

Abstract
CaV1 L-type calcium channels are key to regulating neuronal excitability, with the range of functional roles enhanced by interactions with calmodulin, accessory proteins, or CaMKII that modulate channel activity. In hippocampal pyramidal cells, a prominent elevation of CaV1 activity is apparent in late channel openings that can last for seconds following a depolarizing stimulus train. The current study tested the hypothesis that a reported interaction among CaV1.3 channels, the scaffolding protein densin, and CaMKII could generate a facilitation of channel activity that outlasts a depolarizing stimulus. We found that CaV1.3 but not CaV1.2 channels exhibit a long-duration calcium-dependent facilitation (L-CDF) that lasts up to 8 s following a brief 50 Hz stimulus train, but only when coexpressed with densin and CaMKII. To test the physiological role for CaV1.3 L-CDF, we coexpressed the intermediate-conductance KCa3.1 potassium channel, revealing a strong functional coupling to CaV1.3 channel activity that was accentuated by densin and CaMKII. Moreover, the CaV1.3–densin–CaMKII interaction gave rise to an outward tail current of up to 8 s duration following a depolarizing stimulus in both tsA-201 cells and male rat CA1 pyramidal cells. A slow afterhyperpolarization in pyramidal cells was reduced by a selective block of CaV1 channels by isradipine, a CaMKII blocker, and siRNA knockdown of densin, and spike frequency increased upon selective block of CaV1 channel conductance. The results are important in revealing a CaV1.3–densin–CaMKII interaction that extends the contribution of CaV1.3 calcium influx to a time frame well beyond a brief input train.
SIGNIFICANCE STATEMENT CaV1 L-type calcium channels play a key role in regulating the output of central neurons by providing calcium influx during repetitive inputs. This study identifies a long-duration calcium-dependent facilitation (L-CDF) of CaV1.3 channels that depends on the scaffolding protein densin and CaMKII and that outlasts a depolarizing stimulus by seconds. We further show a tight functional coupling between CaV1.3 calcium influx and the intermediate-conductance KCa3.1 potassium channel that promotes an outward tail current of up to 8 s following a depolarizing stimulus. Tests in CA1 hippocampal pyramidal cells reveal that a slow AHP is reduced by blocking different components of the CaV1.3-densin-CaMKII interaction, identifying an important role for CaV1.3 L-CDF in regulating neuronal excitability.
Keywords: CaV1.2, CaV1.3, CDF, densin, facilitation, KCa3.1
Introduction
L-type calcium channels of the CaV1 family have a key role in providing calcium influx to cortical pyramidal neurons in response to repetitive inputs. The activity of CaV1 channels can in turn be regulated by calcium, accessory subunits, or phosphorylation that tailors their ability to modify membrane excitability. CaV1 channels undergo an immediate regulation when calcium influx increases an association with calmodulin (CaM) to induce a calcium-dependent inactivation (CDI; Ben-Johny and Yue, 2014). Neuronal CaV1.3 C-terminal splice isoforms can act to modify CDI or support calcium-dependent facilitation (CDF), dynamically increasing the range of functions for CaV1.3 channels depending on isoform expression (Koschak et al., 2001; Singh et al., 2008; Jenkins et al., 2010; Koschak, 2010; Moreno et al., 2016). In particular, a long C-terminal splice isoform of CaV1.3 channels found in CA1 hippocampal pyramidal cells can exhibit CDF by associating with the accessory protein densin that acts as a bridge to bind CaMKII (Jenkins et al., 2010). Specifically, the combination of densin and CaMKII reduces the CDI that occurs during repetitive stimuli to increase CaV1.3-mediated calcium influx (Jenkins et al., 2010). Densin is known to localize to dendrites where a facilitation of CaV1.3 channels could increase calcium influx to influence synaptic function and signal processing (Jenkins et al., 2010; Stanika et al., 2016). Yet, a direct functional role for CaV1.3 CDF that involves the densin–CaMKII interaction has not been identified.
One role for CaV1.3 channels in controlling postsynaptic excitability comes from the finding that a calcium-dependent slow afterhyperpolarization (sAHP) of several seconds duration in pyramidal cells is reduced by dihydropyridines (DHPs) and in CaV1.3−/− animals (Lima and Marrion, 2007; Gamelli et al., 2011). Moreover, CaV1 channels in pyramidal cells exhibit late openings following a train of depolarizing stimuli and even a delayed facilitation for up to 6 s that could modify cell excitability (Thibault et al., 1993; Kavalali and Plummer, 1994, 1996; Cloues et al., 1997; Schjott and Plummer, 2000). It is known that CaV1 calcium channels can functionally couple with calcium-gated potassium channels, including KCa1.1 (BK; Berkefeld et al., 2006; Loane et al., 2007; Berkefeld and Fakler, 2008; Vandael et al., 2010) and KCa2.x (SK) channels (Bowden et al., 2001; Adelman et al., 2012; Vandael et al., 2015). A class of calcium-gated potassium channel of 19 pS conductance that is activated by nifedipine-sensitive calcium channels was also reported in CA1 pyramidal cells (Marrion and Tavalin, 1998; Lima and Marrion, 2007). Indeed, the pattern of delayed facilitation of CaV1 channels and the activation of these potassium channels is similar following short trains of depolarization (Cloues et al., 1997; Bowden et al., 2001; Lima and Marrion, 2007; King et al., 2015). Progress in identifying the channels that contribute to this late response comes from the finding that the intermediate-conductance calcium-gated potassium channel KCa3.1 is expressed in pyramidal cells and activates over the time course of the sAHP (King et al., 2015; Turner et al., 2015, 2016). However, we do not know whether CaV1.3 channels can exhibit direct or functional coupling to KCa3.1 channels or whether CaV1 channel facilitation can influence KCa3.1 contributions to the sAHP.
The current study examined the ability for CaV1.3 calcium channels to undergo facilitation that outlasts a depolarizing stimulus, and its ability to activate KCa3.1 channels. We find a form of CaV1.3 CDF that is densin- and CaMKII-dependent that outlasts a depolarizing stimulus by seconds. A strong functional coupling between CaV1.3 and KCa3.1 channels further allows the CaV1.3–densin–CaMKII interaction to accentuate a long-duration outward tail current consistent with the sAHP in CA1 pyramidal cells.
Materials and Methods
Brain slice preparation
Sprague Dawley rats (Charles River) were raised from timed-pregnant dams maintained according to the guidelines of the Canadian Council of Animal Care in an Animal Resource Center of the University of Calgary. Male rats of postnatal day 18 (P18) to P24 age were used to prepare coronal hippocampal brain slices as previously described (King et al., 2015). Briefly, animals were anesthetized by isoflurane inhalation until unresponsive to ear pinch. The brains were rapidly dissected and placed in ice-cold cutting solution composed of the following (in mm): 215 sucrose, 25 NaHCO3, 20 d-glucose, 2.5 KCl, 0.5 CaCl2, 1.25 NaH2PO4, and 3 MgCl2 continuously bubbled with carbogen (95% O2 and 5% CO2) gas to cut 250 μm thick slices by vibratome (VT1200 S, Leica). The slices were allowed to recover for 20–30 min at 37°C and then maintained at room temperature (22–24°C) in artificial CSF (aCSF) composed of the following (in mm): 125 NaCl, 3.25 KCl, 1.5 CaCl2, 1.5 MgCl2, 25 NaHCO3, and 25 d-glucose bubbled with carbogen gas. For recordings, slices were moved to a custom-made recording chamber placed on the stage of a Zeiss Examiner.A1 Microscope and maintained as an interface preparation at 32–33°C.
Hippocampal dissociated cell culture
Low-density dissociated cultures of rat hippocampal neurons were prepared as previously described (Fan et al., 2016). Briefly, P0–P1 rat pups were anesthetized by being placed on ice for 5–10 min, and hippocampi were dissected and dissociated by papain treatment, followed by trituration. The cell suspension was plated on poly-l-lysine- and laminin-coated glass coverslips in 24-well culture dishes at a density of 3500–5300 cells/cm2. After plating cells were allowed to grow on coverslips for 7 d in neuronal growth medium (NGM) composed of free Neurobasal medium, 1 mm Na-pyruvate, 2 mm l-glutamine, 10 mm HEPES, 1% B-27 supplement, 5% FBS, 0.6% glucose, and 1% penicillin-streptomycin. All cell culture chemicals were obtained from Life Technologies.
After 7 d in vitro cultured neurons were transfected with siRNAs against densin (SASI_Rn02_00267099, SASI_Rn01_00094090, Sigma-Aldrich), referred to as densin siRNA-1 and siRNA-2, or with universal negative control siRNA (SIC001, Sigma-Aldrich) using lipofectamine 2000 (Thermo Fisher Scientific). eGFPN1 was cotransfected with the siRNAs to visualize transfected cells. Each well of a 24-well plate was transfected with 50 pm siRNA and 0.1 μg of eGFPN1 using 3 μl of lipofectamine. On the day of transfection, the conditioned NGM was replaced with fresh Neurobasal media devoid of any growth factor and antibiotics. The media was again replaced 3–4 h after transfection with conditioned NGM, and cells were used for experiments 3 d after transfection.
tsA-201 cell cultures
tsA-201 cells were maintained in DMEM supplemented with 10% heat inactivated fetal calf serum and 1% penicillin-streptomycin. The calcium phosphate-based method was used to transiently transfect cDNAs into tsA-201 cells (Engbers et al., 2012). For CaV1.2 channel expression, 2 μg each of human-α1C-PMT2, α2δ1-PMT2, and β1B-PMT2 was used. For CaV1.3 expression, 2 μg each of human-α1D-GFP−, α2δ1-pcDNA, and β1B-pcDNA was used. CaV1.3-α1D-GFP− was a gift from Professor J. Striessnig (University of Innsbruck, Innsbruck, Austria). CaV1.2- and CaV1.3-expressing cells were cotransfected with 100 ng of eGFPN1 to identify transfected cells and 1 μg of KCa3.1-GFP when coexpressed with KCa3.1 channels (King et al., 2015).
For determining the facilitation of CaV1 currents, mCherryC1-CaMKII and densin-GFP were cotransfected along with CaV1 isoforms. Threonine at position 286 in mCherryC1-CaMKII was mutated to alanine to generate an mCherryC1–CaMKII-T286A construct using appropriate forward and reverse PCR primers. DNA sequencing was used to verify the mutation. Human full-length densin-GFP was provided by Professor Hans-Jurgen Kreienkamp (University Medical Center Hamburg-Eppendorf, Hamburg, Germany) (Quitsch et al., 2005) and a rat short CaV1.3 isoform (CaV1.3s) clone was commercially obtained (catalog #49333, Addgene; Xu and Lipscombe, 2001).
Electrophysiology
Cells maintained in a slice preparation or as dissociated culture were visualized using infrared differential interference contrast and whole-cell patch-clamp recordings obtained using a Multiclamp 700B Amplifier, Digidata 1440A Digitizer, and pClamp version 10.5 software. Recordings in tissue slices were specifically directed to CA1 pyramidal cells. All current traces were DC-10 kHz bandpass filtered and digitized at 40 kHz. Glass pipettes with an outer diameter of 1.5 mm (A-M Systems) having 4–8 MΩ resistance were pulled using a P-95 horizontal puller (Sutter Instruments). For whole-cell voltage-clamp recordings, series resistance was compensated to 70% and leak was subtracted off-line in Clampfit 10.5 software. For current-clamp recordings, cells with more than −55 mV resting membrane potential were accepted with a negative bias current of <150 pA applied to maintain a resting potential at approximately −70 mV. After attaining whole-cell configuration, cells were allowed to equilibrate with the internal pipette solution for 5 min before recordings, and cells were rejected for any drift in access resistance of >20%. Stimulating electrodes were concentric bipolar (CBCMX75 [JL2], Frederick Haer) and controlled by a stimulus isolation unit (pulse width, 0.1–0.2 ms; Digitimer).
