

Abstract
Schwann cells (SCs) are myelinating cells of the PNS. Although SCs are known to express different channels and receptors on their surface, little is known about the activation and function of these proteins. Ionotropic glutamate receptors are thought to play an essential role during development of SC lineage and during peripheral nerve injury, so we sought to study their functional properties. We established a novel preparation of living peripheral nerve slices with preserved cellular architecture and used a patch-clamp technique to study AMPA-receptor (AMPAR)-mediated currents in SCs for the first time. We found that the majority of SCs in the nerves dissected from embryonic and neonatal mice of both sexes respond to the application of glutamate with inward current mediated by Ca2+-permeable AMPARs. Using stationary fluctuation analysis (SFA), we demonstrate that single-channel conductance of AMPARs in SCs is 8–11 pS, which is comparable to that in neurons. We further show that, when SCs become myelinating, they downregulate functional AMPARs. This study is the first to demonstrate AMPAR-mediated conductance in SCs of vertebrates, to investigate elementary properties of AMPARs in these cells, and to provide detailed electrophysiological and morphological characterization of SCs at different stages of development.
SIGNIFICANCE STATEMENT We provide several important conceptual and technical advances in research on the PNS. We pioneer the first description of AMPA receptor (AMPAR)-mediated currents in the PNS glia of vertebrates and provide new insights into the properties of AMPAR channels in peripheral glia; for example, their Ca2+ permeability and single-channel conductance. We describe for the first time the electrophysiological and morphological properties of Schwann cells (SCs) at different stages of development and show that functional AMPARs are expressed only in developing, not mature, SCs. Finally, we introduce a preparation of peripheral nerve slices for patch-clamp recordings. This preparation opens new possibilities for studying the physiology of SCs in animal models and in surgical human samples.
Keywords: development, glutamate receptors, patch-clamp, peripheral nerve, Schwann cells
Introduction
Schwann cells (SCs) are myelinating cells of the PNS and are accommodated within the peripheral nerves. Similar to neurons and glial cells of the CNS, they carry a large set of channels and receptors on their surface (Samara et al., 2013). Among the least studied receptors expressed by SCs are the ligand-gated ionotropic receptors for amino acid glutamate, which comprise AMPA receptors (AMPARs), NMDA receptors (NMDARs), and kainate receptors. Discovering the properties of ionotropic glutamate receptors in SCs is very important because these receptors are likely to play an essential role during the development of SC lineage and during peripheral nerve injury, in analogy to the role of ionotropic glutamate receptors in myelinating cells of the CNS under physiological and pathological conditions (Spitzer et al., 2016).
Originally, the idea that SCs express functional ionotropic glutamate receptors came from experiments in the giant axons of squid, in which axonal stimulation caused changes in the membrane potential of SCs that were blocked by inhibitors of ionotropic glutamate receptors (Lieberman et al., 1989; Evans et al., 1991; Lieberman and Sanzenbacher, 1992). Later studies suggested that, in primary cell cultures, mammalian SCs respond to the application of glutamate with a transient increase in intracellular Ca2+ concentration (Fink et al., 1999) and/or ATP release (Liu and Bennett, 2003). Interestingly, only NMDARs, not AMPARs/kainate receptors, were of primary importance in regulation of SC migration in culture (Mantuano et al., 2015; Campana et al., 2017). However, a drawback of cell culture experiments is that the expression of channels and receptors in SCs in vitro develop without many of the natural signals present in vivo. Furthermore, the absence of neurons may result in considerable artifacts. Remarkably, all three types of ionotropic glutamate receptors, NMDARs, AMPARs, and kainate receptors, were described in mammalian SCs using immunohistochemistry and electron microscopy (Demêmes et al., 1995; Kinkelin et al., 2000). However, those studies did not investigate whether glutamate receptors are functional and if they are expressed in all SCs or only in cells at certain stages of development. A recent imaging study pointed to the fact that only NMDARs, not AMPARs, are of functional significance in myelinating SCs because activation of these receptors triggered morphological changes in PNS myelin ex vivo (Christensen et al., 2016). Furthermore, only NMDARs, not AMPARs, were upregulated after sciatic nerve injury in vivo (Mantuano et al., 2015; Campana et al., 2017).
To address the lack of knowledge regarding the functional expression of AMPARs in mammalian SCs, we investigated directly whether ligand binding activates AMPAR-mediated conductance in these glial cells. AMPARs are transmembrane ligand-gated ion channels that allow Na+ and K+ (and in some cases Ca2+) ions to pass through the cell membrane in response to the binding of agonists. However, transmembrane AMPAR-mediated currents have never been recorded directly in SCs of vertebrates and there is no information about the functional properties of these receptors in SCs. The major obstacle for performing such experiments has been the absence of a convenient preparation with preserved cellular architecture; for example, the one analogous to the brain slice. Recently, we have established a new preparation of living peripheral nerve slices for two-photon imaging (Barzan et al., 2016). In the present study, our goal was to investigate AMPAR-mediated currents in SCs using patch-clamp recordings in this preparation. We found that, similar to the oligodendroglial lineage cells of the CNS, developing SCs express functional AMPARs but downregulate them upon maturation. Furthermore, stationary fluctuation analysis demonstrated that single-channel conductance of AMPARs in SCs is comparable to neurons and glial cells of the CNS. This study is the first to describe AMPAR-mediated currents in the PNS glia of vertebrates and to suggest that AMPARs are functionally important for the developing, but not for the mature, myelinating SCs.
Materials and Methods
Animals.
C57BL/6N mice were obtained from Charles River Laboratories and bred in-house. Mice were kept in 12/12 h light/dark cycle and food and water were available ad libitum. All experiments were performed in accordance with the guidelines of the Animal Care and Use Committee at the University of Tübingen.
Surgery.
Pregnant mice were anesthetized using a mixture of oxygen and isoflurane (1.5–2.5%, v/v) and positioned on the heating mat (Happy Hamster, 8 Watts; Fenix). Body temperature was monitored using a rectal thermometer and kept at 36°C. Ointment (Bepanthen Augen-und Nasensalbe; Bayer) was applied to the eyes. The depth of the anesthesia was verified by testing the pedal reflex. The abdominal skin area was disinfected, the abdomen was opened, the uterus with fetuses was exposed, and the fetuses were removed one by one. The mother mouse was then killed by decapitation.
Preparation of sciatic nerve live slices.
Embryonic day 16 (E16)–E18 or postnatal day 0 (P0)–P2 mouse pups of both sexes were killed by decapitation without anesthesia. The ∼2- to 5-mm-long segments of each sciatic nerve were dissected (see Fig. 2A) and embedded at 36°C into 2.5% low-melting agarose dissolved in artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 1.25 NaH2PO4, 1.3 MgSO4, 2.7 KCl, 26 NaHCO3, 2 CaCl2, 2 ascorbic acid, and 18.6 glucose, pH 7.4, osmolality 300 mOsm/kg. Then, 100 μm thin longitudinal nerve slices were prepared on a vibratome (VT1200S; Leica Biosystems) using ice-cold high-Mg2+ ACSF containing the following (in mm): 124 NaCl, 1.25 NaH2PO4, 10 MgSO4, 2.7 KCl, 26 NaHCO3, 2 CaCl2, 2 ascorbic acid, and 18.6 glucose. The slices were placed into a Haas-type interface chamber and maintained at room temperature up to 8 h perfused with normal ACSF gassed with a mixture of 95% O2 and 5% CO2.
Figure 2.

Novel preparation of live sciatic nerve slices for electrophysiological recordings. A, Schematic drawing indicating the part of the sciatic nerve dissected from a mouse pup and cut into longitudinal sections using a vibratome. Blue indicates a sensory axon, red a motor axon, and green a neuron located in the spinal cord. B, DIC image showing one region of the live sciatic nerve slice under the patch-clamp microscope. Stars indicate SCs; arrow indicates direction of the axons in the nerve. C, Time course of CAP recording in the sciatic nerve slice. Each point indicates single amplitude measurement. Bar indicates time period of TTX application. Green letters indicate regions from which the example traces for D were taken. D, Example traces of CAPs recorded before and during TTX application and during TTX washout. Each trace is an average of 11 sweeps. Red arrows indicate time point of electrical stimulation. Stimulation artifacts are blanked for clarity.
Preparation of brain slices.
P13–P19 mice of both sexes were anesthetized with a mixture of oxygen and isoflurane (3%, v/v) and decapitated. Brain slices were prepared and stored as described by us previously (Nagy et al., 2017).
Patch-clamp recordings.
After a minimum of 1 h following the preparation, the individual sciatic nerve slices were transferred to a submerged recording chamber, mounted on a glass slide coated with poly-l-lysine (0.05%; Sigma-Aldrich), and incubated at 35°C for 5 min with 0.1% collagenase (Sigma-Aldrich) dissolved in ACSF. Subsequently, the recording chamber with the slice was mounted on the stage of an upright microscope (FN-1; Nikon) and superfused continuously (∼2 ml/min) with gassed ACSF. Cells for whole-cell voltage-clamp recordings were selected randomly using infrared differential interference contrast video microscopy. Patch pipettes were pulled from borosilicate glass on a vertical puller (PP-830; Narishige) and had a resistance of 5.0–7.0 MΩ when filled with internal solution containing the following (in mm): 125 potassium gluconate, 2 Na2-ATP, 2 MgCl2, 0.5 EGTA, 10 HEPES, 20 KCl, and 3 NaCl, pH 7.3, osmolality 280–290 mOsm/kg. Cells were voltage-clamped at the holding potential Vhold = −80 mV with an EPC-8 amplifier (HEKA); Vhold was corrected for a −13 mV liquid junction potential before seal formation. Liquid junction potential was calculated using JPCalc for Windows (Peter H. Barry, Sydney, Australia). Whole-cell currents in response to voltage steps were low-pass filtered at 10 kHz and digitized with a sampling frequency of 20 kHz (ITC-18; HEKA Instruments). The glutamate-induced currents were low-pass filtered at 3 kHz and digitized with a sampling frequency of 10 kHz. Data acquisition was performed using Recording Artist (written by Rick Gerkin, Arizona State University) running under Igor Pro 6.3 (WaveMetrics). For presentation purposes, the majority of traces shown on the figures were smoothed offline using a boxcar smoothing algorithm (with num value in the range between 3 and 11) in IgorPro. Care was taken that the kinetics of responses were not affected by the smoothing. Patch-clamp recordings of oligodendrocyte precursor cells in the corpus callosum, as well as electrical stimulation of callosal and sciatic nerve axons, were performed as described by us previously (Barzan et al., 2016; Nagy et al., 2017). Unless indicated differently, the drugs were applied via the bath.
Pneumatic application of drugs.
For the application of glutamate, vehicle (ACSF), or sucrose, we used pneumatic drug ejection system with microJECT (PDES; NPI Electronic). Each application pipette had a tip opening diameter of 3–4.5 μm and was positioned at a distance of 18–23 μm (for glutamate application) or 50 μm (for sucrose application) from the recorded cell. Unless indicated differently, the drugs were applied for 30 ms with a pressure intensity of 5 psi. The time interval between drug applications was 40 s. A voltage step of −10 mV was applied at the beginning of each sweep to monitor the dynamics of the series resistance. All recordings of glutamate-induced or ACSF-induced currents were performed in ACSF containing the voltage-gated sodium channel antagonist tetrodotoxin (TTX, 1 μm; Alomone), the NMDAR antagonist 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP, 10 μm; Tocris Bioscience), the metabotropic receptor antagonist (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495, 50 μm; Tocris Bioscience), the GABAA receptor antagonist picrotoxin (100 μm; Sigma-Aldrich), the GABAB receptor antagonist 2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP55845, 5 μm; Tocris Bioscience), the P2 purinergic receptor antagonist 4-[[4-formyl-5-hydroxy-6-methyl-3-[(phosphonooxy)methyl]-2-pyridinyl]azo]-1,3-benzenedisulfonic acid (PPADS, 30 μm; Ascent Scientific), the nicotinic ACh receptor antagonist mecamylamine (50 μm; Sigma-Aldrich), the α7-containing nicotinic ACh receptor antagonist methyllycaconitine (MLA, 200 nm; Sigma-Aldrich), the metabotropic ACh receptor antagonist atropine (10 μm; Sigma-Aldrich), and the allosteric modulator of AMPARs cyclothiazide (CTZ, 100 μm; Ascent Scientific). For negative control experiments, we performed pneumatic application of ACSF to the type 1 or type 2 cells (Fig. 4A, B; n = 6) because these cells typically show response to glutamate. No inward current was observed upon ACSF application.
