

Abstract
Temporal lobe epilepsy (TLE) is the most frequent form of focal epilepsies and is generally associated with malfunctioning of the hippocampal formation. Recently, a preferential loss of parvalbumin (PV) neurons has been observed in the subiculum of TLE patients and in animal models of TLE. To demonstrate a possible causative role of defunct PV neurons in the generation of TLE, we permanently inhibited GABA release selectively from PV neurons of the ventral subiculum by injecting a viral vector expressing tetanus toxin light chain in male mice. Subsequently, mice were subjected to telemetric EEG recording and video monitoring. Eighty-eight percent of the mice presented clusters of spike-wave discharges (C-SWDs; 40.0 ± 9.07/month), and 64% showed spontaneous recurrent seizures (SRSs; 5.3 ± 0.83/month). Mice injected with a control vector presented with neither C-SWDs nor SRSs. No neurodegeneration was observed due to vector injection or SRS. Interestingly, mice that presented with only C-SWDs but no SRSs, developed SRSs upon injection of a subconvulsive dose of pentylenetetrazole after 6 weeks. The initial frequency of SRSs declined by ∼30% after 5 weeks. In contrast to permanent silencing of PV neurons, transient inhibition of GABA release from PV neurons through the designer receptor hM4Di selectively expressed in PV-containing neurons transiently reduced the seizure threshold of the mice but induced neither acute nor recurrent seizures. Our data demonstrate a critical role for perisomatic inhibition mediated by PV-containing interneurons, suggesting that their sustained silencing could be causally involved in the development of TLE.
SIGNIFICANCE STATEMENT Development of temporal lobe epilepsy (TLE) generally takes years after an initial insult during which maladaptation of hippocampal circuitries takes place. In human TLE and in animal models of TLE, parvalbumin neurons are selectively lost in the subiculum, the major output area of the hippocampus. The present experiments demonstrate that specific and sustained inhibition of GABA release from parvalbumin-expressing interneurons (mostly basket cells) in sector CA1/subiculum is sufficient to induce hyperexcitability and spontaneous recurrent seizures in mice. As in patients with nonlesional TLE, these mice developed epilepsy without signs of neurodegeneration. The experiments highlight the importance of the potent inhibitory action mediated by parvalbumin cells in the hippocampus and identify a potential mechanism in the development of TLE.
Keywords: basket cell, epilepsy, epileptogenesis, feedforward inhibition, parvalbumin, subiculum
Introduction
Temporal lobe epilepsy (TLE) is the most frequent form among focal epilepsies. It is characterized by spontaneous recurrent seizures (SRSs) arising from limbic brain structures and is often associated with Ammon's horn sclerosis (Spielmeyer, 1927; Honavar and Meldrum, 1997) and, consequently, with memory deficits and emotional disturbances. TLE may be provoked by an initial insult such as prolonged febrile seizures, head injury, or sustained seizures. It often takes years until TLE becomes manifest (Bragin et al., 2000). During the preceding “silent phase,” neuronal circuitries are altered to permit recurrent hypersynchronization and hyperexcitability displayed as epileptic seizures (Paz and Huguenard, 2015). The underlying cellular and molecular mechanisms mediating this transition from a normal to an epileptic brain are still poorly understood.
For instance, Cossart et al. (2001) proposed that a deficit in dendritic feedback inhibition, presumably due to a loss in oriens-lacunosum moleculare (O-LM) neurons, reduces the seizure threshold, whereas augmented somatic feedforward inhibition limits epileptiform activity (Cossart et al., 2001; Dinocourt et al., 2003). Feedforward inhibition is exerted particularly by fast-spiking parvalbumin (PV)-containing basket cells that establish perisomatic synapses upon pyramidal cells of the hippocampus and subiculum and decisively control their excitatory output (Szabó et al., 2010; Gulyás and Freund, 2015). Thus, a sudden loss of perisomatic inhibition in the hippocampus leads to uninhibited massive bursting of pyramidal cells and the occurrence of fast ripples (Salami et al., 2014; Alexander et al., 2016) and may give rise to the formation of an epileptic focus (Gulyás and Freund, 2015).
Interestingly, PV-containing interneurons are preferentially lost in the subiculum of patients with TLE even without Ammon's horn sclerosis (Andrioli et al., 2007). Also in animal models of TLE, PV-containing interneurons of the subiculum selectively degenerate after an initial status epilepticus (Dinocourt et al., 2003; Knopp et al., 2008; Drexel et al., 2011). Thus, a loss or impairment of PV-containing basket cells could be directly connected to the subsequent development of TLE.
To test this hypothesis, we now permanently inhibited GABA release selectively from PV interneurons of the ventral subiculum of mice and subjected them to continuous telemetric EEG and video monitoring. For silencing PV-containing neurons locally, we injected an adeno-associated viral vector (AAV)-expressing tetanus toxin light chain [TeLC; fused with a green fluorescent protein (GFP) tag] with its reading frame inverted in a flip-excision (FLEX) cassette (termed AAV-TeLC) into the ventral subiculum of transgenic mice expressing Cre-recombinase under the PV promoter (PV-cre mice). Cre-recombinase then inverts the TeLC construct selectively in PV-expressing neurons, allowing permanent transgene expression (Murray et al., 2011). TeLC cleaves vesicle-associated membrane protein 2 (synaptobrevin), thereby permanently inhibiting GABA release (Schiavo et al., 1992). Control mice were injected with a corresponding GFP vector (AAV–GFP). Mice injected with AAV–TeLC developed clusters of spike-wave discharges (C-SWDs) that were often followed by SRSs. Furthermore, to test whether transient inhibition of GABA release would also be sufficient to induce C-SWDs and SRSs, we took advantage of the DREADD (designer receptors exclusively activated by designer drugs) technology (Roth, 2016). We expressed hM4Di receptors specifically in PV-containing interneurons and subsequently inhibited GABA release from these neurons transiently by injection of clozapine N-oxide (CNO), selectively inhibiting neurons containing the DREADD receptors. In contrast to permanent silencing of PV/GABA neurons with TeLC, their transient inhibition did not induce C-SWDs or SRSs.
Materials and Methods
Mice.
Animal experiments were conducted according to national guidelines and European Community laws and were approved by the Committee for Animal Protection of the Austrian Ministry of Science. PV-cre transgenic mice (Pvalbtm1(cre)Arbr; RRID:IMSR_JAX:017320) were originally purchased from The Jackson Laboratory through Charles River and then maintained on a C57BL/6N background. These mice in adulthood express Cre-recombinase under the PV promoter. C57BL/6N wild-type (WT) mice were obtained from Charles River. The mice were housed in groups of 3–5 in single ventilated cages under standard laboratory conditions (12 h light/dark cycle, with lights turned on at 6:30 A.M.) and had access to food and water ad libitum. For the experiments, 10 to 14 weeks old male heterozygous mice (Pvalbtm1(cre)Arbr+/−) were used, which exhibited a similar number of PV-expressing interneurons in the subiculum as WT C57BL/6N mice (data not shown).
Preparation of vectors.
The AAV1/2 vectors contained the genes for TeLC fused with a GFP tag or GFP alone, respectively with its reading frame inverted in a FLEX cassette (AAV-TeLC and AAV-GFP, respectively). Thus, AAV-mediated transgene expression is limited to PV-expressing cells at the injection site. The FLEX- and the AAV2-based vector backbones on a cytomegalovirus enhancer/chicken β-actin promoter were constructed (Murray et al., 2011). Vectors pseudotyped for AAV1 were produced and highly purified as previously described (Mietzsch et al., 2014). Briefly, HEK293 cells were cotransfected with rAAV plasmids pAM-FLEX-TeLC or pAM-FLEX-GFP and pDP1rs helper plasmids by calcium phosphate coprecipitation. Benzonase-treated cleared cell lysates were purified by HPLC on AVB columns with successive dialysis. AAV titers were determined by quantitative PCR as the number of genomic particles per milliliter (gp/ml). Titers were 2.4 × 1010 gp/ml for pAM-FLEX-TeLC, and 1.4 × 1011 gp/ml for pAM-FLEX-GFP.
AAV-FLEX-hM4Di vector.
For experiments using DREADD technology, the recombinant AAV2-hSyn-FLEX-hM4Di-mCherry plasmid deposited by Bryan Roth (University of North Carolina at Chapel Hill, Chapel Hill, NC; Krashes et al., 2011) was obtained from Addgene. It was packaged into AAV2 capsids with a final titer of 2.7 × 1012 gp/ml and was injected at a dilution of 1:4 (6.7 × 1011 gp/ml).
Surgery.
Stereotaxic surgeries were performed as described in detail previously (Jagirdar et al., 2015). Heterozygous male PV-cre transgenic mice (age, 10–14 weeks) were treated with the analgesic drug carprofen (5 mg/kg, s.c.; Rimadyl, Pfizer) 60 min before surgery. We anesthetized the mice with 150 mg/kg, i.p. ketamine (stock solution 50 mg/ml; Ketasol, Ogris Pharma Vertriebs) and maintained anesthesia by applying 1–4% sevoflurane (Sevorane, Abbott) through a veterinary anesthesia mask using oxygen (400 ml/min) as a carrier gas. We placed the mice into a stereotaxic frame (David Kopf Instruments) and opened the skin above the skull. A telemetric EEG transmitter (TA10EA-F20, Data Sciences International) was inserted into a subcutaneous pocket at the right abdominal wall, and the electrode wires were pulled through the subcutaneous channel formed from the pocket to the skull, and the pocket was carefully sutured. Bilateral holes were drilled for AAV vector injection and insertion of a depth electrode [coordinates from bregma (in mm): posterior, 3.8; lateral, 3.5] and for the epidural reference electrode [coordinates from bregma (in mm): posterior, 2.0; lateral, 1.6 mm].
We injected the AAV vectors AAV-TeLC or AAV-GFP (for controls) unilaterally into the left ventral subiculum (3.8 mm posterior; 3.5 mm left; 3.5 ventral). AAV-TeLC or AAV-GFP injections were performed in mice of the same litters on the same days. For telemetric EEG recordings, we implanted a tungsten depth electrode (catalog #577100, Science Products) into the left ventral subiculum (3.8 mm posterior; 3.5 mm left, 3.0 mm ventral) and, as reference electrode, attached a stainless steel screw (M1*2, Hummer und Riess) to the skull in an epidural position (2.0 mm posterior, 1.6 mm right). In some mice, an epidural electrode was set above the ipsilateral dorsal hippocampus instead of the depth electrode. Electrodes were fixed to the skull using dental cement (Paladur, Kulzer). After surgery and during EEG recording, mice were single housed.