Hippocampal tissue slices were maintained at 32–33°C, and dissociated hippocampal cultures and tsA-201 cells were maintained at room temperature (22–24°C). The bathing medium used for hippocampal slices was aCSF (see above), with a similar HEPES-buffered medium for dissociated hippocampal and tsA-201 cell cultures composed of the following (in mm): 120 NaCl, 3 NaHCO3, 3.5 KCl, 1.5 MgCl2, 1.5 CaCl2, 10 HEPES, and 10 d-glucose, pH 7.4 with NaOH. Voltage-clamp recordings in hippocampal slices or dissociated cultures included the synaptic blockers 25 μm APV, 10 μm DNQX, and 50 μm picrotoxin. For current-clamp recordings in CA1 neurons, 100 nm apamin and 10 μm XE-991 was added to the aCSF but without any synaptic blockers. Unless otherwise noted, voltage-clamp and current-clamp recordings in hippocampal cells used an electrolyte composed of the following (in mm): 130 KMeSO4, 0.1 EGTA, 10 HEPES, 7 NaCl, 0.3 MgCl2, 2 ATP, 0.5 GTP, and 5 phosphocreatine, pH 7.3 with KOH, providing an ECl of −75 mV and EK of −97 mV.
Isolating CaV1 calcium current
L-type calcium currents were recorded from tsA-201 cells expressing channel isoforms in isolation and in hippocampal tissue slices. In tsA-201 or CA1 pyramidal cells, the internal electrolyte contained the following (in mm): 110 CsCl, 2 MgCl2, 0.5 EGTA, 10 HEPES, 2 ATP, 0.5 GTP, and 5 phosphocreatine, pH 7.3 with CsOH. The bathing medium for tsA-201 cells contained the potassium channel blockers 2 mm CsCl, 5 mm TEA, 5 mm 4-AP, 100 nm apamin, and 1 μm TRAM-34. For isolating CaV1 currents in hippocampal CA1 neurons, the aCSF contained the same potassium channel blockers and 10 μm XE-991 along with synaptic blockers and 1 μm TTX. For voltage-clamp recordings in CA1 pyramidal cells, the aCSF also contained 200 nm ω-agatoxin IVA, 1 μm ω-conotoxin MVIIC, 200 nm SNX-482, and 1 μm TTA-P2 to block CaV2.1, CaV2.2, CaV2.3, and CaV3.X currents, respectively. To study voltage-dependent facilitation of CaV1.3 currents in tsA-201 cells, 5 mm BaCl2 replaced CaCl2 in the bathing medium.
Isolating KCa3.1 current
KCa3.1 current was recorded in tsA-201 cells bathed in the following medium (in mm): 150 Na gluconate, 4.5 KCl, 1.5 CaCl2, 1.5 MgCl2, 10 HEPES, 2 CsCl, 5 TEA, 5 4-AP, and 100 nm apamin, pH 7.4 with NaOH. Free calcium concentration in the internal electrolyte was adjusted to 1 μm for activation of KCa3.1 currents, composed of the following (in mm): 145 K gluconate, 2 MgCl2, 10 EGTA, 10 HEPES, 8.5 CaCl2, 2 ATP, 0.5 GTP, and 5 phosphocreatine, pH 7.2, adjusted with KOH. Maxchelator software (http://maxchelator.stanford.edu/CaEGTA-NIST.htm) was used to calculate the EGTA and CaCl2 concentrations to maintain 1 μm free Ca2+ in the internal electrolyte. In some cases, KCa3.1 current was blocked by internal infusion of 1 μm TRAM-34 through the electrode using an ALA pipette perfusion system (King et al., 2015).
Isolating IsAHP in hippocampal neurons
The KCa3.1-mediated sAHP current (IsAHP) in hippocampal CA1 neurons in the slice preparation was isolated in voltage clamp by including in the aCSF 2 mm CsCl, 5 mm tetraethyl ammonium chloride (TEA), 5 mm 4-AP, 100 nm apamin, 10 μm XE-991, and 1 μm TTX along with synaptic blockers (above). The external medium used to isolate IsAHP in dissociated cultured neurons included the same potassium channel, sodium channel, and synaptic blockers as for slice preparations (above), and the calcium channel blockers 200 nm ω-agatoxin IVA, 1 μm ω-conotoxin MVIIC, 200 nm SNX-482, and 1 μm TTA-P2 were added to the bath by pressure ejection (Automate Scientific).
Foerster resonance energy transfer
For Foerster resonance energy transfer (FRET) imaging, tsA-201 cells were seeded onto poly-l-lysine-coated 35-mm glass-bottom Petri dishes. Cells were transiently transfected with KCa3.1-GFP, CaV1.3-mKate, and mKate–CaM constructs generated using appropriate sets of restriction enzymes and PCR primer sets followed by DNA sequencing for validation. Cells were then incubated for 36–42 h at 37°C and 5% CO2, washed, and replaced with colorless low K+-based imaging medium composed of the following (in mm): 148 NaCl, 3 KCl, 10 HEPES, 3 CaCl2, 10 glucose, and 1 MgCl2, pH 7.3 at 25°C. To depolarize cells, the low-potassium imaging medium was replaced with high-potassium (50 mm KCl) medium. Cells were examined with a Nikon C1si spectral confocal laser-scanning microscope with a 40×/1.3 numerical aperture oil-immersion objective. For FRET imaging, KCa3.1-GFP was excited at 457 nm, and CaV1.3-mKate or mKate-CaM was excited at 561 nm. Emission spectra of GFP and mKate were recorded at between 400 and 750 nm. The FRET signal was measured every 10 s for 500 s, and ImageTrak software (P.K. Stys, University of Calgary, Calgary, AB, Canada; http://www.ucalgary.ca/styslab/imagetrak; Stirling et al., 2014) was used for processing the images. To separate the fluorescence signals of GFP and mKate, spectral images were linearly unmixed using ImageTrak software collapsing a 32-channel spectral image into a 2-channel image representing the integrated intensities of GFP and mKate fluorescence emissions.
Western blot and coimmunoprecipitation assays
Western blot.
tsA-201 cells expressing densin-GFP in combination with either of two different siRNAs were lysed in a buffer containing the following (in mm): 150 NaCl, 50 Tris, 2.5 EGTA, 0.5% sodium-deoxycholate, 1% NP-40, and 0.001 CaCl2, pH 7.5 at 4°C for 15–30 min with constant rotation. Lysates were centrifuged for 10 min at 12,500 rpm at 4°C, and supernatant was used for gel loading. Eluted lysates were loaded on 10% Tris-glycine gel and resolved using SDS-PAGE. Samples were transferred to 0.2 μm PVDF membrane (Millipore), and Western blot analysis was performed using a rabbit polyclonal anti-GFP (1:1000; Santa Cruz Biotechnology) or a rabbit polyclonal anti-α-tubulin antibody (1:1000; Abcam). Anti-rabbit secondary antibody conjugated to HRP (1:5000; Invitrogen) was used and reacted with ECL solution (Life Technologies) to visualize protein bands.
Coimmunoprecipitation.
Hippocampus was dissected out from adult male rats (weight, ∼250 g) after isoflurane anesthesia and lysed in a buffer containing the following (in mm): 150 NaCl, 50 Tris, 2.5 EGTA, 0.5% sodium-deoxycholate, 1% NP-40, and 0–0.05 CaCl2, pH 7.5 using a hand-held glass homogenizer. The nominal level of free calcium in solutions was calculated using Maxchelator software to conduct coimmunoprecipitations in the presence of different levels of free calcium concentrations ranging from 0 to 50 μm as described in the study by Asmara et al. (2017). Hippocampal lysates were centrifuged at 12,500 rpm for 10 min at 4°C, and supernatants were transferred to new tubes. Solubilized proteins were incubated with 10 μg of rabbit anti-CaV1.2 (ACC-003, Alomone Labs) or rabbit anti-CaV1.3 antibodies (ACC-005, Alomone Labs) with 30 μl of Protein G beads (Life Technologies) while rotating at 4°C overnight. Specificity and cross-reactivity of CaV1.2 and CaV1.3 antibodies used in this study were validated using the CaV1.2- and CaV1.3-transfected tsA-201 cells to verify appropriate labeling and no cross-reactivity. After three rounds of washing with PBS, the coimmunoprecipitates were reconstituted in an equal volume of 2× loading buffer as follows (in mm): 100 Tris, 100 2-mercaptoethanol, 4% SDS, 0.02% bromophenol blue, and 20% glycerol, pH 6.8 followed by incubation at 100°C for 5 min for protein elution from the beads. Eluted samples were loaded on 8% Tris-glycine gel and resolved using SDS-PAGE. Samples were transferred to 0.2 μm PVDF membrane, and Western blot analysis was performed using a mouse monoclonal anti-KCa3.1 (D-5, 1:1000; sc-356265, Santa Cruz Biotechnology).
tsA-201 cells expressing KCa3.1-GFP along with CaV1.2 or CaV1.3 were lysed using the same lysis buffer as for hippocampal lysates. Ten micrograms of mouse monoclonal anti-eGFP (Santa Cruz Biotechnology) antibody was used for pulldown of protein samples. Rabbit anti-CaV1.2 or rabbit anti-CaV1.3 antibodies (1:1000) were used for determining coimmunoprecipitation.
Quantitative PCR
Total RNA from monolayers of hippocampal neurons transfected with either of two densin siRNAs or with control siRNA were extracted using the RNeasy Plus Microkit (Qiagen) following manufacturer instructions. mRNA (100 ng) was converted to cDNA with a QuantiTect Reverse Transcription Kit (Qiagen). Real-time PCR-based quantification of densin (NM_057142) and β-actin (NM_031144) mRNAs were performed using 5′-ATGTACCTCCGGACACCATT-3′/5′-AAGGCTCTGTGACCCCATC-3′ and 5′-AGGCCCCTCTGAACCCTAAG-3′/5′-CCAGAGGCATACAGGGACAAC-3′ forward/reverse primer pairs, respectively (Wheeler et al., 2012), using SyGreen Mix Separate-ROX kit (D-Mark Biosciences). Relative levels of mRNA expression were determined with the 2-ΔΔCt method from Ct (threshold cycle) values obtained from quantitative PCR (Q-PCR) results. β-Actin was used as an internal control for normalization of the Ct values. Data were obtained from three individual cell group transfections, and each sample was run in triplicate for the Q-PCR cycle with equal amounts of cDNA used for all experimental sets. Statistical significance was determined with one-way ANOVA followed by post hoc t tests.
Experimental design and statistical analysis
CaV1 current–voltage (I–V) plots were prepared from the inward peak calcium current amplitudes measured at different voltages. For determining half-activation potential (Vhalf) and reversal potential (Vrev), I–V plots were fitted with a Boltzmann equation using Prism 5 software according to the following:
 |
where Gmax is the maximum CaV1 conductance and k is the slope factor. Calcium-induced decay of CaV1.x current was fitted with the Levenberg–Marquardt biexponential fitting equation in Clampex 10.5 software to determine the inactivation time constants according to the following:
 |
where A is the contribution of each kinetic component (τfast or τslow) of the decay of CaV1.x current and C is the noninactivating component.
CaV1.x-activated KCa3.1 outward tail current areas in tsA-201-transfected cells were measured as the area over the baseline. The IsAHP area in CA1 cells was measured as the area under (in current-clamp mode) or over (in voltage-clamp mode) the baseline. All data were analyzed using Clampfit 10.5 software, and statistical analysis was performed in SigmaPlot 12.5 and Prism 5 software. All figures were prepared using OriginPro 8 software and Adobe illustrator. Normality for datasets was determined using the Shapiro-Wilk test. Student's t tests or one-way ANOVA followed by post hoc paired t test comparisons were used to determine the significance indices unless otherwise indicated.
Results
To compare the properties of facilitation of L-type channel isoforms, we transiently expressed in tsA-201 cells the channel-forming α1 subunits of CaV1.2 or CaV1.3 together with α2δ1 and β1b accessory proteins, which were selected given their dense expression pattern in CA1 pyramidal cells (Hell et al., 1993; Cole et al., 2005; Taylor and Garrido, 2008). Whole-cell voltage-clamp analysis was conducted 48–72 h following transfection in the presence of physiological concentrations of 1.5 mm [Ca]o in the bath.
