

Abstract
Control of Ca2+ flux between the cytosol and intracellular Ca2+ stores is essential for maintaining normal cellular function. It has been well established in both neuronal and non-neuronal cells that stromal interaction molecule 1 (STIM1) initiates and regulates refilling Ca2+ into the ER. Here, we describe a novel, additional role for STIM1, the regulation of free cytosolic Ca2+, and the consequent control of spike firing in neurons. Among central neurons, cerebellar Purkinje neurons express the highest level of STIM1, and they fire continuously in the absence of stimulation, making somatic Ca2+ homeostasis of particular importance. By using Purkinje neuron-specific STIM1 knock-out (STIM1PKO) male mice, we found that the deletion of STIM1 delayed clearance of cytosolic Ca2+ in the soma during ongoing neuronal firing. Deletion of STIM1 also reduced the Purkinje neuronal excitability and impaired intrinsic plasticity without affecting long-term synaptic plasticity. In vestibulo-ocular reflex learning, STIM1PKO male mice showed severe deficits in memory consolidation, whereas they were normal in memory acquisition. Our results suggest that STIM1 is critically involved in the regulation of the neuronal excitability and the intrinsic plasticity of the Purkinje neurons as well as cerebellar memory consolidation.
SIGNIFICANCE STATEMENT Stromal interaction molecule 1 (STIM1), which regulates the refilling of ER Ca2+, has been investigated in several systems including the CNS. In addition to a previous study showing that STIM1 regulates dendritic ER Ca2+ refilling and mGluR1-mediated synaptic transmission, we provide compelling evidence describing a novel role of STIM1 in spike firing Purkinje neurons. We found that STIM1 regulates cytosolic Ca2+ clearance of the soma during spike firing, and the interruption of this cytosolic Ca2+ clearing disrupts neuronal excitability and cerebellar memory consolidation. Our results provide new insights into neuronal functions of STIM1 from single neuronal Ca2+ dynamics to behavior level.
Keywords: cerebellum, intrinsic excitability, intrinsic plasticity, memory consolidation, Purkinje neuron, STIM1
Introduction
Ca2+ flux originating from both the extracellular space and from intracellular stores modifies cytosolic Ca2+ (Verkhratsky, 2005). The endoplasmic reticulum (ER) is the largest intracellular Ca2+ store and contributes to the dynamics of cytosolic Ca2+ by releasing stored Ca2+ through activation of ryanodine receptors (RyRs) and inositol-triphosphate receptors (IP3Rs). These forms of Ca2+ release are allowed only when ER Ca2+ stores are filled. Thus, refilling Ca2+ into the ER is an essential process for appropriate cytosolic Ca2+ dynamics. Stromal interaction molecule (STIM), an ER membrane-bounded protein, plays a central role in the refilling process by sensing Ca2+ concentration in the ER lumen ([Ca2+]ER). In response to low [Ca2+]ER, STIM oligomerizes and initiates the ER store refilling process in which ER and STIM localize in close apposition to the plasma membrane in association with proteins including store-operated Ca2+ channels (SOCs) and sarco/endoplasmic reticulum Ca2+-ATPase (SERCA; Soboloff et al., 2012). While well studied in other cellular systems, the neuronal functions of STIM have just begun to be identified (Hartmann et al., 2014; Sun et al., 2014; Zhang et al., 2015).
The Purkinje neuron (PN) is the central neuron of the cerebellar cortex. It broadly integrates sensory-motor information and provides the sole output of the cerebellar cortex (Dean et al., 2010). The ER in PNs is widely distributed, extending from dendritic spines, where synaptic plasticity occurs, to the soma, where final characteristics of neuronal output are determined (Terasaki et al., 1994). Cellular mechanisms responsible for learning and memory have been explained by the regulation of single neuronal input and output, most well represented by synaptic plasticity and intrinsic plasticity, respectively (Zhang and Linden, 2003; Kandel et al., 2014). Therefore, it would seem to be obvious that STIM in PNs could contribute to cerebellar learning and memory via Ca2+ dynamics involving the ER.
Although PNs have the highest expression of type 1 STIM (STIM1) among all brain regions (Skibinska-Kijek et al., 2009), only one study has reported the role of STIM1 in PNs. Hartmann et al. (2014) proposed STIM1 as a linker of metabotropic glutamate receptor type 1 (mGluR1) to downstream factors. Their model was based on the canonical function of STIM1, which regulates SOCs (TRPC/Orai channels) to refill ER Ca2+ stores, and provided us with the new insight that STIM1 as an mGluR1 linker performs roles in regulating normal synaptic transmission by maintaining ER Ca2+ stores. However, the experiments that suggested this model were performed in dendritic regions and in voltage-clamp (VC; −70 mV) cells where spiking was completely suppressed.
Several types of neurons including PNs are featured to fire spikes spontaneously. Spikes inevitably accompany Ca2+ influx from extracellular space into the soma where action potentials (APs) occur (Raman and Bean, 1999). To maintain spike firing properly, such neurons are equipped to handle this Ca2+ influx. STIM1 is strongly expressed in the soma of PNs (Hartmann et al., 2014; Fig. 1B). In this study, we asked how STIM1 participates in the handling of somatic Ca2+ influx and regulates intrinsic firing of PNs. By using PN-specific STIM1 knock-out (KO; STIM1PKO) mice, we found that STIM1 contributes to SERCA-dependent cytosolic Ca2+ clearance of the soma in firing PNs, which affects intrinsic firing properties. Furthermore, the deletion of STIM1 caused a severe impairment in the intrinsic plasticity of PNs. STIM1PKO mice showed defects in memory consolidation of several types of vestibulo-ocular reflex (VOR) learning, including gain-up, gain-down, and phase reversal. Our study not only complements the roles of STIM1 in cerebellar learning, but also elucidates the function of STIM1 that is unique to neurons.
Figure 1.

STIM1 deletion altered spike-evoked somatic Ca2+ dynamics of PNs. A, Schematic strategy for generating PN-specific STIM1PKO mice. B, STIM1 was completely abolished in STIM1PKO PNs. Calbindin (green), STIM1 (red), merged from left to right. The inset shows a magnified single-cell image. Scale bars, 200 μm. C, Purkinje neuron morphology was not different in both groups. Representative image of PNs in wild-type and STIM1PKO mice. Area of soma and dendrite was the same (wild-type, n = 34; STIM1PKO, n = 25; soma, p = 0.264; dendrite, p = 0.754). Scale bars, 50 μm. D, Ca2+ propagation patterns by different stimulations. Ca2+ measured sites in a single PN (inset). All sites showed Ca2+ transient by 500 ms of depolarization (left). While Ca2+ continuously accumulated in the soma (black), there was almost no response in dendrites (red, blue, and purple) in 24 Hz of AP clamping (right). E, Somatic Ca2+ accumulation under same spontaneous firing rate. STIM1PKO mice showed more rapid accumulation of Ca2+ during 24 Hz of AP clamping (left). At the 30 s point, the changed Ca2+ signal ratio of STIM1PKO was significantly higher than that of wild-type (wild-type, n = 7; STIM1PKO, n = 7; p < 0.001, right). Lines and shaded areas show mean and SEM. F, Somatic Ca2+ transient after 500 ms of depolarization. Representative trace with single exponential curve fitting (left). Decaying τ of STIM1PKO mice was significantly longer than that of wild-type littermates (wild-type, n = 15; STIM1PKO, n = 13; p = 0.008, center) with unchanged peak amplitude (p = 0.554, right). G, Dendritic Ca2+ transient after 500 ms of depolarization. We analyzed confined regions of dendrites (distance from the center of soma, ∼65 μm) to exclude the deviation by spatial location of dendrites. No difference was found in both decaying τ and peak amplitude between STIM1PKO and wild-type littermates (wild-type, n = 10; STIM1PKO, n = 8; decaying τ, p = 0.13; center: peak, p = 0.31, right). Independent-samples t test used for all bar graphs. Error bars denote SEM. **p < 0.01; ***p < 0.001.
Materials and Methods
Animals
STIM1PKO mice were generated by crossing the homozygous PCP2-Cre line (B6.129-Tg(Pcp2-cre)2Mpin/J line from The Jackson Laboratory) with the STIM1-floxed line (C57BL/6 background). The first filial generation (F1) was crossed again with the STIM1-floxed line. Among the second filial generation (F2), male mice that were homozygous for floxed-STIM1 alleles were used for this study. We used male mice in all of the experiments. All procedures were approved by the Institutional Animal Care and Use Committee of Seoul National University College of Medicine.
Immunohistochemistry
Anesthetized 9- to 13-week-old mice were perfused with PBS (Life Technologies) and again with 4% paraformaldehyde (PFA; T&I). Brains were taken out and fixed in 4% PFA overnight. After embedding the tissues in paraffin, we obtained 5-μm-thick sagittal slices on slides by using a microtome (RM2145, Leica). Paraffin was removed with 100% xylene (Junsei Chemical), and xylene was washed out with 99.9%, 95%, 90%, 80%, and 70% ethanol (diluted from absolute ethanol; Sigma-Aldrich), sequentially. Afterward, slides were submerged in distilled water for hydration. Epitope retrieval was performed with heated citrate buffer, pH 6.0 (T&I). Next, slices were blocked with a serum solution containing PBS-T (0.3% Triton X-100) and 5% goat serum (Vector Laboratories) for 1 h at room temperature. The slices were then incubated overnight at 4°C with diluted primary antibodies, anti-STIM1 (rabbit, 1:500; Cell Signaling Technology) and calbindin (mouse, 1:500; Abcam). After washing in PBS, fluorescence-conjugated secondary antibodies, Alexa Fluor-488 and 568 (anti-rabbit, 1:500; anti-mouse, 1:500; Abcam), were used to treated the slices for 1 h at room temperature. Primary antibodies and secondary antibodies were diluted in serum solution and PBS, respectively. Images were acquired and processed using a confocal microscope (A1, Nikon) and NIS-Element software (Nikon).
Electrophysiology
Slice preparation.
Sagittal slices of the cerebellar vermis (250 μm thick) were obtained from 5- to 9-week-old mice using a vibratome (VT1200, Leica) after isoflurane anesthesia and decapitation. The slices were cut with ice-cold cutting solution containing the following (in mm): 75 sucrose, 75 NaCl, 2.5 KCl, 7 MgCl2, 0.5 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 25 glucose, bubbled with 95% O2 and 5% CO2. The slices were immediately moved to artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose bubbled with 95% O2 and 5% CO2. For recovery, slices were incubated at 32°C for 30 min and for a further 1 h at room temperature. All recordings were obtained within 8 h from recovery.
Cell-attached and whole-cell recordings.
Brain slices were placed in a submerged chamber and perfused with ACSF for at least 10 min before recording. Somatic whole-cell VC and current-clamp (CC) recordings were made at 29.5−30°C. We used recording pipettes (3–4 MΩ) filled with the following (in mm): 9 KCl, 10 KOH, 120 K-gluconate, 3.48 MgCl2, 10 HEPES, 4 NaCl, 4 Na2ATP, 0.4 Na3GTP, and 17.5 sucrose, pH 7.25, for testing synaptic plasticity and current-clamp recordings; Cs-methanesulfonate 4 NaCl, 0.5 CaCl2, 10 HEPES, 2 MgATP, and 5 EGTA, pH 7.3, for miniature EPSC (mEPSC) recording, and nothing but 140 Cs-methanesulfonate was replaced by 140 CsCl for miniature IPSC (mIPSC) recording. For cell-attached recordings, experiments were performed at 32–32.5°C by using recording pipettes (4–5 MΩ) filled with the following (in mm): 125 NaCl, 10 HEPES, 3 KCl, and 2 CaCl2. Low Ca2+-ACSF contained a lower Ca2+ concentration (100 μm), and a reduced amount of CaCl2 was substituted with an equivalent amount of MgCl2. Data were acquired using an EPC9 patch-clamp amplifier (HEKA Elektronik) and PatchMaster software (HEKA Elektronik) with a sampling frequency of 20 kHz, and the signals were filtered at 2 kHz. All electrophysiological recordings were acquired in Lobule III–V of cerebellar central vermis, except for the data presented in Figure 5A.
Figure 5.

Attenuated spontaneous and current injection-evoked spike firing in STIM1-deleted PNs. A, Representative traces of spontaneous firing from attached recording in the lobule III–V (left). Spontaneous simple spike firing frequency was considerably reduced in STIM1PKO. Reduced firing frequency of STIM1PKO was not only observed in anterior lobule (III–V; wild-type, n = 55; STIM1PKO, n = 43; p < 0.001), but also lobule X and the flocculus. (wild-type, n = 19; STIM1PKO, n = 30; p < 0.001). B, Distribution plot of corresponding interspike interval (left). CV was significantly increased in STIM1PKO mice (wild-type, n = 31; STIM1PKO, n = 26; p = 0.007), but CV2 was unaffected (p = 0.878). C, Representative trace at 600 pA of current injection. D, Evoked firing rate was decreased in STIM1PKO mice (wild-type, n = 60; STIM1PKO, n = 43; F = 7.221, p = 0.008, left). Bar graph at 600 pA of current injection (p = 0.002, right). E, The amplitude of mAHP significantly increased in STIM1PKO (F = 28.220, p < 0.001, left). Bar graph at 600 pA injection (p < 0.001, right). F, STIM1PKO showed strengthened SFA. Wild-type mice have the shorter duration of 10 spikes than STIM1PKO (left). Instantaneous firing frequency more steeply decreased in STIM1PKO (F = 11.367, p = 0.001, right). One-way ANOVA was used for A. Independent-samples t test was used for B. Two-way repeated-measures ANOVA was used for D–F, and asterisks in bar graphs in A, D, and E were marked by post hoc Tukey's test pairwise comparison. Error bars denote the SEM. **p < 0.01; ***p < 0.001.
Stimulations.
To induce long-term synaptic plasticity in the Purkinje neuron, parallel fibers (PFs) and climbing fibers (CFs) were stimulated by ACSF-containing glass pipettes placed onto the molecular layer (ML) and the granule cell layer (GCL), respectively. PF-stimulated EPSC as a test pulse was evoked every 15 s to avoid any plasticity in whole-cell VC mode. The baseline recording of the EPSC was made for 5–10 min, and the mode was switched to CC mode for plasticity induction. After induction, EPSC amplitude was monitored in VC mode. Recordings were excluded if the series resistance or paired-pulse ratio varied by >20%.
Drugs.
