were then embedded with Entellan (Merck) and allowed to dry overnight before imaging.
Image processing and quantification.
Images were acquired with an AxioPlan 2 microscope (Zeiss) with Planapochromat objectives, using Evolution QEi black and white or Evolution MP color camera (MediaCybernetics) and ImagePro software. The 10× magnification pictures were taken for SN sections, and 2.5× magnification pictures were taken for the striatum sections with fixed exposure times for each channel. The exposure time was selected so the fluorescence signal was not overexposed.
Estimation of neuronal density in the SN.
For each animal, three sections from one cresyl violet- and TH-stained series were used for measuring the neuronal density within the SN as described by Bao et al. (2005) and Huitinga et al. (2000). Briefly, the SN pars compacta (SNpc) structure in the noninjected side of the brain was identified at 2.5× magnification using the color camera on the Axioskop microscope. The anatomical borders of the noninjected SN were defined and outlined using the mouse brain atlas (Paxinos, 2001), and the outlined area was projected in a mirror fashion on to the injected SN, with slight adjustments to fit the anatomy of the contralateral SN. The outlined area was subdivided into a square grid using an Image Pro Plus macro, with each grid field representing one image at 40× magnification. Based on the SD of the number of counted neurons per field, sampling of 35% of the total number of fields was determined sufficient to estimate the neuronal density. Each neuron was identified based on its size and neuronal-like morphology (large and spherical shape) and, most importantly, the presence of a nucleus with a nucleolus. TH+ and TH− neurons were counted separately. The total number of neurons counted in the injected and noninjected SN was corrected for the size of the outlined area and thickness of each section to yield the average neuronal number in cubic millimeters. Because neuronal atrophy decreases the size of neuronal cells to be comparable with (small) glial cells, it potentially eliminates atrophied neurons from the counting criteria. This means that a decrease of neuronal density may also reflect neuronal atrophy in additional to neuronal loss.
Fluorescence intensity and area analysis.
Mouse SNpc was outlined based on the TH staining and anatomical borders in the ImagePro Plus Measure Threshold macro. Additionally, an area just outside the SNpc was outlined to obtain a measurement of the background fluorescence levels. Total fluorescence intensity of TH, GFAP, and Iba1 was measured in both areas, and the background value was subtracted from the fluorescent intensity measured in the SNpc. The total intensity value was then corrected for the size of the outlined area by multiplying the surface area of fluorescence signal, resulting in the average fluorescent intensity value or the integrated optical density. Striatal sections were outlined based on the Hoechst staining and anatomical borders. Here TH, GFAP, and Iba1 fluorescence intensity was also measured and corrected for background and size of the area. Finally, all sections from one series per each animal (8–10 sections) were averaged, resulting in readout of average fluorescence intensity in the SN or the striatum for each animal.
The total area positive for TH immunofluorescence was outlined and measured using ImagePro Plus in the SNpc and in the background area placed above the SN. Only TH signal >3 times the average background fluorescent value of all sections was measured. In case any signal was detected in the background outline, these values were subtracted from the SN area. For each animal, an average of TH+ area per section was calculated, with 8–10 sections measured per animal.
Akt and pAkt image analysis in TH+ SN neurons.
Images of sections stained for TH and Akt were analyzed using ImageJ software (National Institutes of Health). The soma of TH+ cells within the SNpc were identified using standardized thresholding. To exclude any axo-dendritic signal, a particle exclusion was applied. The created mask was then superimposed on to the Akt or the pAKT channel, and the average intensity of the Akt or the pAkt signal was determined. An average of 4 sections were quantified per animal.
Statistical analysis.
Data analysis was performed in the SPSS software (IBM) or in the GraphPad Prism software. All data are expressed as mean ± SEM. Statistical analysis was performed using one-way ANOVA with Tukey's multiple testing correction when comparing between more than two animal groups, unpaired Student's t test when comparing between the bilaterally injected animal groups, or paired Student's t test when comparing SN and striatum within the unilaterally injected animals.
Behavioral testing statistical analysis was performed using a two-way ANOVA with Tukey's multiple testing correction post hoc analysis. A p value <0.05 was considered significant for all analyses.
Study approval.
All animal experimental procedures and postoperative care were performed in accordance with the Institutional Animal Care and Use Committee of the Royal Netherlands Academy of Arts and Sciences.
Results
RGMa and neogenin are expressed by neurons in human postmortem SN
Gene expression analysis on human postmortem SN tissue from PD patients and age-matched controls showed a 2.1-fold increase in RGMa mRNA expression in the PD SN (Bossers et al., 2009). In situ hybridization analysis of the tissue from the same patients used in the gene expression analysis revealed a neuron-specific expression of RGMa mRNA in SN tissue (Fig. 1A–C). Immunohistochemical analysis, using an antibody recognizing the C-terminal fragment of RGMa, revealed a punctate expression of RGMa protein in TH+ neurons and in the surrounding extracellular space, suggesting release of RGMa (Fig. 1D,E). Cellular expression and release of RGMa in the extracellular environment, including the C terminal and N-terminal fragments, has also been shown in multiple sclerosis patients (Demicheva et al., 2015). Human SN neurons also express the RGMa receptor neogenin (Fig. 1F,G).
Figure 1.
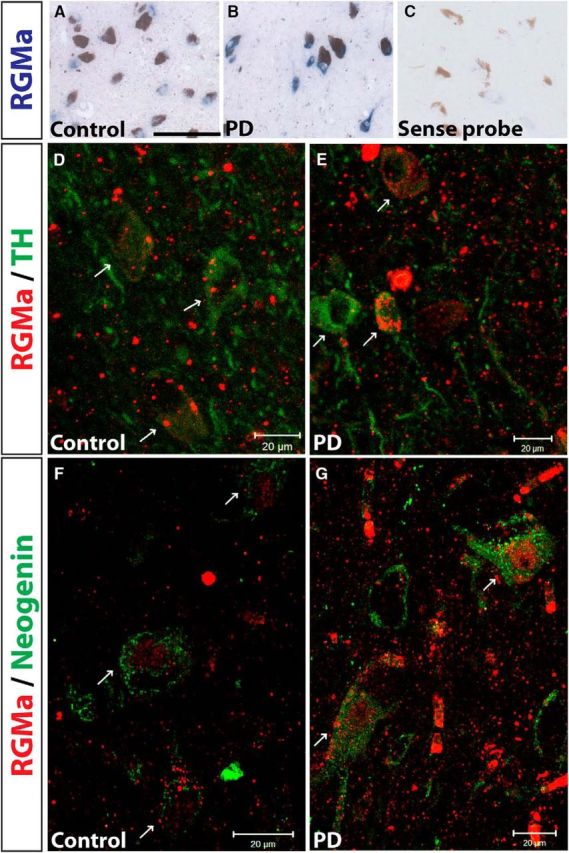
RGMa is expressed in human dopaminergic neurons in the SN of both control subjects and PD patients. A–C, In situ hybridization for human RGMa mRNA (blue staining) is exclusively present in cellular structures morphologically identified as neurons (large cells with a nucleolus in the center of the nucleus) in a control (A) and PD patient (B). Note the absence of staining in the sense probe (C). Most neurons contained neuromelanin (brown pigmentation), indicating their DA phenotype (best visible in C). Sections were used from the following NBB donors: control 00–049 (A), PD 02–064 (B), and control 00–050 (C). Scale bar, 0.1 mm. D, E, Immunofluorescent staining for RGMa protein (red; antibody SC-46482) in the SN of control (D) and PD (E) brains is mainly localized to DA neurons counterstained for TH (green). Arrows point to TH+ neurons also positive for punctate RGMa protein expression. Sections were used from NBB donors: control 98–126 and PD 00–115. F, G, Immunohistochemical staining for RGMA protein (red) and its receptor neogenin (green) in control (F) and PD (G) SN tissue. Arrows point to neogenin-positive neurons also showing punctate RGMa protein expression. Sections were used from NBB donors: control 00–142 and PD 02–003. Scale bar, 20 μm.
AAV-mediated delivery of RGMa results in enhanced RGMa levels in the SN and transport of RGMa to the striatum
Based on the specific neuronal expression of RGMa in human SN, we constructed an AAV vector driving mouse RGMa expression specifically in neurons using a synapsin promotor. AAV-synapsin-driven expression of mouse RGMa protein was validated after transfecting N2A cells with this construct. We detected the full-length membrane bound form (55–49 kDa) and the cleaved membrane bound C-terminal form (33 kDa) of RGMa in the cell lysate, as well as the 30 kDa N-terminal fragment of RGMa in the culture medium of the N2A cells (Fig. 2A). This pattern of expression and release is consistent with the report of Tassew et al. (2012).
Figure 2.

AAV vectors direct RGMa expression and secretion in a neuronal cell line and dose-dependent expression in the mouse brain. A, AAV plasmid-mediated overexpression of mouse RGMa in N2A cells results in the production and secretion of RGMa protein. N2A cells were either untreated (UnTr) or transfected with the expression plasmids pAAV-SYN-GFP or pAAV-SYN-mRGMa. Anti-mouse RGMa antibody (R&D Systems, AF2458) was used to detect mouse RGMa protein in the cell lysate (LYSATE samples) or the culture medium (MEDIUM samples) 3 d after transfection. N2A cells produce mouse RGMa after transfection of the expected molecular weight: multiple bands are visible ∼49–55 kDa representing the full-length form, and one prominent band at 33 kDa representing the cleaved membrane bound C-terminal form. RGMa is also released from the transfected N2A cells, which results in the N-terminal 30 kDa form of RGMa in the medium samples. β-actin was used as loading control. B, Comparison of RGMa protein levels in the SN of mouse injected with a high titer (9.0 × 1012 gc/ml) and low titer (3.0 × 1012 gc.ml). The RGMa fluorescence was twice as high in the high-titer-injected mice compared with the low-titer-injected mice (p < 0.0001, F(3,22) = 41.21). Data are represented in percentages of RGMa fluorescence levels relative to the AAV-Empty RGMa. Tissue from 6 mice was quantified in the high-titer treatment group, and from 7–8 mice in low-titer treatment group. Example images used for the quantification of RGMa are shown in Figure 6B (high-dose experiment) and Figure 7B (low-dose experiment). Statistical analysis was performed using one-way ANOVA with Tukey's post hoc multiple testing correction: ***p < 0.001; ****p < 0.0001. All data are expressed as mean with bars representing SEM.
