

Abstract
The gene patched domain containing 1 (PTCHD1) is mutated in patients with autism spectrum disorders and intellectual disabilities and has been hypothesized to contribute to Sonic hedgehog (Shh) signaling and synapse formation. We identify a panel of Ptchd1-interacting proteins that include postsynaptic density proteins and the retromer complex, revealing a link to critical regulators of dendritic and postsynaptic trafficking. Ptchd1 knock-out (KO) male mice exhibit cognitive alterations, including defects in a novel object recognition task. To test whether Ptchd1 is required for Shh-dependent signaling, we examined two Shh-dependent cell populations that express high levels of Ptchd1 mRNA: cerebellar granule cell precursors and dentate granule cells in the hippocampus. We found that proliferation of these neuronal precursors was not altered significantly in Ptchd1 KO male mice. We used whole-cell electrophysiology and anatomical methods to assess synaptic function in Ptchd1-deficient dentate granule cells. In the absence of Ptchd1, we observed profound disruption in excitatory/inhibitory balance despite normal dendritic spine density on dentate granule cells. These findings support a critical role of the Ptchd1 protein in the dentate gyrus, but indicate that it is not required for structural synapse formation in dentate granule cells or for Shh-dependent neuronal precursor proliferation.
SIGNIFICANCE STATEMENT The mechanisms underlying neuronal and cellular alterations resulting from patched domain containing 1 (Ptchd1) gene mutations are unknown. The results from this study support an association with dendritic trafficking complexes of Ptchd1. Loss-of-function experiments do not support a role in sonic hedgehog-dependent signaling, but reveal a disruption of synaptic transmission in the mouse dentate gyrus. The findings will help to guide ongoing efforts to understand the etiology of neurodevelopmental disorders arising from Ptchd1 deficiency.
Keywords: autism, glutamatergic synapse, mental retardation, retromer, sonic hedgehog
Introduction
Autism-spectrum disorders (ASDs) are a heterogeneous group of neurodevelopmental syndromes. Twin studies indicate a high concordance rate in monozygotic twins, but much lower concordance in dizygotic twins, highlighting a strong genetic contribution to autism risk. Human genetic studies have identified a large number of mutations and copy number variations that may result in an increased risk for ASD (de la Torre-Ubieta et al., 2016). In addition, there are environmental risk factors, including prenatal immune challenges, that correlate with the incidence of ASD (Estes and McAllister, 2015).
Each individual mutation identified thus far appears to account for only a very small fraction of autism cases. One of the more frequently identified ASD risk factors are alterations in the patched domain containing 1 gene (PTCHD1) encoded on the X-chromosome. Studies in male probands with ASD identified multiple different microdeletions in PTCHD1 and missense mutations, which are thought to result in a loss of Ptchd1 protein function (Marshall et al., 2008; Noor et al., 2010). Subsequent studies confirmed an association of common and rare variants with ASD and intellectual disability in several additional patient populations (Filges et al., 2011; Torrico et al., 2015). A phenotypic survey of 23 individuals with PTCHD1 mutations reported variable degrees of intellectual disability and prominent behavioral alterations, with >40% having ASD or ASD-like behaviors (Chaudhry et al., 2015). Moreover, most patients present with mild motor incoordination. Recent studies in Ptchd1 mutant mice reported behavioral alterations, in particular attention-deficit/hyperactivity disorder (ADHD)-like behaviors, gait abnormalities, hyperaggression, inhibitory avoidance, and defects in contextual fear conditioning (Wells et al., 2016; Ung et al., 2017). Moreover, conditional ablation of Ptchd1 from interneurons defined by somatostatin-cre expression recapitulated the ADHD-like phenotypes observed in global Ptchd1 knock-out (KO) mice (Wells et al., 2016). These alterations are accompanied by a loss of burst firing in neurons of the thalamic reticular nucleus, one site where Ptchd1 mRNA expression is particularly high (Wells et al., 2016).
Despite these major advances, the function of the Ptchd1 protein and the molecular mechanisms underlying the phenotypes are unknown. Ptchd1 protein sequences share a sterol-sensing domain with the sonic hedgehog (Shh) receptor Patched (Ptch1) and Nieman–Pick disease, type C1 (NPC1), a cholesterol transport protein. Based on this sequence similarity and transcriptional reporter assays in cell lines, it has been hypothesized that Ptchd1 may contribute to Shh signaling (Noor et al., 2010). However, this hypothesis has not been tested with loss-of-function approaches. Heterozygous or conditional homozygous mutations of Ptch result in Shh-independent activation of the signaling molecule Smoothened. Hyperactivation of Smoothened regulation results in neuronal precursor overproliferation, enlargement of the external germinal layer of the cerebellum, and the development of medulloblastomas (Goodrich et al., 1997; Yang et al., 2008). Recent studies have identified Boc and Gas1 as obligate coreceptors for Shh signaling in the cerebellum. Their loss of function results in reduced Shh-dependent proliferation of precursors (Izzi et al., 2011) and cerebellar abnormalities. Given these well documented functions of the Shh-Ptch1 pathway, we investigated whether neuronal precursor proliferation is altered in Ptchd1 KO mice.
We found that Ptchd1 removal has no effect on neuronal precursor proliferation in the cerebellum or hippocampus of mice. Instead, an unbiased search for Ptchd1-interacting proteins suggested that Ptchd1 interacts with dendritic trafficking complexes. Supporting a role in regulating synaptic function, loss of Ptchd1 results in a disruption in excitatory/inhibitory balance in the dentate gyrus.
Materials and Methods
Mice.
Ptchd1 mutant mice were generated in the C57BL/6N background in collaboration with the Mouse Biology Project at the University of California–Davis. Targeting vector PRPGS00100_C_H03 (KOMP) was electroporated into JM8.N4 ES cell and colonies selected. The targeting cassette is based on the KO first vector design, replacing exon 2 of the Ptchd1 gene. Mice with germline transmission were mated with ROSA26-Flpe females (Jax stock no: 003946), backcrossed over 10 generations with C57BL/6N mice, to remove FRT-flanked genetrap/LacZ sequences, yielding a conditional allele. Germline ablation of exon 2 was created by crossing with CMV-cre mice (Jax stock no: 006054). The “KO first” mice generated by the Mouse Biology Project were deposited at the Mutant Mouse Resource & Research Centers (MMRRC) repository. Genotyping of the KO mice was as follows: the Ptchd1−/y allele was detected by PCR using primers 5′-AACACCATGACTGAAGCAACTTGGG-3′(CSD-Ptchd1-F) with 5′-AGTGCTGAGGAGTTAGCTATGCAGG-3′(CSD-Ptchd1-R). The Ptchd1WT allele was detected using 5′-AACACCATGACTGAAGCAACTTGGG-3′(CSD-Ptchd1-F) with 5′-TCCAGAACTGAGAAATCAGGTTCGC-3′(CSD-Ptchd1-ttR). Only male mice were used for all experiments. Procedures related to animal experimentation were reviewed and approved by the Kantonales Veterinäramt Basel-Stadt.
In situ hybridization and PCR analysis.
In situ hybridizations with chromogenic detection were done using digoxigenin-labeled cRNA probes and were performed as described previously (Schaeren-Wiemers and Gerfin-Moser, 1993). The DNA fragment encoding the Ptchd1 probe contained SP6 and T7 promoters flanking the 5′- or 3′-end, respectively: Ptchd1:CCGCTCTGCTCTAGGATGCTGCGGCAGGTTCTGCACAGGGGCTTGAGGACGTGTTTCTCCCGGCTTGGCCACTTCATTGCCAGTCACCCGGTCTTCTTTGCTTCGGCGCCGGTGCTCATCTCCATCCTGCTCGGCGCCAGCTTCAGCCGCTACCAGGTCGAAGAGAGCGTGGAGCACCTGCTGGCGCCCCAGCACAGCCTAGCCAAGATCGAGCGCAACCTAGTCAACAGCCTCTTCCCGGTCAACCGCTCCAAGCACCGGCTCTACTCGGACCTGCAGACCCCTGGGCGCTACGGCCGGGTCATTGTCACCTCCTACCAGAAAGCCAACATGCTAGACCAACATCACACGGACCTGA.
Templates for in vitro transcription using SP6-polymerase (antisense probe) or T7-polymerase (sense probe) were amplified by PCR using the ISP-SP6-5′ (5′-CTATCGATTTAGGTGACACTATAGAAG-3′) and ISP-T7-3′ (5′-GAATTGTAATACGACTCACTATAGGGA-3′) primers.
FISH was performed following the RNAscope Multiplex Fluorescent Assay protocol (Wang et al., 2012). The assay was performed on 20 μm snap-frozen postnatal day 21 (P21) brain cryostat sections. Sections were cut between bregma −1.46 and −2.40 including the dorsal hippocampus. Ptchd1 transcripts were detected with a commercial Ptchd1 probe (ACD, catalog #489651). A negative control probe (ACD, catalog #320871) was used to assess the specificity of hybridization signals observed. In situ images were acquired on a Widefield Axio Scan Z1 slide scanner.
