

Abstract
Exposure to drug-paired cues causes drug memories to be in a destabilized state and interfering with memory reconsolidation can inhibit relapse. Calpain, a calcium-dependent neutral cysteine protease, is involved in synaptic plasticity and the formation of long-term fear memory. However, the role of calpain in the reconsolidation of drug reward memory is still unknown. In the present study, using a conditioned place preference (CPP) model, we found that exposure to drug-paired contextual stimuli induced the activation of calpain and decreased the expression of glutamate receptor interacting protein 1 (GRIP1) in the nucleus accumbens (NAc) core, but not shell, of male rats. Infusions of calpain inhibitors in the NAc core immediately after retrieval disrupted the reconsolidation of cocaine/morphine cue memory and blocked retrieval-induced calpain activation and GRIP1 degradation. The suppressive effect of calpain inhibitors on the expression of drug-induced CPP lasted for at least 14 d. The inhibition of calpain without retrieval 6 h after retrieval or after exposure to an unpaired context had no effects on the expression of reward memory. Calpain inhibition after retrieval also decreased cocaine seeking in a self-administration model and this effect did not recover spontaneously after 28 d. Moreover, the knock-down of GRIP1 expression in the NAc core by lentivirus-mediated short-hairpin RNA blocked disruption of the reconsolidation of drug cue memories that was induced by calpain inhibitor treatment. These results suggest that calpain activity in the NAc core is crucial for the reconsolidation of drug reward memory via the regulation of GRIP1 expression.
SIGNIFICANCE STATEMENT Calpain plays an important role in synaptic plasticity and long-term memory consolidation, however, its role in the reconsolidation of drug cue memory remains unknown. Using conditioned place preference and self-administration procedures, we found that exposure to drug-paired cues induced the activation of calpain and decreased glutamate receptor interacting protein 1 (GRIP1) expression in the nucleus accumbens (NAc) core. The inhibition of calpain activity in the NAc core immediately after retrieval disrupted the reconsolidation of cocaine/morphine cue memory that was blocked by prior GRIP1 knock-down. Our findings indicate that calpain-GRIP signaling is essential for the restabilization process that is associated with drug cue memory and the inhibition of calpain activity may be a novel strategy for the prevention of drug relapse.
Keywords: calpain, CPP, drug reward memory, GRIP, nucleus accumbens, reconsolidation
Introduction
Drug addiction is a chronic brain disorder that is characterized by compulsive drug seeking and high rates of relapse (Grant et al., 1996; Childress et al., 1999; Dackis and O'Brien, 2001; Kilts et al., 2001). Repeated drug use produces intense and long-lasting drug memories that associate the drug-paired contextual stimuli (Stewart et al., 1984; O'Brien et al., 1992; Nestler, 2001). After reactivation, memory returns to a labile state, termed reconsolidation, during which the memories can be modified or erased by pharmacological interventions (Nader et al., 2000a; Alberini, 2005; Nader and Einarsson, 2010). The disruption of drug memory reconsolidation is considered an important and effective strategy to treat addiction and to prevent relapse (Milton and Everitt, 2010). Although accumulating evidence from both animal and human studies indicates that disruption of the reconsolidation of drug cue memories dampens drug cue-induced craving and relapse to drug-seeking behavior (Lee et al., 2006; Valjent et al., 2006; Zhao et al., 2009; Xue et al., 2012; Ren et al., 2013; Jian et al., 2014; Otis et al., 2015; Sorg et al., 2015; Zhang et al., 2016b), the specific mechanisms that underlie the reconsolidation of drug-paired cue memories are still unclear.
Calpain is a calcium-dependent, neutral cysteine protease with 15 isoforms in the human genome (Wu and Lynch, 2006). μ-Calpain and m-calpain are widely expressed, especially in dendritic spines and postsynaptic density (PSD) (Baudry et al., 2013). The double deletion of both μ-calpain and m-calpain impaired synaptic plasticity in hippocampal slices (Amini et al., 2013). Calpain has a great diversity of substrates, including cytoskeleton proteins, membrane receptors/channels, protein kinases, phosphatases, and scaffolding/anchoring proteins (Wu and Lynch, 2006; Baudry et al., 2013). Calpain overactivation led to the limited cleavage or degradation of key neuronal proteins, such as poly (A)-binding protein-interacting protein 2A (PAIP2A; Khoutorsky et al., 2013) and suprachiasmatic nucleus circadian oscillatory protein (SCOP) during the process of protein synthesis and synaptic plasticity (Shimizu et al., 2007). As a substrate of calpain, glutamate receptor-interacting protein (GRIP) anchors the Glu2/3A subunit of AMPA receptors to the membrane, which is essential for their trafficking and stabilization (Dong et al., 1997). GRIP1 is ubiquitously distributed in synaptosomal membranes and the PSD in neurons (Li et al., 2005) and is involved in dendrite morphogenesis and synaptic plasticity (Hoogenraad et al., 2005). The conditioned deletion of GRIP in the nucleus accumbens (NAc) potentiates the cue-induced reinstatement of cocaine-seeking behavior (Briand et al., 2014). However, the role of calpain–GRIP signaling in the reconsolidation of drug cue memories remains unclear.
The NAc plays a critical role in drug cue memories and the reinstatement of drug seeking (Self, 2004; Day and Carelli, 2007; Pickens et al., 2011; Bossert et al., 2013). In the present study, we hypothesized that calpain activity in the NAc core is crucial for the reconsolidation of drug reward memory via the regulation of GRIP1 expression.
Materials and Methods
Subjects.
Male Sprague Dawley rats (260–280 g) were purchased from the Laboratory Animal Center of Peking University Health Science Center and housed 5 per cage with ad libitum access to food and water on a reverse 12 h/12 h light/dark cycle with controlled temperature (23°C ± 2°C) and humidity (50 ± 5%). All animal procedures were performed in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and were approved by the Biomedical Ethics Committee for animal use and protection of Peking University.
Surgery.
After a period of adaptation, surgery was performed as described previously (Wang et al., 2010; Wu et al., 2011). The rats (300–320 g) were anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and implanted bilaterally with permanent guide cannulae (23 gauge; Plastics One) 1 mm above the NAc core or shell. The cannula was placed at a 16° angle toward the midline to avoid penetration of the lateral ventricle. The coordinates for the NAc core were the following: anterior/posterior, 1.5 mm; medial/lateral, 3.8 mm; dorsal/ventral, 6.0 mm. The coordinates for the NAc shell were the following: anterior/posterior, 1.8 mm; medial/lateral, 3.2 mm; dorsal/ventral, 6.6 mm. These coordinates were based on previous work (Wang et al., 2010; Li et al., 2011; Ding et al., 2013; Ren et al., 2013). A stainless-steel stylet blocker was inserted into each cannula to prevent blockage and infection. All of the rats were allowed to recover for 5–7 d after surgery.
Drugs and injection procedures.
Cocaine HCl and morphine sulfate (10 mg/kg; Qinghai Pharmaceuticals) were dissolved in 0.9% saline and injected 5 min before the conditioned place preference (CPP) training sessions. A calpain inhibitor mixture was prepared as described previously (Shimizu et al., 2007; Khoutorsky et al., 2013). Briefly, calpain inhibitor I, calpain inhibitor III and calpeptin (all from Calbiochem) were dissolved in pure dimethylsulfoxide (DMSO) and further diluted in 10% Cremophor EL (Sigma-Aldrich) in saline so that the drug and vehicle solutions had a final concentration of 2 mm per inhibitor, 18% DMSO, and 8.2% Cremophor EL. Injections were performed with Hamilton syringes that were connected to 30-gauge injectors (Plastics One) that were lowered 1 mm below the guide cannula. Calpain inhibitors (1 μl) or vehicle were infused bilaterally in the NAc core or shell over 2.5 min. The injectors were kept in the cannula for 1 additional minute to allow for drug diffusion into the tissue. The injectors were then removed and the stylets were replaced (Khoutorsky et al., 2013; Han et al., 2016).
Design, construction, and validation of lentiviral vectors for GRIP1 knock-down.
We used a lentiviral vector-based short-hairpin RNA (shRNA) system to knock down the expression of GRIP1 in the NAc core based on the manufacturer's instructions (catalog #RSH051451-LVRH1GP; GeneCopoeia). The hairpin loop sequence was TCAAGAG. The targeted sequence was 5′-GCCACTACTAGTTGAAGTTGC-3′. The primer sequences were 5′-TAATACGACTCACTATAGGG-3′ (forward) and 5′-CTGGAATAGCTCAGAGGC-3′ (reverse). For lentiviral-mediated gene transfer, these sequences were cloned into the hairpin structures under control of the H1 promoter in the psi-LVRH1GP vector.