In the experiments for measuring intrinsic excitability (IE), all of the recordings were performed within the ACSF containing 10 μm 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline and 100 μm picrotoxin to block excitatory and inhibitory synaptic inputs, respectively. Other experiments that required synaptic event were performed with appropriate drug composition. All drugs used in the experiments, except for picrotoxin (Sigma-Aldrich), were purchased from Tocris Bioscience.
Data analysis.
All patch-clamp data, except for the data from mEPSC and mIPSC recordings, were imported and analyzed using Igor Pro (WaveMetrics). The mEPSC and mIPSC data were analyzed using Mini Analysis (Synaptosoft). Pooled data underwent further analysis using software custom built with LabView (National Instruments) and Matlab (MathWorks) routines. Membrane capacitance (Cm) was measured from a current trace of a 5 mV depolarizing voltage step (50 ms duration), and input resistance (Rin) was measured from the end of the voltage trace of 100 pA hyperpolarizing current step (1 s duration). Active membrane properties were analyzed from the action potential train induced by a 600 pA depolarizing current injection (1 s duration). The threshold was determined by measuring the membrane potential where its velocity entered the range of 30–60 mV/ms (Kim et al., 2012). AP amplitude was calculated as a difference between the threshold and positive peak. Upstroke and downstroke were measured as the maximal derivative of the voltage with respect to time (dV/dt) ratio of the rising and falling phases, respectively. The slope of postspike depolarization was measured as the slope between the negative peak of the target spike and the threshold of the next spike that covers the interaction potential window, while the interspike interval (ISI) was assessed as the time difference between positive peaks of the target spike and the next spike that covers both action potentials and interaction potential windows. Fast afterhyperpolarization (fAHP) and medium AHP (mAHP) were measured by subtracting the negative peak of the end of the action potential and the action potential train from both the threshold and −70 mV baseline. The coefficient of variation (CV) and the coefficient of variation of adjacent intervals (CV2) of ISIs were calculated by the method from a previous study (Wulff et al., 2009).
Behavioral tests
Surgical procedure.
Mice at the age of 7–12 weeks were used. A head fixation pedestal was formed with two nuts (M2) and four screws (M1.2 × 5.5). Nuts were placed on bregma and lambda of skull, and screws were implanted between the nuts. Mice were under isoflurane anesthesia during surgery. After surgery, at least 24 h of recovery time was given to mice.
Instrumentation.
The image of the eye of the mouse was taken by CCD camera (IPX-VGA210, IMPERX) with a lens (VS-LD 35, VST) and an infrared (IR) filter (LP830), and was processed into a PC through a camera link grabber board (PCI-1426, National Instruments). IR lighting was generated by IR-LED (DR4–56R-IR85, LVS), and an additional single IR-LED was placed around the camera to generate reference cornea reflex for calibration, which is described below. Optokinetic stimulation was applied by a drum, 50 cm in diameter, mounted on a motor (AKM22E-VBBNR-00, Kollomorgen). A custom-made turntable was also mounted on another motor (D061M-12-1310, Kollomorgen) for vestibular stimulation. Since both stimuli were generated by independent motors, visuo-vestibular mismatch stimulation could be applied. Data acquisition (DAQ) PCI board (PCI-6230, National Instruments) was responsible for the input and output between PC and motion. The acquired image data were processed by several virtual instruments written in LabView (National Instruments).
Recording preparation.
Before every recording, physostigmine salicylate solution (Eserine, Sigma-Aldrich) was treated for pupil dilatation control with brief isoflurane anesthetization. The concentration of Eserine solution was constantly increased from 0.1% to 0.15% and 0.2% because of drug resistance. To effectively washout the side effect of the anesthetic, mice were given a recovery phase for at least 20 min after Eserine treatment. After recovery, mice were restrained in a custom-built animal holder. The holder was placed in the center of the turntable.
Acclimation and calibration.
Acclimation began at least 24 h after surgery. Two sessions of acclimation were performed. During acclimation, the mouse was fixed onto a custom-made restrainer for 15 min without any stimulation. Calibration was performed during the day after 2 d of acclimation. Briefly, the purpose of calibration was to convert linear eye position to angular eye positions. As the results of calibration, we could calculate the radius of the pupil, which is an important value for calculating the gain and phase values of eye movement. The equation was used and the procedure for calibration was performed according to the study by Stahl et al., (2000). At the recording after calibration, the mouse and holder were placed in the position where calibration was performed.
Eye movement recordings.
Three basal ocular–motor responses, which are optokinetic response (OKR), VOR in dark (dVOR), and VOR in light (lVOR), were measured. For OKR, drum stimulation was provided in sinusoidal rotation with ±5° of rotation amplitude. For dVOR and lVOR, turntable stimulation was applied in sinusoidal rotation with ±5° of rotation amplitude. The only difference between dVOR and lVOR was under conditions of light off and on, respectively. Each response was recorded at four different rotating frequencies (0.1, 0.25, 0.5, and 1.0 Hz).
Learning protocols.
Associative visuo-vestibular stimulation was applied to induce dVOR learning at 0.5 Hz frequency. Drum and table simultaneously rotated with ±5° of amplitude in and out of phase. For gain-down and gain-up learning, the protocols contained three 10 min training sessions and four check points (see Figs. 12A, 13A). After daily learning, mice were placed in a completely dark condition for 24 h until the next learning session. Afterward, dVOR was measured again as prelearning check point and another daily learning session began. For phase reversal learning, the dVOR was recorded six times, before learning started and after finishing five learning sessions (see Fig. 13D). While gain-up and gain-down learning keep the protocol until the end of learning, the protocol of phase reversal contained different daily drum stimulation. On the first day, the turntable and drum rotated in phase with 5° of amplitude. The next day, the drum rotated 2.5° more than the turntable. On the third day, the final learning day, the drum rotated 5° than the turntable totally. Caging in the completely dark condition between learning sessions was the same as gain-down and gain-up. Each mouse was trained by only one protocol and did not learn multiple visuo-vestibular stimulations.
Figure 12.

STIM1PKO mice showed memory consolidation deficiency in VOR learning. A, Gain-down learning protocol: 5° of in-phase paired turntable and drum stimulation was applied three times and 10 min each; 0.5 Hz dVOR was recorded in prelearning and postlearning and every interval between learning (arrows in top bar diagram). B, Gain-down learning in short term. In both groups, memory acquired by gain-down learning remained perfect until 30 min and 1 h (wild-type, n = 10; STIM1PKO, n = 10; F = 0.284, p = 0.6). C, Both groups showed the same memory consolidation level for the gain value (right; 30 min, p = 0.063; 1 h, p = 0.796). D, Gain-down learning. STIM1PKO mice could keep up with wild-type mice at the end of the daily learning, but there were considerable differences between two groups in gain values on the next day (wild-type, n = 14; STIM1PKO, n = 14). E, Consolidation levels were significantly reduced in STIM1PKO mice (Day 1–2, p < 0.001; Day 2–3, p = 0.019). Two-way repeated-measures ANOVA was used for B, and D and asterisks were marked by a post hoc Fisher's LSD test pairwise comparison. Asterisks in C and E were marked by Mann–Whitney U test. Error bars denote the SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 13.

STIM1PKO mice showed memory consolidation deficiency in various VOR learning protocols. A, Gain-up learning protocol. Same as gain-down protocol but with out-of-phase paired stimulation. B, Gain-up learning. STIM1PKO mice learned the same as wild-type mice, but they lost almost all of learned memory by 24 h after learning (wild-type, n = 11; STIM1PKO, n = 11). C, Comparison with wild-type littermates, memory consolidation levels are substantially lower in STIM1PKO mice (Day 1–2, p = 0.001; Day 2–3, p = 0.040). D, Phase reversal learning protocol. Five learning sessions for each day. From day 1 to day 3, the rotating amplitude of the drum increases 2.5° each day from 5°. Turntable rotating amplitude was fixed to 5°. E, The normalized gain in phase reversal learning. At the beginning of days 2 and 3, the gain of STIM1PKO mice was significantly higher than that of wild-type mice (wild-type, n = 11; STIM1PKO, n = 11). F, Consolidation levels in gain values. STIM1PKO mice had considerably lower consolidation levels than wild-type mice in daily memory consolidation levels (Day 1–2, p = 0.028; Day 2–3, p = 0.034). G, Phase values in phase reversal learning. There were no differences in the end of entire learning (last point of the third day). Same as the gain graph; there were significant differences in the beginning point. H, Consolidation level in phase values. Wild-type mice have a much higher level of trained memory (Day 2–3, p < 0.001; Day 3–4. p = 0.001). Two-way repeated-measures ANOVA was used for B, E, and G, and asterisks were marked by post hoc Fisher's LSD test pairwise comparison. Asterisks in all bar graphs were marked by Mann–Whitney U test. Error bars denote the SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Data analysis.
The given stimulus and the response were fitted to sine curves. In the fitted curves, the gain value was obtained by calculating the ratio of the response amplitude to the stimulus amplitude. The time lag and the lead of response (Phase) were determined by calculating the phase difference between the two sine curves. For all these procedures, we used a data analysis tool custom-built in LabView. To measure the level of memory consolidation, ratio of the percentage of remaining memory to learned memory was calculated.
Two-photon Ca2+ imaging
Image acquisition.
For two-photon Ca2+ imaging, we used a multiphoton microscope (LSM 7 MP, Zeiss) equipped with a Ti:Sapphire laser (Chameleon Vision II, Coherent). Wide-field images were taken using a CCD camera (UM-300, Uniq Vision). Microscopes were equipped with water-immersion objectives [W Plan-Apochromat 20×/1.0 differential interference contrast (DIC) M27 75 mm, W Plan-Apochromat 63×/1.0 M27; Zeiss]. In this recording, depolarization stimuli were evoked by an EPC-8 amplifier (HEKA Elektronik). Slice preparation was the same as previously described. Cs-containing pipette solution was used for a brief depolarization experiment using the following (in mm): 150 Cs-methanesulfonate, 2 MgCl2*6H2O, 10 HEPES, 2 Na2ATP, and 0.4 Na3GTP with 200 μm Oregon BAPTA 488 Green-1 (OGB-1, Invitrogen) and 25 μm Alexa Fluor-594 hydrazide (Sigma-Aldrich). A K-containing pipette solution, which was used in synaptic plasticity and current-clamp recordings, with added OGB-1 and Alexa Fluor-594 was used for the AP clamping experiment. AP clamping was performed by using PatchMaster software (HEKA Elektronik), and the template of whole-cell spontaneous firing was chosen from the recordings that we had previously obtained from PNs. For sufficient dye diffusion, PNs were dialyzed for at least 30 min after membrane rupture. We obtained simultaneously both Ca2+-sensitive images (by 200 μm OGB-1) and Ca2+-insensitive images (by 25 μm Alexa Fluor-594) via two different optical filters (filter 1, 500–550 nm bandpass filter; filter 2, 575–610 nm bandpass filter).
Image analysis.
We obtained the ratio (r) of OGB-1 signals (G) to Alexa Fluor-594 signals (R; r = G/R). rmin (0.136) and rmax (0.926) were measured using a pipette solution with 200 μm OGB-1 and 25 μm Alexa Fluor-594 containing 10 mm BAPTA or 10 mm CaCl2, respectively. All of our Ca2+ imaging data did not exceed r = 0.8. We also measured the ratios at an intermediate Ca2+ concentration with several combinations of EGTA and CaCl2 (at 32°C: 1 mm EGTA and 0.3 mm CaCl2 for 91.6 nm free Ca2+; 1 mm EGTA and 0.5 mm CaCl2 for 213.2 nm free Ca2+). Ca2+ concentration was calculated using MAXCHELATOR (http://web.stanford.edu/∼cpatton/). We ensured that our data are within the linear range from the rmin (0.136) to r = 0.801 (213.2 nm free Ca2+; linear regression, R2 = 0.964). Data acquisition and analysis were performed using Zen 2010 software (Zeiss) and customized Matlab code.
Real-time quantitative PCR and Western blot
Sampling.
Sagittal slices of the cerebellar vermis (250 μm thick) were obtained from 7-week-old mice. Whole regions of the vermis were microdissected into Purkinje neuronal layer (PNL) plus ML and GCL plus white matter according to the visually detectable borderline between PNL and GCL under an illuminated stereomicroscope (Fig. 2A). To analyze the gene expression in Purkinje neurons while minimizing any contamination from other cell types, we used PNL plus ML only for quantitative PCR (qPCR).
Figure 2.
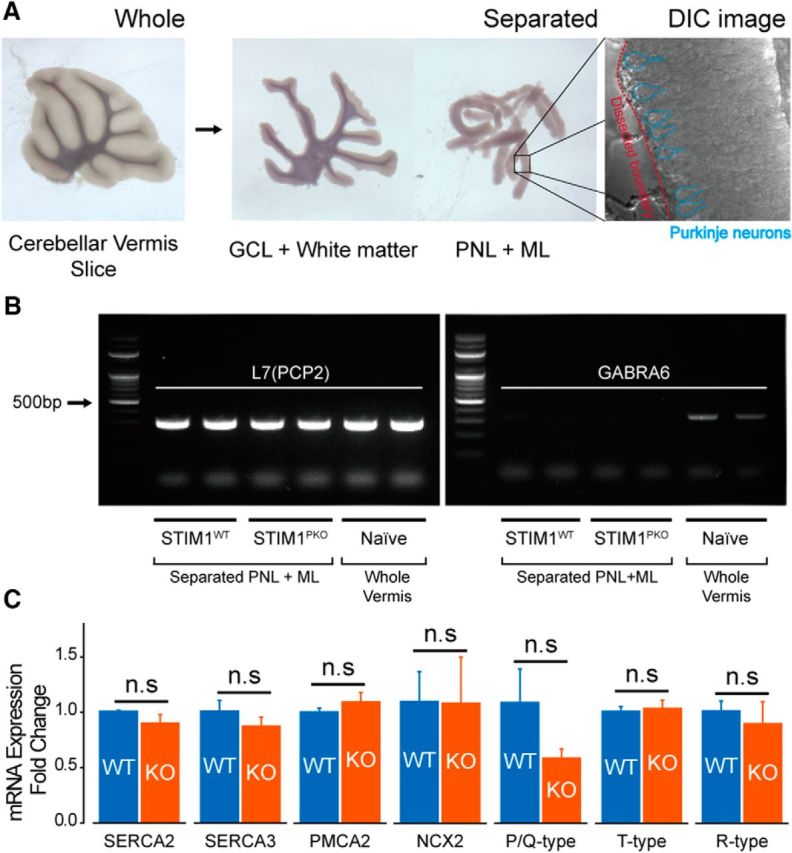
STIM1 deletion did not significantly change the mRNA expression of Ca2+ clearance/influx sources. A, Slices of the cerebellar vermis (left) were microdissected into PNL + ML and GCL + white matter (middle) according to the visually detectable borderline between PNL and GCL under an illuminated surgery microscope. A DIC microscopic image showed that a piece of separated PNL + ML tissue contained PNs (right). B, RT-PCR (30 cycles) with primers of Purkinje neuronal marker [L7(PCP2)] and granule cell marker (GABRA6) showed that separated PNL + ML showed a lack of genomic contents from granule cells. C, qPCR analysis of representative Ca2+ clearance and influx sources of PNs (number of samples from different animals: wild-type, n = 3; STIM1PKO, n = 3). From left to right: SERCA2 (p = 0.700), SERCA3 (p = 0.700), PMCA2 (p = 0.700), NCX2 (p > 0.999), P/Q-type VGCCs (Cav2.1; p = 0.100), T-type VGCCs (Cav3.1; p > 0.999), R-type VGCCs (Cav2.3; p > 0.999). Mann–Whitney U test was used for C. Error bars denote the SEM.
cDNA extraction and qPCR.