Prolonged facilitation of CaV1.3 by densin and CaMKII
Given the role for CaV1.3 calcium channels in regulating cell excitability, we were interested in the basis for a delayed facilitation of CaV1 channel activity that is triggered following brief trains of depolarizing steps in CA1 pyramidal cells (Cloues et al., 1997). Importantly, CaV1.3 channels were reported to exhibit a CDF during high-frequency stimulation through an interaction with the scaffolding protein densin and CaMKII (Jenkins et al., 2010). A PDZ binding domain on densin allows it to associate with the CaV1.3 distal C terminus to enable CaMKII modulation. In this way, the extent of CaV1.3 CDI during a repetitive stimulus train is reduced. To consider the potential for this form of CDF to outlast the stimulus train, we delivered a 10 pulse 50 Hz train of command steps from −70 to 40 mV in tsA-201 cells and tested whole-cell CaV1 current with a single pulse at varying delays (Fig. 1A). Step commands were delivered to the peak current for CaV1.3 (0 mV) or CaV1.2 (10 mV; Fig. 2) using an early test pulse at 20 ms after train stimulus and subsequent test intervals at 500 ms increments to 5–10 s. Separate test of single pulses were delivered at 30 s intervals to allow recovery between stimulus trains.
Figure 1.
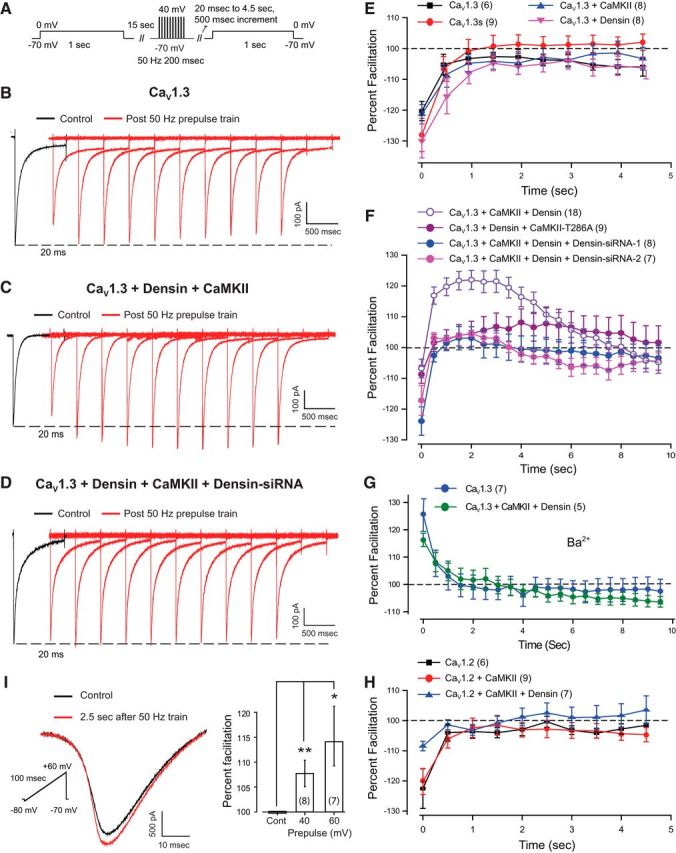
A CaV1.3, densin, and CaMKII interaction generates a prolonged facilitation of calcium current. A, Schematic diagram of step protocols to test CaV1 channel response to a 10 pulse 50 Hz train of depolarizing stimuli in tsA-201 cells in B–D. B–D, Whole-cell recordings of CaV1.3 current evoked by a step to 0 mV before (black trace) and after (red traces) a 10 pulse 50 Hz train of steps to 40 mV from a holding potential of −70 mV. The first test response was evoked at a delay of 20 ms and all subsequent records at 500 ms increments. B, C, CaV1.3 expressed alone exhibits no facilitation following a stimulus train (B), but a pronounced late facilitation after the stimulus train when coexpressed with densin and CaMKII (C). D, Cotransfecting densin–siRNA-2 blocks facilitation after the stimulus train (D). E, No late facilitation is detected in cells expressing CaV1.3, a short isoform CaV1.3s, or CaV1.3 with either CaMKII or densin. F, CaV1.3 expressed in combination with densin and CaMKII exhibits a pronounced late facilitation that lasts up to 8 s following the stimulus train. Facilitation is reduced upon coexpressing a mutant CaMKII construct that cannot undergo autophosphorylation (CaMKII-T286A) or coexpressing either of two densin siRNAs. G, Recordings of CaV1.3 or CaV1.3 coexpressed with densin and CaMKII in the presence of 2 mm Ba2+ indicate a brief voltage-dependent facilitation at 20 ms following a stimulus train but no late facilitation, identifying the CAV1.3–densin–CaMKII interactions in F as an extended form of CDF. H, No late facilitation is detected in cells expressing CaV1.2 or CaV1.2 in combination with CaMKII or densin and CaMKII. I, Whole-cell calcium current in rat CA1 pyramidal cells recorded in the presence of blockers against all CaV channels except L-type (1 μm ω-conotoxin, 200 nm ω-agatoxin, 200 nm SNX-482, and 1 μm TTA-P2). Superimposed traces show L-type current evoked by a 100 ms ramp command tested with or without a preceding 50 Hz stimulus train of square wave steps (200 ms) to 60 mV at a delay of 2.5 s. Bar plots show the mean facilitation of calcium current evoked by a ramp command in the presence or absence of a preceding 50 Hz stimulus train square wave steps (200 ms) to either 40 or 60 mV. Facilitation is expressed as a percentage of the calcium current recorded in the absence of the 50 Hz train (100%, Cont.; p < 0.05, one-way ANOVA followed by post hoc Student's paired t test). Cell numbers are shown in brackets. *p < 0.05; **p < 0.01.
Figure 2.
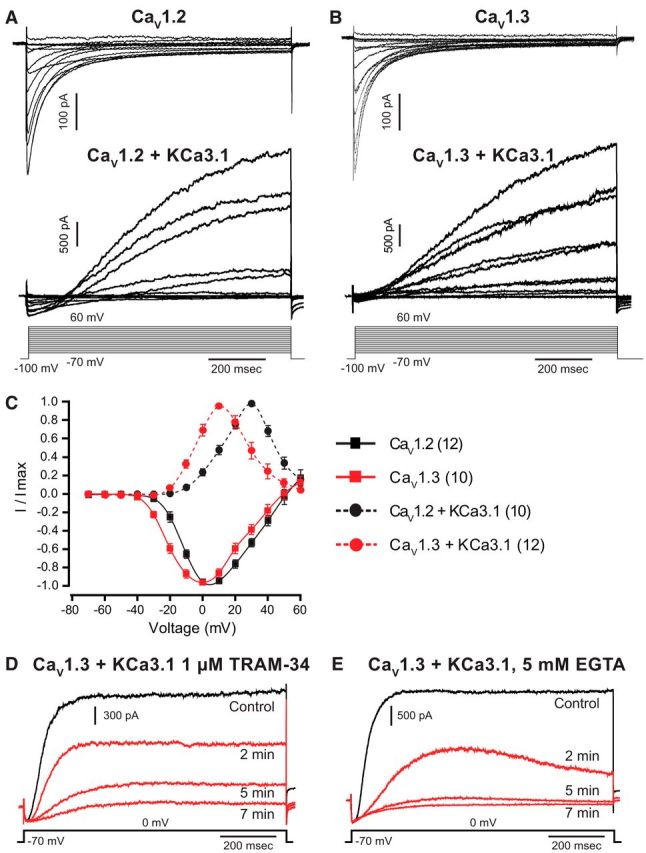
L-type calcium and KCa3.1 potassium channels functionally couple. A, B, Whole-cell currents of CaV1.2 (A) and CaV1.3 (B) calcium channels expressed in tsA-201 cells exhibit pronounced calcium-dependent inactivation. CaV1 channels were coexpressed with α2δ1 and β1b accessory subunits. Coexpression of KCa3.1 channels with CaV1.2 (A) or CaV1.3 (B) result in an inward Ca2+ current followed by KCa3.1-mediated potassium current. C, The current–voltage plots for KCa3.1 channels closely follow the voltage dependency of corresponding CaV1 calcium channel isoforms. Outward currents were evoked from a holding potential of −70 mV and measured at 1 s. D, E, Representative traces of KCa3.1 currents blocked by internal perfusion of 1 μm TRAM-34 (D) or 5 mm EGTA (E) over a 5–7 min time period. Numbers of cells are denoted in brackets in C.
Initial tests in cells expressing CaV1.3 alone revealed that the repetitive stimulus train evoked no facilitation but rather a slight depression of current amplitude that persisted after CDI had developed within the stimulus train (Fig. 1B,E). This depression was most evident at an initial test delay of 20 ms (as expected from the development of CDI during the stimulus train) and then returned to a modest level of depression by 1 s (presumably reflecting recovery from CDI) but persisted at a low level in some cases for at least 5 s (Fig. 1B,E). Similarly, the expression of a short C-terminal isoform of CaV1.3 (CaV1.3s) only exhibited CDI following a 50 Hz stimulus train (Fig. 1E), as expected given that this isoform does not contain the PDZ binding site for densin (Jenkins et al., 2010). A process of CDI was also apparent for CaV1.3 when coexpressed with either densin or CaMKII alone (Fig. 1E). Comparing the percentage facilitation of CaV1.3 current under each of these conditions at 2.5 s following the stimulus train revealed no significant difference (one-way ANOVA, p = 0.412). In contrast, coexpressing the combination of CaV1.3, densin, and CaMKII revealed an initial CDI at 20 ms but a long-lasting facilitation of current amplitude following the stimulus train (Fig. 1C,F) compared with CaV1.3 expressed alone (Fig. 1B,E). Plotting the time course of current amplitude for the CaV1.3–densin–CaMKII combination revealed that facilitation increased to a stable peak value ∼25–30% higher than control by 1.5 s and then decayed over almost 8 s beyond the end of the stimulus train (Fig. 1F; p < 0.01 at 2.5 s post-stimulus train, Student's t test).
Control tests confirmed that coexpression of densin and CaMKII did not shift the voltage dependence of CaV1.3 on I–V plots (data not shown), a result consistent with the report of Jenkins et al. (2010). We further found that coexpressing CaV1.3 and densin together with a CaMKII-T286A mutant construct that cannot exhibit autophosphorylation (Jenkins et al., 2010) greatly reduced the degree of facilitation following the stimulus train (Fig. 1F; p < 0.05 at 2.5 s post-stimulus train, Student's t test). Similarly, coexpressing CaV1.3, densin, and CaMKII with either of two forms of densin siRNA completely blocked facilitation following the stimulus train (Fig. 1F; p < 0.01 for both siRNA-1 and siRNA-2 at 2.5 s post-stimulus train, one-way ANOVA). When recording with 5 mm barium only a short-lasting facilitation was observed in tsA-201 cells coexpressing CaV1.3, CaMKII, and densin (Fig. 1G) compared with this combination in calcium-containing solution (Fig. 1F; p < 0.01 at 2.5 s post-stimulus train, Student's t test). Finally, we found no facilitation for CaV1.2 channels alone or when expressed with CaMKII or with CaMKII and densin (Fig. 1H; p = 0.669, one-way ANOVA). Together, these data establish that a long-duration calcium-dependent facilitation (L-CDF) of CaV1.3 by densin and CaMKII is calcium dependent, reflecting a previously unidentified form of CaV1 facilitation that extends for a period of up to 8 s following a stimulus train.
