

Abstract
Astrocytes can control basal synaptic strength and arteriole tone via their resting Ca2+ activity. However, whether resting astrocyte Ca2+ can adjust to a new steady-state level, with an impact on surrounding brain cells, remains unknown. Using two-photon Ca2+ imaging in male rat acute brain slices of the somatosensory neocortex, we found that theta burst neural activity produced an unexpected long-lasting reduction in astrocyte free Ca2+ in the soma and endfeet. The drop in intracellular Ca2+ was attenuated by antagonists targeting multiple ionotropic and metabotropic glutamate receptors, and intracellular cascades involved Ca2+ stores and nitric oxide. The reduction in astrocyte endfoot Ca2+ was coincident with an increase in arteriole tone, and both the Ca2+ drop and the tone change were prevented by an NMDA receptor antagonist. Astrocyte patch-clamp experiments verified that the glutamate receptors in question were located on astrocytes and that Ca2+ changes within astrocytes were responsible for the long-lasting change in arteriole diameter caused by theta burst neural activity. In astrocytes from animals that lived in an enriched environment, we measured a relatively lower resting Ca2+ level that occluded any further drop in Ca2+ in response to theta burst activity. These data suggest that electrically evoked patterns of neural activity or natural experience can adjust steady-state resting astrocyte Ca2+ and that the effect has an impact on basal arteriole diameter.
SIGNIFICANCE STATEMENT The field of astrocyte-neuron and astrocyte-arteriole interactions is currently in a state of refinement. Experimental evidence ex vivo suggests that direct manipulation of astrocyte-free Ca2+ regulates synaptic signaling and local blood flow control; however, in vivo experiments fail to link synaptically evoked astrocyte Ca2+ transients and immediate changes to various astrocyte-mediated processes. To clarify this discrepancy, we examined a different aspect of astrocyte Ca2+: the resting, steady-state free Ca2+ of astrocytes, its modulation, and its potential role in the tonic regulation of surrounding brain cells. We found that ex vivo or in vivo neural activity induced a long-lasting reduction in resting free astrocyte Ca2+ and that this phenomenon changed arteriole tone.
Keywords: arteriole, astrocyte, calcium, plasticity, theta burst, two-photon
Introduction
Changes in astrocyte free cytosolic Ca2+ are thought to be important for an array of physiological and pathological brain processes. Most investigations have focused on trying to understand large, transient increases in astrocyte Ca2+ that are evoked by neural activity and how these events impart changes to synaptic strength as well as microvascular diameter. However, the functional role of resting Ca2+ activity, or more specifically, the steady-state free cytosolic Ca2+ in astrocytes, is much less explored. In particular, it is unknown whether resting astrocyte Ca2+ can be adjusted to a new level in response to physiological patterns of brain activity with an impact on adjacent brain cells.
Several studies have highlighted the difficulty in linking synaptically evoked astrocyte Ca2+ transients to physiological processes, such as synaptic modulation and local blood flow in vivo due to temporal discrepancies, lack of Ca2+ signals, no involvement of a major Ca2+ release pathway (IP3R2), or unnatural experimental conditions (Agulhon et al., 2010; Nizar et al., 2013; Takata et al., 2013; Bonder and McCarthy, 2014; Paukert et al., 2014; but see Lind et al., 2013; Otsu et al., 2015). Because direct experimental manipulation of free Ca2+ in astrocytes does impact surrounding brain cells using ex vivo preparations (Fellin et al., 2004; Mulligan and MacVicar, 2004; Gordon et al., 2008; Panatier et al., 2011; Rosenegger et al., 2015), perhaps a different way to understand such experiments and their relevance to brain biology is to explore the connection to subtler changes to steady-state free Ca2+ in astrocytes. Changes in resting, steady-state Ca2+ could be congruent with a tonic action of these cells (Rosenegger et al., 2015), in which modulation could occur on relatively slow time scales (Rosenegger and Gordon, 2015). Numerous intracellular proteins have an affinity for Ca2+ around resting cytosolic concentration (∼100 nm) (Carafoli, 1987), including SERCA pumps (Verboomen et al., 1992) and cPLA2 (Pettus et al., 2004), such that the modulation of these proteins could influence astrocyte functions. For example, decreasing intracellular free Ca2+ in astrocytes with high-affinity Ca2+ buffers via patch-clamp decreased basal excitatory synaptic strength in the hippocampus (Panatier et al., 2011). Furthermore, reducing free Ca2+ in astrocyte endfeet caused a localized increase in arteriole tone (Rosenegger et al., 2015). Early work on gliotransmission demonstrated that only a small change in free Ca2+ (84–140 nm) was necessary for astrocyte-derived glutamate release (Parpura and Haydon, 2000). These findings are also interesting in light of data examining resting Ca2+ using quantitative Fluorescence Life Time Imaging in brain slices and in vivo (Zheng et al., 2015). These measurements revealed that astrocyte free Ca2+ decreased with developmental age and that astrocyte neighbors were more likely to have similar levels of resting free Ca2+ compared with astrocytes separated by distance. An interesting possibility to explain the latter is the amount, or type, of previous neural activity experienced by populations of astrocytes in the intact animal. Yet, whether patterns of neural activity can have a long-lasting impact on steady-state astrocyte free Ca2+ remains unexplored.
We used two-photon Ca2+ imaging in acute rat neocortical brain slices to discover that astrocytes exhibited an unexpected long-lasting reduction in the level of resting steady-state Ca2+ in the soma and endfeet following trains of theta burst neural activity. This drop in astrocyte Ca2+ was blocked by individual pharmacological antagonists targeting ionotropic or metabotropic glutamate receptors, and was occluded by previous in vivo experience. The reduction in astrocyte endfoot Ca2+ was accompanied by an increase in arteriole tone, suggesting that basal blood flow may be one functional result of adjusting this factor. Our data highlight a novel brain process whereby plasticity can be conferred via long-lasting changes in steady-state free astrocyte Ca2+.
Materials and Methods
Acute brain slice preparation.
Care and use of animals for this project were performed in accordance with approved guidelines set forth by the University of Calgary Animal Care and Use Committee. Male Sprague Dawley rats (p21-p30) were deeply anesthetized with gaseous isoflurane (5%) and then decapitated using a rodent guillotine. The brain was rapidly surgically removed and submerged for ∼2 min in ice-cold slicing solution (in mm as follows): 119.9 N-methyl-d-glucamine, 2.5 KCl, 25 NaHCO3, 1.0 CaCl2-2H2O, 6.9 MgCl2-6H2O, 1.4 NaH2PO4-H2O, and 20 d-glucose. The brain was then Krazy Glued onto a vibratome tray (Leica Instruments, VT1200S) and resubmerged in ice-cold slicing solution. Acute coronal slices were prepared from the somatosensory cortex (400 μm thick). The slices were incubated for 45 min at 33°C in a recovery chamber filled with ACSF containing the following (in mm): 126 NaCl, 2.5 KCl, 25 NaHCO3, 1.3 CaCl2, 1.2 MgCl2, 1.25 NaH2PO4, and 10 glucose. At all steps, the brain/slices were continuously bubbled with carbogen (95% O2/5% CO2). Low O2 experiments used ACSF bubbled with 30% O2/5% CO2 and balanced N2, which required 45 min preincubation before experimentation.
Loading dyes in astrocytes, neurons, and arterioles.
After recovery from slicing, the tissue was incubated in a 3 ml well at 33°C for 45 min with the calcium indicator Rhodamine-2 acetoxymethyl ester (Rhod-2 AM) (15 μm) and the morphological dye Calcein Green AM (17 μm) (Biotium), 0.2% DMSO, 0.006% pluronic acid, and 0.0002% Cremophore EL. During incubation in the small well, slices received very fine carbogen bubbling using a flexible 34 gauge pipette tip (WPI). This duo labeling allowed us to take a ratio of the changes in the fluorescence of the calcium indicator over that of the changes in the morphological dye. Astrocytes were identified by their bright uptake of Rhod-2 AM and by their perivascular endfeet (Simard et al., 2003; Mulligan and MacVicar, 2004). Rhod-2 and Calcein trace data were independently normalized (F = F1/F0), where F is fluorescence, 1 is any given time point, and 0 is an average baseline value, followed by a ratio of the two (Rhod-2 F/Calcein F). A similar procedure was followed for GCaMP3 and sulforhodamine-101 (SR-101) experiments. SR-101 (10 μm) (Sigma-Aldrich) was bulk loaded for 20 min immediately after slice recovery in a smaller (40 ml) incubation chamber that also received carbogen bubbling. Slices were then transferred back to the main incubation chamber and allowed to rest for an additional hour before imaging to permit complete uptake and stabilization of the SR-101 signal.
For experiments that examined parenchymal arterioles, the animal received a tail vein injection of 15 mg FITC dextran (2000 kDa, Sigma-Aldrich), dissolved in 300 μl of lactated Ringer's solution under isoflurane anesthesia (5% induction, 2% maintenance) before decapitation. Luminal FITC-dextran permitted visualization of the brain microvasculature, and we quantified arteriole diameter as a change in lumen area. For an experiment, a given slice was transferred to a superfusion chamber on the rig and was superfused using a pressure-driven (carbogen) solution delivery system at ∼1.5 ml/min, maintained at room temperature. For experiments examining arterioles, the thromboxane A2 analog U46619 (100 nm) was present continually in the bath to provide constant artificial tone, as described previously (Institoris et al., 2015).
Two-photon fluorescence microscopy.
Slices were imaged using a custom-built two-photon microscope (Rosenegger et al., 2014) fed by a Ti:Sapph laser source (Coherent Ultra II, ∼4W average output at 800 nm, ∼80 MHz). Image data were acquired using MATLAB (2013, RRID:SCR_001622) running an open source scanning microscope control software called ScanImage (version 3.8.1, Howard Hughes Medical Institute/Janelia Farms, RRID:SCR_014307) (Pologruto et al., 2003). Imaging was performed at an excitation wavelength of 850 nm for Rhod-2/Calcein experiments and 920 nm for GCaMP3/SR-101 experiments. The microscope was equipped with a primary dichroic mirror at 695 nm, and green and red fluorescence was split and filtered using a secondary dichroic at 560 nm and two bandpass emission filters: 525–40 nm and 605–70 nm (Chroma Technologies). Time-series images were acquired at 0.98 Hz with a pixel density of 512 × 512 and a field of view size of ∼150 μm. Imaging used a Zeiss 40× water dipping objective lens (NA 1.0, WD 2.5 mm).
Patch-clamping astrocytes.