Figure 4.
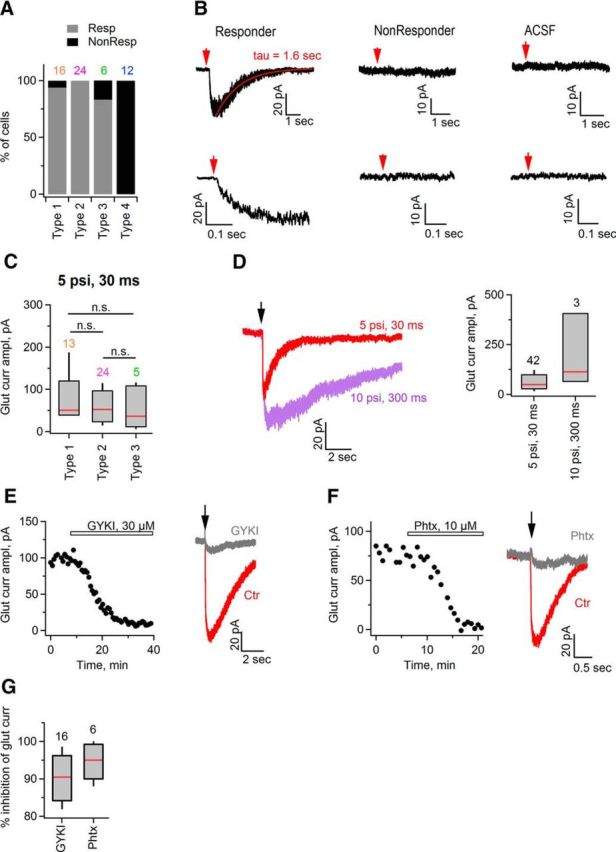
The majority of SCs in the developing mouse sciatic nerve express functional AMPARs. A, Bar graphs summarizing percentage of cells responding (Resp) and nonresponding (NonResp) to pressure-induced application of 1 mm glutamate, among the total number of recorded cells of a given type. B, Top left, Example trace (single sweep) showing inward current induced in a SC upon glutamate (1 mm) application. Top middle, As in top left but for another SC that showed no response to the application of 1 mm glutamate. Top right, As in top left, but ACSF was applied to a recorded SC instead of glutamate. Bottom left, middle, right, Same traces, as in the top panels shown on the expanded timescale for better visualization of the inward current activation. All cells were recorded in a whole-cell mode with a holding potential Vhold = −80 mV. Glutamate was applied for 30 ms with a pressure of 5 psi. Red arrow indicates time point of glutamate application. Red line indicates exponential fit to the decaying phase of the current and tau is the corresponding decay time constant. C, Box plot showing amplitude of glutamate-induced inward current in SCs. Only responders are considered for this graph. In each cell, glutamate was applied several times for 30 ms with a pressure of 5 psi each and the maximal amplitude was taken for the graph. Vhold = −80 mV. Red lines indicate median amplitude. n.s., Not significantly different by the Kruskal–Wallis test. D, Left, Example traces showing inward currents in a SC induced by application of glutamate for 30 ms at 5 psi (red) and for 300 ms at 10 psi (purple). Vhold = −80 mV. The black arrow indicates the time point of glutamate application. Right, Box plot showing that longer duration of glutamate application at higher pressure induces larger inward current in SCs. Red line indicates median value of the amplitude. E, Left, Time course of glutamate-induced inward current in a SC. Glutamate was applied for 30 ms with a pressure of 5 psi, Vhold = −80 mV. Each point indicates single amplitude measurement. Bar indicates time-period of GYKI53655 application. Right, Example traces of inward currents recorded before (red) and during (gray) GYKI53655 application. Black arrow indicates time point of glutamate application. F, As in E but for application of philantotoxin-433 (Phtx). G, Box plot showing percentage of inhibition of glutamate-induced inward current in SC by GYKI (n = 16) and philantotoxin-433 (n = 6). Red line indicates median of the percentage of inhibition. For the box-and-whisker plots throughout the figure, the bottom and top of each box represent 20th and 80th percentiles of the data, respectively, and the whiskers represent 10th and 90th percentiles. Black or colored numbers above the bar graphs indicate the numbers of analyzed cells.
Analysis of patch-clamp recordings.
Only those recordings in which the offset drift by the end of the experiment was smaller than ±5 mV and the change in the Rs was <30% of the original value were considered for analysis. The Rs, input resistance (Rinput), and membrane capacitance were determined as described previously (Kukley et al., 2010). The Rs was between 20 and 40 MΩ in all experiments. To estimate the amplitude of the total (Itotal), and voltage-gated outward K+ currents, we applied a series of depolarizing voltage steps (increment +10 mV, length 400–600 ms, Vhold = −80 mV), and at each command potential (Vcomm), averaged the values of the current amplitude over the 10-ms-long segment located at the end of the voltage step. This procedure was performed on the original sweeps or on the leak-subtracted sweeps for measurements of the amplitude of Itotal or voltage-gated K+ current, respectively. To determine the amplitude of the ohmic leak current (Ileak), we obtained the value of the Ileak at Vcomm = −70 mV and linearly scaled it for each Vcomm. To determine the activation time constant (τactivation) of the outward voltage-gated K+ current at each Vcomm, the initial part of each leak-subtracted sweep was fitted with double-exponential function. τactivation was determined from the fit as follows:
 |
where τ1 and τ2 are the time constants and I1 and I2 are the amplitudes of each exponential component of the outward K+ current. Clustering of SCs was performed using K-means clustering algorithm in IgorPro.
The amplitude of glutamate-induced inward currents was measured manually in IgorPro. Two to 19 sweeps were analyzed in each cell and the median and maximal amplitude were determined. For stationary fluctuation analysis (SFA), two to 17 sweeps were analyzed per cell. Only the 1.6- to 12-s-long decaying phase of the current was considered in each cell. The part of the sweep containing the decaying phase was cut into 200-ms-long segments, as shown in Figure 5A (Cull-Candy and Usowicz, 1989). Eight to 60 segments (median = 20) were obtained in each cell. The current amplitude within each segment was stable. The baseline period before glutamate application was 600–1000 ms long to obtain three to five segments. Mean current amplitude and variance were calculated in each segment and the baseline amplitude and variance were subtracted. The resulting wave containing the amplitude values in each segment was binned with bin = 1 pA and the corresponding values of the variance wave were then obtained in accordance with this binning (Hartveit and Veruki, 2007). The values of the variance were plotted versus the corresponding values of the mean current amplitude in each cell. The resulting variance–mean relationship was fitted with a parabola function in IgorPro. In some cases, the software was not able to fit the parabola function and the linear fit was used. If the data could be fitted with a parabola function, then both single-channel current and the number of channels were determined from the following equation:
 |
Where I is the amplitude of glutamate-induced inward current, i is the single-channel current, σ is the amplitude variance, and N is the number of channels (Traynelis and Jaramillo, 1998). If the data could be fitted with a linear function, then only single-channel current was determined from the following equation:
 |
To estimate the single-channel conductance, the value of the single-channel current was divided by the driving force ΔV = 80 mV. All analysis was performed using custom-written macros in IgorPro.
Figure 5.
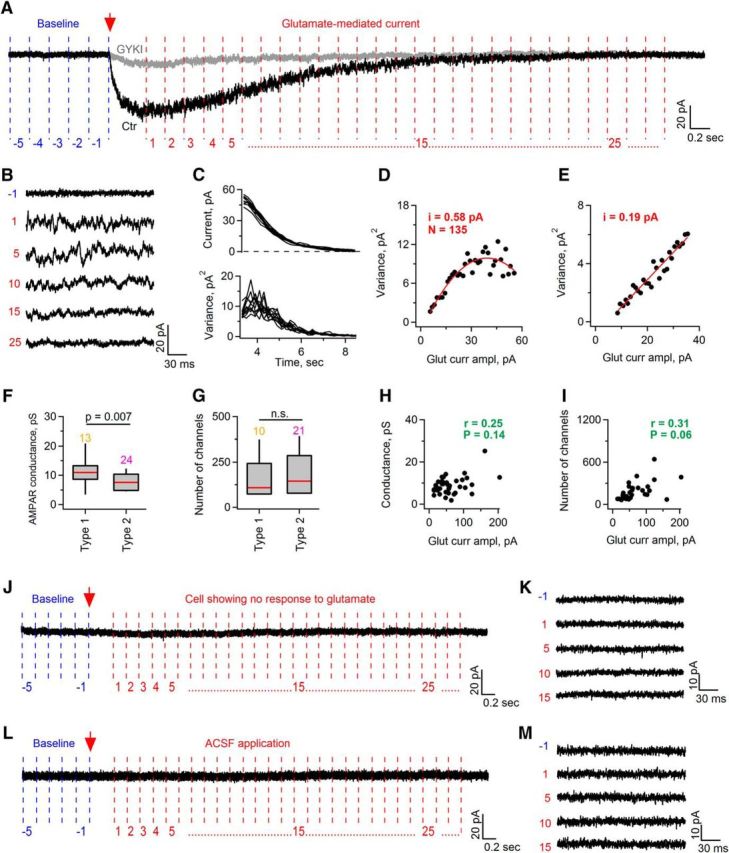
Glutamate application onto SCs induces an increase in single-channel noise. A, Example trace (Ctr, black) showing glutamate-induced inward current in a SC (Vhold = −80 mV). Red arrow indicates time point of glutamate application. Note that noise at the peak and during the decaying phase of the current is much higher than the baseline noise before glutamate application. Also note that GYKI (gray trace) abolishes, not only inward current, but also the increase in the noise. Vertical lines in blue (baseline) and red (glutamate application) denote segments (200 ms long) into which each recorded sweep was cut offline for subsequent SFA. Red and blue digits number the segments of the sweep. B, Examples of 200-ms-long segments of the trace shown in A. Blue and red digits allow locating the position of each segment within the trace shown in A. C, Top, Examples of the mean glutamate-induced current amplitude measured from sequential 200-ms-long segments that were cut as shown in A. Bottom, As at top but for the variance of the current amplitude. x-axis shows time after glutamate application. D, Example showing plot of variance versus mean amplitude of glutamate-induced current in a SC. Variance–mean relationship was fit with parabola function, allowing determination of single-channel current (i) and average number of activated channels (N). E, As in D but for a cell in which the variance–mean relationship was fit with a linear function, allowing determination of single-channel current (i) only. F, Box plot showing average single-channel conductance of AMPARs in SCs. Red line indicates median conductance value. Colored numbers above the bar graphs indicate the numbers of analyzed cells. Mann–Whitney test was used for the statistical analysis. G, As in F but for the number of channels. Only those cells in which the variance–mean relationship was fit with parabola function were considered. Colored numbers above the bar graphs indicate the numbers of analyzed cells. n.s., Not significantly different by the Mann–Whitney test. H, Spearman's rank correlation test did not reveal a correlation between the amplitude of glutamate-induced currents and the conductance value, where r is the Spearman's rank correlation coefficient and p is the p-value. I, Spearman's rank correlation test did not reveal a correlation between the amplitude of glutamate-induced currents and the number of open channels, where r is the Spearman's rank correlation coefficient and p is the p-value. J, K, As in A and B but for a cell showing no response to glutamate. L, M, As in A and B but for ACSF application. For the box-and-whisker plots throughout the figure, the bottom and the top of each box represent 20th and 80th percentiles of the data, respectively, and the whiskers represent 10th and 90th percentiles.