CA1 hippocampal pyramidal cells are known to express CaV1.2 and CaV1.3 calcium channel isoforms and densin (Koschak et al., 2007; Jenkins et al., 2010; Gamelli et al., 2011; Moreno et al., 2016; Stanika et al., 2016). We thus repeated the tests of CaV1 current facilitation in rat CA1 pyramidal cells by recording whole-cell calcium current in the presence of blockers against P/Q-, N-, R-, and T-type channel isoforms (1 μm ω-conotoxin MVIIC, 200 nm ω-agatoxin IVA, 200 nm SNX-482, and 1 μm TTA-P2). Calcium current was further isolated using a set of blockers against other ion channels (see Materials and Methods). To avoid space-clamp issues in pyramidal cells, we evoked calcium current using a short ramp command (100 ms) applied from −80 to 60 mV with or without a preceding 50 Hz train of step commands of 200 ms duration to 40 or 60 mV (Fig. 1I). To allow sufficient settling of membrane current following a stimulus train, we tested for the occurrence of calcium current facilitation at a delay of 2.5 s. These measurements revealed a facilitation that was graded with the step command voltage, reaching a facilitation of 14.1 ± 6.0% of peak calcium current when the stimulus train was delivered using steps to +60 mV (n = 7, p < 0.05; Fig. 1I). While the absolute degree of facilitation was less than that found in tsA-201 cells, CaV1.3 channels are reported to comprise ∼20% of total CaV1 calcium current in pyramidal cells (Hell et al., 1993; Koschak et al., 2007; Moreno et al., 2016). Assuming that only CaV1.3 channels exhibit facilitation in pyramidal cells, the absolute percentage increase in L-type calcium current is thus predicted to be less than that recorded for CaV1.3 expressed in isolation in tsA-201 cells.
CaV1 calcium channels couple to KCa3.1 potassium channels
L-type calcium channels have been previously reported to activate KCa1.1 and KCa2.x calcium-dependent potassium channels (Marrion and Tavalin, 1998; Bowden et al., 2001; Berkefeld et al., 2006; Berkefeld and Fakler, 2008; Babai et al., 2010; Adelman et al., 2012). In some cases, coupling between these channels can be close enough to allow the voltage-dependent properties of the calcium channel to be reflected in activation of the potassium channel (Berkefeld and Fakler, 2008). Given our recent evidence for KCa3.1 expression in CA1 pyramidal cells that activate over the timeframe of a slow AHP (King et al., 2015; Turner et al., 2015), we tested for the potential ability for CaV1 channels to couple to activation of KCa3.1 potassium channels. CaV1.2 and CaV1.3 calcium channels coexpressed with α2δ1 and β1b accessory subunits and recorded in the presence of 1.5 mm [Ca]o revealed inward calcium currents with fast activation and inactivation kinetics (Fig. 2A,B). Voltage step commands from −70 to 60 mV (1 s) in 10 mV increments confirmed that CaV1.2 channels activate at more depolarized membrane potentials than CaV1.3 (Xu and Lipscombe, 2001). The Vhalf for CaV1.2 channels was −9.8 ± 1.4 mV (n = 12), which was significantly greater than the CaV1.3 Vhalf of −16.9 ± 1.9 mV (n = 10; p < 0.007, unpaired Student's t test). CaV1.2-mediated inward currents were initially detected at −30 mV and peaked at 10 mV, while CaV1.3 channels activated at −40 mV and peaked at 0 mV (Fig. 2C). The reversal potential was 56.17 ± 3.6 mV (n = 12) and 51.68 ± 3.0 mV (n = 10) for CaV1.2 and CaV1.3 channels, respectively (p = 0.365, unpaired Student's t test; Fig. 2A,B). There was no significant difference observed between peak current amplitudes (CaV1.2, −215 ± 41 pA at 10 mV, n = 12; CaV1.3, −284 ± 37 pA at 0 mV, n = 15; p = 0.196, unpaired Student's t test). Similarly, there was no statistical difference in tau values for inactivation (CaV1.2 at 10 mV: τ1 = 40.9 ± 3.30 ms; τ2 = 249.7 ± 14.24 ms; n = 12; CaV1.3 at 0 mV: τ1 = 41.3 ± 3.86 ms, p = 0.938; τ2 = 212.1 ± 11.94 ms, p = 0.087; n = 10, unpaired Student's t test).
We then coexpressed CaV1.2 or CaV1.3 channels with KCa3.1. Whole-cell current measurements were again conducted in the presence of 1.5 mm [Ca]o and with identical internal solutions used for recording CaV1 currents. These tests revealed biphasic current responses consisting of an initial inward calcium current followed by outward potassium current (Fig. 2A,B). I–V plots of outward currents revealed that KCa3.1 activation closely followed that of the coexpressed CaV1 channel isoform. Thus, when coexpressed with CaV1.2 channels, KCa3.1 current was initially detected at −10 mV and peaked at 30 mV, but when coexpressed with CaV1.3 channels KCa3.1 current was detected initially at steps to approximately −20 mV and peaked at −10 mV (Fig. 2C). The mean steady-state KCa3.1 current (measured at 1 s) was 1194 ± 442 pA (n = 10) for coexpression of CaV1.2 and KCa3.1, and 2218 ± 423 pA (n = 12) for coexpression of CaV1.3 and KCa3.1 (p = 0.112, unpaired Student's t test). The close correspondence between CaV1.3 calcium conductance and KCa3.1 activation extended to a decrease in outward current for steps more positive than 10 mV (CaV1.3) or 30 mV (CaV1.2), which is consistent with a progressive decrease in CaV1.x conductance as the step command approached the calcium reversal potential (Fig. 2C). The identity of potassium current as KCa3.1 was confirmed by a block by 1 μm TRAM-34, a selective blocker of KCa3.1 channels (Fig. 2D; Wulff et al., 2000, 2007). The reduction of KCa3.1 by TRAM-34 was evident within 2 min of initiating internal infusion through the patch electrode and was complete by 7 min, compared with a relatively stable KCa3.1 current amplitude maintained at 93.2 ± 3.0% (n = 7) of original amplitude for 5–10 min in the control electrolyte, ruling out any influence of washout (Fig. 2D; Wang et al., 2016).
To test whether CaV1.x and KCa3.1 channels interact at the level of a microdomain or a nanodomain, we internally infused through the electrode 5 or 10 mm EGTA or BAPTA to introduce slow versus fast calcium chelating agents, respectively (Augustine et al., 2003). In cells coexpressing CaV1.3 and KCa3.1, we found a 95.0 ± 2.4% (n = 9) block of KCa3.1 current following infusion of 5 mm EGTA (Fig. 2E) and a 98.4 ± 0.9% (n = 6) block by 5 mm BAPTA (data not shown). These results suggest a functional coupling at the microdomain level despite the close association suggested by tsA-201 cell recordings (Fig. 2). Consistent with this, we found that neither CaV1.3 nor CaV1.2 coimmunoprecipitated with KCa3.1 channels from rat brain lysates or from tsA-201 cells expressing channel proteins (data not shown). In addition, coexpressing KCa3.1-GFP and CaV1.3-mKate constructs in tsA-201 cells failed to elicit FRET in response to 457 nm laser excitation (Fig. 3A,B), even though FRET could be detected between KCa3.1-GFP and mKate-CaM as a positive control (Fig. 3C,D). Together, the available evidence is consistent with CaV1.3 and KCa3.1 channels interacting at the level of a microdomain compared with a link at the molecular level that would support a nanodomain interaction. Nonetheless, we cannot rule out a role for unidentified interacting proteins at this time.
Figure 3.
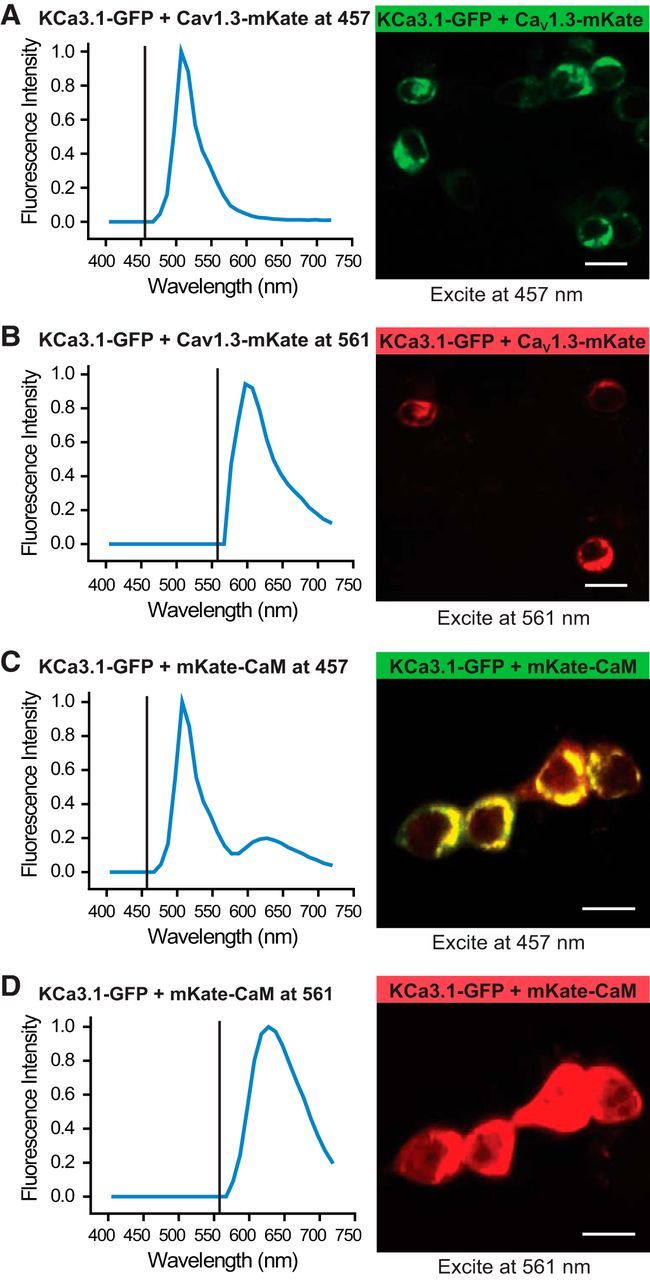
CaV1.3 does not associate with KCa3.1 at a level that supports FRET. Fluorescence confocal images of tsA-201 cells expressing constructs of KCa3.1-GFP (donor molecule) and CaV1.3-mKate (acceptor molecule) excited with a laser at 457 or 561 nm, respectively. Plots show the excitation line (vertical line) and the associated average emission spectra for each condition obtained from 20–30 cells from three to five independent experiments. A, B, Excitation at 457 nm of cells expressing KCa3.1-GFP and CaV1.3-mKate results in only a GFP emission spectra, indicating no FRET. C, In comparison, the expression of KCa3.1-GFP and mKate-CaM as a positive control revealed FRET, as indicated by an emission spectra for mKate (peaks, ∼630 nm; Fig. 3C) when excited at 457 nm. D, Excitation of cells expressing KCa3.1-GFP and mKate-CaM at 561 nm returns only mKate spectra. Scale bars, 20 μm.
CaV1 channels activate a KCa3.1 tail current
It is established that in CA1 cells delivering a depolarizing stimulus as a step command or repetitive train evokes a large outward tail current (IsAHP) of several seconds duration (Lima and Marrion, 2007; Sametsky et al., 2009; Andrade et al., 2012; Chen et al., 2014; King et al., 2015). Specific evidence for a role for CaV1.3 calcium channels in activating the IsAHP is a selective reduction in sAHP amplitude in CaV1.3−/− but not CaV1.2−/− mice (Gamelli et al., 2011). To address the potential for CaV1.x channels to activate KCa3.1 tail currents that could outlast a depolarizing stimulus, we coexpressed these constructs in tsA-201 cells. To more readily compare results obtained in tsA-201 cells to those of CA1 pyramidal cells, we delivered step commands of varying duration to the voltage for peak calcium current activation (0 mV, CaV1.3; 10 mV, CaV1.2). Command steps were delivered from a holding potential of −70 mV for durations of 0–150 ms in 5 ms increments using a 30 s interpulse interval.