EEG recordings and video monitoring.
EEGs were recorded continuously using a telemetry system (Dataquest A.R.T. Gold 4.33 Acquisition, Data Sciences International). For behavioral analysis, continuous video recordings were performed using Axis 221 network cameras (Axis Communications) and infrared illumination (Conrad Electronics) during darkness. EEGs were recorded at a sampling rate of 1000 Hz without a priori filter cutoff and saved on external hard disk drives.
EEG traces of local field potentials were visually inspected by two independent observers using the Dataquest A.R.T. Gold 4.33 Analysis software (Data Sciences International). We defined seizures as EEG segments with continuous activity with an amplitude of at least two times the baseline amplitude, a duration of at least 10 s, and the presence of a postictal depression (EEG signal below baseline amplitude). C-SWDs were defined as series of at least five high-amplitude (at least 2× baseline amplitude) SWDs that were not >60 s apart. We used corresponding synchronized video recordings to analyze behavioral correlates to EEG seizures. At the end of the experiment (6–8 weeks after the initial vector injection), mice were deeply anesthetized (killed) with thiopental (150 mg/kg) and perfused with 4% paraformaldehyde and their brains were dissected out for immunohistochemistry.
Evaluation of motor seizures.
All seizures defined by EEG were also investigated for a possible behavioral correlate by evaluating the synchronized video recordings. Seizure rating was performed in accordance with that of kainic acid-injected rats (Sperk et al., 1983), as follows: stage 1, staring, arrest, chewing; stage 2, unilateral or bilateral tonic movements/seizure; stage 3, rearing without falling; and stage 4, rearing with falling, limbic seizures.
Determination of seizure threshold using a threshold dose of pentylenetetrazole.
Pentylenetetrazole (PTZ; 30 mg/kg in saline) was injected intraperitoneally. This dose was established in a pilot experiment (data not shown). Only 1 of 13 AAV-GFP-injected mice and no WT mice (n = 5) revealed an acute convulsion. Immediately after PTZ injection, the transmitter-implanted mice were placed into cages for continuous video and EEG recordings.
Transient silencing of PV neurons using the DREADD system.
To achieve specific and reversible chemogenetic silencing of PV-expressing interneurons in the subiculum, we used DREADD technology. Twelve heterozygous male PV-cre mice were injected with an AAV-hM4Di vector (1.0 μl) expressing the inhibitory hM4Di receptor [together with a red fluorescent protein (RFP) tag] into the left ventral subiculum using the same coordinates and protocol as for AAV-TeLC injections. Epidural EEG electrodes were set above the ipsilateral hippocampus (2.0 mm posterior and 1.6 mm lateral) and fixed to the skull using dental cement. Fifteen days later, seven mice were injected intraperitoneally with the selective hM4Di agonist clozapine-N-oxide (CNO, Tocris Bioscience; 10 mg/kg, i.p., dissolved in sterile 0.9% NaCl at a concentration of 1.0 mg/ml) and EEGs were inspected for the occurrence of C-SWDs or seizures. AAV-hM4Di vector-injected controls (n = 5) were injected with saline instead of CNO.
To prove the efficacy of DREADD treatment in vivo, we investigated the effect of transient silencing of PV interneurons on the seizure threshold. For this, another 12 AAV-hM4Di vector-injected mice were injected with CNO (10 mg/kg, i.p.; n = 6) or saline (n = 6) after 15 d, and 45 min later with a threshold dose of the GABAA receptor antagonist PTZ (30 mg/kg, i.p.). The interval between CNO and PTZ injections has been chosen based on previous experiments showing a maximal behavioral readout due to inhibition of PV-expressing interneurons in the nucleus accumbens already 30 min after CNO application (Wirtshafter and Stratford, 2016). We then monitored acute seizures and C-SWDs by telemetric EEG recording for 24 h.
Slice preparation and patch-clamp electrophysiology of evoked IPSCs.
To verify a loss of inhibitory input to pyramidal neurons of the subiculum, we applied whole-cell voltage-clamp recordings in acute brain slices also containing the subiculum 2–3 weeks after vector injection. Mice (AAV-TeLC, n = 6; AAV-GFP, n = 5) were anesthetized with isoflurane (Baxter) and decapitated, and their brains were rapidly removed and placed in ice-cold oxygenated (95% O2/5% CO2) artificial CSF (aCSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 26 NaHCO3, 1.25 NaH2PO4, and 10 glucose. Horizontal hippocampal slices at 300 μm thickness were cut on a vibratome (VT1200, Leica Microsystems) and recovered in aCSF (32–34°C) for 1 h before recordings.
For recording evoked IPSCs (eIPSCs), recording pipettes with a final tip resistance of 2–5 MΩ, prepared using a micropipette puller (P-1000 Sutter Instruments) contained the following (in mm): 135.0 CsCl, 10.0 CsOH-HEPES, 0.2 CsOH-EGTA, 2.0 Mg-ATP, 0.3 Na3-GTP, 8.0 NaCl, and 5.0 N-ethyl-lidocaine chloride (Abcam), pH 7.2–7.4, osmolarity of 295–305 mOsm (Murray et al., 2011). Subicular pyramidal neurons were visually identified based upon their anatomical location and pyramidal morphology using an upright microscope (BX51, Olympus Deutschland) equipped with a 40× water-immersion objective, infrared light with differential interference contrast, and a digital camera. eIPSCs were recorded at a holding potential of −70 mV in response to electrical stimulation (10–35 μA; set to the minimum current required to evoke IPSCs with maximal amplitude) delivered using a concentric, bipolar platinum/iridium electrode with a diameter of 2–3 μm (MicroProbes for Life Science) connected to a constant current stimulator (Digitimer). Brain slices were constantly perfused with aCSF (32–34°C) containing glutamate AMPA and NMDA receptor antagonists, 10 μm 6,7-dinitroquinoxaline-2,3-dione (DNQX) and 100 μm dl-2-amino-5-phosphonopentanoic acid (dl-AP5; Abcam), respectively, and 1 μm WIN 55,212-2 mesylate (Tocris Bioscience) was dissolved in aCSF immediately before recordings. Data were filtered at 2.9 kHz and sampled at 10 kHz with an EPC10 patch-clamp amplifier and analyzed using PatchMaster and FitMaster software (HEKA Electronic).
Patch-clamp electrophysiology of miniature IPSCs and EPSCs.
To record miniature IPSCs (mIPSCs) and miniature EPSCs (mEPSCs), we used 3- to 6-month-old male mice (n = 21) and a protective recovery method for slice preparation (Zhao et al., 2011). Briefly, mice were deeply anesthetized and perfused with 40 ml of ice-cold preoxygenated (95% O2/5% CO2) solution containing the following (in mm): 93 N-methyl-d-glucamin-HCl, 30 NaHCO3, 2.5 KCl, 1.2 NaH2PO4, 20 HEPES-NaOH, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2, 8 MgSO4, and 25 glucose, pH 7.4.
Next, brains were rapidly extracted and immersed in the same solution. Subsequently, 300-μm-thick horizontal hippocampal slices were cut on a vibratome (VT1200S, Leica) and transferred to a recovery chamber filled with the same solution (at 32°C) for 12 min. Thereafter, slices were kept (for at least 60 min before the recordings) in a solution containing the following (in mm): 90 NaCl, 26 NaHCO3, 3 KCl, 1.2 NaH2PO4, 20 HEPES-NaOH, 5 Na-ascorbate, 3 Na-pyruvate, 1.5 CaCl2, 2 MgSO4, 0.5 l-glutathione, and 25 glucose, pH 7.4.
For voltage-clamp recordings of mIPSCs, pipettes with a resistance of 3–4.5 MΩ contained the following (in mm): 120 K-gluconate, 6 KCl, 10 HEPES-KOH, 5 EGTA, 4 ATP-Mg, and 0.3 GTP, with pH was adjusted to 7.3 with KOH. For mEPCs recordings the pipette solution contained the following (in mm): 135 KCl, 1 MgCl2, 10 HEPES-KOH, 1 EGTA, 2 Mg-ATP, and 0.3 GTP, at pH 7.4. To record mIPSCs and mEPSCs, pyramidal neurons were voltage clamped at −70 mV in aCSF. The amplitude and frequency of mEPSs and mIPCs were analyzed from a 100 s trace recorded 5 min after the application of tetrodotoxin (1 μm) and 50 μm picrotoxin for mEPSC recordings or 25 μm NBQX plus 50 μm d-AP5 for mIPSCs recordings. Electrophysiological data were analyzed using Clampfit version 10.0 (Molecular Devices), the Mini Analysis Program (Synaptosoft), and SigmaPlot (Systat Software).
Immunohistochemistry.
These studies were performed after terminating recordings (6–8 weeks after vector injection) on free-floating, 4% paraformaldehyde-fixed, 30-μm-thick horizontal sections using indirect peroxidase labeling or by immunofluorescence as described previously (Sperk et al., 2012; Wood et al., 2016). The following antisera were used for immunohistochemistry: monoclonal rat anti-GFP (1:2000; catalog #04404-84, Nacalai Tesque (through GERBU Biotechnik); RRID:AB_10013361); rabbit anti-ΔFosB (1:2000; catalog #sc-48, Santa Cruz Biotechnology; RRID:AB_631515); rabbit anti-PV (1:15,000; catalog #PV 25, Swant; RRID:AB_10000344); rabbit anti-dynorphin (a gift from Dr. Philippe Ciofi, INSERM, Bordeaux, France; Sperk et al., 2012); rabbit anti-somatostatin (1:1000; Sperk and Widmann, 1985); rabbit anti-GABA (1:1000; catalog #A2052, Sigma Aldrich; RRID:AB_477652); and rabbit anti-RFP [1:1000; catalog #600-401-379, Rockland Anitbodies and Assays (through Sanova Pharma); RRID:AB_2209751]. The antibodies were characterized by the supplier or were validated by us in previous experiments by immunocytochemistry (Sperk and Widmann, 1985; Sperk et al., 2012; Wood et al., 2016). In brief, horizontal sections were incubated free floating with 10% normal goat or horse serum (Biomedica) in Tris-HCl-buffered saline (TBS; 50 mm), pH 7.2, containing 0.4% Triton X-100 (TBS-Triton) for 90 min, followed by incubation with the respective primary antisera (at room temperature for 16 h), followed by washing with TBS-Triton. Primary antibodies bound to the respective antigens were then visualized by incubation with horseradish peroxidase (HRP)-coupled secondary antibodies reacting host specific for the primary antiserum (1:250; goat anti-rabbit, catalog #P0448, Dako; RRID:AB_2617138; 1:500; donkey anti-rabbit, catalog #711035152, Jackson ImmunoResearch; RRID:AB_10015282; or 1:500; donkey anti-rat secondary antibodies, catalog #712035153, Jackson ImmunoResearch; RRID:AB_2340639) at room temperature for 150 min. After washing with TBS, HRP bound to the secondary antibodies was revealed with 0.05% diaminobenzidine (DAB; Fluka and Sigma-Aldrich Handels) and 0.005% H2O2 substrate. Sections were washed in TBS, mounted on slides, dehydrated in ethanol series, and coverslipped with Eukitt (Gröpl).