Extracellular recordings and their analysis.
To record the compound action potentials (CAPs), the stimulating and recording glass electrodes were positioned in the nerve slice at a distance of 100–250 μm from each other. Single or paired (interpulse interval = 40 ms) rectangular electrical pulses of 100–250 μs duration were applied each 15–30 s using the isolated pulse stimulator (model 2100; A-M Systems). CAPs were recorded using extracellular amplifier (EXT-10-2F; NPI Electronic), high-pass filtered at 0.3 Hz, and low-pass filtered at 2 kHz. The recordings were digitized with a sampling frequency of 25–50 kHz. Data acquisition was performed as described above.
RNA isolation and RT-PCR.
We used primers and restriction enzymes described previously (Seifert et al., 1997). Total RNA was isolated from sciatic nerves using TRIzol reagent (Life Technologies) according to the manufacturer's protocol. RNA was resuspended with 20 μl of nuclease-free water, its concentration was measured, and RNA was stored at −80°C. RT-PCR was performed using the OneStep RT-PCR Kit (Qiagen). The RT-PCR mixture contained the following: 80–280 ng of total RNA, 1× OneStep RT-PCR buffer, 400 μm dNTP, 0.6 μm concentrations of each primer, 1.2 μl of OneStep RT-PCR Enzyme Mix, 1 unit/μl RNase inhibitor (Promega), and nuclease-free water to the final volume of 20 μl. The following RT-PCR protocol was used: (1) reverse transcription at 50°C, 30 min; (2) initial PCR activation step at 95°C, 15 min; (3) 35 cycles as follows: 94°C, 45 s; 58°C, 45 s; 72°C, 1 min; and (4) final extension at 72°C, 10 min. RT-PCR amplified all four subunits of AMPARs. The primer sequences were as follows: the sense primer 5′-TGTG(TC)AT(AT)GT(GC)TT(TC)GC(CTA)TATTGG-3′; the antisense primer 5′-TGCTCAG(GA)CT(CGT)A(GA)(AG)GCACT(GC)GTCTT-3′. The cDNA from RT-PCR was purified, eluted with 25 μl of nuclease-free water, and its concentration was measured. cDNA was stored at −20°C.
Second PCR amplification and restriction analysis.
Purified cDNA from RT-PCR was used as a template for the second PCR amplification. The PCR mixture contained 40–60 ng of purified cDNA, 1× KAPAHiFi Fidelity buffer with MgCl2 (Kapa Biosystems), 300 μm dNTP mix, 0.3 μm concentrations of each primer, 0.5 U DNA polymerase, and nuclease-free water to the final volume of 25 μl. To separate four subunits of AMPAR (GluA1-GluA4) specifically, the nested primers were used. The sense primers from 5′ to 3′ were as follows: GGACGAGATCAGACAACCAG, GAGAAACACAAAGTAGT, TGACCCACAAAGCCCTCCTG, and GAAGAACCCAGTGACCAGC (for GluA1, GluA2, GluA3, and GluA4 subunits, respectively). The antisense primer 5′-TCGTACCACCATTTG(CT)TTTTCA-3′ was common for four subunits. The PCR protocol was as follows: (1) 95°C, 5 min; (2) 35 cycles as follows: 98°C for 20 s; annealing temperature (θ) for 15 s; 72°C for 30 s; and (3) final extension 72°C for 5 min. θ was 59°C (for GluA1), 42°C (for GluA2), or 56°C (for GluA3, GluA4). The PCR product was analyzed by 1.7% agarose gel electrophoresis (stained with GelRed; Biotium) with the marker φX174 DNA/HaeIII (Thermo Scientific). For restriction analysis, the PCR products from GluA1, GluA2, GluA3, and GluA4 were digested with PstI, SduI, Eco47III, and EcoRI (all from Thermo Scientific), respectively. The digestion products were analyzed by agarose gel electrophoresis (stained with GelRel) with the marker pUC19 DNA/MspI (Hpall) (Thermo Scientific). For positive controls, HEK293 were transfected with pRK5-GluR1i-L497Y-(Ser831-Ser845), pRetroCAG-GluA2(Q583R), pRK5-GluA3 flip, or pCI-EGFPGluA4 using lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Transfected cells were harvested 48 h after transfection and total RNA was isolated with RNA Isolation Kit (Roche). Total RNA containing one of AMPAR subunits was used as a template for a positive control. RNA isolation and PCRs were repeated three times with samples from different nerves/animals. In each case, sciatic nerves were dissected from mouse pups and five to six nerves from three to four animals were pooled.
Immunohistochemistry.
Postrecording immunohistochemical and morphological analysis of cells was possible because, during patch-clamp recordings, the fluorescent dye Alexa Fluor-568 (0.02%; Invitrogen) was included in the pipette solution, so each recorded cell was labeled (one cell per nerve slice). Labeling with immunohistochemical markers was performed on free-floating slices and all steps were performed at room temperature, with the exception of PFA fixation and primary antibody incubation, which were performed at 4°C. The following primary antibodies were used: rat anti-MBP (Abcam, ab7349, 1:250), rabbit anti-IBA1 (WAKO, 019–19741, 1:500), rat anti-CD68 (Serotec, MCA1957, 1:100), and rabbit anti-Sox-10 (DCS Diagnostics, SI058C002, 1:50). The following secondary antibodies were used: goat anti-rat Alexa Fluor-488 (Invitrogen, 1:500) and goat anti-rat biotin-SP-conjugated (1:200; Dianova), followed by application of streptavidin-Cy5 (1:200; Biomol/Rockland), goat anti-rabbit Alexa Fluor-488 (Invitrogen, 1:500), and goat anti-rabbit Cy5 (1:200; Biomol/Rockland). For counterstaining, DAPI (0.2 μg; Sigma-Aldrich) was applied for 5 min. For negative controls, the primary antibody was omitted. The slice was transferred to the glass slide, dried, covered with Vectashield (Vector Laboratories), and sealed with nail polish.
Image acquisition.
A laser scanning microscope (LSM 710; Zeiss) was used for image acquisition. Images were acquired and saved as z-stacks with a 16-bit pixel depth. Each z-stack was 6–43 μm thick and consisted of 12–73 z-slices; the z-step was 0.493–0.587 μm. Pixel size ranged between 0.07 × 0.07 μm and 0.259 × 0.259 μm for different images. Each scan represented a triple-channel (or, in a few cases, quadruple-channel) fluorescence image in which channels were acquired sequentially in ZEN-2009 software using a 40× oil-immersion objective (numerical aperture = 1.3). The following excitation (ex.) laser lines and emission (em.) detection ranges were used: for DAPI, ex. 405 nm, em. 414–490 nm; for Alexa Fluor-488, ex. 488 nm, em. 494–572 nm; for Alexa Fluor-568, ex. 561 nm, em. 572–668 nm; and for Cy5, ex. 633 nm, em. 644–759 nm. The beam splitters for each dye matched the excitation laser lines. The pinhole was either set to 1 Airy unit for each channel or was adjusted between 0.8 and 1.1 μm to create equal optical section thickness for each channel. Laser power was set to 1–2% (5–10% for the infrared laser), detector gain to 630–750, and no digital offset was used. All settings were adjusted such that the final scan (4 averages per line) had a good signal to background noise ratio. All images presented in the figures are representative examples; for presentation purposes, the brightness of some images was adjusted using the “Adjust Local Contrast” function in ImageJ.
Analysis of cell morphology.
Analysis of cell morphology was performed in ImageJ. Each image within the stack was enhanced using ImageJ Contrast Limited Adaptive Histogram Equalization (CLAHE) tool. Plugin parameters were optimized to enhance cell processes without greatly affecting the soma. Afterward, the whole stack was projected onto a single plane using maximum intensity Z-project and the resulting image was converted to an 8 bit grayscale image. The Z-projection was filtered by the median (2 pixel) or Kuwahara (3–5 pixel) filters. Filtered images were binarized using the “Auto Local Threshold” plugin, Phansalkar method. Binarization errors remaining in the image were hand corrected. Corrections included filling of holes, edge restoration/redrawing, and manual separation of processes passing in close proximity to each other, which were binarized as one object. Finally, all objects not belonging to the cell were removed from the image. Soma and processes were separated manually on the binary images based on the changes in thickness. A process was defined as a tubular structure originating from the soma regardless of it length or further branching. We defined a branch as a substructure of a process with a unique termination point (ending). Therefore, a short protrusion that met the above criteria was considered a proper process as much as a larger structure with multiple branches. Measurements of the cell process area and perimeter were performed using the “Wand” tool in legacy mode and the “Analyze Particles” function. Processes and branches were counted manually. To receive an unbiased measure of the thickness of cell structures, the “Local Thickness” plugin was run on the binary images in the silent, calibrated mode (Dougherty and Kunzelmann, 2007) and a color-coded image was created in which the thickest structures are displayed as white and the thinnest structures as dark blue. Cell length and thickness were measured by drawing a freehand selection that was anchored at the beginning of the process on one side of the soma, passed through the soma, and ended at the ending of a process on the opposite side (see Fig. 8H). Cell asymmetry was calculated by dividing the length of the shorter bipolar process by the length of the longer bipolar process. Changes to cell thickness along length were coded as changes to pixel intensities in a plot profile. To compensate for differences in pixel size between original images, the profile-derived thickness measurements were transferred into IgorPro and rescaled through linear extrapolation to 10,000 data points. The technique created nearly identical scaling for all measurements with differences of no more than 0.07 μm. Finally, data were transferred into an Excel spreadsheet and aligned to the center of the soma, starting with shorter of the measured processes and averaged within a cell type.
Figure 8.
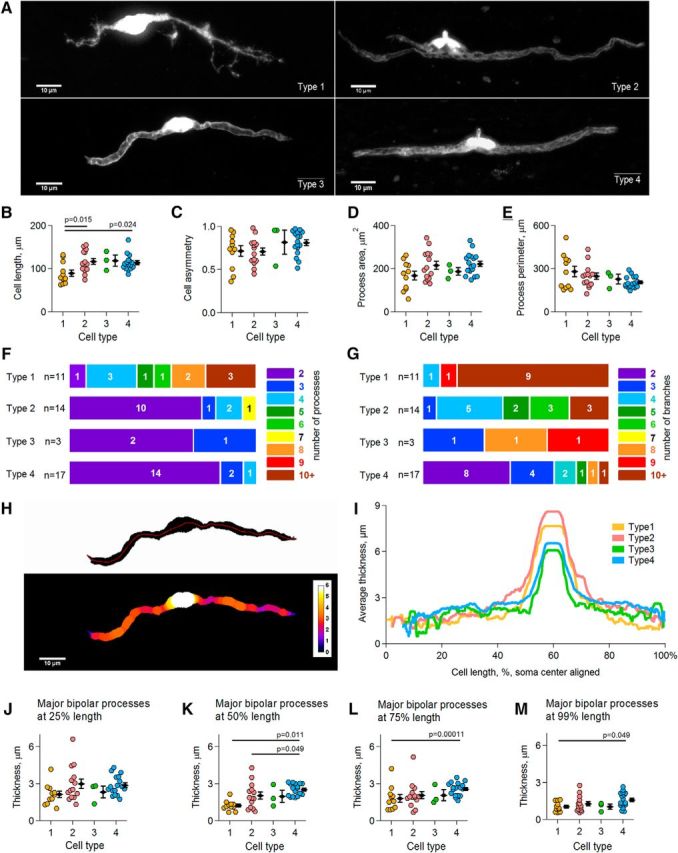
Morphology of electrophysiologically distinct SC types. A, Representative examples of different SC types. Each image is a maximum intensity projection. See extended Figure 8-1 for type 1, Figure 8-2 for type 2, Figure 8-3 for type 3, and Figure 8-4 for type 4 for images showing maximum intensity projection of each individual cell included into the analysis. B–E, Scatter plots showing cell length, processes asymmetry ratio, total area of all processes, and perimeter of all processes, respectively, for each cell type. Each colored point represents the measurement for an individual cell. Black diamonds represent mean ± SEM within a group of a given cell type. p is the p-value in the post hoc Gabriel test. F, Summary of the number of processes per cell for each cell type. Colors and the colored numbers right from the color scale represent the number of processes per cell. The width of each colored box represents the percentage of cells with a given count of processes. White numbers within colored boxes indicate the exact number of cells with a given number of processes, where n is the number of analyzed cells of each type. G, As in F but for the number of process branches per cell. H, Graphical representation of the cell thickness. Top, Binary image of a type 3 cell shown in A. Red line indicates the track along which cell thickness was measured. Bottom, Same cell but with a thickness color code overlay. White indicates the thickest and dark blue the thinnest structures. I, Mean cell thickness for each cell type measured along the tracks as indicated in H. J, Scatter plot showing cell thickness measured at 25% of each major bipolar process length and averaged between the two processes. Each colored point represents the measurement for an individual cell. Black diamonds represent mean ± SEM within a group of a given cell type. K–M, As in J but for the cell thickness measured at 50%, 75%, and 99% of process length, respectively. p is the p-value in the post hoc Dunn's test (K–L) or the Gabriel test (M).