These tests revealed large outward current following step commands in cells coexpressing CaV1.3 and KCa3.1, with the amplitude and duration of tail currents increasing directly with the duration of the preceding command step (Fig. 4A). While a KCa3.1 outward current was also recorded following a step command when coexpressed with CaV1.2, the outward current was greatly reduced compared with CaV1.3 (Fig. 4C,D). By comparison, expressing KCa3.1 channels in isolation revealed no corresponding tail current following the same series of step commands (Fig. 4A), as was the case for CaV1.2 or CaV1.3 expressed in isolation (n = 6, CaV1.2; n = 8, CaV1.3; data not shown). To compare the relative efficacy of KCa3.1 activation by CaV1.2 versus CaV1.3 channels, the area of the outward tail current was normalized to the preceding peak calcium current in response to 100 ms step pulses. This measure showed that the outward tail current area was increased by a factor of ∼6.5× in cells coexpressing KCa3.1 and CaV1.3 channels (n = 10) compared with cells coexpressing KCa3.1 and CaV1.2 channels (n = 9; p < 0.05; Fig. 4B–D). The greater activation of KCa3.1 tail current by CaV1.3 was further reflected in the duration of the outward current, with KCa3.1- and CaV1.3-expressing cells producing 3.2 ± 0.8 s (n = 10) of tail current compared with only 0.5 ± 0.15 s (n = 9) of tail current observed in CaV1.2- and KCa3.1-expressing cells (Fig. 4D).
Figure 4.
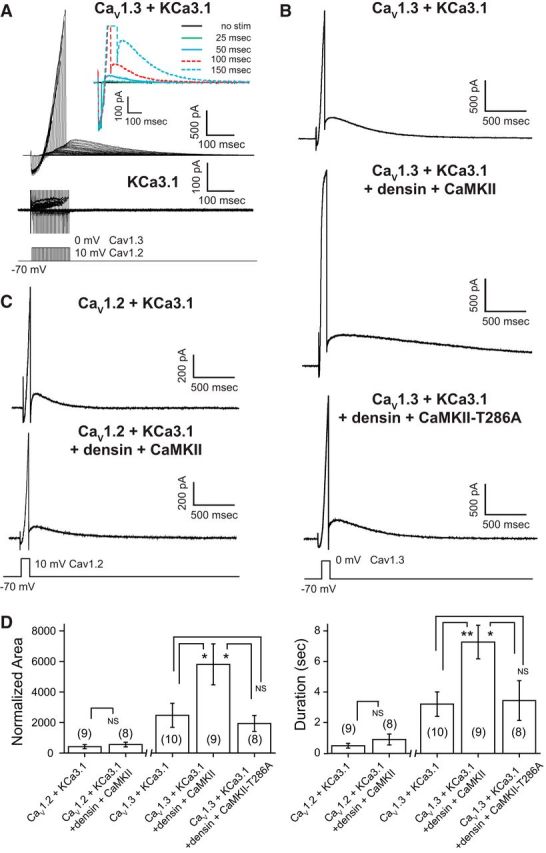
CaV1.3 channels evoke a long outward tail current when coexpressed with KCa3.1. A, A long outward tail current is preferentially evoked in tsA-201 cells coexpressing CaV1.3 and KCa3.1 channels. Inset of a magnified view of tail currents reveals a graded activation of the tail current in direct relation to the duration of a step command. However, no tail currents are detected in cells expressing KCa3.1 in isolation (bottom records). B, Coexpression of CaV1.3 and KCa3.1 along with densin and CaMKII reveals larger outward tail currents of up to 6 s in response to a 100 ms depolarizing step compared with cells expressing CaV1.3 and KCa3.1 alone. Larger tail currents are not observed in cells expressing the autophosphorylation mutant CaMKII-T286A along with densin, CaV1.3, and KCa3.1. C, Outward tail currents recorded in CaV1.2- and KCa3.1-expressing cells are comparatively small in amplitude and are not augmented by the coexpression of densin and CaMKII. D, Bar plots of the mean area and duration of tail current activated by a 100 ms step. All values are normalized to the preceding inward peak calcium current. CaV1.3 channels are more effective than CaV1.2 in generating an outward tail current that is further augmented by densin and autophosphorylated CaMKII. A one-way ANOVA followed by post hoc unpaired Student's t test was performed between the CaV1.3 groups for area (p < 0.05) and duration (p < 0.05) and an unpaired Student's t test between CaV1.2 groups (NS). NS, Not significant. *p < 0.05; **p < 0.01. Cell numbers are shown in brackets in D.
CaV1.3 channel facilitation augments KCa3.1 tail current
The effect of CaV1 current facilitation on calcium influx could be important to regulating CaV1 channel-mediated activation of afterhyperpolarizations that control spike output in CA1 cells, where CaV1.3, CaV1.2, and densin are all expressed (Jenkins et al., 2010; Gamelli et al., 2011; Stanika et al., 2016; Wang et al., 2017). To determine the effect of densin- and CaMKII-induced CaV1 channel facilitation on KCa3.1 outward tail currents, we coexpressed CaV1.3 and KCa3.1 or CaV1.2 and KCa3.1 together with densin and CaMKII.
We found that coexpressing CaV1.3 and KCa3.1 together with densin and CaMKII dramatically increased the outward KCa3.1 tail current following a step command compared with cells coexpressing only CaV1.3 and KCa3.1 (n = 9, p < 0.05; Fig. 4B,D). Importantly, coexpressing CaV1.3 with densin and CaMKII did not change CaV1.3 peak current amplitude (220.9 ± 24.4 pA, n = 9; p = 0.150, unpaired Student's t test) or voltage dependence (n = 7), as reported previously (Jenkins et al., 2010). A separate set of experiments established that coexpressing CaV1.3, densin, and CaMKII did not generate a calcium tail current that could account for the longer KCa3.1 tail current (n = 7; data not shown). Similarly, coexpressing KCa3.1 with densin and CaMKII did not alter KCa3.1 activation on I–V plots (n = 7) or produce an outward tail current (n = 6; data not shown). By comparison, coexpressing CaV1.2 with KCa3.1, densin, and CaMKII did not promote a significant increase in outward tail current compared with CaV1.2 and KCa3.1 (n = 8, p = 0.495; Fig. 4C,D). Finally, to determine whether the densin–CaMKII-mediated increase in KCa3.1 tail current requires autophosphorylation of CaMKII, we coexpressed CaV1.3, KCa3.1, and densin with the CaMKII-T286A mutant CaMKII construct lacking the autophosphorylation site (Liraz et al., 2009; Jenkins et al., 2010). These tests established that facilitation of the KCa3.1 outward current was lost in cells expressing CaMKII-T286A (Fig. 4B,D).
Together, these data establish that CaV1.3 and KCa3.1 interact in a manner that activates a long-lasting outward tail current that is much greater than that for CaV1.2 and that is facilitated by the coexpression of densin and activated CaMKII.
L-type current facilitation and the sAHP in CA1 pyramidal cells
A functional coupling between CaV1.3 and KCa3.1 that is facilitated by densin and CaMKII could be of direct significance to the generation of an sAHP in CA1 pyramidal cells. The calcium-dependent IsAHP has been proposed to reflect the activation of KCa2.x isoforms (Bowden et al., 2001; Lima and Marrion, 2007), KCa3.1 (King et al., 2015; Turner et al., 2016), and the Na-K pump (Gulledge et al., 2013), with cell-specific contributions by KCNQ channels (Tzingounis et al., 2010; Chen et al., 2014; Soh et al., 2014). Restricting analyses to non-KCa2.x channel isoforms is straightforward given their sensitivity to low levels of apamin, the block of KCNQ channels by XE-991, and Na-K pump activity reduced by recording at 32°C (Gulledge et al., 2013). The IsAHP in CA1 pyramidal cells is insensitive to a range of calcium channel blockers but is reduced by dihydropyridines, suggesting a primary role for CaV1 channels in activating the sAHP (Moyer et al., 1992; Lima and Marrion, 2007). However, pharmacological tests using dihyropyridines are complicated by a comparable sensitivity of CaV1 and KCa3.1 channels to block by dihydropyridines. For instance, the IC50 for nimodipine is 0.135 μm for CaV1.2 and 2.7 μm for CaV1.3, with KCa3.1 exhibiting a Kd of 1 μm for nimodipine (Catterall et al., 2005; Wulff et al., 2007; Huang et al., 2013). By comparison, the typical concentration used experimentally to block CaV1 channels is 10 μm nimodipine.
To identify a drug concentration that could reduce CaV1.x current without affecting KCa3.1, we first applied a range of concentrations of dihydropyridines on whole-cell recordings in tsA-201 cells using an internal electrolyte containing 1 μm calcium. KCa3.1 was evoked using a 500 ms ramp command from −110 to 50 mV, evoking a current that reversed at EK (−70.68 ± 0.22 mV, n = 10) and exhibited only 6.7 ± 1.6% rundown after 10–15 min of whole-cell recording (n = 5). To confirm the expression as KCa3.1, we bath applied the KCa3.1 blocker TRAM-34 (1 μm), which reduced KCa3.1 current by ∼90% within 3–5 min (n = 6; p < 0.001; Fig. 5A). We then bath applied different L-type channel blockers at concentrations typically used experimentally. These tests confirmed that KCa3.1 current measured at 50 mV was significantly reduced by 30–45% by the DHPs nifedipine (10 μm) and nimodipine (10 μm), and to a similar extent by the phenylalkylamine verapamil (100 μm; Fig. 5A). Importantly, isradipine applied at 500 nm had no significant direct effect on KCa3.1 current after 10–15 min of exposure (n = 6, p = 0.364; Fig. 5A,B). We then expressed CaV1.2 or CaV1.3 channels to evoke calcium current with a 100 ms ramp command from −80 to 60 mV, finding that 500 nm isradipine provided a substantial block of CaV1.2 (64.9 ± 4.4%, n = 7, p < 0.001) or CaV1.3 (54.7 ± 4.6%, n = 6, p < 0.001) currents (Fig. 5C).
Figure 5.

L-type channels play a key role in evoking the CA1 pyramidal cell IsAHP. A–C, The effects of L-type calcium channel blockers on KCa3.1 whole-cell current recorded in tsA-201 cells with 1 μm calcium in the electrode and evoked by a ramp command as shown in B. Bar plots show a differential block of KCa3.1 current by L-type blockers (A). Representative recordings of KCa3.1 current in B and C illustrate a complete block by 1 μm TRAM-34 but no effect of 500 nm isradipine, a dose sufficient to provide substantial block of whole-cell L-type calcium current in tsA-201 cells expressing either CaV1.2 or CaV1.3 cDNA or in CA1 pyramidal cells (C). D, E, The IsAHP in rat CA1 hippocampal pyramidal cells evoked by a depolarizing step command of 200 ms to 40 mV. The IsAHP is only slightly reduced by a medium containing blockers against all CaV channels except L-type (1 μm ω-conotoxin MVIIC, 200 nm ω-agatoxin IVA, 200 nm SNX-482, 1 μm TTA-P2). The remaining presumed L-type channel-activated IsAHP is significantly reduced by 1 μm TRAM-34. Recordings in the bottom part of D are from a separate cell in which 500 nm isradipine was applied in the presence of the non-L-type CaV channel blockers, revealing a significant reduction of IsAHP. *p < 0.05; **p < 0.01; ***p < 0.001. NS, Not significant. Cell numbers are shown in brackets in A, C, and E.
We then prepared rat hippocampal tissue slices and obtained whole-cell recordings to evoke the IsAHP using a 200 ms step command from −100 to 40 mV. To focus on the potential interactions between CaV1.x and KCa3.1 channels, all IsAHP recordings were conducted in the presence of apamin (100 nm) and XE-991 (10 μm), and recordings were conducted at 32°C. We then perfused a set of calcium channel blockers to reduce the activity of all channel isoforms except L-type calcium channels (1 μm ω-conotoxin MVIIC, 200 nm ω-agatoxin IVA, 200 nm SNX-482, and 1 μm TTA-P2), revealing a slight but nonsignificant reduction in IsAHP (Fig. 5D). Internal infusion of 1 μm TRAM-34 through the electrode to block KCa3.1 channels then reduced the remaining IsAHP to ∼15% of its original amplitude (Fig. 5D,E). In a separate set of tests, we recorded the IsAHP in the presence of the non-L-type calcium channel blockers (as described above) and then perfused 500 nm isradipine to block CaV1 channels, which reduced the IsAHP by ∼40% within 10 min (Fig. 5D,E). The incomplete block of the IsAHP by isradipine is taken to reflect only a partial block of CaV1 channels at this relatively low concentration (Helton et al., 2005) but is one we could ensure had no direct effects on KCa3.1 (Fig. 5B,C). These tests are important in providing pharmacological evidence that L-type calcium channels contribute to activating at least the KCa3.1-mediated component of the IsAHP in rat CA1 pyramidal cells.