Double immunofluorescence was performed as described previously (Wood et al., 2016). Two sections per animal were anatomically matched to those from the other animals (n = 29) and processed for GABA and GFP, somatostatin and GFP, or PV and GFP. The same antibodies as described above were used at the same concentrations (at room temperature for 16 h).
For double labeling GFP and PV, the secondary reaction was performed by simultaneous incubation with a donkey anti-rat antibody coupled to Alexa Fluor 488 (1:500; Thermo Fisher Scientific) for GFP and with an HRP-coupled goat anti-rabbit antibody (1:250; catalog #P0448, Dako; RRID:AB_2617138) for PV at room temperature for 120 min. The HRP-coupled antibody was then further reacted with TSA-Cy3 (homemade; Lumiprobe; 1:100 in 50 mm PBS and 0.005% H2O2) at room temperature for 5 min.
Double labeling of GABA and GFP.
GFP was labeled in the same way as described above, and GABA was reacted with the HRP-coupled goat–anti-rabbit antibody (see above) at room temperature for 120 min. The HRP-coupled antibody was then incubated with TSA-AMCA (1:100 in 50 mm PBS, 0.02% H2O2; Thermo Fisher Scientific) at room temperature for 5 min. For concomitant labeling of somatostatin and GFP, we used the HRP-coupled donkey anti-rabbit antibody (1:500; catalog #711035152, Jackson ImmunoResearch; RRID:AB_10015282) and the anti-rat antibody coupled to Alexa Fluor 488 (1:500; Invitrogen), respectively, as secondary antibodies. The HRP-coupled donkey anti-rabbit antibody was then further reacted with TSA-Cy3 as described above.
For identifying expression sites of hM4Di in PV-containing interneurons after AAV-hM4Di injection, double labeling for the RFP tag of the hM4Di vector and for PV was performed also using the primary antibodies described above. As secondary antibodies biotinylated donkey anti-mouse (1:200; Vectastain PK 4002, Szabo Scandic; RRID:AB_2336811) and HRP-coupled goat anti-rabbit antibodies (1:250; catalog #P0448, Dako; RRID:AB_2617138) were used (at room temperature for 120 min). Biotinylated donkey anti-mouse antibodies were reacted with streptavidin (1:100; Dylight Streptavidin 649 SA 5649, both Vector Laboratories and Szabo Scandic; RRID:AB_2336421; at room temperature for 120 min) and goat anti-rabbit antibodies with TSA-Cy3 (homemade, Lumiprobe; 1:100 in 50 mm PBS, 0.005% H2O2; at room temperature for 5 min).
For double labeling of caspase 3 and GFP, we incubated three sections obtained at different levels of the ventral hippocampus (∼240 μm apart) from four AAV-TeLC mice injected with a rabbit anti-caspase 3 antibody (1:500; catalog #9661, Cell Signaling Technology; RRID:AB_2341188) together with the monoclonal rat anti-GFP antibody (1:2000; catalog #04404–84, Nacalai Tesque; RRID:AB_10013361) at room temperature for 16 h. For detection of the caspase 3 antibody, we used a biotinylated goat anti-rabbit antibody (1:200; Vectastain PK 4001, Szabo Scandic; RRID:AB_2336810; at room temperature for 120 min), which was then reacted with streptavidin (1:100; Dylight Streptavidin 649 SA 5649, RRID:AB_2336421, both Vector Laboratories and Szabo Scandic) at room temperature for 120 min. For detecting the GFP antibody, an Alexa Fluor 488-coupled antibody was used (1:500; Invitrogen; RRID:AB_221477) concomitantly with the biotinylated goat anti-rabbit antibody at room temperature for 120 min. We used hippocampal sections from mice unilaterally injected with kainic acid (350 pmol/70 nl; Jagirdar et al., 2015) as positive controls for identifying caspase 3-positive degenerating neurons (data not shown).
All double immunofluorescence-labeled sections were mounted on fluorescence-free glass slides and covered in 86% glycerol and 2.5% DABCO (D27802, Sigma-Aldrich) and analyzed by confocal microscopy.
Semiquantitative analysis of immunohistochemical data.
To determine the number of PV-labeled neurons in the subiculum of wild-type mice (n = 5) and of heterozygous PV-cre mice (n = 5), cell numbers in the intermediate to ventral subiculum were counted on 30-μm-thick horizontal sections reacted for PV (two matched sections per mouse) at 40× primary magnification. Values obtained from left and right hemispheres and from different sections were averaged and expressed as neurons per region (3473 neurons were analyzed).
For counting neurons coexpressing TeLC/GFP together with GABA, PV, or somatostatin at the site of AAV vector injection, microphotographs of 30-μm-thick horizontal double immunofluorescence-labeled sections (29 mice, two sections per mouse) were taken at 20× primary magnification using a fluorescence microscope (Imager.M1, Carl Zeiss). Images were imported into NIH ImageJ 1.51d (National Institutes of Health, Bethesda, MD), and photographs of the individual channels were displayed side by side. The area evaluated was defined by the region containing GFP-positive neurons (directly affected by the vector injection). Using the Cell Counter plugin, the numbers of single-labeled and double-labeled cells were determined. In total, 10,590 labeled neurons (GFP, 2724; GABA, 4966; PV, 1114; somatostatin, 1786) were analyzed for identifying colabeling with other markers.
To identify neurons stimulated during spontaneous seizures, we labeled sections for ΔFosB accumulating in activated neurons (Morris et al., 2000). To determine numbers of intensely labeled ΔFosB-expressing neurons, 8-bit microphotographs of individual subregions of the in the hippocampal formation were taken from Ni-DAB-stained, 30-μm-thick horizontal immune-labeled sections (two sections per mouse, 34 mice in total) at 10× magnification and constant illumination. Microphotographs were imported into NIH ImageJ, an unspecific background (measured in the alveus or fimbria) was measured and subtracted from the gray values of the pixels, and a common threshold was set for all images to obtain binary images displaying black neurons on white background. The ImageJ program function Analyze Particles was used to determine the numbers of intensely labeled neurons in the outlined regions of interest (parameters: size, 5–25 μm; circularity, 0.3–1). Due to the dense packing of granule cells in the dentate gyrus, these neurons were counted manually. The area of each outlined region was measured, and the number of cells per square millimeter were then calculated. Values from both hemispheres and individual sections were averaged.
Statistical analyses.
All statistical analyses were performed using GraphPad Prism statistical software (version 5.0f; GraphPad Software). The Fisher's exact test was used for analysis of the effect of low-dose PTZ in controls (AAV-GFP) and AAV-TeLC-injected mice. The Kruskal–Wallis test with Dunn's multiple-comparison post hoc test was used for comparing multiple groups with one control group (evaluation of ΔFosB-positive neurons). Two-way repeated-measures ANOVA was used to analyze electrophysiological data, and an unpaired Student's t test was used for analyzing mean differences between two groups. A p value of <0.05 was considered as statistically significant.
Results
TeLC expression is selective for PV interneurons
AAV-TeLC injection resulted in TeLC expression in 61.4 ± 2.43% of PV-containing interneurons at the injection site in the subiculum (total number of neurons counted, 1114 PV cells; Fig. 1A,C,G–L). Variable expression of TeLC was also detected in the sector CA1, presubiculum, and parasubiculum and occasionally in parts of the entorhinal cortex and dentate gyrus (Fig. 1A). Double immunofluorescence labeling for GFP (the tag for TeLC) was evaluated in 25 mice (total number of GFP-positive neurons, 2724) and revealed 95.7 ± 0.68%, 83.2 ± 1.27%, and 23.7 ± 1.61% colabeling with GABA (Fig. 1D–F), PV (Fig. 1G–L), or somatostatin (data not shown), respectively (Fig. 1M), indicating a high specificity of transfection/viral transduction for the PV-containing subpopulations of GABAergic basket and axo-axonic cells. Approximately 15–30% of PV-expressing neurons also contain somatostatin (Jinno and Kosaka, 2000; Klausberger and Somogyi, 2008; Fig. 1M), which is supportive of the quantitative data ratio we obtained above. These neurons represent a subpopulation of O-LM and bistratified cells (Fig. 1O) coexpressing PV; they were therefore also affected by the TeLC vector. In reverse, the majority of PV-expressing neurons expressed TeLC (60%), whereas only minor populations of GABA and of somatostatin-immunoreactive neurons expressed TeLC (∼18% and 15%, respectively; Fig. 1N). This again underlines the specificity of our experimental manipulation using the TeLC vector. Considering that majorities of GABA and of somatostatin neurons do not contain PV, their lack of labeling confirms the specificity of vector delivery.
Figure 1.