Experimental design and statistical analysis.
The data acquisition was randomized. Each cell was recorded in a different nerve slice. The numbers of cells and animals used for the analysis are indicated in the text. Statistical analysis was performed using SPSS, including tests for normal distribution and homoscedasticity. For the analysis of the electrophysiological data, because the data points were not distributed normally, we used the Mann–Whitney test (when two groups were compared) or the Kruskal–Wallis test (when more than two groups were compared). The correlation between the two variables was assessed using the Spearman rank correlation test in IgorPro because the relationship between the variables was expected to be monotonic rather than linear. For the analysis of the morphological data, if the data were normally distributed, we used one-way ANOVA and the post hoc Gabriel test. If the data were not normally distributed, we used the Kruskal–Wallis and the post hoc Dunn's test. The results of the statistical analysis are presented in the text or in the figures.
Results
Transcripts for all AMPAR subunits are present in the developing mouse sciatic nerve
First, we tested whether RNA for each AMPAR subunit is present in the developing mouse sciatic nerve by performing PCR. We were able to detect the transcripts for GluA2, GluA3, and GluA4 in all trials and the transcripts for GluA1 in 33% of trials (n = 3 trials, 5–6 nerves from 3–4 animals were pooled for each trial; Fig. 1A,B). To verify that each band corresponds to the subunit of interest, the PCR products were digested with the restriction enzymes specific for a given subunit. Digestion yielded two fragments of the expected length for each subunit (Fig. 1A,B). All four subunits of the AMPARs were detected in the positive control samples and no bands were observed in the negative control samples (Fig. 1A,B). These findings indicate that GluA1, GluA2, GluA3, and GluA4 subunits of AMPARs are present in the developing mouse sciatic nerve and are expressed by glial cells and/or axons.
Figure 1.

Detection of AMPAR subunits in the mouse sciatic nerve. A, cDNA for GluA1–GluA4 subunits obtained after the second round of PCR was analyzed with electrophoresis in agarose gel. An example gel is shown. The predicted size of the PCR products was as follows: 632 bp for GluA1, 628 bp for GluA2, 630 bp for GluA3, and 626 bp for GluA4. A1, A2, A3, and A4 represent GluA1, GluA2, GluA3, and GluA4, respectively. M denotes the φX174 DNA/HaeIII molecular weight marker. Band size for the marker is indicated on the left. B, Products of GluA1, GluA2, GluA3, and GluA4 obtained after the second round of PCR were digested with restriction enzymes specific for each subunit: PstI (275 and 357 bp) for GluA1, SduI (270 and 358 bp) for GluA2, Eco47III (229 and 401 bp) for GluA3, and EcoRI (289 and 337 bp) for GluA4. An example gel is shown. cDNA for each subunits was cut into two fragments of the predicted size. A1, A2, A3, and A4 represent GluA1, GluA2, GluA3, and GluA4, respectively. M denotes the pUC19 DNA/MspI (Hpall) molecular weight marker. Band size for the marker is indicated on the left. Throughout the figure, for positive controls, total RNA was extracted from HEK293 cells transfected with a plasmid encoding GluA1, GluA2, GluA3, or GluA4; this RNA was used as a template. For a negative control, the template was omitted. During RT-PCR, second-round PCR, and digestion, the samples of positive and negative control were always processed in parallel with the samples of RNA extracted from the sciatic nerves.
Novel preparation of living peripheral nerve slices for patch-clamp recordings
We next performed patch-clamp recordings from SCs in near physiological conditions, in order to characterize the SCs electrophysiologically and to test whether AMPAR-mediated currents can be triggered by agonist application. We used a widely described technical approach used for preparation of live brain slices and set up a preparation of live longitudinal slices of mouse sciatic nerve (Barzan et al., 2016). For this, we dissected the nerves from E16–E18 or P0–P2 mouse pups, embedded them into agarose, and cut longitudinal slices 100 μm thick (Fig. 2A). Glial cells and axons could be visualized clearly in these slices using differential interference contrast (DIC) videomicroscopy with a 60× water objective (Fig. 2B). To evaluate the quality of our preparation, in selected experiments, we stained live slices with propidium iodide, a dye that binds to a DNA but is membrane impermeable and thus can only be detected in dead/damaged cells. We counted very few cells labeled by propidium iodide in our slices (data not shown), indicating that the majority of glial cells are intact. To verify that axons are viable in our preparation, we stimulated them electrically and performed recordings of compound action potentials. Compound action potentials could be easily recorded in sciatic nerve slices over a long period of time (>40 min), were blocked by sodium channel inhibitor TTX (1 μm, n = 4 slices from 4 animals), and recovered after washout of TTX (Fig. 2C,D). Therefore, we were convinced that cellular components are preserved in our new preparation of sciatic nerve slices. We used this preparation throughout this study to investigate functional expression of ion channels and AMPARs in SCs.
SCs in the developing mouse sciatic nerve differ in their passive membrane properties and voltage-gated K+ currents
SCs in culture and in teased nerve preparations are diverse in their intrinsic membrane properties and expression of voltage-gated and ungated ion channels, which may depend on the age of the animal and myelination status (Chiu, 1991; Baker, 2002). Therefore, we aimed to find out whether SCs in our preparation were electrophysiologically distinct or a homogeneous population. We applied a series of depolarizing voltage steps (increment +10 mV, Vhold = −80 mV) to each SC in the whole-cell voltage-clamp mode and evaluated cell capacitance, leak currents, and voltage-gated outward K+ currents. We then performed simple cluster analysis using the K-means clustering algorithm on two independent parameters: cell capacitance and the ratio between the amplitudes of the total persistent outward K+ current and the leak current (Itotal/Ileak). We chose to use Itotal/Ileak rather than membrane resistance because Itotal/Ileak can be calculated for different voltage steps; that is, Vcomm = +20 mV (Fig. 3A), Vcomm = +10 mV, 0 mV, and −10 mV (data not shown), and the respective clustering results can be compared, thus making the clustering analysis more solid. In addition, preliminary inspection of our data showed that Itotal/Ileak is a good parameter with which to classify the cells. We found that recorded SCs fell into three distinct clusters: (1) a cluster of cells (75% of all cells, i.e., 149 cells in 113 animals of 197 cells recorded in 135 animals) showing small capacitance (median value 26 pF) and Itotal/Ileak ≫ 1 (Fig. 3A–C) that we called “small nonleaky”; (2) a cluster of cells (5% of all cells, i.e., 9 cells in 9 animals of 197 cells recorded in 135 animals) showing intermediate capacitance values (median 598 pF) and Itotal/Ileak ∼ 1 (Fig. 3A–C) that we called “intermediate leaky”; and (3) a cluster of cells (20% of all cells, i.e., 39 cells in 28 animals of 197 cells recorded in 135 animals) showing large capacitance (median value 1662 pF) and Itotal/Ileak ∼ 1 (Fig. 3A–C) that we called “large leaky.” If the ratio Itotal/Ileak is close to 1, as it is the case in intermediate and large leaky SCs, then the cells express no/little voltage-gated K+ channels and their outward currents dominate by ohmic leak currents. If the ratio Itotal/Ileak is significantly larger than 1 at the Vcomm more positive than −30 mV, as it is the case in small nonleaky SCs, then voltage-gated K+ currents strongly contribute to the total outward current. Therefore, we next investigated whether all small nonleaky cells are uniform with respect to the amplitude and properties of their outward voltage-gated K+ currents. We again used the K-means clustering algorithm, but in this case, on the following two parameters: weighted activation time constant of the outward K+ currents and amplitude of the outward K+ currents (both parameters measured after leak subtraction). As above, clustering was performed using data recorded at different command potentials, i.e., Vcomm = +20 mV (Fig. 3D), +10 mV, 0 mV, and −10 mV (data not shown), and the respective clustering results were compared. We found that the group of small nonleaky SCs contained three clusters of cells: (1) cells with slow-activating outward K+ currents of small amplitude (τactivation 8–41 ms, amplitude 39–145 pA, depending on the Vcomm; 57 cells in 46 animals; Fig. 3D–F) that we called type 1 cells; (2) cells with fast-activating outward K+ currents of large amplitude (τactivation 3–7 ms, amplitude 309–781 pA depending on the Vcomm; 67 cells in 59 animals; Fig. 3D–F) that we called type 2 cells, and (3) cells that classified as type 1 at some Vcomm and as type 2 at other Vcomm (21 cells in 21 animals; Fig. 3D) that we called type 1/2 cells but did not further consider them within Figure 3.
Figure 3.