L-type current facilitation of sAHP and spike output in CA1 pyramidal cells
The sAHP has an established role of contributing to spike accommodation in CA1 pyramidal cells (Lancaster and Nicoll, 1987; Chen et al., 2014; King et al., 2015). To test the functional contribution of L-type calcium channels to the sAHP and spike accommodation, we activated spike discharge in CA1 cells using postsynaptic current injection in the presence of 100 nm apamin and 10 μm XE-991 to block KCa2.x and KCNQ channels, respectively. Applying 500 nm isradipine to reduce CaV1 channel current significantly increased the total number and frequency of spikes evoked (n = 6; p < 0.05, Wilcoxon signed rank test; Fig. 6A,B).
Figure 6.

L-type channels control hippocampal CA1 cell excitability by modulating spike accommodation and the sAHP. A, B, Current-evoked spike discharge in rat CA1 hippocampal pyramidal cells in the presence of 100 nm apamin and 10 μm XE-991 to block SK and KV7 potassium channels, respectively. Bath application of 500 nm isradipine reduced the interspike interval and increased the total spike frequency and number. C, Whole-cell recordings of sAHP in CA1 pyramidal cells evoked by a suprathreshold 5 pulse, 50 Hz stimulus train to stratum radiatum inputs indicates that the sAHP is reduced by 500 nm israpidine. Inset shows in another cell that isradipine does not block the smaller sAHP that follows a subthreshold train of synaptic inputs. D, Bar plots of the mean values of sAHP indicate a significant reduction of sAHP by 500 nm isradipine when the sAHP is evoked by suprathreshold but not subthreshold stratum radiatum inputs (C). *p < 0.05, Wilcoxon signed rank test. Cell numbers are shown in brackets in B and D.
A large sAHP that is insensitive to apamin and XE-991 can also be evoked following a short train of 50 Hz stimuli to stratum radiatum inputs (King et al., 2015), the same pattern of stimulus found here to facilitate CaV1.3 current. Single-channel recordings have shown that high voltage-activated calcium channels are preferentially evoked in response to synaptic stimulation that evokes spike discharge (Magee and Johnston, 1995). Previous work also established that the sAHP following a short synaptic input train is larger for suprathreshold versus subthreshold levels of activation (Wu et al., 2004). We therefore compared the sensitivity of the sAHP evoked following a 5 pulse 50 Hz train of synaptic stimuli to isradipine. We first examined the sAHP following a 50 Hz train and confirmed that suprathreshold synaptic activation evoked an sAHP that was ∼13× larger in terms of area and ∼10× longer duration compared with subthreshold synaptic inputs (Fig. 6C,D). To test the contribution of L-type channels to subthreshold and suprathreshold inputs, we applied 500 nm isradipine. Control tests confirmed that this concentration of isradipine did not affect the baseline amplitude of stratum radiatum-evoked subthreshold EPSPs (6.5 ± 1.3 mV, control; 5.9 ± 1.4 mV, isradipine 500 μm; n = 8; p = 0.148, Wilcoxon signed rank test). We found that 500 nm isradipine significantly reduced the sAHP area following a train of suprathreshold synaptic stimuli to 51.41 ± 6.68% of its original size (n = 6; p < 0.03, Wilcoxon signed rank test). In contrast, isradipine had no significant effect on the sAHP evoked by subthreshold synaptic stimuli (Fig. 6C,D; n = 8; p = 0.547, Wilcoxon signed rank test).
While we cannot rule out the potential influence of isradipine on other calcium channel isoforms, these data are consistent with a role for CaV1 calcium channel-mediated activation of KCa3.1 channels in producing spike accommodation and the sAHP that follows a train of suprathreshold synaptic inputs.
Densin and CaMKII contribute to the IsAHP
The tests conducted in tsA-201 cells established a role for densin and CaMKII in facilitating CaV1.3 current and producing a long KCa3.1-mediated outward tail current in response to a brief step command (Figs. 2, 4). Hippocampal CA1 pyramidal cells are known to express densin (Apperson et al., 1996; Jenkins et al., 2010). To test the potential influence of densin on the IsAHP, we cultured hippocampal neurons to perform knock-down experiments. Whole-cell recordings were obtained from cultured neurons in the presence of 100 nm apamin and 10 μm XE-991 to isolate KCa3.1 currents. Under voltage clamp, delivering short trains of 10 step commands to 40 mV (for 5 ms) at 50 Hz evoked an IsAHP with an area of 10,710 ± 3016 pA × ms (n = 10; Fig. 7A). To test the role of densin in the sAHP, we transfected cultured neurons with either of two different siRNAs against densin and recorded the IsAHP in cells recorded 3–5 d following transfection. These tests revealed that densin siRNA reduced the IsAHP to 30–40% of the value found in nontransfected cells (n = 8–12; Fig. 7B). By comparison, no significant reduction in the IsAHP was found in cells transfected with scrambled control siRNA (n = 10, p = 0.653; Fig. 7B). Finally, we established through Western blot analysis that densin protein levels were significantly reduced (n = 3; p < 0.001, one-way ANOVA) in lysates of tsA-201 cells coexpressing a densin–GFP construct and either form of densin siRNA (Fig. 7C). Similarly, Q-PCR analysis revealed a 25–32% decrease in mRNA following exposure to either form of densin siRNA in hippocampal cultures (Fig. 7D), a value that approximates the transfection rate of cultured neurons using lipofectamine.
Figure 7.
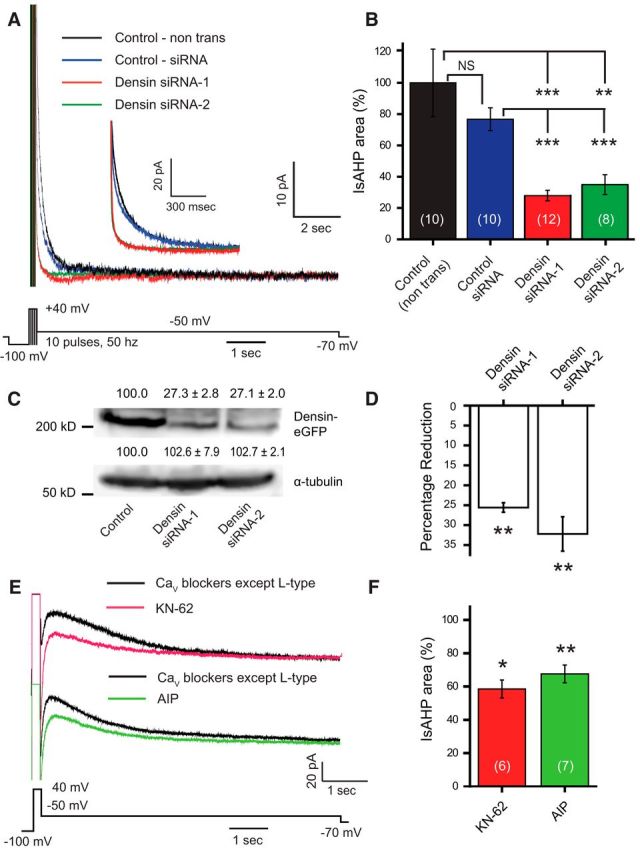
The IsAHP in hippocampal neurons is regulated by densin and CaMKII. A, Whole-cell recordings of IsAHP evoked by a series of brief pulses to 40 mV in dissociated hippocampal pyramidal neurons in the presence of 100 nm apamin and 10 μm XE-991 to block SK and Kv7 potassium channels, respectively. Pulse trains consisted of 10 5 ms pulses at 50 Hz. Inset shows an enlarged view of the early component of the IsAHP recorded under the indicated conditions. B, The area of the IsAHP is significantly reduced in cells pretreated with either of two siRNAs directed against the postsynaptic scaffolding protein densin compared with nontransfected cells (control) or with cells transfected with a universal control siRNA (one-way ANOVA; p < 0.001). C, Representative Western blot of protein levels of densin or the loading control α-tubulin prepared from lysates of tsA-201 cells expressing densin-GFP with either of two forms of densin siRNA. Proteins were detected using antibodies against GFP or α-tubulin and the density was quantified (using ImageJ) with mean percentages relative to cells expressing only densin-GFP shown above each lane (n = 3; one-way ANOVA, p < 0.001). The levels of densin are greatly reduced in tsA-201 cells cotransfected with either siRNA-1 or siRNA-2 compared with controls. D, Percentage reduction of densin mRNA levels in cultured hippocampal neurons transfected with either of two densin siRNAs compared with cells transfected with control siRNA. The Ct values of densin mRNA from each group are normalized with the Ct value of β-actin as the internal control. Both siRNAs significantly reduced densin mRNA levels in hippocampal neurons compared with neurons transfected with control siRNA (n = 3; one-way ANOVA, p < 0.01). E, Whole-cell recordings of IsAHP in rat CA1 pyramidal cells evoked in a medium containing blockers against all CaV channels except L-type (1 μm ω-conotoxin MVIIC, 200 nm ω-agatoxin IVA, 200 nm SNX-482, and 1 μm TTA-P2). The remaining presumed L-type channel-activated IsAHP is reduced by the CaMKII inhibitors KN-62 and AIP. KN-62 (10 μm) was bath applied, and AIP (20 μm) was internally infused through the electrode. F, Bar plots of mean values for IsAHP area in CA1 pyramidal cells indicate a significant reduction upon the application of CaMKII inhibitors KN-62 or AIP relative to traces obtained before the drug application (as in E; paired Student's t test). *p < 0.05; **p < 0.01; ***p < 0.001. NS, Not significant. Cell numbers are shown in brackets in B and F.
To test the potential role of CaMKII in generating the IsAHP, we recorded from rat CA1 pyramidal cells in the slice preparation and used a brief step command to 40 mV (200 ms) to evoke the IsAHP (Fig. 7E). As before, these recordings were conducted in a medium containing blockers against all CaV channels except L-type channels (1 μm ω-conotoxin MVIIC, 200 nm ω-agatoxin IVA, 200 nm SNX-482, and 1 μm TTA-P2), together with 100 nm apamin and 10 μm XE-991. We then exposed the cells to blockers of CaMKII autophosphorylation using KN-62 (10 μm) in the bathing medium or autocamtide-2-related inhibitory peptide (AIP; 20 μm) through internal infusion through the electrode. In both cases, the IsAHP was significantly reduced by interfering with CaMKII activation (Fig. 7E,F). This is consistent with a previous report in which αCaMKII-T286A knock-in mice showed a significant decrease in the Schaeffer collateral-evoked sAHP and an increased excitability of CA1 neurons compared with wild-type animals (Sametsky et al., 2009). However, it is important to note that these studies cannot distinguish between direct actions of CaMKII on CaV1.2 channels (Blaich et al., 2010) or CaV1.3 channels (Jenkins et al., 2010).
Together, these tests reveal that the CaV1-mediated activation of KCa3.1 to contribute to the IsAHP is augmented by the actions of densin and CaMKII in rat CA1 pyramidal cells.
Discussion
The current study examined the nature of CaV1.3 facilitation in response to a brief train of stimuli and the degree to which this can produce a physiologically relevant output in the activation of KCa3.1 channels. The data reveal a new form of CaV1.3-specific CDF that long outlasts a depolarizing stimulus train through a CaV1.3–densin–CaMKII interaction. This interaction further reveals the triggering of a long-duration KCa3.1-mediated tail current consistent with activation of a slow AHP that is found in many central neurons.