Astrocytes were targeted for patch clamp at a depth between ∼25 and 40 μm from the surface of the slice, and in arteriole tone experiments, were within ∼30–50 μm from the blood vessel under study. A Giga-Ohm seal was maintained for 5 min before obtaining a whole-cell configuration, followed by 15 min of astrocyte network filling to allow adequate internal solution diffusion into the astrocyte network. This time also allowed for any patch-related effects on arteriole diameter to subside (Zonta et al., 2003). The intracellular solution contained AlexaFluor-488 sodium hydrazide (100–200 μm) to (1) visualize the extent of patch solution diffusion throughout the astrocyte network and (2) serve as a morphological indicator for performing a ratio against Rhod-2. Control intracellular solution contained the following (in mm): 108 potassium gluconate, 8 KCl, 8 sodium gluconate, 2 MgCl2, 10 HEPES, 0.1 potassium EGTA, 4 ATP, and 0.3 sodium GTP. Experiments targeting G-protein signaling removed GTP and added 2 mm GDP-β-S. Experiments targeting NMDA receptors added 10 μm MK-801. Astrocyte Ca2+ clamp intracellular solution contained the following (in mm): 10 potassium BAPTA, 4.38 CaCl2, 96 potassium gluconate, 8 sodium gluconate, 2 MgCl2, 10 HEPES, 4 potassium ATP, and 0.3 sodium GTP. All internal solutions were corrected for osmolarity to ∼285 mosmol and corrected for pH with KOH to 7.2. The astrocyte cell type was confirmed by a low input resistance (10–20 mΩ), extensive dye transfer between coupled cells via gap junctions, and visibly loaded endfeet apposed to microvasculature. Astrocytes were voltage-clamped at −85 mV.
Astrocyte GCaMP3 mice.
To achieve a genetically encoded Ca2+ indicator in astrocytes, we crossed GLAST-Cre-ERT mice (RRID:IMSR_Jax:012586) with LSL-GCaMP3 mice (RRID:IMSR_Jax:014538). Male mice positive for both inserts received three tamoxifen injections intraperitoneally (100 mg/kg corn oil vehicle) on consecutive days starting between p16 and p30. Animals were allowed at least 3 weeks for recombination to take place before acute brain slices were made.
Enriched environment experiments.
For the enriched animal group, 7 Sprague Dawley rats from the age of p21 to p42–45 lived communally in an enriched environment. The environment was ∼2 feet × 1.5 feet × 1.5 feet in size, multileveled, and contain a running wheel, tunnels, ladders, hammocks, and shelters, as well as free access to food and water. For the deprived group, 5 Sprague Dawley rats were individually housed for the same period of time with minimal added elements for stimulation (basic cage bedding and some paper materials for a nest). To compare the resting Ca2+ signal in astrocytes between the enriched and deprived groups, we bulk loaded astrocytes with Rhod-2 AM and Calcein AM, and also introduced a sharp electrode (resistance ∼25 mΩ) filled with a known concentration of AlexaFluor-488 hydrazide sodium salt (200 μm) (Invitrogen). A z stack incorporating several astrocytes and the pipette was taken. The x, y, z coordinates of both the pipette and the z stack relative to the surface of the slice were kept constant across experiments. At any given focal plane, we first took the ratio of the astrocyte soma Rhod-2 signal to the Calcein signal. We then took this value (Rhod-2/Calcein) and performed a second ratio over the signal coming from the Alexa-488 patch pipette (explanation in Results). All astrocyte values from a given z stack were then averaged to arrive at the measurement for a given n; thus, n indicates a z stack, and each z stack was acquired from a naive brain slice.
Theta burst stimulation.
A concentric bipolar electrode (FHC) coupled to a Grass S88X stimulator was used to provide electrical stimulation to the cortical tissue. A single theta burst trial used trains of 100 Hz each lasting 0.05 s, repeated at a rate of 4 Hz for a total of 50 s after 45 s of baseline at 0.8, 1.0, or 1.2 V using a pulse duration of 1 ms. Successive theta burst trials were separated by 5 min, and voltages of stimulation were applied in steps, increasing from 0.8 to 1.0 to 1.2 V. Imaging was performed in neocortical layers 1 and 2. The concentric bipolar electrode was placed ∼250 μm lateral (keeping in the same cortical layer) to the area being imaged.
Data collection and statistics.
A given astrocyte was deemed to have shown a change in steady-state resting Ca2+ when the post theta burst Rhod-2/Calcein ratio was >3 SDs from baseline fluctuation. In a given experiment, if more than one astrocyte was imaged in the field of view and they each showed the same directional change in Ca2+, the normalized ratios from each were averaged together for a n of 1. Each individual experiment was performed on a naive brain slice and the number of slices was used for statistical n, with the animal number in each group always being ≥3. Imaging data were stored on a computer for off-line analysis using ImageJ (RRID:SCR_003070) and GraphPad Prism (version 6, RRID:SCR_002798). Experimental values are presented as mean ± SEM, statistical analyses were performed using two-tailed Student's t test (paired or unpaired as appropriate) or a one-way ANOVA when comparing multiple groups. Correlations were performed to determine the relationship between two variables. p < 0.05 was accepted as statistically significant, and the Pearson correlation coefficient (r) was used to determine the tendency of variables to vary together.
Pharmacological agents.
The 50 μm dl-APV, 5 μm 3-((2-methyl-4-thiazolyl)ethynyl)pyridine (MTEP), 2 μm LY456236 (6-methoxy-N-(4-methoxyphenyl)-4-quinazolinamine hydrochloride), 10 μm CNQX, 500 nm CGP-54626 hydrochloride ((2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid hydrochloride), 10 μm NPLA (N5-[imino(propylamino)methyl]-l-ornithine hydrochloride), 10 μm (+)-MK-801 maleate ((5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate) were all purchased from Tocris Bioscience. The 2 mm GDP-β-S (guanosine 5′-[β-thio]diphosphate trilithium salt) was purchased from Sigma-Aldrich. CNQX and LY456236 were dissolved in DMSO (final concentration of DMSO in the bath <0.1%), and all other pharmacological agents were dissolved in ACSF. Slices were preincubated with a given pharmacological antagonist for 20 min before we conducted a theta burst stimulation and Ca2+ imaging experiment.
Results
Theta burst neural activity decreased steady-state free Ca2+ in astrocytes
To test whether patterns of neural activity could induce changes to resting astrocyte Ca2+, we electrically evoked afferent neural activity using theta burst patterns, well known to cause enduring changes in synaptic strength (Collins, 1994; Hess and Donoghue, 1994; Castro-Alamancos et al., 1995), near astrocytes in neocortical layers 1 and 2. We employed two-photon Ca2+ imaging of astrocytes using Rhod-2 AM. This Ca2+ probe was selected because of its relatively high-affinity, excellent two-photon absorbance cross-section (So et al., 2000) and because it is readily taken up by astrocytes (Mulligan and MacVicar, 2004). However, because only relative changes in intracellular free Ca2+ are quantified by Rhod-2 using standard two-photon fluorescence and because astrocytes can undergo cell volume changes during afferent activity due to elevations in external K+ (Florence et al., 2012), we bulk coloaded astrocytes with Calcein Green AM. This duo labeling allowed us to take the ratio of the Ca2+ indicator fluorescence over that of the changes in the morphological dye. This helped correct for any changes in the Rhod-2 signal that were caused by a decrease in dye concentration from cell swelling and/or other optical changes occurring in the tissue over time (Fig. 1A,B). In response to 50 s of theta burst activity, using the lowest voltage of stimulation from which we detected Ca2+ signals (0.8 V), we observed that a fraction of astrocytes (56%, 14 of 25 astrocytes, 8 slices) exhibited an anticipated increase in Ca2+ during the evoked synaptic activity (1.22 ± 0.18) but subsequently displayed an unexpected decrease in the resting free Ca2+ signal (determined by the Rhod-2/Calcein ratio) that persisted for the remainder of the imaging session (0.92 ± 0.02, p < 0.0001; Fig. 1C–E). This change in Ca2+ appeared to achieve a new, lower, steady-state level and could be clearly observed in the somata and major astrocyte processes (Fig. 1C). In contrast, the remaining fraction of astrocytes exhibited neither the evoked increase (1.02 ± 0.01) nor a change after the cessation of stimulation (1.01 ± 0.01). To test whether this apparent drop in astrocyte Ca2+ was an artifact caused by an unexpected increase in Calcein Green fluorescence, we plotted the Calcein data alone and found no long-term increase that would account for the decreased Rhod-2/Calcein ratio (0.98 ± 0.01, p < 0.09; Fig. 1D, inset). To determine whether the decrease in steady-state astrocyte Ca2+ could be enhanced by increasing the stimulation voltage, we tested 1.0 and 1.2 V using the same pattern and duration of electrical stimulation. Notably, the magnitude of the drop in Ca2+ was not significantly enhanced with stimulation voltage (0.8 V: 0.92 ± 0.02, n = 8; 1.0 V: 0.92 ± 0.01, n = 18; 1.2 V: 0.91 ± 0.02, n = 22; Fig. 1E); however, we found that the fraction of astrocytes that exhibited the drop in Ca2+ increased (0.8 V: 56% n = 14 astrocytes; 1.0 V: 62% n = 18 astrocytes; 1.2 V: 71% n = 22 astrocytes; Fig. 1D, right). This is consistent with the idea that higher voltages of stimulation recruited more afferent neural fibers and thus affected more astrocytes in the field of view.
Figure 1.
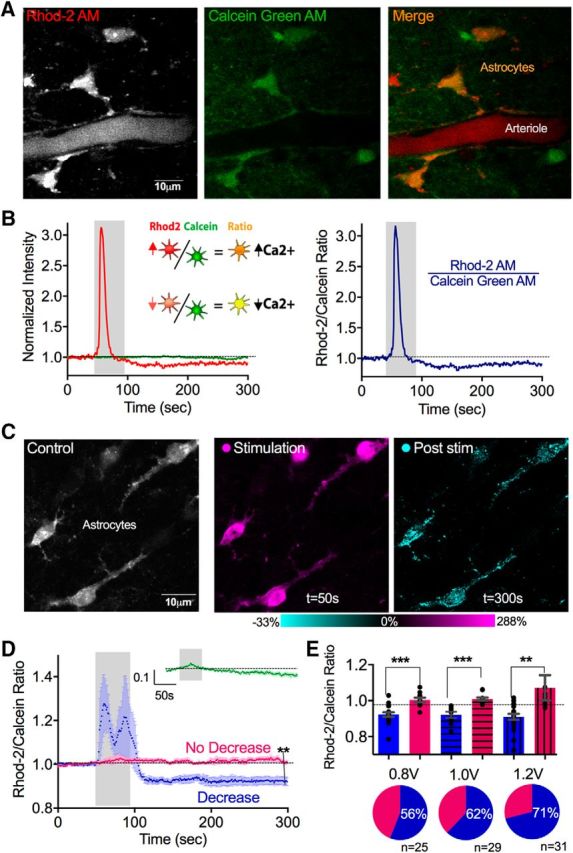
Theta burst neural activity caused a long-lasting reduction in resting free cytosolic Ca2+ in astrocytes. A, Two-photon images of neocortical astrocytes coloaded with the Ca2+ indicator Rhod-2 AM and the morphological dye Calcein Green AM. B, Representative Rhod-2 and Calcein measurements to a single bout of theta burst neural afferent electrical stimulation. Left, Separate Rhod-2 (red) and Calcien (green) traces. Right, Rhod-2/Calcein ratio. C, Images of neocortical astrocytes (left, gray) depicting the transient increase in Ca2+ detected during theta burst activity (middle, magenta), and the drop in Ca2+ in somata and major processes measured following the cessation of theta burst (right, cyan). D, Ca2+ measurements (Rhod-2/Calcein ratio) with evoked theta burst neural activity (0.8 V stimulation) showing two populations of astrocytes: ones that exhibit a significant, transient increase followed by a significant sustained decrease in Ca2+ (blue) and ones that show little response (pink). Inset green trace, Time course summary data of only the Calcein measurement. E, Top, Summary changes in baseline Ca2+ of astrocytes that showed the drop in Ca2+ (blue) and astrocytes that did not show the drop (pink) at different electrical stimulation voltages. Bottom, Pie charts depicting the percentage of cells exhibiting the drop in Ca2+ at different stimulation voltages. **p < 0.01. ***p < 0.001.