Increased RGMa protein levels were observed both after high- and low-titer stereotactic injection of the AAV-RGMa vector in the SN. As expected, the injection of high-titer virus resulted in 2 times higher RGMa protein levels compared with the low-titer viral injection (Fig. 2B; p < 0.0001, F(3,22) = 41.21). High-titer AAV-RGMa injection resulted in an increase in the protein levels in both the SN and the striatum of the bilateral and unilateral AAV-RGMa-injected animals (Fig. 3A). Similarly, in the low-titer treatment paradigm, increased RGMa protein levels were observed both in the SN and the striatum of the AAV-RGMa-injected animals (Fig. 3B), but not in the AAV-Empty-, AAV-GFP-, or saline-injected animals. RGMa was expressed in DA neurons as well as extracellularly, confirming the release of the protein. Example images used for the quantification of RGMa are shown in Figure 6B (high-dose experiment) and Figure 7B (low-dose experiment). These results are indicative of successful AAV-mediated overexpression of RGMa in the SN.
Figure 3.
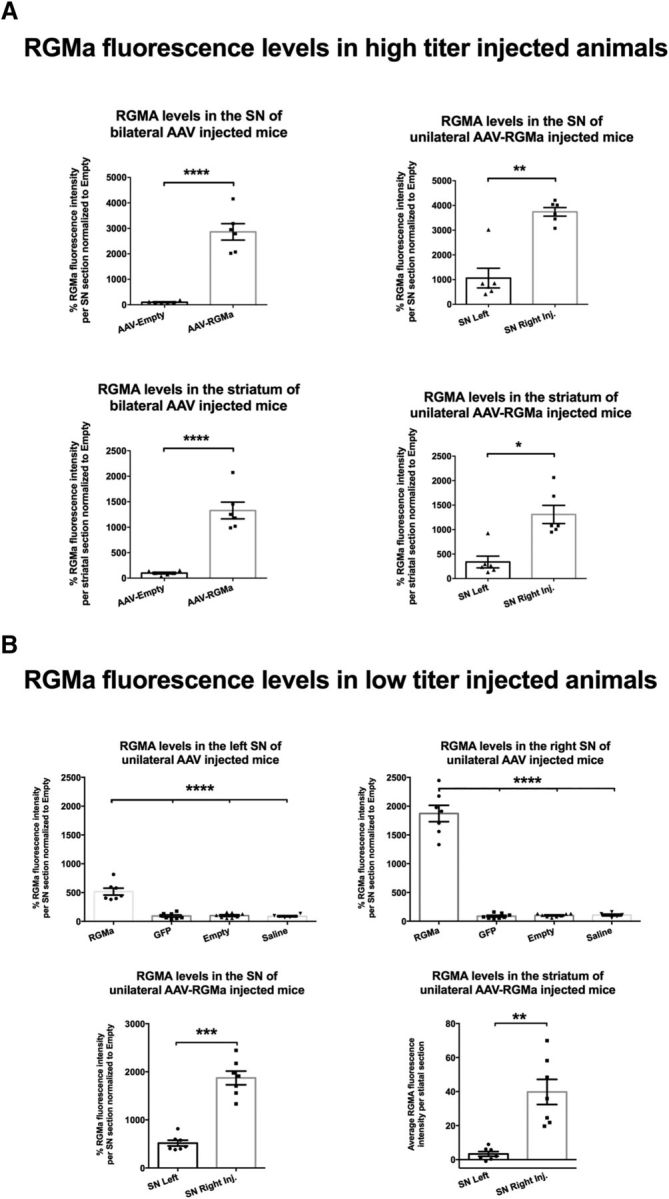
AAV vectors drive RGMa expression and secretion in the mouse brain. A, Quantification of the percentage of RGMa fluorescence intensity in SN and the striatum of the bilateral and unilateral high-titer-injected animals. RGMa protein levels were significantly increased in the injected SN compared with AAV-Empty-injected (p < 0.0001) and noninjected SN (p = 0.003). Similarly, striatal RGMa levels were significantly higher in the bilaterally AAV-RGMa-injected animals compared with the AAV-Empty-injected animals (p < 0.0001) and in the ipsilateral striatum versus the contralateral striatum of the unilaterally AAV-RGMa-injected animals (p = 0.0126). B, Quantification of RGMA fluorescence intensity in SN and striatum in low-titer AAV-RGMa-injected animals. The percentage of RGMa fluorescence intensity was significantly increased in the right SN injected with AAV-RGMa and, to a smaller extent, in the left noninjected SN, compared with all control groups (GFP, saline, or Empty, SN right, p < 0.0001, F(3,25) = 159.0; SN left, p < 0.0001, F(3,25) = 42.9). Both striatal (p = 0.001) and SN (p = 0.0001) levels of RGMa were significantly higher on the right transduced side of the nigrostriatal tract. Data are represented in percentages of fluorescence relative to the AAV-Empty RGMa fluorescence. For representative images of the RGMa immunohistochemical stained mouse SN and striatum, see Figures 6B, E and 7B, E. Tissue from 6 mice was measured in the high-titer treatment group, and from 7–8 mice in low-titer treatment group. Statistical analysis was performed using Student's t test and one-way ANOVA with Tukey's post hoc multiple testing correction: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. All data are expressed as mean with bars representing SEM.
Figure 6.
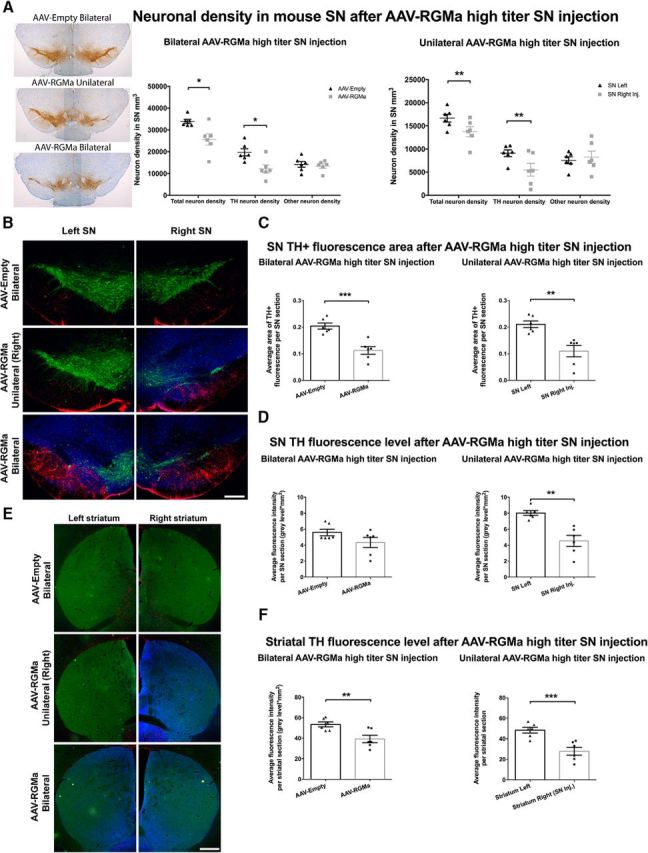
High-dose AAV-mediated overexpression of RGMa in the mouse SN induces a decline in the number of DA neurons and affects TH expression in the surviving neurons. A, Quantification of the neuronal density in the SN in AAV-Empty (bilateral), AAV-RGMa (unilateral), and AAV-RGMa (bilateral) injected SN. Bilateral RGMa overexpression resulted in a decrease in both the total (24%, p = 0.0136) and TH+ (38%, p = 0.012) neuronal density in the SN compared with the AAV-Empty bilateral injected SN (total SN neuron count combined from both SN). Unilateral AAV-RGMa-mediated overexpression also induced a significant decrease in total number of neurons (18%, p = 0.004) and in TH+ neurons (40%, p = 0.0072) in the right-injected SN compared with the left, noninjected SN. Other (TH−) neuronal profiles were not affected by RGMa overexpression. B, Immunohistochemical staining for TH (green), GFAP (red), and RGMa (blue) in mouse SN injected with AAV-Empty (bilateral), AAV-RGMA (unilateral), and AAV-RGMA (bilateral). C, Quantification of TH fluorescence-positive area in the SN. Bilateral overexpression of RGMa in the SN is associated with a decrease of TH+ area in the SN compared with bilateral AAV-Empty-injected SN (p = 0.0005; values from both left and right SN are pooled). TH fluorescence area in the right SN of animals unilaterally injected with AAV-RGMa is also decreased compared with the left, noninjected SN (p = 0.0026). D, Quantification of TH fluorescence levels in the SN. Despite the TH+ neuronal loss, TH fluorescence intensity is not significantly different in the SN of animals bilaterally injected with AAV-RGMa compared with the AAV-Empty-injected animals (p = 0.1261). In the unilaterally AAV-RGMa-injected animals, the left, noninjected SN shows a significant increase in TH fluorescence intensity compared with the right injected SN (p = 0.006), suggesting a compensatory mechanism. E, Immunohistochemical staining for TH (green), GFAP (red), and RGMa (blue; antibody SC-46482) in the striatum following the transduction of the SN with AAV-Empty (bilateral), AAV-RGMa (unilateral), and AAV-RGMa (bilateral). RGMa protein is shown to be transported from the transduced SN to the nigrostriatal projection target sites in the striatum. F, Quantification of TH fluorescence levels in the striatum. Overexpression of RGMa in the SN is associated with a decrease in TH fluorescence intensity in the striatum in bilateral RGMa injection paradigm compared with the AAV-Empty-injected animals (p = 0.0085). Similarly, the striatal TH intensity levels are decreased in the right striatum of the right SN-injected animals compared with the left striatum (p = 0.0004). Each treatment group includes 6 animals per quantification. Statistical analysis was performed using Student's t test: *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars: B, 0.25 mm; E, 0.5 mm. All data are expressed as mean with bars representing SEM.