Total RNA was isolated from PNL plus ML tissues using the RNeasy Mini Kit (QIAGEN) according to the manufacturer instructions. To eliminate genomic DNA, DNase I (QIAGEN) was treated. A total of 350 ng of total RNA per sample was reverse transcribed into cDNA using SuperScript III First-Strand (Invitrogen). qPCR was performed using the CFX Connect system (Bio-Rad) with SYBR Premix (TaKaRa). Primers were designed by using Primer3 (http://primer3.ut.ee/) or were selected from the Harvard Primer Bank (http://pga.mgh.harvard.edu/primerbank/index.html). Primer sequences are available upon request. The mRNA expression levels of STIM1PKO relative to wild type were normalized according to the ΔΔCt method. All ΔCt values were normalized by Ct value of GAPDH.
Western blot.
Tissues were lysed with 1% Triton X-100, 300 mm NaCl, 50 mm Tris-HCl, 500 mm NaF, 200 Mm Na3VO4, protease inhibitor cocktail (P8340, Sigma-Aldrich) and phosphatase inhibitors (P5726 and P0044, Sigma-Aldrich). After quantification using BCA assay (Pierce BCA kit, Life Technologies), an equal amount of protein was loaded onto SDS-PAGE 8–12% acrylamide gels. After blocking using 5% skim milk in TBS-T (TBS with 0.1% Tween 20), blots were incubated overnight with primary antibodies at 4°C. The primary antibodies used were as follows: monoclonal mouse anti-calbindin (1:1000; CB955, Abcam); polyclonal goat anti-parvalbumin (1:1000; Swant); and monoclonal mouse anti-α-tubulin (1:10,000; TU-02, Santa Cruz Biotechnology). After washing with PBS-T, blots were then incubated for 1 h with secondary antibodies. The secondary antibodies used were as follows: HRP-conjugated goat anti-mouse IgG (1:1000-10000; Bio-Rad); HRP-conjugated goat anti-rabbit (1:1000; Bio-Rad); and HRP-conjugated rabbit anti-goat IgG (1:2000; Bio-Rad). Images were acquired using an Imager 600 (GE Healthcare) and were stored in JPG and TIFF file format. Protein quantification was performed using ImageJ software (National Institutes of Health), and each band density was normalized to α-tubulin in the same lane.
Statistics
Extracted data were managed with Origin 8.5 (OriginLab), with which simple statistics and curve fitting were carried out. Further statistical analysis was performed using SPSS Statistics 21 (IBM) and SigmaPlot version 12 (Systat Software). In parametric cases, an independent-samples t test was used and obtained two-tailed p values for comparing two groups, and ANOVA was used for comparison of more than three groups. In nonparametric cases, a Mann–Whitney U test was used.
Results
To generate PN-specific STIM1 KO (PCP2Cre;STIM1flox/flox, STIM1PKO) mice, we selectively removed loxP-floxed exon 6 (which is translated into the transmembrane domain of STIM1) of the Stim1 gene using the PCP2(L7)-Cre transgenic mouse line (Fig. 1A). Immunostaining showed that STIM1 protein was completely abolished only in PNs (Fig. 1B). Immunoreactivity of granule cell layers for STIM1 remained in both groups, showing that STIM1 knockout was confined to PNs (Fig. 1B, middle). We asked whether STIM1 deletion might result in morphological changes that could affect electrophysiological properties of neurons by comparing the shape and size of PNs in STIM1PKO mice with those in wild-type littermates (STIM1flox/flox). There was no significant difference in the size of the soma and in dendrite growth between PNs of two groups (wild-type, n = 34; STIM1PKO, n = 25; soma: wild-type, 416.710 ± 12.410 μm2; STIM1PKO, 396.365 ± 12.533 μm2, p = 0.254; dendrite: wild-type, 14345.2 ± 470.737 μm2; STIM1PKO, 14,140.2 ± 406.102 μm2; p = 0.743, independent-samples t test; Fig. 1C).
Altered cytosolic Ca2+ dynamics of STIM1-deleted PN soma
Pacemaking activity, which generates simple spikes spontaneously without synaptic input, is a distinguishing feature of PNs. Every single AP is accompanied by the opening of voltage-gated Ca2+ channels (VGCCs; Raman and Bean, 1999), and ER Ca2+ stores could be consistently restored by these abundant Ca2+ sources in excitable neurons. Therefore, the functional significance of STIM1 in spontaneously firing neurons may be different from that in nonexcitable cells, of which Ca2+ stores are mainly restored via SOCs. A previous study showed that dendritic Ca2+ level was not affected by simple spike firing because Na+ and K+ channels distributed in the soma and dendrites of PNs limit the back-propagation of simple spikes to their dendrites (Stuart and Häusser, 1994; Brenowitz et al., 2006; Ohtsuki et al., 2012; Masoli et al., 2015). Thus, we focused on somatic Ca2+ dynamics of firing PNs. The observation of Ca2+ transients by every single AP is an ideal way to identify the effect of STIM1 on somatic Ca2+ dynamics during neuronal firing; however, the duration of a single AP of PNs is too short (<1 ms) to detect the Ca2+ kinetics. Therefore, we observed the Ca2+ kinetics of clustered APs. We adopted the action potential-shaped voltage-clamp (AP clamp) technique, which applies voltage commands from real recorded APs. By using this technique, we were able to compare cytosolic Ca2+ kinetics between two groups during the firing of APs that were exactly the same. Since more frequent firing of APs causes more chances of Ca2+ influx into the cytosol, we had to fix the same firing rate of APs in both groups to compare the difference in Ca2+ dynamics during repetitive firing. We chose 24 Hz spontaneous firing of APs from our current-clamp recording as a template for the AP clamp. To confirm that the AP clamp voltage stimulus does not evoke a dendritic Ca2+ response, we compared Ca2+ signals between prolonged depolarization and spike-like depolarization by AP clamp (Fig. 1D). While prolonged depolarization (−70 to 0 mV, 500 ms) could evoke both somatic and dendritic Ca2+ transients (Fig. 1D, left), AP clamp voltage stimulus could evoke only somatic Ca2+ transients (Fig. 1D, right). In accordance with a previous study (Brenowitz et al., 2006), these results confirmed that simple spikes induce negligible effects on dendritic Ca2+ signals. In the AP clamp recordings, cytosolic Ca2+ of the soma is accumulated much more rapidly in STIM1PKO PNs than in wild type (wild type, 1.492 ± 0.058, n = 7; STIM1PKO, 1.932 ± 0.056, n = 7; p < 0.001, independent-samples t test; Fig. 1E). Either increased Ca2+ influx or delayed Ca2+ decay can account for this overaccumulation of cytosolic Ca2+ in STIM1PKO PNs. We measured the cytosolic Ca2+ transient of the soma after depolarizing voltage step (500 ms; −70 to 0 mV) in both wild-type and STIM1PKO PNs to validate which Ca2+ kinetics is affected by STIM1. The decaying kinetics of Ca2+ following depolarization was significantly delayed in STIM1PKO PNs without a difference in peak value of the Ca2+ transient (wild type, n = 15; STIM1PKO, n = 13; τdecay: wild-type, 3.434 ± 0.305 ms; STIM1PKO, 5.232 ± 0.583 ms, p < 0.001; peak: wild-type, 2.665 ± 0.167; STIM1PKO, 2.503 ± 0.217, p = 0.554, independent-samples t test; Fig. 1F).
To examine whether STIM1 deletion also affects dendritic calcium signaling, we recorded dendritic Ca2+ transients following depolarization (Fig. 1G). There was no difference in the decaying kinetics or amplitudes of dendritic Ca2+ transients between wild-type and STIM1PKO PNs, suggesting that STIM1 deletion does not affect dendritic calcium dynamics by depolarization (wild type, n = 10; STIM1PKO, n = 8; τdecay: wild type, 0.722 ± 0.038 ms; STIM1PKO, 0.644 ± 0.026 ms, p = 0.131; peak: wild-type, 1.858 ± 0.079; STIM1PKO, 1.982 ± 0.089, p = 0.312, independent-samples t test; Fig. 1G). Previous studies showed that STIM1 inhibits L-type VGCC (Cav1.2; Park et al., 2010; Wang et al., 2010). Unlike other cortical neurons, mature PNs express P/Q-type VGCCs (Cav2.1) as the predominant high voltage-activated VGCC, instead of the L-type VGCC (Usowicz et al., 1992). So far, there has been no report on whether STIM1 could also inhibit P/Q-type VGCCs. Our data suggest that STIM1 deletion did not affect the amount of Ca2+ influx through P/Q-type VGCCs.
We performed qPCR analyses to quantitatively compare the transcript levels of several Ca2+ clearance/influx sources mainly expressed in PNs, such as SERCA2/3, plasma-membrane Ca2+-ATPase 2 (PMCA2), Na+–Ca2+ exchanger 2 (NCX2), and VGCCs [P/Q-type (Cav2.1), T-type (Cav3.1) and R-type (Cav2.3)]. Tissues containing PNL and ML were microdissected to specifically measure the mRNA levels in PNs (Fig. 2A). We confirmed the specificity of tissue dissection by using reverse transcription PCR (RT-PCR) with primers for a Purkinje neuronal marker [L7(PCP2)] and a granule cell marker [GABAA receptor α6 subunit (GABRA6); Boyden et al., 2006; Fig. 2B]. Wild-type and STIM1PKO mice showed comparable mRNA levels for those genes (wild-type, n = 3; STIM1PKO, n = 3; SERCA2: wild-type, 1.007 ± 0.012; STIM1PKO, 0.090 ± 0.075, p = 0.700; SERCA3: wild-type, 1.010 ± 0.098; STIM1PKO, 0.087 ± 0.082, p = 0.700; PMCA2: wild-type, 1.004 ± 0.033; STIM1PKO, 1.094 ± 0.085, p = 0.700; NCX2: wild-type, 1.100 ± 0.266; STIM1PKO, 1.087 ± 0.417, p > 0.999; P/Q-type VGCC: wild-type, 1.091 ± 0.303; STIM1PKO, 0.588 ± 0.084, p = 0.100; T-type VGCC: wild-type, 1.012 ± 0.043; STIM1PKO, 1.037 ± 0.076, p > 0.999; R-type VGCC: wild-type, 1.017 ± 0.089; STIM1PKO, 0.900 ± 0.200, p > 0.999; Mann–Whitney U test; Fig. 2C). However, it may be worthy to note that we observed a fairly clear tendency of decreased mRNA expression level of P/Q-type VGCCs in the STIM1PKO group even though it did not reach a statistically significant level. In discordance with the qPCR result, our Ca2+ imaging data showed no significant difference of Ca2+ influx by depolarization between the two groups (Fig. 1F,G). These results indicated that gene expression levels could not explain the alterations in Ca2+ signals in STIM1PKO PNs.
Since PNs were well known for their high endogenous Ca2+ buffering (Fierro and Llano, 1996), delayed decaying kinetics of STIM1PKO PNs could be caused by alterations in the capacity of endogenous Ca2+ buffering. We measured the protein expression levels of calbindin D28K and parvalbumin, two representative Ca2+ buffers in PNs (Schmidt et al., 2003). There was a tendency toward decreased calbindin D28K and parvalbumin expression in STIM1PKO PNs, but it was not statistically significant (calbindin D28K: wild-type, 1.026 ± 0.117, n = 6; STIM1PKO, 0.757 ± 0.066, n = 5, p = 0.092; parvalbumin: wild-type, 1.324 ± 0.147, n = 6; STIM1PKO, 1.105 ± 0.116, n = 5, p = 0.286, independent-samples t test; Fig. 3A,B). We also quantified the basal levels of Ca2+ signals at −70 mV holding potential. The basal Ca2+ level of STIM1PKO PNs was slightly increased but was not statistically significant either (wild-type: 0.216 ± 0.024, n = 15; STIM1PKO: 0.295 ± 0.034, n = 13, p = 0.07, independent-samples t test; Fig. 3C).
Figure 3.

STIM1 deletion did not significantly change Ca2+ buffers and basal Ca2+ level. A, Representative Western blot bands for calbindin D28K and parvalbumin. B, There was no significant difference in the abundance of calbindin D28K and parvalbumin (wild-type, n = 6; STIM1PKO, n = 5; calbindin D28K, p = 0.092; parvalbumin, p = 0.286). Each protein expression level was normalized by α-tubulin in the same lane. C, There was no significant difference in basal Ca2+ signals at −70 mV holding potential (wild-type, n = 15; STIM1PKO, n = 13; p = 0.07). Independent-samples t test used for B and C. Error bars denote the SEM.