Rhod-2 is positively charged and may be transported out of the cell or sequestered into mitochondria (Minta et al., 1989). To test the possibility that the long-lasting decrease in the Rhod-2/Calcein ratio that we observed was due to Rhod-2 trafficking, we repeated the above experiment using the genetically encoded Ca2+ indicator GCaMP3 in Cre-Lox mice. As a ∼50 kDa cytosolic protein, GCaMP3 is impervious to ion/solute carriers. As the glutamate transporter EAAT1 (GLAST) is specific to astrocytes in the cortex (Slezak et al., 2007), we crossed conditional GLAST-CreERT mice with LSL-GCaMP3 mice to achieve GCaMP3 expression in astrocytes following tamoxifen administration (Tran and Gordon, 2015). In an analogous manner to our Rhod-2 experiments, GCaMP3 brain slices were bulk loaded with SR-101 to counterstain astrocytes with a red morphological dye so that we could perform a GCaMP3/SR-101 ratio. Similar to our observations in the Rhod-2/Calcein experiments, we found that a single bout of theta burst stimulation (1.2 V) caused a significant reduction in the GCaMP3/SR-101 fluorescence ratio in the soma and major processes (0.75 ± 0.06, p < 0.004, n = 6; Fig. 2A,B,D). Furthermore, this effect occurred in some (55%), but not all (1.03 ± 0.06, n = 5, p < 0.76; Fig. 2B), imaging experiments that were conducted on individual astrocytes expressing GCaMP3. This suggested a bona fide decrease in resting Ca2+ that was not the result of Rhod-2 trafficking. GCaMP3 also permitted us to compare Ca2+ measurements from the astrocyte arbor to the soma, a feat that was not possible with Rhod-2 due to potential mixed neuronal and astrocyte Ca2+ signals in the neuropil. Approximately half of the astrocyte arbors examined showed little change following theta burst neural activity (54% n = 7, 0.97 ± 0.03; Fig. 2C,D), whereas the other arbors showed a variable increase (1.14 ± 0.06, n = 6; Fig. 2C,D) and the difference was only significant at limited time points between the two populations. Collectively, these data suggest that astrocyte somata and major processes, rather than the fine arbor, can achieve a new, lower, steady-state resting Ca2+ level in response to patterns of neural activity that are typical for inducing long-lasting changes in neural function.
Figure 2.

The decrease in astrocyte steady-state Ca2+ occurs in the soma and major processes, but not in the fine arbor. A, Image of a neocortical astrocyte expressing the genetically encoded Ca2+ indicator GCaMP3 (left), coloaded with the morphological dye SR-101 (middle) and merge (right). Image histogram maximums were decreased, saturating the somata, to visualize the astrocyte arbor. B, Representative somata Ca2+ measurements (GCaMP3/SR-101 ratio) (left) and summary traces (right) in response to evoked theta burst neural activity (1.2 V stimulation) showing astrocytes that exhibited a transient increase followed by a decrease in Ca2+ (blue), and ones that show little response (pink). C, Summary of changes in baseline Ca2+ in astrocyte somata following stimulation. D, Representative arbor Ca2+ measurements (GCaMP3/SR-101 ratio) (left) and summary traces (right) in response to evoked theta burst neural activity (1.2 V stimulation) showing a different pattern than that of somata Ca2+ measurements. E, Summary of changes in baseline Ca2+ in astrocyte arbor following stimulation. **p < 0.01.
Multiple glutamate receptors mediated the decrease in free astrocyte Ca2+
We next explored which neurotransmitters and ligand-gated plasma membrane receptors and channels could be involved in this phenomenon. For instance, LTP relies on a number of different receptors for glutamate, including both ionotropic (AMPAR and NMDAR) (Collingridge et al., 1983) and metabotropic (mGluR1 and mGluR5) varieties (Aiba et al., 1994; Lu et al., 1997). We first set out to determine whether we could mimic the long-lasting decrease in steady-state astrocyte Ca2+ with glutamate alone. To test this idea, we spritzed l-glutamate from a patch pipette positioned 50 μm over the surface of the slice above the imaging region (100 μm, 2 min). This method was chosen because it evoked a similar magnitude and duration of astrocyte soma Ca2+ transient (1.32 ± 0.21; Fig. 3A) to that produced by electrical theta burst stimulation. Consistent with a principle role for glutamate, this treatment resulted in a long-lasting reduction in the Rhod-2/Calcein ratio (0.86 ± 0.07, p < 0.04, n = 7; Fig. 3A).
Figure 3.

Ionotropic and metabotropic glutamate receptors cooperatively mediate the drop in resting astrocyte Ca2+. A, Left, Summary Ca2+ time course (Rhod-2/Calcein ratio) showing that glutamate puff triggers the decrease in astrocyte somata free Ca2+ in the absence of theta burst stimulation. Inset, Representative trace from a single astrocyte. Right, Summary bar graph of baseline and maximum decrease in Ca2+ with glutamate puff. B, Left, Summary Ca2+ time course in response to three successive bouts of ramping voltages of theta burst stimulation. Inset, Representative trace from a single astrocyte. Right, Summary bar graph of decreases in astrocyte Ca2+ in response to successive bouts of ramping voltages of theta burst stimulation. C–G, Summary of Ca2+ time course in response to three successive bouts of theta burst at increasing voltage of stimulation in the presence of CNQX (A, blue), dl-APV (B, red), MTEP (C, magenta), LY456236 (D, orange), and CGP-54626 (G, cyan) compared with the control dataset (green). H, Summary bar graph of decreases in astrocyte resting Ca2+ following the 1.2 V stimulation after successive theta burst stimulation in control conditions and in the presence of various pharmacological antagonists. I, Correlation plot with regression line showing that there is only a weak relationship between the magnitude of the evoked astrocyte Ca2+ transient and the astrocyte Ca2+ decrease following theta burst synaptic activity. Error bars represent SEM. *p < 0.05. **p < 0.01. ***p < 0.001.
Because only a fraction of astrocytes exhibited the reduction in free cytosolic Ca2+ in response to theta burst stimulation, this posed a potential challenge in testing the efficacy of pharmacological antagonists targeting glutamate receptors. For instance, when we pooled the data from all astrocytes (cells that exhibited the drop in Ca2+ and those that did not) for the 0.8 V stimulation, the Ca2+ signal did not decrease significantly from baseline (0.97 ± 0.02, p = 0.23). To increase the magnitude and fidelity of the decrease in Ca2+, which would allow us to analyze all the astrocytes collectively, we used successive trains of theta burst stimulation with ramping voltages (0.8, 1.0, and 1.2 V), each separated by 5 min. When normalized to the baseline before the first stimulation, we found that this procedure generated a large enough decrease in the Rhod-2/Calcein ratio that, when the data from all the astrocytes were pooled together, allowed us to perform pharmacological antagonist experiments (1.0 V: 0.91 ± 0.02, p = 0.014; 1.2 V: 0.85 ± 0.03, p = 0.0028, n = 11; Fig. 3B).
Using this protocol, we first tested the contribution of ionotropic non-NMDA receptors with the competitive antagonist CNQX (10 μm). In the presence of CNQX, the decrease in steady-state astrocyte Ca2+ evoked by consecutive bouts of theta burst was largely abolished (0.97 ± 0.03, p = 0.019 at 900 s compared with no antagonist, n = 7; Fig. 3C,H). Because postsynaptic AMPA receptor activity regulates the opening of NMDA receptors by facilitating the removal of the voltage-dependent Mg2+ pore block, we pondered whether the block of the effect by CNQX was upstream of the involvement of NMDA receptors. Supporting this idea, experiments performed in the presence of the competitive NMDA receptor antagonist dl-APV (50 μm) similarly eliminated the decrease in the Rhod-2/Calcein ratio caused by repetitive theta burst stimulations (0.97 ± 0.01, p = 0.0077, n = 8; Fig. 3D,H). We then explored whether Group I mGluRs could be involved in this long-term change in resting astrocyte cytosolic Ca2+. For example, mGluR5 activity on astrocytes is linked to the efficacy of postsynaptic LTP via the release of glial-derived d-serine, a coagonist of NMDA receptors along with l-glutamate (Henneberger et al., 2010). Interestingly, the competitive mGluR5 antagonist MTEP (5 μm) potently blocked the decrease in astrocyte Ca2+ (0.98 ± 0.01, p = 0.003, n = 8; Fig. 3E,H). Furthermore, the presence of the mGluR1 antagonist LY456236 (10 μm) also prevented the decrease in astrocyte Ca2+ (0.99 ± 0.04, p = 0.013, n = 7; Fig. 3F,H). These results suggest that multiple ionotropic and metabotropic glutamate receptors act cooperatively to mediate the reduction in steady-state astrocyte Ca2+.
Because of the involvement of multiple glutamatergic receptors in producing the drop in steady-state free astrocyte Ca2+, we pondered what pathways lacked a role in this phenomenon. It was not possible to interpret the involvement of GABAA receptors using picrotoxin (50 μm), or the endocannabinoid system by targeting CB1 receptors with AM251 (10 μm), because theta burst stimulation in the presence of either of these compounds led to excessive excitation of the tissue. This was evident because Ca2+ escalated in both neurons and astrocytes without recovery (data not shown). This was not surprising due to the fact that these receptors are key constituents of major inhibitory control mechanisms in the brain. Although we found that, in the presence of the GABAB antagonist CGP-54626 (500 nm), the drop in astrocyte Ca2+ that was caused by theta burst stimulation was unaffected, suggesting no role for the metabotropic GABA pathway in this phenomenon (0.88 ± 0.01, p = 0.49, n = 8; Fig. 3G,H).
Surprisingly, GABAB antagonism fully prevented the evoked Ca2+ transients in astrocytes during theta burst activity (control 1.2 V: 1.39 ± 0.24; CPG-54626: 0.99 ± 0.003, p = 0.0008). This suggested that evoked astrocyte transients and steady-state free Ca2+ can be differentially regulated. We noted that all of the glutamate receptor blockers blunted the evoked Ca2+ transients in astrocytes elicited by theta burst synaptic activity (control 1.2 V: 1.39 ± 0.24; CNQX: 0.98 ± 0.02, p = 0.001; LY456236: 1.00 ± 0.004, p = 0.01; MTEP: 1.00 ± 0.01, p = 0.04), although dl-APV did not significantly affect them (1.05 ± 0.03, p = 0.24). Given some of the differential effects of antagonism on the evoked Ca2+ transient and the drop in free Ca2+, we hypothesized that the degree of astrocyte activation during theta burst activity would not predict the degree of the astrocyte drop in Ca2+. Indeed, when examining individual astrocytes in the control state, there was only a weak correlation between the peak of the evoked Ca2+ transient and the maximal decrease in resting Ca2+ (r = −0.21, p = 0.36; Fig. 3I). In fact, some astrocytes showed a clear drop in Ca2+ after theta burst, without exhibiting any Ca2+ transients during theta burst.