Figure 7.
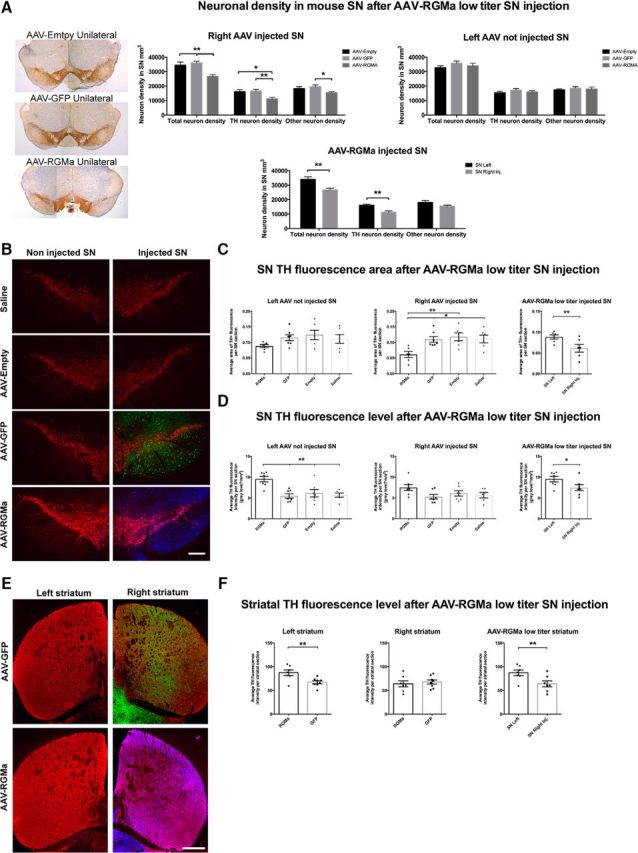
Low-dose AAV-mediated overexpression of RGMa induced a decline in DA neurons and increase in TH protein levels indicative of a compensation mechanism. A, Quantification of the neuronal density in AAV-Empty, AAV-GFP, and AAV-RGMa-injected and noninjected SN. RGMa overexpression resulted in a decrease in both the total (23% and 26%) and TH+ (30% and 33%) neuronal density in the AAV-RGMa-injected SN compared with the AAV-Empty and AAV-GFP injected SN (total neurons: p = 0.0015, F(2,19) = 9.318; TH neurons: p = 0.0041, F(2,19) = 7.449). TH− neurons were generally not affected by RGMa overexpression, with an exception compared with AAV-GFP-injected SN (p = 0.0488, F(2,19) = 3.555). Total and TH+ neuronal density was significantly decreased in the AAV-RGMa-injected SN compared with the not injected SN (p = 0.0029 and p = 0.0018, respectively). B, Immunohistochemical staining for TH (red), GFP (green), and RGMa (blue) in the SN injected with saline, AAV-Empty, AAV-GFP, or AAV-RGMA virus. C, Quantification of TH+ area in the SN. The TH+ area was significantly decreased in the SN injected with AAV-RGMa compared with all control groups (p = 0.0063, F(3,21) = 5.44) as well as to the contralateral (noninjected) SN of the RGMa-treated animals (p = 0.0073). D, Quantification of TH fluorescence levels in the SN. Overexpression of RGMa in the SN is associated with an increase in TH fluorescence in the noninjected contralateral SN compared with all control groups (p = 0.0006, F(3,23) = 8.3). TH fluorescence intensity in the AAV-RGMa-injected SN is not altered compared with the control treatment groups. E, Immunohistochemical staining for TH (red), GFP (green), and RGMa (blue; antibody SC-46482) in the striatum following the transduction of the SN with AAV-GFP or AAV-RGMa. Both GFP and RGMa protein is transported from the injected (right) SN to the nigrostriatal projection target sites in the right striatum. F, Quantification of TH fluorescence intensity in the striatum. Overexpression of RGMa in the SN is associated with an increase in TH fluorescence intensity in the left contralateral striatum compared with the AAV-GFP control striatum (p = 0.0082) and compared with the right AAV-RGMa striatum (p = 0.0037). TH fluorescent levels in the right striatum were not different in the AAV-RGMa group compared with all three control treatment groups (p = 0.6139). Each treatment group includes 7–8 animals per quantification, with the saline group containing 6 animals. Statistical analysis was performed using one-way ANOVA with Tukey's post hoc multiple testing correction and Student's t test: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. All data are expressed as mean with bars representing SEM. Scale bars: B, 0.25 mm; E, 0.5 mm.
RGMa overexpression in the SN induces severe motor impairments
We next investigated whether overexpression of RGMa in the SN affects motor performance in mice. Animals injected with a high dose of AAV-RGMa (at a titer of 9.0 × 1012 gc/ml) either unilaterally or bilaterally in the SN developed severe motor deficits over time (Fig. 4A–D). No motor deficits were observed in the control animals, bilaterally injected with titer matched AAV-Empty virus. The most striking behavioral deficit was revealed by the grid test, which is designed to measure accuracy of front paw placement and is significantly correlated with striatal DA levels (Tillerson and Miller, 2003). Both unilaterally and bilaterally AAV-RGMa-injected animals performed progressively worse in this test (p < 0.0001, F(2,270) = 46.52) compared with the AAV-Empty control group. By 2.5 weeks after injection, motor deficits started to be evident in the animals overexpressing RGMa and they progressively worsened until the end of the experiment at week 18.5 (Fig. 4A). The narrow beam test (Drucker-Colín and García-Hernández, 1991; Fleming et al., 2004) showed a significant increase in the number of hindlimb errors made by the unilaterally AAV-RGMa-injected animals (p < 0.0001, F(2,270) = 49.97), already starting 2 weeks after surgery (Fig. 4B).
Figure 4.

High-dose AAV-mediated overexpression of RGMa in the mouse SN results in a progressive and severe induction of motor deficits. A, Grid test performance showed a progressive increase of front paw placement errors in both unilateral (red) and bilateral (purple) RGMa-overexpressing mice compared with the AAV-Empty-injected animals (blue) (p < 0.0001, F(2,270) = 46.52). Starting at week 2.5 after surgery, RGMa-overexpressing mice displayed significantly higher error rates compared with the control with variations at different time points (see colored asterisks). B, Hindlimb placement measurements on the narrow beam test showed significant increase in the number of hindpaw placement errors between the injection groups (p < 0.0001, F(2,270) = 49.97) with post hoc test indicating unilaterally RGMa-injected mice to develop an increase in hindlimb slips compared with the AAV-Empty-injected animals (red asterisks). C, Preference of bilateral or unilateral front paw use was measured by a cylinder test. The ratio of single right paw use during rearing over total rearing events was determined (p < 0.0001, F(2,240) = 46.96). Both bilateral and unilateral RGMa-overexpressing mice showed increased right paw use at different time points compared with the AAV-Empty control group. D, The swing test revealed a significant difference between the injected groups (p < 0.0001, F(2,195) = 57.19), with an increase in the rotation preference toward the left in animals injected unilaterally with AAV-RGMa compared with AAV-Empty animals. Animals receiving a bilateral injection of AAV-RGMa or AAV-Empty did not develop a swing preference over time. E, Increased events of tremor were observed during the time of the experiment in the RGMa-overexpressing mice (p < 0.0001, F(2,240) = 55.58), with both unilaterally and bilaterally injected RGMa animals compared with the AAV-Empty-injected animals starting from week 4.5 onwards. F, Mice bilaterally overexpressing RGMa showed a significant decrease in body weight toward the end of the experiment (p < 0.0001, F(2,360) = 42.86). For all tests, week 0 is the baseline measurement performed 2 d before the AAV injection. Each treatment group consists of 6 animals. Statistical analysis was performed with a two-way ANOVA with Tukey's post hoc multiple testing correction: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Within each panel, different colored asterisks indicate significance between different treatment groups compared with the AAV-Empty group: black asterisks, both RGMa groups; red asterisks, unilateral RGMa group; purple asterisks, bilateral RGMa group. All data are expressed as mean with bars representing SEM.
In the cylinder test (Liu et al., 1999; Ulusoy et al., 2009), mice overexpressing RGMa either unilaterally or bilaterally developed a significant preference of single right paw use during rearing behavior (p < 0.0001, F(2,240) = 46.96; Fig. 4C). Additionally, the bilaterally treated RGMa group also showed a significant increase (p < 0.0001, F(2,240) = 18.84) in the use of their left paw over the use of both paws, suggestive of a dysfunction in coordinated use of both forepaws in this group (data not shown). This was not the case for the unilaterally injected animals; as expected, single left paw use was not increased compared with use of both paws together indicative of the contralateral effects of injection of AAV-RGMa in the right SN. Animals injected with AAV-Empty did not develop a preference in single paw use. The swing test (Roghani et al., 2002) revealed a significant preference in turning toward the left (contralateral to the side of the lesion) in unilaterally AAV-RGMa-injected animals (p < 0.0001, F(2,195) = 57.19; Fig. 4D). As expected, the bilateral RGMa group did not develop a side preference in the swing test given that both SN were targeted. Similarly, AAV-Empty-injected animals did not develop a side preference in the swing test.