AAV-TeLC induces selective expression of TeLC in PV-containing interneurons in the subiculum of PV-cre mice. A, Distribution of TeLC (immunoreactivity for the GFP tag) after unilateral injection of AAV-TeLC into the subiculum of PV-cre mice and after EEG monitoring for up to 8 weeks. B, C, The overall distribution of PV-containing interneurons and fibers in the subiculum of C57BL/6N WT mice (B) is similar to the distribution of TeLC-expressing neurons in AAV-TeLC-injected mice (C). A–C, Representative images for WT mice (B; n = 5) and AAV-TeLC-injected mice (A, C; n = 25). See Table 1 for regional distribution of TeLC expression in individual animals. D–L, TeLC is specifically expressed in PV-containing interneurons. D–F, Only a subpopulation of GABA neurons (blue) expressed TeLC (green; white arrows in F indicate double-labeled neurons). The majority of GABA-containing neurons did not express TeLC (red arrows in F). G–L, The majority of PV-containing neurons were positive for TeLC/GFP at the site of AAV-TeLC injection, shown at high magnification (G–I) and low magnification (J–L; white arrows indicate some of the double-labeled cells). M, N, Cell counts of double-labeled cells (in total 2724 GFP-positive neurons were evaluated in 25 mice). M, More than 95% of TeLC-expressing cells contain GABA, 80% PV and ∼20% also somatostatin (interneurons containing somatostatin and PV; Jinno and Kosaka, 2000). N, In reverse, ∼60% of PV neurons express TeLC at the injection site, whereas <20% of GABA-expressing neurons and ∼15% of SOM-expressing interneurons are TeLC/GFP positive. O, Scheme of PV-containing interneurons in the subiculum. The major population comprises PV-containing basket cells forming perisomatic synapses on pyramidal cells (PCs). Minor populations are axo-axonic cells forming synapses on PC axon initial segments and subpopulations of somatostatin-containing O-LM cells and bistratified cells mediating feedback inhibition through PC dendrites. DG, Dentate gyrus; PaS, parasubiculum; PrS, presubiculum; Sub, subiculum; oml, outer molecular layer; iml, inner molecular layer; pcl, pyramidal cell layer. The dashed line in B indicates the border between the subiculum and sector CA1. Scale bars: A, 600 μm; C (for B, C), 300 μm; I (for D–I), 25 μm; L (for J–L), 100 μm.
Electrophysiology
To verify a loss in perisomatic inhibition of pyramidal cells by TeLC injection, we used whole-cell voltage-clamp recordings in slices of the subiculum 2–3 weeks after vector injection. We observed only an insignificant reduction in eIPSCs but no change in either mIPSCs or mEPSCs (Fig. 2). However, when we applied the CB1 receptor agonist WIN 55,212–2 to suppress GABA release from non-PV, cholecystokinin (CCK)-containing basket cells we observed, consistent with a previous report on CA1 neurons (Murray et al., 2011), that the amplitude of the WIN 55,212–2-insensitive component of the eIPSC (representing inhibition mediated by PV-containing basket cells) was significantly reduced in the subiculum of AAV-TeLC-injected mice compared with AAV-GFP-injected controls (Fig. 2A–E; treatment: F(1,20) = 4.72, p < 0.05; time: F(8,160) = 7.74, p < 0.01; interaction: F(8,160) = 0.82, p > 0.05). Together, our experiments show a significant reduction in eIPSC caused by silencing of PV neurons, while the inhibitory tone of other interneurons (notably that of CCK-containing basket cells) is not affected. Importantly, in addition neither mEPSCs nor mIPSCs are altered after vector injection.
Figure 2.

TeLC expression in PV-containing interneurons attenuates inhibitory input at synapses on pyramidal neurons of the subiculum. To quantify the loss of inhibition, 2–3 weeks after AAV-TeLC or AAV-GFP injection (time when C-SWDs had become manifest), whole-cell voltage-clamp recordings from pyramidal neurons of the subiculum were obtained and eIPSCs were recorded in the presence of glutamate receptor antagonists (DNQX, 10 μm; dl-AP5, 100 μm). In the subiculum, eIPSCs originate from various types of local interneurons, including CCK- and PV-containing neurons. To partially isolate the eIPSC component from PV-containing neurons, the CB1 agonist WIN 55,212–2 (1 μm) was bath applied to selectively suppress GABA release from CCK-containing neurons (known to express the CB1 receptor), as previously described for pyramidal neurons of sector CA1 (Glickfeld et al., 2008; Murray et al., 2011). A, B, The baseline maximal amplitude of eIPSCs and the stimulation intensity required to evoke maximal responses was not different between groups (p = 0.286 and p = 0.651, respectively; t test). C, Two-way repeated-measures ANOVA revealed that the amplitude of eIPSCs in the presence of WIN 55,212–2 was significantly reduced in AAV-TeLC-injected mice compared with AAV-GFP-injected mice (treatment: F(1,20) = 9.83, p = 0.0052; time: F(8,160) = 5.97, p < 0.0001). D, Importantly, the WIN 55,212-2-insensitive component of the eIPSC was significantly larger in mice injected with AAV-GFP compared with those injected with AAV-TeLC (treatment: F(1,20) = 4.72, p = 0.0419; time: F(8,160) = 7.74, p < 0.0001). E, Specifically, WIN 55,212–2 reduced the amplitude of eIPSCs by 33% in AAV-GFP-injected mice, which is consistent with previous reports (Glickfeld et al., 2008), while mice injected with AAV-TeLC exhibited a 59% reduction in the amplitude of eIPSCs. Thus, TeLC expression in PV-containing neurons results in a significant reduction of inhibitory input to subicular pyramidal neurons (n = 10 cells from 6 AAV-TeLC-injected mice; n = 12 cells from 5 AAV-GFP-injected mice). Values are given as the mean ± SEM. F, The representative traces of the mIPSC recordings of subiculum pyramidal neurons from AAV-GFP (black) and AAV-TeLC-injected (gray) mice. G, Visualization of subiculum pyramidal neurons (magenta color) after patch clamping (arrows) with a solution containing biocytin. PV-positive neurons in the subiculum are visible in green because of stereotaxic injection of a Cre-driven AAV-GFP construct. H–I, Analysis of mEPSC (H) and mIPSC (I) amplitudes (H, I, left) and frequencies (H, I, right) in subiculum pyramidal neurons from Pvalbtm1(cre)Arbr mice injected with AAV-GFP (black bars, n = 10 cells for mEPSC recordings; n = 9 cells for mIPSC recordings) and AAV-TeLC (red bars, n = 11 cells for mEPSC recordings; n = 10 cells for mIPSC recordings). Data are not significantly different between the two experimental groups.
Development of recurrent C-SWDs and SRSs after AAV-TeLC injection
We then performed telemetric EEG recordings for up to 57 d in freely moving mice injected with AAV-TeLC (n = 25) or AAV-GFP (n = 9) into the subiculum (Fig. 3). At day 16, 22 of the 25 mice (88%) exhibited unprovoked C-SWDs (Figs. 3B,D). On average, these C-SWDs consisted of 42.1 ± 3.14 SWDs (Fig. 3G) and probably reflect a preictal state or subthreshold epileptic activity (Table 1). Among these mice, 16 (representing 64% of all AAV-TeLC-injected mice or 72.7% of mice with C-SWDs) developed SRSs (range, 1–19; median, 5.5 SRSs/30 d; Fig. 3E,F,H). SRSs developed in parallel with C-SWDs, although six mice did not present with SRSs by day 42 after AAV-TeLC injection despite showing C-SWDs (Fig. 3B). As summarized in Table 2, these SRS were mostly (89.5% of 102 total SRSs recorded) preceded by preictal C-SWDs (Fig. 3E), had a mean duration of 24.6 ± 0.62 s, and were followed by postictal depression of the EEG (Fig. 3F,H). None of the AAV-GFP-injected mice exhibited C-SWDs or SRSs during the 6–8 week recording period (Fig. 3C). Three mice (12%) developed neither C-SWDs nor SRSs (Fig. 3B).
Figure 3.
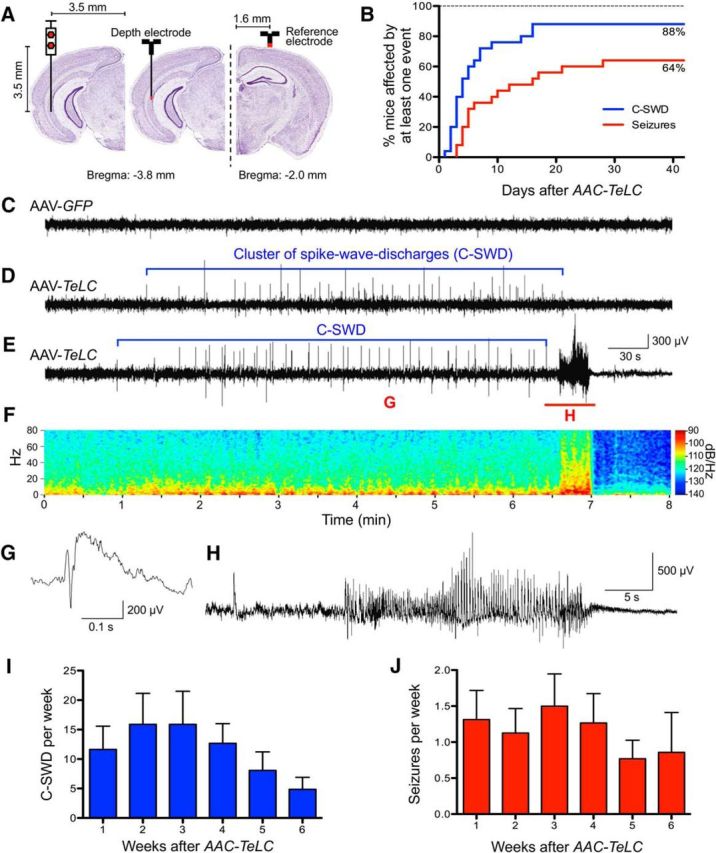
Permanent silencing of PV neurons by unilateral AAV-TeLC injection into the ventral subiculum of PV-cre mice results in SRSs. A, Scheme of electrode positions and anatomical site of vector injection. B, Cumulative presentation of C-SWDs and of SRSs. Eighty-eight percent of mice developed C-SWDs by day 16, and 64% of mice presented at least one SRS by day 28. C–E, Representative 8 min EEG traces obtained in mice 18 d after AAV-GFP (C) or AAV-TeLC (D, E) injection. Whereas AAV-GFP-injected mice showed an unchanged EEG, AAV-TeLC-injected mice presented either C-SWDs only (D) or C-SWDs and SRSs (16 of 25 mice; E). The intermittent C-SWDs lasted ∼5 min and were detected in 22 of 25 AAV-TeLC-injected mice (D). E, Approximately 90% of SRSs occurred immediately after preictal C-SWDs. F, Magnitude spectrum (0–80 Hz, logarithmic power scale) of the EEG recording in E. Note the profound and long-lasting post-seizure depression of EEG amplitude in all frequency bands. G, H, High-resolution EEG traces of a preictal single SWD (G, duration, ∼300 ms; marked as “G” in E) and of a spontaneous tonic-clonic seizure (H; marked as “H” in E). E, F, H, EEG seizures lasted ∼25 s and were always accompanied by generalized tonic–clonic motor seizures with loss of posture (stage 3 to 4). I, J, Mean C-SWDs (±SEM; I) and mean SRSs (±SEM) presented per week (J). Note the reduction in C-SWDs and seizure frequencies after 5–6 weeks.