For qPCR analysis, brain tissues were dissected in ice-cold PBS, microdissected hippocampal subregions were isolated according to the protocol of Hagihara et al. (2009). Tissue was homogenized in TRIzol reagent (Sigma-Aldrich), followed by the addition of chloroform. After homogenization, samples were centrifuged for 15 min at 16,000 × g. The supernatant was used for RNA purification with the RNAeasy mini plus kit (Qiagen) following the manufacturer's instructions. One microgram of total RNA was reverse transcribed using random hexamers and ImProm-II Reverse Transcriptase (Promega). qPCR was performed on a StepOne plus qPCR system (Applied Biosystems). Gene expression assays were done with the Power SYBR Green Master Mix (Applied Biosystems) and the comparative CT method. cDNA levels were normalized to the housekeeping gene Gapdh cDNA. Primer sequences used with Power SYBR Green PCR Mastermix were as follows: Ptchd1: 5′-CAAGATCGAGCGCAACCTAG-3′ and 5′-ATGTTGGCTTTCTGGTAGGAG-3′; Gapdh: 5′-CTCGTGTGGATTTGGAGAG-3′ and 5′-AGTTCTCGAACTTCCTCCT-3′; Hprt: 5′-GATGAACCAGGTATGACCTAGATTTG-3′ and 5′-ATGGCCTCCCATCTCCTTCAT-3′; TDO2: 5′-ATGAGTGGGTGCCCGTTTG-3′ and 5′-GGCTCTGTTTACACCAGTTTGAG-3′; DSP: 5′-GCTGAAGAACACTCTAGCCCA-3′ and 5′-ACTGCTGTTTCCTCTGAGACA-3′; Mrg1b: 5′-CTGGCGAGATCACGATGACG-3′ and 5′-AAGCTACGCTGTTGTCTAACC-3′. Primers used to confirm Ptchd1 exon 2 deletion were as follows: forward primer in Exon1: 5′-CCAACATGCTAGACCAACATCACA-3′; reverse primer in Exon3: 5′-TTTATGGTTCCAGCCTTGTGTTCA-3′.
Antibodies, cDNA expression vectors, Western blots.
Polyclonal anti-Ptchd1 antibodies were raised in guinea pigs using synthetic peptides derived from the following Ptchd1 sequences: DFQKTSRVSERYLVT and VDIDSTRVVDQITTV. Peptides were conjugated to keyhole limpet hemocyanin (Eurogentec). Antisera and affinity-purified antibodies were tested on Western blot applications; immunohistochemical applications were inconclusive. For Western blots, the following commercial antibodies were used: mouse anti-actin (JLA20-S; DSHB) 1:3000, mouse anti-GFP (Abcam, catalog #ab38689) 1:500 mouse anti-PSD95(DLG4) (NeuroMab, catalog #75-028) 1:3000, rabbit anti-VPS35 (Bethyl Laboratories, catalog #A304-727A) 1:200, and mouse anti-vGlut1 (NeuroMab, catalog #75-066) 1:3000. For immunohistochemistry assays the following antibodies were used: rat anti-BrdU (Abcam, catalog #ab6326) 1:50 and rabbit anti-phospho-histone 3 (PH3) (Millipore, catalog #06-570) 1:1000.
Immunoblotting was done with HRP-conjugated secondary antibodies (Jackson ImmunoResearch) and Pierce ECL Western blotting substrate. Signals were acquired using a digital imager (Bio-Rad, ChemiDoc MP Imaging System). The pEGFP-C1-Ptchd1 vector used for overexpression assays was created with the cDNA MG219209 from OriGene.
BrdU incorporation assays and diolistic labeling.
P4 Ptchd1−/y males and littermate wild-type (WT) controls were weighed and then injected intraperitoneally with 100 mg/kg BrdU. After 30 min, mice were killed and brains were transferred to fixative (4% paraformaldehyde, 30% sucrose in PBS) and incubated overnight at 4°C. On the next day, brains were frozen at −80°C and 30 μm cryosections were cut for immunohistochemistry. For the adult neurogenesis assay, P60 Ptchd1−/y males and WT littermate controls were used and analyzed 4 d after injection of BrdU. Sections underwent an antigen retrieval step before the standard immunostaining protocol: in brief, sections were wash 3 times in PBS, placed in 0.1% Triton X-100 in PBS for 20 min, transferred to 2 m HCl for 20 min at 37°C, neutralized with borate buffer (0.1 m, pH 8.5) 3 times for 10 min, and washed in PBS 3 times for 10 min. A standard immunostaining protocol was then applied for BrdU and PH3 detection. Quantitative assessment of BrdU-positive cells was performed as described previously (Han et al., 2008; Li et al., 2013). Briefly, confocal image stacks were acquired from consecutive sagittal sections of cerebellum lobule VI and dorsal dentate gyrus. An anatomically equivalent block of 300–400 μm thickness was analyzed for each animal (10–15 30 μm anatomical sections per mouse, with each section imaged as a confocal image stack). For the adult neurogenesis experiment, the neurogenic subgranular zone of the dentate gyrus was analyzed for BrdU- and PH3-positive cells. For the P4 developmental hippocampus experiment, the entire DG area was analyzed.
Diolistic labeling (Gan et al., 2000) was performed according to a previously published protocol (Staffend and Meisel, 2011). Images were acquired on an inverted LSM700 confocal microscope (Zeiss) using 10×, 20×, or 63× Apochromat objectives and controlled by Zen 2010 software. For spine counting, confocal image stacks were acquired of granule cell dendritic segments in the distal molecular layer of the dorsal dentate gyrus. Spines were counted per 30 μm dendritic segment by evaluating the 3D information of the image stacks. Headed and nonheaded spines were distinguished manually. Images for display were assembled using Adobe Photoshop and Illustrator software. All quantitative assessments were performed by an investigator blinded to the genotype.
Shh binding assay.
We modified a protocol from Cheng and Flanagan (2001). In brief, 12 mm glass coverslips were coated with 100 μg/ml poly-l-lysine (Sigma-Aldrich) for 2 h and then washed twice with water. MEF or COS7 cells were plated on uncoated or PLL-coated 12 mm glass coverslips, respectively, in 24-well plates. Cells were transfected with pEGFP-C1-Ptch1 + Casp9-DN-HA (HA-tagged dominant-negative caspase 9, to prevent apoptosis; Mille et al., 2009), pEGFP-C1-Ptchd1, or pEGFP-C1-NPC1 using Lipofectamine 3000 according to the manufacturer's instructions. Forty-eight hours later, the medium was removed and the cells washed twice with binding buffer A (BBA: DMEM + 10% FBS + 0.1% NaN2 + 50 mm HEPES). A concentration of 500 nm Shh-Fc, diluted in BBA, was then added to the cells and incubated for 1 h at room temperature. Cells were then washed once in BBA and 3 times in PBS (containing Ca2+ and Mg2+), followed by fixation with 4% PFA for 10 min at room temperature. Shh-Fc was detected with standard immunostaining protocols using Cy3-conjugated donkey anti-human-Fc, diluted 1:1000 in PBS with 1% normal donkey serum and 0.1% Triton X-100, for 1.5 h at room temperature. The coverslips were imaged on a Leica DM6B microscope with a Hammamatsu Orca Flash 4.0 camera using Leica LASX software. Shh-Fc binding to Ptch-expressing cells results in a distinct cell surface staining pattern. The percentage of cells exhibiting binding was scored by an investigator blinded to the identity of the transfected cDNA. The assay was conducted three times, twice with COS7 cells and once with mouse embryonic fibroblast cells, yielding similar results. Surface expression of overexpressed proteins was confirmed by surface biotinylation and revealed similar surface levels of Ptch and Ptchd1 proteins.
Biochemical interaction assays.
cDNA encoding fusion peptide sequences used for pull-down analysis were inserted in frame in a pET9d-His6-glutathione-S-transferase (GST) vector. Plasmids were transformed in BL21 Rosetta E. coli. Bacteria were grown to reach an optical density of 0.8. Expression was induced with 200 μm IPTG for 2.5 h at 25°C. Bacteria were pelleted at 5000 × g at 4°C for 20 min, lysed, and purified on Protino Ni-TED 2000 columns (Machery-Nagel). Purified proteins were dialyzed overnight at 4°C against storage buffer (PBS, 2 mm EDTA, 10% glycerol, 0.1 mm AEBSF, 0.1 mm TCEP). For affinity purification of binding partners, Ptchd1 peptides were bound to glutathione Sepharose 4 fast flow beads (GE Healthcare, catalog #17-5132–01) following the manufacturer's instructions. For the input fractions, adult male mouse brains (n = 3) were lysed and homogenized in ∼1.5 ml/g in an ice-cold glass homogenizer and centrifuged at 800 × g at 4°C for 10 min. The supernatant was transferred to a new tube and centrifuged at 16,000 × g at 4°C for 15 min. The resulting supernatant was used as the “soluble fraction input”; the pellet, resuspended in lysis buffer, was used as “membrane fraction Input.” GST beads were washed three times with PBS before adding the input fractions. Beads were incubated with protein extracts for 2 h at room temperature, then washed again 3 times with 0.1% Triton X-100 in PBS, followed by elution with 2% sodium deoxycolate in 130 μl. Eluted samples were trypsin digested and prepared for mass spectrometry (MS) following standard protocol.