In conclusion, all motor behavioral tests revealed motor impairments in mice overexpressing RGMa in the SN, but to a different degree depending on whether RGMa was delivered unilaterally or bilaterally. Additionally, a tremor assessment indicated significantly more tremor events in AAV-RGMa-injected animals (p < 0.0001, F(2,240) = 55.58; Fig. 4E). Bilateral RGMa overexpression had a negative impact on body weight, but this was only evident from 12.5 weeks on during the experiment (p < 0.0001, F(2,360) = 42.86; Fig. 4F), and can thus not be responsible for the behavioral changes that are observed at earlier time points.
Low-dose RGMa overexpression induces subtle behavioral deficits
We show above that overexpression of RGMa in the SN induces severe motor deficits. To test the potency of the RGMa effect, we hypothesized that administration of a lower dose of RGMa would result in the development of more subtle motor deficits in mice and therefore mimic more closely the gradual progressive nature of the clinical symptoms in PD. To this end, mice were injected with a viral vector titer of 3.0 × 1012 gc/ml, which is 3 times lower than the high-dose RGMa-overexpressing experiment described above. Animals injected with this lower titer of AAV-RGMa virus in the right SN developed more subtle progressive behavioral motor deficits as measured in the grid and cylinder tests (Fig. 5). No motor deficits were observed in the control groups injected with either saline or titer-matched AAV-Empty or AAV-GFP viruses. The most striking behavioral deficit was revealed by the grid test in which RGMa-overexpressing animals performed progressively worse (p < 0.0001, F(3,400) = 61.31; Fig. 5A), compared with the three control groups. The dysfunction started at 3.5 weeks and remained present until the end of the experiment at week 12 (Fig. 5A). At week 12, RGMa animals showed some degree of recovery but still performed significantly worse than the AAV-GFP-injected animals.
Figure 5.

Low-dose AAV-mediated overexpression of RGMa in the mouse SN results in a mild but progressive induction of motor behavioral deficits. A, A significant increase in front paw placement errors was observed in the grid test in the RGMa group (red) compared with the three control groups (AAV-Empty, blue; AAV-GFP, green; and saline, black) (p < 0.0001, F(3,400) = 61.31). Post hoc testing showed an increase in error rates in RGMa-overexpressing mice from 3.5 weeks after surgery compared with all three control groups (black asterisks) with some variation at different testing time points (red asterisks). B, Hindlimb placement was measured by the narrow beam test. Mice treated with AAV-RGMa showed small but significant increased error rate in this test compared with the controls (p = 0.0002, F(3,416) = 6.586). C, Preference of bilateral or unilateral front paw use was measured in a cylinder test. The ratio of right paw use over total rearing events was determined. RGMa-overexpressing mice progressively increased their preference for only right paw use compared with control groups (p < 0.0001, F(3,375) = 12.37). Post hoc testing indicated more right paw use in RGMa animals compared with either one or two control treatment groups (red asterisks) or to all control groups (black asterisk) starting from 8 weeks after surgery. D, The swing test revealed no significant differences in the rotation preference of the mice between any of the treatment groups. E, Tremor was observed in RGMa-overexpressing mice (p < 0.0001, F(3,416) = 20.71) starting at 3.5 weeks after surgery. F, None of the treatment groups showed any significant differences in weight gain. For all tests, week 0 is the baseline measurement performed 2 d before the AAV injection. Each treatment group includes 7–8 animals per quantification, with the saline group containing 6 animals. Statistical analysis was performed with a two-way ANOVA with Tukey's post hoc multiple testing correction: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. All data are expressed as mean with bars representing SEM.
In the cylinder test, during the course of the experiment, mice overexpressing RGMa developed a significant preference for single right paw use during rearing compared with the control-injected animals (p < 0.0001, F(3,375) = 12.37; Fig. 5C). This suggests a dysfunction of the left front paw, caused by low-dose RGMa overexpression in the contralateral SN.
The narrow beam test revealed small but significant impairment in hindlimb placement after low-dose RGMa expression (p = 0.0002, F(3,416) = 6.586; Fig. 5B). In contrast to the high-dose RGMa experiment, the swing test did not reveal a preference in turning behavior in any of the treatment groups in the low-dose experiment (Fig. 5D). A tremor assessment indicated more positive events in the AAV-RGMa-injected animals starting 3.5 weeks after surgery (p < 0.0001, F(3,416) = 20.71; Fig. 5E). Finally, unilateral low-dose RGMa overexpression had no impact on body weight (Fig. 5F). In summary, the extent of motor impairment in mice injected with the low-titer AAV-RGMa virus is less dramatic than in mice injected with a high-titer AAV-RGMa virus.
RGMa decreases neuronal density in the SN
We next investigated the effects of RGMa overexpression on SN neuronal integrity. Neuronal density was determined in cresyl violet- and TH-stained sections. High levels of bilateral RGMa overexpression in the SN resulted in 24% reduction in the total neuronal densities (TH+ and other neurons combined, p = 0.0136) and in 38% reduction of TH+ neuronal densities (p = 0.012), compared with the AAV-Empty bilaterally injected control mice (Fig. 6A). Unilateral high-titer AAV-RGMa injection into the right SN resulted in a significant decrease of 18% in the total neuronal density (p = 0.004) and a 40% decrease of TH+ neuronal density compared with the left noninjected SN (p = 0.0072; Fig. 6A). Importantly, TH− neuron numbers were not affected by RGMa overexpression in either of the experimental groups (p = 0.67, p = 0.53).
Similarly, in the low-titer RGMa-overexpressing experiment, a 22% decrease in the total neuron numbers (p = 0.0029) and a 30% decrease in the TH+ neuron numbers (p = 0.0018) were revealed in the injected SN in animals overexpressing RGMa compared with the contralateral noninjected SN at 12 weeks after injection (Fig. 7A). In animals injected with AAV-Empty or AAV-GFP, no changes in neuronal density were observed (Fig. 7A). Overexpression of RGMa in the SN compared with the AAV-Empty- and AAV-GFP-injected SN resulted in a significant decrease in the total neuronal densities (23% and 26%, respectively, p = 0.0015, F(2,19) = 9.318) and in the total density of TH+ neurons (30% and 33%, respectively, p = 0.0041, F(2,19) = 7.449) (Fig. 7A). As seen in the high-dose experiments, RGMa overexpression did not affect the density of the TH− neurons within the SN to a large extent, except compared with AAV-GFP injected SN, showing a small, yet significant, decrease in the density of TH− neurons after RGMa overexpression (p = 0.0488, F(2,19) = 3.555; Fig. 7A).
RGMa induced degeneration of TH+ neurons and upregulation of TH in surviving DA neurons
RGMa overexpression in the mouse SN was accompanied by a significant decrease in TH+ area within the SN compared with the AAV-Empty-injected SN (p = 0.0005; Fig. 6B,C) and compared with the noninjected SN in the unilateral treatment group (p = 0.0026; Fig. 6B,C). We also quantified the TH fluorescence intensity levels over the entire SN. To our surprise, the TH fluorescence levels in the AAV-RGMa-injected SN of the bilateral treated animals were not significantly decreased compared with the AAV-Empty control animals (Fig. 6D). This, in conjunction with the decrease in TH+ neurons observed in these treatment conditions (Fig. 6A), is suggestive of a compensatory increase in TH expression in the surviving DA neurons of the injected SN. Moreover, the TH fluorescence levels in the noninjected SN of the unilateral AAV-RGMa-treated animals were significantly increased compared with the injected right SN (p = 0.006) This is, again, suggestive of a compensatory effect, this time in the DA neurons of the contralateral (noninjected) SN, as a result of TH depletion in the injected SN in unilateral AAV-RGMa-treated animals.
Striatal TH fluorescence levels showed a significant decrease in the right striatum of the animals overexpressing RGMa unilaterally compared with the left striatum (p = 0.0004; Fig. 6E,F), and in both striata in the animals overexpressing RGMa bilaterally compared with the AAV-Empty animals (p = 0.0085; Fig. 6E,F). The decrease in striatal TH levels in RGMa-injected SN (unilaterally or bilaterally) supports the observed behavioral deficits in these animals. Although the TH levels of the contralateral SN in unilaterally AAV-RGMa-injected animals were increased compared with TH levels in the injected SN, this did not translate to a similar increase in striatal TH levels. The presumed compensatory effect occurring in the contralateral, noninjected, SN of unilaterally treated animals seems to be limited to the SN.
Comparable with the high-dose, low-dose RGMa overexpression also resulted in a significant decrease of the TH+ area in the injected SN compared with the noninjected SN (p = 0.0073) and the AAV-Empty, AAV-GFP, and saline-injected SN (p = 0.0063, F(3,21) = 5.44; Fig. 7B,C). The noninjected SN of the RGMa-treated mice showed a trend toward a decrease in TH+ area compared with the control treatment noninjected SN, which is most likely caused by a minor spread of the viral vector to the contralateral SN (Fig. 7B and Fig. 3B). Similar to the high-dose experiment, we did not see a decrease in the intensity of the TH signal in AAV-RGMa-injected animals. In contrast, the TH fluorescence levels in the contralateral (noninjected) SN of animals treated with AAV-RGMa were strongly increased compared with all of the control groups (p = 0.0006, F(3,23) = 8.3) and compared with the injected SN (p = 0.038; Fig. 7D). In contrast to the high-dose experiment, this increase was also observed in the striatum: TH fluorescence levels of nigrostriatal axons and terminals in the left striatum (the projection area of the left, noninjected SN neurons) were significantly increased in AAV-RGMa-treated animals compared with the right striatum (p = 0.0037) or to the left striatum of the AAV-GFP-treated animals (p = 0.0082; Fig. 7E,F). The right striatum, the projection area of the right, injected SN, did not show a significant decrease in TH fluorescence levels as a result of RGMa overexpression compared with the GFP-transduced nigrostriatal terminals (p = 0.6139). These observations suggest that surviving DA neurons in the injected SN are increasing their TH production as a possible compensatory mechanism to overcome the progressive decrease in DA levels within the nigrostriatal system. This may explain why the behavioral deficits in the low-dose paradigm (Fig. 5) are not as severe as in the high-dose paradigm (Fig. 4). Additionally, it is important to note that, in the low-dose experiment, TH levels were measured in the striatum at the end of the experiment (at week 12) when some behavioral deficits show signs of recovery (Fig. 5A). Animals treated with a high dose of AAV-RGMa do not display a recovery in motor function (Fig. 4), consistent with the decreased levels of TH fluorescence measured in the striatum of these animals at the end of the high-dose experiment (week 18.5; Fig. 6E,F). Perhaps analysis of the TH levels in the striatum at an earlier stage after low-dose RGMa administration (e.g., between week 7 to 9) would have resulted in a detectible decrease in TH levels consistent with this worsened level of behavioral ability observed at those time points.