CPP.
The CPP procedure was performed using an unbiased, counterbalanced protocol as described previously (Li et al., 2010; Jian et al., 2014). The apparatus for CPP conditioning consisted of 10 identical three-chamber polyvinyl chloride (PVC) boxes. The boxes had two larger chambers (27.9 cm length × 21.0 cm width × 20.9 cm height) that differed in their floor texture (bar or grid, respectively) and the houselights on the walls. The two larger chambers were separated by a smaller chamber (12.1 cm length × 21.0 cm width × 20.9 cm height, with a smooth PVC floor). Baseline preference was assessed by placing the rats in the center chamber of the CPP apparatus and allowing them to explore all three chambers freely for 15 min. Rats that showed a strong unconditioned preference for either side chamber (i.e., >540 s) were excluded from the experiments. On the following days, the rats were trained for 8 consecutive days with alternating injections of cocaine (10 mg/kg, i.p.) or saline (1 ml/kg, i.p.) and were confined to the conditioning chambers for 45 min after each injection before being returned to their home cages. Tests for the expression of cocaine-induced CPP were identical to the initial baseline preference assessment and were performed on different days after training. The training and testing procedures for morphine-induced CPP were identical with the exception that morphine (10 mg/kg, s.c.) was injected instead of cocaine. For the retrieval of drug cue memories, the rats were reexposed to the drug-paired context for 10 min. To test the specificity of the effects of the calpain inhibitor on drug cue memories, the rats were exposed to an unpaired context (black-and-white checkered walls and ceramic tile floor) for 10 min. Exposure to the unpaired context was expected to provide a similar behavioral experience to drug-paired context without drug cue memory reactivation. The CPP score was defined as the time (in seconds) spent in the cocaine-paired chamber minus the time spent in the saline-paired chamber during the CPP tests.
Intravenous cocaine self-administration.
After 1 week of acclimation, jugular catheters were implanted and the rats were singly housed. After recovery, the rats were trained to self-administer cocaine (0.75 mg/kg/infusion) for 10 consecutive days (3 h/d, 3 1 h sessions daily separated by 5 min) in operant chambers (AniLab Software and Instruments). A fixed-ratio 1 (FR1) schedule of reinforcement was used, with a 40 s timeout period after each infusion. Each session began with illumination of the house light, which remained on for the entire session. The number of cocaine infusions was limited to 20/h. At the end of the training phase, the groups in the different experimental conditions were matched for cocaine intake during training.
The retrieval session was conducted 24 h after the last cocaine self-administration training session for 15 min. Nose-poke responses led to a 5 s tone–light cue under an FR1 schedule without cocaine delivery. The cue-induced cocaine seeking test and spontaneous recovery test conditions were the same as during training, with the exception that cocaine was no longer available. The duration of the test was 1 h.
Tissue sample preparation.
The procedure was similar to previous reports (Ren et al., 2013; Chai et al., 2014). Briefly, the rats were decapitated. NAc core and shell tissue was homogenized (10–15 s × 3, 5 s interval) with an electrical disperser (Wiggenhauser) after being lysed in RIPA lysis buffer with protease and phosphatase inhibitors (Beyotime Biotechnology; 50 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1% sodium orthovanadate, 2.5 mm sodium pyrophosphate, 1 mm ethylenediaminetetraacetic acid, 0.5 μg/ml leupeptin, and 1 mm phenylmethanesulfonyl fluoride) for 30 min. Subsequently, the homogenate was centrifuged at 10,000 × g for 20 min and the supernatant was collected. All of the above procedures were performed at low temperature (0–4°C). The protein concentration was determined using the BCA assay kit (Applygen Technologies) and then normalized by diluting the samples with RIPA lysis buffer.
Western blot assays.
The Western blot procedures were based on our previous studies (Li et al., 2011; Zhang et al., 2016a). Loading buffer (5×) (Applygen Technologies) was added to each sample before boiling for 5 min. The proteins were loaded onto 8% SDS-PAGE and electrophoretically transferred to Immobilon-P transfer membranes (Millipore). The blots were blocked for 2 h with blocking buffer (5% bovine serum albumin in TBST) at room temperature. The membranes were then incubated overnight at 4°C with the following primary antibodies: spectrin breakdown protein (SBDP, 1:500; Santa Cruz Biotechnology), GRIP1 (1:500; Santa Cruz Biotechnology), calpain (1:500; Abcam), and β-actin (1:1000; Santa Cruz Biotechnology). After washing, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody (goat anti-mouse IgG for β-actin and goat anti-rabbit IgG for the others; 1:5000; Santa Cruz Biotechnology) for 50 min at room temperature. After washing 4 times in TBST, the blots were detected by super signal enhanced chemiluminescence substrate (detection reagents 1 and 2, 1:1 ratio; Applygen Technologies) and visualized using the Sygene Bio Image system. Band intensities were quantified using Quantity One 4.4.0 software (Bio-Rad). The optical density of specific proteins was normalized to β-actin. The Western blot data are presented as a percentage of the naive group and each individual experiment had its own naive group.
Histology.
The animals were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.) and transcardially perfused with 0.01 m PBS, followed by 4% paraformaldehyde in 0.2 m phosphate buffer. The brain was extracted, postfixed overnight at 4°C, and cryoprotected in 30% sucrose in 0.2 m phosphate buffer. The cannula placements were confirmed in 40-μm-thick sections using Nissl staining under light microscopy. Rats with misplaced cannulae were excluded from the statistical analysis. The locations of the cannula tips are shown in Figure 1. For eGFP expression in lentiviral vector-injected animals, consecutive 10 μm coronal brain sections were examined using an Olympus BX53 fluorescence microscope (Jian et al., 2014; Xue et al., 2014).
Figure 1.

Schematic representation of the injection sites and photomicrographs of representative cannula placements in the NAc core (A) and shell (B).
Experimental design.
In the present study, rats were trained with drug-induced CPP for 8 d and recieved CPP formation test on day 9 (see “CPP” section). At first, we evaluated the effect of the retrieval of drug cue memories (cocaine and morphine) on calpain activity and GRIP1 expression in the NAc core and shell. Second, we tested the role of calpain activity in the NAc core on the reconsolidation of drug cue memories. Here, we used four conditions to confirm whether the effect of calpain inhibitors on reconsolidation was temporally specific and retrieval dependent, rats received an infusion of calpain inhibitors or vehicle in the NAc core in the following conditions: (1) without retrieval, (2) 6 h after retrieval, (3) before Test2 on day 10, and (4) after reexposed in an unpaired context for 10 min. Third, we investigated the long-term effect of calpain inhibition in the NAc core on the expression of drug-induced CPP. We used GRIP1 knock-down rats to determine whether prior knock-down of GRIP1 could block the disruption of the reconsolidation of drug cue memories by calpain inhibition. Finally, we explored the effect of the inhibition of calpain activity in the NAc core on cue-induced cocaine-seeking behavior in a self-administration model (see details in Results).
Statistical analysis.
The data are expressed as mean ± SEM. The statistical analyses were performed using one-way or two-way ANOVA with appropriate within-subjects and between-subjects factors for each experiment (see details in Results), followed by Tukey's post hoc test (see Results for details). Values of p < 0.05 were considered statistically significant.
Results
Effect of drug memory retrieval on calpain activity and GRIP1 expression in the NAc core and shell
We first determined the effect of the retrieval of cocaine cue memories on calpain activity and GRIP1 expression in the NAc core and shell. As shown in Figure 2A, 6 groups of rats (n = 8–9 per group) were trained for cocaine-induced CPP for 8 d and then underwent the CPP test on day 9 (Test 1). On day 10, 5 groups were confined to the cocaine-paired context for 10 min to reactivate the cocaine cue memories and then were decapitated 10 min, 30 min, 1 h, 2 h, or 6 h later. Another group of rats that were confined to their home cages without undergoing the retrieval trial was decapitated on day 10. The rats in the naive group that was subjected only to daily handling in their home cages were decapitated at the same time as the no-retrieval group. The tissues of the NAc core and shell were removed for the further detection of SBDP and GRIP1 expression. We used SBDP (molecular size = 140–145 kDa), a substrate of calpain, as an index of calpain activity, in which higher SBDP expression reflects calpain activation (Andres et al., 2013). Repeated-measures ANOVA of CPP scores with test (baseline and Test 1) as the within-subjects factor and group (no retrieval, 10 min, 30 min, 1 h, 2 h, and 6 h) as the between-subjects factor revealed a significant effect of test (F(1,44) = 242.063, p < 0.001; Fig. 2B), but no effect of group (F(5,44) = 0.033, p = 0.999; Fig. 2B). One-way ANOVA of the Western blot data revealed that SBDP expression increased in the NAc core 30 min after retrieval and returned to baseline 1 h after retrieval (F(5,44) = 18.267, p < 0.001; Fig. 2C). No significant difference in SBDP expression was found in the NAc shell (Fig. 2D). The expression of GRIP1 decreased in the NAc core 30 min after retrieval and returned to baseline 1 h after retrieval (F(5,44) = 4.255, p = 0.003; Fig. 2C). No significant difference in GRIP1 expression was found in the NAc shell (Fig. 2D). Another two groups of rats were used to determine the expression of calpain protein levels in the NAc core after the retrieval of cocaine cue memories. One-way ANOVA was used to analyze the Western blot data, which revealed no significant difference in calpain protein expression between groups (F(1,11) = 0.119, p = 0.737; Fig. 2F) and indicated that cocaine memory retrieval increased calpain activity, but not calpain protein levels, in the NAc core.