STIM1-mediated cytosolic Ca2+ dynamics of soma depends on SERCA
Delayed decaying kinetics indicates delayed clearance of Ca2+ from the cytosol after Ca2+ influx. Among various molecules associated with STIM1-driven ER Ca2+ store refilling (Soboloff et al., 2012), SERCA is the most appropriate candidate responsible for cytosolic Ca2+ clearance of PNs (Fierro et al., 1998). Previous studies demonstrated that SERCA, a final executor of the refilling process, is assisted by STIM1 (Krapivinsky et al., 2011). We tested the effect of blocking SERCA by cyclopiazonic acid (CPA) on somatic Ca2+ dynamics. We applied CPA during 24 Hz AP clamping in both groups. The accumulation of Ca2+ of wild-type PNs with CPA was similar to that of STIM1PKO with CPA at all time points. There were no significant differences among the three groups (wild-type with CPA, STIM1PKO with CPA, and STIM1PKO without CPA) at late time points [wild-type with CPA, n = 7; STIM1PKO, n = 7; STIM1PKO with CPA, n = 8; 30 s, wild-type with CPA, 1.958 ± 0.060; STIM1PKO with CPA, 2.041 ± 0.123; wild-type with CPA vs STIM1PKO, p = 0.831; STIM1PKO vs STIM1PKO with CPA, p = 0.359; wild-type with CPA vs STIM1PKO with CPA, p = 0.484, post hoc Fisher's least significant difference (LSD) test after one-way ANOVA; Fig. 4A]. We also compared the effect of SERCA blocking on somatic Ca2+ dynamics in 500 ms depolarization. CPA application delayed the decay kinetics of cytosolic Ca2+ more in both groups (wild-type with CPA, n = 9; STIM1PKO with CPA, n = 9; τdecay: wild-type with CPA, 6.858 ± 0.507 ms; STIM1PKO with CPA, 8.186 ± 0.837 ms; wild-type vs wild-type with CPA, p < 0.001; STIM1PKO vs STIM1PKO with CPA, p < 0.001; peak: wild-type with CPA, 1.922 ± 0.239; STIM1PKO with CPA, 2.073 ± 0.117, post hoc Fisher's LSD test after one-way ANOVA; Fig. 4B). However, decaying kinetics delayed by CPA of wild-type was not significantly different from that of STIM1PKO PNs (p = 0.132, post hoc Fisher LSD test after one-way ANOVA; Fig. 4B). These results indicate that STIM1 deletion caused hypofunction of SERCA-dependent cytosolic Ca2+ clearance.
Figure 4.

Blocking SERCA reproduced abnormal Ca2+ dynamics observed in STIM1PKO mice. A, CPA (30 μm) elevated the level of accumulated Ca2+ in wild-type mice to that of STIM1PKO mice. Under the same firing rate as achieved by AP clamping, Ca2+ more rapidly accumulated after CPA treatment (top). At the 30 s point, the changed Ca2+ signal ratio of wild-type mice was significantly lower than that of CPA-treated groups and STIM1PKO mice (wild-type, n = 7; STIM1PKO, n = 7; wild-type + CPA, n = 7; STIM1PKO + CPA, n = 8; F = 8.589, p < 0.001, bottom). Lines and shaded areas show the mean and SEM. B, Prolonged decaying τ of Ca2+ transient by CPA treatment. Representative traces with fitted single exponential curves (left). Decaying τ became significantly longer in CPA-treated groups (wild-type, n = 15; STIM1PKO, n = 13; wild-type + CPA, n = 9; STIM1PKO + CPA, n = 9; F = 14.492, p < 0.001, top right). Peak values were not different among all groups (F = 0.562, p < 0.643, bottom right). Drug-untreated data from wild-type and STIM1PKO mice were pooled from Figure 1. One-way ANOVA with post hoc Fisher's LSD test for statistics. Error bars denote the SEM. *p < 0.05; ***p < 0.001.
Altered neuronal excitability in STIM1PKO
To examine the functional consequence of STIM1 deletion in PNs, we measured the simple spike firing rate of STIM1PKO mice against that of wild-type mice while blocking both excitatory and inhibitory synaptic input by using cell-attached recording. PNs of STIM1PKO mice showed significantly lower firing rates in anterior lobules of cerebellar central vermis (Lobule III–V: wild-type, 37.308 ± 1.454 Hz, n = 55; STIM1PKO, 22.039 ± 1.344 Hz, n = 43; p < 0.001, post hoc Tukey's test after one-way ANOVA; Fig. 5A). It is well known that the firing rates of PNs correlate with localized zebrin (aldolase C) expression patterns (Zhou et al., 2014). To confirm whether reduced firing rates of STIM1PKO PNs depend on the location of PNs, we recorded the firing rates of PNs in lobule X and the flocculus, which were well known as purely zebrin-positive regions (Zhou et al., 2014). The firing rate is also significantly reduced in STIM1PKO PNs compared with wild-type PNs in these zebrin-positive regions (Lobule X and the flocculus, wild-type, 25.407 ± 1.629 Hz, n = 19; STIM1PKO, 15.222 ± 0.958 Hz, n = 30; p < 0.001, post hoc Tukey after one-way ANOVA; Fig. 5A), demonstrating that the reduction of firing rates in STIM1PKO PNs does not depend on the regional specificity by zebrin expression patterns. We also compared the regularity of PN firing. STIM1PKO PNs showed a larger CV for ISIs than in wild-type littermates (wild-type, 0.054 ± 0.002, n = 31; STIM1PKO 0.066 ± 0.004, n = 26, p = 0.007, independent-samples t test; Fig. 5B, left). However, CV2 values from both groups showed no significant difference (wild-type, 0.065 ± 0.003; STIM1PKO 0.066 ± 0.004, p = 0.878, independent-samples t test; Fig. 5B, right).
To investigate the details of spike properties, we injected step currents into PNs in current-clamp recording (wild-type, n = 60; STIM1PKO, n = 43; Fig. 5C–F). The firing frequency versus injected current (f–I) curve showed the reduced excitability of PNs in STIM1PKO mice [curve: p = 0.008, two-way repeated-measures (RM) ANOVA; +600 pA: wild-type, 69.133 ± 1.304; STIM1PKO, 61.860 ± 2.361; p = 0.002, post hoc Tukey's test after two-way RM ANOVA; Fig. 5D] with considerably increased mAHP following spike trains (curve: p < 0.001, two-way RM ANOVA; +600 pA: wild-type, 8.520 ± 0.255; STIM1PKO, 10.882 ± 0.344, p < 0.001, post hoc Tukey's test after two-way repeated-measures ANOVA; Fig. 5E). The properties of a single spike such as voltage threshold or amplitude were similar between the two groups (Table 1); however, spike frequency adaptation (SFA) was more prominent in STIM1PKO mice (p = 0.001, two-way repeated-measures ANOVA; Fig. 5F). Altered SFA indicates that STIM1 deletion changed the excitability of PNs qualitatively as well as quantitatively (Benda and Herz, 2003; Pozzorini et al., 2013). Among several mechanisms explaining SFA, the Ca2+-activated K+ current (KCa current) has been known as a determinant of mAHP in PNs (Belmeguenai et al., 2010). Based on both experimental data and theoretical modeling, previous studies have shown that the KCa-dependent SFA mechanism accompanies intracellular Ca2+ dynamics, including both Ca2+ flux to the cytosol and Ca2+ removal from the cytosol (Benda and Herz, 2003).
Table 1.
Passive and active membrane properties of wild-type and STIM1PKO mice
| Spike | Wild-type mice | STIM1PKO mice | p Value | |
|---|---|---|---|---|
| Cm (pF) | 644.9 ± 12.7 | 634.5 ± 14.0 | 0.586 | |
| Rin (MΩ) | 43.9 ± 1.1 | 38.1 ± 1.0 | <0.001*** | |
| First spike latency (ms) | 16.5 ± 0.5 | 18.0 ± 0.9 | 0.137 | |
| Rheobase (pA) | 261.1 ± 9.1 | 281.6 ± 11.2 | 0.159 | |
| Threshold (mV) | 1st spike | −47.0 ± 0.3 | −46.0 ± 0.5 | 0.080 |
| 25th spike | −43.1 ± 0.4 | −41.9 ± 0.5 | 0.070 | |
| AP amplitude (mV) | 1st spike | 82.0 ± 1.0 | 82.4 ± 1.1 | 0.775 |
| 25th spike | 70.6 ± 1.0 | 71.4 ± 1.0 | 0.545 | |
| FWHM (ms) | 1st spike | 0.205 ± 0.004 | 0.195 ± 0.003 | 0.031* |
| 25th spike | 0.226 ± 0.005 | 0.211 ± 0.003 | 0.012* | |
| Upstroke (V/s) | 1st spike | 542.6 ± 10.1 | 570.1 ± 10.3 | 0.059 |
| 25th spike | 421.0 ± 9.8 | 448.5 ± 8.8 | 0.039* | |
| Downstroke (V/s) | 1st spike | −452.9 ± 10.9 | −466.0 ± 9.6 | 0.368 |
| 25th spike | −368.8 ± 10.3 | −385.9 ± 9.1 | 0.214 | |
| Slope of postspike depolarization (V/s) | 1st spike | 1.69 ± 0.03 | 1.62 ± 0.05 | 0.234 |
| 25th spike | 1.19 ± 0.03 | 1.01 ± 0.04 | <0.001*** | |
| ISI (ms) | 1st spike | 10.4 ± 0.2 | 10.7 ± 0.3 | 0.402 |
| 25th spike | 14.4 ± 0.3 | 17.3 ± 0.8 | <0.001*** | |
| fAHP (mV) | 1st spike | 14.6 ± 0.3 | 14.3 ± 0.4 | 0.581 |
| 25th spike | 15.8 ± 0.3 | 15.9 ± 0.4 | 0.830 | |
| mAHP (mV) | 8.5 ± 0.3 | 10.9 ± 0.3 | <0.001*** |
Several parameters, including Rin, slope of postspike depolarization, ISI, and mAHP were significantly different in STIM1PKO mice compared to wild-type littermates (p < 0.001). Active properties of the 25th spike showed more differences than those of the 1st spike. Slope of postspike depolarization, especially, were remarkably lowered in STIM1PKO mice, which could explain the increase of ISI despite the shortened action potential waveform (p < 0.05). Up-stroke and down-stroke, rising and falling phase of action potential, respectively. Data were from the experiment performed in Figure 5. Data are expressed as the mean ± SEM.
*p < 0.05, independent-samples t test.
***p < 0.001, independent-samples t test.
In AP-clamp recordings, we assumed that normal PNs and STIM1-deleted PNs fire equally, more Ca2+ in the cytosol of STIM1PKO PNs accumulated during repetitive firing of APs in that condition (Fig. 1E). However, in actual PN firing, STIM1PKO PNs fire more slowly than wild-type PNs (Fig. 5A). If STIM1PKO PNs are not able to efficiently remove Ca2+ influx by a single AP from the cytosol, PNs should engage more negative feedback mechanisms to maintain the homeostasis of cytosolic Ca2+ concentration ([Ca2+]i). KCa current, which reduces the chances of Ca2+ influx by decelerating firing rates of neurons, is one of these feedback mechanisms. Reduced excitability in STIM1PKO PNs could be explained by the enhanced negative feedback mechanisms such as KCa current, which is recruited more by excessive cytosolic Ca2+ in STIM1-deleted PNs to prevent overaccumulation of cytosolic Ca2+.
Global effects of STIM1 deletion on Ca2+-dependent ionic currents
To determine whether the altered Ca2+ dynamics causes the changes in excitability of STIM1-deleted PNs, we recorded spontaneous firing while replacing most of the extracellular Ca2+ with Mg2+ to exclude Ca2+ effects on firing (wild-type, n = 15; STIM1PKO, n = 14; Fig. 6A). Lowering extracellular Ca2+ levels made the firing rates of the two groups equal and narrowed the gap between the two groups recovered under reperfusion of normal ACSF (baseline: wild-type, 28.165 ± 1.640 Hz; STIM1PKO, 16.212 ± 1.430 Hz, p < 0.001; peak: wild-type, 127.097 ± 9.701 Hz; STIM1PKO, 127.969 ± 8.140 Hz, p = 0.949; recovery: wild-type, 26.919 ± 2.490 Hz; STIM1PKO, 16.202 ± 1.397 Hz; p < 0.001, Mann–Whitney U test; Fig. 6B). These results confirmed that the difference in simple spike firing was attributed to Ca2+ dynamics. The changed Ca2+ dynamics could affect diverse kinds of Ca2+-dependent ionic currents. The broad effects of the KCa current on the excitability of PNs have been well established in previous studies (Edgerton and Reinhart, 2003; Benton et al., 2013), and, as mentioned above, altered parameters of STIM1PKO PNs, including increased mAHP and SFA, have been known to be related to KCa current (Benda and Herz, 2003; Belmeguenai et al., 2010). We tested the effects of KCa current on the firing of PNs in both groups by blocking or potentiating small and big conductance KCa channels [small-potassium (SK) and big-potassium (BK) channels, respectively]. During cell-attached recording, the application of apamin, a selective inhibitor of SK channels, was not able to narrow the gap in firing rates between the two groups (Fig. 6C). Additionally, two kinds of BK channel-selective blockers, iberiotoxin and paxilline, also could not narrow the gap in firing rates (Fig. 6D,E). Both groups fired equally while blocking both SK and BK channels (wild-type, n = 7; STIM1PKO, n = 9; baseline: wild-type, 36.973 ± 3.833 Hz; STIM1PKO, 24.756 ± 1.799 Hz; p = 0.003; maximum: wild-type, 151.429 ± 20.300 Hz; STIM1PKO, 123.333 ± 14.337 Hz; p = 0.351, Mann–Whitney U test; Fig. 6F). However, in both groups, the application of an apamine–paxilline cocktail induced burst firing of PNs (Fig. 6F), which is involved in dendritic Ca2+ signals (Brenowitz et al., 2006), so we could not precisely estimate the contribution of KCa channels to simple spike firing and somatic Ca2+ signals. 1-EBIO, a nonselective activator of KCa channels (Benton et al., 2013), decreased the firing rates of both groups to the same level without changing the firing pattern (wild-type, n = 7; STIM1PKO, n = 15; baseline: wild-type, 28.824 ± 2.554 Hz; STIM1PKO, 20.111 ± 1.010 Hz, p = 0.004; 25 min: wild-type, 11.673 ± 0.931 Hz; STIM1PKO, 9.772 ± 0.637 Hz; p = 0.078, Mann–Whitney U test; Fig. 6G). These results indicate that the alteration of PN activity in STIM1PKO cannot be explained by the modulation of a single channel, but several channels may be globally affected by changes in intracellular Ca2+ environments.
Figure 6.
