

Abstract
Polysialic acid is a glycan modification of the neural cell adhesion molecule (NCAM) produced by the polysialyltransferases ST8SIA2 and ST8SIA4. Polysialic acid has been detected in multiple sclerosis plaques, but its beneficial or adverse role in remyelination is elusive. Here, we show that, despite a developmental delay, myelination at the onset and during cuprizone-induced demyelination was unaffected in male Ncam1−/− or St8sia2−/− mice. However, remyelination, restoration of oligodendrocyte densities, and motor recovery after the cessation of cuprizone treatment were compromised. Impaired differentiation of NCAM- or ST8SIA2-negative oligodendrocyte precursors suggested an underlying cell-autonomous mechanism. In contrast, premature differentiation in ST8SIA4-negative cultures explained the accelerated remyelination previously observed in St8sia4−/− mice. mRNA profiling during differentiation of human stem cell-derived and primary murine oligodendrocytes indicated that the opposing roles of ST8SIA2 and ST8SIA4 arise from sequential expression. We also provide evidence that potentiation of ST8SIA2 by 9-cis-retinoic acid and artificial polysialylation of oligodendrocyte precursors by a bacterial polysialyltransferase are mechanisms to promote oligodendrocytic differentiation. Thus, differential targeting of polysialyltransferases and polysialic acid engineering are promising strategies to advance the treatment of demyelinating diseases.
SIGNIFICANCE STATEMENT The beneficial or adverse role of polysialic acid (polySia) in myelin repair is a long-standing question. As a modification of the neural cell adhesion molecule (NCAM), polySia is produced by the polysialyltransferases ST8SIA2 and ST8SIA4. Here we demonstrate that NCAM and ST8SIA2 promote oligodendrocyte differentiation and myelin repair as well as motor recovery after cuprizone-induced demyelination. In contrast, ST8SIA4 delays oligodendrocyte differentiation, explaining its adverse role in remyelination. These opposing roles of the polysialyltransferases are based on different expression profiles. 9-cis-retinoic acid enhances ST8SIA2 expression, providing a mechanism for understanding how it supports oligodendrocyte differentiation and remyelination. Furthermore, artificial polysialylation of the cell surface promotes oligodendrocyte differentiation. Thus, boosting ST8SIA2 and engineering of polySia are promising strategies for improving myelin repair.
Keywords: demyelinating diseases, developmental myelination, oligodendrocyte precursors, polysialic acid, polysialyltransferases, PSA-NCAM
Introduction
Demyelination and loss of oligodendrocytes in the CNS are hallmark features of devastating diseases such as multiple sclerosis and leukodystrophies (Trapp and Nave, 2008; Franklin and Ffrench-Constant, 2008; Köhler, 2010). In multiple sclerosis, remyelination can occur spontaneously but often fails or is incomplete. Reasons might be a lack of oligodendrocyte precursor cells (OPCs) or insufficient recruitment of OPCs to demyelinated lesions, but impaired differentiation of OPCs into remyelinating oligodendrocytes is considered the most likely explanation for the failure of remyelination (Kuhlmann et al., 2008; Franklin and Gallo, 2014). Some of the underlying mechanisms are decoded, but a better understanding of these processes will promote strategies to improve remyelination.
Polysialic acid (polySia) is a negatively charged cell surface glycan present on developing axons and OPCs (Schnaar et al., 2014). In vitro and in vivo data suggest that polySia plays a dual role during oligodendrocyte development. Its presence promotes motility, chemotactic migration, and recruitment of OPCs (Wang et al., 1994; Barral-Moran et al., 2003; Zhang et al., 2004; Glaser et al., 2007) but inhibits differentiation into myelinating oligodendrocytes (Decker et al., 2000, 2002; Franceschini et al., 2004). Developmental downregulation of polySia before myelination appears to be a prerequisite for efficient myelin formation and maintenance (Trotter et al., 1989; Bartsch et al., 1990; Nait Oumesmar et al., 1995; Charles et al., 2000; Fewou et al., 2007; Jakovcevski et al., 2007; Bakhti et al., 2013). Re-expression of polySia is associated with demyelinated axons in chronic multiple sclerosis lesions but not with remyelinated axons in shadow plaques. Therefore, polySia has been suggested to act as an inhibitor of remyelination (Charles et al., 2002). However, demyelinated multiple sclerosis lesions also harbor polySia-positive progenitors of the oligodendrocyte lineage (Nait Oumesmar et al., 2007).
PolySia is produced by the polysialyltransferases (polySTs) ST8SIA2 and ST8SIA4. During brain development, these enzymes show overlapping but clearly distinct expression profiles and cell type-specific contributions to polysialylation (Schnaar et al., 2014). Both polySTs modify the neural cell adhesion molecule (NCAM; gene name Ncam1), which is by far the major polySia carrier in neurons and OPCs. Additionally, polysialylation of the synaptic cell adhesion molecule (SynCAM 1; gene name Cadm1) by ST8SIA2 has been described in a subpopulation of mainly NCAM-negative murine OPCs as well as in stem-cell derived human OPCs (Werneburg et al., 2015a,b).
Oral administration of cuprizone is widely used as a highly reproducible model to investigate the pathobiology of demyelination and remyelination in the brain without the influence of the peripheral immune system (Matsushima and Morell, 2001; Kipp et al., 2009; Skripuletz et al., 2011). Scrutinizing the idea of an inhibitory role of polySia during remyelination, accelerated recovery from cuprizone-induced demyelination was detected in the corpus callosum of ST8SIA4-negative mice (Koutsoudaki et al., 2010). In contrast to ST8SIA4, the contributions of NCAM and ST8SIA2 to CNS remyelination have not yet been analyzed, but a recent study indicates that ST8SIA2 promotes oligodendrocyte differentiation and is implicated in myelin maintenance (Szewczyk et al., 2017).
Here, we comparatively analyze developmental myelination and cuprizone-induced demyelination and remyelination in Ncam1−/− and St8sia2−/− mice, which is correlated to motor coordination monitored by the rotarod test (Franco-Pons et al., 2007). We demonstrate a congruent delay of developmental myelination, remyelination, and oligodendrocyte recovery in the absence of NCAM or ST8SIA2, accompanied by a prolonged decline of motor function. The difference between the current results and the improved remyelination in ST8SIA4-negative mice (Koutsoudaki et al., 2010) was followed up by comparing in vitro differentiation of Ncam1−/−, St8sia2−/−, and St8sia4−/− OPCs. In addition, we studied the time course of polyST expression, its modulation by 9-cis-retinoic acid (9cRA), a potent promoter of OPC differentiation and remyelination (Huang et al., 2011), and the impact of enzymatically engineered polysialylation on the differentiation of wild-type, NCAM-deficient, or polyST-deficient OPCs.
Materials and Methods
Animals.
Ncam1−/−, St8sia2−/−, and St8sia4−/− mice were backcrossed to the C57BL/6J genetic background as described previously (Werneburg et al., 2015b). Cohorts of wild-type and knock-out mice were generated by mating +/+ and −/− mice obtained from heterozygous founder colonies. Protocols for animal use were in compliance with the German Animal Welfare Act and approved by the local authorities (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, permission no. 33.9-42502-04-11/0574).
Cuprizone treatment.
Experimental demyelination was induced by feeding male Ncam1−/− or St8sia2−/− mice and their respective wild-type littermates a diet containing 0.2% cuprizone (bis-cyclohexanone oxaldihydrazone, Sigma-Aldrich) mixed into a standard rodent chow (Koutsoudaki et al., 2010). Cuprizone feeding started at postnatal day 56 (P56) and was maintained for 5 weeks to induce profound demyelination. Spontaneous remyelination was enabled by withdrawing cuprizone from the diet. During experimentally induced demyelination and remyelination, mice were observed daily and their body weight was determined twice a week.
Histology and immunohistochemistry.