Figure 2.

Retrieval of cocaine cue memories induced increased calpain activity and decreased GRIP1 expression in the NAc core, but not shell. A, Experimental timeline. B, CPP scores in rats in the baseline and test phases. All six groups of rats acquired equivalent cocaine-induced CPP. *p < 0.05 compared with baseline test. C, SBDP and GRIP1 levels and representative Western blots in the NAc core after the retrieval of cocaine cue memories. SBDP increased significantly 30 min after the retrieval trial and then returned to baseline 1 h after the retrieval trial. The expression of GRIP1 decreased significantly 30 min after the retrieval trial and then returned to baseline 1 h after the retrieval trial. D, SBDP and GRIP1 levels and representative Western blots in the NAc shell after the retrieval of cocaine cue memories. No significant changes in SBDP or GRIP1 levels were observed after the retrieval trial. The data are expressed as a percentage (mean ± SEM) of SBDP and GRIP1 levels relative to naive rats (n = 8–9 per group). *p < 0.05 compared with the no retrieval (NoR) group. E, Experimental timeline. F, Expression of calpain protein in the NAc core. No significant changes in calpain protein levels were observed after the retrieval of cocaine cue memories. The data are expressed as a percentage (mean ± SEM) of calpain levels relative to naive rats (n = 6–7 per group).
We then used an identical procedure to study the effect of retrieval of morphine cue memories on calpain activity and GRIP1 expression in the NAc core and shell (Fig. 3A). Repeated-measures ANOVA of CPP scores in Test 1 revealed a significant effect of test (F(1,24) = 294.908, p < 0.001; Fig. 3B), but no effect of group (F(5,24) = 0.038, p = 0.999; Fig. 3B). One-way ANOVA of the Western blot data revealed that SBDP expression increased in the NAc core 30 min and 1 h after retrieval and returned to baseline 2 h after retrieval (F(5,24) = 6.459, p = 0.001; Fig. 3C). No significant difference in SBDP expression was found in the NAc shell (Fig. 3D). GRIP1 expression decreased in the NAc core 30 min after retrieval and returned to baseline 1 h after retrieval (F(5,24) = 11.207, p < 0.001; Fig. 3C). No significant difference in GRIP1 expression was found in the NAc shell (Fig. 3D). These results indicate that the retrieval of drug cue memories increased the activity of calpain and decreased the expression of GRIP1 in the NAc core, but not shell.
Figure 3.

Retrieval of morphine cue memories increased calpain activity and decreased GRIP1 expression in the NAc core, but not shell. A, Experimental timeline. B, CPP scores in rats before and after CPP training. All six groups acquired equivalent morphine-induced CPP. *p < 0.05 compared with baseline test. C, SBDP and GRIP1 levels and representative Western blots in the NAc core after the retrieval of morphine cue memories. SBDP levels increased significantly 30 min and 1 h after exposure and then returned to baseline. GRIP1 was degraded significantly 30 min after exposure and then returned to baseline. D, SBDP and GRIP1 levels and representative Western blots in the NAc shell after the retrieval of morphine cue memories. No significant changes in SBDP or GRIP1 levels were observed after the retrieval of morphine cue memories. The data are expressed as a percentage (mean ± SEM) of SBDP and GRIP1 levels relative to naive rats (n = 5 per group). *p < 0.05 compared with the no retrieval (NoR) group.
Effect of calpain inhibition in the NAc core and shell on the reconsolidation of drug cue memories
We found that the retrieval of drug cue memories increased calpain activity. We studied the effect of infusions of calpain inhibitors in the NAc core and shell on the reconsolidation of cocaine-induced CPP. As shown in Figure 4A, all of the rats underwent 8 d of CPP training and Test 1 was performed on day 9. Vehicle or a mixture of three calpain inhibitors (i.e., calpeptin, calpain inhibitor I, and calpain inhibitor III) was microinjected in the NAc core or shell immediately after reexposure to the cocaine-paired context on day 10. Test 2 of CPP was performed 24 h later (on day 11). All of the rats were decapitated 30 min after Test 2 for the subsequent determination of SBDP and GRIP1 expression (n = 11–12 per group). Repeated-measures ANOVA of CPP scores with treatment (calpain inhibitors and vehicle) as the between-subjects factor and test (baseline, Test 1, and Test 2) as the within-subjects factor revealed significant effects of test (F(2,42) = 77.314, p < 0.001; Fig. 4B) and treatment (F(1,21) = 9.015, p = 0.007; Fig. 4B) and a significant test × treatment interaction (F(2,42) = 14.989, p < 0.001; Fig. 4B). Post hoc analysis revealed that the rats in both groups (treated with calpain inhibitors or vehicle) acquired CPP after CPP training (p < 0.001; Fig. 4B). No significant difference was found between groups in Test 1 (p = 0.950; Fig. 4B). After the drug infusion, CPP scores in rats that received calpain inhibitor infusions decreased significantly in Test 2 (p < 0.001; Fig. 4B). One-way ANOVA revealed that SBDP levels decreased significantly (F(1,21) = 74.522, p < 0.001; Fig. 4C) and GRIP1 levels increased (F(1,21) = 16.569, p = 0.001; Fig. 4C) in the calpain-inhibitor-treated group.
Figure 4.

Intra-NAc core, but not shell, infusion of calpain inhibitors immediately after retrieval impaired the reconsolidation of cocaine cue memories. A, Experimental timeline. B, Microinjection of calpain inhibitors in the NAc core immediately after retrieval impaired the reconsolidation of cocaine cue memories (n = 11–12 per group). *p < 0.05 compared with CPP scores in Test 2 in the vehicle group and compared with CPP scores in Test 1 in the calpain inhibitor group. C, Protein levels of SBDP and GRIP1 and representative Western blots in the NAc core 30 min after Test 2. The data are expressed as a percentage of SBDP and GRIP1 levels relative to naive control rats (n = 11–12 per group). Calpain inhibitors decreased SBDP levels and increased GRIP1 levels after CPP Test 2. *p < 0.05 compared with vehicle group. D, Calpain inhibitor infusion in the NAc shell had no effect on the reconsolidation of cocaine cue memories (n = 10–12 per group). E, Experimental timeline. F, CPP scores in rats before and after CPP training. All 4 groups acquired equivalent cocaine-induced CPP (n = 6 per group). G, Inhibition of calpain activity in the NAc core immediately after retrieval blocked the increase in calpain activity and decrease in GRIP1 expression that were induced by retrieval (n = 6 per group). *p < 0.05 compared with the no retrieval (NoR) + vehicle group. The data are expressed as mean ± SEM. Ret, Retrieval.
Two groups of rats (n = 10–12 per group) were infused with calpain inhibitors or vehicle in the NAc shell immediately after the retrieval of cocaine cue memories and then underwent the CPP test 24 h later (Fig. 4D). Repeated-measures ANOVA of CPP scores revealed a significant effect of test (F(2,40) = 44.117, p < 0.001; Fig. 4D), but not treatment (F(1,20) = 0.175, p = 0.680; Fig. 4D), and no test × treatment interaction (F(2,40) = 0.418, p = 0.662; Fig. 4D), indicating that the infusion of calpain inhibitors in the NAc shell immediately after retrieval had no effect on the reconsolidation of cocaine cue memories.