Glutamate receptors functionally localized to astrocytes
To determine whether the glutamate receptors implicated in the theta burst induced decrease in astrocyte free Ca2+ were functionally expressed by astrocytes, we performed a series of astrocyte patch-clamp experiments. To test the role of astrocytic G-protein signaling, which would be downstream of mGluR1 and mGluR5 activation, we whole-cell patch-loaded the astrocyte network for 15 min with the G-protein inhibitor GDP-β-S (2 mm). Given the small molecular of weight of GDP-β-S, it should readily pass to neighboring astrocytes via gap junctions, enabling us to analyzed Ca2+ activity in astrocytes adjacent to the patched cell (Fig. 4A) and avoid patch-related side effects (Zheng et al., 2015). We found that GDP-β-S not only prevented the decrease in astrocyte free Ca2+ in response to a single 50 s bout of theta burst (1.2 V) but instead resulted in a sustained elevation of resting Ca2+ (1.08 ± 0.06, n = 15; Fig. 4B–D). Importantly, a similar fraction of astrocytes (77%, 20 of 26) from control patch experiments using standard internal solution still displayed the drop in free Ca2+ following theta burst (0.91 ± 0.01, p > 0.001, n = 13; Fig. 4B–D). This was different from GDP-β-S (p = 0.02), suggesting that the patching processes itself did not disrupt the ability of astrocytes adjacent to the patched cell to exhibit the phenomenon.
Figure 4.
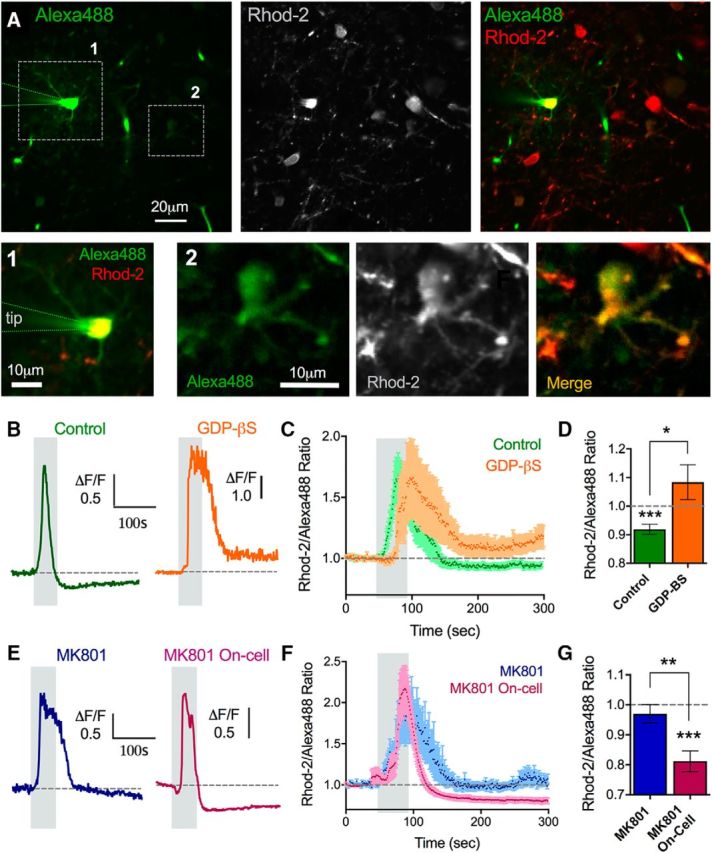
Glutamate receptors functionally localize to astrocytes. A, Two-photon images showing a whole-cell patched astrocyte and the astrocyte network filled with Alexa-488 (left), costained with Rhod-2 (middle) and the merge (right). Alexa images were gamma filtered at 0.8 to view the less bright coupled astrocytes. 1, Patched astrocyte zoomed in (left image). 2, An astrocyte distant to the patched cell that filled with the Alexa-488 via gap junctional coupling (right 3 images). Ca2+ measurements were made in astrocyte neighbors to the patched cell, such as ROI 2. B, Representative traces of the astrocyte Ca2+ response to theta burst stimulation during a control astrocyte network infusion (green, left) or GDP-β-S loading into the astrocyte network (orange, right). C, Summary traces of the same experiments in B. D, Summary of the peak Ca2+ changes. E, Representative traces of the astrocyte Ca2+ response to theta burst stimulation during the infusion of MK-801 into the astrocyte network (blue, left) or a MK-801 on-cell control (no whole-cell event) (pink, right). F, Summary traces of the same experiments in E. G, Summary of the peak Ca2+ changes. Error bars represent SEM. *p < 0.05. **p < 0.01. ***p < 0.001.
Next, we tested for astrocytic NMDA receptors via patch-loading MK-801 (10 μm), the activity-dependent NMDA receptor pore blocker, into the astrocyte network and again analyzed Ca2+ in astrocytes adjacent to the patched cell subsequent to theta burst activity. We found that the decrease in astrocyte Ca2+ was abolished with intracellular MK-801 (0.97 ± 0.03, p > 0.05, n = 7; Fig. 4E–G). We also performed an experiment to control for the possibility that, during the patching process, MK-801 was spritzed into the extracellular space, possibly blocking neural NMDARs. Although the 20 min from patch sealing to theta burst stimulation would likely permit washout of the extracellular MK-801, we performed the same MK-801 patching experiment but without going whole-cell on the astrocyte (remaining in Giga-Ohm seal configuration). Here, MK-801 was still spritzed into the external milieu but did not enter astrocytes. In this experiment, we observed a robust drop in astrocyte Ca2+ after theta burst stimulation (0.80 ± 0.03, p < 0.001, n = 9; Fig. 4E–G), which was different from intracellular MK-801 (p = 0.002). Furthermore, the Ca2+ drop occurred in a large fraction of astrocytes (80%, 20 of 25), similar to control stimulations in the absence of the patch pipette. This suggested that MK-801 inside astrocytes was necessary to block the effect and that any remaining extracellular trace of MK-801 was not effective on neurons or astrocytes. Although we quantified the fraction of astrocytes that exhibited the drop in resting Ca2+ in control patch and MK-801 on-cell patch experiments above, the summary data for each group represented all astrocytes pooled together for comparisons. Collectively, these data support the idea that the metabotropic receptors and NMDA receptors implicated in the decrease in astrocyte Ca2+ are located on astrocytes.
Decreased astrocyte steady-state Ca2+ partially involved NO and internal stores
NMDA receptor opening is tightly coupled with the production of nitric oxide (NO) via the NOS1 enzyme (Brenman et al., 1996; Doyle et al., 1996). NO affects spontaneous astrocyte Ca2+ signals and astrocyte Ca2+ stores (Bal-Price et al., 2002; Li et al., 2003). Based on this, we tested the hypothesis that NOS1 activity contributed to the decrease in astrocyte free Ca2+ caused by successive theta burst stimulation. In the presence of the selective NOS1 antagonist NPLA (10 μm), we found that the decrease in astrocyte Ca2+ was partially, but significantly, reduced compared with control (0.93 ± 0.01, p = 0.02, n = 7; Fig. 5C,D). This indicated an important role for NO derived from NOS1 activity.
Figure 5.
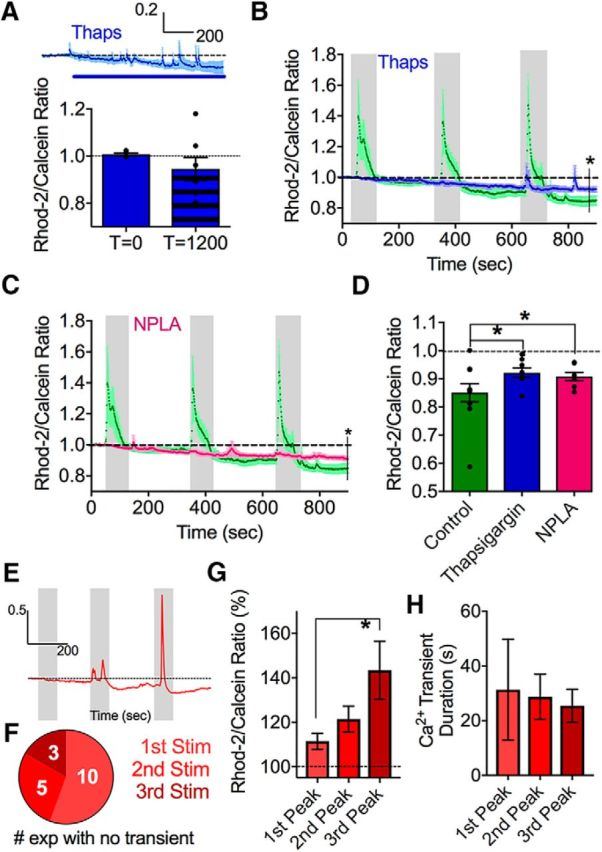
The drop in astrocyte resting Ca2+ partially involves NO and internal Ca2+ stores. A, A modest effect of the SERCA pump inhibitor thapsigargin on steady-state intracellular astrocyte Ca2+. Summary time course (above). Blue line indicates thapsigargin bath application. Bar graph (below) represents measurements at 0 and 1200 s. B, Summary of Ca2+ time course in response to successive bouts of theta burst stimulation in the presence of thapsigargin (blue) compared with control (green). C, Summary of Ca2+ time course in response to successive bouts of theta burst stimulation in the presence of the NOS1 enzyme antagonist NPLA (pink) compared with the control dataset (green). D, Summary bar graph after the 1.2 V stimulation comparing control, NPLA, and thapsigargin conditions. E, Representative trace showing how the evoked Ca2+ transients in astrocytes become larger with three successive theta burst stimulation at the same voltage (1 V). F, Pie chart showing that the chance of observing an evoked astrocyte Ca2+ transient increases with successive theta burst stimulations of a total of 16 experiments. G, Summary data of the evoked peak amplitude of astrocyte Ca2+ transients from the experiments shown in in E, F; failures omitted. H, Summary data of the duration of the evoked astrocyte Ca2+ transients from E, F; failures omitted. Error bars represent SEM. *p < 0.05.
We next tested the involvement of internal Ca2+ stores in this phenomenon. Stored endoplasmic reticulum (ER) Ca2+ plays a significant role in regulating the free cytosolic Ca2+ concentration in many cell types (Carafoli, 1987). Astrocytes express IP3Rs (Sherwood et al., 2017) and SERCA pumps (Beck et al., 2004) on the ER for the release and reuptake of Ca2+ to and from the cytosol, respectively. Additionally, NO can increase SERCA pump activity (Cohen et al., 1999; Trepakova et al., 1999), which may be one method through which NO is working to decrease steady-state Ca2+ in astrocytes. Thus, we used the potent SERCA pump inhibitor thapsigargin (1 μm) and monitored astrocyte Ca2+ during its application onto the slice. We found that thapsigargin only marginally decreased resting Ca2+ over 20 min (0.95 ± 0.05, p = 0.056, n = 8; Fig. 5A), suggesting that Ca2+ stores do not have a large impact on maintaining resting cytosolic Ca2+ in quiescent astrocytes over short time frames. In a separate experiment, started after 30 min of thapsigargin preincubation, we performed consecutive theta burst stimulation trials and found a partial, but significant, attenuation of the drop in steady-state astrocyte Ca2+ (0.92 ± 0.02, p = 0.04, n = 8; Fig. 5B,D). As the release of stored Ca2+, triggered by Gq-coupled metabotropic receptors, is a major pathway in astrocytes, we could confirm that internal stores were emptied by thapsigargin as theta burst activity failed to evoke Ca2+ transients (1.00 ± 0.01, p = 0.01, n = 8; Fig. 5B). These data demonstrate an important role for NO and intracellular Ca2+ stores in the theta burst-triggered decrease in resting astrocyte free Ca2+.