At the indicated time points, mice were deeply anesthetized and killed by transcardial perfusion (Schiff et al., 2011). Brains were dissected, post-fixed overnight in 4% PFA and embedded in paraffin. Histology and immunohistochemistry were performed on coronal, 7-μm-thick serial sections as described previously (Gudi et al., 2009) with the following modifications: Luxol fast blue (LFB) staining was performed with cresyl violet according to Klüver and Barrera (1953). Antigen retrieval was performed for 10 min in a steam oven using 10 mm citrate buffer, pH 6.0, with 0.05% Tween-20. Sections were stained using the Mouse on Mouse Basic Immunodetection Kit (Vector Laboratories) according to the manufacturer instructions. The following primary antibodies were used: anti-APC (adenomatous polyposis coli) mouse IgG2b (1:200; catalog #ab16794, Abcam); anti-APP (amyloid precursor protein) mouse IgG1 (1:1000; catalog #MAB348, Millipore); anti-cleaved caspase-3 rabbit (clone 5A1E; 1:200; catalog #9664S, Cell Signaling Technology); polyclonal rabbit anti-GFAP (glial fibrillary acidic protein; 1:100; catalog #G9269, Sigma-Aldrich); polyclonal rabbit anti-Iba1 (ionized calcium-binding adapter molecule 1; 1:200; catalog #019-19741, Wako); polyclonal rabbit anti-Ki-67 (SP6; 1:200; catalog #RM-9106, Thermo Fisher Scientific); anti-myelin basic protein (MBP) mouse IgG2b (1:500; catalog #SMI-99P-100, Covance); polyclonal rabbit anti-NG2 chondroitin sulfate proteoglycan (1:400; AB5320, Millipore); polyclonal rabbit anti-Nogo-A (neurite outgrowth inhibitor A; 1:750; catalog #AB5888, Millipore); polyclonal rabbit anti-Olig2 (1:500; AB9610, Millipore); and anti-myelin proteolipid protein (PLP) mouse IgG2a (1:500; catalog #MCA839G, Serotec). After washing, sections were incubated with biotinylated secondary antibodies (1:500), followed by peroxidase-coupled avidin–biotin complex (all from Vector Laboratories). Reactivity was visualized using diaminobenzidine (DAB; Vector Laboratories). Slides were mounted with Vitro-Clud (R. Langenbrinck). All sections for one experiment were processed in parallel and incubated with DAB for the exact same time.
Proliferation or apoptosis of OPCs was addressed by double-immunofluorescence staining of Olig2 with Ki-67-specific rat IgG2a (clone TEC-3; 1:50; M7249, Dako), or by combining Olig2 immunofluorescence with terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), as described previously (Schiff et al., 2011). Antigen retrieval was performed for 20 min, and primary antibodies were detected with Alexa Fluor-488- and Alexa Fluor-568-conjugated secondary antibodies (Invitrogen).
Purification, differentiation, and treatment of primary OPCs.
Primary OPCs were purified from mixed glial cultures as described previously (Werneburg et al., 2015b). After adherence onto poly-l-lysine-coated glass coverslips for 2 h, differentiation of OPCs into mature oligodendrocytes was induced by cultivating the cells in neurobasal medium supplemented with 2 mm GlutaMAX and 2% B27 (Thermo Fisher Scientific). Where indicated, 30 ng/ml T3 (triiodothyronine; catalog #T6397, Sigma-Aldrich) or 10 nm 9cRA (R4643, Sigma-Aldrich) was added.
Engineering of polysialic acid.
As previously described (Keys et al., 2014), polyST from Neisseria meningitidis serogroup B (NmB) was expressed, purified, desalted, and stored at −80°C as a 2 mg/ml stock solution with 5% glycerol. A solution of 1.2 mg/ml NmB polyST and 1 mm CMP-activated sialic acid (Nacalai Tesque) was added to either polySia-NCAM-positive or polySia-negative, NCAM-positive CHO cell clones (wild-type clone C6 or polysialylation-deficient clone 2A10; Eckhardt et al., 1995) or to primary OPC cultures before differentiation. Cells were incubated for 1 h to induce cell surface polysialylation independent of the presence of NCAM, as shown previously (El Maarouf et al., 2012).
Differentiation of human stem cell-derived OPCs.
Human embryonic stem cells ES03 (ES Cell International and National Stem Cell Bank) were maintained and differentiated into OPCs and mature oligodendrocytes as described previously (Werneburg et al., 2015a).
Immunocytochemistry, immunoprecipitation, and immunoblotting.
Immunocytochemistry, immunoprecipitation, and immunoblotting were performed as described previously (Werneburg et al., 2015b).
Image acquisition and cell counting.
Microscopy was performed using an Axio Observer.Z1 Microscope equipped with a differential interference contrast (DIC) module, an ApoTome module for optical sectioning with structured illumination, and ZEN 2012 software (all from Carl Zeiss Microimaging). DAB-stained sections were imaged with DIC, and immunofluorescence-stained sections or cell cultures were imaged using ApoTome technology. Micrographs covering the entire corpus callosum were acquired using the MosaiX module of the ZEN software. For each staining, identical settings were used to gather images of all experimental groups.
For evaluation, all micrographs were coded and randomized to ensure blinding of the experimental conditions. Counting of immunopositive cells was performed by a trained examiner. On sections, cell densities were determined by counting frames sized 0.005–0.02 mm2 randomly placed within the corpus callosum close to the midline. In cell cultures, randomly chosen frames of 0.15 mm2 were evaluated.
Evaluation of myelination.
A reliable scoring system was used to evaluate developmental myelination as well as demyelination and remyelination (Lindner et al., 2008). Per brain, at least two sections were examined with a light microscope (CX31, Olympus) by two independent blinded observers and categorized on a scale from 0 (complete demyelination) to 4 (normal myelin).
Motor performance.
Motor performance was evaluated using an accelerating rotarod treadmill (IITC). Mice were placed on the stationary rod and rotation accelerated within 180 s from an initial speed of 5 rpm to a final speed of 40 rpm. The maximum duration of each trial was 240 s. On each day of testing, the maximum latency to fall time was determined in three trails for each animal with an intertrial interval of ∼5 min. Starting on P53, all mice were trained on 3 consecutive days. Cuprizone treatment started the next day (P56) and lasted for 5 weeks, followed by 3 weeks of recovery. Throughout the 8 weeks, motor performance was assessed once every 7 days, always at the same time of day and by one trained experimenter, who was blinded to the genotypes of the mice. After the last day of testing, mice were killed and their brains were prepared for myelin analysis.
RNA isolation and quantitative PCR.
Total RNA was extracted using the NucleoSpin RNA II kit (Macherey-Nagel), reverse transcribed, and subjected to quantitative real-time RT-PCR of murine and human St8sia2/ST8SIA2 or St8sia4/ST8SIA4 as described previously (Werneburg et al., 2015a,b).
Statistical analysis.
Data are presented as the mean ± SEM. Numbers of replicates are indicated in each figure legend. Statistical analyses were performed using GraphPad Prism 7 software (GraphPad Software). The unpaired Student's t test was used to assess differences between two groups. One-way ANOVA followed by Fisher's least significance difference (LSD) post-tests were performed to compare multiple independent groups. When one animal or cultures originating from one animal were tested or analyzed at different time points, two-tailed Student's t tests for two groups and, where appropriate, one-way or two-way ANOVAs for multiple groups were performed for each time point separately. Comparisons between one control and multiple test groups were performed using Dunett's multiple-comparison post-test (one-way ANOVA) and comparisons of treatment by the Bonferroni post-test (two-way ANOVA).
Results
Loss of NCAM and ST8SIA2 cause a comparable delay of developmental myelination
Myelin was visualized by the standard LFB myelin stain and by immunohistochemistry for PLP and MBP. Despite some variation between the different staining methods, the NCAM- and the ST8SIA2-negative mice displayed a comparable delay of myelin development at P8 and P21 when compared with respective Ncam1+/+ and St8sia2+/+ animals (Fig. 1). The analysis of myelin development was complemented by quantification of NG2-positive OPCs in the corpus callosum (Table 1). Slightly reduced OPC densities were detected in Ncam1−/− and St8sia2−/− animals at P8 and P21, respectively. At P56, however, these differences were no longer detectable. Thus, at the beginning of cuprizone treatment the myelination of Ncam1−/− and St8sia2−/− animals was comparable to that in the wild-type animals.
Figure 1.