We then used 4 groups of rats (n = 6 per group) to determine whether infusions of calpain inhibitors in the NAc core immediately after cocaine reward memory retrieval blocked the changes in calpain activity and GRIP1 expression. After cocaine-induced CPP training, two groups of rats were microinjected with calpain inhibitors or vehicle in the NAc core immediately after the retrieval of cocaine cue memories and then were decapitated 30 min after retrieval on day 10. Another two groups of rats received calpain inhibitors or vehicle without undergoing retrieval and then were decapitated 30 min later on day 10 (Fig. 4E). Tissues of the NAc core were then collected for Western blot. Repeated-measures ANOVA with group (no retrieval + vehicle, no retrieval + calpain inhibitors, retrieval + vehicle, and retrieval + calpain inhibitors) as the between-subjects factor and test (baseline and Test 1) as the within-subjects factor revealed a significant effect of test (F(1,20) = 190.288, p < 0.001; Fig. 4F), but no effect of group (F(3,20) = 0.082, p = 0.969; Fig. 4F), indicating that all of the rats acquired CPP. One-way ANOVA of the Western blot data revealed significant effects of group on the levels of SBDP (F(3,20) = 20.873, p < 0.001; Fig. 4G) and GRIP1 (F(3,20) = 16.057, p < 0.001; Fig. 4G). Post hoc analysis revealed that SBDP levels in vehicle-treated rats increased significantly (p < 0.001; Fig. 4G) and GRIP1 levels decreased significantly (p < 0.001; Fig. 4G) 30 min after retrieval. Treatment with calpain inhibitors blocked these changes. These results indicate that the inhibition of calpain activation in the NAc core, but not shell, immediately after retrieval impaired the reconsolidation of cocaine cue memories.
Effects of the inhibition of calpain activity in the NAc core without retrieval 6 h after retrieval or after unpaired context exposure on the expression of cocaine cue memories
We further evaluated whether the effects of calpain inhibition in the NAc core on the reconsolidation of cocaine cue memories are retrieval dependent. Two groups of rats were trained for cocaine-induced CPP and then received an infusion of calpain inhibitors or vehicle in the NAc core on day 10 without undergoing the retrieval trial (Fig. 5A). Repeated-measures ANOVA of CPP scores revealed a main effect of test (F(2,38) = 42.367, p < 0.001; Fig. 5B), but no effect of treatment (F(1,19) = 0.065, p = 0.802; Fig. 5B) and no test × treatment interaction (F(2,38) = 0.038, p = 0.963; Fig. 5B). The inhibition of calpain activity in the NAc core had no effects on the subsequent expression of cocaine-induced CPP when the rats did not undergo the retrieval trial, indicating that the disruptive effect of calpain inhibition in the NAc core on the subsequent expression of cocaine-induced CPP was retrieval dependent.
Figure 5.

Calpain inhibition in the NAc core without retrieval or 6 h after retrieval or after unpaired context exposure had no effect on the expression of cocaine cue memories. A, B, Microinjection of calpain inhibitors without memory retrieval did not disrupt cocaine reward memories (n = 10–11 per group). C, D, Inhibition of calpain activity in the NAc core 6 h after memory retrieval had no effect on cocaine-induced CPP (n = 10 per group). E, F, Calpain inhibitor infusion in the NAc core had no effect on the retrieval of cocaine cue memories itself (n = 11 per group). G, H, Microinjection of calpain inhibitors in the NAc core after exposure to the unpaired context had no effect on cocaine cue memories (n = 7–9 per group). The data are expressed as mean ± SEM.
We then tested whether the effects of calpain inhibition in the NAc core on the subsequent expression of cocaine cue memories is temporally limited. Two groups of rats were trained for cocaine-induced CPP and then received an infusion of calpain inhibitors or vehicle in the NAc core 6 h after the retrieval trial on day 10 (Fig. 5C). Repeated-measures ANOVA of CPP scores revealed a significant effect of test (F(2,36) = 62.123, p < 0.001; Fig. 5D), but no effect of treatment (F(1,18) = 0.089, p = 0.769; Fig. 5D) and no test × treatment interaction (F(2,36) = 0.443, p = 0.646; Fig. 5D), indicating that the effects of calpain inhibition in the NAc core on the reconsolidation of cocaine cue memories was temporally limited.
Next, we determined the effect of calpain inhibition in the NAc core on the expression of cocaine-induced CPP. Two groups of rats were trained for cocaine-induced CPP and then received an infusion of calpain inhibitors or vehicle in the NAc core before CPP Test 2 on day 10 (Fig. 5E). Repeated-measures ANOVA of CPP scores revealed a main effect of test (F(2,40) = 73.547, p < 0.001; Fig. 5F), but no effect of treatment (F(1,20) < 0.001, p = 0.994; Fig. 5F) and no test × treatment interaction (F(2,40) = 0.037, p = 0.964; Fig. 5F), indicating that calpain inhibition in the NAc core had no effect on the retrieval of cocaine cue memories itself.
Finally, to exclude possible effects on nonspecific arousal, two groups of rats were trained for cocaine-induced CPP and then placed in an unpaired context for 10 min before receiving an infusion of calpain inhibitors or vehicle in the NAc core. Repeated-measures ANOVA of CPP scores revealed a significant effect of test (F(2,20) = 61.648, p < 0.001; Fig. 5H), but no effect of treatment (F(1,10) = 0.035, p = 0.856; Fig. 5H) and no test × treatment interaction (F(2,20) = 0.117, p = 0.891; Fig. 5H). These results indicate that the disruptive effect of calpain inhibition in the NAc core on the reconsolidation of cocaine cue memories was both retrieval dependent and temporally specific.
Long-term effect of the inhibition of calpain activity in the NAc core immediately after retrieval on the reconsolidation of drug cue memories
Suppression of the expression of drug cue memories by disrupting the reconsolidation process is resistant to spontaneous recovery and noncontingent drug-priming-induced reinstatement (Wu et al., 2011; Ding et al., 2013; Jian et al., 2014). Therefore, we explored the long-lasting effect of calpain inhibition in the NAc core immediately after retrieval on the expression of drug-induced CPP. As shown in Figure 6A, two groups of rats underwent cocaine/morphine-induced CPP training and Test 1 was performed on day 9. The rats received vehicle or calpain inhibitors in the NAc core immediately after reexposure to the drug-paired context on day 10 and CPP Test 2 was performed on day 11. After Test 2, the rats were kept in their home cages for 14 d and then CPP Test 3 was performed on day 25. On day 26, the rats were given a priming injection of cocaine (10 mg/kg, i.p.) or morphine (5 mg/kg, s.c.) 5 min before the priming test (Fig. 6A). For cocaine-induced CPP, repeated-measures ANOVA of CPP scores revealed main effects of test (F(4,80) = 14.002, p < 0.001; Fig. 6B) and treatment (F(1,20) = 21.344, p < 0.001; Fig. 6B) and a significant test × treatment interaction (F(4,80) = 5.989, p = 0.001; Fig. 6B). Post hoc analysis revealed that CPP scores in the calpain inhibitor group decreased significantly compared with the vehicle group in Test 3 and the priming test (p = 0.002 and p = 0.001; Fig. 6B). We also observed the same inhibitory effect of calpain inhibition on the reconsolidation of morphine-induced CPP (Fig. 6C). Repeated-measures ANOVA of CPP scores revealed main effects of test (F(4,68) = 34.672, p < 0.001; Fig. 6C) and treatment (F(1,17) = 18.565, p < 0.001; Fig. 6C) and a significant test × treatment interaction (F(4,68) = 8.500, p < 0.001; Fig. 6C). Post hoc analysis revealed that CPP scores in the calpain inhibitor group decreased significantly compared with the vehicle group in Test 3 and the priming test (p = 0.002 and p < 0.001; Fig. 6C). These results indicate that suppression of the response to drug-paired cues that was caused by calpain inhibition in the NAc core immediately after the retrieval trial lasted at least 14 d and this inhibitory effect was insensitive to the reinstatement that was induced by the priming injection.
Figure 6.

Calpain inhibition in the NAc core immediately after drug memory retrieval had a long-lasting suppressive effect on the expression of drug-induced CPP. A, Experimental timeline. The inhibitory effect of calpain inhibition on cocaine-induced CPP (B) and morphine-induced CPP (C) lasted at least 2 weeks after drug administration and was not reinstated by cocaine (10 mg/kg, i.p.) or morphine (5 mg/kg, s.c.) priming. *p < 0.05 compared with CPP scores in vehicle group in the same test. The data are expressed as mean ± SEM (B: n = 10–12 per group; C: n = 9–10 per group).