With enhanced SERCA pump activity as a potential mechanism responsible for the decrease in resting free Ca2+ in astrocytes after theta burst, to examine this possibility further, we tested whether internal Ca2+ stores were holding more Ca2+ after theta burst activity, as indicated by larger evoked Ca2+ transients. To do this, we applied three identical bouts of theta burst stimulation (all at the same voltage [1 V] and duration [50 s]) each separated by 5 min, and found that the amplitude of the evoked astrocyte Ca2+ transient increased with each successive bout (first stimulation: 11.4 ± 3.66%, n = 12; third stimulation: 43.4 ± 13.03%; n = 15, p = 0.04; Fig. 5E,G), and that the chance of detecting a Ca2+ transient also increased (Fig. 5F). Despite larger transients in response to successive bouts of theta burst, the duration of the Ca2+ transients remained unchanged (first stimulation: 25.46 ± 6.04 s, n = 6; third stimulation: 31.33 ± 18.5 s, n = 13, p > 0.05; Fig. 5H). These findings are consistent with the idea that astrocyte Ca2+ stores are holding more Ca2+ after theta burst.
In vivo plasticity induced the drop in astrocyte Ca2+
If plasticity-inducing forms of neural activity are important for determining the resting free astrocyte Ca2+ concentration, we hypothesized that previous in vivo plasticity might produce changes to astrocyte Ca2+ that can be measured ex vivo. To test this, we compared neocortical brain slices from rats that lived in an enriched environment versus those from a deprived environment. Our rationale was that an enriched environment should elicit widespread structural and functional plastic changes in neurons and astrocytes, contrasting with minimal strengthening processes in the brains of animals receiving little external stimulation. For ∼3 weeks, enriched animals lived communally (n = 7) in a large, dynamic environment, whereas deprived animals (n = 5) were singly housed in standard cages with some bedding but without toys or structures (see Materials and Methods). First, we aimed to determine whether we could detect relative differences in the resting Ca2+ signal between astrocytes of the two conditions. To do this, we performed z stacks of astrocytes and for every individual cell, we took the ratio of Rhod-2 to Calcein and then subsequently took a ratio of this value to the fluorescence signal from a known concentration of Alexa-488 contained within a patch pipette embedded in the z stack (Fig. 6A). Taking the Rhod-2/Calcein ratio for each astrocyte was important to control for differences in dye loading at different depths in the tissue, varying degrees of dye excitation in distinct regions (i.e., an astrocyte in optically clear layer 1 vs optically dense layer 2), and to control for slice-to-slice discrepancies in loading efficiency. In line with this idea, we found a general relationship between the brightness of the Rhod-2 signal and the brightness of the Calcein signal in single astrocytes when examining many astrocytes across different control z stacks (r = 0.42; Fig. 6B). The second ratio to the known concentration of Alexa-488 was critical to have a consistent standardized value that would ultimately allow comparison of the resting Ca2+ signal between the enriched and deprived groups. Using this approach, we found a significantly lower resting Ca2+ signal in the enriched animals' astrocytes compared with that of the deprived animals' astrocytes (enriched: 0.018 ± 0.002, n = 25; deprived: 0.029 ± 0.005, n = 14, p = 0.036; Fig. 6C,D). As a control, we quantified the raw Calcein measures and the raw Alexa pipette measures and found that they were not different between the deprived and enriched animals (enriched Alexa-488: 124.4 ± 14.34, n = 25; deprived Alexa-488: 120.6 ± 17.47, n = 14, p > 0.05; enriched Calcein: 48.34 ± 5.14, n = 25; deprived Calcein: 49.56 ± 4.88, n = 14, p > 0.05), whereas the raw Rhod-2 signals were on average lower in the enriched animals' astrocytes (enriched: 103.3 ± 18.4 a.u.; deprived: 145.8 ± 24.6 a.u., p = 0.03).
Figure 6.
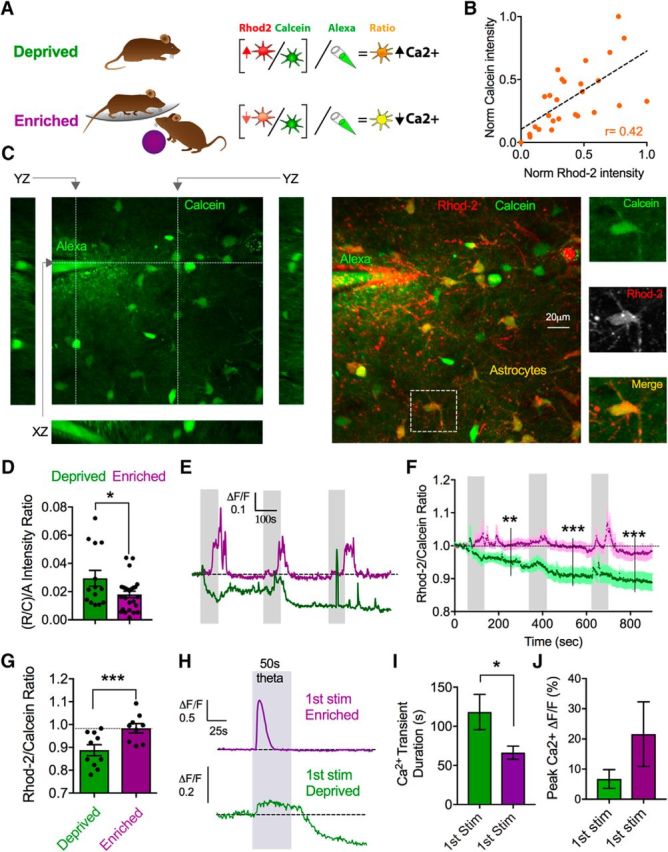
Experience decreases astrocyte resting Ca2+ and limits further changes in response to neural activity ex vivo. A, Diagram representing the deprived and enriched experimental groups and the imaging/analysis strategy to compare resting Ca2+, involving an additional ratio to a reference Alexa-488 pipette. B, Correlation plot of the relationship between baseline Calcein loading and baseline Rhod-2 loading in single astrocytes in control animals. C, z stack images of Calcein and Rhod-2 loaded cortical tissue incorporating a reference pipette containing Alexa-488 dye (200 μm). Images represent xy, yz, and xz views (left), merge xy Rhod-2 and Calcein view (middle), as well as a closeups of an astrocyte (right) indicated by a dashed-line box. D, Summary data comparing the baseline (Rhod-2/Calcein)/Alexa ratio in astrocytes from the deprived (green) or enriched (purple) experimental groups. E, Representative somata Ca2+ measurements (Rhod-2/Calcein ratio) in response to three successive theta burst stimulations from enriched (purple) and deprived (green) animals. F, G, Summary traces (F) and maximum effect after the third stimulation (G) of the same experiment in E. H, Representative traces of the first theta burst stimulation showing the magnitude and the duration of the evoked Ca2+ transient in astrocyte from enriched or deprived animals. I, J, Ca2+ signal duration (I) and magnitude (J) summary data from the experiment in H; failures omitted. Error bars represent SEM. *p < 0.05. **p < 0.01. ***p < 0.001.
Next, we tested the effect of electrical theta burst stimulation in these groups to observe any changes to resting astrocyte Ca2+ levels. We reasoned that, if previous in vivo plasticity caused astrocytes to lower their resting free Ca2+ concentration, this may reduce or occlude our ability to induce further reductions in Ca2+ ex vivo. Indeed, successive theta burst stimulation failed to cause any reduction in the Rhod-2/Calcein ratio in enriched animals (0.98 ± 0.02, p = 0.52, n = 9; Fig. 6E–G). Further evidence supporting this hypothesis was that deprived animals displayed a marked reduction in resting astrocyte Ca2+ following theta burst stimulation (0.89 ± 0.02, p = 0.008, n = 10; Fig. 6E–G).
Finally, we examined the properties of the evoked astrocyte Ca2+ transients between the enriched and deprived groups during the first bout of theta burst stimulation. This was to test the hypothesis that enrichment would enhance the synaptically evoked astrocyte Ca2+ transient, consistent with more efficacious intracellular Ca2+ store dynamics. Notably, astrocyte Ca2+ signals in the enriched group were significantly shorter in duration compared with the deprived group (first stimulation enriched: 66.25 ± 8.40 s, n = 8; first stimulation deprived: 118.3 ± 22.52 s, n = 8, p = 0.04; Fig. 6H,I), and enriched animals displayed a nonsignificant trend toward larger amplitude Ca2+ transients (first stimulation enriched: 21.6 ± 10.7%, first stimulation deprived: 6.7 ± 3.1%, p > 0.05; Fig. 6H,J). These data suggested that enriched astrocytes restored the free Ca2+ level faster after a Ca2+ transient and that their internal stores may hold more Ca2+. Collectively, these data demonstrate that previous experience can elicit long-lasting changes to astrocytic resting free Ca2+ levels, synaptically evoked Ca2+ transients, and quell further decreases in resting Ca2+ in response to subsequent neural activity.
A drop in astrocyte endfeet Ca2+ increased arteriole tone
We next explored a functional implication of lowering astrocyte resting Ca2+. Experiments that introduced a Ca2+ buffer into astrocytes through a patch pipette demonstrated that decreasing endfoot free Ca2+ caused an increase in arteriole tone (Rosenegger et al., 2015). Based on this observation, we explored the possibility that theta burst neural activity also decreased free astrocyte endfoot Ca2+, resulting in a change in arteriole diameter. Because astrocyte endfeet are distinct and functionally separate cellular compartments, it is not obvious that alterations in soma Ca2+ will have an impact on the endfoot Ca2+ level. We performed a single 1.2 V theta burst stimulation protocol while imaging astrocytes, their endfeet, and an associated penetrating arteriole in the neocortex. We also extended the time course of image acquisition to observe how long astrocyte Ca2+ may be perturbed for and to determine whether there were any long-lasting changes to arteriole diameter. Similar to our observations of astrocyte somatic Ca2+, we found that astrocyte endfeet showed an evoked increase in Ca2+ during theta burst stimulation (1.28 ± 0.12) that was followed by a drop in the resting Ca2+ signal that achieved a new, lower, steady-state level (0.92 ± 0.02, p = 0.006, n = 16). Also similar, we observed endfeet with a clear decrease in resting Ca2+ (0.88 ± 0.02, n = 11, 69% of endfeet of a total 48 endfeet examined) versus endfeet that did not change (1.00 ± 0.02, n = 5, 31%; p = 0.011; Fig. 7E). Regarding the arteriole, we observed an expected vasodilation (less tone) (1.13 ± 0.03; Fig. 7A–C) during theta burst neural activity, the brain slice equivalent to functional hyperemia in vivo. Interestingly, this was followed by a slowly developing, long-lasting vasoconstriction (more tone) (0.75 ± 0.04, p = 0.002, n = 11; Fig. 7A–C).
Figure 7.