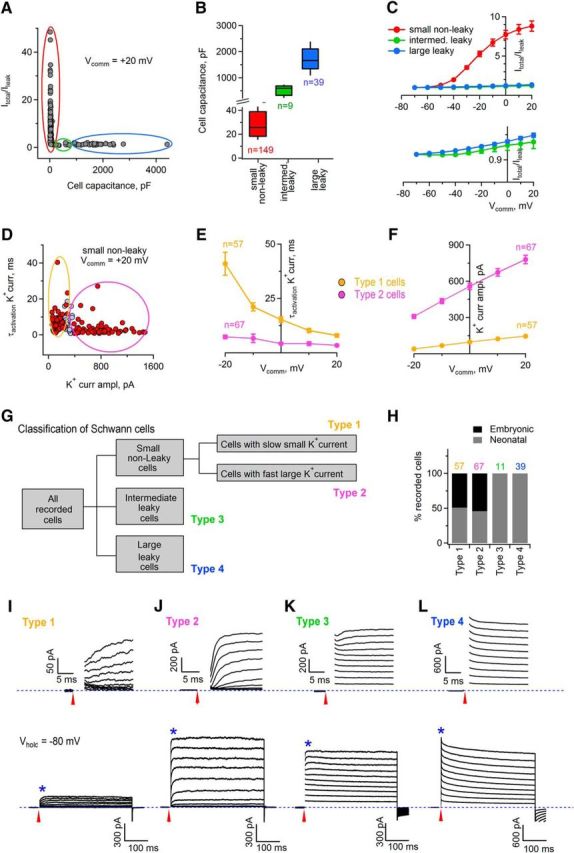
Electrophysiologically distinct cell populations can be identified within the recorded SCs. A, K-means clustering algorithm revealed that SCs fell into three clusters. Clustering was based on two parameters: ratio between total outward current and leak current (Itotal/Ileak), and cell capacitance. Red (n = 149 cells), green (n = 9 cells), and blue (n = 39 cells) circles denote three different clusters. Vcomm indicates command potential at which the amplitude of total and leak currents was measured. B, Box plot showing cell capacitance of small nonleaky cells (corresponding to red cluster in A), intermediate leaky cells (corresponding to green cluster in A), and large leaky cells (corresponding to blue cluster in A). The black lines within the boxes indicate the median value of cell capacitance. The bottom and the top of each box represent 20th and 80th percentiles of the data, respectively; the whiskers represent 10th and 90th percentiles. C, Top, Graph showing Itotal/Ileak for small nonleaky cells (corresponding to red cluster in A), intermediate leaky cells (corresponding to green cluster in A), and large leaky cells (corresponding to blue cluster in A). Vcomm indicates command potentials at which the amplitude of the total and leak currents was measured. Bottom, Itotal/Ileak ratio for intermediate and large leaky cells shown at the expanded scale. D, K-means clustering algorithm revealed that small nonleaky cells fell into two clusters. Clustering was based on two parameters: weighted time constant of activation of outward voltage-gated K+ current and amplitude of this current. Yellow (n = 57 cells) and magenta (n = 67 cells) circles denote two different clusters; violet points (n = 21 cells) indicate cells of the intermediate cluster. Vcomm indicates command potential at which the time constant and the current amplitude were measured. E, Graphs showing the weighted time constant of activation of the outward voltage-gated K+ currents for two different clusters of cells: type 1 cells corresponding to yellow cluster in D and type 2 cells corresponding to magenta cluster in D. Vcomm indicates command potentials at which the values were measured. F, Graphs showing the amplitude of the outward voltage-gated K+ currents for two different clusters of cells: type 1 cells corresponding to yellow cluster in D and type 2 cells corresponding to magenta cluster in D. Vcomm indicates command potentials at which the values were measured. G, Classification of cells responding to glutamate based on the parameters described in A–F. Intermediate leaky cells and large leaky cells are called type 3 and type 4 cells, respectively. H, Percentage of type 1, type 2, type 3, and type 4 SCs recorded in the embryonic and neonatal nerves. Colored numbers above the bar graphs indicate the numbers of analyzed cells. I, Bottom, Example of a current response of a type 1 SC to a series of depolarizing voltage steps applied from Vhold = −80 mV (blue dotted line) with an increment of +10 mV. Top, Region marked by the star on the bottom panel is shown on the expanded scale allowing for visualization of current activation. The currents are overlaid and the capacitive transients are blanked for clarity. Red arrows indicate the time point when a voltage step started. J, As in I but for type 2 SCs. K, As in I but for type 3 SCs. L, As in I but for type 4 SCs. Note the difference between the scale bars in I–L.
In summary, SCs in the developing mouse sciatic nerve are electrophysiologically distinct and encompass at least four different subpopulations of cells (Fig. 3G–L): “small nonleaky” cells, which could be further subdivided into cells with slow-activating small voltage-gated outward K+ current (type 1 cells, 30% of all recorded SCs) and cells with large fast-activating voltage-gated outward K+ current (type 2 cells, 35% of all recorded SCs), “intermediate leaky” cells (which we called type 3 cells, 5% of all recorded SCs), and “large leaky” cells (which we called type 4 cells, 20% of all recorded SCs). Remarkably, although the ratio Itotal/Ileak was close to 1 in both type 3 and type 4 cells, the current response of type 3 cells to a series of depolarizing voltage steps appeared different from the current response of type 4 cells: in type 3 cells, few voltage-gated channels were activated at the Vcomm more positive than 0 mV, whereas in type 4 cells, no voltage-gated channels were activated immediately after the voltage step at any of the tested Vcomm (Fig. 3K,L). Therefore, type 3 cells are electrophysiologically similar to variably rectifying glia of the CNS (Zhou and Kimelberg, 2000), whereas type 4 cells are closer to the protoplasmic astrocytes and oligodendrocytes (Kukley et al., 2010). Interestingly, type 1 and type 2 SCs could be found both in the E16–E18 and P0–P2 sciatic nerves, whereas type 3 and type 4 cells could be found only in the neonatal nerves (Fig. 3H), suggesting that type 3 and type 4 cells represent a more advanced developmental stage of SC lineage than type 1 and type 2 cells.
SCs in the developing mouse sciatic nerve differ in their expression of functional AMPARs
To test for the presence of functional AMPARs in SCs, we combined whole-cell voltage-clamp recordings of SCs in sciatic nerve slices with a fast pressure-induced application of glutamate. Cells were identified within the nerve slice visually using DIC videomicroscopy and selected for patch-clamp recordings randomly. To exclude indirect involvement of other receptors expressed by SCs, all recordings were performed in the presence of the following blockers in the bath: the sodium channel blocker TTX (1 μm), the NMDAR antagonist CPP (10 μm), the metabotropic glutamate receptor antagonist LY341495 (50 μm), the GABAA receptor antagonist picrotoxin (100 μm), the GABAB receptor antagonist CGP (5 μm), the nonselective nicotinic acetylcholine receptor antagonist mecamylamine (50–100 μm), the α7-containing nicotinic acetylcholine receptor antagonist MLA (200 nm), the muscarinic acetylcholine receptor antagonist atropine (10 μm), and the ATP receptor antagonist PPADS (30 μm). In addition, CTZ (100 μm), a positive allosteric modulator of AMPARs that reduces their rapid desensitization (Yamada and Tang, 1993), was included into the bath solution. At holding potential Vh = −80 mV, which is close to the resting potential of the SCs, application of glutamate (1 mm) elicited an inward current in 94% of type 1 cells (15 of 16 cells in 14 animals), 100% of type 2 cells (24 cells in 23 animals), and 83% of type 3 cells (5 of 6 cells in 6 animals; Fig. 4A,B). No response to glutamate was detected in type 4 cells (12 cells in 12 animals; Fig. 4A,B). Application of ACSF did not result in an inward current in SCs (6 cells in 4 animals; Fig. 4B). The values of the median current amplitude in cells responding to glutamate were as follows: 51 pA in type 1 cells (13 cells in 12 animals), 52 pA in type 2 cells (24 cells in 23 animals), and 36 pA in type 3 cells (5 cells in 5 animals; Fig. 4C). There was no difference in this parameter between the cell types (Kruskal–Wallis test, H(2) = 1.871, p = 0.392). Remarkably, application of glutamate for a longer time interval and/or with a higher pressure resulted in a larger current in SCs (42 cells in 36 animals for 5 psi 30 ms, 3 cells in 2 animals for 10 psi 300 ms; Fig. 4D), indicating that not all AMPARs were activated during “30 ms at 5 psi” paradigm. However, larger pressure and/or longer times of drug application resulted in less stable recordings and we were concerned that this may prevent reliable analysis of the effects of antagonists and of a single-channel noise. Therefore, we used the paradigm “30 ms at 5 psi” throughout the study.
SCs showed relatively broad range of amplitudes of glutamate-induced current (Fig. 4C). At the same time, they differed in their input resistance and total membrane capacitance (Fig. 3A–C). Therefore, we wondered whether smaller glutamate-mediated current amplitude may be the result of a smaller cell size or if it may be a technical artifact caused by a higher “leakiness” of the cell. We performed a Spearman rank correlation test and found that there was no correlation between the current amplitude and (1) the input resistance (r = 0.09, p = 0.57), (2) the cell capacitance (r = 0.13, p = 0.40), or (3) the series resistance (r = −0.07, p = 0.67). Therefore, it is likely that SCs in the developing mouse sciatic nerve express different amount of functional AMPARs on their membrane.
To substantiate that inward current in SCs responding to glutamate application is indeed mediated by AMPARs, we tested its sensitivity to GYKI53655, a noncompetitive AMPAR antagonist (Paternain et al., 1995). In each experiment, we first performed repeated (each 40 s) application of 1 mm glutamate to a recorded cell to verify that the amplitude of the inward current remains stable over time (Fig. 4E) and then perfused GYKI53655 (30 μm; Abcam) via the bath. GYKI53655 inhibited the glutamate-induced current by 91% in all cells tested (n = 16 cells in 15 animals; Fig. 4E,G), indicating that this current indeed involves activation of AMPARs. Furthermore, glutamate-induced inward current was reduced by 95% upon bath application of a specific inhibitor of Ca2+-permeable AMPARs philanthotoxin-433 (10 μm; Sigma-Aldrich; Washburn and Dingledine, 1996) in all cells tested (n = 6 cells in 6 animals; Fig. 4F,G). These findings indicate that AMPARs in SCs contain few/no edited GluA2 subunits and allow influx of Ca2+ ions upon activation. We want to emphasize that this result does not contradict our PCR data (Fig. 1) because the primers that we used for the PCR do not distinguish between edited (Ca2+-impermeable) and unedited (Ca2+-permeable) GluA2 subunits of AMPARs. Therefore, it is likely that the GluA2 subunit detected in the sciatic nerve by the PCR is unedited.
Glutamate application onto SCs is accompanied by an increase in single-channel noise.
We noticed that pressure-induced glutamate application not only triggered inward current in SCs, but was also associated with a robust increase in the baseline noise (Figs. 4B, 5A). Noise increase was inhibited by the application of 30 μm GYKI53655 (Fig. 5A), indicating that it originates from the opening of AMPAR channels. To obtain estimates of channel characteristics, we performed SFA (Cull-Candy and Usowicz, 1989). Single sweeps containing glutamate-induced current were broken into 200-ms-long segments. Because current decayed to baseline very slowly, the amplitude within each segment remained stable and it was possible to perform SFA. The noise level was much higher in the segments constituting the peak and the decaying phase of the current than in the segments before glutamate application (Fig. 5A,B), indicating that glutamate triggers openings of the individual AMPAR channels in SCs. Mean current amplitude and current variance were analyzed on the segments building the decaying phase of the current (Fig. 5A,C). Amplitude and variance of the background noise were analyzed on 200-ms-long segments before glutamate application and were subtracted. Variance was displayed as a function of the mean current amplitude and could be fitted with a parabola in the majority of cells (n = 31 cells in 28 animals of 37 cells in 32 animals; Fig. 5D) or with a line in the remaining cells (n = 6 cells in 6 animals of 37 cells in 32 animals; Fig. 5E). Median value of a single-channel conductance of AMPARs estimated from the fits was 30% larger in type 1 cells compared with type 2 cells (Mann–Whitney test, U = 73.00, p = 0.007): 10.9 pS in type 1 cells (13 cells in 12 animals), and 7.6 pS in type 2 cells (24 cells in 23 animals; Fig. 5F). Median values of the number of channels contributing to the AMPAR-mediated currents could be estimated only for the SCs in which the variance–mean relationship was fitted with a parabola. These values were as follows: 109 channels for type 1 cells (n = 10 cells in 9 animals) and 145 channels for type 2 cells (n = 21 cells in 21 animals); there was no statistically significant difference between the groups (Mann–Whitney test, U = 123.00, p = 0.466; Fig. 5G). No correlation was observed between the AMPAR channel conductance and the median inward current amplitude (Fig. 5H) or the number of channels (Fig. 5I) using the Spearman rank correlation test. However, in the latter case, the p-value was close to 0.05 (Fig. 5I), suggesting that larger glutamate-mediated currents observed in some SCs but not others (Fig. 4C) can be explained by the activation of a higher number of AMPAR channels rather than by the activation of AMPARs of a higher single-channel conductance.
We observed no increase in the baseline noise in the majority of SCs (10 of 12) showing no inward current upon glutamate application (Fig. 5J,K) or in the cells onto which the ACSF was applied (n = 6 cells in 6 animals; Fig. 5L,M).
Can synaptic currents mediated by AMPARs be elicited in SCs?