Effect of the inhibition of calpain activity in the NAc core on the reconsolidation of cocaine cue memories in GRIP1 knock-down rats
We found that the retrieval of drug cue memories decreased GRIP1 expression, which was blocked by the infusion of calpain inhibitors in the NAc immediately after retrieval (Figs. 2, 3, 4). A previous study reported that calpain mediated the degradation of GRIP and calpain activation disrupted the interaction between GRIP and the GluA2 subunit of AMPA receptors (Lu et al., 2001). Therefore, we hypothesized that disruption of the reconsolidation of drug cue memories by calpain inhibition in the NAc core may be mediated by an increase in GRIP1 expression. We tested the effect of GRIP1 knock-down before calpain inhibition in the NAc core on the reconsolidation of cocaine cue memories. We first confirmed the efficiency of GRIP1 knock-down by lentivirus-mediated shRNA in the NAc core (Fig. 7A). The rats that were treated with lentivirus vector (LV)-shGRIP1 or LV-scramble in the NAc core were decapitated 8 or 15 d later (n = 5 per group). One-way ANOVA revealed that GRIP1 expression decreased significantly 8 d (F(1,8) = 44.174, p < 0.001; Fig. 7B) and 15 d (F(1,8) = 32.792, p < 0.001; Fig. 7B) after the LV-shGRIP1 injection compared with the LV-scramble group.
Figure 7.
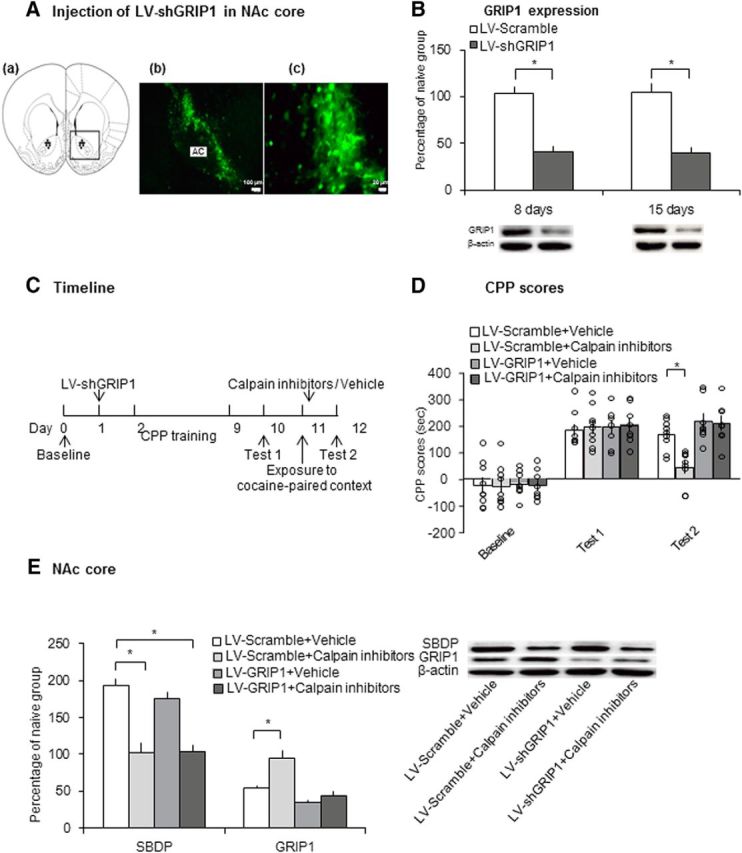
Knock-down of GRIP1 before retrieval blocked impairment of the reconsolidation of cocaine cue memories that was induced by calpain inhibitors. A, Representative photographs of eGFP (green). Left, 4× magnification, scale bar, 100 μm. Right, 40× magnification, scale bar, 20 μm. B, GRIP1 expression and representative Western blots in the NAc core 8 and 15 d after lentivirus injection (n = 5 per group). *p < 0.05 compared with LV-scramble group. C, Experimental timeline. D, Calpain inhibitor infusion in the NAc core immediately after retrieval impaired the reconsolidation of cocaine cue memories only in LV-scramble-treated rats (n = 8–9 per group). *p < 0.05 compared with CPP scores in Test 2 in the LV-scramble + vehicle group. E, Protein levels of SBDP and GRIP1 and representative Western blots in the NAc core after Test 2. The data are expressed as a percentage of SBDP or GRIP1 relative to naive control rats (n = 8–9 per group). Calpain inhibitors blocked the increase of SBDP in both the LV-scramble and LV-shGRIP1 groups, but had no effect on the expression of GRIP1 in LV-shGRIP1 rats. *p < 0.05 compared with LV-scramble + vehicle group. The data are expressed as mean ± SEM. AC, Anterior commissure.
We next evaluated whether the prior knock-down of GRIP1 blocked the impairment of reconsolidation of cocaine cue memories that was induced by calpain inhibition in the NAc core. Four groups of rats were used according to a 2 (LV-scramble and LV-shGRIP1) × 2 (calpain inhibitors and vehicle) factorial design. LV-scramble and LV-shGRIP1 were infused in the NAc core 24 h after the CPP baseline test. All of the rats then underwent cocaine-induced CPP training. On day 10, all of the rats were tested for the expression of CPP (Test 1). Calpain inhibitors or vehicle were microinjected in the NAc core immediately after exposure to the cocaine-paired context on day 11. All of the rats then underwent CPP Test 2 on day 12 and were decapitated 30 min later for Western blot analysis (Fig. 7C). The two-way repeated-measures ANOVA of CPP scores with test as the within-subjects factor (baseline, Test 1, and Test 2) and lentivirus (LV-scramble and LV-shGRIP1) and treatment (calpain inhibitors and vehicle) as the between-subjects factors revealed a significant test × lentivirus × treatment interaction (F(2,60) = 3.271, p = 0.045; Fig. 7D). Post hoc analysis revealed that the calpain inhibitors reduced CPP scores Test 2 significantly compared with vehicle in LV-scramble-treated rats (p = 0.002; Fig. 7D). In LV-shGRIP1-treated rats, calpain inhibition in the NAc core immediately after retrieval had no effect on the reconsolidation of cocaine cue memories (p = 0.133; Fig. 7D).
The two-way ANOVA of the Western blot data with lentivirus (LV-scramble and LV-shGRIP1) and drug (calpain inhibitors and vehicle) as the between-subjects factors revealed a significant main effect of drug (F(1,29) = 59.253, p < 0.001; Fig. 7E) on SBDP levels, but no effect of lentivirus (F(1,29) = 0.586, p = 0.450; Fig. 7E) and no lentivirus × drug interaction (F(1,29) = 0.791, p = 0.381; Fig. 7E). ANOVA also revealed significant effects of lentivirus (F(1,29) = 28.934, p < 0.001; Fig. 7E) and drug (F(1,29) = 14.553, p = 0.001; Fig. 7E) on GRIP1 expression and a significant lentivirus × drug interaction (F(1,29) = 6.140, p = 0.019; Fig. 7E). Post hoc analysis revealed that calpain inhibition blocked the retrieval-induced increase in SBDP expression and the decrease in GRIP1 expression significantly compared with vehicle in LV-scramble-treated rats. The prior microinjection of LV-shGRIP1 blocked the increase in GRIP1 expression in the calpain-inhibitor-treated group (p < 0.001; Fig. 7E). These results indicate that impairment of the reconsolidation of cocaine cue memories that was induced by calpain inhibition in the NAc core was mediated by an increase in GRIP1 expression.
Effect of the inhibition of calpain activity in the NAc core on cue-induced cocaine-seeking behavior
Finally, we assessed the effect of calpain inhibition in the NAc core on the reconsolidation of cocaine-associated cue memories in a self-administration model (Fig. 8A). We trained the rats to self-administer intravenous cocaine for 10 d (Fig. 8B). We used the following four groups of rats to test the effects of calpain inhibition after retrieval on cocaine-seeking behavior: (1) no retrieval + vehicle, (2) no retrieval + calpain inhibitors, (3) retrieval + vehicle, and (4) retrieval + calpain inhibitors. On day 11, we injected calpain inhibitors or vehicle in the NAc core immediately after the 15 min retrieval session or no retrieval. One day later, we tested cue-induced cocaine seeking. Two-way ANOVA revealed a significant condition (retrieval and no retrieval) × treatment (calpain inhibitors and vehicle) interaction in the cue-induced cocaine-seeking test (F(1,22) = 25.163, p < 0.001; Fig. 8C) and spontaneous recovery test after 28 d (F(1,20) = 19.563, p < 0.001; Fig. 8D). Post hoc analysis revealed that active nose-poke responses in the group of rats that received an injection of calpain inhibitors in the NAc core immediately after memory retrieval decreased significantly compared with the other three groups (p < 0.001; Fig. 8C) and this effect did not recover spontaneously after 28 d (p = 0.005; Fig. 8D), indicating that calpain inhibition in the NAc core immediately after memory retrieval disrupted the reconsolidation of cocaine-associated cue memories and this effect lasted at least 28 d.