RGMa overexpression induces gliosis
To investigate whether RGMa overexpression has an effect on the glial response in the SN, we analyzed the expression of the astrocytic marker GFAP and the active microglial marker Iba1 at 18.5 weeks after injection in the high-dose paradigm (Fig. 8A–C). Both GFAP and Iba1 fluorescence levels were increased in SN injected either unilaterally (GFAP, p = 0.0004; Iba1, p = 0.04; Fig. 8B) or bilaterally (GFAP, p = 0.005; Iba1, p = 0.001; Fig. 8C) with AAV-RGMa compared with the noninjected SN or the injected SN of the AAV-Empty animals, respectively.
Figure 8.

High-dose AAV-mediated overexpression of RGMa in the mouse SN induced a gliotic response. A, Immunohistochemical staining for TH (green) and Iba1 (red) in the SN injected with high-titer AAV-Empty bilaterally, AAV-RGMa unilaterally, and AAV-RGMa bilaterally. Scale bar, 0.25 mm. Immunohistochemical stanining for GFAP (red) in the injected SN has been visualized in Fig. 6B. B, Quantification of fluorescence signal in the SN of mice receiving a unilateral injection of high-titer AAV-RGMa. GFAP and Iba1 fluorescence intensities are increased in the AAV-RGMa-injected (right) SN compared with the noninjected (left) SN. C, Quantification of fluorescence signal in the SN of mice receiving a bilateral injection of high-titer AAV-Empty or the AAV-RGMa viral vector. GFAP and Iba1 intensities are increased in the AAV-RGMa-injected SN compared with the AAV-Empty-injected animals. Fluorescence intensities from right and left SN were averaged across both SN to provide the total fluorescence intensity per entire SN in each bilateral AAV treatment. D, Quantification of fluorescence signal in the striatum of mice receiving a unilateral injection of high-titer AAV-RGMa in the SN. GFAP fluorescence intensity is slightly decreased in the right (injected) striatum compared with the left (p = 0.027). We observed no differences in Iba1 levels between the two hemispheres. E, Quantification of fluorescence signal in the striatum of mice receiving a bilateral injection of high-titer AAV-Empty or AAV-RGMa virus in the SN. We observed no differences in GFAP fluorescence intensities between the two groups. Iba1 fluorescence intensity is slightly decreased in the striatum of AAV-RGMa-injected mice compared with the AAV-Empty-injected animals (p = 0.015). Fluorescence intensities from right and left striata were averaged across both striata to provide the total fluorescence intensity per AAV treatment. Each treatment group includes 6 animals per quantification. Statistical analysis was performed using Student's t test: *p < 0.05; **p < 0.01; ***p < 0.001. All data are expressed as mean with bars representing SEM.
Overall, no substantial microgliosis was observed in the striatum of mice overexpressing RGMa in the SN, although there was a small but significant decrease in GFAP signal in the right striatum of animals in the unilateral injection paradigm (p = 0.027; Fig. 8D) and a small significant decrease of Iba1 signal in the striatum of animals overexpressing RGMa bilaterally (p = 0.015; Fig. 8E).
Furthermore, to investigate whether low-dose RGMa overexpression has an effect on the glial response, we analyzed the expression of GFAP and Iba1 in these animals 12 weeks after injection (Fig. 9A–C). Similarly to the high-titer experiment, GFAP fluorescence levels were increased in SN injected with AAV-RGMa compared with both the noninjected SN (p < 0.0001) and the injected SN of the AAV-GFP animals (p = 0.0020, F(3,25) = 6.583; Fig. 9A,B). Additionally, we found an induced activation of microglia in AAV-RGMa-injected SN compared with the noninjected SN (p < 0.0001; Fig. 9A,C) as well compared with AAV-Empty and saline control treatment groups (p < 0.0001, F(3,25) = 19.66). No microgliosis was seen in the striatum of mice injected in the SN with AAV-RGMa (data not shown).
Figure 9.

Low-dose AAV-mediated overexpression of RGMa induced a gliotic response. A, Immunohistochemical staining for TH (green), GFAP (red), and Iba1 (blue) in mouse SN injected with saline, AAV-Empty, AAV-GFP, or AAV-RGMA. The two panels represent the injected and noninjected SN for each animal. Because of a limitation of the available number of fluorescence channels, two series of SN tissue were used for animals injected with AAV-GFP: one to stain for GFAP and one to stain for Iba1. B, Quantification of GFAP fluorescence signal in the SN 12 weeks after injection. Overexpression of RGMa in the SN is associated with an increase in GFAP fluorescence intensity in the injected SN compared with the AAV-GFP group (p = 0.0020, F(3,25) = 6.583) and to the noninjected SN in AAV-RGMa-treated animals (p < 0.0001). GFAP fluorescence is decreased in the noninjected SN of AAV-GFP-treated animals compared with the noninjected SN of the AAV-RGMa, AAV-Empty, and saline-treated groups (p < 0.0001, F(3,25) = 32.97). C, Quantification of Iba1 fluorescence signal in the SN 12 weeks after injection. Overexpression of RGMa in the SN is associated with an increase in Iba1 fluorescence intensity in the injected SN compared with the contralateral noninjected SN in RGMA-treated animals (p < 0.0001), as well as to the injected SN of the AAV-Empty and saline-treated animals (p < 0.0001, F(3,25) = 19.66). Iba1 fluorescence is significantly increased in both SN of animals injected with AAV-GFP compared with both SN of AAV-RGMa, AAV-Empty, and saline-treated animals (not injected SN, p < 0.0001, F(3,25) = 35.26; injected SN, p < 0.0001, F(3,25) = 19.66). AAV-RGMa treatment group consists of 7 animals, AAV-GFP and AAV-Empty consist of 8 animals per group, and the saline treatment group contains 6 animals. Statistical analysis was performed using one-way ANOVA with Tukey's post hoc multiple testing correction and Student's t test: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Scale bars, 0.25 mm. All data are expressed as mean with bars representing SEM.
In addition to RGMa-associated SN gliosis, an increase in microglial activation was seen in AAV-GFP-injected animals in both injected (p < 0.0001, F(3,25) = 19.66) and noninjected SN 12 weeks after injection (p < 0.0001, F(3,25) = 35.26; Fig. 9A–C), suggesting an immune response to the GFP protein, as previously reported by others (Klein et al., 2006; Yang et al., 2016). AAV-GFP-injected animals also showed a significant decrease in GFAP fluorescence intensity in the noninjected SN compared with the other control treatment groups (p < 0.0001, F(3,25) = 32.97; Fig. 9A,B).
RGMa expression in the SN does not decrease phosphoAKT in DA neurons
Akt is an antiapoptotic factor, which has been shown to be dephosporylated in neurons that are degenerating following exposure to RGMa derived from Th17 cells in culture (Tanabe and Yamashita, 2014). To determine whether RGMa induces DA neuron degeneration via pAKT, we examined the phosphorylation state of Akt using pAKT specific antibodies in the DA neurons following overexpression of RGMa. The levels of total AKT protein and pAKT were not different in DA neurons identified by TH immunohistochemistry (Fig. 10). These data suggest that phosphorylation of AKT is not affected by RGMa and that this cell death pathway is not involved in RGMa-induced DA neuron degeneration.
Figure 10.

Akt and pAkt levels in SN DA neurons upon high-dose RGMa overexpression. A, Quantification of Akt and pAkt fluorescence signal in the SN DA neurons. Overexpression of RGMa in the SN is not associated with an increase in either the total Akt or the Ser473 phosphorylated Akt (pAkt) fluorescence intensity in the DA neurons of the AAV-RGMa-injected animals compared with AAV-Empty group (p = 0.174 and p = 0.333, respectively). Total Akt analysis was performed on 6 animals in the AAV-Empty-injected group and 4 animals in the AAV-RGMa-injected group. PhosphoAkt analysis was performed on 6 animals in the AAV-Empty-injected group and 5 animals in the AAV-RGMa-injected group. B, Quantification of pAkt fluorescence signal in the SN DA neurons corrected for the total Akt signal. Overexpression of RGMa in the SN is not associated with an increase in pAkt fluorescence intensity in the DA neurons of the AAV-RGMa-injected animals compared with AAV-Empty group (p = 0.895). PhosphoAkt analysis was performed on 6 animals in the AAV-Empty-injected group and 4 animals in the AAV-RGMa-injected group. Statistical analysis was performed using Student's t test. All data are expressed as mean with bars representing SEM.
Discussion
The upregulation of RGMa in the SN of PD patients and its role in axon repulsion and neuronal survival prompted us to study the consequences of increased RGMa levels in the mesencephalic dopaminergic system of mice. We show that AAV-mediated overexpression of RGMa by midbrain dopaminergic neurons induced degeneration of these neurons resulting in a movement disorder typical for striatal DA deficiency. RGMa overexpression also induced strong astroglial and microglial activation. These data implicate RGMa in the pathophysiology of PD and suggest that targeting RGMa signaling could have therapeutic potential for the treatment of PD.