We next wondered whether AMPARs in SCs can be activated by the neurotransmitter glutamate, which is synaptically released from sciatic nerve axons, in analogy to the situation in oligodendrocyte precursor cells (OPCs) within the optic nerve and the brain white matter structures (Kukley et al., 2007; Ziskin et al., 2007). We combined whole-cell voltage-clamp recordings of SCs in sciatic nerve slices with the following paradigms: (1) local pressure-induced application of a hypertonic solution (sucrose, 500 mm), (2) bath application of ruthenium red (100 μm), (3) bath application of α-latrotoxin (5 nm), (4) bath application of pardaxin (2 μm), or (5) electrical stimulation of sciatic nerve axons with single or paired pulses or trains. Paradigms 1–4 are known to trigger an increase in the frequency of spontaneous synaptic currents in neurons and OPCs (Rosenmund and Stevens, 1996; Bergles et al., 2000; Kukley et al., 2007; Ziskin et al., 2007), whereas electrical stimulation of axons (paradigm 5) is a widely used technique to elicit phasic synaptic currents in neurons and OPCs. We used type 1, type 2, type 1/2, and type 3 SCs for these experiments because only these cell types express AMPARs (Fig. 4A). Neither of the experimental paradigms triggered synaptic currents in SCs: n = 8 cells in 6 animals for sucrose (Fig. 6A); n = 5 cells in 5 animals for ruthenium red (Fig. 6B); n = 9 cells in 7 animals for α-latrotoxin (data not shown); n = 3 cells in 2 animals for pardaxin (data not shown); and n = 14 cells in 12 animals for electrical stimulation (Fig. 6C). To verify that our experimental approaches were effective, we recorded OPCs in the corpus callosum and applied sucrose (n = 3 cells in 3 animals), ruthenium red (n = 3 cells in 2 animals), or stimulated callosal axons (n = 3 cells in 3 animals) electrically. In each case, synaptic currents could be easily elicited in the OPCs (Fig. 6A–C) and the values of their amplitude and/or frequency were similar to those reported by us previously (Kukley et al., 2007, 2010; Nagy et al., 2017). Therefore, in our preparation of sciatic nerve slices dissected from E18 and P2 mouse pups, peripheral nerve axons do not communicate with developing SCs via synaptically released glutamate.
Figure 6.

Synaptic AMPAR-mediated currents were not observed in SCs. A, Left, Representative example trace recorded from a SC in the sciatic nerve slice (Vhold = −80 mV) during and after focal application of 500 mm sucrose. Middle, Representative example trace recorded from an OPC in the corpus callosum (Vhold = −80 mV). Note the barrage of synaptic events in the OPC and the absence of synaptic events in the SC. Asterisks indicate two synaptic events shown on the expanded time scale on the right panel. B, Left, Representative example trace recorded from a SC in the sciatic nerve slice (Vhold = −80 mV) during bath perfusion of ruthenium red (RR, 100 μm). Middle, Representative example trace recorded from an OPC in the corpus callosum (Vhold = −80 mV). Note that synaptic events occur in the OPC but not in the SC. Asterisks indicate two synaptic events shown on the expanded time scale on the right panel. C, Top left, Representative example trace recorded from a SC in the sciatic nerve slice (Vhold = −80 mV) during electrical stimulation of sciatic nerve axons with a single pulse. Black arrowhead indicates time point of stimulation. Top middle, Same cell but an example trace during train stimulation of sciatic nerve axons with 10 pulses at 100 Hz is shown. The horizontal bar above the trace indicates the time points of stimulation. Top right, Same cell but an example trace recorded after cessation of train stimulation is shown. The vertical black bar indicates the end of the train stimulation. Bottom left, middle, and right, As top panels but for two OPCs recorded in the corpus callosum (Vhold = −80 mV). Note that clear phasic events during the train and delayed events after the train can be recorded in the OPC, not in the SC. Stars indicate delayed events; a dashed line indicates delayed release. Stimulation artifacts are blanked for clarity.
Do SCs expressing functional AMPARs build myelin sheathes?
In the CNS, OPCs express AMPARs and receive glutamatergic synaptic input from neurons, but AMPAR expression and axon–OPC synaptic signaling decrease significantly when OPCs start to differentiate (Kukley et al., 2010). We hypothesized that axon–glia synaptic communication follows the same developmental scheme in the PNS and the absence of synaptic currents in SCs may be related to the fact that these cells have already commenced differentiation into the myelinating cells. To test this hypothesis, we studied the expression of myelin basic protein (MBP) in the recorded SCs and investigated their morphological appearance. Because each cell was filled with a fluorescent dye during recordings, we could perform post hoc immunohistochemical and structural analysis using either the cells to which we had previously applied glutamate or the cells that were not subjected to the glutamate application but showed a profile of voltage-gated/ungated K+ currents typical for type 1, type 2, type 3, or type 4 cells (Figs. 3I–L, 7A–C). Neither of the tested type 1 cells (n = 3 cells in 3 animals), type 2 cells (n = 3 cells in 2 animals), or type1/2 cells (n = 2 cells in 2 animals) were stained positively for MBP (Fig. 7A,B,D,E,G,H,K,L), although in one type 2 cell, a small segment of one process showed labeling for MBP (data not shown). Conversely, all tested type 4 cells (n = 5 cells in 5 animals) showed positive labeling for MBP (Fig. 7C,F,I,L), indicating that they are involved in the myelination process. We could not perform immunolabeling of type 3 cells for MBP because these cells were encountered in our preparation only very rarely. These cells most likely represent a short-lived developmental stage of the SC lineage (e.g., a promyelinating cell) or a cell type that is present in very low numbers early postnatally (e.g., a nonmyelinating SC). All tested type 1 cells (n = 11 cells in 8 animals), type 2 cells (n = 5 cells in 4 animals), type 3 cells (n = 2 cells in 2 animals), and type 4 cells (n = 3 cells in 2 animals) appeared negative for the microglial marker Iba1 (data not shown). We also verified whether type 1 and type 2 cells belong to the SC lineage using postrecording labeling for Sox-10. Seventy-one percent of type 1 cells (5 of 7 cells in 6 animals) and 78% of type 2 cells (7 of 9 cells in 9 animals) appeared positive for Sox-10 (data not shown), although the labeling was weak, probably because Sox-10 is a small protein that was partially washed out during patch-clamp recordings.
Figure 7.
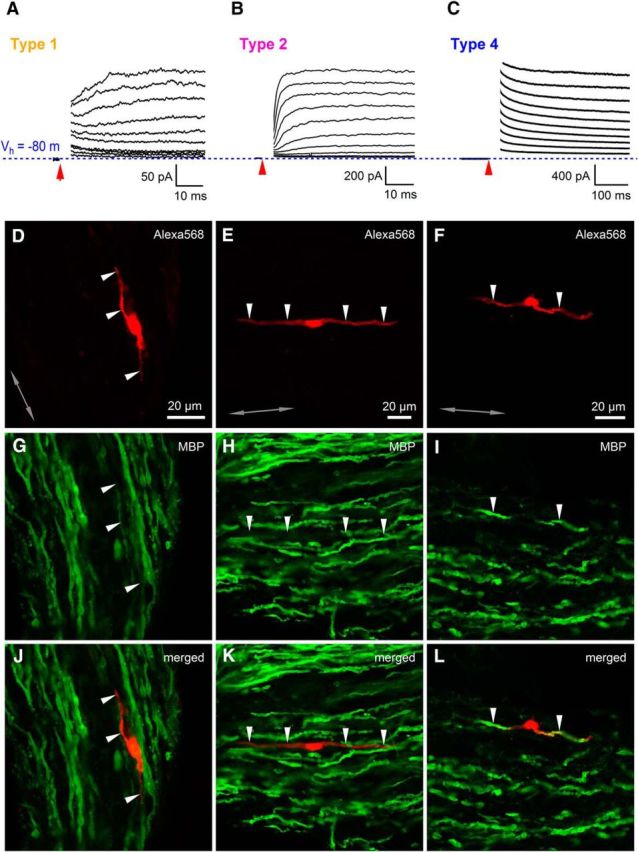
Only SCs without response to glutamate are involved in myelination of axons. A, Example of a current response to a series of depolarizing voltage steps applied from Vhold = −80 mV (blue dotted line) with an increment of +10 mV for a type 1 cell filled with Alexa Fluor-568 (D) and stained for MBP (G, J). The currents are overlaid and the capacitive transients are blanked for clarity. Red arrow indicates the time point when a voltage step started. B, As in A but for a type 2 cell. C, As in A but for a type 4 cell. Note the difference between the scale bars in A–C. D–F, Type 1, type 2, and type 4 cells filled with Alexa Fluor-568 during patch-clamp recordings in the sciatic nerve slice (red, single confocal plane). Arrowheads point to the processes of the cells. G–I, Postrecording immunohistochemistry for MBP (green; Cy5 in G and H or Alexa Fluor-488 in I). The confocal plane and the position of the arrowheads are the same as in D–F. J–L, Overlay of red and green channels shown in D–I. Note that the recorded cells of type 1 and type 2 are negative for MBP, whereas the recorded cell of type 4 is positive for MBP. Gray double-ended arrow indicates the orientation of the axons in the nerve.
We next analyzed the morphology of the recorded SCs. The majority of cells showed a bipolar morphology with two major bipolar processes (Fig. 8A). Type 1 cells (n = 11 cells in 11 animals) were shorter than either type 2 (n = 14 cells in 12 animals) or type 4 cells (n = 17 cells in 15 animals) (one-way ANOVA: F(2,39) = 5.215, p = 0.010; post hoc Gabriel test: type 1 vs type 2, p = 0.015; type 1 vs type 4, p = 0.024; type 2 vs type 4, p = 0.981; Fig. 8B). In the majority of the cells, one of the two major bipolar processes was clearly longer than the other; that is, the cell asymmetry ratio was ≪1 (Fig. 8C) and this parameter was not different between the cell types (one-way ANOVA: F(2,39) = 1.930, p = 0.159). In addition, the total area and the perimeter of the processes were comparable between the groups (Fig. 8D,E: one-way ANOVA: F(2,39) = 2.903, p = 0.067 for Fig. 8D; one-way ANOVA: F(2,39) = 1.323, p = 0.278 for Figure 8E). The subsequent detailed analysis showed that type 1 cells, and to a lesser extent type 2 cells, had a higher number of processes than type 3 and 4 cells (Fig. 8F) and the processes of type 1 and 2 cells branched more extensively than the processes of type 3 and 4 cells (Fig. 8G). Remarkably, 82% of type 4 cells possessed only two bipolar processes (Fig. 8F) that were oriented along the axonal axis and looked like myelin sheathes (Fig. 8A). These findings suggest that type 3 and type 4 cells are more mature cells that are already involved in myelination, whereas type 1 and type 2 cells are less mature and some of them are probably even migrating cells and/or cells searching for a target axon to establish contact with. We also analyzed the thickness of the cells (Fig. 8H, I) and found that, close to the soma (i.e., at 25% of process length), the thickness of the processes was similar in all cells (Fig. 8J: one-way ANOVA: F(2,39) = 2.417, p = 0.102). However, the mid- and terminal parts of the processes (i.e., at 50–99% of process length) were thicker in type 4 cells than in type 1 or type 2 cells (Fig. 8K–M). At 50% of process length (Fig. 8K): Kruskal–Wallis test: H(2) = 10.085, p = 0.006 and post hoc Dunn's test: type 1 versus type 2, p = 1.000; type 1 versus type 4, p = 0.011; type 2 versus type 4, p = 0.049. At 75% of process length (Fig. 8L): Kruskal–Wallis test: H(2) = 17.092, p = 0.00019 and post hoc Dunn's test: type 1 versus type 2, p = 0.102; type 1 versus type 4, p = 0.00011; type 2 versus type 4, p = 0.121. At 99% of process length (Fig. 8M): one-way ANOVA: F(2,39) = 3.246, p = 0.050 and post hoc Gabriel test: type 1 versus type 2, p = 0.695; type 1 versus type 4, p = 0.049; type 2 versus type 4, p = 0.326. These findings suggest that the structural composition of the processes differs between type 4 versus type 1 and type 2 cells. This is largely expected because type 4 cells are myelinating, whereas type 1 and type 2 cells are not (Fig. 7). Interestingly, we noticed a nonuniform filling of the thick bipolar processes with a fluorescent dye in many type 3 cells and all type 4 cells (Fig. 8A). Because type 4 cells (and probably also type 3 cells) are myelinating (Fig. 7), it is likely that the dye in these cells was distributed only within the noncompact myelin; that is, the “myelin channel” (Velumian et al., 2011; Snaidero et al., 2014), whereas the developing compact myelin lacked the dye, thus creating the appearance of a fluorescent channel within the filled cells.