Figure 8.

Calpain inhibition in the NAc core after retrieval decreased cue-induced cocaine craving and spontaneous recovery. A, Experimental timeline. B, Total number of infusions across the acquisition of cocaine self-administration. C, Number of responses at the active nose-poke operandum in the cue-induced cocaine-seeking test that was conducted 24 h after calpain inhibitor or vehicle injection in the NAc core after the retrieval session. D, Number of active responses during the spontaneous recovery test that was conducted 28 d later (n = 6–8 per group). *p < 0.05 compared with retrieval + vehicle group. The data are expressed as mean ± SEM.
Discussion
The present study used rat models of CPP and self-administration to explore the effects of calpain inhibition in the NAc core and shell on the reconsolidation of drug cue memories. The retrieval of either cocaine or morphine cue memories activated calpain and decreased the subsequent expression of GRIP1 in the NAc core, but not shell. The inhibition of calpain activity impaired the reconsolidation of drug cue memories and blocked the changes in calpain activity and GRIP1 expression that were induced by memory retrieval. Furthermore, suppression of the expression of cocaine cue memories that was induced by calpain inhibition lasted at least 14 d and was not recovered by a cocaine-priming injection. Calpain inhibition in the NAc core after retrieval also decreased cue-induced cocaine-seeking behavior in the self-administration model. Furthermore, the knock-down of GRIP1 expression by infusing lentivirus-mediated shRNA in the NAc core before retrieval blocked impairment of the reconsolidation of cocaine cue memories that was induced by calpain inhibition. Altogether, our results indicate that the proteolysis of GRIP by calpain may play an important role in the reconsolidation of drug cue memories.
Role of calpain in synaptic plasticity and learning and memory
Calpains are calcium-dependent cysteine proteases that are distributed widely in mammalian tissues (Briz and Baudry, 2016). Previous studies have shown that calpain plays an important role in synaptic plasticity, memory, neuronal migration and differentiation, and neurodegeneration (Wu and Lynch, 2006; Wang et al., 2013; Wang et al., 2014; Baudry and Bi, 2016). In neurons, calpains are activated by calcium influx after N-methyl-D-aspartate receptor activation, tetanic stimulation, and KCl-induced depolarization (Vanderklish et al., 1995; Vanderklish et al., 2000). Calpains have been shown to be necessary for the formation of long-term potentiation (LTP) through several processes, such as the regulation of protein synthesis and reorganization of the actin cytoskeleton. The calpain-mediated degradation of phosphatase and tensin homolog (PTEN) contributes to brain-derived neurotrophic factor-mediated mTOR activation, which is important for synaptic plasticity (Briz et al., 2013). Calpain can also regulate protein synthesis during synaptic plasticity through the degradation of other substrates such as PAIP2A, cytoplasmic polyadenylation element-binding protein 3 (CPEB3), and GRIP1 and affect LTP (Hoogenraad et al., 2005; Wang and Huang, 2012; Khoutorsky et al., 2013). Calpain also affects reorganization of the actin cytoskeleton that occurs during LTP by regulating RhoA, spectrin, and myristoylated alanine-rich C kinase substrate (Murakoshi et al., 2011; Trovò et al., 2013; Zhu et al., 2015). The double deletion of μ-calpain and m-calpain impaired dendritic morphology and LTP (Amini et al., 2013).
Synaptic plasticity is the basis of memory. Increasing evidence indicates that calpain plays an important role in learning and memory (Shimizu et al., 2007; Khoutorsky et al., 2013; Baudry and Bi, 2016). Calpain inhibitors impaired long-term memory, but not short-term memory, and calpain-1 knock-out mice exhibited impaired fear memory (Zhu et al., 2015). Furthermore, recent studies showed that fear memory was affected by calpain by regulating the level of extracellular signal-regulated kinase phosphorylation (Liu et al., 2016). Addictive substances such as cocaine and morphine induce LTP in excitatory synapses, which contributes to reinforced drug-seeking behavior and maladaptive memories (Ungless et al., 2001; Saal et al., 2003; Fole et al., 2015). In the present study, calpain inhibition immediately after retrieval impaired the expression of cocaine- and morphine-induced CPP and decreased cue-induced cocaine-seeking behavior, indicating that calpain activity in the NAc core is critical for the reconsolidation of drug cue memories.
Specificity of actions of calpain activity in the NAc core on drug cue memories
The NAc is involved in the formation and reconsolidation of drug-associated memories (Ren et al., 2013; White et al., 2016). We found that the retrieval of cocaine or morphine cue memories increased calpain activity and decreased GRIP1 expression in the NAc core, but not shell (Figs. 2, 3), which is consistent with our previous studies of the role of the NAc core in drug cue memories or drug-seeking behavior (Wang et al., 2010; Ding et al., 2013; Ren et al., 2013). The NAc core and shell are neuroanatomically heterogeneous structures with distinct afferent and efferent connections (Voorn et al., 1989; Zahm and Brog, 1992), which may underlie the dissociable role in the conditioned reinforcing effects of drug-associated cues. Furthermore, consistent with previous studies (Nader et al., 2000b; Zhang et al., 2016b), we found that the intra-NAc core infusion of calpain inhibitors 6 h after retrieval or without retrieval had no effects on the expression of cocaine-induced CPP. We also performed control experiments to confirm the specificity of the effects of calpain inhibitors on cocaine cue memory. We found that cocaine cue memories were intact when calpain inhibitors were injected after the rats were exposed to the unpaired context, which may exclude general nonspecific effects of calpain inhibitors such as arousal. Altogether, our results indicate a critical role for calpain activity in the NAc core in the reconsolidation of drug cue memories and the suppressive effect of calpain inhibitors on reconsolidation is anatomically and temporally specific and retrieval dependent.
Role of calpain–GRIP signaling in drug memory
Calpain activity is required for the degradation of GRIP (Lu et al., 2001). The anchoring of the Glu2/3A subunit of AMPA receptors to the membrane is mediated by GRIP, which is essential for the trafficking and stabilization of AMPA receptors and is involved in dendrite morphogenesis, synaptic plasticity, and homeostatic plasticity (Dong et al., 1997; Hoogenraad et al., 2005; Tan et al., 2015). The selective deletion of GRIP in the NAc potentiates the cue-induced reinstatement of cocaine seeking (Briand et al., 2014). A recent study found that the Grip1 gene was associated with alcohol preference in a bidirectionally selected rat model (Lo et al., 2016). Notably, we found that GRIP1 expression decreased after the retrieval of drug cue memories, which was blocked by calpain inhibitors. The knock-down of GRIP1 in the NAc core before CPP training had no effect on the acquisition of cocaine-induced CPP, which is consistent with a previous study finding that GRIP deletion did not affect the acquisition of operant food responding or self-administration (Briand et al., 2014). In addition, we found that GRIP1 knock-down by lentivirus-mediated shRNA in the NAc core blocked suppression of the expression of cocaine-induced CPP by an infusion of calpain inhibitors immediately after retrieval. These findings suggest that GRIP is involved in the process of drug cue memory reconsolidation that is regulated by calpain activity in the NAc core. However, some compensatory changes may occur after long-term GRIP knock-down that contribute to suppression of the effects of calpain inhibitors on drug cue memories.
Memory retrieval results in the exchange of GluA2-containing calcium-impermeable AMPA receptors to GluA2-lacking calcium-permeable AMPARs, indicating that GluA2 endocytosis may be critical to fear memory retrieval (Hong et al., 2013). An in vitro study found that calpain activation resulted in the disruption of GRIP binding to the GluA2 subunit of AMPA receptors and may play an important role in synaptic plasticity (Lu et al., 2001). The involvement of the GRIP1–GluA2 interaction in the reconsolidation of drug reward memory that is modulated by calpain activity requires further investigation.
Concluding remarks
In summary, calpain–GRIP1 signaling in the NAc core is required for the reconsolidation of drug cue memories in a reactivation- and time-dependent manner. The maladaptive memories of drug-associated stimuli have been suggested to be responsible for relapse in both animal models and humans. The present study provides new evidence of the role of calpain–GRIP signaling in the NAc in the reconsolidation of drug cue memories and may also open new opportunities for novel therapies for the treatment of drug addiction and the prevention of relapse.