Myelin development is slightly delayed in Ncam1−/− and St8sia2−/− mice. A, Representative examples of LFB, PLP, and MBP staining in the corpus callosum of wild-type (+/+), Ncam1−/−, and St8sia2−/− mice at P21, compared with respective myelin stains at P56 (shown only for wild-type mice). Scale bar, 100 μm. B, Scoring of LFB, PLP, and MBP staining in the corpus callosum of Ncam1 and St8sia2 wild-type (+/+) and knock-out (−/−) mice during postnatal development. A score of 0 represents no myelin, a score of 4 corresponds to normal myelination of postnatal day 56 wild-type mice. Data are the mean ± SEM of n = 4 animals per genotype and time point. One-way ANOVA indicated significant differences (p < 0.05), and Fisher's LSD post-test was applied for pairwise comparisons between +/+ and −/− for each age. *p < 0.05; **p < 0.01.
Table 1.
NG2 cell quantification in the corpus callosum during developmental myelination
| Postnatal day | NG2 cells/mm2 |
Fisher's LSD | NG2 cells/mm2 |
Fisher's LSD | ||
|---|---|---|---|---|---|---|
| Ncam1+/+ | Ncam1−/− | St8sia2+/+ | St8sia2−/− | |||
| 2 | 517 ± 35 | 499 ± 34 | n.s. | 552 ± 23 | 535 ± 22 | n.s. |
| 8 | 493 ± 32 | 446 ± 14 | n.s. | 526 ± 40 | 462 ± 7 | p < 0.01 |
| 21 | 306 ± 16 | 254 ± 17 | p < 0.1 | 258 ± 17 | 223 ± 11 | p < 0.1 |
| 56 | 467 ± 50 | 443 ± 63 | n.s. | 472 ± 14 | 467 ± 35 | n.s. |
Values are the mean ± SD, unless otherwise indicated; n = 3 each.
Remyelination is impaired in Ncam1−/− and St8sia2−/− mice
As expected, 5 weeks of cuprizone treatment caused a prominent loss of myelin in the corpus callosum of Ncam1+/+ and St8sia2+/+ control mice, followed by a rapid onset of remyelination during the first week after cuprizone withdrawal (Fig. 2). In Ncam1−/− and St8sia2−/− mice, the course of demyelination was unaltered. However, the extent of remyelination during the first week of recovery was significantly reduced.
Figure 2.

Reduced remyelination in Ncam1−/− and St8sia2−/− mice. A, Representative examples of LFB, PLP, and MBP staining in the corpus callosum of wild-type mice 5 weeks after the onset of cuprizone treatment (5w: +/+), compared with respective myelin stains of wild-type (+/+), Ncam1−/−, and St8sia2−/− mice at week 6 (6w; i.e., 1 week after cuprizone withdrawal). Scale bar, 100 μm. B, Scoring of LFB, PLP, and MBP staining in the corpus callosum of Ncam1 and St8sia2 wild-type (+/+) and knock-out (−/−) mice at weeks 4 and 5 of cuprizone treatment and at week 0.5 and 1 of recovery after cuprizone withdrawal (i.e., week 5.5 and 6 after the onset of treatment). A score of 0 represents no myelin, and a score of 4 corresponds to normal myelination of wild-type mice at the beginning of cuprizone treatment (week 0 = postnatal day 56). Data are the mean ± SEM of n = 3 animals per genotype for week 0 and n = 5–7 per genotype for all other time points. One-way ANOVA indicated significant differences (p < 0.05), and a Fisher's LSD post-test was applied for pairwise comparisons between +/+ and −/− for each time point. *p < 0.05; **p < 0.01; ***p < 0.001.
Consistent with previous studies (Koutsoudaki et al., 2010; Salinas Tejedor et al., 2015), a transient increase of NG2-positive OPCs and a strong decrease of Nogo-A- or APC-positive mature oligodendrocytes was detected in the corpus callosum of all cuprizone-treated mice at week 4 and week 5 (Fig. 3A,B). This was accompanied by corresponding increases of proliferative (Ki-67-positive) and apoptotic (TUNEL-positive) OLIG2-labeled cells (Fig. 3C). In Ncam1−/− and St8sia2−/− mice, the reappearance of mature, Nogo-A- and APC-positive oligodendrocytes during remyelination was significantly reduced (Fig. 3B; 5.5 and 6 weeks). In contrast, NG2-positive OPCs were only slightly reduced in Ncam1−/− mice after 5 weeks of cuprizone treatment, and densities of Ki-67- or TUNEL-positive OLIG2-positive cells were not detectably affected in any of the two knock-out lines (Fig. 3B,C). The latter was confirmed by using another apoptotic marker, activated (cleaved) caspase-3. Moreover, the transient increase of acute axonal damage as judged by APP-positive axonal swellings (Lindner et al., 2009) as well as the numbers of Iba1-positive microglia/macrophages or GFAP-positive astrocytes were not affected by genotype (Fig. 3C).
Figure 3.
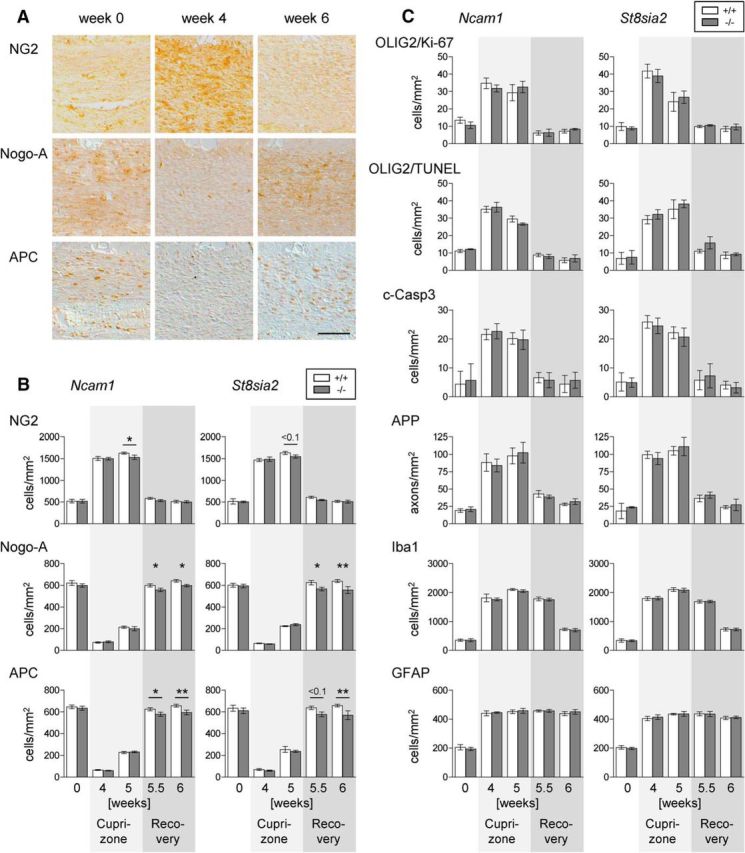
Reduced oligodendrocytes during remyelination in Ncam1−/− and St8sia2−/− mice. A, Representative examples of NG2, Nogo-A, and APC staining in the corpus callosum of wild-type mice at the onset (week 0), after 4 weeks of cuprizone treatment (week 4), and after 1 week of recovery after cuprizone withdrawal (week 6 after the onset of treatment). Scale bar, 100 μm. B, Densities of cells positive for NG2, Nogo-A, or APC. C, Densities of cells double positive for OLIG2 and Ki-67 or OLIG2 and TUNEL, of APP-positive axonal swellings, and of cells positive for cleaved caspase-3 (c-Casp3), Iba1, or GFAP in the corpus callosum of Ncam1 and St8sia2 wild-type (+/+) and knock-out (−/−) mice at weeks 4 and 5 of cuprizone treatment and at weeks 0.5 and 1 of recovery after cuprizone withdrawal (weeks 5.5 and 6 after the onset of treatment). Data are the mean ± SEM of n = 3 animals per genotype for week 0 and n = 5–7 per genotype for all other time points. One-way ANOVA indicated significant differences (p < 0.05), and a Fisher's LSD post-test was applied for pairwise comparisons between +/+ and −/− for each time point. p < 0.1 where indicated; *p < 0.05; **p < 0.01.