Morphology of individual SCs included into the analysis. Images showing maximum intensity projection of each individual cell included into the analysis: Type 1. Download Figure 8-1, TIF file (21.6MB, tif)
Morphology of individual SCs included into the analysis. Images showing maximum intensity projection of each individual cell included into the analysis: Type 2. Download Figure 8-2, TIF file (21.5MB, tif)
Morphology of individual SCs included into the analysis. Images showing maximum intensity projection of each individual cell included into the analysis: Type 3. Download Figure 8-3, TIF file (7.2MB, tif)
Morphology of individual SCs included into the analysis. Images showing maximum intensity projection of each individual cell included into the analysis: Type 4. Download Figure 8-4, TIF file (19.2MB, tif)
Remarkably, the group of type 2 cells showed the highest variability of nearly all analyzed morphological parameters (data not shown). Further, in 64% of type 2 cells, we noticed a nonuniform filling of the thick bipolar processes with the dye, similar as that which occurred in type 3 and 4 cells. This may indicate that some of type 2 cells are on the way to myelination; that is, they started to reduce the amount of cytoplasm (and thus appeared to be nonuniformly filled with the dye), but did not yet express enough MBP to be detectable by immunohistochemistry (Fig. 7).
Together, our data suggest that type 1 cells are nonmyelinating SCs; the group of type 2 cells encompasses cells of multiple stages on the way to myelination, probably nonmyelinating, promyelinating, and those starting to myelinate; type 4 cells (and likely also type 3 cells) are more mature cells involved in the myelination process.
Discussion
The information available so far regarding ionotropic glutamate receptors in SCs of vertebrates mainly relies on cell culture studies and a few experiments on nerve injury (Lyons et al., 1994; Fink et al., 1999; Liu and Bennett, 2003; Mantuano et al., 2015; Campana et al., 2017). However, the expression of channels and receptors in SCs grown in culture can be affected by loss of tissue architecture and/or by substances in the medium to which SCs are not exposed under physiological conditions. Furthermore, SCs of certain developmental stages may be lost during cell isolation before culturing and/or myelinating SCs may not develop properly in culture. Physical connections established between axons and developing SCs in the intact nerve also get lost during cell culture preparation. To address these problems, we established a preparation of live sciatic nerve slices (Barzan et al., 2016) using a widely described technical approach used for the preparation of live brain slices. Using this novel PNS preparation, we demonstrated that the majority of periaxonal SCs in the embryonic and neonatal mouse sciatic nerves express functional AMPARs and respond to the application of glutamate with inward current. Our findings are further substantiated by the SFA showing that the median value of the single-channel conductance of AMPARs in SCs is 8–11 pS, which is comparable to that in neurons (Cull-Candy et al., 1988; Jonas et al., 1993; Benke et al., 1998) and type-2 cerebellar astrocytes (Usowicz et al., 1989; Wyllie and Cull-Candy, 1994), but is lower than single-channel conductance of AMPAR channels in the OPCs (Zonouzi et al., 2011). Notably, non-NMDA glutamate channels in cerebellar type 2 astrocytes exhibit multiple conductance levels in the range of 6, 10–11, 16–20, 32–40, and 45–52 pS in response to the application of glutamate receptor agonists in outside-out patches (Usowicz et al., 1989; Wyllie and Cull-Candy, 1994). Because we estimated the conductance of AMPAR channels in SCs from the macroscopic currents, which reflect the behavior of all active AMPA channels in the membrane, we only know the “weighted” average conductance of all the multiple levels. However, our estimates strongly favor the idea that AMPAR channels in SCs give only small- and medium-level conductance openings (<30 pS) in response to glutamate application and show no/few openings to the larger levels. In this respect, SCs differ from the glial cells of the CNS in which non-NMDA channels give rise to >30 pS events in addition to the events of the smaller amplitudes (Usowicz et al., 1989; Wyllie and Cull-Candy, 1994; Zonouzi et al., 2011). The variations in the single-channel conductance of AMPARs observed in our study (Fig. 5F) are likely related to the fact that SCs express AMPARs of different subunit composition. In addition, the location of AMPARs may vary in different SCs. Because we probably had not activated all AMPARs on a given cell using pressure-induced glutamate application (Fig. 4D), it is possible that, in some cells, we targeted only a subset of AMPARs of a given subunit composition and/or activated their opening to one rather than multiple conductance levels.
Our findings suggest that developing, not mature, SCs express functional AMPARs during physiological conditions. The evidence for this in our study is fourfold. First, SCs responding to glutamate showed small or intermediate values of the membrane capacitance (Fig. 3B), indicating that their membrane area is (relatively) small. This is expected only for the cells that do not generate a large amount of myelin membrane. In contrast, cells that did not respond to glutamate application (Fig. 4A,B) had large membrane capacitance because they are involved in myelination (Figs. 7, 8). Second, cells responding to glutamate were found in the embryonic and neonatal nerves, whereas the majority of nonresponding cells were found in the neonatal nerves (Figs. 3H, 4A). Third, SCs responding to glutamate did not show MBP labeling, whereas nonresponding cells were positive for MBP (Fig. 7). Fourth, cells responding to glutamate possessed a higher number of processes than nonresponding cells; their processes branched more and were generally thinner, especially at large distances from the cell soma (Fig. 8). This morphological complexity, together with a lack of labeling for MBP, suggests that cells responding to glutamate are more immature than nonresponding cells. Our results favor the idea that functional AMPARs are present mainly in the developing SCs, so AMPAR-mediated signaling is of particular importance during the development of the SC lineage.
How do AMPARs in the developing SCs get activated and what is their functional role? SCs within the peripheral nerves are in intimate contact with axons and thus are ideally placed to respond to axonal activity. Mammalian peripheral axons, in turn, appear capable of releasing the neurotransmitter glutamate from their shafts when stimulated electrically or magnetically (Wheeler et al., 1966; DeFeudis, 1971; Weinreich and Hammerschlag, 1975; Wieraszko and Ahmed, 2009). It is feasible that the released neurotransmitter is detected by AMPARs in SCs, giving the axons a possibility to signal to SCs in an activity-dependent fashion. In the CNS, such axon–glia signaling takes place between the axons and the OPCs (Kriegler and Chiu, 1993; Kukley et al., 2007; Ziskin et al., 2007). Intriguingly, neurons build synapses with the OPCs, and action potentials propagating through the white matter and the optic nerve trigger fast vesicular glutamate release at axon–OPC synapses, which is detected by AMPARs in the OPCs. In the present study, we tested the hypothesis that similar signaling may exist in the peripheral nerves. We applied several electrophysiological and pharmacological approaches that are widely used to trigger synaptic currents in neurons and OPCs, but were not able to elicit synaptic currents in SCs. The tested SCs were type 1 and type 2 cells, which usually show response to glutamate. Furthermore, the cells were compact (membrane capacitance <50 pF) and had membrane resistance >200 MΩ (Fig. 3A–C), which is appropriate for the detection of small synaptic events. Therefore, it is unlikely that we missed synaptic currents for technical reasons. Based on our morphological analysis, type 2 cells likely encompass a heterogeneous population of cells and some of them may have commenced myelination. Indeed, in one type 2 cell, we observed that a small segment of a process was positively labeled with MBP, indicating that the cell had already started expressing this myelin protein. Therefore, the type 2 cells may be “analogous” to the premyelinating/young oligodendrocytes of the CNS, which are not involved or loosely involved in synaptic signaling with neurons (Kukley et al., 2010). This could explain why we were not able to record synaptic currents in type 2 cells. However, our data suggest that type 1 cells represent less mature cells of the SC lineage and may be “analogous” to the OPCs. If functional expression of AMPARs in SCs and synaptic axon–glia communication in the peripheral nerve follow the same developmental scheme as in the oligodendrocyte lineage cells (Kukley et al., 2010), then we expected to record synaptic currents in type 1 cells, but this was not the case. The absence of synaptic currents in the developing SCs, which are positioned close to the axons and express AMPARs, indicates that glutamatergic axons in the PNS do not establish synapses with SCs. Interestingly, a recent study investigating communication between axons of dorsal root ganglion (DRG) neurons and the OPCs in culture demonstrated that, although DRG axons are capable of vesicular glutamate release upon electrical stimulation and the processes of OPCs are located in close contact with the DRG axons, axon–OPC synaptic currents do not occur in this system (Wake et al., 2015). These findings suggest that DRG axons may not use vesicular glutamate release for synaptic communication with glial cells and in this regard are different from the CNS axons. Because DRG axons are the primary candidates for glutamate release in the sciatic nerve, the findings of Wake et al. (2015) may explain why we have not observed glutamatergic synaptic currents in developing SCs.
It would be of future interest to find out how AMPARs are distributed within the SCs and whether these receptors are clustered. If AMPARs are located too far away from the axonal sites of vesicular glutamate release, then their activation may involve other mechanisms such as axonal efflux of glutamate via a carrier or passive leak, vesicular nonsynaptic release, or a nonvesicular mechanism (Kriegler and Chiu, 1993). In addition, it is also plausible that glutamate is released from SCs themselves and binds to auto-AMPARs or AMPARs on the neighboring SCs. It is important to consider that absence of receptor clustering in SCs may represent an obstacle for detecting AMPAR-mediated currents upon glutamate release with the patch-clamp technique because the amplitude of these currents may be too small to be distinguished reliably from the electrical noise. However, even AMPAR-mediated currents of small amplitude may play an important role for the physiology of SCs in vivo because activation of these ionotropic receptors is expected to generate changes in ion concentration within the cells, in particular Ca2+, which can be further amplified by Ca2+-induced Ca2+ release from the internal stores or other mechanisms. Future experiments involving inducible deletion of multiple subunits of AMPARs and/or modification of Ca2+ permeability of AMPARs in SCs at different stages of development should help to clarify whether these receptors contribute to the regulation of migration, survival, proliferation, and/or differentiation of the developing SCs, in analogy to the suggested role of glutamatergic signaling for the progenitors of neurons and myelinating glia of the CNS (Yuan et al., 1998; Ghiani et al., 1999; Jansson and Åkerman, 2014; Luhmann et al., 2015).
Footnotes
This work was supported by German Research Foundation (Grants EXC-307 and KU2569/1-1 to M.K.) and by the Christiane-Nüsslein-Volhard Foundation (N.F.). We thank Stephen Traynelis (Emory University, Atlanta, GA), Rolf Sprengel (MPI for Medical Research), and Ingrid Ehrlich (HIH/CIN Tübingen, Germany) for providing plasmids; Rick Gerkin (Arizona State University, Phoenix, AZ) for help with the acquisition software; Thorsten Schmidt (Institute for Medical Genetics, Tübingen, Germany) for help with setting up PCRs; Balint Nagy (CIN, Tübingen, Germany) for help with statistical analysis; Julia Fitzgerald (HIH, Tübingen, Germany) for proofreading the manuscript; Daniela Eiβler (CIN, Tübingen, Germany) for excellent technical assistance; and members of the Kukley laboratory for helpful comments on the manuscript.