MS analysis and label-free quantification.
Samples were subjected to LC–MS analysis in triplicate using a dual pressure LTQ-Orbitrap Elite MS (Thermo Fisher Scientific) connected to an electrospray ion source (Thermo Fisher Scientific) as described previously (Glatter et al., 2012). Peptide separation was performed on an EASY nLC-1000 system (Thermo Fisher Scientific) equipped with an RP-HPLC column (75 μm × 30 cm) packed in-house with C18 resin (ReproSil-Pur C18-AQ, 1.9 μm resin; Dr. Maisch GmbH). A linear gradient from 95% solvent A (0.15% formic acid, 2% acetonitrile) and 5% solvent B (98% acetonitrile, 0.15% formic acid) to 28% solvent B over 90 min at a flow rate of 0.2 μl/min was used. Data acquisition mode was set to obtain one high-resolution MS scan in the FT part of the MS at a resolution of 240,000 full-width at half-maximum (at m/z 400), followed by 20 MS/MS scans in the linear ion trap of the most intense ions (TOP20) using rapid scan speed. Unassigned and singly charged ions were excluded from analysis and dynamic exclusion duration was set to 30 s.
MS1-based label-free quantification of MS data was performed using Progenesis QI software (Nonlinear Dynamics version 2.0).
MS raw files were imported into Progenesis QI software and analyzed using the default parameter settings. MS/MS data were exported from the software in “mgf” format and searched with a target/decoy strategy against a database containing forward and reverse sequences of the proteome from Mus musculus (UniProt, 33,984 entries) using MASCOT (version 2.4.1). Search criteria required full tryptic specificity allowing for three missed cleavages. Carbamidomethylation of cysteine was specified in Mascot as a fixed modification. Oxidation of methionine and acetyl of the N terminus were specified in Mascot as variable modifications. Mass tolerance was set to 10 ppm for precursor ions and 0.6 Da for fragment ions. The peptide and protein false discovery rate was set to 1%. Results from the database search were imported into Progenesis QI and the resulting peptide measurement list containing peak area values of identified peptides was exported. Processing and statistical evaluation of peptide and protein quantities between samples was performed using SafeQuant (Glatter et al., 2012). Normalized peptide and protein intensities from SafeQuant analysis were used to calculate intensity ratios, log2 ratios, and q values between experimental conditions for identified proteins.
Synaptosome preparation.
Adult male mouse brain was homogenized with a glass homogenizer in 20 μl/mg tissue containing the following: 0.32 m sucrose, 1 mm NaHCO3, 1 mm MgCl2, 0,5 mm CaCl2 and complete EDTA-free protease inhibitor (Roche). The homogenate was centrifuged at 1400 × g for 10 min. The supernatant was further centrifuged at 16,000 × g for 20 min at 4°C, resulting in the crude synaptosomal pellet and a cytosolic supernatant. The pellet was resuspended in 1 mm NaHCO3 with 0.32 m sucrose and loaded on top of a sucrose step gradient (1 m, 1.2 m sucrose) and centrifuged at 82,500 × g for 2 h. Synaptosomes were collected at the interface between the 1.0 m and 1.2 m sucrose layers. The synaptosome fraction was extracted for 15 min at 4°C in 25 mm Tris-HCl, pH 8.1, 0.32 m sucrose, 1% Triton X-100, and centrifuged at 32,800 × g for 30 min to yield a Triton X-100-soluble and -insoluble (postsynaptic density, PSD) fraction.
Mouse behavior analysis.
Behavioral testing was done as described previously (Leger et al., 2013; Nguyen et al., 2016). WT littermate control and Ptchd1−/y male mice aged between 7 and 9 weeks were used. Behavioral tests were done on three mouse cohorts (total n = 10 Ptchd1−/y, n = 8 WT littermates). Mice were first tested in an open-field arena (50 × 50 × 25 cm) and then the object recognition test was initiated on the following day in the same arena. Before each behavioral test, mice were allowed to habituate to the behavioral room for at least 30 min. After each testing, the arena and objects were cleaned with 70% ethanol. Explorative behavior in the open field was recorded for 7 min with a BASLER Ethovision Camera (Noldus). The distance traveled and mobility state were quantified using Ethovision 10 software (Noldus). The user-defined thresholds for definition of the high mobility state were set to 60% of changed pixel area. The open-field test served as a habituation phase for the novel object recognition test. For the novel object recognition test, recordings were acquired with a Canon camera (LEGRIA HFG10). Animals were allowed to explore 2 identical small Falcon tissue culture flasks (50 ml volume) filled with sand for 5 min and then returned to their home cages. After a 1 h intertrial interval, one flask was replaced with a tower of Lego bricks and duration of interaction was assessed in a second 5 min trial. Exploration time for each object was measured manually by an investigator blinded to the genotype of the mouse. If a mouse exhibited <3 s exploring one object during the initial exposure, it was excluded from the analysis (this was the case for 2 KO animals). Object exploration was defined as the orientation of the mouse snout toward the object and sniffing or touching with the snout within 2 cm distance. Leaning, climbing, or looking over or biting the objects were not considered as exploration time. The position of the objects in the test was counterbalanced between the animals in a group.
Electrophysiology.
Horizontal hippocampal slices (300 μm) were prepared from both hemispheres of age-matched mice (P21–P24) anesthetized with intraperitoneal injection of ketamine/xylazine (100 and 10 mg/kg, respectively). Mice were transcardially perfused with an ice-cold oxygenated (95% O2/5% CO2) N-methyl-d-glucamine (NMDG)-based dissection buffer (Ting et al., 2014) containing the following (in mm): 93 NMDG, 93 HCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 25 glucose, 20 HEPES, 5 Na-ascorbate, 3 Na-pyruvate, 2 thio-urea, 10 MgSO4, and 0.5 CaCl2, pH 7.4, 310 mOsm. Slices were cut with a vibratome (VT1200S; Leica) in oxygenated NMDG dissection buffer. Slices were transferred to NMDG-based oxygenated dissection buffer at 32°C for 15 min and then transferred to room temperature artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 2 CaCl2, 2 MgSO4, 24 NaHCO3, 5 HEPES, and 13 glucose, pH 7.4, 310 mOsm. Slices incubated at room temperature at least 1 h to allow for recovery, then transferred to the recording chamber and perfused (1.5–2.0 ml/min) with oxygenated ACSF at room temperature. Somatic whole-cell recordings were made from granule cells in the dentate gyrus, which were voltage clamped with a Multiclamp 700B amplifier (Molecular Devices) and identified using infrared-differential interference contrast video microscopy digitized by Digidata 1440a (Molecular Devices). Patch pipettes (4–8 MΩ) were filled with voltage clamp (Vc) solution for paired-pulse facilitation, excitation/inhibition ratio, and spontaneous EPSC (sEPSC)/spontaneous IPSC (sIPSC) experiments. The solution contained the following (in mm): 135 CsMeSO3, 10 HEPES, 8 NaCl, 0.3 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 5 QX-314, and 0.1 spermine, 295 mOsm. EPSCs were measured by voltage clamping granule cells to −70 mV, which is near the reversal potential for inhibition. IPSCs were measured by voltage clamping granule cells to 0 mV, which is near the reversal potential for excitation. Data were filtered at 2 kHz, digitized at 10 kHz, and analyzed with Clampfit 10.6 (Molecular Devices). Perforant path afferents were stimulated with a small glass bipolar electrode (Master-9 and ISO-Flex Stimulator; A.M.P.I.). Short-term facilitation was induced with five stimuli of equal intensity presented at variable interstimulus intervals ranging from 25 to 200 ms. Peak amplitudes measured from the responses from stimulations delivered at various frequencies were quantified and normalized to the first peak amplitude in a train. Responses from at least 10 sweeps for each stimulation frequency were quantified to generate the facilitation ratios. AMPAR/NMDAR ratios were recorded in the presence of 10 μm bicuculline (Tocris Bioscience). The average of the maximal peak from 10 evoked EPSCs voltage clamped at −70 mV and the average of the maximal peak of 10 evoked EPSCs voltage-clamped at +40 mV (50 ms after the maximal peak recorded at −70 mV) were used to quantify AMPAR/NMDAR ratios. Then, 2.5 min of recorded granule cell neurons voltage clamped at −70 mV for sEPSC and 0 mV for sIPSC were analyzed with Axograph X software (version 1.5.4) to determine the amplitude and frequency of sEPSCs and sIPSCs. The average of the maximal peak from 10 evoked EPSCs voltage clamped at −70 mV and the average of the maximal peak of 10 evoked EPSCs voltage-clamped at 0 mV were used to quantify excitation/inhibition ratios.