Footnotes
This work was supported by the Natural Science Foundation of China (Grants 81225009, U1402226, 31571099, and 81521063), the National Basic Research Program of China (Grants 2015CB553503 and 2015CB856400), and Beijing Municipal Science and Technology Commission (Grants Z161100002616006).
The authors declare no competing financial interests.
References
- Alberini CM. (2005) Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trends Neurosci 28:51–56. 10.1016/j.tins.2004.11.001 [DOI] [PubMed] [Google Scholar]
- Amini M, Ma CL, Farazifard R, Zhu G, Zhang Y, Vanderluit J, Zoltewicz JS, Hage F, Savitt JM, Lagace DC, Slack RS, Beique JC, Baudry M, Greer PA, Bergeron R, Park DS (2013) Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP, and promotes neuronal survival following injury. J Neurosci 33:5773–5784. 10.1523/JNEUROSCI.4247-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres AL, Regev L, Phi L, Seese RR, Chen Y, Gall CM, Baram TZ (2013) NMDA receptor activation and calpain contribute to disruption of dendritic spines by the stress neuropeptide CRH. J Neurosci 33:16945–16960. 10.1523/JNEUROSCI.1445-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Bi X (2016) Calpain-1 and calpain-2: the Yin and Yang of synaptic plasticity and neurodegeneration. Trends Neurosci 39:235–245. 10.1016/j.tins.2016.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Chou MM, Bi X (2013) Targeting calpain in synaptic plasticity. Expert Opin Ther Targets 17:579–592. 10.1517/14728222.2013.766169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert JM, Marchant NJ, Calu DJ, Shaham Y (2013) The reinstatement model of drug relapse: recent neurobiological findings, emerging research topics, and translational research. Psychopharmacology (Berl) 229:453–476. 10.1007/s00213-013-3120-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Kimmey BA, Ortinski PI, Huganir RL, Pierce RC (2014) Disruption of glutamate receptor-interacting protein in nucleus accumbens enhances vulnerability to cocaine relapse. Neuropsychopharmacology 39:759–769. 10.1038/npp.2013.265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briz V, Baudry M (2016) Calpains: master regulators of synaptic plasticity. Neuroscientist. In press. 10.1177/1073858416649178 [DOI] [PubMed] [Google Scholar]
- Briz V, Hsu YT, Li Y, Lee E, Bi X, Baudry M (2013) Calpain-2-mediated PTEN degradation contributes to BDNF-induced stimulation of dendritic protein synthesis. J Neurosci 33:4317–4328. 10.1523/JNEUROSCI.4907-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai N, Liu JF, Xue YX, Yang C, Yan W, Wang HM, Luo YX, Shi HS, Wang JS, Bao YP, Meng SQ, Ding ZB, Wang XY, Lu L (2014) Delayed noradrenergic activation in the dorsal hippocampus promotes the long-term persistence of extinguished fear. Neuropsychopharmacology 39:1933–1945. 10.1038/npp.2014.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O'Brien CP (1999) Limbic activation during cue-induced cocaine craving. Am J Psychiatry 156:11–18. 10.1176/ajp.156.1.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dackis CA, O'Brien CP (2001) Cocaine dependence: a disease of the brain's reward centers. J Subst Abuse Treat 21:111–117. 10.1016/S0740-5472(01)00192-1 [DOI] [PubMed] [Google Scholar]
- Day JJ, Carelli RM (2007) The nucleus accumbens and Pavlovian reward learning. Neuroscientist 13:148–159. 10.1177/1073858406295854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZB, Wu P, Luo YX, Shi HS, Shen HW, Wang SJ, Lu L (2013) Region-specific role of Rac in nucleus accumbens core and basolateral amygdala in consolidation and reconsolidation of cocaine-associated cue memory in rats. Psychopharmacology (Berl) 228:427–437. 10.1007/s00213-013-3050-8 [DOI] [PubMed] [Google Scholar]
- Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL (1997) GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature 386:279–284. 10.1038/386279a0 [DOI] [PubMed] [Google Scholar]
- Fole A, Martin M, Morales L, Del Olmo N (2015) Effects of chronic cocaine treatment during adolescence in Lewis and Fischer-344 rats: novel location recognition impairment and changes in synaptic plasticity in adulthood. Neurobiol Learn Mem 123:179–186. 10.1016/j.nlm.2015.06.001 [DOI] [PubMed] [Google Scholar]
- Grant S, London ED, Newlin DB, Villemagne VL, Liu X, Contoreggi C, Phillips RL, Kimes AS, Margolin A (1996) Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci U S A 93:12040–12045. 10.1073/pnas.93.21.12040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Luo Y, Sun J, Ding Z, Liu J, Yan W, Jian M, Xue Y, Shi J, Wang JS, Lu L (2016) AMPK signaling in the dorsal hippocampus negatively regulates contextual fear memory formation. Neuropsychopharmacology 41:1849–1864. 10.1038/npp.2015.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong I, Kim J, Kim J, Lee S, Ko HG, Nader K, Kaang BK, Tsien RW, Choi S (2013) AMPA receptor exchange underlies transient memory destabilization on retrieval. Proc Natl Acad Sci U S A 110:8218–8223. 10.1073/pnas.1305235110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Milstein AD, Ethell IM, Henkemeyer M, Sheng M (2005) GRIP1 controls dendrite morphogenesis by regulating EphB receptor trafficking. Nat Neurosci 8:906–915. 10.1038/nn1487 [DOI] [PubMed] [Google Scholar]
- Jian M, Luo YX, Xue YX, Han Y, Shi HS, Liu JF, Yan W, Wu P, Meng SQ, Deng JH, Shen HW, Shi J, Lu L (2014) eIF2α dephosphorylation in basolateral amygdala mediates reconsolidation of drug memory. J Neurosci 34:10010–10021. 10.1523/JNEUROSCI.0934-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoutorsky A, Yanagiya A, Gkogkas CG, Fabian MR, Prager-Khoutorsky M, Cao R, Gamache K, Bouthiette F, Parsyan A, Sorge RE, Mogil JS, Nader K, Lacaille JC, Sonenberg N (2013) Control of synaptic plasticity and memory via suppression of poly(A)-binding protein. Neuron 78:298–311. 10.1016/j.neuron.2013.02.025 [DOI] [PubMed] [Google Scholar]
- Kilts CD, Schweitzer JB, Quinn CK, Gross RE, Faber TL, Muhammad F, Ely TD, Hoffman JM, Drexler KP (2001) Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry 58:334–341. 10.1001/archpsyc.58.4.334 [DOI] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ (2006) Cue-induced cocaine seeking and relapse are reduced by disruption of drug memory reconsolidation. J Neurosci 26:5881–5887. 10.1523/JNEUROSCI.0323-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FQ, Xue YX, Wang JS, Fang Q, Li YQ, Zhu WL, He YY, Liu JF, Xue LF, Shaham Y, Lu L (2010) Basolateral amygdala cdk5 activity mediates consolidation and reconsolidation of memories for cocaine cues. J Neurosci 30:10351–10359. 10.1523/JNEUROSCI.2112-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RW, Serwanski DR, Miralles CP, Li X, Charych E, Riquelme R, Huganir RL, de Blas AL (2005) GRIP1 in GABAergic synapses. J Comp Neurol 488:11–27. 10.1002/cne.20566 [DOI] [PubMed] [Google Scholar]
- Li YQ, Xue YX, He YY, Li FQ, Xue LF, Xu CM, Sacktor TC, Shaham Y, Lu L (2011) Inhibition of PKMzeta in nucleus accumbens core abolishes long-term drug reward memory. J Neurosci 31:5436–5446. 10.1523/JNEUROSCI.5884-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang Y, Zhu G, Sun J, Bi X, Baudry M (2016) A calpain-2 selective inhibitor enhances learning and memory by prolonging ERK activation. Neuropharmacology 105:471–477. 10.1016/j.neuropharm.2016.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CL, Lossie AC, Liang T, Liu Y, Xuei X, Lumeng L, Zhou FC, Muir WM (2016) High resolution genomic scans reveal genetic architecture controlling alcohol preference in bidirectionally selected rat model. PLoS Genet 12:e1006178. 10.1371/journal.pgen.1006178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Wyszynski M, Sheng M, Baudry M (2001) Proteolysis of glutamate receptor-interacting protein by calpain in rat brain: implications for synaptic plasticity. J Neurochem 77:1553–1560. 10.1046/j.1471-4159.2001.00359.x [DOI] [PubMed] [Google Scholar]