The authors declare no competing financial interests.
References
- Baker MD. (2002) Electrophysiology of mammalian Schwann cells. Prog Biophys Mol Biol 78:83–103. 10.1016/S0079-6107(02)00007-X [DOI] [PubMed] [Google Scholar]
- Barzan R, Pfeiffer F, Kukley M (2016) N- and L-type voltage-gated calcium channels mediate fast calcium transients in axonal shafts of mouse peripheral nerve. Front Cell Neurosci 10:135. 10.3389/fncel.2016.00135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benke TA, Lüthi A, Isaac JT, Collingridge GL (1998) Modulation of AMPA receptor unitary conductance by synaptic activity. Nature 393:793–797. 10.1038/31709 [DOI] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, Jahr CE (2000) Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405:187–191. 10.1038/35012083 [DOI] [PubMed] [Google Scholar]
- Campana WM, Mantuano E, Azmoon P, Henry K, Banki MA, Kim JH, Pizzo DP, Gonias SL (2017) Ionotropic glutamate receptors activate cell signaling in response to glutamate in Schwann cells. FASEB J 31:1744–1755. 10.1096/fj.201601121R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SY. (1991) Functions and distribution of voltage-gated sodium and potassium channels in mammalian Schwann cells. Glia 4:541–558. 10.1002/glia.440040602 [DOI] [PubMed] [Google Scholar]
- Christensen PC, Welch NC, Brideau C, Stys PK (2016) Functional ionotropic glutamate receptors on peripheral axons and myelin. Muscle Nerve 54:451–459. 10.1002/mus.25078 [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Usowicz MM (1989) Whole-cell current noise produced by excitatory and inhibitory amino acids in large cerebellar neurones of the rat. J Physiol 415:533–553. 10.1113/jphysiol.1989.sp017735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Howe JR, Ogden DC (1988) Noise and single channels activated by excitatory amino acids in rat cerebellar granule neurones. J Physiol 400:189–222. 10.1113/jphysiol.1988.sp017117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFeudis FV. (1971) Effects of electrical stimulation on the efflux of L-glutamate from peripheral nerve in vitro. Exp Neurol 30:291–296. 10.1016/S0014-4886(71)80008-0 [DOI] [PubMed] [Google Scholar]
- Demêmes D, Lleixa A, Dechesne CJ (1995) Cellular and subcellular localization of AMPA-selective glutamate receptors in the mammalian peripheral vestibular system. Brain Res 671:83–94. 10.1016/0006-8993(94)01322-9 [DOI] [PubMed] [Google Scholar]
- Dougherty R, Kunzelmann K (2007) Computing local thickness of 3D structures with ImageJ. Microsc Microanal 13(S02), 1678–1679. 10.1017/S1431927607074430 [DOI] [Google Scholar]
- Evans PD, Reale V, Merzon RM, Villegas J (1991) Mechanisms of axon-Schwann cell signaling in the squid nerve fiber. Ann N Y Acad Sci 633:434–447. 10.1111/j.1749-6632.1991.tb15634.x [DOI] [PubMed] [Google Scholar]
- Fink T, Davey DF, Ansselin AD (1999) Glutaminergic and adrenergic receptors expressed on adult guinea pig Schwann cells in vitro. Can J Physiol Pharmacol 77:204–210. 10.1139/y99-008 [DOI] [PubMed] [Google Scholar]
- Ghiani CA, Yuan X, Eisen AM, Knutson PL, DePinho RA, McBain CJ, Gallo V (1999) Voltage-activated K+ channels and membrane depolarization regulate accumulation of the cyclin-dependent kinase inhibitors p27(Kip1) and p21(CIP1) in glial progenitor cells. J Neurosci 19:5380–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartveit E, Veruki ML (2007) Studying properties of neurotransmitter receptors by non-stationary noise analysis of spontaneous postsynaptic currents and agonist-evoked responses in outside-out patches. Nat Protoc 2:434–448. 10.1038/nprot.2007.47 [DOI] [PubMed] [Google Scholar]
- Jansson LC, Åkerman KE (2014) The role of glutamate and its receptors in the proliferation, migration, differentiation and survival of neural progenitor cells. J Neural Transm 121:819–836. 10.1007/s00702-014-1174-6 [DOI] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B (1993) Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol 472:615–663. 10.1113/jphysiol.1993.sp019965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkelin I, Bröcker EB, Koltzenburg M, Carlton SM (2000) Localization of ionotropic glutamate receptors in peripheral axons of human skin. Neurosci Lett 283:149–152. 10.1016/S0304-3940(00)00944-7 [DOI] [PubMed] [Google Scholar]
- Kriegler S, Chiu SY (1993) Calcium signaling of glial cells along mammalian axons. J Neurosci 13:4229–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E, Dietrich D (2007) Vesicular glutamate release from axons in white matter. Nat Neurosci 10:311–320. 10.1038/nn1850 [DOI] [PubMed] [Google Scholar]
- Kukley M, Nishiyama A, Dietrich D (2010) The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J Neurosci 30:8320–8331. 10.1523/JNEUROSCI.0854-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman EM, Sanzenbacher E (1992) Mechanisms of glutamate activation of axon-to-Schwann cell signaling in the squid. Neuroscience 47:931–939. 10.1016/0306-4522(92)90041-Y [DOI] [PubMed] [Google Scholar]
- Lieberman EM, Abbott NJ, Hassan S (1989) Evidence that glutamate mediates axon-to-Schwann cell signaling in the squid. Glia 2:94–102. 10.1002/glia.440020205 [DOI] [PubMed] [Google Scholar]
- Liu GJ, Bennett MR (2003) ATP secretion from nerve trunks and Schwann cells mediated by glutamate. Neuroreport 14:2079–2083. 10.1097/00001756-200311140-00014 [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Fukuda A, Kilb W (2015) Control of cortical neuronal migration by glutamate and GABA. Front Cell Neurosci 9:4. 10.3389/fncel.2015.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SA, Morell P, McCarthy KD (1994) Schwann cells exhibit P2Y purinergic receptors that regulate intracellular calcium and are up-regulated by cyclic AMP analogues. J Neurochem 63:552–560. [DOI] [PubMed] [Google Scholar]
- Mantuano E, Lam MS, Shibayama M, Campana WM, Gonias SL (2015) The NMDA receptor functions independently and as an LRP1 co-receptor to promote Schwann cell survival and migration. J Cell Sci 128:3478–3488. 10.1242/jcs.173765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy B, Hovhannisyan A, Barzan R, Chen TJ, Kukley M (2017) Different patterns of neuronal activity trigger distinct responses of oligodendrocyte precursor cells in the corpus callosum. PLoS Biol 15:e2001993. 10.1371/journal.pbio.2001993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternain AV, Morales M, Lerma J (1995) Selective antagonism of AMPA receptors unmasks kainate receptor-mediated responses in hippocampal neurons. Neuron 14:185–189. 10.1016/0896-6273(95)90253-8 [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. 10.1016/S0896-6273(00)80146-4 [DOI] [PubMed] [Google Scholar]
- Samara C, Poirot O, Domènech-Estévez E, Chrast R (2013) Neuronal activity in the hub of extrasynaptic Schwann cell-axon interactions. Front Cell Neurosci 7:228. 10.3389/fncel.2013.00228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert G, Rehn L, Weber M, Steinhäuser C (1997) AMPA receptor subunits expressed by single astrocytes in the juvenile mouse hippocampus. Brain Res Mol Brain Res 47:286–294. 10.1016/S0169-328X(97)00059-4 [DOI] [PubMed] [Google Scholar]
- Snaidero N, Möbius W, Czopka T, Hekking LH, Mathisen C, Verkleij D, Goebbels S, Edgar J, Merkler D, Lyons DA, Nave KA, Simons M (2014) Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 156:277–290. 10.1016/j.cell.2013.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer S, Volbracht K, Lundgaard I, Káradóttir RT (2016) Glutamate signalling: A multifaceted modulator of oligodendrocyte lineage cells in health and disease. Neuropharmacology 110:574–585. 10.1016/j.neuropharm.2016.06.014 [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Jaramillo F (1998) Getting the most out of noise in the central nervous system. Trends Neurosci 21:137–145. 10.1016/S0166-2236(98)01238-7 [DOI] [PubMed] [Google Scholar]
- Usowicz MM, Gallo V, Cull-Candy SG (1989) Multiple conductance channels in type-2 cerebellar astrocytes activated by excitatory amino acids. Nature 339:380–383. 10.1038/339380a0 [DOI] [PubMed] [Google Scholar]
- Velumian AA, Samoilova M, Fehlings MG (2011) Visualization of cytoplasmic diffusion within living myelin sheaths of CNS white matter axons using microinjection of the fluorescent dye Lucifer yellow. Neuroimage 56:27–34. 10.1016/j.neuroimage.2010.11.022 [DOI] [PubMed] [Google Scholar]
- Wake H, Ortiz FC, Woo DH, Lee PR, Angulo MC, Fields RD (2015) Nonsynaptic junctions on myelinating glia promote preferential myelination of electrically active axons. Nat Commun 6:7844. 10.1038/ncomms8844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MS, Dingledine R (1996) Block of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by polyamines and polyamine toxins. J Pharmacol Exp Ther 278:669–678. [PubMed] [Google Scholar]
- Weinreich D, Hammerschlag R (1975) Nerve impulse-enhanced release of amino acids from non-synaptic regions of peripheral and central nerve trunks of bullfrog. Brain Res 84:137–142. 10.1016/0006-8993(75)90807-0 [DOI] [PubMed] [Google Scholar]
- Wheeler DD, Boyarsky LL, Brooks WH (1966) The release of amino acids from nerve during stimulation. J Cell Physiol 67:141–147. 10.1002/jcp.1040670116 [DOI] [PubMed] [Google Scholar]
- Wieraszko A, Ahmed Z (2009) Axonal release of glutamate analog, d-2,3–3H-Aspartic acid and l-14C-proline from segments of sciatic nerve following electrical and magnetic stimulation. Neurosci Lett 458:19–22. 10.1016/j.neulet.2009.04.024 [DOI] [PubMed] [Google Scholar]
- Wyllie DJ, Cull-Candy SG (1994) A comparison of non-NMDA receptor channels in type-2 astrocytes and granule cells from rat cerebellum. J Physiol 475:95–114. 10.1113/jphysiol.1994.sp020052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KA, Tang CM (1993) Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J Neurosci 13:3904–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Eisen AM, McBain CJ, Gallo V (1998) A role for glutamate and its receptors in the regulation of oligodendrocyte development in cerebellar tissue slices. Development 125:2901–2914. [DOI] [PubMed] [Google Scholar]
- Zhou M, Kimelberg HK (2000) Freshly isolated astrocytes from rat hippocampus show two distinct current patterns and different [K(+)](o) uptake capabilities. J Neurophysiol 84:2746–2757. [DOI] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE (2007) Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci 10:321–330. 10.1038/nn1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonouzi M, Renzi M, Farrant M, Cull-Candy SG (2011) Bidirectional plasticity of calcium-permeable AMPA receptors in oligodendrocyte lineage cells. Nat Neurosci 14:1430–1438. 10.1038/nn.2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Morphology of individual SCs included into the analysis. Images showing maximum intensity projection of each individual cell included into the analysis: Type 1. Download Figure 8-1, TIF file (21.6MB, tif)
Morphology of individual SCs included into the analysis. Images showing maximum intensity projection of each individual cell included into the analysis: Type 2. Download Figure 8-2, TIF file (21.5MB, tif)
Morphology of individual SCs included into the analysis. Images showing maximum intensity projection of each individual cell included into the analysis: Type 3. Download Figure 8-3, TIF file (7.2MB, tif)
Morphology of individual SCs included into the analysis. Images showing maximum intensity projection of each individual cell included into the analysis: Type 4. Download Figure 8-4, TIF file (19.2MB, tif)