Statistical analysis and experimental design.
Statistical analysis was done with Prism software (GraphPad). Images from immunohistochemical and dye-labeling experiments were analyzed in Fiji (ImageJ) with manual cell or spine counting. Sample sizes are chosen based on previous examples from the literature and variation observed in the experiment. Exclusion criteria were animal weights that differed from the population mean by at least 20% or severe behavioral alterations observed in single animals before the experiment. No animals were excluded in the present study. Blinding was applied as stated for each experimental procedure below.
The qRT-PCR analysis was performed with n = 3 mouse brain tissues for each time point and brain area. Data were analyzed with multiple-comparisons one-way ANOVA. p-values (Tukey's multiple-comparisons test) are shown in Figure 1E.
Figure 1.
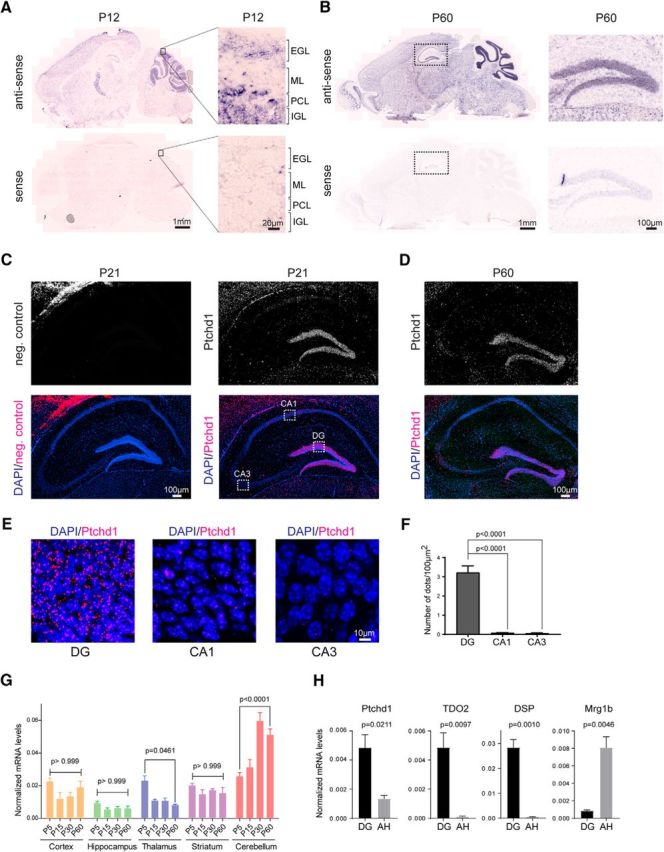
Ptchd1 mRNA distribution in the mouse brain. A, Ptchd1 in situ hybridization on mouse brain sagittal sections at P12 with antisense (up) and sense probes (down). High-magnification view of the developing cerebellum is shown on the right. ML, Molecular layer; PCL, Purkinje cell layer; IGL, internal cell layer. B, Ptchd1 in situ hybridization at P60 with with antisense (up) and sense probes (down). High-magnification view of the dentate gyrus is shown on the right. C, FISH of mouse P21 hippocampus with negative control probe set (left) and Ptchd1 probe set (right). DAPI is shown in blue; Ptchd1 mRNA in magenta. D, FISH of mouse P60 hippocampus with Ptchd1 probe set; nuclei are stained with DAPI in blue; Ptchd1 mRNA in magenta. E, High-magnification view of the dentate gyrus (DG), CA1, and CA3 of the hippocampus (P21 WT mouse coronal section). F, Quantification of fluorescence signal measured as number of dots in whole DG, CA1, and CA3 area (n = 3 P21 mice, n = 22 coronal sections both hemispheres analyzed; mean ± SEM; Student's t test). G, Quantitative RT-PCR assay of Ptchd1 mRNA expression levels during brain development and across different brain regions (n = 3 mice per age, mean ± SEM, one-way ANOVA, Tukey's multiple-comparisons test). H, Quantitative RT-PCR assay of Ptchd1 mRNA expression levels in microdissected dentate gyrus (DG) and Ammon's horn (AH). TDO2 and DSP serve as control to confirm DG tissue enrichment. Mrg1b serves as control to confirm isolation of AH tissue (n = 3 P21 mice, mean ± SEM; Student's t test).
FISH quantification was done with n = 3 WT mice and n = 22 hippocampi (11 coronal sections with 6–8 hippocampi per animal).
The developmental BrdU analysis was performed with n = 5 mice per genotype [for external granule cell layer (EGL), n = 65 Ptchd1−/y, 61 WT sections; for the hippocampal study n = 61 Ptchd1−/y, 61 WT sections]. Cells positive for either BrdU or PH3 were normalized to the EGL area. The P4 hippocampal analysis was normalized to the dentate gyrus area. Cell densities assessed per section were averaged per animal and genotype differences were analyzed with an unpaired t test using the number of animals as “n.”. p-values are shown in Figure 3, B and D.
Figure 3.

Proliferation of neuronal precursors in developing Ptchd1−/y mice. A, WT and Ptchd1−/y (KO) mice were injected with 100 mg/kg BrdU at P4 and analyzed 30 min later. Cerebellar sections were stained for BrdU, PH3, and DAPI. B, Quantification of BrdU- and PH3-positive cell density in the EGL. Cell counts are normalized to the EGL area. Shown are means ± SD from n = 5 mice per genotype; n = 65 Ptchd1−/y, 61 WT sections (light dots); solid dots are means of cell density per animal; unpaired t test. C, Hippocampal sections of P4 WT and Ptchd1−/y (KO) mice stained for BrdU, DAPI. Insets show enlargements of the area marked with a dashed box. D, Quantification of BrdU- and PH3-positive cell density in the DG. Cell density was normalized to the DG area. Shown are means ± SD from n = 5 mice per genotype; n = 61 Ptchd1−/y, 61 WT sections (light dots); solid dots are means of cell density per animal; unpaired t test.
The adult neurogenesis analysis was done with n = 5 mice per genotype (n = 78 Ptchd1−/y sections; n = 80 WT sections). All BrdU/PH3 scoring was done blinded to genotype. Cells in the subgranular zone positive for either BrdU or PH3 were normalized to the length of the subgranular zone. Sections were averaged per animal and genotype differences were analyzed with an unpaired t test; p-values are shown in Figure 4B.
Figure 4.

Adult neurogenesis in Ptchd1−/y mice and Shh binding to Ptchd1. A, WT and Ptchd1−/y (KO) mice were injected with 100 mg/kg BrdU at P60 and analyzed 4 d later (P64). Hippocampal sections of WT and Ptchd1−/y (KO) mice were stained for the DAPI (blue), BrdU (green), and PH3 (red). Insets show enlargements of the area marked with a dashed box. B, Quantification of BrdU- and PH3-positive cell density in the subgranular zone (SGZ) of the dentate gyrus. Cell numbers were normalized to SGZ length. Shown are means ± of n = 156 Ptchd1−/y, 160 WT dentate gyrus regions analyzed (light dots) from n = 5 mice per genotype (solid dots are means of cell density per animal), unpaired t test. C, Shh binding assay on COS cells overexpressing Ptch1-GFP, NPC1-GFP, or Ptchd1-GFP fusions. GFP expression, Shh-Fc immunostaining, and DAPI are shown. D, Quantification of Shh-Fc binding to COS7 cells (n ≥ 103 cells) and to MEF cells (n ≥ 40 cells).
Shh-Fc binding quantification was analyzed on n ≥ 103 COS7 cells and n ≥ 40 MEF cells.
In the diolistic experiments, image acquisition and spine counting analysis were done by an investigator blinded to genotype. For P21, n = 4 Ptchd1−/y and n = 4 WT; n = 44 anatomical sections per genotype. For P60, n = 5 Ptchd1−/y and n = 4 WT; Ptchd1−/y n = 49, WT n = 42 sections. Spine densities per dendritic segment were averaged per animal and genotype differences were analyzed with an unpaired t test; p-values are shown in Figure 6B.
Figure 6.