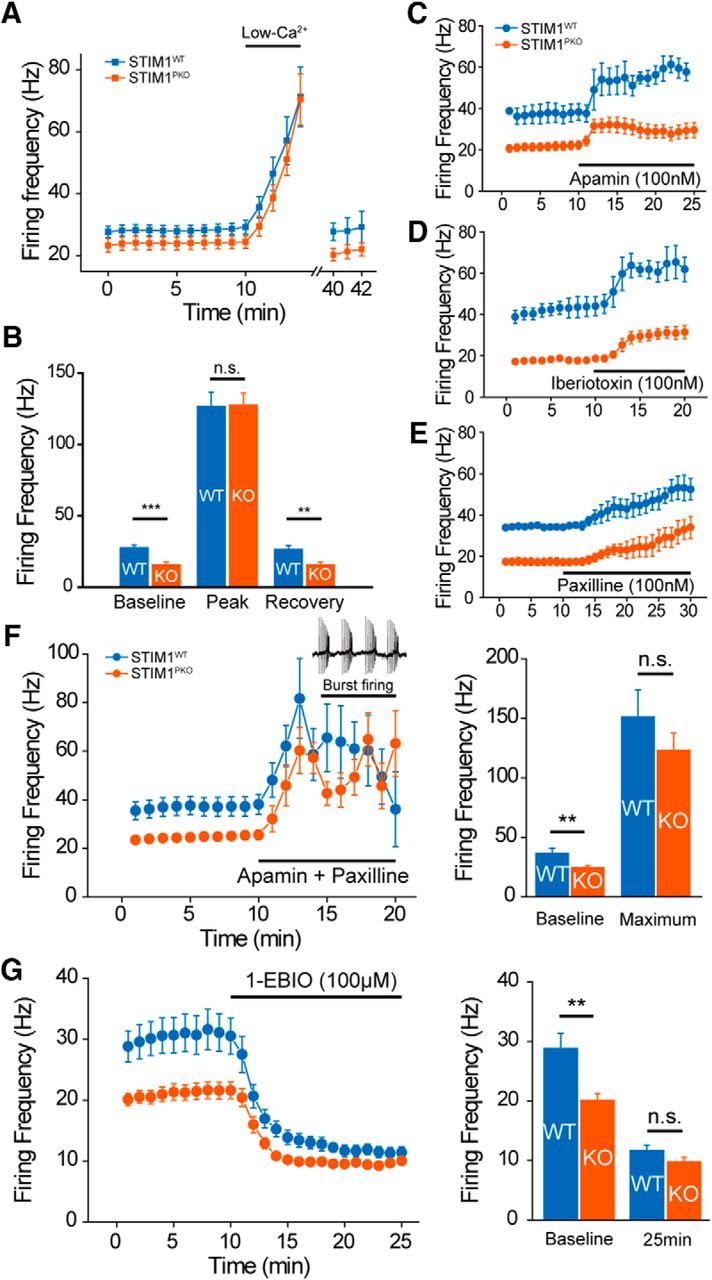
Altered excitability of STIM1PKO PNs required activation of Ca2+-activated K+ channels. A, Continuous cell-attached recording during the application of ACSF containing low Ca2+ concentration. Under low extracellular Ca2+ concentration (100 μm) firing rates of both groups increased and reached a similar peak point within 5 min. When extracellular Ca2+ concentration returned to normal (2 mm), the firing rate slowly recovered to the initial level (wild-type, n = 15; STIM1PKO, n = 14). B, Bar graph of low-Ca2+ contained ACSF application (right). Though PNs of STIM1PKO mice had significantly lower baseline firing rates than wild-type littermates at baseline (p < 0.001), the firing rates finally met at the peak (p = 0.949). When the firing rate recovered under normal ACSF, the gap between wild-type and STIM1PKO mice was also recovered (p = 0.003). C, Blocking SK channel by apamin (100 nm) increases the firing rates of both groups of PNs (wild-type, n = 3; STIM1PKO, n = 13). D, E, Blocking BK channel also raised spontaneous firing rate in both groups. D, Application of iberiotoxin (100 nm; wild-type, n = 3; STIM1PKO, n = 4). E, Paxilline, a broader spectrum blocker that is able to block iberiotoxin-insensitive BK currents (100 nm; wild-type, n = 2; STIM1PKO, n = 6) were not able to reduce the firing rate difference between two groups. F, Blocking both SK and BK channels with the cocktail of apamin (100 nm) and paxilline (100 nm). The mixture of drugs narrows the gap in firing rates between wild-type and STIM1PKO mice (wild-type, n = 7; STIM1PKO, n = 9). Although blocking both SK and BK channels suddenly changes the firing pattern (from tonic firing to burst firing), the firing frequencies of both groups became similar before pattern change (left). Bar graph in two time points (right). Baseline is before drug treatment, and maximum is the maximum firing rate before pattern change. In the comparison of maximum firing rates, there was no statistical significance between wild-type and STIM1PKO (baseline, p = 0.003; maximum, p = 0.351). G, Potentiating KCa channels with 1-EBIO (100 μm). 1-EBIO treatment decreased the difference between wild-type and STIM1PKO mice without changing the firing pattern (left; wild-type, n = 7; STIM1PKO, n = 15). The bar graph show the baseline and 15 min after drug application (right). At 15 min after 1-EBIO treatment, statistical significance between wild-type and STIM1PKO mice disappeared (baseline, p = 0.004; 25 min, p = 0.078). Asterisks in all bar graphs were marked by the Mann–Whitney U test. Error bars denote the SEM. **p < 0.01; ***p < 0.001.
Alterations in excitability depend on STIM1–SERCA interaction
STIM1 interacts with various molecules to maintain ER Ca2+ stores (Soboloff et al., 2012). STIM1 recruits SOCs to bring Ca2+ from extracellular space to cytosol, and then SERCA uptakes cytosolic Ca2+ into ER stores. Stored Ca2+ can be released to the cytosol by the influx of Ca2+, which is called Ca2+-induced Ca2+ release (CICR), which is mediated by RyRs. During cell-attached recording, pharmacological blocking of RyRs had no effect on spontaneous firing rates in both groups (Fig. 7). These results showed that CICR may not be involved in the reduced firing rates of STIM1-deleted PNs.
Figure 7.

Blocking Ca2+ release from ER store did not affect the spontaneous firing of PNs. A, Ca2+ transient after treating caffeine. Large Ca2+ transient was recorded by caffeine treatment (80 mm, puff application, 5 s, 5 psi) before the application of dantrolene (50 μm; black). After 20 min of bath application of the drug, Ca2+ response after caffeine treatment was reduced (red), and the response was abolished after 40 min of dantrolene application (blue). B, Blocking ryanodine receptor by dantrolene (50 μm) was not able to change the firing rate of PNs in both groups (wild-type, n = 6; STIM1PKO, n = 2).
We already showed that STIM1 deletion in PNs decreased the SERCA-dependent cytosolic Ca2+ clearance in the soma (Fig. 4). Few studies have revealed that sequestration of cytosolic Ca2+ by SERCA can influence the neuronal excitability (Cueni et al., 2008), but the pacemaking activity of PNs has never been studied in this regard. We examined whether SERCA-dependent Ca2+ signals could affect pacemaking activity. Blocking SERCA by CPA decreased the spontaneous firing rates of wild-type PNs to the level of STIM1PKO PNs. However, importantly, CPA treatment did not change the firing rates of STIM1PKO PNs (wild-type, n = 11; STIM1PKO, n = 9; inset Δfrequency: wild-type, −8.862 ± 1.616 Hz; STIM1PKO, −1.141 ± 0.979 Hz; p = 0.001, Mann–Whitney U test; Fig. 8A, bar graph; Table 2). To examine whether CPA affects SERCA in the soma or in dendrites, we also recorded the spontaneous firing rates during local CPA puffing onto the soma or dendrites of PNs. Only CPA puffing onto the soma (n = 4) significantly decreased the firing rates, whereas CPA puffing onto the dendrites (n = 4) did not decrease firing rates in wild-type slices. Consistent with the bath application experiment, CPA puffing onto the soma (n = 4) did not also alter firing rates in STIM1PKO (reduced firing rate: wild-type soma, 74.84 ± 10.69; wild-type dendrite, 103.7 ± 5.792; STIM1PKO soma, 105.5 ± 3.500; p = 0.027, one-way ANOVA; Fig. 8B). These data indicate that SERCAs in the soma have critical roles in regulating firing rates. We injected step currents under bath application of CPA. The drug reduced the gain of the f–I curve and increased both mAHP and SFA of wild-type PNs to the level of STIM1PKO PNs. Furthermore, CPA administration did not lead to significant changes in the gain of the f–I curve, mAHP, and SFA of STIM1PKO PNs (Fig. 8C–E; Table 2). These data strongly indicate that the reduced excitability of STIM1PKO PNs involved a SERCA-dependent mechanism. The results from blocking RyRs (Fig. 7) suggest that the altered excitability was not caused by the CPA-induced depletion of ER Ca2+ store and the consequent lack of Ca2+ release from the ER. Instead, the Ca2+ uptake process of SERCA known to remove and buffer cytosolic Ca2+ (Fierro et al., 1998; Higgins et al., 2006) could be a crucial step in STIM1-dependent regulation of the excitability. Our data suggest that STIM1 deletion causes less efficient SERCA-dependent cytosolic Ca2+ clearing, and delayed decaying kinetics of somatic Ca2+ enhances Ca2+-dependent ionic currents, which decrease the excitability of PNs to limit Ca2+ influx.
Figure 8.
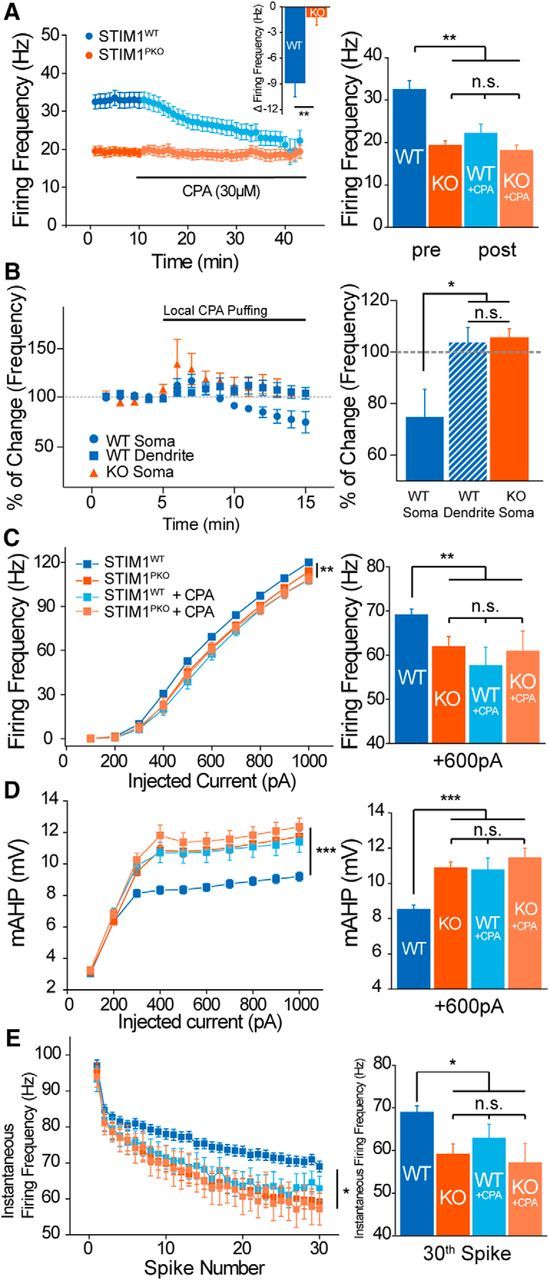
Blocking SERCA reproduced the attenuated intrinsic excitability of STIM1-deleted PNs. A, CPA treatment during the attached recording. CPA (30 μm) decreases firing rates of wild-type PNs to the level of STIM1PKO PNs, while PNs of STIM1PKO seem unaffected by CPA (wild-type, n = 11; STIM1PKO, n = 9). Reduced firing frequency of wild-type PNs was significantly larger than that of STIM1PKO PNs (p = 0.001, inset). Comparing initial firing rates to 30 min after CPA treatment, the initial firing rates of wild-type PNs are remarkably higher than others. B, Somatic SERCAs are important for the regulation of spontaneous firing rates. Local puffing of CPA (600 μm, 4 psi) onto the soma (n = 4, circle, blue) caused a reduction of firing rates, but there was no change in local CPA puffing onto dendrites of wild-type PNs (n = 4, square, blue) and the soma of STIM1PKO PNs (n = 4, triangle, red). After 10 min of treatment, the wild-type soma treatment group only showed significant reduction of firing rates (F = 5.556, p = 0.027). C, 30 min of CPA (30 μm) preincubation reduces the evoked firing rate of wild-type littermates. Wild-type littermates showed significantly higher evoked firing frequency than other groups (wild-type, n = 60; STIM1PKO, n = 43; wild-type + CPA, n = 17; STIM1PKO + CPA, n = 13). At the 600 pA point, the differences between wild-type mice and others are obvious. D, mAHP after preincubation of CPA. Only wild-type littermates were affected by the drug, and mAHP of wild-type littermates became same as other groups. The representative bar graph at 600 pA of current injection shows that wild-type mice had considerably smaller mAHP than other groups. E, CPA strengthened the SFA of wild-type PNs. The instantaneous firing frequency at the 30th spike was significantly lower in the other three groups than wild-type littermates. Data from drug-untreated wild-type and STIM1PKO mice, except A and B, were pooled from Figure 5. One-way ANOVA was used for B, and other details of data and statistics see also Table 2. Error bars denote the SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Table 2.
Statistical table of CPA treatment in Figure 8
| Figure 8A | Fig. 8C | Fig. 8D | Fig. 8E | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Firing frequency (Hz) |
Firing frequency (Hz) |
mAHP (mV) |
Firing frequency (Hz) |
|||||||
| n | Mean | SEM | n | Mean | SEM | Mean | SEM | Mean | SEM | |
| WT | 11 | 32.876 | 1.957 | 60 | 69.133 | 1.304 | 8.520 | 0.255 | 68.967 | 1.510 |
| KO | 9 | 19.300 | 1.121 | 43 | 61.860 | 2.361 | 10.882 | 0.344 | 59.146 | 2.411 |
| WT + CPA | 11 | 24.538 | 2.099 | 17 | 57.588 | 4.168 | 10.762 | 0.669 | 62.909 | 3.312 |
| KO + CPA | 9 | 18.472 | 1.318 | 13 | 60.846 | 4.578 | 11.452 | 0.544 | 57.143 | 4.479 |
| One-way ANOVA |
Two-way RM ANOVA |
|||||||
|---|---|---|---|---|---|---|---|---|
| F | p | F | p | F | p | F | p | |
| Statistics | ||||||||
| WT vs KO | 14.338 | <0.001 | 7.221 | 0.008 | 28.11 | <0.001 | 11.367 | 0.001 |
| WT vs WT + CPA | 12.021 | <0.001 | 12.619 | <0.001 | 5.491 | 0.022 | ||
| WT vs KO + CPA | 8.939 | 0.004 | 21.503 | <0.001 | 6.519 | 0.013 | ||
| KO vs WT + CPA | 0.81 | 0.372 | 0.00163 | 0.968 | 0.109 | 0.743 | ||
| KO vs KO + CPA | 0.51 | 0.478 | 1.25 | 0.269 | 0.0199 | 0.888 | ||
| WT + CPA vs KO + CPA | 0.00749 | 0.932 | 0.734 | 0.399 | 0.154 | 0.698 | ||
| p value (post hoc Tukey's test) | ||||||||
| WT vs KO | <0.001 | 0.002 | <0.001 | <0.001 | ||||
| WT vs WT + CPA | 0.007 | <0.001 | <0.001 | 0.007 | ||||
| WT vs KO + CPA | <0.001 | 0.005 | <0.001 | 0.003 | ||||
| KO vs WT + CPA | 0.177 | 0.276 | 0.848 | |||||
| KO vs KO + CPA | 0.989 | 0.631 | 0.373 | |||||
| WT + CPA vs KO + CPA | 0.092 | 0.683 | 0.400 | |||||
First row, Mean and SEM values of bar graphs in each panel; second row, F and p values of each pair after ANOVA, but the values after one-way ANOVA were comparisons of the whole group; third row, p values of bar graphs in each panel after post hoc Tukey's test.