RGMa induced degeneration of mouse SN: similarities to PD etiology
The effects of overexpression of RGMa in the mouse midbrain bear several striking commonalities with the neuropathology of PD. Upregulation of RGMa in the adult mouse SN, both at high and low levels, induced progressive movement deficits, albeit at different severity, due to DA loss in the striatum (Tillerson and Miller, 2003), mimicking the clinical motor symptoms of PD patients. Additionally, we observed a preferential loss of TH+ neurons in the targeted SN, whereas TH− neurons were unaffected. This TH+ neuronal loss was observed in both high- and low-dose RGMa-overexpressing animals. Selective degeneration of TH+ neurons and the sparing of TH− neuronal populations, including calbindin-positive neurons in the SN, is a hallmark of the disease (Yamada et al., 1990; Kordower et al., 2013) and has also been observed in other animal models for PD, such as MPTP-treated monkeys (Lavoie and Parent, 1991; German et al., 1992). Interestingly, in unilateral high-dose RGMa-expressing animals, the striatal TH levels on the injected side were decreased compared with the contralateral side, whereas TH levels in the noninjected SN were increased compared with the injected side. This may suggest that the unaffected SN is compensating for loss of DA signaling in the contralateral nigrostriatal pathway by upregulating TH protein levels as a means to increase DA production. Furthermore, in low-dose RGMa-overexpressing mice, in which slightly less severe DA neuron loss is observed, TH protein levels were also increased in the noninjected SN and the nonaffected striatum, suggestive of the development of a compensatory mechanism in spared DA neurons. Such a compensatory mechanism, aimed to combat the diminishing DA levels in the nigrostriatal pathway, may explain the observed trend toward a less severe behavioral phenotype and some degree of functional recovery as observed in these low-dose RGMa animals in the grid test 12 weeks after surgery. A similar mechanism has been previously described in low-dose MPTP-treated asymptomatic primates where surviving dopaminergic neurons enhanced their DA release into the striatum (Perez et al., 2008). Moreover, in a “low-dose” synucleinopathy model for PD, striatal TH expression returned to baseline levels following an initial decrease, correlating with a rescue of behavioral deficits (Koprich et al., 2011).
To further investigate the commonalities between RGMa-induced cellular changes in the mouse nigrostriatal system and neuropathology in PD, we studied another pathological feature that is prominent in PD: glial reactivity (Langston et al., 1999; Hanisch, 2002; Barcia et al., 2003; Hirsch et al., 2003; McGeer and McGeer, 2008; Mosley et al., 2012). RGMa induced both astrogliosis and microgliosis in the mouse SN. In PD, gliosis may play a crucial role in the progression of neuronal degeneration by increasing the release of cytokines and chemokines from activated glia cells (Hanisch, 2002; Barcia et al., 2003; Hirsch et al., 2003; Mosley et al., 2012). Indeed, treatments that prevent microglia activation in the MPTP mouse model (Schintu et al., 2009) or reduce proinflammatory cytokine release in the 6-OHDA rat model (Smith et al., 2015) have been shown to promote dopaminergic neuron survival in vivo.
Possible mechanisms of RGMa-induced neurodegeneration
Based on the known functions of RGMa, we discuss two scenarios how the current neurodegenerative phenotype might develop. Following neuron-specific overexpression of RGMa in the SN, we observed elevated RGMa protein levels in the SN and in the ipsilateral striatum indicative of RGMa protein transport via nigrostriatal projections. The increased levels of RGMa may have significant implications for RGMa neogenin signaling within the nigrostriatal system.
In the first scenario, the increased levels of RGMa protein in the striatum may induce repulsive signaling in the dopaminergic nigrostriatal projections, leading to synaptic loss, axonal retraction, and finally atrophy and/or neuronal death in the SN. RGMa-induced axonal repulsion occurs when RGMa signals through neogenin to induce growth cone collapse (Wilson and Key, 2007; Yamashita et al., 2007). RhoA/Rho-kinase and PKC become activated, and downstream myosin II phosphorylation leads to a reduction of F-actin in growth cones and subsequent axonal collapse (Conrad et al., 2007; Kubo et al., 2008). Furthermore, RGMa acting through neogenin can also inhibit outgrowth and repulsion of neurites of cortical neurons by inducing growth cone collapse (Endo and Yamashita, 2009). The induction of retrograde neuronal death and/or atrophy would be in support of the dying back hypothesis (Dauer and Przedborski, 2003; Cheng et al., 2010; Maday, 2016; Tagliaferro and Burke, 2016), stating that the primary neurodegenerative event in PD is the loss of dopaminergic nigrostriatal presynaptic terminals followed by the subsequent retraction of axons, degeneration of dopaminergic neurons in the SN, and induction of a glial response.
In the second scenario, increased RGMa expression in neurons would lead to local RGMa signaling in the SN where it could interact with neogenin expressed on DA neurons and/or attract and activate immune cells, which express neogenin. In the human brain, RGMa is associated with DA neurons and is present in extracellular deposits, whereas after forced overexpression in the mouse RGMa is expressed in DA neurons and in a diffuse extracellular pattern. This suggest that RGMa can act as a short-range as well as a long-range cue in the SN. RGMa neogenin signaling in immune cells can lead to the production of proinflammatory cytokines, which consequently can induce neuronal toxicity. The role of the immune system in terms of a pathological feature of PD has gained much attention in recent years (for review, see Kannarkat et al., 2013). There is evidence of increased CD4+ and CD8+ T-cell numbers in the SN of PD patients (Brochard et al., 2009), and an increase in CD4+ T-cell infiltration into the SN in the MPTP model has also been observed (Reynolds et al., 2010). Neogenin is expressed by various non-neuronal cells, including CD4+ T cells and macrophages in a mouse MS model (Muramatsu et al., 2011) and by CD3+ T cells in the brain and the spinal cord of MS patients (Muramatsu et al., 2011). Furthermore, T-cell recruitment to active lesion areas in the MS mouse model has been shown to be mediated through RGMa binding to its receptor neogenin (Muramatsu et al., 2011; Tanabe and Yamashita, 2014). Therefore, RGMa released by dopaminergic neurons may result in the activation of immune cells and induce cytokine and chemokine production, which leads to neuronal stress and neurotoxicity. Consequently, RGMa overexpression-mediated recruitment of these proinflammatory cells could induce neuronal degeneration in the mouse SN.
In conclusion, we identified RGMa as a new key player in dopaminergic neuron degeneration in the adult SN. The data suggest that RGMa may have a crucial role in the development of PD pathology. A recent pilot study reports that levodopa increased RGMa levels in blood plasma of PD patients, which may further inhibit mechanisms of neuronal survival (Müller et al., 2016). Various axon guidance cues, with important roles during neuronal development, are now being linked to the pathophysiology of neurodegenerative diseases, including semaphorins, ephrins, slits, and netrins and their respective receptors (Van Battum et al., 2015; for review, see Korecka et al., 2016). Changes in the expression levels or expression patterns of these axon guidance cues might induce alterations in the neuronal circuitry, thereby contributing to the development of the neuropathology. The current findings have implications for therapy development as interfering with the function of this specific guidance cue may be beneficial to the survival of DA neurons. Given the fact that the use of anti-RGMa antibodies in disease models, such as MS (Tanabe and Yamashita, 2014; Demicheva et al., 2015) or stroke (Shabanzadeh et al., 2015), has led to functional improvements, our findings point to RGMa as a promising therapeutic target for PD and RGMa monoclonal antibodies may be applied as a disease-modifying treatment for PD.
Footnotes
This work was supported by the Stichting ParkinsonFonds, Top Institute Pharma, and Stichting Vrienden van het Herseninstituut. We thank Prof. Deniz Kirik (Lund University, Lund, Sweden) for providing the pAAV2Sna-SW and pTRUF20B-SEW plasmids.
The authors declare no competing financial interests.