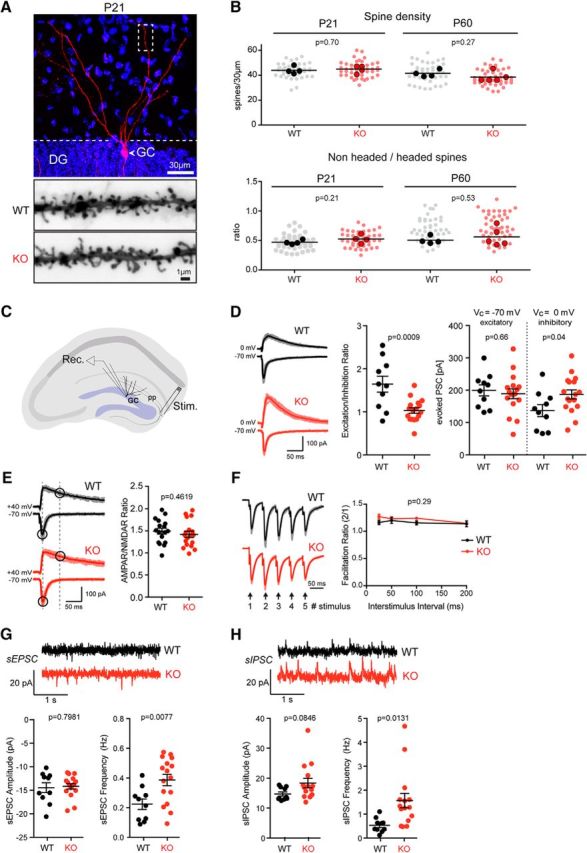
Morphology and physiology of dentate gyrus granule cells in Ptchd1−/y mice. A, Diolistic labeling (red) of a hippocampal dentate granule cell (GC) in a P21 Ptchd1−/y (KO) mouse (DAPI labeling of nuclei in blue). The bottom panel shows an example of a P21 WT and KO DG granule cell distal dendrite. B, Spine density (per 30 μm dendrite segment) and morphology (headed vs nonheaded spines) were quantified for P21 and adult (P60) WT and Ptchd1−/y (KO) mice. Means of 4 WT and 4 Ptchd1−/y (P21) and 4 WT and 5 Ptchd1−/y (P60) mice are displayed (lines and solid dots); mean and unpaired Student's t test. The light dots represent values for individual dendritic segments scored (P21: Ptchd1−/y n = 44, WT n = 44; P60: Ptchd1−/y n = 49, WT n = 42). C, Schematic showing the configuration of whole-cell voltage-clamp recordings made from acute hippocampal slices of P21–P24 mice. Postsynaptic responses in GC neurons were measured after stimulation of the perforant pathway (pp). D, Representative traces of evoked EPSCs and IPSCs recorded from granule cells in acute hippocampal slices show disrupted excitation/inhibition balance in Ptchd1−/y (KO) granule cells (WT, n = 10; KO, n = 17; n = 3 animals for each genotype; mean ± SEM, unpaired t test). E, Representative traces of evoked EPSCs used to calculate AMPAR/NMDAR ratios (WT, n = 17; KO, n = 18; n = 3 animals for each genotype, unpaired t test). F, Normalized evoked postsynaptic responses of WT or Ptchd1−/y GC neurons to five stimuli delivered to the pp with a 50 interstimulus interval, The facilitation ratio of the second EPSC amplitude normalized to the first EPSC amplitude shows normal facilitation across various interstimulus intervals in Ptchd1−/y (KO) GC neurons (stimulus: third/first, p = 0.18; fourth/first, p = 0.43; fifth/first, p = 0.64; WT, n = 8; KO, n = 16; n = 3 animals for each genotype, two-way repeated-measures ANOVA with multiple comparisons). G, Representative traces of sEPSCs from WT or Ptchd1−/y GC neurons that were voltage clamped at −70 mV, sEPSC frequency (WT, 0.22 ± 0.04 Hz; KO, 0.39 ± 0.04 Hz), but not sEPSC amplitude (WT, 0.22 ± 0.04 Hz; KO, 0.39 ± 0.04 Hz), is increased in Ptchd1−/y GC neurons (WT, n = 10; KO, n = 16; n = 3 animals for each genotype, unpaired t test). H, Representative traces of sIPSCs from WT or Ptchd1−/y GC neurons that were voltage clamped at 0 mV. sIPSC frequency (WT, 0.53 ± 0.09 Hz; KO, 1.56 ± 0.3 Hz), but not sIPSC amplitudes (WT, 14.72 ± 0.67 pA; KO, 18.35 ± 1.57), is significantly increased in Ptchd1−/y GC neurons (WT, n = 10; KO, n = 15; n = 3 animals for each genotype, unpaired t test).
The open-field behavioral data were obtained with automated video-tracking software Ethovision 10. The data were analyzed with an unpaired t test; p-values are shown in Figure 7.
Figure 7.

Ptchd1−/y mouse behavioral analysis. A, Open-field track visualization of a KO and a WT mouse. B, Distance traveled in the open-field arena and high mobility state (n = 9 Ptchd1−/y mice; n = 5 WT mice) analyzed by mean, unpaired t test. C, Interaction time and discrimination ratio between familiar object and novel object presented 1 and 24 h after initial exposure (n = 10 Ptchd1−/y mice; n = 8 WT mice), paired t test. D, Habituation curve to familiar object over time (n = 10 Ptchd1−/y mice; n = 8 WT mice) shown as mean ± SEM as shown by two-way ANOVA.
All behavioral tests were analyzed by an investigator blinded to the genotype. For the novel object recognition test, the initial exploration time was analyzed with an unpaired t test. Interaction times with objects A and B were analyzed with a repeated-measures two-way ANOVA. Discrimination ratios were analyzed with an unpaired t test. The habituation time to object A was analyzed with a repeated-measures two-way ANOVA (see Fig. 7G).
The electrophysiological characterization was performed with n ≥ 10 cells and n = 3 mice per genotype by an investigator blinded to genotype. An unpaired t test was used to assess significance in excitation/inhibition ratios, sEPSC, and sIPSC data. A two-way repeated-measures ANOVA with a Sidak's multiple-comparisons test was used to assess significance in short-term plasticity experiments.
Results
Developmental regulation of Ptchd1 expression in the mouse brain
To identify cell populations in the mouse brain with high Ptchd1 expression, we used in situ hybridization with a probe directed against the 5′-UTR (exon 1) of mouse Ptchd1. In developing mouse brain (P12), we observed significant hybridization signals across the entire brain, with high signals in the thalamic reticular nucleus and the cerebellum. In the cerebellum, there was significant expression in the external germinal layer, a site where granule cell precursors proliferate before they initiate their migration toward the internal granular layer (Fig. 1A). In adult mice (P60) we found highest hybridization signals in the internal granular layer of the cerebellum, the reticular nucleus of the thalamus, and the dentate gyrus of the hippocampus, consistent with a previous report (Wells et al., 2016; Fig. 1B). Previous work reported synaptic deficits in the hippocampus of Ptchd1 mutant mice (Ung et al., 2017). Therefore, we sought to obtain additional, more quantitative insights into Ptchd1 expression in this brain area. FISH with Ptchd1-specific probes revealed high Ptchd1 mRNA expression in dentate granule cells of 21-d-old mice, whereas expression in the CA1–3 pyramidal cells was very low (Fig. 1C,E,F). Similarly, high hybridization signals were observed in the dentate gyrus of adult (P60) mice (Fig. 1D).
qPCR measurements for Ptchd1 mRNA across multiple brain areas confirmed broad expression and developmental regulation of the Ptchd1 transcript (Fig. 1G). In the cerebellum, there was a 2- to 3-fold upregulation of Ptchd1 relative to Gapdh from P15–P30 (Fig. 1G; a similar upregulation was observed when Hprt was used as a normalizer). To further confirm differential expression of Ptchd1 mRNA across the principal cell types of the hippocampal circuit, we microdissected dentate gyrus and Ammon's horn from P21 mice and performed qPCR analyses (Fig. 1H). Again, we observed high expression of Ptchd1 mRNA in dentate gyrus and low expression in Ammon's horn preparations. Together, these observations support significant expression of Ptchd1 mRNAs in the developing and adult brain, and particularly prominent expression in the thalamic reticular nucleus, hippocampal dentate granule cells, and cerebellar granule neurons.
To examine essential functions of Ptchd1, we generated a conditional KO allele in which exon 2 is flanked by loxP sites (Fig. 2A). We then generated a germline Ptchd1 mutant (Ptchd1−/y) by crossing with cre-deleter mice. Using RT-PCR, we observed a loss of exon 2-containing sequences and detected transcripts containing exon 1 sequences joined to exon 3 (Fig. 2B). Due to a frame shift, these transcripts carry a translational stop codon 93 aa into exon 3, resulting in the loss of 9 of the 12 transmembrane domains and disruption of the sterol-sensing domain of the Ptchd1 protein. To examine the impact of exon 2 deletion on Ptchd1 protein expression, we raised polyclonal antibodies and probed Ptchd1 protein levels in Western blots. In transfected HEK293 cells, Ptchd1 antibodies specifically recognized EGFP-tagged Ptchd1 protein of 120 kDa (Fig. 2C). In mice, we detected a 95 kDa protein in lysates from adult mouse WT hippocampus and cerebellum that was absent in male Ptchd1−/y mice (Fig. 2D). Using the same antibody, we further confirmed broad Ptchd1 protein expression and high expression in the mouse cerebellum (Fig. 2D).
Figure 2.