Facilitation of CaV1.3 current
CaV1.3 channels exhibit several splice isoforms with varying lengths of the C terminus that can determine the contribution to CDI or CDF. The current study focused on a long C-terminal isoform of CaV1.3 that contains an interacting site for densin that acts as a bridge for CaMKII to reduce cumulative inactivation due to CDI during a stimulus train (Jenkins et al., 2010). We now find that this CaV1.3–densin–CaMKII interaction supports a late facilitation process that extends beyond a brief 50 Hz depolarizing stimulus by up to 8 s. The late facilitation was absent in barium-containing medium, revealing the CaV1.3–densin–CaMKII interaction as a new form of L-CDF that follows a depolarizing stimulus. While we also observed CaV1.3 CDI during a stimulus train, test pulses following the stimulus suggest that the majority of this process recovers within 500 ms in cells expressing only CaV1.3 (Fig. 1B). The long C-terminal isoform also contains proximal and distal regulatory domains that interfere with the binding of CaM to the IQ domain to reduce CDI (Singh et al., 2008; Bock et al., 2011). These regulatory domains do not account for L-CDF given the select increase of current amplitude and the lack of change in the rate of CDI in the facilitated responses. Alternative splicing can also give rise to short C-terminal isoforms, with a CaV1.3s producing a form of CDF through a cooperative gating mechanism that arises from a link between CaV1.3 C termini (Moreno et al., 2016). However, there is no apparent C-terminal linkage or cooperative gating in the long CaV1.3 isoform (Moreno et al., 2016). Instead, L-CDF required coexpression of CaV1.3, densin, CaMKII, and autophosphorylation of CaMKII (Fig. 1), while no L-CDF was apparent for the CaV1.3s isoform.
Such an extended form of L-CDF has rarely been reported for other calcium channels. CaV2.1 (P/Q-type) channels exhibit facilitation following a preconditioning pulse that is calmodulin dependent and CaMKII independent and only persists for 1.5 s (DeMaria et al., 2001; Lee et al., 2002). CaV1.2 channels of cardiac myocytes exhibit CaMKII-dependent facilitation upon phosphorylation of two amino acid residues on the C terminus (Blaich et al., 2010), but this has not been reported for neuronal CaV1.2. In expression systems CaV1.2 channels can exhibit a voltage-dependent facilitation (Bourinet et al., 1994; Lee et al., 2006) or a CaMKII-dependent facilitation, but the latter only when amino acids responsible for CDI are mutated (Hudmon et al., 2005; Christel and Lee, 2012). These mechanisms are distinct from CaV1.3 L-CDF given that densin does not facilitate CaV1.2 and the CaV1.3 C terminus lacks CaMKII binding sites (Jenkins et al., 2010; Wang et al., 2017). Consistent with this, we found no facilitation for CaV1.2 when coexpressed with densin and/or CaMKII, or for CaV1.3 when coexpressed with CaMKII. Therefore, the CaV1.3–densin–CaMKII interaction that supports L-CDF is distinct from the molecular basis for facilitation reported for other high voltage-activated calcium channels.
CaV1.3–KCa3.1 coupling
Calcium channels can couple to potassium channels at the molecular or functional level. A functional coupling is signified for channel separations of 50–200 nm that can be blocked by the relatively slow calcium buffer EGTA (Augustine et al., 2003). A link at the molecular level can position channels within a distance of 50 nm to give rise to a nanodomain interaction that is sensitive to BAPTA. KCa3.1 channels have been shown to couple at the nanodomain level to CaV3.2 T-type calcium channels in cerebellar Purkinje cells, where they mediate a slow AHP (Engbers et al., 2012) and maintain axonal spike conduction (Gründemann and Clark, 2015). KCa3.1 channels in enteric neurons are activated by a calcium increase by CaV2.2 (N-type) calcium channels and internal release to mediate a slow AHP (Smith et al., 2003; Neylon et al., 2004).
In the current study, we found a close correspondence between CaV1.3 calcium influx and KCa3.1 activation that could reflect a microdomain interaction, as judged by a block by EGTA and a lack of coimmunoprecipitation and FRET. The ability for CaV1 channels to activate KCa3.1 channels in CA1 pyramidal cells was established using a 500 μm dose of isradipine that restricted actions to CaV1 calcium channels and by a TRAM-34-sensitive block of the IsAHP evoked under conditions that isolated CaV1 channels (Fig. 5D,E). This dose of isradipine further increased spike output during direct current injections and highlighted the dependence of a CaV1-mediated sAHP on spike discharge using subthreshold versus suprathreshold synaptic inputs (Fig. 6). These data reveal a functional coupling between israpidine-sensitive CaV1 channels and KCa3.1 that depends on spike-activated calcium influx. The dependence of the IsAHP on densin and CaMKII autophosphorylation is also consistent with a CaV1.3–KCa3.1 interaction in pyramidal cells.
It is important to acknowledge previous evidence for CaV1.2 calcium channel activation of KCa2.1 (SK1) channels in pyramidal cells, as supported by colocalization of CaV1.2 and KCa2.1 immunolabels and activation of a 10 pS potassium channel (Bowden et al., 2001). We cannot rule out a potential role for this channel pair in the sAHP, but our recordings were conducted in 100 nm apamin to block all KCa2.x isoforms. Another potential means for CaV1.x channels to activate KCa3.1 is a cooperative gating mode detected between CaV1.3s or between CaV1.2 channels that can increase calcium current and thus potentially KCa3.1 activation (Dixon et al., 2015; Moreno et al., 2016). We predict that any such facilitation would be distinct from the L-CDF reported here, given that CaV1.3s lacks a densin binding site. Our tests on CaV1.3s in fact revealed no L-CDF following a 50 Hz stimulus train (Fig. 1E) and no significant difference in the CaV1.3s-mediated KCa3.1 tail current compared with the long C-terminal isoform of CaV1.3 in the absence of densin and CaMKII (data not shown).
CaV1.3 L-CDF, IsAHP, and KCa3.1
L-type channels in hippocampal pyramidal cells are known to exhibit delayed openings following depolarizing steps that can outlast the stimulus, referred to as repolarization openings (Thibault et al., 1993) or Lp versus Ls channel subtypes (Kavalali and Plummer, 1994, 1996; Schjott and Plummer, 2000). However, this channel activity lasts <500 ms following a stimulus, even in the presence of DHP agonists, and thus potentially within the time of a calcium tail current. By comparison, the duration of the CaV1.3-activated sAHP is consistent with a delayed facilitation of L-type channel activity in pyramidal cells (Cloues et al., 1997). Here L-type channel openings gave rise to an ensemble average that peaked within 500 ms after the train and decayed over ∼4 s (Lima and Marrion, 2007). However, the recordings that documented delayed facilitation were conducted in 10 mm barium, whereas the L-CDF detected here in relation to densin and CaMKII was blocked in 5 mm barium-containing medium (Fig. 1G). It thus appears that the delayed facilitation reported earlier (Cloues et al., 1997; Lee et al., 2002) reflects a voltage-dependent process that presumably works in concert with that of L-CDF under conditions yet to be defined.
Studies to identify the channels that underlie the sAHP in pyramidal cells have been long underway (Lancaster and Nicoll, 1987; Lima and Marrion, 2007; Tzingounis et al., 2010; Andrade et al., 2012; Gulledge et al., 2013; Chen et al., 2014; King et al., 2015; Turner et al., 2016; Wang et al., 2016). Of interest to the procedures we used to record the sAHP are reports of presumed SK potassium channels where ensemble averages recapitulated the sAHP following a stimulus train (Bowden et al., 2001; Lima and Marrion, 2007). These potassium channels were even shown to exhibit a close coupling in time to L-type channel openings within single-patch recordings (Marrion and Tavalin, 1998). Unfortunately, the pharmacological tools available to test these potassium channels were limited at the time. Many of the properties of these channels would appear to match that of TRAM-34-sensitive KCa3.1 potassium channels recorded following a train of synaptic stimuli (King et al., 2015). We would thus interpret at least some of the channel recordings presented previously (Cloues et al., 1997; Marrion and Tavalin, 1998; Bowden et al., 2001; Lima and Marrion, 2007) as corresponding to KCa3.1 channels.
The current study thus identifies a new form of CaV1.3 channel CDF that long outlasts a depolarizing stimulus due to interactions with the accessory protein densin and CaMKII. One functional output of this interaction is the generation of a long outward tail current following a stimulus train that helps account for the activation of calcium-gated potassium channels underlying the sAHP in CA1 pyramidal cells. Given the widespread expression of these proteins in cortical neurons, this form of L-CDF could be functionally relevant to many different cell types.
Footnotes
This work was supported by a National Science and Engineering Research Council Discovery Grant (R.W.T.). Further support was provided by an Eyes High Postdoctoral Fellowship (University of Calgary; to G.S.) and Alberta Innovates–Health Solutions (AI-HS) Fellowships (to G.S. and F.-X.Z.). R.W.T. and G.W.Z. are AI-HS Scientists, and G.W.Z. holds a Canada Research Chair. We thank M. Kruskic, L. Chen, and Dr. F. Visser for expert technical assistance in immunocytochemistry, cell culture preparations, and quantitative PCR experiments.
The authors declare no competing financial interests.