Table 1.
Summary of mice injected with AAV-TeLC or with AAV-GFP, the anatomical distribution of TeLC expression, and the total number of seizures observed
| No. | Injection: AAV-FLEX- | Sub/CA1 | CA3 | PrS | PaS | DG | EC | C-SWD | Number of seizures | Days recorded |
|---|---|---|---|---|---|---|---|---|---|---|
| Group 1: AAV-FLEX-TeLC injected, no seizures | ||||||||||
| 1 | TeLC | ++ | + | ++ | ++ | + | ++ | − | 0 | 28 |
| 2 | TeLC | ++ | + | − | 0 | 32 | ||||
| 3 | TeLC | +++ | ++ | − | 0 | 48 | ||||
| 4 | TeLC | +++ | + | ++ | +++ | ++ | ++ | + | 0 | 55 |
| 5 | TeLC | +++ | +++ | + | ++ | +++ | + | 0 | 56 | |
| 6 | TeLC | ++ | + | 0 | 48 | |||||
| 7 | TeLC | ++ | + | 0 | 55 | |||||
| 8 | TeLC | +++ | ++ | ++ | +++ | +++ | ++ | + | 0 | 56 |
| 9 | TeLC | ++ | + | ++ | + | + | 0 | 55 | ||
| Group 2: AAV-FLEX-TeLC injected, with seizures | ||||||||||
| 10 | TeLC | +++ | + | + | + | 1 | 35 | |||
| 11 | TeLC | ++ | + | ++ | ++ | ++ | + | 3 | 56 | |
| 12 | TeLC | +++ | + | +++ | + | + | + | 19 | 47 | |
| 13 | TeLC | ++ | ++ | +++ | +++ | ++ | + | 5 | 30 | |
| 14 | TeLC | +++ | + | + | 2 | 47 | ||||
| 15 | TeLC | +++ | + | ++ | + | 5 | 47 | |||
| 16 | TeLC | +++ | + | + | 2 | 18 | ||||
| 17 | TeLC | +++ | + | + | + | + | 6 | 32 | ||
| 18 | TeLC | +++ | + | ++ | +++ | +++ | + | 12 | 56 | |
| 19 | TeLC | ++ | ++ | + | 9 | 30 | ||||
| 20 | TeLC | +++ | + | + | + | 9 | 40 | |||
| 21 | TeLC | ++ | + | + | 6 | 30 | ||||
| 22 | TeLC | +++ | + | ++ | + | 9 | 25 | |||
| 23 | TeLC | ++ | + | 2 | 38 | |||||
| 24 | TeLC | ++ | + | 5 | 28 | |||||
| 25 | TeLC | +++ | + | ++ | + | 7 | 34 | |||
| Group 3: AAV-FLEX-GFP injected | ||||||||||
| 26 | GFP | ++ | + | +++ | − | 0 | 56 | |||
| 27 | GFP | ++ | + | + | +++ | +++ | +++ | − | 0 | 55 |
| 28 | GFP | +++ | ++ | +++ | + | + | − | 0 | 47 | |
| 29 | GFP | n.e. | − | 0 | 55 | |||||
| 30 | GFP | n.e. | − | 0 | 43 | |||||
| 31 | GFP | +++ | ++ | +++ | +++ | ++ | ++ | − | 0 | 57 |
| 32 | GFP | ++ | − | 0 | 54 | |||||
| 33 | GFP | ++ | + | − | 0 | 52 | ||||
| 34 | GFP | ++ | ++ | ++ | +++ | ++ | + | − | 0 | 52 |
| Group 4: AAV-FLEX-TeLC injected and 10 d later with PTZ | ||||||||||
| 35 | TeLC + PTZ | ++ | + | + | 14 | 28 | ||||
| 36 | TeLC + PTZ | ++ | + | 6 | 53 | |||||
| 37 | TeLC + PTZ | ++ | + | + | + | 43 se | 34 | |||
| 38 | TeLC + PTZ | ++ | + | + | 9 | 53 | ||||
| 39 | TeLC + PTZ | ++ | + | 9 | 52 | |||||
| 40 | TeLC + PTZ | ++ | + | + | 7 | 52 | ||||
Injections were performed unilaterally into the ventral subiculum, and mice were subjected to telemetric EEG and video monitoring. Group 1 (1–9), Mice that revealed no spontaneous seizures, but (except 1–3) C-SWDs. Six of these mice (4–9) that had experienced C-SWDs but were seizure free at day 42 (Fig. 3) were then injected with a threshold dose of PTZ resulting in acute and then recurrent spontaneous seizures (Fig. 6). Group 2 (10–25), 16 of 25 AAV-TeLC-injected mice (10–25) revealed spontaneous recurrent seizures and C-SWDs. Group 3 (26–34), none of nine AAV-GFP-injected mice exhibited recurrent seizures and were considered as controls. The fact that these mice also did not present C-SWDs argues against an unspecific effect of vector injection. Group 4 (35–40), these mice were injected with AAV-TeLC and after 10 d with PTZ. They were seizure free after AAV-TeLC injection but exhibited acute and then spontaneous recurrent C-SWDs and seizures after PTZ injection. Mouse 37 died from a status epilepticus on day 34. Expression of GFP (tag for AAV-TeLC) was most prominent in the subiculum/sector CA1, often extending to the presubiculum and parasubiculum, and, in some instances to the entorhinal cortex or to the dentate gyrus. se, Status epilepticus; PrS, presubiculum; PaS, parasubiculum; EC, entorhinal cortex; DG, dentate gyrus; n.e., not evaluated.
Table 2.
Characteristics of spontaneous recurrent seizures and C-SWDs
Spontaneous recurrent seizures
|
C-SWDs
|
Data are presented as the mean ± SEM.
Importantly, all SRSs recorded by EEG were accompanied by motor seizures. Video recordings revealed that 93% of EEG seizures could also be evaluated for the behavior of the animals. Among these, 100% of mice revealed generalized limbic motor seizures with rearing (stage 3–4).
Figure 3, I and J, shows the frequencies of C-SWDs and of SRSs during the 6 week period after AAV-TeLC injection, respectively. Although the occurrence of both C-SWDs and SRSs varied between animals, it was relatively equal, on average amounting to 1.5 seizures and 15 C-SWDs per animal and per week. Notably, the frequency of C-SWDs and SRSs tended to decline by weeks 5 and 6.
Neither AAV-TeLC injection nor recurrent seizures caused neurodegeneration
After EEG monitoring (6–7 weeks after vector injection), the mice were killed and their brains processed by immunohistochemistry for PV, GABA, somatostatin, caspase 3, ΔFosB, or by Nissl staining. Importantly, cell counts at the injection site revealed neither a loss in PV-containing neurons (expressing TeLC) nor in GABA-immunoreactive neurons in AAV-TeLC-injected mice that had presented only with C-SWDs or C-SWDs and SRSs compared with uninjected controls or AAV-GFP-injected mice (Fig. 4A,B). Since immunohistochemistry was performed after concluding EEG monitoring (6–8 weeks after vector injection; Table 1), also cell losses induced by SRSs can be excluded. Only a few caspase 3-positive neurons were seen in close vicinity to the needle track 3 and 10 d after AAV-TeLC or AAV-GFP injection (Fig. 4F), indicating minimal neurodegeneration due to the injection procedure per se. Furthermore, no changes in Nissl staining (Figs. 4C,D) or mossy fiber sprouting, an indicator for seizure-induced loss of mossy cells in the dentate hilus (Houser et al., 1990; Pirker et al., 2001; Fig. 4E), were observed in any of the AAV-TeLC-injected mice. Together, these data indicate that neither AAV-TeLC injections per se nor chronic silencing or SRSs caused damage of the transfected PV neurons, despite inhibiting the release of GABA.
Figure 4.