A drop in endfoot Ca2+ correlates with increased arteriole tone. Ai–Aiii, Two-photon images showing astrocytes (Rhod-2 and Calcein) and a cortical arteriole (FITC green) before (t = 0 s) and after (t = 1200 s) a single bout of theta burst stimulation (1.2 V). Closeup images of the astrocyte somata (Ai), endfoot (Aii), and arteriole lumen (Aiii) show the drop in astrocyte Ca2+ and the vasoconstriction that occurred following theta burst stimulation. B, Summary time course data of average arteriole diameter changes in response to theta burst activity in 95% O2 (green) and in 30% O2 (orange). Inset, Duration of the evoked vasodilation in the two oxygen conditions. C, Summary bar graph showing the maximum dilations and constrictions determined from individual experiments (not the average trace) in the two oxygen conditions. D, E, Summary traces of astrocyte endfoot Ca2+ in response to theta burst activity in 30% O2 (D) and in 95% O2 (E). Inset, Pie charts represent the fraction of astrocytes decreasing or not changing in each condition. E, Correlation plot of the change in arteriole tone after theta burst stimulation versus the peak evoked Ca2+ transient in astrocytes during theta burst stimulation (r = 0.01). F, Correlation plot of the change in arteriole tone after theta burst stimulation versus the evoked peak astrocyte Ca2+ signal during theta burst. G, Correlation plot of the change in arteriole tone after theta burst stimulation versus the decrease in astrocyte resting Ca2+ observed after theta burst stimulation (r = −0.54). Error bars represent SEM. *p < 0.05.
Next, we repeated this experiment under more physiological concentrations of oxygen. Standard brain slice conditions are hyperoxic, which facilitates vasoconstriction cell pathways over vasodilation (Gordon et al., 2008). Thus, we aerated our solutions with 30% O2 (instead of 95%), which provides close to physiological levels of oxygen at the depths we image and enhances vasodilation to brief (1 s), intense (50 Hz) afferent activity (Institoris et al., 2015). We found that one 1.2 V, 50 s bout of theta burst caused a similar magnitude reduction in astrocyte soma and endfoot steady-state free Ca2+ (endfoot: 0.90 ± 0.01, p < 0.0001 to baseline, 32 endfeet, n = 11 slices; Fig. 7D,E). However, fewer astrocytes exhibited this decrease (46%) compared with 95% O2 (69%) (Fig. 7D,E), which was likely due to increased extracellular adenosine tone and a suppression of excitatory synaptic transmission in low O2 (Gordon et al., 2008). The arteriole also showed a similar profile in 30% O2 compared with 95% O2; however, as expected from the lower O2 microenvironment, the stimulations evoked larger vasodilations (1.25 ± 0.04, p = 0.04, n = 11; Fig. 7B,C), with a trend to longer-lasting dilations (315.8 ± 76.23 s, p > 0.05, n = 11; Fig. 7B, inset), but with no significant difference in the magnitude of the post theta burst vasoconstriction (0.72 ± 0.046, p > 0.05, n = 11; Fig. 7B,C). Notably, in several of our control 95% O2 experiments, we observed the increase in arteriole tone in the absence of any detectable evoked Ca2+ transients in the endfeet during theta burst stimulation. Indeed, plotting the relationship between peak evoked astrocyte Ca2+ and the degree of increased tone demonstrated no correlation (r = 0.01; Fig. 7F). Conversely, plotting the degree of the endfoot Ca2+ drop to the degree of the tone change revealed a moderate correlation (r = −0.54, p = 0.044; Fig. 7G).
From our previous pharmacology experiments, we achieved the most consistent block of the drop in somatic astrocyte Ca2+ with NMDAR blockade. Thus, we examined endfoot-arteriole dynamics in the presence of dl-APV. These experiments were performed in standard 95% O2. In response to a single electrical theta burst stimulation (1.2 V), we found that dl-APV abolished the drop in endfoot Ca2+ (0.98 ± 0.02, p = 0.03, n = 10; Fig. 8A) along with the increase in arteriole tone (0.97 ± 0.03, p = 0.04, n = 10; Fig. 8B), without significantly affecting the initial evoked Ca2+ transient in endfeet (1.16 ± 0.08, p > 0.05; Fig. 8A). To directly test the role of endfoot Ca2+ in mediating the post theta burst increase in arteriole tone, we 'clamped' Ca2+ in the astrocyte network surrounding an arteriole using whole-cell patch clamp. By using an internal solution based on BAPTA plus CaCl2 (see Materials and Methods), a free concentration of Ca2+ of 100 nm (approximate resting value in astrocytes) (Zheng et al., 2015) can be maintained in equilibrium with the buffer despite natural increases or decreases in cytosolic Ca2+. Under astrocyte Ca2+ clamp, we found that the vasoconstriction observed after theta burst was significantly attenuated (0.93 ± 0.014, p = 0.006, n = 6; Fig. 8C–E). Importantly, the decrease in endfoot Ca2+ we found previously was also blocked, whereby endfeet exhibited a relatively flat Rhod-2/Calcein ratio during and after theta burst under Ca2+ clamp conditions (1.00 ± 0.04, p = 0.02, n = 6; Fig. 8F). These data suggest that NMDARs and changes to free astrocyte Ca2+ are responsible for the long-lasting tone increase caused by theta burst.
Figure 8.
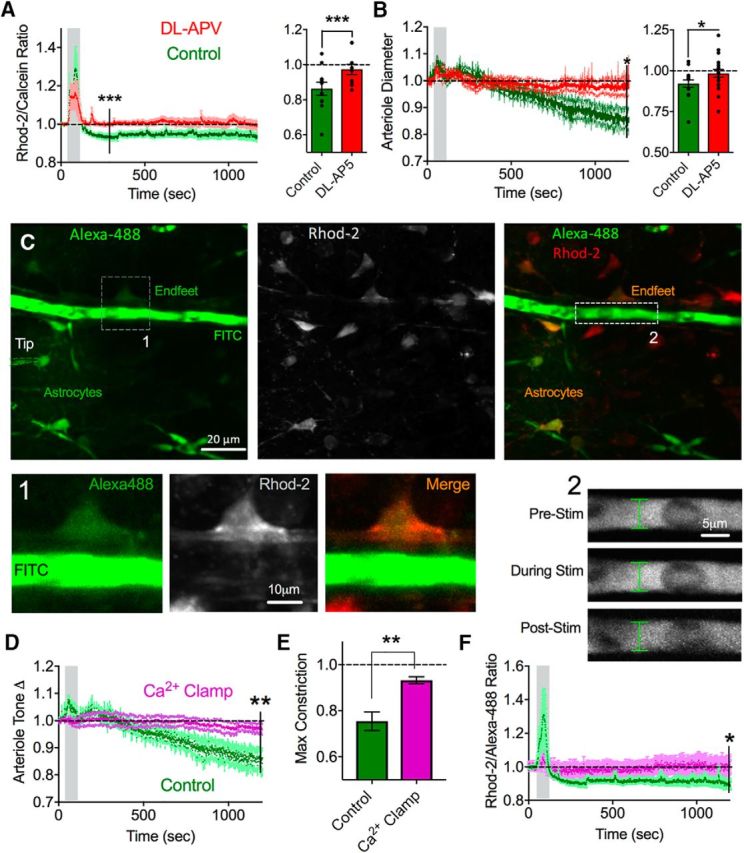
NMDAR antagonism or astrocyte Ca2+ clamp prevent the arteriole response to theta burst. A, Left, Summary endfoot Ca2+ average traces in response to theta burst in control (green) and in the presence of dl-APV (red). Right, Summary bar graph of maximum decrease in Ca2+ in the same experiments. Values were from individual experiment maximum decreases. B, Left, Summary of relative changes in arteriole tone in response to theta burst stimulation in control or in dl-APV. Right, Summary bar graph of the maximum vasoconstrictions in the same experiment. C, Representative images showing astrocyte patch-clamp setup for endfoot Ca2+ clamp. Left, Whole-cell astrocyte filling the astrocyte syncytium, including endfeet abutting an arteriole, with Alexa-488 dye. FITC dextran filled the lumen of the arteriole. Astrocytes were prestained with Rhod-2 AM (middle). Merged images (right). 1, Zoomed-in images of an arteriole lumen filled with FITC dextran and endfoot filled with Alexa-488 costained with Rhod-2. Green channel images were gamma filtered at 0.7 to better visualize relatively faint endfeet. 2, Zoomed-in images of arteriole diameter at varying time points in the stimulation period during endfoot Ca2+ clamp. D, Summary of relative changes to arteriole tone in the presence of Ca2+ clamp (pink) compared with control (green). E, Summary bar graph of maximum vasoconstriction in response to theta burst stimulation for control and Ca2+ clamp conditions. F, Summary endfoot Ca2+ time course in response to theta burst stimulation under endfoot Ca2+ clamp (pink) or control (green). Error bars represent SEM. *p < 0.05. **p < 0.01.
Discussion
The involvement of four different types of glutamate receptors mediating the drop in astrocyte free Ca2+ is perhaps not surprising given that a wealth of data implicate these same plasmalemma proteins in LTP, and that we used a common LTP-inducing protocol: theta burst stimulation. Although there is an essential role of AMPARs and NMDARs in LTP and in learning and memory under most circumstances (Park et al., 2014), Group I mGluRs are also important for LTP (Aiba et al., 1994; Bortolotto et al., 1994; Lu et al., 1997), but their contribution depends on the intensity of the induction protocol (Wilsch et al., 1998; Raymond and Redman, 2002). The involvement of mGluR1 and mGluR5 in the effects we observed likely related to the relatively long duration and multiple bouts of theta burst stimulation we applied. Astrocytes express mGluRs, and these proteins are involved in generating large Ca2+ transients in response to nearby neuronal activity (Pasti et al., 1997; Bezzi et al., 1998). Evidence points to mGluR3 and mGluR5 in neocortical and hippocampal astrocytes (Schools and Kimelberg, 1999), with increasing proportion of mGluR3 after 2 weeks of age (Sun et al., 2013). Although astrocytic mGluR5 is thought to participate in LTP through the release of d-serine, a coagonist of the NMDAR (Henneberger et al., 2010), there is also mGluR5 expressed in neuronal dendrites (Sun et al., 2013). Because our experiments were performed from p21 to p30, inhibition of the drop in astrocyte Ca2+ using an mGluR5 antagonist may reflect residual levels of mGluR5 in astrocytes at this age. Along this line, we observed little correlation between the evoked Ca2+ in astrocytes and the degree of the Ca2+ drop in astrocytes following stimulation. This either indicates a Ca2+-independent role of mGluR5 on astrocytes, such as via G-protein α-subunit switching (Daaka et al., 1997), or that neuronal mGluR1 and mGluR5 mediate the effect on astrocytes through a retrograde transmitter. Although the rationale of our GDP-β-S experiment was to test the role of astrocytic mGluR1 and mGluR5, this treatment inhibits cell-wide G-protein signaling and thus is not a direct test for these glutamatergic metabotropic receptors. Given the attenuation of the effect with mGluR antagonists, coupled with a block using intracellular GDP-β-S, the most parsimonious explanation is that the mGluRs were located on astrocytes. However, we cannot rule out other, constitutively active metabotropic receptors on astrocytes that keep steady-state free Ca2+ low. In this scenario, GDP-β-S may have inhibited this process, leading to an elevation in resting Ca2+ that dominated over a neural mGluR effect on resting astrocyte free Ca2+. Regarding potential retrograde transmitters hailing from neurons, endocannabinoids are an interesting candidate for this role (Navarrete and Araque, 2008), but unfortunately we were unable to explore CB1 receptors due to excessive excitation of the cortical tissue during theta burst in the presence of the CB1R antagonist AM251. Nevertheless, due to the similarity in the receptor complement involved in this phenomenon to LTP, we anticipate that astrocytes exhibit this change along with LTP in nearby synapses in vivo. This idea was supported by the fact that we could occlude the electrically evoked drop in astrocyte Ca2+ by previous in vivo experience. This also suggests that the drop in astrocyte Ca2+ is long-lasting, as we only confirmed the sustained decrease up to ∼20 min from our longest imaging time series.