References
- Adelman JP, Maylie J, Sah P (2012) Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol 74:245–269. 10.1146/annurev-physiol-020911-153336 [DOI] [PubMed] [Google Scholar]
- Andrade R, Foehring RC, Tzingounis AV (2012) The calcium-activated slow AHP: cutting through the Gordian knot. Front Cell Neurosci 6:47. 10.3389/fncel.2012.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apperson ML, Moon IS, Kennedy MB (1996) Characterization of densin-180, a new brain-specific synaptic protein of the O-sialoglycoprotein family. J Neurosci 16:6839–6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asmara H, Micu I, Rizwan AP, Sahu G, Simms BA, Zhang FX, Engbers JDT, Stys PK, Zamponi GW, Turner RW (2017) A T-type channel-calmodulin complex triggers alphaCaMKII activation. Mol Brain 10:37. 10.1186/s13041-017-0317-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K (2003) Local calcium signaling in neurons. Neuron 40:331–346. 10.1016/S0896-6273(03)00639-1 [DOI] [PubMed] [Google Scholar]
- Babai N, Kanevsky N, Dascal N, Rozanski GJ, Singh DP, Fatma N, Thoreson WB (2010) Anion-sensitive regions of L-type CaV1.2 calcium channels expressed in HEK293 cells. PLoS One 5:e8602. 10.1371/journal.pone.0008602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Johny M, Yue DT (2014) Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J Gen Physiol 143:679–692. 10.1085/jgp.201311153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Fakler B (2008) Repolarizing responses of BKCa-Cav complexes are distinctly shaped by their Cav subunits. J Neurosci 28:8238–8245. 10.1523/JNEUROSCI.2274-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, Klugbauer N, Reisinger E, Bischofberger J, Oliver D, Knaus HG, Schulte U, Fakler B (2006) BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 314:615–620. 10.1126/science.1132915 [DOI] [PubMed] [Google Scholar]
- Blaich A, Welling A, Fischer S, Wegener JW, Köstner K, Hofmann F, Moosmang S (2010) Facilitation of murine cardiac L-type Ca(v)1.2 channel is modulated by calmodulin kinase II-dependent phosphorylation of S1512 and S1570. Proc Natl Acad Sci U S A 107:10285–10289. 10.1073/pnas.0914287107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock G, Gebhart M, Scharinger A, Jangsangthong W, Busquet P, Poggiani C, Sartori S, Mangoni ME, Sinnegger-Brauns MJ, Herzig S, Striessnig J, Koschak A (2011) Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J Biol Chem 286:42736–42748. 10.1074/jbc.M111.269951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Charnet P, Tomlinson WJ, Stea A, Snutch TP, Nargeot J (1994) Voltage-dependent facilitation of a neuronal alpha 1C L-type calcium channel. EMBO J 13:5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden SE, Fletcher S, Loane DJ, Marrion NV (2001) Somatic colocalization of rat SK1 and D class (Cav1.2) L-type calcium channels in rat CA1 hippocampal pyramidal neurons. J Neurosci 21:RC175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J (2005) International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev 57:411–425. 10.1124/pr.57.4.5 [DOI] [PubMed] [Google Scholar]
- Chen S, Benninger F, Yaari Y (2014) Role of small conductance Ca2+-activated K+ channels in controlling CA1 pyramidal cell excitability. J Neurosci 34:8219–8230. 10.1523/JNEUROSCI.0936-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christel C, Lee A (2012) Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim Biophys Acta 1820:1243–1252. 10.1016/j.bbagen.2011.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloues RK, Tavalin SJ, Marrion NV (1997) Beta-adrenergic stimulation selectively inhibits long-lasting L-type calcium channel facilitation in hippocampal pyramidal neurons. J Neurosci 17:6493–6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RL, Lechner SM, Williams ME, Prodanovich P, Bleicher L, Varney MA, Gu G (2005) Differential distribution of voltage-gated calcium channel alpha-2 delta (alpha2delta) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J Comp Neurol 491:246–269. 10.1002/cne.20693 [DOI] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT (2001) Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411:484–489. 10.1038/35078091 [DOI] [PubMed] [Google Scholar]
- Dixon RE, Moreno CM, Yuan C, Opitz-Araya X, Binder MD, Navedo MF, Santana LF (2015) Graded Ca(2)(+)/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. Elife 4:e05608. 10.7554/eLife.05608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engbers JD, Anderson D, Asmara H, Rehak R, Mehaffey WH, Hameed S, McKay BE, Kruskic M, Zamponi GW, Turner RW (2012) Intermediate conductance calcium-activated potassium channels modulate summation of parallel fiber input in cerebellar Purkinje cells. Proc Natl Acad Sci U S A 109:2601–2606. 10.1073/pnas.1115024109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Stemkowski PL, Gandini MA, Black SA, Zhang Z, Souza IA, Chen L, Zamponi GW (2016) Reduced hyperpolarization-activated current contributes to enhanced intrinsic excitability in cultured hippocampal neurons from PrP(−/−) mice. Front Cell Neurosci 10:74. 10.3389/fncel.2016.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamelli AE, McKinney BC, White JA, Murphy GG (2011) Deletion of the L-type calcium channel Ca(V) 1.3 but not Ca(V) 1.2 results in a diminished sAHP in mouse CA1 pyramidal neurons. Hippocampus 21:133–141. 10.1002/hipo.20728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründemann J, Clark BA (2015) Calcium-activated potassium channels at nodes of Ranvier secure axonal spike propagation. Cell Rep 12:1715–1722. 10.1016/j.celrep.2015.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Dasari S, Onoue K, Stephens EK, Hasse JM, Avesar D (2013) A sodium-pump-mediated afterhyperpolarization in pyramidal neurons. J Neurosci 33:13025–13041. 10.1523/JNEUROSCI.0220-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA (1993) Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol 123:949–962. 10.1083/jcb.123.4.949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton TD, Xu W, Lipscombe D (2005) Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci 25:10247–10251. 10.1523/JNEUROSCI.1089-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Yu D, Soong TW (2013) C-terminal alternative splicing of CaV1.3 channels distinctively modulates their dihydropyridine sensitivity. Mol Pharmacol 84:643–653. 10.1124/mol.113.087155 [DOI] [PubMed] [Google Scholar]
- Hudmon A, Lebel E, Roy H, Sik A, Schulman H, Waxham MN, De Koninck P (2005) A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J Neurosci 25:6971–6983. 10.1523/JNEUROSCI.4698-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, Usachev YM, Obermair GJ, Colbran RJ, Lee A (2010) Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J Neurosci 30:5125–5135. 10.1523/JNEUROSCI.4367-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Plummer MR (1994) Selective potentiation of a novel calcium channel in rat hippocampal neurones. J Physiol 480:475–484. 10.1113/jphysiol.1994.sp020376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Plummer MR (1996) Multiple voltage-dependent mechanisms potentiate calcium channel activity in hippocampal neurons. J Neurosci 16:1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King B, Rizwan AP, Asmara H, Heath NC, Engbers JD, Dykstra S, Bartoletti TM, Hameed S, Zamponi GW, Turner RW (2015) IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Rep 11:175–182. 10.1016/j.celrep.2015.03.026 [DOI] [PubMed] [Google Scholar]
- Koschak A. (2010) Impact of gating modulation in CaV1.3 L-type calcium channels. Channels 4:523–525. 10.4161/chan.4.6.12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J (2001) alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem 276:22100–22106. 10.1074/jbc.M101469200 [DOI] [PubMed] [Google Scholar]
- Koschak A, Obermair GJ, Pivotto F, Sinnegger-Brauns MJ, Striessnig J, Pietrobon D (2007) Molecular nature of anomalous L-type calcium channels in mouse cerebellar granule cells. J Neurosci 27:3855–3863. 10.1523/JNEUROSCI.4028-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA (1987) Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol 389:187–203. 10.1113/jphysiol.1987.sp016653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Westenbroek RE, Haeseleer F, Palczewski K, Scheuer T, Catterall WA (2002) Differential modulation of Ca(v)2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci 5:210–217. 10.1038/nn805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, Karl R, Moosmang S, Lenhardt P, Klugbauer N, Hofmann F, Kleppisch T, Welling A (2006) Calmodulin kinase II is involved in voltage-dependent facilitation of the L-type Cav1.2 calcium channel: identification of the phosphorylation sites. J Biol Chem 281:25560–25567. 10.1074/jbc.M508661200 [DOI] [PubMed] [Google Scholar]
- Lima PA, Marrion NV (2007) Mechanisms underlying activation of the slow AHP in rat hippocampal neurons. Brain Res 1150:74–82. 10.1016/j.brainres.2007.02.067 [DOI] [PubMed] [Google Scholar]
- Liraz O, Rosenblum K, Barkai E (2009) CAMKII activation is not required for maintenance of learning-induced enhancement of neuronal excitability. PLoS One 4:e4289. 10.1371/journal.pone.0004289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Lima PA, Marrion NV (2007) Co-assembly of N-type Ca2+ and BK channels underlies functional coupling in rat brain. J Cell Sci 120:985–995. 10.1242/jcs.03399 [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D (1995) Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science 268:301–304. 10.1126/science.7716525 [DOI] [PubMed] [Google Scholar]
- Marrion NV, Tavalin SJ (1998) Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395:900–905. 10.1038/27674 [DOI] [PubMed] [Google Scholar]
- Moreno CM, Dixon RE, Tajada S, Yuan C, Opitz-Araya X, Binder MD, Santana LF (2016) Ca(2+) entry into neurons is facilitated by cooperative gating of clustered CaV1.3 channels. Elife 5:e15744. 10.7554/eLife.15744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR Jr, Thompson LT, Black JP, Disterhoft JF (1992) Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol 68:2100–2109. [DOI] [PubMed] [Google Scholar]
- Neylon CB, D'Souza T, Reinhart PH (2004) Protein kinase A inhibits intermediate conductance Ca2+-activated K+ channels expressed in Xenopus oocytes. Pflugers Arch 448:613–620. 10.1007/s00424-004-1302-5 [DOI] [PubMed] [Google Scholar]
- Quitsch A, Berhörster K, Liew CW, Richter D, Kreienkamp HJ (2005) Postsynaptic shank antagonizes dendrite branching induced by the leucine-rich repeat protein densin-180. J Neurosci 25:479–487. 10.1523/JNEUROSCI.2699-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sametsky EA, Disterhoft JF, Ohno M (2009) Autophosphorylation of alphaCaMKII downregulates excitability of CA1 pyramidal neurons following synaptic stimulation. Neurobiol Learn Mem 92:120–123. 10.1016/j.nlm.2009.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schjott JM, Plummer MR (2000) Sustained activation of hippocampal Lp-type voltage-gated calcium channels by tetanic stimulation. J Neurosci 20:4786–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Gebhart M, Fritsch R, Sinnegger-Brauns MJ, Poggiani C, Hoda JC, Engel J, Romanin C, Striessnig J, Koschak A (2008) Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J Biol Chem 283:20733–20744. 10.1074/jbc.M802254200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TK, Kang SH, Vanden Berghe P (2003) Calcium channels in enteric neurons. Curr Opin Pharmacol 3:588–593. 10.1016/j.coph.2003.10.001 [DOI] [PubMed] [Google Scholar]
- Soh H, Pant R, LoTurco JJ, Tzingounis AV (2014) Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. J Neurosci 34:5311–5321. 10.1523/JNEUROSCI.3919-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika R, Campiglio M, Pinggera A, Lee A, Striessnig J, Flucher BE, Obermair GJ (2016) Splice variants of the CaV1.3 L-type calcium channel regulate dendritic spine morphology. Sci Rep 6:34528. 10.1038/srep34528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP, Cummins K, Mishra M, Teo W, Yong VW, Stys P (2014) Toll-like receptor 2-mediated alternative activation of microglia is protective after spinal cord injury. Brain 137:707–723. 10.1093/brain/awt341 [DOI] [PubMed] [Google Scholar]
- Taylor CP, Garrido R (2008) Immunostaining of rat brain, spinal cord, sensory neurons and skeletal muscle for calcium channel alpha2-delta (alpha2-delta) type 1 protein. Neuroscience 155:510–521. 10.1016/j.neuroscience.2008.05.053 [DOI] [PubMed] [Google Scholar]
- Thibault O, Porter NM, Landfield PW (1993) Low Ba2+ and Ca2+ induce a sustained high probability of repolarization openings of L-type Ca2+ channels in hippocampal neurons: physiological implications. Proc Natl Acad Sci U S A 90:11792–11796. 10.1073/pnas.90.24.11792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RW, Kruskic M, Teves M, Scheidl-Yee T, Hameed S, Zamponi GW (2015) Neuronal expression of the intermediate conductance calcium-activated potassium channel KCa3.1 in the mammalian central nervous system. Pflugers Arch 467:311–328. 10.1007/s00424-014-1523-1 [DOI] [PubMed] [Google Scholar]
- Turner RW, Asmara H, Engbers JD, Miclat J, Rizwan AP, Sahu G, Zamponi GW (2016) Assessing the role of IKCa channels in generating the sAHP of CA1 hippocampal pyramidal cells. Channels 10:313–319. 10.1080/19336950.2016.1161988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, Jentsch TJ (2010) The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc Natl Acad Sci U S A 107:10232–10237. 10.1073/pnas.1004644107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandael DH, Marcantoni A, Mahapatra S, Caro A, Ruth P, Zuccotti A, Knipper M, Carbone E (2010) Ca(v)1.3 and BK channels for timing and regulating cell firing. Mol Neurobiol 42:185–198. 10.1007/s12035-010-8151-3 [DOI] [PubMed] [Google Scholar]
- Vandael DH, Marcantoni A, Carbone E (2015) Cav1.3 Channels as Key Regulators of Neuron-Like Firings and Catecholamine Release in Chromaffin Cells. Curr Mol Pharmacol 8:149–161. 10.2174/1874467208666150507105443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Mateos-Aparicio P, Hönigsperger C, Raghuram V, Wu WW, Ridder MC, Sah P, Maylie J, Storm JF, Adelman JP (2016) IK1 channels do not contribute to the slow afterhyperpolarization in pyramidal neurons. Elife 5:e11206. 10.7554/eLife.11206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Stanika RI, Wang X, Hagen J, Kennedy MB, Obermair GJ, Colbran RJ, Lee A (2017) Densin-180 controls the trafficking and signaling of L-type voltage-gated Cav1.2 Ca2+ channels at excitatory synapses. J Neurosci 37:4679–4691. 10.1523/JNEUROSCI.2583-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW (2012) Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 149:1112–1124. 10.1016/j.cell.2012.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Disterhoft JF (2004) Slow afterhyperpolarization governs the development of NMDA receptor-dependent afterdepolarization in CA1 pyramidal neurons during synaptic stimulation. J Neurophysiol 92:2346–2356. 10.1152/jn.00977.2003 [DOI] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG (2000) Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci U S A 97:8151–8156. 10.1073/pnas.97.14.8151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V (2007) Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem 14:1437–1457. 10.2174/092986707780831186 [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D (2001) Neuronal CaV1.3α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21:5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]