Ptchd1 KO mouse generation and validation. A, Ptchd1 KO strategy, in which the Ptchd1 gene was modified by replacing exon 2 with a cassette containing a gene trap lacZ-Neo cassette (engrailed 2 splicing acceptor) flanked by Frt sites and additional loxP sites flanking Exon 2. Conditional Ptchd1 floxed animals were obtained by breeding to Flip deleter mice, resulting in an excision of the trap cassette. The Ptchd1 KO allele was generated by crossing Ptchd1 floxed animals with CMVcre deleter mice. B, RT-PCR of WT and Ptchd1−/y cDNA showing successful removal of exon 2 and splicing of exon 1 into exon 3, resulting into a frame shift. Splice junctions were confirmed by DNA sequencing. C, Specificity of a Ptchd1-specific antibody was confirmed by Western blot analysis of HEK293T cells overexpressing a Ptchd1-EGFP fusion protein. D, Successful ablation of Ptchd1 protein in KO animals was verified by Western blot analysis of hippocampal tissue lysate (left) and cerebellar tissue lysate (middle). Right, Ptchd1 protein relative expression levels across brain regions in adult P60 mouse. Similar results were obtained in three independent experiments.
Proliferation of neuronal precursors in Ptchd1−/y mice
Ptchd1 has been suggested to act as an Shh receptor (Noor et al., 2010). During early postnatal development, cerebellar granule cell precursors proliferate in response to Purkinje-cell-derived Shh (Wechsler-Reya and Scott, 1999) and homozygous or heterozygous KO of the Shh receptor Patched 1 (Ptch1) results in granule cell precursor overproliferation (Goodrich et al., 1997; Yang et al., 2008). Given the high expression of Ptchd1 in granule cell precursors during early postnatal development, we tested whether precursor proliferation was altered in Ptchd1−/y mice. We injected mice at P4, a time when Shh-dependent proliferation is high, with 100 mg/kg BrdU and then probed the number of BrdU-positive cells 30 min after injection. Using this protocol, the density of BrdU-positive cells and the density of cells immunopositive for the proliferation marker PH3 were unchanged in Ptchd1−/y mice (Fig. 3A,B).
A second neuronal precursor population that is dependent on Shh signaling during development is the granule cells in the dentate gyrus of the hippocampus (Han et al., 2008; Li et al., 2013). Therefore, we quantified BrdU- and PH3-positive cells in WT and Ptchd1−/y mice. Similar to the cerebellum, BrdU incorporation and density of PH3-positive cells was not changed significantly (Fig. 3C,D). We then tested BrdU incorporation in adult (P60) dentate gyrus because Shh has also been implicated in the maintenance of adult neural stem cells in this area (Lai et al., 2003). However, in Ptchd1−/y mice, we did not detect a significant change in the density of PH3- or BrdU-positive cells 4 d after BrdU injection (Fig. 4A,B).
Our results thus far suggested that the absence of Ptchd1 has no dramatic effect on neuronal precursor proliferation in vivo in cell populations that undergo Shh-dependent proliferation. To determine directly whether Ptchd1 may interact with Shh, we tested whether Shh can bind in vitro to cells expressing Ptchd1 protein. We observed robust binding of Shh to COS7 and mouse embryonic fibroblast (MEF) cells expressing Ptch1, but no binding to cells expressing Ptchd1 nor NPC1 under identical experimental conditions (Fig. 4C). Therefore, 50% of COS7 cells and 78% of MEFs expressing Ptch1 exhibited clearly recognizable Shh surface binding, whereas none of the cells expressing Ptchd1, GFP, or NPC1 did (Fig. 4D). In aggregate, these experiments suggest that expression of Ptchd1 is not essential for the Shh-dependent proliferation or maintenance of neuronal precursors in the developing cerebellum or the postnatal dentate gyrus.
Ptchd1 cytoplasmic tail interacts with scaffolding proteins and the retromer complex
Ptchd1 is predicted to form a 12 pass transmembrane protein with a PDZ-binding motif at the C-terminal tail (Fig. 5A). To identify proteins that may contribute to Ptchd1 localization or function, we performed affinity purifications from whole-brain extracts with the 43 aa cytoplasmic C terminus of Ptchd1 either including (WT) or lacking (ΔPDZ) the last four residues that contain the PDZ-binding motif. As a negative control, we used a protein containing an extracellular sequence of Ptchd1 (ECD; Fig. 5A,B). Proteins retained on hexahistidine–GST fusions of these baits were identified by shotgun MS and specificity of the interaction was assessed by comparison with the ECD control protein (Fig. 5C,D; for complete MS data, see Figure 5-1). Quantitative analysis identified a number of proteins that were enriched on the WT C terminus, whereas other candidate binding partners were enriched on both WT and ΔPDZ tails. Twelve of 13 proteins recovered preferentially on the WT Ptchd1 tail compared with the ΔPDZ mutant-containing PDZ domains, including common components of the PSD (Dlg1-4, Magi1 and 3, and Lin7). Interestingly, we recovered three components of the retromer complex involved in regulating dendritic protein trafficking between endosomal compartments and the plasma membrane (Choy et al., 2014): Sorting-nexin 27 (which contains a PDZ domain) was recovered selectively on the WT Ptchd1 tail and VPS26B and VPS35, which interacted with both the WT and the ΔPDZ tail. In addition, there was a notable number of ribosomal proteins recovered both on WT and ΔPDZ bait proteins (RL5, RL7, RL8, RL18A, RL27, RL31, RL32, RL37, and Rpl31). Whether these ribosomal proteins represent physiological binding partners or if they are recruited nonspecifically through the stretch of highly charged amino acids in the Ptchd1 cytoplasmic tail remains to be explored. Using Western blotting, we validated our MS approach confirming the binding of Dgl4 (also known as PSD95) and VPS35 to the recombinant Ptchd1 C terminus. Endogenous DLG4 and VPS35 from membrane and soluble mouse brain fractions were recovered specifically on the GST fusion proteins containing the cytoplasmic but not the extracellular, negative control sequences of Ptchd1 (Fig. 5E). Binding of DLG4 was dependent on the PDZ-binding motif in Ptchd1, whereas VPS35 binding was not (Fig. 5E).
Figure 5.
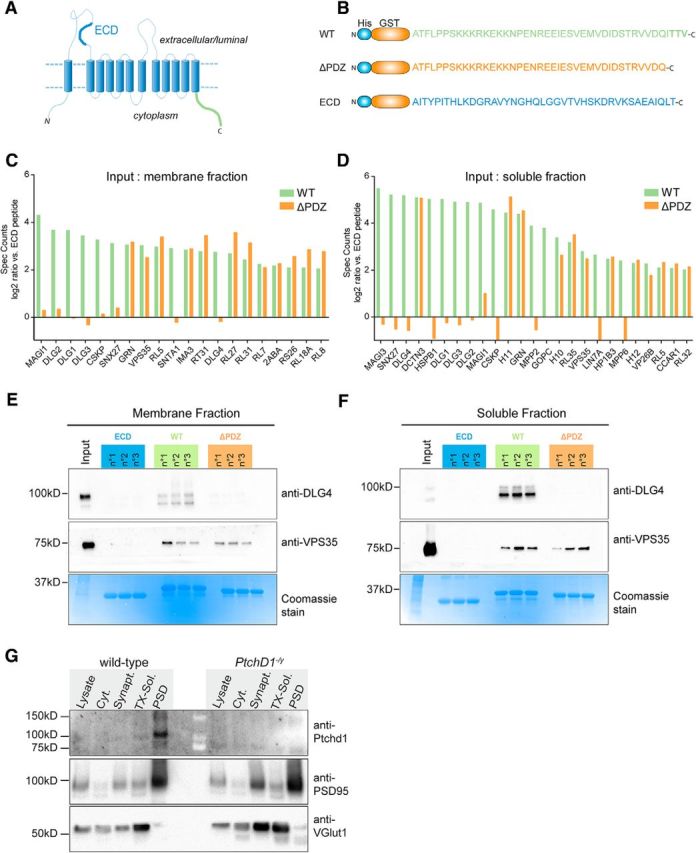
Ptchd1 cytoplasmic tail interacts with scaffolding proteins and retromer complex. A, Schematic representation of predicted Ptchd1 protein topology. B, Three recombinant hexahistidine-GST fusion proteins used for the pull-down analysis: WT, Ptchd1 C-terminal sequence (green); ΔPDZ, C-terminal lacking the predicted PDZ binding motif (orange); and ECD, negative control sequence from the first extracellular loop of the Ptchd1 (blue). C, Quantitative shotgun MS data of proteins recovered in Ptchd1 pull-down assays using a soluble brain membrane fraction as input. D, Quantitative shotgun MS data of proteins recovered in Ptchd1 pull-down assays using a soluble brain protein fraction as input. All proteins identified with at least 3 peptides, 400 spectra counts, and enriched at least 4-fold on the WT Ptchd1 bait compared with the negative control (His-GST-ECD) are displayed. The graphs show enrichment (log2 ratio) compared with the ECD fraction (n = 3 purifications, each measured in duplicate). The complete data from the MS analysis is shown in the extended data (Figure 5-1). E, F, Validation of Ptchd1-DLG4 (PSD95) and Ptchd1-VPS35 interaction by Western blotting. Ptchd- interacting proteins were isolated by GST pull-down with ECD, WT, and ΔPDZ baits from adult mouse membrane fractions or soluble brain protein extracts. The bottom panel shows the respective protein fractions of the pull-down assay labeled with Coomassie dye. G, Synaptosome preparation from WT and Ptchd1−/y brains probed with Ptchd1, Dlg4/PSD95, and vGlut1 antibodies. Twenty micrograms of total protein analyzed from total lysate, cytosolic, synaptosome, Triton X-100-soluble synaptosome, and Triton X-100-insoluble synaptosome (PSD) fractions are shown. Molecular weight markers are indicated in kDa.