- Milton AL, Everitt BJ (2010) The psychological and neurochemical mechanisms of drug memory reconsolidation: implications for the treatment of addiction. Eur J Neurosci 31:2308–2319. 10.1111/j.1460-9568.2010.07249.x [DOI] [PubMed] [Google Scholar]
- Murakoshi H, Wang H, Yasuda R (2011) Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472:100–104. 10.1038/nature09823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Einarsson EO (2010) Memory reconsolidation: an update. Ann N Y Acad Sci 1191:27–41. 10.1111/j.1749-6632.2010.05443.x [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE (2000a) The labile nature of consolidation theory. Nat Rev Neurosci 1:216–219. 10.1038/35044580 [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE (2000b) Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406:722–726. 10.1038/35021052 [DOI] [PubMed] [Google Scholar]
- Nestler EJ. (2001) Neurobiology. Total recall–the memory of addiction. Science 292:2266–2267. 10.1126/science.1063024 [DOI] [PubMed] [Google Scholar]
- O'Brien CP, Childress AR, McLellan AT, Ehrman R (1992) Classical conditioning in drug-dependent humans. Ann N Y Acad Sci 654:400–415. 10.1111/j.1749-6632.1992.tb25984.x [DOI] [PubMed] [Google Scholar]
- Otis JM, Werner CT, Mueller D (2015) Noradrenergic regulation of fear and drug-associated memory reconsolidation. Neuropsychopharmacology 40:793–803. 10.1038/npp.2014.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickens CL, Airavaara M, Theberge F, Fanous S, Hope BT, Shaham Y (2011) Neurobiology of the incubation of drug craving. Trends Neurosci 34:411–420. 10.1016/j.tins.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren ZY, Liu MM, Xue YX, Ding ZB, Xue LF, Zhai SD, Lu L (2013) A critical role for protein degradation in the nucleus accumbens core in cocaine reward memory. Neuropsychopharmacology 38:778–790. 10.1038/npp.2012.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC (2003) Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37:577–582. 10.1016/S0896-6273(03)00021-7 [DOI] [PubMed] [Google Scholar]
- Self DW. (2004) Regulation of drug-taking and -seeking behaviors by neuroadaptations in the mesolimbic dopamine system. Neuropharmacology 47:242–255. 10.1016/j.neuropharm.2004.07.005 [DOI] [PubMed] [Google Scholar]
- Shimizu K, Phan T, Mansuy IM, Storm DR (2007) Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell 128:1219–1229. 10.1016/j.cell.2006.12.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg BA, Todd RP, Slaker M, Churchill L (2015) Anisomycin in the medial prefrontal cortex reduces reconsolidation of cocaine-associated memories in the rat self-administration model. Neuropharmacology 92:25–33. 10.1016/j.neuropharm.2014.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J, de Wit H, Eikelboom R (1984) Role of unconditioned and conditioned drug effects in the self-administration of opiates and stimulants. Psychol Rev 91:251–268. 10.1037/0033-295X.91.2.251 [DOI] [PubMed] [Google Scholar]
- Tan HL, Queenan BN, Huganir RL (2015) GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proc Natl Acad Sci U S A 112:10026–10031. 10.1073/pnas.1512786112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trovò L, Ahmed T, Callaerts-Vegh Z, Buzzi A, Bagni C, Chuah M, Vandendriessche T, D'Hooge R, Balschun D, Dotti CG (2013) Low hippocampal PI(4,5)P(2) contributes to reduced cognition in old mice as a result of loss of MARCKS. Nat Neurosci 16:449–455. 10.1038/nn.3342 [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411:583–587. 10.1038/35079077 [DOI] [PubMed] [Google Scholar]
- Valjent E, Corbillé AG, Bertran-Gonzalez J, Hervé D, Girault JA (2006) Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc Natl Acad Sci U S A 103:2932–2937. 10.1073/pnas.0511030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderklish PW, Krushel LA, Holst BH, Gally JA, Crossin KL, Edelman GM (2000) Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc Natl Acad Sci U S A 97:2253–2258. 10.1073/pnas.040565597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderklish P, Saido TC, Gall C, Arai A, Lynch G (1995) Proteolysis of spectrin by calpain accompanies theta-burst stimulation in cultured hippocampal slices. Brain Res Mol Brain Res 32:25–35. 10.1016/0169-328X(95)00057-Y [DOI] [PubMed] [Google Scholar]
- Voorn P, Gerfen CR, Groenewegen HJ (1989) Compartmental organization of the ventral striatum of the rat: immunohistochemical distribution of enkephalin, substance P, dopamine, and calcium-binding protein. J Comp Neurol 289:189–201. 10.1002/cne.902890202 [DOI] [PubMed] [Google Scholar]
- Wang CF, Huang YS (2012) Calpain 2 activated through N-methyl-D-aspartic acid receptor signaling cleaves CPEB3 and abrogates CPEB3-repressed translation in neurons. Mol Cell Biol 32:3321–3332. 10.1128/MCB.00296-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Luo YX, He YY, Li FQ, Shi HS, Xue LF, Xue YX, Lu L (2010) Nucleus accumbens core mammalian target of rapamycin signaling pathway is critical for cue-induced reinstatement of cocaine seeking in rats. J Neurosci 30:12632–12641. 10.1523/JNEUROSCI.1264-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Briz V, Chishti A, Bi X, Baudry M (2013) Distinct roles for mu-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration. J Neurosci 33:18880–18892. 10.1523/JNEUROSCI.3293-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhu G, Briz V, Hsu YT, Bi X, Baudry M (2014) A molecular brake controls the magnitude of long-term potentiation. Nat Commun 5:3051. 10.1038/ncomms4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AO, Kramár EA, López AJ, Kwapis JL, Doan J, Saldana D, Davatolhagh MF, Alaghband Y, Blurton-Jones M, Matheos DP, Wood MA (2016) BDNF rescues BAF53b-dependent synaptic plasticity and cocaine-associated memory in the nucleus accumbens. Nat Commun 7:11725. 10.1038/ncomms11725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Lynch DR (2006) Calpain and synaptic function. Mol Neurobiol 33:215–236. 10.1385/MN:33:3:215 [DOI] [PubMed] [Google Scholar]
- Wu P, Xue YX, Ding ZB, Xue LF, Xu CM, Lu L (2011) Glycogen synthase kinase 3beta in the basolateral amygdala is critical for the reconsolidation of cocaine reward memory. J Neurochem 118:113–125. 10.1111/j.1471-4159.2011.07277.x [DOI] [PubMed] [Google Scholar]
- Xue YX, Luo YX, Wu P, Shi HS, Xue LF, Chen C, Zhu WL, Ding ZB, Bao YP, Shi J, Epstein DH, Shaham Y, Lu L (2012) A memory retrieval-extinction procedure to prevent drug craving and relapse. Science 336:241–245. 10.1126/science.1215070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue YX, Xue LF, Liu JF, He J, Deng JH, Sun SC, Han HB, Luo YX, Xu LZ, Wu P, Lu L (2014) Depletion of perineuronal nets in the amygdala to enhance the erasure of drug memories. J Neurosci 34:6647–6658. 10.1523/JNEUROSCI.5390-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahm DS, Brog JS (1992) On the significance of subterritories in the “accumbens” part of the rat ventral striatum. Neuroscience 50:751–767. 10.1016/0306-4522(92)90202-D [DOI] [PubMed] [Google Scholar]
- Zhang RX, Han Y, Chen C, Xu LZ, Li JL, Chen N, Sun CY, Chen WH, Zhu WL, Shi J, Lu L (2016a) EphB2 in the medial prefrontal cortex regulates vulnerability to stress. Neuropsychopharmacology 41:2541–2556. 10.1038/npp.2016.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xue Y, Meng S, Luo Y, Liang J, Li J, Ai S, Sun C, Shen H, Zhu W, Wu P, Lu L, Shi J (2016b) Inhibition of lactate transport erases drug memory and prevents drug relapse. Biol Psychiatry 79:928–939. 10.1016/j.biopsych.2015.07.007 [DOI] [PubMed] [Google Scholar]
- Zhao LY, Zhang XL, Shi J, Epstein DH, Lu L (2009) Psychosocial stress after reactivation of drug-related memory impairs later recall in abstinent heroin addicts. Psychopharmacology (Berl) 203:599–608. 10.1007/s00213-008-1406-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Liu Y, Wang Y, Bi X, Baudry M (2015) Different patterns of electrical activity lead to long-term potentiation by activating different intracellular pathways. J Neurosci 35:621–633. 10.1523/JNEUROSCI.2193-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]