References
- Bao AM, Hestiantoro A, Van Someren EJ, Swaab DF, Zhou JN (2005) Colocalization of corticotropin-releasing hormone and oestrogen receptor-alpha in the paraventricular nucleus of the hypothalamus in mood disorders. Brain 128:1301–1313. 10.1093/brain/awh448 [DOI] [PubMed] [Google Scholar]
- Barcia C, Fernández Barreiro A, Poza M, Herrero MT (2003) Parkinson's disease and inflammatory changes. Neurotox Res 5:411–418. 10.1007/BF03033170 [DOI] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A (2013) NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossers K, Meerhoff G, Balesar R, van Dongen JW, Kruse CG, Swaab DF, Verhaagen J (2009) Analysis of gene expression in Parkinson's disease: possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain Pathol 19:91–107. 10.1111/j.1750-3639.2008.00171.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. 10.1016/S0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119:182–192. 10.1172/JCI36470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capelli LP, Krepischi AC, Gurgel-Giannetti J, Mendes MF, Rodrigues T, Varela MC, Koiffmann CP, Rosenberg C (2012) Deletion of the RMGA and CHD2 genes in a child with epilepsy and mental deficiency. Eur J Med Genet 55:132–134. 10.1016/j.ejmg.2011.10.004 [DOI] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67:715–725. 10.1002/ana.21995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SJ, Bradford D, Cooper HM (2007) Neogenin: a multi-functional receptor regulating diverse developmental processes. Int J Biochem Cell Biol 39:1569–1575. 10.1016/j.biocel.2006.11.009 [DOI] [PubMed] [Google Scholar]
- Conrad S, Genth H, Hofmann F, Just I, Skutella T (2007) Neogenin-RGMa signaling at the growth cone is bone morphogenetic protein-independent and involves RhoA, ROCK, and PKC. J Biol Chem 282:16423–16433. 10.1074/jbc.M610901200 [DOI] [PubMed] [Google Scholar]
- Cooper-Knock J, Kirby J, Ferraiuolo L, Heath PR, Rattray M, Shaw PJ (2012) Gene expression profiling in human neurodegenerative disease. Nat Rev Neurol 8:518–530. 10.1038/nrneurol.2012.156 [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39:889–909. 10.1016/S0896-6273(03)00568-3 [DOI] [PubMed] [Google Scholar]
- Demicheva E, Cui YF, Bardwell P, Barghorn S, Kron M, Meyer AH, Schmidt M, Gerlach B, Leddy M, Barlow E, O'Connor E, Choi CH, Huang L, Veldman GM, Rus H, Shabanzadeh AP, Tassew NG, Monnier PP, Müller T, Calabresi PA, et al. (2015) Targeting repulsive guidance molecule A to promote regeneration and neuroprotection in multiple sclerosis. Cell Rep 10:1887–1898. 10.1016/j.celrep.2015.02.048 [DOI] [PubMed] [Google Scholar]
- Drucker-Colín R, García-Hernández F (1991) A new motor test sensitive to aging and dopaminergic function. J Neurosci Methods 39:153–161. 10.1016/0165-0270(91)90081-A [DOI] [PubMed] [Google Scholar]
- Edwards YJ, Beecham GW, Scott WK, Khuri S, Bademci G, Tekin D, Martin ER, Jiang Z, Mash DC, ffrench-Mullen J, Pericak-Vance MA, Tsinoremas N, Vance JM (2011) Identifying consensus disease pathways in Parkinson's disease using an integrative systems biology approach. PLoS One 6:e16917. 10.1371/journal.pone.0016917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M, Yamashita T (2009) Inactivation of Ras by p120GAP via focal adhesion kinase dephosphorylation mediates RGMa-induced growth cone collapse. J Neurosci 29:6649–6662. 10.1523/JNEUROSCI.0927-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF (2004) Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci 24:9434–9440. 10.1523/JNEUROSCI.3080-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM (2002) Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A 99:11854–11859. 10.1073/pnas.182412299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, Manaye KF, Sonsalla PK, Brooks BA (1992) Midbrain dopaminergic cell loss in Parkinson's disease and MPTP-induced parkinsonism: sparing of calbindin-D28k-containing cells. Ann N Y Acad Sci 648:42–62. 10.1111/j.1749-6632.1992.tb24523.x [DOI] [PubMed] [Google Scholar]
- Gorell JM, Peterson EL, Rybicki BA, Johnson CC (2004) Multiple risk factors for Parkinson's disease. J Neurol Sci 217:169–174. 10.1016/j.jns.2003.09.014 [DOI] [PubMed] [Google Scholar]
- Hanisch UK. (2002) Microglia as a source and target of cytokines. Glia 40:140–155. 10.1002/glia.10161 [DOI] [PubMed] [Google Scholar]
- Hata K, Fujitani M, Yasuda Y, Doya H, Saito T, Yamagishi S, Mueller BK, Yamashita T (2006) RGMa inhibition promotes axonal growth and recovery after spinal cord injury. J Cell Biol 173:47–58. 10.1083/jcb.200508143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermens WT, ter Brake O, Dijkhuizen PA, Sonnemans MA, Grimm D, Kleinschmidt JA, Verhaagen J (1999) Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum Gene Ther 10:1885–1891. 10.1089/10430349950017563 [DOI] [PubMed] [Google Scholar]
- Hernandez DG, Reed X, Singleton AB (2016) Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 139 [Suppl 1]: 59–74. 10.1111/jnc.13593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Breidert T, Rousselet E, Hunot S, Hartmann A, Michel PP (2003) The role of glial reaction and inflammation in Parkinson's disease. Ann N Y Acad Sci 991:214–228. 10.1111/j.1749-6632.2003.tb07478.x [DOI] [PubMed] [Google Scholar]
- Huitinga I, van der Cammen M, Salm L, Erkut Z, van Dam A, Tilders F, Swaab D (2000) IL-1beta immunoreactive neurons in the human hypothalamus: reduced numbers in multiple sclerosis. J Neuroimmunol 107:8–20. 10.1016/S0165-5728(00)00248-4 [DOI] [PubMed] [Google Scholar]
- Jankovic J. (2008) Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79:368–376. 10.1136/jnnp.2007.131045 [DOI] [PubMed] [Google Scholar]
- Kalia LV, Lang AE (2015) Parkinson's disease. Lancet 386:896–912. 10.1016/S0140-6736(14)61393-3 [DOI] [PubMed] [Google Scholar]
- Kannarkat GT, Boss JM, Tansey MG (2013) The role of innate and adaptive immunity in Parkinson's disease. J Parkinsons Dis 3:493–514. 10.3233/JPD-130250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RL, Dayton RD, Leidenheimer NJ, Jansen K, Golde TE, Zweig RM (2006) Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol Ther 13:517–527. 10.1016/j.ymthe.2005.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeberle PD, Tura A, Tassew NG, Schlichter LC, Monnier PP (2010) The repulsive guidance molecule, RGMa, promotes retinal ganglion cell survival in vitro and in vivo. Neuroscience 169:495–504. 10.1016/j.neuroscience.2010.04.079 [DOI] [PubMed] [Google Scholar]
- Koprich JB, Johnston TH, Huot P, Reyes MG, Espinosa M, Brotchie JM (2011) Progressive neurodegeneration or endogenous compensation in an animal model of Parkinson's disease produced by decreasing doses of alpha-synuclein. PLoS One 6:e17698. 10.1371/journal.pone.0017698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 136:2419–2431. 10.1093/brain/awt192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korecka JA, Levy S, Isacson O (2016) In vivo modeling of neuronal function, axonal impairment and connectivity in neurodegenerative and neuropsychiatric disorders using induced pluripotent stem cells. Mol Cell Neurosci 73:3–12. 10.1016/j.mcn.2015.12.004 [DOI] [PubMed] [Google Scholar]
- Kubo T, Endo M, Hata K, Taniguchi J, Kitajo K, Tomura S, Yamaguchi A, Mueller BK, Yamashita T (2008) Myosin IIA is required for neurite outgrowth inhibition produced by repulsive guidance molecule. J Neurochem 105:113–126. 10.1111/j.1471-4159.2007.05125.x [DOI] [PubMed] [Google Scholar]
- Kubo T, Tokita S, Yamashita T (2012) Repulsive guidance molecule-a and demyelination: implications for multiple sclerosis. J Neuroimmune Pharmacol 7:524–528. 10.1007/s11481-011-9334-z [DOI] [PubMed] [Google Scholar]
- Kumaran R, Cookson MR (2015) Pathways to Parkinsonism Redux: convergent pathobiological mechanisms in genetics of Parkinson's disease. Hum Mol Genet 24:R32–R44. 10.1093/hmg/ddv236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lah GJ, Key B (2012) Dual roles of the chemorepellent axon guidance molecule RGMa in establishing pioneering axon tracts and neural fate decisions in embryonic vertebrate forebrain. Dev Neurobiol 72:1458–1470. 10.1002/dneu.22010 [DOI] [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D (1999) Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol 46:598–605. [DOI] [PubMed] [Google Scholar]
- Lavoie B, Parent A (1991) Dopaminergic neurons expressing calbindin in normal and parkinsonian monkeys. Neuroreport 2:601–604. 10.1097/00001756-199110000-00012 [DOI] [PubMed] [Google Scholar]
- Lesnick TG, Papapetropoulos S, Mash DC, Ffrench-Mullen J, Shehadeh L, de Andrade M, Henley JR, Rocca WA, Ahlskog JE, Maraganore DM (2007) A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet 3:e98. 10.1371/journal.pgen.0030098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Lesnick TG, Maraganore DM, Isacson O (2009) Axon guidance and synaptic maintenance: preclinical markers for neurodegenerative disease and therapeutics. Trends Neurosci 32:142–149. 10.1016/j.tins.2008.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kim D, Himes BT, Chow SY, Schallert T, Murray M, Tessler A, Fischer I (1999) Transplants of fibroblasts genetically modified to express BDNF promote regeneration of adult rat rubrospinal axons and recovery of forelimb function. J Neurosci 19:4370–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S. (2016) Mechanisms of neuronal homeostasis: autophagy in the axon. Brain Res 1649:143–150. 10.1016/j.brainres.2016.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga E, Chédotal A (2004) Repulsive guidance molecule/neogenin: a novel ligand-receptor system playing multiple roles in neural development. Dev Growth Differ 46:481–486. 10.1111/j.1440-169x.2004.00768.x [DOI] [PubMed] [Google Scholar]