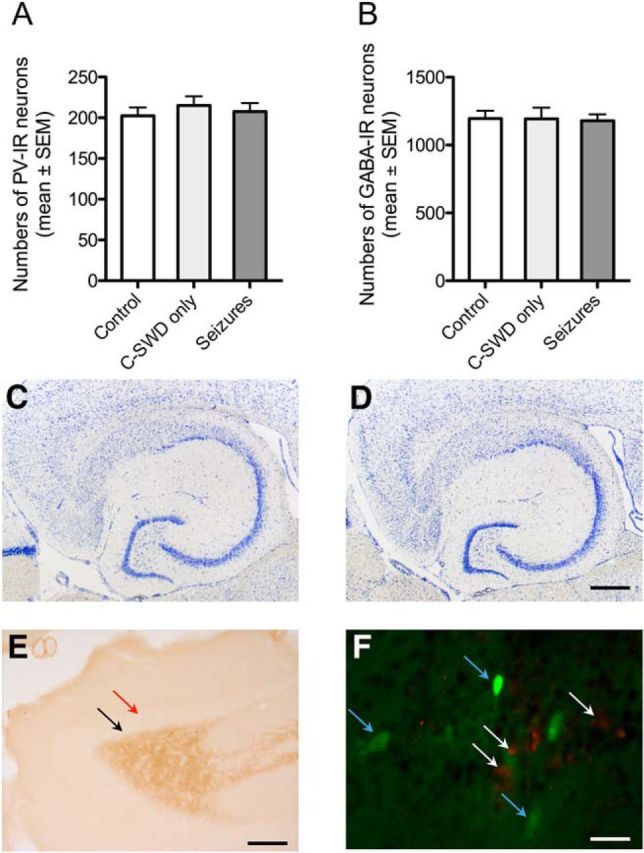
AAV-TeLC injection or related seizures did not induce signs of neurodegeneration. A, B, AAV-TeLC did not affect cell numbers of PV neurons at the injection site. Numbers (mean ± SEM) of PV-positive and GABA-positive neurons were reduced neither in the subiculum of mice that had experienced C-SWD only (n = 6) nor in mice presenting SRSs after AAV-TeLC injection (n = 16). Cell counts were performed 6–8 weeks after AAV-TeLC injection (and monitoring). Numbers are shown for the injected subiculum; they did not differ from those in the contralateral subiculum (data not shown). Controls: uninjected mice (n = 6); data were not different from those for AAV-GFP-injected mice (n = 9; data not shown). C, D, Representative Nissl stains in brain slices at the level of the ventral hippocampus of mice with SRS after AAV-TeLC injection (n = 16; C), and after AAV-GFP injection (seizure free; n = 9; D). E, To test for possible mossy fiber sprouting as a consequence of loss in highly vulnerable mossy cells, we labeled hippocampal mossy fibers by immunohistochemistry for dynorphin. Although mossy fibers were strongly positive for dynorphin, no immunoreactivity was observed in the inner molecular layer of the dentate gyrus, which would be indicative for sprouted mossy fibers (red arrow in E) in AAV-TeLC-injected mice that had experienced SRSs for 6–8 weeks. The black arrow (E) indicates the area of the granule cell layer that is also devoid of dynorphin immunoreactivity (representative for 16 mice). F, Double labeling for the apoptosis marker caspase 3 (red cells, white arrows) and for TeLC/GFP (green cells, blue arrows) at the injection site of AAV-TeLC 10 d after injection. Labeling was performed in three horizontal sections obtained at different dorsoventral levels (∼240 μm apart from each other) from four mice killed 3 or 10 d after AAV-TeLC injection. Zero to maximally five apoptotic cells per section were detected close to the needle track. Note the intact GFP-positive cells expressing TeLC but not caspase 3 in close vicinity to presumably apoptotic cells. As positive controls, we investigated caspase 3 expression in the dorsal hippocampus of mice injected locally with kainic acid 2 and 10 d before (Jagirdar et al., 2015) and observed caspase 3-positive cells at both intervals (data not shown). Scale bars: D (for C, D), 500 μm; E, 200 μm; F, 20 μm.
To determine the anatomical sites of seizure propagation, we investigated expression of the transcription factor ΔFosB after EEG monitoring. ΔFosB accumulates in neurons activated by seizure activity (Morris et al., 2000). Whereas ΔFosB was only barely expressed in controls and in mice that exhibited only C-SWDs (Fig. 5A,D), it prominently labeled neurons in the hippocampus, subiculum, and entorhinal and perirhinal cortices after frequent SRSs (Fig. 5B,C,E–J), corroborating a role for these temporal lobe areas in seizure propagation.
Figure 5.

Expression of ΔFosB in the hippocampal formation after spontaneous seizures induced by AAV-TeLC injection. To identify neurons stimulated during spontaneous seizures, sections of the ventral hippocampus were obtained after EEG monitoring (up to 8 weeks after AAV-TeLC or AAV-GFP injection) and labeled for ΔFosB-immunoreactivity, a cellular marker accumulating in activated neurons (Morris et al., 2000). A, D, Only very faint ΔFosB labeling was detected in some neurons of the hippocampal formation (A), including the subiculum (D; higher magnification) in AAV-GFP-injected mice (no EEG alterations) and in AAV-TeLC-injected mice with C-SWDs but no SRSs (data not shown). B, C, E, F, ΔFosB was clearly expressed in the neurons of mice that experienced one to six SRSs (B) and high numbers of ΔFosB-positive neurons were present throughout the hippocampal formation and the deep and superficial layers of the entorhinal and perirhinal cortices of mice that had experienced between 9 and 19 SRSs after AAV-TeLC (C, E, F). E, Higher magnification of the subiculum of these mice. C, F, Note the extremely strong expression of ΔFosB in the granule cell layer of these mice (C), sometimes particularly concentrated in the inner and outer portions of the granule cell layer (F). G–J, Semiquantitative estimates of ΔFosB-positive cells using the ImageJ program were done in blinded fashion and are depicted in sector CA3 (G), granule cells of the dentate gyrus (H), the subiculum (I), and layers V to VI of the perirhinal cortex after injection of AAV-GFP (GFP; n = 10) or AAV-TeLC (TeLC; J). AAV-TeLC-injected mice had experienced at most C-SWDs (TeLC sw; n = 9) or 1–6 (TeLC rs+; n = 10) or more frequent (9–19) SRSs (TeLC rs++, n = 5). Scale bars: C (for A–C), 500 μm; F (for D–F), 100 μm. Similar increases in numbers of ΔFosB-positive neurons were also obtained for sector CA1, the superficial layers of the perirhinal cortex (PRC), and the deep and superficial entorhinal cortices (data not shown). Statistical analyses were conducted using the Kruskal–Wallis test with Dunn's multiple-comparison post hoc test (all groups compared with controls; *p < 0.05; **p < 0.01; ***p < 0.001 vs AAV-GFP group). Values are given as the mean ± SEM. CA1 and CA3, Hippocampal sectors CA1 and CA3; DG, dentate gyrus; EC, entorhinal cortex; PRC, perirhinal cortex; Sub, subiculum.
Seizure threshold is decreased also in mice exhibiting C-SWDs only
We then hypothesized that mice presenting C-SWDs but no SRSs may already have a lowered seizure threshold and thus be prone to epileptic seizures. To test this, we injected six mice that had been seizure free for 6 weeks (despite presenting C-SWDs: 1–27 per 6 weeks; mean, 11.5 ± 4.87 seizures; n = 69), with a threshold dose of the GABAA receptor antagonist PTZ (30 mg/kg, i.p.). This treatment provoked acute tonic–clonic seizures in all TeLC vector-injected mice, but only in one of seven AAV-GFP-injected controls and in none of five WT mice (p = 0.0047; Fig. 6A–C). The EEGs of the mice was monitored for an additional 14 d. Importantly, during this time all mice revealed recurrent C-SWDs (data not shown) and SRSs (0.8 ± 0.25 per mouse and per week; Fig. 6A).
Figure 6.

The seizure threshold is reduced in mice exposing C-SWDs but not SRSs after AAV-TeLC. Seizure threshold was tested with a threshold dose of PTZ (30 mg/kg, i.p.) in PV-cre mice that did not develop SRSs after AAV-TeLC. A, PTZ induces acute (red bar) and then recurrent seizures and C-SWDs (data not shown) in mice that were seizure free for 6 weeks after AAV-TeLC. Bars represent the mean number of seizures (±SEM) per day. The red bar depicts acute PTZ-induced seizures, and the gray bars show SRSs. B, Representative EEG traces after PTZ in mice initially injected with AAV-GFP (top trace) and AAV-TeLC (bottom trace), respectively. C, After PTZ, seizures were observed in all AAV-TeLC-injected mice (n = 6) but in only 1 of 7 AAV-GFP-injected controls. D, Injection of a threshold dose of PTZ in still seizure-free mice (n = 6) 10 d after AAV-TeLC provoked one acute seizure (red bar) and subsequently SRSs (gray bars) and C-SWDs (data not shown), but not in AAV-GFP-injected controls (data not shown).
The seizure threshold is decreased already shortly after silencing of PV-containing interneurons
From these experiments, the question arose of whether the decrease in seizure threshold was a consequence of silencing PV-containing interneurons or was induced (as in kindling) by repeated C-SWDs experienced over a prolonged period of time. We therefore challenged another group of six mice (still seizure free and only 2.3 ± 0.84 C-SWD/mouse/10 d) with the threshold dose of PTZ already 10 d after TeLC vector injection (at this interval, the vector had already resulted in full expression of TeLC). All mice responded with acute and thereafter with recurrent seizures (mean 9.0 ± 3.55/30 d, n = 6; Fig. 6D). This demonstrates that the inhibition of GABA release from PV-containing interneurons of the subiculum/CA1 per se is sufficient to lower the seizure threshold and also induces a preictal state in mice that exposed (only a few) C-SWDs.
Transient inhibition of PV-expressing interneurons is not sufficient to induce acute or spontaneous seizures
At this point, we asked whether already transient inhibition of GABA release from PV-containing interneurons would be sufficient to induce C-SWDs and SRSs. We therefore injected PV-cre mice into the ventral subiculum with AAV vector-expressing hM4Di receptors (AAV-hM4Di), which are exclusively activated by designer drugs (DREADD; Roth, 2016). This treatment resulted in specific expression of the inhibitory hM4Di-DREADD in PV neurons (Fig. 7A–E). Fifteen days later, we temporarily inhibited virally transduced PV-containing interneurons by injecting the mice with CNO. Neither hM4Di expression in PV neurons per se nor transient silencing of PV neurons at the injection site for a period as short as 1–2 h induced C-SWDs or SRSs during the subsequent 5 d (n = 7; Fig. 7F). This confirmed that only sustained, but not transient, inhibition of GABA release from PV-containing neurons is capable of inducing seizures/epilepsy.
Figure 7.

Only transient inhibition of PV neurons in the subiculum. A, Distribution of hM4Di/RFP expressed after AAV-hM4Di injection. B, hM4Di is found in neurons and dendrites at the injection site. C–E, At the injection site, the majority of PV neurons was also labeled for RFP (a tag for hM4Di). F, PV-cre mice injected with AAV-hM4Di (n = 7) were treated after 12 d with saline (not indicated) and after 15 d with CNO (10 mg/kg, i.p.). Neither treatment resulted in acute or spontaneous seizures (EEG recordings for 5 d after CNO). G, To investigate whether CNO treatment was efficient, we injected additional 12 mice with CNO or saline on day 15 after AAV-hM4Di and 45 min later with a threshold dose of PTZ (30 mg/kg, i.p.). EEGs were monitored for a further 24 h. All CNO- and PTZ-injected mice (n = 6) showed acute C-SWDs (4.2 ± 1.14 per 24 h) and two mice showed acute seizures (one and three seizures, respectively) during the initial 2 h, indicating that GABAergic transmission is reduced after CNO injection (right bars). Six AAV-hM4Di-injected mice (n = 6) treated concomitantly with saline (instead of CNO) and later with PTZ revealed neither C-SWDs nor seizures (left bars). CA1 and CA3, Hippocampal sectors CA1 and CA3; DG, dentate gyrus; EC, entorhinal cortex; Sub, subiculum. Scale bars: A, 500 μm; E (for B–E), 25 μm.