Loss of ST8SIA2 may not only affect polySia synthesis on NCAM but would also completely prevent the polysialylation of SynCAM 1 in OPCs (Rollenhagen et al., 2012; Werneburg et al., 2015b). To address a possible contribution of polySia-SynCAM 1 to remyelination, we analyzed brains from wild-type and Ncam1−/− mice after 4 and 5 weeks of cuprizone treatment (i.e., the stages with the highest numbers of NG2-positive OPCs). Using the established repertoire of polySia immunoprecipitation followed by Western blot analysis of SynCAM1 and vice versa (Galuska et al., 2010; Werneburg et al., 2015b), we were not able to detect polySia-SynCAM 1. In contrast, polySia-SynCAM 1 was readily detected in brain extracts from postnatal day 1 Ncam1−/− mice, which were used as a positive control (data not shown). Likewise, immunohistochemistry on paraffin sections yielded the previously described polySia signals in the brain of Ncam1−/− mice at postnatal day 1 (Galuska et al., 2010), but, despite extensive screening of corpus callosum and other brain areas, no polySia signals were found in the brains of Ncam1−/− mice after 4 or 5 weeks of cuprizone treatment (data not shown).
Deficits in remyelination affect locomotor recovery
Locomotor performance and motor coordination were tested in a rotarod apparatus by measuring the time mice were able to walk on a progressively accelerating rotating rod before falling down. To control for a general decline of fitness, changes in body weight were controlled over the 8 weeks of the experiment. The typical reduction of body weight during cuprizone treatment (Stidworthy et al., 2003) overlapped with the normal age-dependent increase, but weight changes in Ncam1−/− and St8sia2−/− mice were indistinguishable from those in the respective wild-type controls (data not shown). All animals improved their motor skills during a 3 d training period before the cuprizone treatment (Fig. 4A). Consistent with previous reports (Franco-Pons et al., 2007), the performance in the control groups declined between 3 and 5 weeks of cuprizone treatment and only incomplete recovery was observed. A prolonged decline in Ncam1−/− and St8sia2−/− mice led to a significantly worse performance at weeks 6 and 7, but within 3 weeks after the end of cuprizone treatment (week 8) both recovered to the level of the control groups (Fig. 4A). This was unexpected, but the assessment of the myelin status revealed that myelination was also indistinguishable between the knock-out and control groups at the end of the rotarod tests (Fig. 4B). Hence, the delay of motor recovery corresponds to the delay of remyelination in Ncam1−/− and St8sia2−/− mice.
Figure 4.

Motor recovery is slightly delayed in Ncam1−/− and St8sia2−/− mice. A, The fall latency of mice walking on an accelerating rotating rod (rotarod) was determined for cohorts of Ncam1 and St8sia2 wild-type (+/+) and knock-out (−/−) mice during 3 d of training before cuprizone treatment (days 1–3), 5 weeks of cuprizone treatment (weeks 1–5), and 3 weeks of recovery (weeks 6–8). B, Scoring of LFB, PLP, and MBP staining in the corpus callosum of rotarod-trained Ncam1 and St8sia2 wild-type (+/+) and knock-out (−/−) mice after the end of the experiment (8 weeks). Data are the mean ± SEM of n = 12–16 animals (A) or n = 4 animals per genotype (B). Two-tailed t tests were applied for pairwise comparisons between +/+ and −/− for each time point. *p < 0.05; **p < 0.01).
9-cis-retinoic acid amplifies the divergent expression of St8sia2 and St8sia4 during OPC differentiation
The identical negative impact of Ncam1 or St8sia2 deficiency indicates that remyelination requires ST8SIA2-dependent polysialylation of NCAM rather than SynCAM 1, the only other known ST8SIA2 acceptor substrate, which has been detected in a confined subset of OPCs (Werneburg et al., 2015b). As demonstrated before, human and murine OPCs use ST8SIA2 and ST8SIA4 to produce polySia-NCAM at the cell surface (Werneburg et al., 2015a,b). However, in clear contrast to the previously observed acceleration of remyelination in St8sia4−/− mice, which was associated with a faster recovery of mature oligodendrocytes and therefore has been assigned to enhanced OPC differentiation (Koutsoudaki et al., 2010), the reduced reappearance of Nogo-A- and APC-positive mature oligodendrocytes during remyelination suggests a delay of OPC differentiation in Ncam1−/− and St8sia2−/− mice. This poses the question of possible differences in ST8SIA2 and ST8SIA4 expression patterns and of the role of NCAM polysialylation by ST8SIA2 and ST8SIA4 during OPC differentiation.
PolyST expression levels during in vitro differentiation of human embryonic stem cell-derived and primary murine OPCs were analyzed by quantitative PCR (Fig. 5). For human embryonic stem cell-derived OPCs, the in vitro differentiation protocol started with cultures containing >90% PDGF receptor α and OLIG2-double-positive cells (OPC stage; day 0 = day 98 of in vitro differentiation starting from the stem cell stage) and resulted in cultures with predominantly branched MBP-positive, but NG2- and polySia-negative, oligodendrocytes at day 34 (Werneburg et al., 2015a). As shown in Figure 5A, mRNA levels of ST8SIA2 and ST8SIA4 increased from day 0 to day 12, followed by significant downregulation until day 34 of human OPC differentiation. Compared with human OPCs, mouse OPCs differentiate much faster (Hu et al., 2009). Therefore, and to achieve a higher temporal resolution, the course of polyST expression during in vitro differentiation of mouse OPCs was studied on a much smaller time scale. As depicted in Figure 5B, this analysis revealed clearly distinct mRNA expression profiles of St8sia2 and St8sia4. During differentiation in the absence of mitogens or differentiation-promoting reagents like thyroid hormone or retinoic acid (Barres et al., 1994), St8sia2 mRNA levels increased significantly between day 0 and day 3 of in vitro differentiation (Fig. 5B, left, open circles). In contrast, St8sia4 levels remained unaltered until day 3 but exhibited a significant, more than twofold, increase between day 3 and day 6 (Fig. 5B, right, open circles). In a parallel set of cultures, we studied the impact of 9cRA on polyST expression during OPC differentiation, because 9cRA promotes OPC differentiation and remyelination, and because its specific receptor, retinoid X receptor gamma (Rxrg), is upregulated in demyelinated lesions and influences OPC differentiation and remyelination in the same manner (Huang et al., 2011). Moreover, ST8SIA2 and ST8SIA4 are differentially regulated during retinoic acid-induced differentiation of human neuroblastoma cells (Seidenfaden and Hildebrandt, 2001). The presence of 9cRA during OPC differentiation caused a massive increase of St8sia2 at day 3 and St8sia4 at day 6, but, importantly, the distinct temporal expression pattern with an early peak of St8sia2 and a late increase of St8sia4 mRNA was maintained (Fig. 5B, closed circles).
Figure 5.

Differences of polyST expression profiles during OPC differentiation are amplified by 9-cis-retinoic acid. A, ST8SIA2 and ST8SIA4 mRNA levels during in vitro differentiation of human embryonic stem cell-derived (ESd) OPCs (see text for details). B, St8sia2 and St8sia4 mRNA levels during in vitro differentiation of mouse OPCs cultured under control conditions (ctrl) or in the presence of 9-cis-retinoic acid (RA). For each mRNA species, data are expressed relative to the mean expression level at day 0, which was set to 1. Values represent the mean ± SEM from n = 3 independent replicates for each time point. One-way ANOVA indicated significant differences (p < 0.05), and a Fisher's LSD post-test was applied for pairwise comparisons between different time points (*p < 0.05; **p < 0.01; ***p < 0.001) or between ctrl and RA-treated cultures of the same time point (##p < 0.01). rel., Relative.