Excel tables with complete dataset from mass-spectrometric analysis of Ptchd1-binding proteins. Sheet 1 contains data for pull-downs from cytosolic protein fraction. Sheet 2 contains data for pull-down from solubilized membrane fraction. Log2 ratios, q and p values shown for GST_WT and GST_deltaPDZ peptides are calculated over the control peptide form the extracellular domain of Ptchd1 (GST_ECD). Download Figure 5-1, XLSX file (639.5KB, xlsx)
Considering the association with PSD proteins and retromer, we tested whether endogenous Ptchd1 protein is concentrated in PSD preparations from mouse brain. Synaptosome fractionations revealed an enrichment of Ptchd1 in the PSD obtained from adult mouse brain (Fig. 5F; Ptchd1−/y brains were analyzed to control for antibody specificity). These results demonstrate a biochemical interaction of Ptchd1 with postsynaptic trafficking proteins in the mouse brain.
Loss of Ptchd1 disrupts synaptic transmission in the dentate gyrus
Given the high expression of Ptchd1 in dentate granule cells and its association with the PSD proteins, we tested whether the density and differentiation of glutamatergic spine synapses might be altered in the hippocampus of Ptchd1−/y mice. Dentate granule cells in perfusion-fixed tissue from adult and P21 Ptchd1−/y mice were labeled by diolistics (Gan et al., 2000) and then spine density and morphology were assessed (Fig. 6A,B). Using this approach, we did not detect any difference in the overall density of dendritic protrusions in Ptchd1−/y neurons (Fig. 6B). Further, the fraction of headed, spine-like protrusions was unaltered (Fig. 6B). Therefore, Ptchd1 is dispensable for the formation of dendritic spines in dentate granule cells at P21 and in adult mice.
To determine whether Ptchd1 enrichment in dentate granule cells is associated with synaptic dysfunction, we examined synaptic transmission using whole-cell recordings in acute hippocampal slices from P21–P24 mice. EPSCs and IPSCs were evoked by stimulation of the perforant pathway (Fig. 6C). Interestingly, Ptchd1−/y dentate granule cells exhibited a profound reduction in the excitation/inhibition ratio (Fig. 6D). AMPAR:NMDAR ratios and paired-pulse ratios of evoked glutamatergic transmission were unchanged (Fig. 6E,F). We hypothesized that the reduced excitation/inhibition ratio may have been caused by an increase in inhibition or a decrease in excitation, so we examined basal activity by measuring sEPSCs or sIPSCs. Surprisingly, we observed a significant increase in the frequency of both spontaneous excitatory and inhibitory events in Ptchd1−/y dentate granule cells (Fig. 6G,H). Together, these data suggest that loss of Ptchd1 results in severe alterations in synaptic function in the dentate gyrus.
Ptchd1−/y mice show deficits in hippocampus-related behavioral tasks
Considering the alterations in synaptic transmission, we further explored behavioral phenotypes in our new line of Ptchd1 KO mice. In open-field tests, Ptchd1−/y mice covered the same distance as littermate controls; however, the mutants frequently exhibited bouts of high mobility largely due to jumping behaviors (Fig. 7B). We then used an object recognition test, which in part depends on hippocampal function (Cohen and Stackman, 2015). Animals are exposed to novel and familiar objects in a test arena. The time exploring the objects is then scored 1 and 24 h after the initial exposure. Although WT mice showed a clear preference for novel over familiar objects, we observed a marked loss of short-term (1 h) and long-term (24 h) memory in Ptchd1−/y mice (Fig. 7C): notably, the mean object interaction time during the first (acquisition) trial was the same for WT and mutant mice (Fig. 7D). During the retest, WT mice showed decreased interaction with the familiar object and preference for the novel object. In contrast, Ptchd1−/y mice did not exhibit significant habituation to the familiar object in the 1 or 24 h retest (Fig. 7D), thus resulting in a lack of discrimination of novel and familiar stimuli. These results indicate a cognitive deficit in this line of Ptchd1−/y mice.
Discussion
In this study, we generated and characterized a new Ptchd1 KO mouse line to explore the cellular and molecular functions of the Ptchd1 protein. Previous work reported a specific impairment in the thalamic reticular nucleus of Ptchd1−/y mice and ADHD-like behavioral deficits (Wells et al., 2016). These deficits are linked to an impairment in SK-channel conductance and a function of Ptchd1 in somatostatin-positive interneurons. However, it remains unknown whether Ptchd1 loss-of-function results in these changes due to a modification of neuronal cell fate, precursor proliferation, or other cellular functions in postmitotic neurons.
An important goal of our work was testing whether loss of Ptchd1 results in Shh signaling-related phenotypes in vivo. The hypothesis that Ptchd1 acts as a Shh receptor is based on the presence of a sterol-sensing domain and reporter assays in which exogenous Ptchd1 was overexpressed (Noor et al., 2010). A major function for Shh-Ptch1 signaling in the postnatal brain is the control of neuronal precursor proliferation in the developing cerebellum, developing dentate granule cells, and adult neural precursors in the dentate gyrus. Therefore, we tested precursor proliferation and maintenance in the hippocampal dentate gyrus and the external germinal layer of the cerebellum. However, we did not observe significant changes in Ptchd1 mutants. It is possible that the presence of Ptch1 compensates for a loss of Ptchd1, thus obscuring a potential phenotype. We consider this unlikely for the following reasons: (1) heterozygous deletion of Ptch1 results in precursor overproliferation and medulloblastomas in the mouse cerebellum, indicating that even partial loss of Ptch1-signaling results in severe Shh-related phenotypes; and (2) we did not observe binding of recombinant Shh to Ptchd1 under conditions in which an interaction with Ptch1 was readily detectable. Notably, Ptchd1-like proteins are also found in nonmammalian species, including C. elegans, which do not encode recognizable Shh orthologs in their genomes (C. elegans Sequencing Consortium, 1998). Therefore, we favor the interpretation that Ptchd1 has different, Shh-independent functions in the developing and adult mouse brain.
Our unbiased proteomic approach to identifying Ptchd1-interacting proteins confirmed binding to PDZ-domain-containing scaffolding molecules (Ung et al., 2017). This was expected due to the C-terminal consensus sequence ITTV for PDZ domain interactions. In addition, we discovered an unexpected interaction with the retromer complex consisting of VPS35, VPS26B, and SNX27. SNX27 contains a PDZ domain and its interaction depends on the C-terminal PDZ-binding motif of Ptchd1. However, VPS35 and VPS26B were also recovered on the ΔPDZ bait protein, highlighting multiple interactions sites for Ptchd1 and the retromer complex. SNX27 and VPS35 have been implicated in membrane protein trafficking between dendritic endosomes and the plasma membrane, in particular postsynaptic glutamate receptors and β-adrenergic receptors (Wang et al., 2013; Choy et al., 2014). Therefore, retromer may be involved in trafficking of Ptchd1 between endosomal and cell surface compartments including the postsynaptic compartment.
Supporting a link to synaptic trafficking proteins, we observed a critical role of Ptchd1 in the excitatory/inhibitory balance in dentate granule cells. The frequency of spontaneous excitatory and inhibitory events in dentate granule cells was increased. Previous work demonstrated alterations in SK channel activity in somatostatin-positive interneurons in the thalamus (Wells et al., 2016). Therefore, the alterations in spontaneous excitatory and inhibitory events in the dentate gyrus may result from Ptchd1-dependent phenotypes in glutamatergic and GABAergic interneurons. Determining the cellular and/or synaptic source of this electrophysiological phenotype will require conditional ablation of Ptchd1 from specific cell populations in the dentate gyrus.
In Ptchd1−/y dentate gyrus, we did not observe alterations in facilitation ratios at glutamatergic synapses or any loss of postsynaptic spine structures. This was surprising because a recent report indicated a 33% reduction in excitatory synapse density in Ptchd1−/y hippocampus (Ung et al., 2017). One difference in our study is the focus on dentate granule cells, which express the highest level of Ptchd1 mRNA in the hippocampus. In contrast, Ung et al. (2017) focused on the CA region, which, according to our expression analysis, expresses only very low levels of Ptchd1 transcripts. Another difference between the studies is the mutant allele used. Similar to Wells et al. (2016), we used a germline deletion of exon 2. Ung et al. (2017) used a mutant allele where exon 2 is replaced with a gene trap cassette. However, we consider it more likely that there is a differential requirement for Ptchd1 in synapse development across cell types with high and low Ptchd1 expression levels such as dentate granule and CA1 cells. Moreover, some phenotypes may emerge as secondary effects due to functional alterations in upstream circuit elements. Analysis of conditional mutants may resolve this in the future.