- Matsunaga E, Tauszig-Delamasure S, Monnier PP, Mueller BK, Strittmatter SM, Mehlen P, Chédotal A (2004) RGM and its receptor neogenin regulate neuronal survival. Nat Cell Biol 6:749–755. 10.1038/ncb1157 [DOI] [PubMed] [Google Scholar]
- Matsunaga E, Nakamura H, Chédotal A (2006) Repulsive guidance molecule plays multiple roles in neuronal differentiation and axon guidance. J Neurosci 26:6082–6088. 10.1523/JNEUROSCI.4556-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG (2008) Glial reactions in Parkinson's disease. Mov Disord 23:474–483. 10.1002/mds.21751 [DOI] [PubMed] [Google Scholar]
- Meredith GE, Kang UJ (2006) Behavioral models of Parkinson's disease in rodents: a new look at an old problem. Mov Disord 21:1595–1606. 10.1002/mds.21010 [DOI] [PubMed] [Google Scholar]
- Metzger M, Conrad S, Skutella T, Just L (2007) RGMa inhibits neurite outgrowth of neuronal progenitors from murine enteric nervous system via the neogenin receptor in vitro. J Neurochem 103:2665–2678. 10.1111/j.1471-4159.2007.04994.x [DOI] [PubMed] [Google Scholar]
- Miñones-Moyano E, Porta S, Escaramís G, Rabionet R, Iraola S, Kagerbauer B, Espinosa-Parrilla Y, Ferrer I, Estivill X, Martí E (2011) MicroRNA profiling of Parkinson's disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet 20:3067–3078. 10.1093/hmg/ddr210 [DOI] [PubMed] [Google Scholar]
- Monnier PP, Sierra A, Macchi P, Deitinghoff L, Andersen JS, Mann M, Flad M, Hornberger MR, Stahl B, Bonhoeffer F, Mueller BK (2002) RGM is a repulsive guidance molecule for retinal axons. Nature 419:392–395. 10.1038/nature01041 [DOI] [PubMed] [Google Scholar]
- Mosley RL, Hutter-Saunders JA, Stone DK, Gendelman HE (2012) Inflammation and adaptive immunity in Parkinson's disease. Cold Spring Harbor Perspect Med 2:a009381. 10.1101/cshperspect.a009381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller BK, Yamashita T, Schaffar G, Mueller R (2006) The role of repulsive guidance molecules in the embryonic and adult vertebrate central nervous system. Philos Trans R Soc Lond B Biol Sci 361:1513–1529. 10.1098/rstb.2006.1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller T, Trommer I, Muhlack S, Mueller BK (2016) Levodopa increases oxidative stress and repulsive guidance molecule A levels: a pilot study in patients with Parkinson's disease. J Neural Transm (Vienna) 123:401–406. 10.1007/s00702-016-1519-4 [DOI] [PubMed] [Google Scholar]
- Muramatsu R, Kubo T, Mori M, Nakamura Y, Fujita Y, Akutsu T, Okuno T, Taniguchi J, Kumanogoh A, Yoshida M, Mochizuki H, Kuwabara S, Yamashita T (2011) RGMa modulates T cell responses and is involved in autoimmune encephalomyelitis. Nat Med 17:488–494. 10.1038/nm.2321 [DOI] [PubMed] [Google Scholar]
- Niederkofler V, Salie R, Sigrist M, Arber S (2004) Repulsive guidance molecule (RGM) gene function is required for neural tube closure but not retinal topography in the mouse visual system. J Neurosci 24:808–818. 10.1523/JNEUROSCI.4610-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohra R, Beyeen AD, Guo JP, Khademi M, Sundqvist E, Hedreul MT, Sellebjerg F, Smestad C, Oturai AB, Harbo HF, Wallström E, Hillert J, Alfredsson L, Kockum I, Jagodic M, Lorentzen J, Olsson T (2010) RGMA and IL21R show association with experimental inflammation and multiple sclerosis. Genes Immun 11:279–293. 10.1038/gene.2009.111 [DOI] [PubMed] [Google Scholar]
- Olanow CW, Stern MB, Sethi K (2009) The scientific and clinical basis for the treatment of Parkinson disease. Neurology 72 [Suppl 4]:1–136. [DOI] [PubMed] [Google Scholar]
- Paxinos AF. (2001) The mouse brain in stereotaxic coordinates: San Diego: Academic. [Google Scholar]
- Perez XA, Parameswaran N, Huang LZ, O'Leary KT, Quik M (2008) Pre-synaptic dopaminergic compensation after moderate nigrostriatal damage in non-human primates. J Neurochem 105:1861–1872. 10.1111/j.1471-4159.2008.05268.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE (2010) Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson's disease. J Immunol 184:2261–2271. 10.4049/jimmunol.0901852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Pan P, Parkkila S (2007) Expression studies of neogenin and its ligand hemojuvelin in mouse tissues. J Histochem Cytochem 55:85–96. 10.1369/jhc.6A7031.2006 [DOI] [PubMed] [Google Scholar]
- Roghani M, Behzadi G, Baluchnejadmojarad T (2002) Efficacy of elevated body swing test in the early model of Parkinson's disease in rat. Physiol Behav 76:507–510. 10.1016/S0031-9384(02)00753-9 [DOI] [PubMed] [Google Scholar]
- Samad TA, Srinivasan A, Karchewski LA, Jeong SJ, Campagna JA, Ji RR, Fabrizio DA, Zhang Y, Lin HY, Bell E, Woolf CJ (2004) DRAGON: a member of the repulsive guidance molecule-related family of neuronal- and muscle-expressed membrane proteins is regulated by DRG11 and has neuronal adhesive properties. J Neurosci 24:2027–2036. 10.1523/JNEUROSCI.4115-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schintu N, Frau L, Ibba M, Caboni P, Garau A, Carboni E, Carta AR (2009) PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson's disease. Eur J Neurosci 29:954–963. 10.1111/j.1460-9568.2009.06657.x [DOI] [PubMed] [Google Scholar]
- Schwab JM, Conrad S, Monnier PP, Julien S, Mueller BK, Schluesener HJ (2005a) Spinal cord injury-induced lesional expression of the repulsive guidance molecule (RGM). Eur J Neurosci 21:1569–1576. 10.1111/j.1460-9568.2005.03962.x [DOI] [PubMed] [Google Scholar]
- Schwab JM, Monnier PP, Schluesener HJ, Conrad S, Beschorner R, Chen L, Meyermann R, Mueller BK (2005b) Central nervous system injury-induced repulsive guidance molecule expression in the adult human brain. Arch Neurol 62:1561–1568. 10.1001/archneur.62.10.1561 [DOI] [PubMed] [Google Scholar]
- Shabanzadeh AP, Tassew NG, Szydlowska K, Tymianski M, Banerjee P, Vigouroux RJ, Eubanks JH, Huang L, Geraerts M, Koeberle PD, Mueller BK, Monnier PP (2015) Uncoupling Neogenin association with lipid rafts promotes neuronal survival and functional recovery after stroke. Cell Death Dis 6:e1744. 10.1038/cddis.2015.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Rocha EM, Rooney T, Barneoud P, McLean JR, Beagan J, Osborn T, Coimbra M, Luo Y, Hallett PJ, Isacson O (2015) A Nurr1 agonist causes neuroprotection in a Parkinson's disease lesion model primed with the toll-like receptor 3 dsRNA inflammatory stimulant poly(I:C). PLoS One 10:e0121072. 10.1371/journal.pone.0121072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan BS, Doostzadeh J, Absalan F, Mohandessi S, Jalili R, Bigdeli S, Wang J, Mahadevan J, Lee CL, Davis RW, William Langston J, Ronaghi M (2009) Whole genome survey of coding SNPs reveals a reproducible pathway determinant of Parkinson disease. Hum Mutation 30:228–238. 10.1002/humu.20840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland GT, Matigian NA, Chalk AM, Anderson MJ, Silburn PA, Mackay-Sim A, Wells CA, Mellick GD (2009) A cross-study transcriptional analysis of Parkinson's disease. PLoS One 4:e4955. 10.1371/journal.pone.0004955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliaferro P, Burke RE (2016) Retrograde axonal degeneration in Parkinson disease. J Parkinsons Dis 6:1–15. 10.3233/JPD-150769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe S, Yamashita T (2014) Repulsive guidance molecule-a is involved in Th17-cell-induced neurodegeneration in autoimmune encephalomyelitis. Cell Rep 9:1459–1470. 10.1016/j.celrep.2014.10.038 [DOI] [PubMed] [Google Scholar]
- Tassew NG, Charish J, Seidah NG, Monnier PP (2012) SKI-1 and Furin generate multiple RGMa fragments that regulate axonal growth. Dev Cell 22:391–402. 10.1016/j.devcel.2011.11.022 [DOI] [PubMed] [Google Scholar]
- Tillerson JL, Miller GW (2003) Grid performance test to measure behavioral impairment in the MPTP-treated-mouse model of parkinsonism. J Neurosci Methods 123:189–200. 10.1016/S0165-0270(02)00360-6 [DOI] [PubMed] [Google Scholar]
- Ulusoy A, Sahin G, Björklund T, Aebischer P, Kirik D (2009) Dose optimization for long-term rAAV-mediated RNA interference in the nigrostriatal projection neurons. Mol Ther 17:1574–1584. 10.1038/mt.2009.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Battum EY, Brignani S, Pasterkamp RJ (2015) Axon guidance proteins in neurological disorders. Lancet Neurol 14:532–546. 10.1016/S1474-4422(14)70257-1 [DOI] [PubMed] [Google Scholar]
- van den Heuvel DM, Hellemons AJ, Pasterkamp RJ (2013) Spatiotemporal expression of repulsive guidance molecules (RGMs) and their receptor neogenin in the mouse brain. PLoS One 8:e55828. 10.1371/journal.pone.0055828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NH, Key B (2007) Neogenin: one receptor, many functions. Int J Biochem Cell Biol 39:874–878. 10.1016/j.biocel.2006.10.023 [DOI] [PubMed] [Google Scholar]
- Yamada T, McGeer PL, Baimbridge KG, McGeer EG (1990) Relative sparing in Parkinson's disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res 526:303–307. 10.1016/0006-8993(90)91236-A [DOI] [PubMed] [Google Scholar]
- Yamashita T, Mueller BK, Hata K (2007) Neogenin and repulsive guidance molecule signaling in the central nervous system. Curr Opin Neurobiol 17:29–34. 10.1016/j.conb.2006.12.001 [DOI] [PubMed] [Google Scholar]
- Yang C, Hao F, He J, Lu T, Klein RL, Zhao LR, Duan WM (2016) Sequential adeno-associated viral vector serotype 9-green fluorescent protein gene transfer causes massive inflammation and intense immune response in rat striatum. Hum Gene Ther 27:528–543. 10.1089/hum.2015.083 [DOI] [PubMed] [Google Scholar]
- Yoshida J, Kubo T, Yamashita T (2008) Inhibition of branching and spine maturation by repulsive guidance molecule in cultured cortical neurons. Biochem Biophys Res Commun 372:725–729. 10.1016/j.bbrc.2008.05.124 [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N (1999) Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6:973–985. 10.1038/sj.gt.3300938 [DOI] [PubMed] [Google Scholar]