Loss of NCAM or ST8SIA2 impairs but loss of ST8SIA4 promotes OPC differentiation
The impact of NCAM, ST8SIA2, and ST8SIA4 on OPC differentiation was studied by monitoring the changing composition of OPC cultures from wild-type and knock-out mice over 3 d in T3-supplemented differentiation medium. OPCs were marked by NG2, immature, and differentiated oligodendrocytes by the O4 antibody, and mature oligodendrocytes were marked by MBP (Fig. 6A). Compared with wild-type cultures, the appearance of MBP-positive oligodendrocytes at the expense of NG2-positive OPCs was significantly delayed in the Ncam1−/− and St8sia2−/− cultures but were accelerated in St8sia4−/− cultures (Fig. 6B). Accordingly, O4-positive cells were diminished in Ncam1−/− and St8sia2−/− but were augmented in St8sia4−/− cultures at day 0 and 1. Inversely to the altered numbers of MBP-positive cells, the subfraction of NG2- and O4-double-positive cells was enhanced in Ncam1−/− and St8sia2−/− cultures but was diminished in St8sia4−/− cultures at day 2, whereas O4- and MBP-double-positive cells were reduced in Ncam1−/− and St8sia2−/− cultures at days 2 and 3 and were increased in St8sia4−/− cultures at day 2. Thus, impaired in vitro differentiation of NCAM- or ST8SIA2-negative OPCs contrasted with premature appearance of MBP-positive oligodendrocytes in ST8SIA4-negative cultures.
Figure 6.
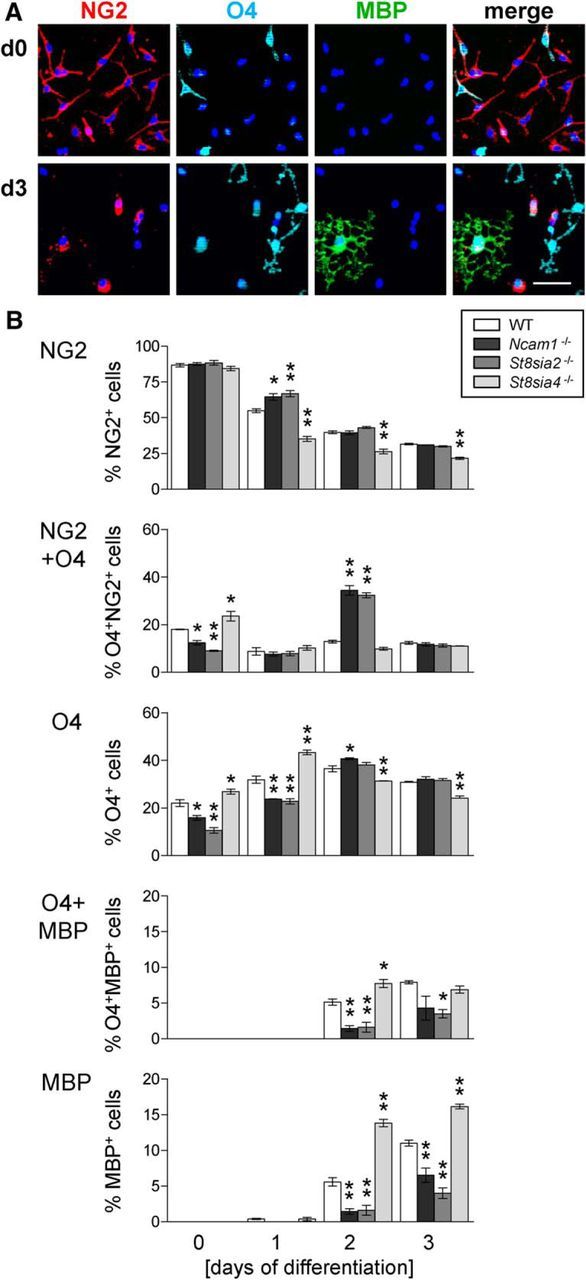
OPC differentiation is delayed in Ncam1−/− and St8sia2−/− cultures but is accelerated in St8sia4−/− cultures. A, Representative examples of OPC cultures stained for NG2, O4, and MBP at day 0 (d0) and d3 of in vitro differentiation in T3 medium. DAPI (blue) was used as a nuclear counterstain. Scale bar, 50 μm. B, Percentages of cells positive for NG2, O4, or MBP and of cells double positive for NG2 and O4 or O4 and MBP in OPC cultures derived from wild-type (WT), Ncam1−/−, St8sia2−/−, or St8sia4−/− mice after 0, 1, 2, and 3 d of in vitro differentiation. Per genotype, OPCs from three animals were prepared independently, and each of the OPC pools was split into cultures that were differentiated for 0, 1, 2, and 3 d, respectively. After triple immunofluorescence staining, marker-positive cells were evaluated by the counting of 13–15 randomly selected frames per culture. Data are the mean ± SEM of n = 3 cultures per genotype and time point. For each time point, one-way ANOVA was applied, and for p < 0.05 pairwise comparisons between WT mice and each of the −/− groups were performed by Dunett's multiple-comparison post-test. *p < 0.05; **p < 0.01.
PolySia engineering promotes OPC differentiation and overcomes deficits caused by loss of NCAM or ST8SIA2
The identical negative impact of Ncam1 or St8sia2 deficiency indicated that remyelination and OPC differentiation require polysialylation of NCAM by ST8SIA2. In principle, polySia on NCAM can modulate NCAM-specific interactions or exert NCAM-independent functions by shaping the properties of the cell surface or by interacting with polySia-binding factors (Schnaar et al., 2014). Artificially enhanced polysialylation can be achieved by polySTs of bacteria such as NmB, producing a capsular polysaccharide that is chemically identical to the mammalian polySia (El Maarouf et al., 2012; Keys et al., 2014). As shown before, the addition of the NmB polyST and activated sialic acid (CMP-Neu5Ac) as its donor substrate is sufficient to synthesize polySia directly on the surface of various cell types (El Maarouf et al., 2012). Using NCAM-negative cells, the previous study indicated that NmB polyST is able to generate polySia on proteins other than NCAM. To corroborate this, we performed Western blot analyses of polySia-NCAM-positive or polySia-negative, NCAM-positive CHO cell clones (Eckhardt et al., 1995) treated for just 1 h with NmB polyST. For both clones, this yielded a polySia-immunoreactive smear extending from the position of the polySia-NCAM signal down to lower molecular weights. The broadly dispersed signals indicate that not just NCAM, but also numerous other proteins were polysialylated (data not shown). Hence, polysialylation by NmB polyST is independent from a specific protein scaffold and implemented in the presence or absence of NCAM.
When added to OPC cultures for 1 h before differentiation, NG2-positive precursors were significantly reduced and the more differentiated O4-positive cells were increased in wild-type, Ncam1−/− and St8sia2−/− cultures, but not in St8sia4−/− cultures, after 1 d of differentiation (Fig. 7, top two panels). Inversely, after 3 days the percentage of MBP-positive cells was increased twofold to fourfold in polyST-treated compared with untreated wild-type, Ncam1−/−, and St8sia2−/− cultures but was only 50% higher in the St8sia4−/− cultures, in which the numbers of MBP cells were already increased in the absence of polyST (Fig. 7, bottom). Thus, artificial polysialylation of the cell surface promotes OPC differentiation independent of NCAM expression and rescues the deficits observed in Ncam1−/− and St8sia2−/− cultures. Furthermore, the increase to similar levels of MBP-positive cells after treatment with NmB polyST indicates that acutely altered polySia levels and not developmental deficits lead to the observed changes of differentiation in OPC cultures derived from Ncam1- or polyST-deficient mice.
Figure 7.

Enforced polysialylation of the cell surface strongly accelerates OPC differentiation. Percentages of cells positive for NG2, O4, and MBP in OPC cultures derived from wild-type (WT), Ncam1−/−, St8sia2−/−, and St8sia4−/− mice after 1 and 3 d of in vitro differentiation. Before differentiation, cultures were treated for 1 h with (+) or without (−) NmB polyST, as indicated. OPC pools were prepared as described for Figure 6 and split into pairs of polyST+ and polyST− cultures that were differentiated for 1 or 3 d, respectively. After triple-immunofluorescence staining, marker-positive cells were evaluated by the counting of 10–15 randomly selected frames per culture. Data are the mean ± SEM of n = 3 cultures per genotype and time point. For each time point, two-way ANOVA was applied (p < 0.05 for treatment, each), and comparisons between the polyST+ and polyST− pairs of cultures were performed by the Bonferroni post-test: *p < 0.05; **p < 0.01; ***p < 0.001. Importantly, note that the polyST treatment was performed in parallel with the experiment shown in Figure 6, and the same values for untreated cultures (polyST−) were used in Figures 6 and 7. The separation into two graphs is for the sole purpose of clear presentation.