Preserved basal synaptic transmission in STIM1PKO
There was no difference between two groups in basal synaptic transmissions, including mEPSC (wild-type, n = 8; STIM1PKO, n = 11; amplitude: wild-type, 17.405 ± 0.761 pA; STIM1PKO, 16.211 ± 0.696 pA, p = 0.206; frequency: wild-type, 3.412 ± 0.681 Hz; STIM1PKO, 3.204 ± 0.506 Hz; p = 1.000, Mann–Whitney U test; Fig. 9A) and mIPSC (wild-type, n = 5; STIM1PKO, n = 7; amplitude: wild-type, 40.225 ± 1.381 pA; STIM1PKO, 42.800 ± 4.111 pA; p = 0.639; frequency: wild-type, 2.287 ± 0.503 Hz; STIM1PKO, 2.913 ± 0.745 Hz; p = 0.876, Mann–Whitney U test; Fig. 9B). We also recorded strong excitatory synaptic transmission by CF, which is essential for long-term depression (LTD) induction (Coesmans et al., 2004), as well as for the induction of other forms of PN output besides simple spikes, complex spikes (CSs). STIM1PKO mice had no differences in properties of CSs compared with wild-type littermates (wild-type, n = 43; STIM1PKO, n = 34; spikelets: wild-type, 4.468 ± 0.142; STIM1PKO, 4.500 ± 0.165; p = 0.884; CS duration: wild-type, 9.517 ± 0.535; STIM1PKO, 9.486 ± 0.511; p = 0.968, independent-samples t test; Fig. 9C,D).
Figure 9.

Basal synaptic transmission and climbing fiber activity remained intact in PNs of STIM1PKO mice. A, Miniature EPSCs. Representative trace of mEPSCs recorded from wild-type and STIM1PKO (top). Cumulative distribution plots of frequency (bottom left) and amplitude (bottom right). In both frequency and amplitude, there was no statistical significance between both groups (wild-type, n = 8; STIM1PKO, n = 11; amplitude, p = 0.206; frequency, p = 1.000). B, Miniature IPSCs. Data are presented same as in A, and there were no differences between wild-type and STIM1PKO mice (wild-type, n = 5; STIM1PKO, n = 7; amplitude, p = 0.639; frequency, p = 0.876). C, The number of CS spikelets in both groups was the same. Representative trace of CSs induced by CF stimulation from whole-cell recording (left). Average number (p = 0.884, center) and probability (right) of the spikelets were almost the same (wild-type, n = 30; STIM1PKO, n = 34). D, Properties of each spikelet showed no statistically significant differences between both groups. The peak amplitude of each spikelet was not significantly different between wild-type and STIM1PKO mice (first spike, p = 0.935; second spike, p = 0.792; third spike, p = 0.145; left). The times when each spikelet appeared (first spike, p = 0.412; second spike, p = 0.385; third spike, p = 0.494; fourth spike, p = 0.724; top, right) and the total spike duration (p = 0.968, bottom, right) were also unchanged. A Mann–Whitney U test was used for bar graphs in A and B. An independent-samples t test was used for C and D. Error bars denote the SEM.
Intrinsic plasticity, but not synaptic plasticity, was impaired in STIM1PKO mice
LTD and long-term potentiation (LTP) at glutamatergic synapses between PF inputs and PNs have been considered as critical cellular mechanisms underlying cerebellar learning and memory (Ito, 1982; De Zeeuw et al., 1998; Hansel et al., 2006; Jörntell and Hansel, 2006). It has been well known that LTD induction requires Ca2+ release from the ER store through IP3Rs (Inoue et al., 1998). Considering the ER Ca2+ refilling by STIM1, we examined whether STIM1 deletion affects long-term synaptic plasticity. Interestingly, STIM1PKO mice showed levels of LTP (wild-type, n = 11; STIM1PKO, n = 8; p = 0.952, two-way repeated-measures ANOVA; Fig. 10A) and LTD (wild-type, n = 10; STIM1PKO, n = 11; p = 0.653, two-way repeated-measures ANOVA; Fig. 10B) comparable to those of their wild-type littermates. We hypothesize that sufficient Ca2+ stores to induce synaptic plasticity may be filled by other ER store refilling processes, for instance, Ca2+ influx through VGCCs (further referred to in the Discussion section).
Figure 10.

Intrinsic plasticity, but not synaptic plasticity, was impaired in STIM1PKO mice. A, LTP was successfully induced in both groups (wild-type, n = 11; STIM1PKO, n = 8). Representative trace of EPSCs at preinduction and postinduction (left). Normalized EPSCs of both groups were the same in the entire time period (F = 0.004, p = 0.952, center) with unchanged paired-pulse ratio (right). B, LTD was also successfully induced in both groups with no differences (wild-type, n = 10; STIM1PKO, n = 11; F = 0.208, p = 0.653). Data were presented as in A. C, Intrinsic plasticity was abolished in STIM1PKO mice. While LTP-IE was induced by the LTP induction protocol (1 Hz, 300 times, 5 min) in the wild-type group, this plasticity was unable to be induced in STIM1PKO mice. Two-way repeated-measures ANOVA was used for plasticity graphs in all panels. An independent-samples t test was used for the bar graph in C. Error bars denote the SEM. *p < 0.05; **p < 0.01.
In addition to synaptic plasticity, intrinsic plasticity has been suggested to be critical for learning and memory (Schonewille et al., 2010; Peter et al., 2016). We investigated whether STIM1 deletion could also affect intrinsic plasticity as well as basal intrinsic excitability. We recorded LTP of IE (LTP-IE) after LTP induction (1 Hz PF stimulation for 5 min; Belmeguenai et al., 2010; Peter et al., 2016). Interestingly, LTP-IE was significantly impaired in STIM1PKO PNs compared with that in wild-type PNs (wild-type, n = 9; STIM1PKO, n = 9; p = 0.009, two-way repeated-measures ANOVA; Fig. 10C). These results indicate that STIM1 is required for intrinsic plasticity but not for synaptic plasticity.
Motor memory consolidation deficiency in STIM1PKO mice
The previous study (Hartmann et al., 2014) showed that STIM1PKO mice have defects in two types of motor learning tests, an elevated beam-balancing test and a rotarod test. However, an elevated beam-balancing test (Piot-Grosjean et al., 2001; Urakawa et al., 2007) or rotarod test (Rothwell et al., 2014; Scholz et al., 2015) reflects the function of various brain regions other than the cerebellum. Thus, these behavioral tests could not elucidate which specific steps of cerebellar learning are interrupted in STIM1PKO mice, and more appropriate behavioral tests are required to specify the rules of STIM1 in cerebellar circuits related to learning.
The VOR is a reflex movement of eyes that makes for the proper location of images on the retina during head movement. This reflex is characterized by its well known neural circuits involving the cerebellar cortex (Boyden et al., 2004; Ito, 2013). The adaptive learning of VOR is achieved by plastic changes in neural circuits including the cerebellar cortex and vestibular nuclei (VNs) in the brainstem. As the sole output of the cerebellar cortex, PNs compute and integrate two different kinds of sensory stimuli [vestibular stimuli via mossy fibers (MFs) and retinal slip via CF] and provide instructive signals for learning to vestibular nuclei. Thus, the VOR has been regarded as a suitable behavioral model to identify the cerebellar learning mechanisms at the neural circuit level, along with the eye-blink conditioning (Kloth et al., 2015).
Mice were prepared with a head-fixed pedestal for the test, and three different basal oculomotor responses were tested before the learning: OKR, dVOR, and lVOR (Fig. 11A). STIM1PKO mice showed the same levels as wild-type mice in these parameters, and these results indicate that STIM1PKO mice have normal visual and vestibular functions compared with wild-type mice (wild-type, n = 47; STIM1PKO, n = 39; OKR: gain, p = 0.253; phase, p = 0.203; dVOR: gain, p = 0.391; phase, p = 0.737; lVOR: gain, p = 0.052, phase: p = 0.611, two-way repeated-measures ANOVA; Fig. 11B–D).
Figure 11.
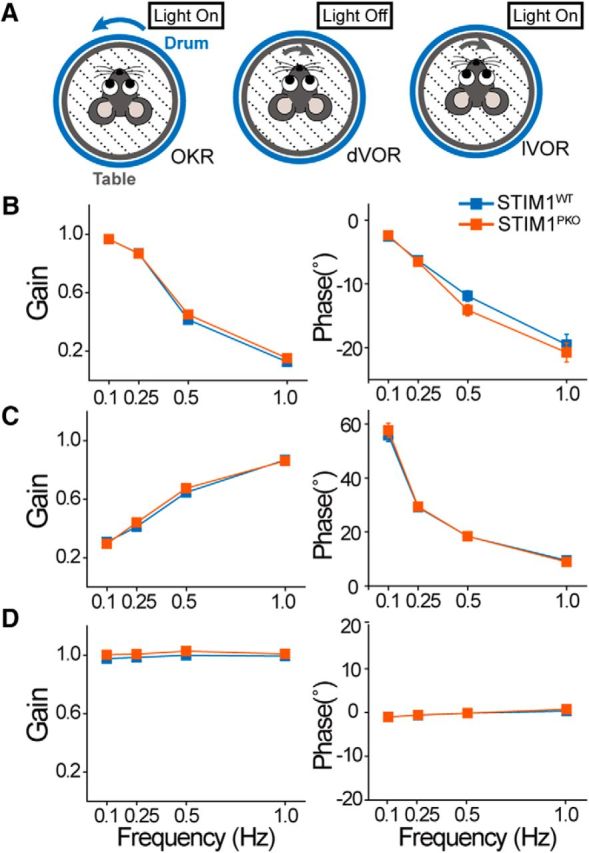
The basal ocular–motor performance of STIM1PKO mice was comparable to that of wild-type mice. A, Recording procedures. OKR, dVOR, and lVOR. B, OKR responses in different drum rotating frequencies. Gain and phase values were not significantly different from each other group (wild-type, n = 47; STIM1PKO, n = 39; gain: F = 1.324, p = 0.253; phase: F = 1.649, p = 0.203). C, dVOR responses in different drum rotation frequencies. Gain and phase values were not significantly different between groups (gain: F = 0.743, p = 0.391; phase: F = 0.114, p = 0.737). D, lVOR responses in different drum rotation frequencies. Gain and phase values were not significantly different between groups (gain: F = 3.873, p = 0.052; phase: F = 0.261, p = 0.611).
Visual stimuli in the same direction of vestibular stimuli induce a decrease of VOR gain (the ratio of eye velocities to head velocities; gain-down learning), whereas visual stimuli in the opposite direction of vestibular stimuli induce an increase of VOR gain (gain-up learning; Boyden et al., 2004). Adaptation of the VOR also occurs in the phase difference between the eye and head velocities as well as VOR gain (Wulff et al., 2009). We applied the gain-down protocol (Fig. 12A), and STIM1PKO mice successfully decreased gain to the same extent as wild-type littermates during learning sessions (wild-type, n = 11; STIM1PKO, n = 10; p = 0.6, two-way repeated-measures ANOVA; Fig. 12B). The memory of reduced gain was fully retained until 30 min or 1 h after the learning sessions in both groups (+30 min: wild-type, 100.053 ± 4.318%; STIM1PKO, 109.698 ± 2.478%; p = 0.063; +1 h: wild-type, 114.380 ± 3.575%; STIM1PKO, 115.485 ± 6.868%; p = 0.796, Mann–Whitney U test; Fig. 12C). To determine the effect of STIM1PKO on long-term memory retention, we extended learning procedure to 4 d with three 24 h intervals. STIM1PKO mice had much lower levels of retained memory than wild-type mice 24 h after the first day of learning (day 1–2; Fig. 12D). In a further 2 d of learning, STIM1PKO mice were able to catch up to the level of daily learning of wild-type mice but forgot most of the newly acquired memory on the next day (day 2–4; Fig. 12D). Therefore, the memory consolidation level (percentage change brought forward from learned memory of the previous day; Wulff et al., 2009) of STIM1PKO mice was considerably lower than that of the wild-type mice on both day 1–2 and day 2–3 intervals (wild-type, n = 14; STIM1PKO, n = 14; day 1–2: wild-type, 65.013 ± 5.817%; STIM1PKO, 25.014 ± 5.943%, p < 0.001, day 2–3: wild-type, 66.891 ± 5.225%; STIM1PKO, 44.015 ± 6.963%; p = 0.019, Mann–Whitney U test; Fig. 12E). Because there are mouse models showing defects only in specific types of eye movement learning paradigms (Boyden et al., 2006; Hansel et al., 2006), the gain-up learning protocol (Fig. 13A) was applied to check whether the same phenomenon occurs in that learning. As seen in gain-down learning, STIM1PKO mice had daily learning ability comparable to that of wild-type mice but lost most of what they had learned before 24 h, again showing reduced memory consolidation of learning (wild-type, n = 11; STIM1PKO, n = 10; day 1–2: wild-type, 84.291 ± 9.483%; STIM1PKO, 22.284 ± 9.842%; p = 0.001; day 2–3: wild-type, 67.069 ± 13.510%; STIM1PKO, 32.161 ± 7.585%; p = 0.040, Mann–Whitney U test; Fig. 13B,C). We asked whether the defect of STIM1PKO mice in memory consolidation exists not only in the amplitude (gain) of VOR, but also in the timing (phase). Both groups were tested in phase reversal learning, which reduces the gain on day 1 and afterward shifts the phase of VOR on days 2 and 3 (Fig. 13D; Wulff et al. (2009)). The protocol of the first-day learning was similar to that of gain-down learning but with two more learning sessions. During the learning on day 1, both groups performed the gain-down learning equally well without differences in both gain and phase values (day 1: wild-type, n = 11; STIM1PKO, n = 11; Fig. 13E,G), but after 24 h, the level of memory consolidation in gain was significantly different between two groups (day 1–2: wild-type, 63.899 ± 5.485%; STIM1PKO, 44.884 ± 5.567%; p = 0.028, Mann–Whitney U test; Fig. 13F). These results confirmed the defect of STIM1PKO mice by showing that the same phenomenon occurs even in more intensive learning. The phase was shifted from the second day of learning, and the memory consolidation deficiency of STIM1PKO mice was also reproduced in the phase learning (day 2–4; Fig. 13G; day 1–2: wild-type, 57.544 ± 8.372%; STIM1PKO, 15.367 ± 5.760%; p < 0.001; day 2–3: wild-type, 83.535 ± 6.823%; STIM1PKO, 32.796 ± 9.242%; p = 0.001, Mann–Whitney U test; Fig. 13H). On the third day of learning, the direction of learning in wild-type mice was changed from gain-down-bound to gain-up-bound (day 3; Fig. 13E), as previously reported (Wulff et al., 2009), since the phase of VOR was completely reversed as a consequence of accrued memory by repetitive phase shifting (day 3; Fig. 13G). However, STIM1PKO mice were not able to change the direction of alterations in gain due to the loss of the memory (day 3; Fig. 13E,G). Together, these results show that PN-specific STIM1 deletion significantly impairs the consolidation of cerebellar memory without affecting the acquisition.