A survey of 23 subjects with PTCHD1 mutations reported intellectual disability, ASD, and motor alterations as phenotypes that were shared across significant subgroups of these patients (Chaudhry et al., 2015). Moreover, several patients exhibited ADHD and hyperactivity. The increase in high-mobility bouts and jumping of our Ptchd1−/y mice observed in open-field assays is reminiscent of the hyperactivity phenotype reported previously for additional mouse models (Wells et al., 2016; Ung et al., 2017). Conditional ablation of Ptchd1 in somatostatin-positive interneurons replicates the hyperactivity phenotype of the mice, but did not replicate the learning deficits seen for global KO mice in an inhibitory avoidance task (Wells et al., 2016). The defect in objection recognition memory identified in our mouse line highlights a further cognitive deficit in this rodent model. Future work will be aimed at illuminating whether this defect is due to hippocampal dysfunction.
Footnotes
This work was supported by the National Center of Competence in Research Synapsy, the Swiss National Science Foundation (P.S.), and European Autism Interventions–A Multicentre Study for Developing New Medications (EU-AIMS), which receives support from the Innovative Medicines Initiative Joint Undertaking. A.M.G was supported by an EMBO long-term fellowship (ALTF-70-2015) cofunded by LTFCOFUND2013 (Grant GA-2013-609409) and an EMBO advanced long-term fellowship (Grant aALTF-760-2016). Work done in the Charron laboratory was supported by grants from the Canadian Institutes of Health Research and the Canada Foundation for Innovation. F.C. holds the Canada Research Chair in Developmental Neurobiology. We thank Caroline Bornmann and Laetitia Burkle for excellent technical assistance, Harald Witte for construction of a Ptchd1 expression vector and for establishing the diolistic labeling method, Bernhard Bettler for generously sharing equipment, Patrick Mehlen for sharing the dominant negative caspase 9 construct, Tevye Stachniak for useful discussion and the Imaging Core Facility and Proteomic Core Facility of the Biozentrum for expert assistance.
The authors declare no competing financial interests.
References
- Chaudhry A, et al. (2015) Phenotypic spectrum associated with PTCHD1 deletions and truncating mutations includes intellectual disability and autism spectrum disorder. Clin Genet 88:224–233. 10.1111/cge.12482 [DOI] [PubMed] [Google Scholar]
- Cheng HJ, Flanagan JG (2001) Cloning and characterization of RTK ligands using receptor-alkaline phosphatase fusion proteins. Methods Mol Biol 124:313–334. [DOI] [PubMed] [Google Scholar]
- Choy RW, Park M, Temkin P, Herring BE, Marley A, Nicoll RA, von Zastrow M (2014) Retromer mediates a discrete route of local membrane delivery to dendrites. Neuron 82:55–62. 10.1016/j.neuron.2014.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Stackman RW Jr (2015) Assessing rodent hippocampal involvement in the novel object recognition task: a review. Behav Brain Res. 285:105–117. 10.1016/j.bbr.2014.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282:2012–2018. 10.1126/science.282.5396.2012 [DOI] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Won H, Stein JL, Geschwind DH (2016) Advancing the understanding of autism disease mechanisms through genetics. Nat Med 22:345–361. 10.1038/nm.4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes ML, McAllister AK (2015) Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat Rev Neurosci 16:469–486. 10.1038/nrn3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filges I, Röthlisberger B, Blattner A, Boesch N, Demougin P, Wenzel F, Huber AR, Heinimann K, Weber P, Miny P (2011) Deletion in Xp22.11: PTCHD1 is a candidate gene for X-linked intellectual disability with or without autism. Clin Genet 79:79–85. 10.1111/j.1399-0004.2010.01590.x [DOI] [PubMed] [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW (2000) Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron 27:219–225. 10.1016/S0896-6273(00)00031-3 [DOI] [PubMed] [Google Scholar]
- Glatter T, Ludwig C, Ahrné E, Aebersold R, Heck AJ, Schmidt A (2012) Large-scale quantitative assessment of different in-solution protein digestion protocols reveals superior cleavage efficiency of tandem Lys-C/trypsin proteolysis over trypsin digestion. J Proteome Res 11:5145–5156. 10.1021/pr300273g [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenković L, Higgins KM, Scott MP (1997) Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277:1109–1113. 10.1126/science.277.5329.1109 [DOI] [PubMed] [Google Scholar]
- Hagihara H, Toyama K, Yamasaki N, Miyakawa T (2009) Dissection of hippocampal dentate gyrus from adult mouse. J Vis Exp 33: pii: 1543. 10.3791/1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A (2008) Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci 11:277–284. 10.1038/nn2059 [DOI] [PubMed] [Google Scholar]
- Izzi L, Lévesque M, Morin S, Laniel D, Wilkes BC, Mille F, Krauss RS, McMahon AP, Allen BL, Charron F (2011) Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev Cell 20:788–801. 10.1016/j.devcel.2011.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai K, Kaspar BK, Gage FH, Schaffer DV (2003) Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci 6:21–27. [DOI] [PubMed] [Google Scholar]
- Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, Freret T (2013) Object recognition test in mice. Nat Protoc 8:2531–2537. 10.1038/nprot.2013.155 [DOI] [PubMed] [Google Scholar]
- Li G, Fang L, Fernández G, Pleasure SJ (2013) The ventral hippocampus is the embryonic origin for adult neural stem cells in the dentate gyrus. Neuron 78:658–672. 10.1016/j.neuron.2013.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, et al. (2008) Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 82:477–488. 10.1016/j.ajhg.2007.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mille F, Thibert C, Fombonne J, Rama N, Guix C, Hayashi H, Corset V, Reed JC, Mehlen P (2009) The Patched dependence receptor triggers apoptosis through a DRAL-caspase-9 complex. Nat Cell Biol 11:739–746. 10.1038/ncb1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TM, Schreiner D, Xiao L, Traunmüller L, Bornmann C, Scheiffele P (2016) An alternative splicing switch shapes neurexin repertoires in principal neurons versus interneurons in the mouse hippocampus. eLife 5: pii: e22757. 10.7554/eLife.22757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor A, et al. (2010) Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Sci Transl Med 2:49ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A (1993) A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100:431–440. 10.1007/bf00267823 [DOI] [PubMed] [Google Scholar]
- Staffend NA, Meisel RL (2011) DiOlistic labeling in fixed brain slices: phenotype, morphology, and dendritic spines. Curr Protoc Neurosci Chapter 2:Unit 2.13. 10.1002/0471142301.ns0213s55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, Feng G (2014) Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol 1183:221–242. 10.1007/978-1-4939-1096-0_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrico B, Fernàndez-Castillo N, Hervás A, Milà M, Salgado M, Rueda I, Buitelaar JK, Rommelse N, Oerlemans AM, Bralten J, Freitag CM, Reif A, Battaglia A, Mazzone L, Maestrini E, Cormand B, Toma C (2015) Contribution of common and rare variants of the PTCHD1 gene to autism spectrum disorders and intellectual disability. Eur J Hum Genet 23:1694–1701. 10.1038/ejhg.2015.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ung DC, Iacono G, Meziane H, Blanchard E, Papon M-A, Selten M, van Rhijn J-R, Montjean R, Rucci J, Martin S, Fleet A, Birling M-C, Marouillat S, Roepman R, Selloum M, Lux A, Thepault R-A, Hamel P, Mittal K, et al. (2017) Ptchd1 deficiency induces excitatory synaptic and cognitive dysfunctions in mouse. Mol Psychiatry. Advance online publication. doi: 10.1038/mp.2017.39. 10.1038/mp.2017.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y (2012) RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 14:22–29. 10.1016/j.jmoldx.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. (2013) Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat Med 19:473–480. 10.1038/nm.3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22:103–114. 10.1016/S0896-6273(00)80682-0 [DOI] [PubMed] [Google Scholar]
- Wells MF, Wimmer RD, Schmitt LI, Feng G, Halassa MM (2016) Thalamic reticular impairment underlies attention deficit in Ptchd1(Y/-) mice. Nature 532:58–63. 10.1038/nature17427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schüller U, Machold R, Fishell G, Rowitch DH, Wainwright BJ, Wechsler-Reya RJ (2008) Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 14:135–145. 10.1016/j.ccr.2008.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Excel tables with complete dataset from mass-spectrometric analysis of Ptchd1-binding proteins. Sheet 1 contains data for pull-downs from cytosolic protein fraction. Sheet 2 contains data for pull-down from solubilized membrane fraction. Log2 ratios, q and p values shown for GST_WT and GST_deltaPDZ peptides are calculated over the control peptide form the extracellular domain of Ptchd1 (GST_ECD). Download Figure 5-1, XLSX file (639.5KB, xlsx)