Discussion
This study demonstrates that the absence of NCAM or ST8SIA2 causes a congruent delay of developmental myelination as well as impaired remyelination and oligodendrocyte recovery after cuprizone-induced demyelination. The deficits of myelin repair after cuprizone withdrawal are accompanied by a prolonged decline of motor function. Together, the data indicate that the ST8SIA2-mediated polysialylation of NCAM is necessary for efficient remyelination and for the differentiation of OPCs into MBP-positive oligodendrocytes. However, even in the absence of NCAM, polysialylation of the cell surface is sufficient to promote oligodendrocyte differentiation and to rescue the deficits caused by the absence of NCAM or ST8SIA2.
The impaired in vitro differentiation of St8sia2-deficient OPCs corroborates findings of our previous study, which focused on deficits in myelin maintenance manifesting in increasingly thinner myelin sheaths of 3- to 8-month-old St8sia2−/− mice (Szewczyk et al., 2017). However, the previous study did not investigate whether the loss of ST8SIA2 exerts its effects by affecting polySia synthesis on NCAM or on SynCAM 1 as the second carrier of polySia in OPCs (Werneburg et al., 2015b). In contrast to NCAM, SynCAM 1 is exclusively polysialylated by ST8SIA2 (Rollenhagen et al., 2012; Werneburg et al., 2015b). Therefore, an NCAM-independent role of polySia-SynCAM 1 in OPC differentiation and remyelination would have provided a rationale for the divergent impact of ST8SIA2 and ST8ISIA4 on these processes. Yet, despite considerable efforts, we could not detect polySia-SynCAM 1 in brains of cuprizone-treated mice. Together with the congruent negative impact of Ncam1 or St8sia2 deficiency, this clearly argues that ST8SIA2-dependent polysialylation of NCAM, not SynCAM 1, is required for OPC differentiation in vitro and during myelin repair.
In contrast to these findings in Ncam1- or St8sia2-deficient mice, we demonstrated previously that the loss of ST8SIA4 promotes remyelination (Koutsoudaki et al., 2010). Strikingly, the divergent impact of ST8SIA2 and ST8SIA4 on remyelination in vivo is mirrored by opposing roles of the two enzymes during the cell-autonomous process of OPC differentiation in vitro. As discussed in the previous paragraph, the differentiation-promoting effect of ST8SIA2 is based on the polysialylation of NCAM. Paradoxically, the inhibitory effect of ST8SIA4 on OPC differentiation also depends on polysialylation of NCAM, because NCAM is the only substrate for ST8SIA4 in these cells, as shown by the absence of polySia from OPCs derived from Ncam1 and St8sia2 double-deficient mice (Werneburg et al., 2015b). Hence, it seems enigmatic how the two enzymes can exert opposite effects, but the sequence of expression with ST8SIA2 at early stages and ST8SIA4 later during OPC differentiation provides a reasonable explanation. Accordingly, polysialylation of NCAM during the early phase of OPC differentiation is mainly driven by ST8SIA2. Altered receptor tyrosine kinase signaling, particularly increased levels and activity of PDGF receptor α, have been observed in St8sia2-deficient OPCs, and this may be the underlying mechanism for how the loss of ST8SIA2 inhibits differentiation (Szewczyk et al., 2017). Considering the equally enhanced differentiation of NCAM-positive and NCAM-negative OPCs in response to artificial polysialylation with NmB polyST, an NCAM-independent role of polySia can be envisaged (e.g., in modulating the binding of soluble ligands), as shown in various other cellular systems (Schnaar et al., 2014). During later stages of oligodendrocytic differentiation, the polysialylation of NCAM is taken over by ST8SIA4 before polySia has to be downregulated to enable myelin sheath formation. In the absence of interactions with axons, the mechanisms underlying the inhibitory role of polySia at this stage remain to be studied. It also will be interesting to explore the regulatory mechanisms underlying the temporal expression patterns of the polySTs during OPC differentiation, but this will be challenging because so far, the transcriptional regulation of these enzymes is largely unknown.
The inhibitory effect of ST8SIA4 on OPC differentiation supports the previous assumption that improved differentiation of OPCs into myelinating oligodendrocytes causes the enhanced remyelination observed in St8sia4−/− mice (Koutsoudaki et al., 2010). Reversely, impaired OPC differentiation is regarded as a major reason for the failure of remyelination. Hence, the reduced differentiation of NCAM- or ST8SIA2-negative OPCs in vitro is in agreement with compromised remyelination in Ncam1−/− and St8sia2−/− mice. A causal relation is further supported by the observation that not only myelin, but also the densities of mature Nogo-A- or APC-positive cells, are markedly reduced in remyelinating Ncam1−/− and St8sia2−/− mice, whereas NG2-positive cells are less affected. Moreover, the transiently increased densities of proliferating or apoptotic cells of the oligodendrocyte lineage and of microglia/macrophages, astrocytes, or damaged axons are not detectably affected. Together, these data converge into the coherent picture that impaired or enhanced generation of myelinating oligodendrocytes from OPCs cause delayed or accelerated remyelination in Ncam1−/− and St8sia2−/− or St8sia4−/− mice, respectively.
Recent studies highlight the intricate reciprocal interactions among myelination, motor cortex activity, and fine tuning of motor performance (Gibson et al., 2014; McKenzie et al., 2014; Xiao et al., 2016). Consistently, studies that relate the progress of remyelination to motor coordination demonstrate that the extent of demyelination correlates with the severity of motor dysfunction (Franco-Pons et al., 2007; Skripuletz et al., 2015; Okazaki et al., 2016). The concomitant delay of remyelination and motor recovery in Ncam1−/− and St8sia2−/− mice as well as the return of both parameters to wild-type levels after 3 weeks of recovery, as shown in the current study, provide further support for a causal relationship between myelination and motor performance.
Because of the prominent role of 9cRA in promoting OPC differentiation and remyelination in vitro and in vivo (Huang et al., 2011), we studied its impact on polyST expression during OPC differentiation. Remarkably, we could demonstrate that 9cRA regulates St8sia2 and St8sia4 differentially. The expression of both polySTs is strongly upregulated under 9cRA treatment, but St8sia2 RNA levels peak well before the St8sia4 transcripts start to rise, so that the sequential expression pattern of the two polySTs, as observed without differentiation-promoting reagents, is maintained. In conjunction with the impairments of OPC differentiation and remyelination in the absence of ST8SIA2, the strong but still temporary augmentation of St8sia2 expression by 9cRA implicates that the improved differentiation of OPCs cultured in the presence of 9cRA, as shown by Huang et al. (2011), is based on a transient transcriptional upregulation of St8sia2. Combined, the data suggest that 9cRA promotes OPC differentiation and remyelination by increasing the levels of ST8SIA2 during early stages of oligodendrocytic differentiation. Notably, the data on polyST expression during the differentiation of human embryonic stem cell-derived OPCs, although preliminary and on a coarse time scale, indicate that human oligodendrogenesis also involves a significant upregulation and downregulation of both enzymes. Thus, a selective targeting of polySTs, as achieved by 9cRA, could be an efficient therapeutic strategy to improve myelin repair in human diseases.
PolySia engineering by the application of NmB polyST to cultured OPCs before differentiation not only overcomes the differentiation deficits caused by the loss of Ncam and St8sia2, but also overrides the effect of St8sia4 deficiency. As shown in the mouse model, injection of bacterial polyST into the brain leads to cell surface polysialylation within <3 h and has no obvious adverse consequences (El Maarouf et al., 2012). As corroborated by the results of the current study, NmB polyST adds polySia mainly not to NCAM but to numerous other cell surface proteins, implicating that the overall polysialylation of the cell surface and not altered functions of a specific protein affect cellular behavior. Importantly, bacterial polyST induces polysialylation in gray and white matter transiently. Under the conditions of the former study, the engineered polySia was present for ∼2–3 weeks, depending on the site of injection (El Maarouf et al., 2012). Therefore, the prominent effects of engineered polySia on the differentiation of wild-type as well as differentiation-compromised OPCs in vitro provide proof of principle that enforced polysialylation of the cell surface holds the potential to promote myelin repair by overcoming impaired differentiation of OPCs into remyelinating oligodendrocytes.