To verify that GABA release was indeed transiently inhibited after injecting CNO into the transgenic mice expressing hM4Di, we injected another group of mice 15 d after AAV-hM4Di injection with CNO (n = 6) or with saline (n = 6) and 45 min later with a threshold dose of PTZ (30 mg/kg), and recorded their EEGs for an additional 24 h. Now, all six mice injected with CNO and PTZ presented C-SWDs (4.2 ± 1.14/24 h) and two of six mice showed acute convulsions after PTZ injection (Fig. 7G). In contrast, none of the AAV-hM4Di mice injected with saline (instead of CNO) revealed C-SWDs or seizures upon PTZ injection. This experiment documented that CNO indeed had transiently impaired GABA release. Thus, the inhibition of PV neurons in the ventral subiculum/CA1 by CNO may transiently lower seizure threshold but is per se not sufficient to induce acute convulsions.
Discussion
Our present experiments build on previous findings reporting selective losses in PV-containing interneurons in the subiculum of patients with TLE and in animal models of TLE (Andrioli et al., 2007; Knopp et al., 2008; Drexel et al., 2011). We now directly interfered with the activity of PV-containing basket cells, a central component of the hippocampal circuitry mediating feedforward inhibition upon pyramidal cells. We demonstrate that anatomically restricted inhibition of GABA release from PV-containing interneurons in the subiculum, notably without signs of neurodegeneration, induces recurrent C-SWDs that progress to SRSs in ∼73% of the mice. Recurrent C-SWDs reflect a permanent decrease in seizure threshold since a threshold dose of the GABAA receptor antagonist PTZ already has precipitated acute convulsions followed by SRSs (epilepsy). Importantly, in contrast to the permanent inhibition of GABA release from PV-containing neurons (by expressing TeLC), only transient silencing (for ∼1 to 2 h) of the same neurons using DREADD (Roth, 2016; Wirtshafter and Stratford, 2016) was not sufficient to induce SRSs, clearly documenting that only prolonged silencing of PV/GABA neurons results in SRSs or a preictal state in which recurrent seizure activity can be provoked.
AAV-TeLC injections into the subiculum selectively targeted PV-containing interneurons as revealed by double-label immunohistochemistry. The injections were not exclusively restricted to the subiculum, but often also affected some PV-containing interneurons in sector CA1 and the presubiculum and parasubiculum or, in a few cases, the dentate gyrus. Therefore, we cannot exclude a contribution of PV neurons silenced to some extent also in other hippocampal subfields. The slight variations in targeting individual areas of the hippocampus were due to the anatomical situation apparent when injecting into the ventral hippocampus. We have chosen the ventral hippocampus as a target area because of the much larger presentation of the subiculum in the temporal part of the hippocampus and the presumed higher relevance of the ventral hippocampus for epileptogenesis (Becker et al., 1997).
As expected, TeLC expression was found in several classes of interneurons of the subiculum known to contain PV (Fig. 1O). The most prominent population comprises PV-containing basket cells forming perisomatic synapses on pyramidal cells (Lee et al., 2014). However, axo-axonic cells targeting the axon initial segment of pyramidal cells and somatostatin-containing O-LM cells and bistratified neurons, both impinging on pyramidal cell dendrites (Klausberger and Somogyi, 2008), also contain PV and were therefore affected by the AAV-TeLC injections. The major populations of neurons expressing TeLC, however, were basket and axo-axonic cells, and only ∼15% of the somatostatin-positive O-LM and bistratified cells expressed TeLC.
Fast-spiking PV-containing basket cells exert extremely potent, but presumably transient feedforward inhibition upon the perisomatic region of pyramidal cells, whereas O-LM and bistratified cells mediate enduring feedback inhibition on pyramidal cell dendrites (Freund and Katona, 2007; Gulyás and Freund, 2015). On the other hand, axo-axonic cells may also exert an excitatory function by a locally reversed chloride gradient at their synapses (Szabadics et al., 2006).
PV-containing interneurons clearly have a prominent role in protecting the brain from initiation of spontaneous seizures. Thus, onset of epileptic activity rapidly induces an inhibitory barrage by activation of fast-spiking PV-containing basket cells but not of somatostatin-containing O-LM cells (Cammarota et al., 2013; Lee et al., 2014; Toyoda et al., 2015). In the pilocarpine model of TLE, PV interneurons, but not O-LM cells, have become activated in all hippocampal subfields (most prevalently in the subiculum) several hours before the onset of spontaneous seizures (Toyoda et al., 2015). A transient drop in their activity may eventually allow seizure initiation (Toyoda et al., 2015). Our present experiments are in line with these findings by showing that focal silencing of PV neurons unilaterally in the ventral subiculum was sufficient to induce lasting SRSs in the absence of cellular signs of neurodegeneration.
Despite the significant effects of vector injections on the behavior of the animals, neither overall eIPSCs or evoked EPSCs nor mIPSCs or mEPSCs were affected in subicular pyramidal cells from slices of AAV-TeLC-injected mice. This is rather surprising but is consistent with the previous data from the study by Murray et al. (2011). There is no clear explanation for this. However, in the presence of the cannabinoid receptor 1 (CB1) agonist WIN 55,212–2 selectively suppressing GABA release from CCK/CB1-containing basket cells (Takács et al., 2015), we observed a significant reduction of the amplitude of eIPSCs (by 33%). This reduction was almost twice as high as in AAV-GFP-injected mice (59%), reflecting the component of CCK/CB1-containing basket cells. These experiments indicate that both types of basket cells exert a primary inhibitory action upon pyramidal cells of CA1 and the subiculum (Takács et al., 2015). Silencing PV-containing interneurons, however, seems to affect only the inhibitory tone exerted by these neurons but is already sufficient to induce SRSs in the mice.
It is important to note that only irreversible silencing of PV neurons by TeLC expression, but not a transient inhibition (for ∼1 to 2 h) through the DREADD system, results in C-SWDs or SRSs. However, when transient inhibition of GABA release from PV cells of the ventral subiculum was coinciding with systemic application of a threshold dose of PTZ, C-SWDs or even acute seizures developed rapidly. Thus, PV neurons in the subiculum are crucial protectors during excitatory challenge and limit the spread of epileptic activity, while they may be transiently dispensable under basal conditions.
The frequency of SRSs is modest (approximately one to two per week) but still beyond the range of seizure frequency in most TLE patients. It was striking, however, that the frequency of SRSs appeared to decline after several weeks. The underlying mechanisms are unclear. One may consider a variety of mechanisms responsible for this, such as the fading of seizure frequency reflecting readaptation of the disturbed neuronal circuitry. One such mechanism may include the sprouting of inhibitory neurons. Thus, the sprouting of somatostatin terminals (presumably O-LM cells) terminating in the outer molecular layers of the dentate gyrus and of the subiculum has been observed in the kainic acid model of TLE (Drexel et al., 2012; Peng et al., 2013), in kindling (Botterill et al., 2017), and in TLE patients (Mathern et al., 1995; Furtinger et al., 2001). Other mechanisms may include a reduction of cannabinoid signaling affecting GABA release from CCK basket cells or a compensatory increase in the activity of PV neurons not affected by the AAV-TeLC injection (e.g., contralateral to the injection site). We observed such a compensatory activation of PV neurons (as indicated by the increased expression of PV mRNA) in surviving PV interneurons of the subiculum of kainic acid-treated rats (Drexel et al., 2011). SRSs could also cause a rapid increase in the expression of neuroprotective neuropeptides mostly inhibiting glutamate release as observed in human TLE and in respective models recently (Mathern et al., 1995; Patrylo et al., 1999; Furtinger et al., 2001; Pirker et al., 2001; Vezzani and Sperk, 2004; Drexel et al., 2012).
Thus, Ledri et al. (2014) recently used selective optogenetic activation of somatostatin- and PV-containing interneurons to investigate their effect on epileptiform activity in hippocampal slices. They demonstrated that the activation of either dendritic (mediated by somatostatin neurons) or perisomatic inhibition (mediated by PV-containing basket cells) suppressed epileptiform activity, although global activation of all GABA neurons was even more effective, indicating important roles for both mechanisms in maintaining physiological transmission (Ledri et al., 2014). We are currently investigating such mechanisms that may be involved in the late reduction in the frequency of SRSs.
Together, our experiments demonstrate a pivotal role of PV-containing basket cells for controlling the excitability of pyramidal neurons in the subiculum. We provide experimental evidence that selective permanent inhibition of PV-containing interneurons may reduce perisomatic feedforward inhibition in vivo, resulting in a decrease in seizure threshold with the development of C-SWDs and SRSs (i.e., epilepsy) without signs of neurodegeneration. In contrast, transient inhibition of PV-containing basket cells requires an additional excitatory challenge for evoking C-SWDs or seizures.
Footnotes
The work was supported by the Austrian Science Fund (Project P 26680) and the European Research Council (Grant ERC-2015-AdG-695136, T.H.). We thank Anneliese Bukovac for technical assistance, and Jonas Blecher for help with evaluation of EEGs.
The authors declare no competing financial interests.