Free cytosolic Ca2+ can be sequestered by intracellular stores or extruded by a variety of membrane pumps, transporters, and exchangers. Notably, NMDA receptors physically link to the production of NO through NOS1 (Brenman et al., 1996; Doyle et al., 1996) and NO has been shown to increase the affinity of SERCA pumps for Ca2+, lowering free cytosolic Ca2+ in a variety of cells (Cohen et al., 1999; Trepakova et al., 1999). Importantly, astrocytes show marked transcription of SERCA pump mRNA (Cahoy et al., 2008). Furthermore, SERCA pumps can load ER stores efficiently in astrocytes without typical capacitative Ca2+ entry (Grimaldi, 2006), suggesting that they could serve as a key protein in setting the resting Ca2+ concentration. As we could partially block the drop in free Ca2+ either with a selective NOS1 inhibitor NPLA or by emptying ER stores via blocking SERCA pump activity (thapsigargin), we propose that one way in which NMDA receptors confer their action on astrocyte resting Ca2+ is through NO and a subsequent enhancement of SERCA pump activity. Other Ca2+ handling proteins, with some evidence of regulating resting Ca2+ in astrocytes, that would be interesting to explore in future work are plasma membrane cation ATPases (Reyes et al., 2012), Na+/Ca2+ exchangers (Rojas et al., 2004; Reyes et al., 2012; Turovsky et al., 2016), and TRPA1 channels (Shigetomi et al., 2011), although recent work argues against a significant role for TRPA1 in Ca2+ influx (Rungta et al., 2016).
Our measurements focused largely on the astrocyte soma and endfoot, as a similar drop in resting Ca2+ in the astrocyte arbor was less clear. Astrocytes possess a broad repertoire of Ca2+ activities (Grosche et al., 1999; Nett et al., 2002; Volterra et al., 2014) with most events occurring as microdomain Ca2+ transients in the fine processes (Shigetomi et al., 2013; Rungta et al., 2016). As we did not measure microdomain transients in our study, it is possible that the modest changes we observed in resting Ca2+ in the arbor was the result of more dramatic changes in microdomain Ca2+ activity that influenced the resting level. This could be properly examined using membrane targeted Lck-GCaMPs instead of cytosolic GCaMPs (Shigetomi et al., 2013). One potential reason that astrocytes lower their resting free Ca2+ concentration (other than modulating vessel tone, below) is to generate larger Ca2+ transients in response to subsequent neural activity. This could occur due to a higher Ca2+ concentration in the stores from enhanced SERCA pump activity, a greater amount of Ca2+ release into the cytosol, and faster Ca2+ clearing mechanisms. Our analysis showing that successive theta burst stimulation increases the magnitude of evoked Ca2+ transients, and the fact that enriched animals display faster decaying Ca2+ transients, are consistent with enhanced Ca2+ store release and refilling mechanisms after plasticity. Larger, spontaneous Ca2+ transients from external Ca2+ influx would also be possible given a lower resting baseline (Srinivasan et al., 2015; Rungta et al., 2016).
The extent to which astrocyte endfeet are involved in the regulation of arteriole diameter is unclear when comparing in vitro and in vivo data. Elevations in astrocyte endfoot Ca2+ in vitro have been unequivocally shown to affect arteriole diameter (Mulligan and MacVicar, 2004; Straub et al., 2006; Gordon et al., 2008), whereas in vivo data suggest that transient changes in blood flow are too rapid to be the product of astrocyte Ca2+ signals (Nizar et al., 2013; Bonder and McCarthy, 2014; but see Lind et al., 2013; Otsu et al., 2015). Evidence from slices show that Ca2+ clamping astrocytes around arterioles, to prevent synaptically evoked Ca2+ transients in these cells, fails to block activity-dependent vasodilations (Mishra et al., 2016). Instead, precapillaries may be the site of astrocyte action (Biesecker et al., 2016; Mishra et al., 2016). However, another means of arteriole control by glia was described in the retina (Kur and Newman, 2014) and in the neocortex (Rosenegger et al., 2015). The concept is that astrocytes use their resting Ca2+ activity to control diameter tonically in a manner free of acute changes in neural activity and independent of large transient increases in astrocyte Ca2+ (Rosenegger et al., 2015). Consistent with this, here we have found that a drop in free astrocyte endfoot Ca2+ caused by theta burst activity correlated with an increase in arteriole tone (vasoconstriction), which did not necessitate the evoked astrocyte Ca2+ transient. Furthermore, Ca2+ clamping many astrocytes around the arteriole at ∼100 nm free Ca2+ prevented the Ca2+ drop caused by theta burst and blocked the ensuing vasoconstriction. The vasoconstriction following theta burst may be counterintuitive, even in 30% O2; however, there are a few potential explanations: (1) the tone increase could extend the dynamic range for blood flow increases in response to subsequent bouts of neuronal activity (Blanco et al., 2008), or the long-lasting increase in arteriole tone, which could lower O2 in vivo, is a signal to spur on subsequent angiogenesis (Wittko-Schneider et al., 2014; Tata et al., 2015) to meet the new and increased energy demands of the region after NMDAR-dependent plasticity. The fact that the phenomena we described were blocked by NMDAR antagonism is interesting in light of work suggesting that Ca2+-dependent release of astrocyte-derived d-serine promotes arteriole dilation via vascular endothelial NMDARs (Stobart et al., 2013). If this mechanism operated tonically, perhaps the vasoconstriction we observed was the result of less d-serine release from astrocytes consequent to the drop in endfoot Ca2+. Given an important role for NMDARs in our effect combined with our data showing that in vivo plasticity changes astrocyte Ca2+, it would be interesting to explore how experience affects arteriole regulation in future work. This could have implications for how synapses are metabolically fueled after learning events, and how neurovascular diseases develop as a consequence of metabolic dysregulation (Zlokovic, 2011; Iadecola, 2013).
Footnotes
This work was supported by the Canada Institutes of Health Research. G.R.G. was supported by Canada Research Chairs and the Heart and Stroke Foundation Alberta. E.M.F.M. was supported by Alberta Innovates Health Solutions, Canada Institutes of Health Research, and the Hotchkiss Brain Institute. C.M.-R. was supported by Campus Alberta Neuroscience and Alberta Innovates Health Solutions. We thank the developers and distributers of ScanImage software through the Howard Hughes Medical Institute/Janelia Farms Open Source License.
The authors declare no competing financial interests.
References
- Agulhon C, Fiacco TA, McCarthy KD (2010) Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science 327:1250–1254. 10.1126/science.1184821 [DOI] [PubMed] [Google Scholar]
- Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S (1994) Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell 79:365–375. 10.1016/0092-8674(94)90204-6 [DOI] [PubMed] [Google Scholar]
- Bal-Price A, Moneer Z, Brown GC (2002) Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia 40:312–323. 10.1002/glia.10124 [DOI] [PubMed] [Google Scholar]
- Beck A, Nieden RZ, Schneider HP, Deitmer JW (2004) Calcium release from intracellular stores in rodent astrocytes and neurons in situ. Cell Calcium 35:47–58. 10.1016/S0143-4160(03)00171-4 [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A (1998) Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391:281–285. 10.1038/34651 [DOI] [PubMed] [Google Scholar]
- Biesecker KR, Srienc AI, Shimoda AM, Agarwal A, Bergles DE, Kofuji P, Newman EA (2016) Glial cell calcium signaling mediates capillary regulation of blood flow in the retina. J Neurosci 36:9435–9445. 10.1523/JNEUROSCI.1782-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco VM, Stern JE, Filosa JA (2008) Tone-dependent vascular responses to astrocyte-derived signals. Am J Physiol Heart Circ Physiol 294:H2855–H2863. 10.1152/ajpheart.91451.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder DE, McCarthy KD (2014) Astrocytic Gq-GPCR-linked IP3R-dependent Ca2+ signaling does not mediate neurovascular coupling in mouse visual cortex in vivo. J Neurosci 34:13139–13150. 10.1523/JNEUROSCI.2591-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortolotto ZA, Bashir ZI, Davies CH, Collingridge GL (1994) A molecular switch activated by metabotropic glutamate receptors regulates induction of long-term potentiation. Nature 368:740–743. 10.1038/368740a0 [DOI] [PubMed] [Google Scholar]
- Brenman JE, Christopherson KS, Craven SE, McGee AW, Bredt DS (1996) Cloning and characterization of postsynaptic density 93, a nitric oxide synthase interacting protein. J Neurosci 16:7407–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278. 10.1523/JNEUROSCI.4178-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E. (1987) Intracellular calcium homeostasis. Annu Rev Biochem 56:395–433. 10.1146/annurev.bi.56.070187.002143 [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Donoghue JP, Connors BW (1995) Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. J Neurosci 15:5324–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RA, Weisbrod RM, Gericke M, Yaghoubi M, Bierl C, Bolotina VM (1999) Mechanism of nitric oxide-induced vasodilatation: refilling of intracellular stores by sarcoplasmic reticulum Ca2+ ATPase and inhibition of store-operated Ca2+ influx. Circ Res 84:210–219. 10.1161/01.RES.84.2.210 [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H (1983) Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol 334:33–46. 10.1113/jphysiol.1983.sp014478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GG. (1994) The characteristics and pharmacology of olfactory cortical LTP induced by theta burst high frequency stimulation and 1S,3R-ACPD. Neuropharmacology 33:87–95. 10.1016/0028-3908(94)90101-5 [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ (1997) Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 390:88–91. 10.1038/36362 [DOI] [PubMed] [Google Scholar]
- Doyle C, Hölscher C, Rowan MJ, Anwyl R (1996) The selective neuronal NO synthase inhibitor 7-nitro-indazole blocks both long-term potentiation and depotentiation of field EPSPs in rat hippocampal CA1 in vivo. J Neurosci 16:418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G (2004) Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43:729–743. 10.1016/j.neuron.2004.08.011 [DOI] [PubMed] [Google Scholar]
- Florence CM, Baillie LD, Mulligan SJ (2012) Dynamic volume changes in astrocytes are an intrinsic phenomenon mediated by bicarbonate ion flux. PLoS One 7:e51124. 10.1371/journal.pone.0051124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Choi HB, Rungta RL, Ellis-Davies GC, MacVicar BA (2008) Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456:745–749. 10.1038/nature07525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi M. (2006) Astrocytes refill intracellular Ca2+ stores in the absence of cytoplasmic [Ca2+] elevation: a functional rather than a structural ability. J Neurosci Res 84:1738–1749. 10.1002/jnr.21064 [DOI] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Möller T, Verkhratsky A, Reichenbach A, Kettenmann H (1999) Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci 2:139–143. 10.1038/5692 [DOI] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA (2010) Long-term potentiation depends on release of D-serine from astrocytes. Nature 463:232–236. 10.1038/nature08673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess G, Donoghue JP (1994) Long-term potentiation of horizontal connections provides a mechanism to reorganize cortical motor maps. J Neurophysiol 71:2543–2547. [DOI] [PubMed] [Google Scholar]