In conclusion, we have shown here that polysialylation of NCAM by ST8SIA2 is needed for oligodendrogenesis, remyelination, and recovery of motor function after cuprizone-induced demyelination. The different time course of St8sia2 and St8Sia4 mRNA expression during the early stages of OPC differentiation provides a rationale for the opposing cell-autonomous impact of the two polySTs on the differentiation process in vitro and on myelin repair in vivo. Finally, the data disclose that the augmentation of ST8SIA2 by 9cRA and enzymatic engineering of polySia are promising therapeutic strategies to improve regeneration in demyelinating disorders.
Footnotes
Parts of this work were supported by Federal Ministry of Education and Research (Germany) Grant 01EW1106/NeuConnect in the frame of ERA-NET NEURON, funding from the Deutsche Forschungsgemeinschaft to H.H. (Grant Hi 678/8–1), and the Cluster of Excellence REBIRTH. We thank Birgit Weinhold, Kerstin Flächsig-Schulz, and Ulrike Peters-Bernard for help with mouse maintenance and breeding; Kirsten Haastert-Talini and Jennifer Metzen for support with the rotoarod test; and Carla Seegers, Sabrina Kubinski, and Nadine Zumbrägel for their contributions to immunostaining and cell counting.
The authors declare no competing financial interests.
References
- Bakhti M, Snaidero N, Schneider D, Aggarwal S, Möbius W, Janshoff A, Eckhardt M, Nave KA, Simons M (2013) Loss of electrostatic cell-surface repulsion mediates myelin membrane adhesion and compaction in the central nervous system. Proc Natl Acad Sci U S A 110:3143–3148. 10.1073/pnas.1220104110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral-Moran MJ, Calaora V, Vutskits L, Wang C, Zhang H, Durbec P, Rougon G, Kiss JZ (2003) Oligodendrocyte progenitor migration in response to injury of glial monolayers requires the polysialic neural cell-adhesion molecule. J Neurosci Res 72:679–690. 10.1002/jnr.10627 [DOI] [PubMed] [Google Scholar]
- Barres BA, Lazar MA, Raff MC (1994) A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development 120:1097–1108. [DOI] [PubMed] [Google Scholar]
- Bartsch U, Kirchhoff F, Schachner M (1990) Highly sialylated N-CAM is expressed in adult mouse optic nerve and retina. J Neurocytol 19:550–565. 10.1007/BF01257243 [DOI] [PubMed] [Google Scholar]
- Charles P, Hernandez MP, Stankoff B, Aigrot MS, Colin C, Rougon G, Zalc B, Lubetzki C (2000) Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc Natl Acad Sci U S A 97:7585–7590. 10.1073/pnas.100076197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Reynolds R, Seilhean D, Rougon G, Aigrot MS, Niezgoda A, Zalc B, Lubetzki C (2002) Re-expression of PSA-NCAM by demyelinated axons: an inhibitor of remyelination in multiple sclerosis? Brain 125:1972–1979. 10.1093/brain/awf216 [DOI] [PubMed] [Google Scholar]
- Decker L, Avellana-Adalid V, Nait-Oumesmar B, Durbec P, Baron-Van Evercooren A (2000) Oligodendrocyte precursor migration and differentiation: combined effects of PSA residues, growth factors, and substrates. Mol Cell Neurosci 16:422–439. 10.1006/mcne.2000.0885 [DOI] [PubMed] [Google Scholar]
- Decker L, Durbec P, Rougon G, Baron-Van Evercooren A (2002) Loss of polysialic residues accelerates CNS neural precursor differentiation in pathological conditions. Mol Cell Neurosci 19:225–238. 10.1006/mcne.2001.1072 [DOI] [PubMed] [Google Scholar]
- Eckhardt M, Mühlenhoff M, Bethe A, Koopman J, Frosch M, Gerardy-Schahn R (1995) Molecular characterization of eukaryotic polysialyltransferase-1. Nature 373:715–718. 10.1038/373715a0 [DOI] [PubMed] [Google Scholar]
- El Maarouf A, Moyo-Lee Yaw D, Lindhout T, Pearse DD, Wakarchuk W, Rutishauser U (2012) Enzymatic engineering of polysialic acid on cells in vitro and in vivo using a purified bacterial polysialyltransferase. J Biol Chem 287:32770–32779. 10.1074/jbc.M112.377614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewou SN, Ramakrishnan H, Büssow H, Gieselmann V, Eckhardt M (2007) Down-regulation of polysialic acid is required for efficient myelin formation. J Biol Chem 282:16700–16711. 10.1074/jbc.M610797200 [DOI] [PubMed] [Google Scholar]
- Franceschini I, Vitry S, Padilla F, Casanova P, Tham TN, Fukuda M, Rougon G, Durbec P, Dubois-Dalcq M (2004) Migrating and myelinating potential of neural precursors engineered to overexpress PSA-NCAM. Mol Cell Neurosci 27:151–162. 10.1016/j.mcn.2004.05.006 [DOI] [PubMed] [Google Scholar]
- Franco-Pons N, Torrente M, Colomina MT, Vilella E (2007) Behavioral deficits in the cuprizone-induced murine model of demyelination/remyelination. Toxicol Lett 169:205–213. 10.1016/j.toxlet.2007.01.010 [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Ffrench-Constant C (2008) Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci 9:839–855. 10.1038/nrn2480 [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Gallo V (2014) The translational biology of remyelination: past, present, and future. Glia 62:1905–1915. 10.1002/glia.22622 [DOI] [PubMed] [Google Scholar]
- Galuska SP, Rollenhagen M, Kaup M, Eggers K, Oltmann-Norden I, Schiff M, Hartmann M, Weinhold B, Hildebrandt H, Geyer R, Mühlenhoff M, Geyer H (2010) Synaptic cell adhesion molecule SynCAM 1 is a target for polysialylation in postnatal mouse brain. Proc Natl Acad Sci U S A 107:10250–10255. 10.1073/pnas.0912103107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, Barres BA, Woo PJ, Vogel H, Monje M (2014) Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344: 1252304. 10.1126/science.1252304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser T, Brose C, Franceschini I, Hamann K, Smorodchenko A, Zipp F, Dubois-Dalcq M, Brüstle O (2007) Neural cell adhesion molecule polysialylation enhances the sensitivity of embryonic stem cell-derived neural precursors to migration guidance cues. Stem Cells 25:3016–3025. 10.1634/stemcells.2007-0218 [DOI] [PubMed] [Google Scholar]
- Gudi V, Moharregh-Khiabani D, Skripuletz T, Koutsoudaki PN, Kotsiari A, Skuljec J, Trebst C, Stangel M (2009) Regional differences between grey and white matter in cuprizone induced demyelination. Brain Res 1283:127–138. 10.1016/j.brainres.2009.06.005 [DOI] [PubMed] [Google Scholar]
- Hu BY, Du ZW, Zhang SC (2009) Differentiation of human oligodendrocytes from pluripotent stem cells. Nat Protoc 4:1614–1622. 10.1038/nprot.2009.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JK, Jarjour AA, Oumesmar BN, Kerninon C, Williams A, Krezel W, Kagechika H, Bauer J, Zhao C, Baron-Van Evercooren A, Chambon P, Ffrench-Constant C, Franklin RJM (2011) Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci 14:45–53. 10.1038/nn.2702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski I, Mo Z, Zecevic N (2007) Down-regulation of the axonal polysialic acid-neural cell adhesion molecule expression coincides with the onset of myelination in the human fetal forebrain. Neuroscience 149:328–337. 10.1016/j.neuroscience.2007.07.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keys TG, Fuchs HL, Ehrit J, Alves J, Freiberger F, Gerardy-Schahn R (2014) Engineering the product profile of a polysialyltransferase. Nat Chem Biol 10:437–442. 10.1038/nchembio.1501 [DOI] [PubMed] [Google Scholar]