References
- Alexander A, Maroso M, Soltesz I (2016) Organization and control of epileptic circuits in temporal lobe epilepsy. Prog Brain Res 226:127–154. 10.1016/bs.pbr.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrioli A, Alonso-Nanclares L, Arellano JI, DeFelipe J (2007) Quantitative analysis of parvalbumin-immunoreactive cells in the human epileptic hippocampus. Neuroscience 149:131–143. 10.1016/j.neuroscience.2007.07.029 [DOI] [PubMed] [Google Scholar]
- Becker A, Letzel K, Letzel U, Grecksch G (1997) Kindling of the dorsal and the ventral hippocampus: effects on learning performance in rats. Physiol Behav 62:1265–1271. 10.1016/S0031-9384(97)00303-X [DOI] [PubMed] [Google Scholar]
- Botterill JJ, Nogovitsyn N, Caruncho HJ, Kalynchuk LE (2017) Selective plasticity of hippocampal GABAergic interneuron populations following kindling of different brain regions. J Comp Neurol 525:389–406. 10.1002/cne.24071 [DOI] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Engel J Jr (2000) Chronic epileptogenesis requires development of a network of pathologically interconnected neuron clusters: a hypothesis. Epilepsia 41 [Suppl 6]:S144–S152. 10.1111/j.1528-1157.2000.tb01573.x [DOI] [PubMed] [Google Scholar]
- Cammarota M, Losi G, Chiavegato A, Zonta M, Carmignoto G (2013) Fast spiking interneuron control of seizure propagation in a cortical slice model of focal epilepsy. J Physiol 591:807–822. 10.1113/jphysiol.2012.238154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben-Ari Y, Esclapez M, Bernard C (2001) Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci 4:52–62. 10.1038/82900 [DOI] [PubMed] [Google Scholar]
- Dinocourt C, Petanjek Z, Freund TF, Ben-Ari Y, Esclapez M (2003) Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures. J Comp Neurol 459:407–425. 10.1002/cne.10622 [DOI] [PubMed] [Google Scholar]
- Drexel M, Preidt AP, Kirchmair E, Sperk G (2011) Parvalbumin interneurons and calretinin fibers arising from the thalamic nucleus reuniens degenerate in the subiculum after kainic acid-induced seizures. Neuroscience 189:316–329. 10.1016/j.neuroscience.2011.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexel M, Kirchmair E, Wieselthaler-Hölzl A, Preidt AP, Sperk G (2012) Somatostatin and neuropeptide Y neurons undergo different plasticity in parahippocampal regions in kainic acid-induced epilepsy. J Neuropathol Exp Neurol 71:312–329. 10.1097/NEN.0b013e31824d9882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I (2007) Perisomatic inhibition. Neuron 56:33–42. 10.1016/j.neuron.2007.09.012 [DOI] [PubMed] [Google Scholar]
- Furtinger S, Pirker S, Czech T, Baumgartner C, Ransmayr G, Sperk G (2001) Plasticity of Y1 and Y2 receptors and neuropeptide Y fibers in patients with temporal lobe epilepsy. J Neurosci 21:5804–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Atallah BV, Scanziani M (2008) Complementary modulation of somatic inhibition by opioids and cannabinoids. J Neurosci 28:1824–1832. 10.1523/JNEUROSCI.4700-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyás AI, Freund TT (2015) Generation of physiological and pathological high frequency oscillations: the role of perisomatic inhibition in sharp-wave ripple and interictal spike generation. Curr Opin Neurobiol 31:26–32. 10.1016/j.conb.2014.07.020 [DOI] [PubMed] [Google Scholar]
- Honavar M, Meldrum BS (1997) Epilepsy. In: Greenfield's neuropathology (Graham DI, Lantos PL, eds), pp 931–962. New York: Oxford UP. [Google Scholar]
- Houser CR, Miyashiro JE, Swartz BE, Walsh GO, Rich JR, Delgado-Escueta AV (1990) Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J Neurosci 10:267–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagirdar R, Drexel M, Kirchmair E, Tasan RO, Sperk G (2015) Rapid changes in expression of class I and IV histone deacetylases during epileptogenesis in mouse models of temporal lobe epilepsy. Exp Neurol 273:92–104. 10.1016/j.expneurol.2015.07.026 [DOI] [PubMed] [Google Scholar]
- Jinno S, Kosaka T (2000) Colocalization of parvalbumin and somatostatin-like immunoreactivity in the mouse hippocampus: quantitative analysis with optical dissector. J Comp Neurol 428:377–388. 10.1002/1096-9861(20001218)428:3<377::AID-CNE1>3.0.CO;2-L [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P (2008) Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321:53–57. 10.1126/science.1149381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopp A, Frahm C, Fidzinski P, Witte OW, Behr J (2008) Loss of GABAergic neurons in the subiculum and its functional implications in temporal lobe epilepsy. Brain 131:1516–1527. 10.1093/brain/awn095 [DOI] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, Lowell BB (2011) Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 121:1424–1428. 10.1172/JCI46229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledri M, Madsen MG, Nikitidou L, Kirik D, Kokaia M (2014) Global optogenetic activation of inhibitory interneurons during epileptiform activity. J Neurosci 34:3364–3377. 10.1523/JNEUROSCI.2734-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Marchionni I, Bezaire M, Varga C, Danielson N, Lovett-Barron M, Losonczy A, Soltesz I (2014) Parvalbumin-positive basket cells differentiate among hippocampal pyramidal cells. Neuron 82:1129–1144. 10.1016/j.neuron.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Pretorius JK, Leite JP (1995) Reactive synaptogenesis and neuron densities for neuropeptide Y, somatostatin, and glutamate decarboxylase immunoreactivity in the epileptogenic human fascia dentata. J Neurosci 15:3990–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietzsch M, Grasse S, Zurawski C, Weger S, Bennett A, Agbandje-McKenna M, Muzyczka N, Zolotukhin S, Heilbronn R (2014) OneBac: platform for scalable and high-titer production of adeno-associated virus serotype 1–12 vectors for gene therapy. Hum Gene Ther 25:212–222. 10.1089/hum.2013.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris TA, Jafari N, DeLorenzo RJ (2000) Chronic DeltaFosB expression and increased AP-1 transcription factor binding are associated with the long term plasticity changes in epilepsy. Brain Res Mol Brain Res 79:138–149. 10.1016/S0169-328X(00)00112-1 [DOI] [PubMed] [Google Scholar]
- Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, Bartos M, Wisden W, Wulff P (2011) Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci 14:297–299. 10.1038/nn.2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrylo PR, van den Pol AN, Spencer DD, Williamson A (1999) NPY inhibits glutamatergic excitation in the epileptic human dentate gyrus. J Neurophysiol 82:478–483. [DOI] [PubMed] [Google Scholar]
- Paz JT, Huguenard JR (2015) Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat Neurosci 18:351–359. 10.1038/nn.3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Zhang N, Wei W, Huang CS, Cetina Y, Otis TS, Houser CR (2013) A reorganized GABAergic circuit in a model of epilepsy: evidence from optogenetic labeling and stimulation of somatostatin interneurons. J Neurosci 33:14392–14405. 10.1523/JNEUROSCI.2045-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Czech T, Baumgartner C, Maier H, Novak K, Fürtinger S, Fischer-Colbrie R, Sperk G (2001) Chromogranins as markers of altered hippocampal circuitry in temporal lobe epilepsy. Ann Neurol 50:216–226. 10.1002/ana.1079 [DOI] [PubMed] [Google Scholar]
- Roth BL. (2016) DREADDs for neuroscientists. Neuron 89:683–694. 10.1016/j.neuron.2016.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salami P, Lévesque M, Benini R, Behr C, Gotman J, Avoli M (2014) Dynamics of interictal spikes and high-frequency oscillations during epileptogenesis in temporal lobe epilepsy. Neurobiol Dis 67:97–106. 10.1016/j.nbd.2014.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C (1992) Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359:832–835. 10.1038/359832a0 [DOI] [PubMed] [Google Scholar]
- Sperk G, Widmann R (1985) Somatostatin precursor in the rat striatum: changes after local injection of kainic acid. J Neurochem 45:1441–1447. 10.1111/j.1471-4159.1985.tb07211.x [DOI] [PubMed] [Google Scholar]
- Sperk G, Lassmann H, Baran H, Kish SJ, Seitelberger F, Hornykiewicz O (1983) Kainic acid induced seizures: neurochemical and histopathological changes. Neuroscience 10:1301–1315. 10.1016/0306-4522(83)90113-6 [DOI] [PubMed] [Google Scholar]
- Sperk G, Wieselthaler-Hölzl A, Pirker S, Tasan R, Strasser SS, Drexel M, Pifl C, Marschalek J, Ortler M, Trinka E, Heitmair-Wietzorrek K, Ciofi P, Feucht M, Baumgartner C, Czech T (2012) Glutamate decarboxylase 67 is expressed in hippocampal mossy fibers of temporal lobe epilepsy patients. Hippocampus 22:590–603. 10.1002/hipo.20923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielmeyer W. (1927) Die pathogenese des epileptischen krampfes. Z Ges Neurol Psychiatr 109:501–520. 10.1007/BF02870249 [DOI] [Google Scholar]
- Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G (2006) Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311:233–235. 10.1126/science.1121325 [DOI] [PubMed] [Google Scholar]
- Szabó GG, Holderith N, Gulyás AI, Freund TF, Hájos N (2010) Distinct synaptic properties of perisomatic inhibitory cell types and their different modulation by cholinergic receptor activation in the CA3 region of the mouse hippocampus. Eur J Neurosci 31:2234–2246. 10.1111/j.1460-9568.2010.07292.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takács VT, Szőnyi A, Freund TF, Nyiri G, Gulyás AI (2015) Quantitative ultrastructural analysis of basket and axo-axonic cell terminals in the mouse hippocampus. Brain Struct Funct 220:919–940. 10.1007/s00429-013-0692-6 [DOI] [PubMed] [Google Scholar]
- Toyoda I, Fujita S, Thamattoor AK, Buckmaster PS (2015) Unit activity of hippocampal interneurons before spontaneous seizures in an animal model of temporal lobe epilepsy. J Neurosci 35:6600–6618. 10.1523/JNEUROSCI.4786-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Sperk G (2004) Overexpression of NPY and Y2 receptors in epileptic brain tissue: an endogenous neuroprotective mechanism in temporal lobe epilepsy. Neuropeptides 38:245–252. 10.1016/j.npep.2004.05.004 [DOI] [PubMed] [Google Scholar]
- Wirtshafter D, Stratford TR (2016) Chemogenetic inhibition of cells in the paramedian midbrain tegmentum increases locomotor activity in rats. Brain Res 1632:98–106. 10.1016/j.brainres.2015.12.014 [DOI] [PubMed] [Google Scholar]
- Wood J, Verma D, Lach G, Bonaventure P, Herzog H, Sperk G, Tasan RO (2016) Structure and function of the amygdaloid NPY system: NPY Y2 receptors regulate excitatory and inhibitory synaptic transmission in the centromedial amygdala. Brain Struct Funct 221:3373–3391. 10.1007/s00429-015-1107-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Ting JT, Atallah HE, Qiu L, Tan J, Gloss B, Augustine GJ, Deisseroth K, Luo M, Graybiel AM, Feng G (2011) Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat Methods 8:745–752. 10.1038/nmeth.1668 [DOI] [PMC free article] [PubMed] [Google Scholar]