- Iadecola C. (2013) The pathobiology of vascular dementia. Neuron 80:844–866. 10.1016/j.neuron.2013.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institoris Á, Rosenegger DG, Gordon GR (2015) Arteriole dilation to synaptic activation that is sub-threshold to astrocyte endfoot Ca2+ transients. J Cereb Blood Flow Metab 35:1411–1415. 10.1038/jcbfm.2015.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kur J, Newman EA (2014) Purinergic control of vascular tone in the retina. J Physiol 592:491–504. 10.1113/jphysiol.2013.267294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Sul JY, Haydon PG (2003) A calcium-induced calcium influx factor, nitric oxide, modulates the refilling of calcium stores in astrocytes. J Neurosci 23:10302–10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind BL, Brazhe AR, Jessen SB, Tan FC, Lauritzen MJ (2013) Rapid stimulus-evoked astrocyte Ca2+ elevations and hemodynamic responses in mouse somatosensory cortex in vivo. Proc Natl Acad Sci U S A 110:E4678–E4687. 10.1073/pnas.1310065110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC (1997) Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci 17:5196–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minta A, Kao JP, Tsien RY (1989) Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J Biol Chem 264:8171–8178. [PubMed] [Google Scholar]
- Mishra A, Reynolds JP, Chen Y, Gourine AV, Rusakov DA, Attwell D (2016) Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci 19:1619–1627. 10.1038/nn.4428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA (2004) Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431:195–199. 10.1038/nature02827 [DOI] [PubMed] [Google Scholar]
- Navarrete M, Araque A (2008) Endocannabinoids mediate neuron-astrocyte communication. Neuron 57:883–893. 10.1016/j.neuron.2008.01.029 [DOI] [PubMed] [Google Scholar]
- Nett WJ, Oloff SH, McCarthy KD (2002) Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J Neurophysiol 87:528–537. 10.1152/jn.00268.2001 [DOI] [PubMed] [Google Scholar]
- Nizar K, Uhlirova H, Tian P, Saisan PA, Cheng Q, Reznichenko L, Weldy KL, Steed TC, Sridhar VB, MacDonald CL, Cui J, Gratiy SL, Sakadzić S, Boas DA, Beka TI, Einevoll GT, Chen J, Masliah E, Dale AM, Silva GA, et al. (2013) In vivo stimulus-induced vasodilation occurs without IP3 receptor activation and may precede astrocytic calcium increase. J Neurosci 33:8411–8422. 10.1523/JNEUROSCI.3285-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, Pfrieger FW, Bergles DE, Charpak S (2015) Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci 18:210–218. 10.1038/nn.3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Vallée J, Haber M, Murai KK, Lacaille JC, Robitaille R (2011) Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146:785–798. 10.1016/j.cell.2011.07.022 [DOI] [PubMed] [Google Scholar]
- Park P, Volianskis A, Sanderson TM, Bortolotto ZA, Jane DE, Zhuo M, Kaang BK, Collingridge GL (2014) NMDA receptor-dependent long-term potentiation comprises a family of temporally overlapping forms of synaptic plasticity that are induced by different patterns of stimulation. Philos Trans R Soc Lond B Biol Sci 369:20130131. 10.1098/rstb.2013.0131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Haydon PG (2000) Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci U S A 97:8629–8634. 10.1073/pnas.97.15.8629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G (1997) Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci 17:7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paukert M, Agarwal A, Cha J, Doze VA, Kang JU, Bergles DE (2014) Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 82:1263–1270. 10.1016/j.neuron.2014.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettus BJ, Bielawska A, Subramanian P, Wijesinghe DS, Maceyka M, Leslie CC, Evans JH, Freiberg J, Roddy P, Hannun YA, Chalfant CE (2004) Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J Biol Chem 279:11320–11326. 10.1074/jbc.M309262200 [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K (2003) ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng Online 2:13. 10.1186/1475-925X-2-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ (2002) Different calcium sources are narrowly tuned to the induction of different forms of LTP. J Neurophysiol 88:249–255. [DOI] [PubMed] [Google Scholar]
- Reyes RC, Verkhratsky A, Parpura V (2012) Plasmalemmal Na+/Ca2+ exchanger modulates Ca2+-dependent exocytotic release of glutamate from rat cortical astrocytes. ASN Neuro 4:e00075. 10.1042/AN20110059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas H, Ramos M, Dipolo R (2004) A genistein-sensitive Na+/Ca2+ exchange is responsible for the resting [Ca2+]i and most of the Ca2+ plasma membrane fluxes in stimulated rat cerebellar type 1 astrocytes. Jpn J Physiol 54:249–262. 10.2170/jjphysiol.54.249 [DOI] [PubMed] [Google Scholar]
- Rosenegger DG, Gordon GR (2015) A slow or modulatory role of astrocytes in neurovascular coupling. Microcirculation 22:197–203. 10.1111/micc.12184 [DOI] [PubMed] [Google Scholar]
- Rosenegger DG, Tran CH, LeDue J, Zhou N, Gordon GR (2014) A high performance, cost-effective, open-source microscope for scanning two-photon microscopy that is modular and readily adaptable. PLoS One 9:e110475. 10.1371/journal.pone.0110475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenegger DG, Tran CH, Wamsteeker Cusulin JI, Gordon GR (2015) Tonic local brain blood flow control by astrocytes independent of phasic neurovascular coupling. J Neurosci 35:13463–13474. 10.1523/JNEUROSCI.1780-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungta RL, Bernier LP, Dissing-Olesen L, Groten CJ, LeDue JM, Ko R, Drissler S, MacVicar BA (2016) Ca(2+) transients in astrocyte fine processes occur via Ca(2+) influx in the adult mouse hippocampus. Glia 64:2093–2103. 10.1002/glia.23042 [DOI] [PubMed] [Google Scholar]
- Schools GP, Kimelberg HK (1999) mGluR3 and mGluR5 are the predominant metabotropic glutamate receptor mRNAs expressed in hippocampal astrocytes acutely isolated from young rats. J Neurosci Res 58:533–543. [DOI] [PubMed] [Google Scholar]
- Sherwood MW, Arizono M, Hisatsune C, Bannai H, Ebisui E, Sherwood JL, Panatier A, Oliet SH, Mikoshiba K (2017) Astrocytic IP3 Rs: contribution to Ca(2+) signalling and hippocampal LTP. Glia 65:502–513. 10.1002/glia.23107 [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Tong X, Kwan KY, Corey DP, Khakh BS (2011) TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat Neurosci 15:70–80. 10.1038/nn.3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S, Xu J, Sofroniew MV, Ellisman MH, Khakh BS (2013) Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol 141:633–647. 10.1085/jgp.201210949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M (2003) Signaling at the gliovascular interface. J Neurosci 23:9254–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slezak M, Göritz C, Niemiec A, Frisén J, Chambon P, Metzger D, Pfrieger FW (2007) Transgenic mice for conditional gene manipulation in astroglial cells. Glia 55:1565–1576. 10.1002/glia.20570 [DOI] [PubMed] [Google Scholar]
- So PT, Dong CY, Masters BR, Berland KM (2000) Two-photon excitation fluorescence microscopy. Annu Rev Biomed Eng 2:399–429. 10.1146/annurev.bioeng.2.1.399 [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, Khakh BS (2015) Ca(2+) signaling in astrocytes from Ip3r2(−/−) mice in brain slices and during startle responses in vivo. Nat Neurosci 18:708–717. 10.1038/nn.4001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobart JL, Lu L, Anderson HD, Mori H, Anderson CM (2013) Astrocyte-induced cortical vasodilation is mediated by d-serine and endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 110:3249–3254. 10.1073/pnas.1215929110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub SV, Bonev AD, Wilkerson MK, Nelson MT (2006) Dynamic inositol trisphosphate-mediated calcium signals within astrocytic endfeet underlie vasodilation of cerebral arterioles. J Gen Physiol 128:659–669. 10.1085/jgp.200609650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, McConnell E, Pare JF, Xu Q, Chen M, Peng W, Lovatt D, Han X, Smith Y, Nedergaard M (2013) Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 339:197–200. 10.1126/science.1226740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata N, Nagai T, Ozawa K, Oe Y, Mikoshiba K, Hirase H (2013) Cerebral blood flow modulation by basal forebrain or whisker stimulation can occur independently of large cytosolic Ca2+ signaling in astrocytes. PLoS One 8:e66525. 10.1371/journal.pone.0066525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata M, Ruhrberg C, Fantin A (2015) Vascularisation of the central nervous system. Mech Dev 138:26–36. 10.1016/j.mod.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CH, Gordon GR (2015) Acute two-photon imaging of the neurovascular unit in the cortex of active mice. Front Cell Neurosci 9:11. 10.3389/fncel.2015.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepakova ES, Cohen RA, Bolotina VM (1999) Nitric oxide inhibits capacitative cation influx in human platelets by promoting sarcoplasmic/endoplasmic reticulum Ca2+-ATPase-dependent refilling of Ca2+ stores. Circ Res 84:201–209. 10.1161/01.RES.84.2.201 [DOI] [PubMed] [Google Scholar]
- Turovsky E, Theparambil SM, Kasymov V, Deitmer JW, Del Arroyo AG, Ackland GL, Corneveaux JJ, Allen AN, Huentelman MJ, Kasparov S, Marina N, Gourine AV (2016) Mechanisms of CO2/H+ sensitivity of astrocytes. J Neurosci 36:10750–10758. 10.1523/JNEUROSCI.1281-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verboomen H, Wuytack F, De Smedt H, Himpens B, Casteels R (1992) Functional difference between SERCA2a and SERCA2b Ca2+ pumps and their modulation by phospholamban. Biochem J 286:591–595. 10.1042/bj2860591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volterra A, Liaudet N, Savtchouk I (2014) Astrocyte Ca2 signalling: an unexpected complexity. Nat Rev Neurosci 15:327–335. 10.1038/nrn3725 [DOI] [PubMed] [Google Scholar]
- Wilsch VW, Behnisch T, Jäger T, Reymann KG, Balschun D (1998) When are class I metabotropic glutamate receptors necessary for long-term potentiation? J Neurosci 18:6071–6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittko-Schneider IM, Schneider FT, Plate KH (2014) Cerebral angiogenesis during development: who is conducting the orchestra? Methods Mol Biol 1135:3–20. 10.1007/978-1-4939-0320-7_1 [DOI] [PubMed] [Google Scholar]
- Zheng K, Bard L, Reynolds JP, King C, Jensen TP, Gourine AV, Rusakov DA (2015) Time-resolved imaging reveals heterogeneous landscapes of nanomolar Ca(2+) in neurons and astroglia. Neuron 88:277–288. 10.1016/j.neuron.2015.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. (2011) Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci 12:723–738. 10.1038/nrn3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozza TN, Carmignoto G (2003) Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6:43–50. 10.1038/nn980 [DOI] [PubMed] [Google Scholar]