- Kipp M, Clarner T, Dang J, Copray S, Beyer C (2009) The cuprizone animal model: new insights into an old story. Acta Neuropathol 118:723–736. 10.1007/s00401-009-0591-3 [DOI] [PubMed] [Google Scholar]
- Klüver H, Barrera E (1953) A method for the combined staining of cells and fibers in the nervous system. J Neuropathol Exp Neurol 12:400–403. 10.1097/00005072-195312040-00008 [DOI] [PubMed] [Google Scholar]
- Köhler W. (2010) Leukodystrophies with late disease onset: an update. Curr Opin Neurol 23:234–241. 10.1097/WCO.0b013e328338313a [DOI] [PubMed] [Google Scholar]
- Koutsoudaki PN, Hildebrandt H, Gudi V, Skripuletz T, ˘kuljec J, Stangel M (2010) Remyelination after cuprizone induced demyelination is accelerated in mice deficient in the polysialic acid synthesizing enzyme St8siaIV. Neuroscience 171:235–244. 10.1016/j.neuroscience.2010.08.070 [DOI] [PubMed] [Google Scholar]
- Kuhlmann T, Miron V, Cuo Q, Wegner C, Antel J, Brück W (2008) Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131:1749–1758. 10.1093/brain/awn096 [DOI] [PubMed] [Google Scholar]
- Lindner M, Heine S, Haastert K, Garde N, Fokuhl J, Linsmeier F, Grothe C, Baumgärtner W, Stangel M (2008) Sequential myelin protein expression during remyelination reveals fast and efficient repair after central nervous system demyelination. Neuropathol Appl Neurobiol 34:105–114. 10.1111/j.1365-2990.2007.00879.x [DOI] [PubMed] [Google Scholar]
- Lindner M, Fokuhl J, Linsmeier F, Trebst C, Stangel M (2009) Chronic toxic demyelination in the central nervous system leads to axonal damage despite remyelination. Neurosci Lett 453:120–125. 10.1016/j.neulet.2009.02.004 [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Morell P (2001) The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol 11:107–116. 10.1111/j.1750-3639.2001.tb00385.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, Richardson WD (2014) Motor skill learning requires active central myelination. Science 346:318–322. 10.1126/science.1254960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nait Oumesmar B, Vignais L, Duhamel-Clérin E, Avellana-Adalid V, Rougon G, Baron-Van Evercooren A (1995) Expression of the highly polysialylated neural cell adhesion molecule during postnatal myelination and following chemically induced demyelination of the adult mouse spinal cord. Eur J Neurosci 7:480–491. 10.1111/j.1460-9568.1995.tb00344.x [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Picard-Riera N, Kerninon C, Decker L, Seilhean D, Höglinger GU, Hirsch EC, Reynolds R, Baron-Van Evercooren A (2007) Activation of the subventricular zone in multiple sclerosis: evidence for early glial progenitors. Proc Natl Acad Sci U S A 104:4694–4699. 10.1073/pnas.0606835104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki R, Doi T, Hayakawa K, Morioka K, Imamura O, Takishima K, Hamanoue M, Sawada Y, Nagao M, Tanaka S, Ogata T (2016) The crucial role of Erk2 in demyelinating inflammation in the central nervous system. J Neuroinflammation 13:235–0690. 10.1186/s12974-016-0690-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen M, Kuckuck S, Ulm C, Hartmann M, Galuska SP, Geyer R, Geyer H, Mühlenhoff M (2012) Polysialylation of the synaptic cell adhesion molecule 1 (SynCAM 1) depends exclusively on the polysialyltransferase ST8SiaII in vivo. J Biol Chem 287:35170–35180. 10.1074/jbc.M112.375642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas Tejedor L, Gudi V, Kucman V, Pul R, Gingele S, Sühs KW, Stangel M, Skripuletz T (2015) Oligodendroglial markers in the cuprizone model of CNS de- and remyelination. Histol Histopathol 30:1455–1464. 10.14670/HH-11-640 [DOI] [PubMed] [Google Scholar]
- Schiff M, Röckle I, Burkhardt H, Weinhold B, Hildebrandt H (2011) Thalamocortical pathfinding defects precede degeneration of the reticular thalamic nucleus in polysialic acid-deficient mice. J Neurosci 31:1302–1312. 10.1523/JNEUROSCI.5609-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaar RL, Gerardy-Schahn R, Hildebrandt H (2014) Sialic acids in the brain: gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol Rev 94:461–518. 10.1152/physrev.00033.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenfaden R, Hildebrandt H (2001) Retinoic acid-induced changes in NCAM polysialylation and polysialyltransferase mRNA expression of human neuroblastoma cells. J Neurobiol 46:11–28. [DOI] [PubMed] [Google Scholar]
- Skripuletz T, Gudi V, Hackstette D, Stangel M (2011) De- and remyelination in the CNS white and grey matter induced by cuprizone: the old, the new, and the unexpected. Histol Histopathol 26:1585–1597. 10.14670/HH-26.1585 [DOI] [PubMed] [Google Scholar]
- Skripuletz T, Manzel A, Gropengießer K, Schäfer N, Gudi V, Singh V, Salinas Tejedor L, Jörg S, Hammer A, Voss E, Vulinovic F, Degen D, Wolf R, Lee DH, Pul R, Moharregh-Khiabani D, Baumgärtner W, Gold R, Linker RA, Stangel M (2015) Pivotal role of choline metabolites in remyelination. Brain 138:398–413. 10.1093/brain/awu358 [DOI] [PubMed] [Google Scholar]
- Stidworthy MF, Genoud S, Suter U, Mantei N, Franklin RJ (2003) Quantifying the early stages of remyelination following cuprizone-induced demyelination. Brain Pathol 13:329–339. 10.1111/j.1750-3639.2003.tb00032.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk LM, Brozko N, Nagalski A, Röckle I, Werneburg S, Hildebrandt H, Wisniewska MB, Kuznicki J (2017) ST8SIA2 promotes oligodendrocyte differentiation and the integrity of myelin and axons. Glia 65:34–49. 10.1002/glia.23048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nave KA (2008) Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 31:247–269. 10.1146/annurev.neuro.30.051606.094313 [DOI] [PubMed] [Google Scholar]
- Trotter J, Bitter-Suermann D, Schachner M (1989) Differentiation-regulated loss of the polysialylated embryonic form and expression of the different polypeptides of the neural cell adhesion molecule by cultured oligodendrocytes and myelin. J Neurosci Res 22:369–383. 10.1002/jnr.490220402 [DOI] [PubMed] [Google Scholar]
- Wang C, Rougon G, Kiss JZ (1994) Requirement of polysialic acid for the migration of the O-2A glial progenitor cell from neurohypophyseal explants. J Neurosci 14:4446–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werneburg S, Buettner FF, Mühlenhoff M, Hildebrandt H (2015a) Polysialic acid modification of the synaptic cell adhesion molecule SynCAM 1 in human embryonic stem cell-derived oligodendrocyte precursor cells. Stem Cell Res 14:339–346. 10.1016/j.scr.2015.03.001 [DOI] [PubMed] [Google Scholar]
- Werneburg S, Mühlenhoff M, Stangel M, Hildebrandt H (2015b) Polysialic acid on SynCAM 1 in NG2 cells and on neuropilin-2 in microglia is confined to intracellular pools that are rapidly depleted upon stimulation. Glia 63:1240–1255. 10.1002/glia.22815 [DOI] [PubMed] [Google Scholar]
- Xiao L, Ohayon D, McKenzie IA, Sinclair-Wilson A, Wright JL, Fudge AD, Emery B, Li H, Richardson WD (2016) Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat Neurosci 19:1210–1217. 10.1038/nn.4351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Vutskits L, Calaora V, Durbec P, Kiss JZ (2004) A role for the polysialic acid-neural cell adhesion molecule in PDGF-induced chemotaxis of oligodendrocyte precursor cells. J Cell Sci 117:93–103. 10.1242/jcs.00827 [DOI] [PubMed] [Google Scholar]