Discussion
Most previous studies on STIM function including studies in the nervous system have focused on its canonical function in ER Ca2+ store refilling (Soboloff et al., 2012; Hartmann et al., 2014; Sun et al., 2014; Kraft, 2015; Zhang et al., 2015). Unlike other cell types where there are limited routes for Ca2+ entry, excitable neurons can frequently bring extracellular Ca2+ into intracellular space via VGCCs (Park et al., 2010; Harraz and Altier, 2014). In contrast to previous studies, our experiments have highlighted the novel function of STIM1 in firing PNs (Fig. 14). We observed STIM1-SERCA-dependent cytosolic Ca2+ kinetics of PN soma where the neuronal output of PNs is determined and demonstrated how this Ca2+ signal has influence on the PN excitability, including pacemaking activity. As a previous study about bursting neurons in nucleus reticularis thalami (nRT; Cueni et al., 2008) showed, the sequestration of cytosolic Ca2+ by SERCA is the significant factor for neuronal excitability, and we newly found that STIM1 mediates the Ca2+ sequestration and subsequently regulates neuronal excitability. It cannot yet be answered whether this sequestration is important for neuronal excitability only in high-frequency firing neurons, such as PNs and nRT neurons, or likewise in relatively slow-firing neurons such as cortical pyramidal neurons. However, it would be obvious that the relationship between this sequestration and neuronal excitability is critical for intracellular Ca2+ homeostasis in high-frequency firing neurons where Ca2+ influx via VGCC is more frequent.
Figure 14.

Schematic representation of STIM1 functions in the soma and dendrites of PNs. Left, A previous study (Hartmann et al., 2014) suggested that STIM1 is important for maintaining dendritic ER Ca2+ stores at rest. The regulation of dendritic Ca2+ homeostasis by STIM1 is essential for mGluR1-dependent synaptic transmission. Right, Our study described a novel, additional STIM1 function: the regulation of the somatic Ca2+ dynamics of firing neurons. The regulation of somatic Ca2+ dynamics, and thereby Ca2+-sensitive ion channels, by STIM1 contributes to normal neuronal firing. Solid lines indicate well established interactions, and dashed lines indicate interactions not fully described in detail. Arrows indicate activation pathways, and a blunt end indicates the inhibition pathway.
In addition to its role in the soma, STIM1 in dendrites could also participate in the regulation of PN output through mGluR1-mediated slow current (Hartmann et al., 2014). Although the physiological role of mGluR1-mediated slow current in PN output is not clear, this current evokes prolonged depolarization of PNs after fast AMPA-mediated depolarization (Kim et al., 2003; Hartmann et al., 2008). Since, in the voltage-clamp condition, this current is missing in STIM1PKO PNs, STIM1-null PNs may be impaired in the regulation of spikes induced by mGluR1-mediated prolonged depolarization in response to intense glutamatergic synaptic transmission, which remains to be investigated further.
We observed severe deficiency of memory consolidation in STIM1PKO mice, and these phenomena were not limited to a specific protocol, but occurred globally. It implies that the STIM1 deletion in PNs may affect the common pathway for cerebellar memory consolidation. STIM1-deleted PNs exhibited alterations in the neuronal excitability and its plasticity. Our results accord well with those of previous studies demonstrating that normal output of PNs is crucial for neuronal information transfer from the cerebellar cortex to the brainstem for successful memory consolidation (Wulff et al., 2009; Yamazaki et al., 2015).
Cytosolic Ca2+ clearance and buffering by STIM1-assisted SERCA
There are several molecular mechanisms that extrude cytosolic Ca2+ increased by depolarization, including SERCA, PMCA, NCX, and mitochondrial calcium uptake (Fierro et al., 1998). Among these mechanisms, SERCA and PMCA are known to be regulated by STIM1 combined with an adaptor protein called POST (partner of STIM1; Krapivinsky et al., 2011). Store depletion promotes the STIM1–POST complex to bind SERCA, keeping SERCA close to Ca2+ entry sites on the plasma membrane to promote ER Ca2+ refilling (Manjarrés et al., 2010, 2011; Krapivinsky et al., 2011). The STIM1–POST complex also inhibits PMCA activity, which removes cytosolic Ca2+ to extracellular space (Ritchie et al., 2012). With respect to cytosolic Ca2+ clearance, STIM1 seems to play opposite roles at the same time. However, a previous study showed that SERCA contributes more to cytosolic Ca2+ clearance than PMCA, especially in high [Ca2+]i conditions in PNs (Fierro et al., 1998). Thus, we deduce that the net effect of STIM1 on [Ca2+]i after depolarization works toward reducing the cytoplasmic concentration, and our data support this inference. There is no evidence that the STIM1–POST complex draws SERCA to the vicinity of VGCCs (distinct from SOCs). However, our data support this possibility because STIM1 does not affect the pumping activity of SERCA itself (Manjarrés et al., 2011). Delayed cytosolic Ca2+ decay and overaccumulated cytosolic Ca2+ during repetitive firing (Figs. 1, 4) suggest that STIM1 could contribute to the coupling between SERCA and Ca2+ entry through VGCCs. Moreover, SERCA itself has Ca2+ buffering activity (Higgins et al., 2006), which affects AHP and SFA. If STIM1 locates SERCA near VGCCs, SERCA could buffer depolarization-evoked Ca2+ influx more quickly. Proper localization of SERCA by STIM1 could optimize Ca2+ clearing and buffering function of SERCA, preventing excessive cytosolic Ca2+ during repetitive firing.
Intact LTP/LTD induction and related complementary Ca2+ refilling
Since the induction of postsynaptic LTP is known to be largely reliant on presynaptically released nitric oxide independently of postsynaptic Ca2+ (Lev-Ram et al., 2002), it seems plausible that LTP induction of STIM1PKO PNs is intact (Fig. 10A). To reach the required [Ca2+]i for inducing LTD, Ca2+ is driven by three major sources: influx from extracellular milieu through CF-induced VGCCs (Konnerth et al., 1992) or NMDA receptors located postsynaptically at CF–PN synapses (Piochon et al., 2010), and Ca2+ efflux via IP3R (Inoue et al., 1998). Given the result of Hartmann et al. (2014), which revealed the necessity of STIM1 for the spontaneous refilling of ER Ca2+ stores in PNs at resting potential, STIM1 might affect the induction of LTD. Contrary to this expectation, LTD was normally induced in STIM1PKO PNs (Fig. 10B). Depolarization-induced Ca2+ influx through VGCCs could compensate the mGluR1–STIM1-dependent inward current by transiently supercharging ER Ca2+ stores, as shown in the study by Hartmann et al. (2014). Because this compensatory refilling is too transitory (diminished within 3 min; data not shown), temporal coincidence between Ca2+ release from ER and prior depolarization is necessary for the successful induction of LTD. Hartmann et al. (2014) showed that STIM1PKO PNs had a defect in IP3R-dependent Ca2+ release at resting potential (−70 mV); however, PNs in vivo have pacemaking activity and are constantly depolarized. As mentioned above, spontaneous simple spike firing cannot evoke Ca2+ transience in dendrites (Brenowitz et al., 2006), but ER networks in PNs are continuously connected from the soma to dendrites (Terasaki et al., 1994). Therefore, Ca2+ introduced by spontaneous firing in the soma could replenish ER Ca2+ stores in dendrites. Moreover, strong depolarization evoked by CF during LTD induction might also perform this function. A recent study directly measured ER Ca2+ stores that were supplemented by 1 Hz CF stimulation as the same frequency we used in LTD induction (Okubo et al., 2015). As a result, Ca2+ release through IP3R needed for LTD induction could be sustained in vivo.
Memory consolidation of cerebellar learning and possible mechanisms
Many previous VOR studies have suggested that synaptic plasticity between PF and PN is crucial for memory acquisition (De Zeeuw et al., 1998; Hansel et al., 2006; Schonewille et al., 2010). Moreover, this acquired memory is stored instantly in the cerebellar cortex, where PF–PN synapses exist (Kassardjian et al., 2005; Shutoh et al., 2006; Okamoto et al., 2011). A recent study suggested that PF–PN plasticity in the cerebellar flocculus, which is known as the main region involved in cerebellum-dependent eye movement learning, has specific timing window for the induction different from that in the cerebellar vermis (Suvrathan et al., 2016). In light of the finding by Suvrathan et al. (2016), we speculate that STIM1PKO mice may also show intact synaptic plasticity in the flocculus since memory acquisition in VOR was normal in STIM1PKO mice (Figs. 10, 12, 13). Intact short-term (∼1 h) memory was also observed in STIM1PKO mice (Fig. 12B,C), which is consistent with the time range of synaptic plasticity that we recorded. However, examining whether synaptic plasticity is maintained for 24 h remains to be further investigated. It is interesting that STIM1PKO mice show severe impairment in intrinsic plasticity. Since intrinsic plasticity is induced by the same protocol for inducing synaptic plasticity, both forms of plasticity may occur together during memory acquisition. Many mouse models had deficits in both forms of plasticity as well as behavioral deficits in both acquisition and consolidation (Schonewille et al., 2010; Peter et al., 2016). To our knowledge, STIM1PKO is the first mouse model to show a selective deficit in intrinsic plasticity with intact synaptic plasticity in the cerebellum. Together with behavior data showing a deficit in memory consolidation, but not in acquisition, our results strongly suggest that intrinsic plasticity is a critical mechanism for memory consolidation in VOR learning. We speculate that new information may be acquired through PF–PN plasticity, which is subsequently consolidated in VNs through intrinsic plasticity.
In addition to intrinsic plasticity, the consolidation step may also require proper firing properties and patterns, which were significantly altered in STIM1PKO mice. PN output relays two types of neuronal information coding, rate coding and temporal coding (De Zeeuw et al., 2011). Previous studies showed that intrinsic plasticity is important for cerebellar learning because it alters both types of coding (Johansson et al., 2015; Grasselli et al., 2016). STIM1-deleted PNs showed the innate alterations both in rate (firing frequency) and temporal (CV and SFA) parameters. Diverse kinds of synaptic plasticity require highly precise timing for the coincidence of presynaptic and postsynaptic neuronal activity (Bi and Rubin, 2005). Several studies suggested that plasticity occurring beyond the cerebellar cortex is important for the consolidation of motor memory, such as MF–VN plasticity (Porrill and Dean, 2007; Ito, 2013; Yamazaki et al., 2015). PN output provides instructive signals to MF–VN plasticity (Pugh and Raman, 2006; Yamazaki et al., 2015). If PN output is transferred improperly and consequently induces temporal discord with postsynaptic activity, it could be possible that MF–VN plasticity is impaired and memory consolidation of the cerebellum is interrupted.
Footnotes
The authors declare no competing financial interests.
This work was supported by grants (the Medical Research Center, Grant 2012R1A5A2A44671346; Small Grant for Exploratory Research, Grant 2016R1D1A1A02937282; Global Ph.D. Fellowship Program, Grant 2013H1A2A1034318; and the Medical Researchers Support Program) from the National Research Foundation of Korea. We thank Professor Sascha du Lac (Johns Hopkins University) and her research group for helping with the installation of the VOR test system. We also thank Geehoon Chung, Jewel Park, Junho Hwang, Sang Bin Hong, and Ji Han Kim for comments on a prior version of the manuscript and for correcting errors in this manuscript.
References
- Belmeguenai A, Hosy E, Bengtsson F, Pedroarena CM, Piochon C, Teuling E, He Q, Ohtsuki G, De Jeu MT, Elgersma Y, De Zeeuw CI, Jörntell H, Hansel C (2010) Intrinsic plasticity complements long-term potentiation in parallel fiber input gain control in cerebellar Purkinje cells. J Neurosci 30:13630–13643. 10.1523/JNEUROSCI.3226-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benda J, Herz AV (2003) A universal model for spike-frequency adaptation. Neural Comput 15:2523–2564. 10.1162/089976603322385063 [DOI] [PubMed] [Google Scholar]
- Benton MD, Lewis AH, Bant JS, Raman IM (2013) Iberiotoxin-sensitive and -insensitive BK currents in Purkinje neuron somata. J Neurophysiol 109:2528–2541. 10.1152/jn.00127.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Rubin J (2005) Timing in synaptic plasticity: from detection to integration. Trends Neurosci 28:222–228. 10.1016/j.tins.2005.02.002 [DOI] [PubMed] [Google Scholar]
- Boyden ES, Katoh A, Raymond JL (2004) Cerebellum-dependent learning: the role of multiple plasticity mechanisms. Annu Rev Neurosci 27:581–609. 10.1146/annurev.neuro.27.070203.144238 [DOI] [PubMed] [Google Scholar]
- Boyden ES, Katoh A, Pyle JL, Chatila TA, Tsien RW, Raymond JL (2006) Selective engagement of plasticity mechanisms for motor memory storage. Neuron 51:823–834. 10.1016/j.neuron.2006.08.026 [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Best AR, Regehr WG (2006) Sustained elevation of dendritic calcium evokes widespread endocannabinoid release and suppression of synapses onto cerebellar Purkinje cells. J Neurosci 26:6841–6850. 10.1523/JNEUROSCI.1280-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coesmans M, Weber JT, De Zeeuw CI, Hansel C (2004) Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron 44:691–700. 10.1016/j.neuron.2004.10.031 [DOI] [PubMed] [Google Scholar]
- Cueni L, Canepari M, Luján R, Emmenegger Y, Watanabe M, Bond CT, Franken P, Adelman JP, Lüthi A (2008) T-type Ca2+ channels, SK2 channels and SERCAs gate sleep-related oscillations in thalamic dendrites. Nat Neurosci 11:683–692. 10.1038/nn.2124 [DOI] [PubMed] [Google Scholar]
- Dean P, Porrill J, Ekerot CF, Jörntell H (2010) The cerebellar microcircuit as an adaptive filter: experimental and computational evidence. Nat Rev Neurosci 11:30–43. 10.1038/nrn2756 [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, Oberdick J (1998) Expression of a Protein Kinase C Inhibitor in Purkinje Cells Blocks Cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron 20:495–508. 10.1016/S0896-6273(00)80990-3 [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Hoebeek FE, Bosman LW, Schonewille M, Witter L, Koekkoek SK (2011) Spatiotemporal firing patterns in the cerebellum. Nat Rev Neurosci 12:327–344. 10.1038/nrn3011 [DOI] [PubMed] [Google Scholar]
- Edgerton JR, Reinhart PH (2003) Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol 548:53–69. 10.1113/jphysiol.2002.027854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, Llano I (1996) High endogenous calcium buffering in Purkinje cells from rat cerebellar slices. J Physiol 496:617–625. 10.1113/jphysiol.1996.sp021713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, DiPolo R, Llano I (1998) Intracellular calcium clearance in Purkinje cell somata from rat cerebellar slices. J Physiol 510:499–512. 10.1111/j.1469-7793.1998.499bk.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasselli G, He Q, Wan V, Adelman JP, Ohtsuki G, Hansel C (2016) Activity-dependent plasticity of spike pauses in cerebellar Purkinje cells. Cell Rep 14:2546–2553. 10.1016/j.celrep.2016.02.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel C, de Jeu M, Belmeguenai A, Houtman SH, Buitendijk GH, Andreev D, De Zeeuw CI, Elgersma Y (2006) αCaMKII is essential for cerebellar LTD and motor learning. Neuron 51:835–843. 10.1016/j.neuron.2006.08.013 [DOI] [PubMed] [Google Scholar]
- Harraz OF, Altier C (2014) STIM1-mediated bidirectional regulation of Ca(2+) entry through voltage-gated calcium channels (VGCC) and calcium-release activated channels (CRAC). Front Cell Neurosci 8:43. 10.3389/fncel.2014.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Dragicevic E, Adelsberger H, Henning HA, Sumser M, Abramowitz J, Blum R, Dietrich A, Freichel M, Flockerzi V, Birnbaumer L, Konnerth A (2008) TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 59:392–398. 10.1016/j.neuron.2008.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann J, Karl RM, Alexander RP, Adelsberger H, Brill MS, Rühlmann C, Ansel A, Sakimura K, Baba Y, Kurosaki T, Misgeld T, Konnerth A (2014) STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 82:635–644. 10.1016/j.neuron.2014.03.027 [DOI] [PubMed] [Google Scholar]
- Higgins ER, Cannell MB, Sneyd J (2006) A buffering SERCA pump in models of calcium dynamics. Biophys J 91:151–163. 10.1529/biophysj.105.075747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Kato K, Kohda K, Mikoshiba K (1998) Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci 18:5366–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. (1982) Cerebellar control of the vestibulo-ocular reflex—around the flocculus hypothesis. Annu Rev Neurosci 5:275–296. 10.1146/annurev.ne.05.030182.001423 [DOI] [PubMed] [Google Scholar]
- Ito M. (2013) Error detection and representation in the olivo-cerebellar system. Front Neural Circuits 7:1. 10.3389/fncir.2013.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson F, Carlsson HA, Rasmussen A, Yeo CH, Hesslow G (2015) Activation of a temporal memory in Purkinje cells by the mGluR7 receptor. Cell Rep 13:1741–1746. 10.1016/j.celrep.2015.10.047 [DOI] [PubMed] [Google Scholar]
- Jörntell H, Hansel C (2006) Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron 52:227–238. 10.1016/j.neuron.2006.09.032 [DOI] [PubMed] [Google Scholar]
- Kandel ER, Dudai Y, Mayford MR (2014) The molecular and systems biology of memory. Cell 157:163–186. 10.1016/j.cell.2014.03.001 [DOI] [PubMed] [Google Scholar]
- Kassardjian CD, Tan YF, Chung JY, Heskin R, Peterson MJ, Broussard DM (2005) The site of a motor memory shifts with consolidation. J Neurosci 25:7979–7985. 10.1523/JNEUROSCI.2215-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Oh SH, Lee JH, Chang SO, Kim J, Kim SJ (2012) Lobule-specific membrane excitability of cerebellar Purkinje cells. J Physiol 590:273–288. 10.1113/jphysiol.2011.221846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ (2003) Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 426:285–291. 10.1038/nature02162 [DOI] [PubMed] [Google Scholar]
- Kloth AD, Badura A, Li A, Cherskov A, Connolly SG, Giovannucci A, Bangash MA, Grasselli G, Peñagarikano O, Piochon C, Tsai PT, Geschwind DH, Hansel C, Sahin M, Takumi T, Worley PF, Wang SS (2015) Cerebellar associative sensory learning defects in five mouse autism models. Elife 4:e06085. 10.7554/eLife.06085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Dreessen J, Augustine GJ (1992) Brief dendritic calcium signals initiate long-lasting synaptic depression in cerebellar Purkinje cells. Proc Natl Acad Sci U S A 89:7051–7055. 10.1073/pnas.89.15.7051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft R. (2015) STIM and ORAI proteins in the nervous system. Channels (Austin) 9:245–252. 10.1080/19336950.2015.1071747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Stotz SC, Manasian Y, Clapham DE (2011) POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc Natl Acad Sci U S A 108:19234–19239. 10.1073/pnas.1117231108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Ram V, Wong ST, Storm DR, Tsien RY (2002) A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc Natl Acad Sci U S A 99:8389–8393. 10.1073/pnas.122206399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjarrés IM, Rodríguez-García A, Alonso MT, García-Sancho J (2010) The sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) is the third element in capacitative calcium entry. Cell Calcium 47:412–418. 10.1016/j.ceca.2010.03.001 [DOI] [PubMed] [Google Scholar]
- Manjarrés IM, Alonso MT, García-Sancho J (2011) Calcium entry-calcium refilling (CECR) coupling between store-operated Ca2+ entry and sarco/endoplasmic reticulum Ca2+-ATPase. Cell Calcium 49:153–161. 10.1016/j.ceca.2011.01.007 [DOI] [PubMed] [Google Scholar]
- Masoli S, Solinas S, D'Angelo E (2015) Action potential processing in a detailed Purkinje cell model reveals a critical role for axonal compartmentalization. Front Cell Neurosci 9:47. 10.3389/fncel.2015.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki G, Piochon C, Adelman JP, Hansel C (2012) SK2 channel modulation contributes to compartment-specific dendritic plasticity in cerebellar Purkinje cells. Neuron 75:108–120. 10.1016/j.neuron.2012.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Endo S, Shirao T, Nagao S (2011) Role of cerebellar cortical protein synthesis in transfer of memory trace of cerebellum-dependent motor learning. J Neurosci 31:8958–8966. 10.1523/JNEUROSCI.1151-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okubo Y, Suzuki J, Kanemaru K, Nakamura N, Shibata T, Iino M (2015) Visualization of Ca2+ filling mechanisms upon synaptic inputs in the endoplasmic reticulum of cerebellar Purkinje cells. J Neurosci 35:15837–15846. 10.1523/JNEUROSCI.3487-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Shcheglovitov A, Dolmetsch R (2010) The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science (New York) 330:101–105. 10.1126/science.1191027 [DOI] [PubMed] [Google Scholar]
- Peter S, Ten Brinke MM, Stedehouder J, Reinelt CM, Wu B, Zhou H, Zhou K, Boele HJ, Kushner SA, Lee MG, Schmeisser MJ, Boeckers TM, Schonewille M, Hoebeek FE, De Zeeuw CI (2016) Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat Commun 7:12627. 10.1038/ncomms12627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piot-Grosjean O, Wahl F, Gobbo O, Stutzmann JM (2001) Assessment of sensorimotor and cognitive deficits induced by a moderate traumatic injury in the right parietal cortex of the rat. Neurobiol Dis 8:1082–1093. 10.1006/nbdi.2001.0450 [DOI] [PubMed] [Google Scholar]
- Piochon C, Levenes C, Ohtsuki G, Hansel C (2010) Purkinje cell NMDA receptors assume a key role in synaptic gain control in the mature cerebellum. J Neurosci 30:15330–15335. 10.1523/JNEUROSCI.4344-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrill J, Dean P (2007) Cerebellar motor learning: when is cortical plasticity not enough? PLoS Comput Biol 3:e197. 10.1371/journal.pcbi.0030197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzorini C, Naud R, Mensi S, Gerstner W (2013) Temporal whitening by power-law adaptation in neocortical neurons. Nat Neurosci 16:942–948. 10.1038/nn.3431 [DOI] [PubMed] [Google Scholar]
- Pugh JR, Raman IM (2006) Potentiation of mossy fiber EPSCs in the cerebellar nuclei by NMDA receptor activation followed by postinhibitory rebound current. Neuron 51:113–123. 10.1016/j.neuron.2006.05.021 [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP (1999) Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci 19:1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie MF, Samakai E, Soboloff J (2012) STIM1 is required for attenuation of PMCA-mediated Ca2+ clearance during T-cell activation. EMBO J 31:1123–1133. 10.1038/emboj.2011.495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Südhof TC (2014) Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158:198–212. 10.1016/j.cell.2014.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Stiefel KM, Racay P, Schwaller B, Eilers J (2003) Mutational analysis of dendritic Ca2+ kinetics in rodent Purkinje cells: role of parvalbumin and calbindin D28k. J Physiol 551:13–32. 10.1113/jphysiol.2002.035824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Niibori Y, Frankland PW, Lerch JP (2015) Rotarod training in mice is associated with changes in brain structure observable with multimodal MRI. Neuroimage 107:182–189. 10.1016/j.neuroimage.2014.12.003 [DOI] [PubMed] [Google Scholar]
- Schonewille M, Belmeguenai A, Koekkoek SK, Houtman SH, Boele HJ, van Beugen BJ, Gao Z, Badura A, Ohtsuki G, Amerika WE, Hosy E, Hoebeek FE, Elgersma Y, Hansel C, De Zeeuw CI (2010) Purkinje cell-specific knockout of the protein phosphatase PP2B impairs potentiation and cerebellar motor learning. Neuron 67:618–628. 10.1016/j.neuron.2010.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutoh F, Ohki M, Kitazawa H, Itohara S, Nagao S (2006) Memory trace of motor learning shifts transsynaptically from cerebellar cortex to nuclei for consolidation. Neuroscience 139:767–777. 10.1016/j.neuroscience.2005.12.035 [DOI] [PubMed] [Google Scholar]
- Skibinska-Kijek A, Wisniewska MB, Gruszczynska-Biegala J, Methner A, Kuznicki J (2009) Immunolocalization of STIM1 in the mouse brain. Acta Neurobiol Exp (Wars) 69:413–428. [DOI] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, Gill DL (2012) STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol 13:549–565. 10.1038/nrm3414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl JS, van Alphen AM, De Zeeuw CI (2000) A comparison of video and magnetic search coil recordings of mouse eye movements. J Neurosci Methods 99:101–110. 10.1016/S0165-0270(00)00218-1 [DOI] [PubMed] [Google Scholar]
- Stuart G, Häusser M (1994) Initiation and spread of sodium action potentials in cerebellar Purkinje cells. Neuron 13:703–712. 10.1016/0896-6273(94)90037-X [DOI] [PubMed] [Google Scholar]
- Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL 3rd, Bezprozvanny I (2014) Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82:79–93. 10.1016/j.neuron.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvrathan A, Payne HL, Raymond JL (2016) Timing rules for synaptic plasticity matched to behavioral function. Neuron 92:959–967. 10.1016/j.neuron.2016.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M, Slater NT, Fein A, Schmidek A, Reese TS (1994) Continuous network of endoplasmic reticulum in cerebellar Purkinje neurons. Proc Natl Acad Sci U S A 91:7510–7514. 10.1073/pnas.91.16.7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urakawa S, Hida H, Masuda T, Misumi S, Kim TS, Nishino H (2007) Environmental enrichment brings a beneficial effect on beam walking and enhances the migration of doublecortin-positive cells following striatal lesions in rats. Neuroscience 144:920–933. 10.1016/j.neuroscience.2006.10.038 [DOI] [PubMed] [Google Scholar]
- Usowicz MM, Sugimori M, Cherksey B, Llinás R (1992) P-type calcium channels in the somata and dendrites of adult cerebellar Purkinje cells. Neuron 9:1185–1199. 10.1016/0896-6273(92)90076-P [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 85:201–279. 10.1152/physrev.00004.2004 [DOI] [PubMed] [Google Scholar]
- Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL (2010) The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science 330:105–109. 10.1126/science.1191086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff P, Schonewille M, Renzi M, Viltono L, Sassoè-Pognetto M, Badura A, Gao Z, Hoebeek FE, van Dorp S, Wisden W, Farrant M, De Zeeuw CI (2009) Synaptic inhibition of Purkinje cells mediates consolidation of vestibulo-cerebellar motor learning. Nat Neurosci 12:1042–1049. 10.1038/nn.2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Nagao S, Lennon W, Tanaka S (2015) Modeling memory consolidation during posttraining periods in cerebellovestibular learning. Proc Natl Acad Sci U S A 112:3541–3546. 10.1073/pnas.1413798112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wu L, Pchitskaya E, Zakharova O, Saito T, Saido T, Bezprozvanny I (2015) Neuronal store-operated calcium entry and mushroom spine loss in amyloid precursor protein knock-in mouse model of Alzheimer's disease. J Neurosci 35:13275–13286. 10.1523/JNEUROSCI.1034-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ (2003) The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci 4:885–900. 10.1038/nrn1248 [DOI] [PubMed] [Google Scholar]
- Zhou H, Lin Z, Voges K, Ju C, Gao Z, Bosman LW, Ruigrok TJ, Hoebeek FE, De Zeeuw CI, Schonewille M (2014) Cerebellar modules operate at different frequencies. Elife 3:e02536. 10.7554/eLife.02536 [DOI] [PMC free article] [PubMed] [Google Scholar]