

Abstract
GABAB receptors are the G-protein coupled receptors for the main inhibitory neurotransmitter in the brain, GABA. GABAB receptors were shown to associate with homo-oligomers of auxiliary KCTD8, KCTD12, KCTD12b, and KCTD16 subunits (named after their T1 K+-channel tetramerization domain) that regulate G-protein signaling of the receptor. Here we provide evidence that GABAB receptors also associate with hetero-oligomers of KCTD subunits. Coimmunoprecipitation experiments indicate that two-thirds of the KCTD16 proteins in the hippocampus of adult mice associate with KCTD12. We show that the KCTD proteins hetero-oligomerize through self-interacting T1 and H1 homology domains. Bioluminescence resonance energy transfer measurements in live cells reveal that KCTD12/KCTD16 hetero-oligomers associate with both the receptor and the G-protein. Electrophysiological experiments demonstrate that KCTD12/KCTD16 hetero-oligomers impart unique kinetic properties on G-protein-activated Kir3 currents. During prolonged receptor activation (one min) KCTD12/KCTD16 hetero-oligomers produce moderately desensitizing fast deactivating K+ currents, whereas KCTD12 and KCTD16 homo-oligomers produce strongly desensitizing fast deactivating currents and nondesensitizing slowly deactivating currents, respectively. During short activation (2 s) KCTD12/KCTD16 hetero-oligomers produce nondesensitizing slowly deactivating currents. Electrophysiological recordings from hippocampal neurons of KCTD knock-out mice are consistent with these findings and indicate that KCTD12/KCTD16 hetero-oligomers increase the duration of slow IPSCs. In summary, our data demonstrate that simultaneous assembly of distinct KCTDs at the receptor increases the molecular and functional repertoire of native GABAB receptors and modulates physiologically induced K+ current responses in the hippocampus.
SIGNIFICANCE STATEMENT The KCTD proteins 8, 12, and 16 are auxiliary subunits of GABAB receptors that differentially regulate G-protein signaling of the receptor. The KCTD proteins are generally assumed to function as homo-oligomers. Here we show that the KCTD proteins also assemble hetero-oligomers in all possible dual combinations. Experiments in live cells demonstrate that KCTD hetero-oligomers form at least tetramers and that these tetramers directly interact with the receptor and the G-protein. KCTD12/KCTD16 hetero-oligomers impart unique kinetic properties to GABAB receptor-induced Kir3 currents in heterologous cells. KCTD12/KCTD16 hetero-oligomers are abundant in the hippocampus, where they prolong the duration of slow IPSCs in pyramidal cells. Our data therefore support that KCTD hetero-oligomers modulate physiologically induced K+ current responses in the brain.
Keywords: G-protein coupled receptor, GABA-B, GPCR, KCTD12, KCTD16, Kir3
Introduction
GABAB receptors are the G-protein coupled receptors (GPCRs) for the inhibitory neurotransmitter GABA. GABAB receptors are expressed throughout the brain and influence synaptic transmission by regulating the activity of ion channels and adenylate cyclases (Couve et al., 2000; Chalifoux and Carter, 2011; Gassmann and Bettler, 2012). Postsynaptic GABAB receptors activate inwardly rectifying K+ channels (Kir3; also known as G-protein-activated inwardly rectifying K+ channels [GIRK]) that generate slow IPSCs (sIPSCs) (De Koninck and Mody, 1997; Lüscher et al., 1997; Lüscher and Slesinger, 2010; Booker et al., 2013). GABAB receptors comprise principal and auxiliary subunits that assemble into molecularly and functionally distinct receptor subtypes (Schwenk et al., 2010, 2016; Gassmann and Bettler, 2012). The principal subunits GABAB1a, GABAB1b, and GABAB2 are seven-transmembrane proteins and generate fully functional heterodimeric GABAB(1a,2) and GABAB(1b,2) receptors (Möhler and Fritschy, 1999; Pin et al., 2004; Monnier et al., 2011). The auxiliary subunits KCTD8, KCTD12, KCTD12b, and KCTD16 (herein collectively referred to as KCTDs) are cytosolic proteins that bind as homo-tetramers (Schwenk et al., 2010; Correale et al., 2013) or homo-pentamers (Smaldone et al., 2016) to the GABAB2 subunit (Bartoi et al., 2010; Schwenk et al., 2010). KCTD8, KCTD12, KCTD12b, and KCTD16 comprise a clade of a larger family of KCTD proteins that all contain a T1 domain, which in voltage-gated K+ channels mediates tetramerization (Liu et al., 2013; Skoblov et al., 2013). The T1 domain of the KCTDs interacts with the C-terminal intracellular domain of the GABAB2 subunit, in which tyrosine-902 (Y902) is mandatory for interaction (Schwenk et al., 2010; Correale et al., 2013). The KCTDs additionally contain a conserved H1 homology domain, which in KCTD12 induces a pronounced desensitization of the GABAB receptor response by uncoupling the βγ subunits of the activated G-protein from the effector channel (Seddik et al., 2012; Turecek et al., 2014). Selectively KCTD8 and KCTD16 feature a conserved H2 homology domain that prevents the receptor desensitization induced by the H1 domain (Seddik et al., 2012).
The KCTDs exhibit overlapping expression patterns in the brain (Schwenk et al., 2010; Metz et al., 2011; Hayasaki et al., 2012) and, in principle, could form hetero-oligomers in various neuronal populations. Here we demonstrate that KCTD12 and KCTD16 indeed form hetero-oligomers through self-association of their T1 and H1 domains. KCTD12/KCTD16 hetero-oligomers directly bind to the receptor-associated G-protein and confer unique desensitization and deactivation kinetics to GABAB receptor-induced K+ currents. In hippocampal neurons, KCTD12/KCTD16 hetero-oligomers increase the duration of GABAB receptor-induced sIPSCs. In summary, our data show that KCTD hetero-oligomers contribute to functionally distinct GABAB receptor responses in the brain.
Materials and Methods
Mice.
Kctd12−/− (RRID:MGI:5756048) and Kctd16−/− knock-out mice and control littermates of either sex were generated as described previously (Metz et al., 2011; Cathomas et al., 2015). Kctd12/16−/− double knock-out mice of either sex were obtained from mating of Kctd12−/− and Kctd16−/− mice. All experiments with mice were subjected to institutional review and approved by the Veterinary Office of Basel-Stadt.
Cell lines and cultured hippocampal neurons.
Human Embryonic Kidney 293 (HEK293) cells were from ATCC (RRID: CVCL_0045) and maintained in DMEM supplemented with GlutaMAX (Invitrogen) and 10% FCS in a humidified atmosphere (5% CO2) at 37°C. CHO-K1 cells (RRID: CVCL_0214), stably expressing human GABAB1b and rat GABAB2 (Urwyler et al., 2001) were maintained in DMEM with 500 μm l-glutamine, 40 μg/ml l-proline, 0.5 mg/ml G418, 0.25 mg/ml zeocine, and 10% FCS. Cells were transfected at 70%–90% confluency in 6- or 24-well plates or in 10 cm dishes using Lipofectamine 2000 (Invitrogen), X-tremeGENE (Roche), or polyethylenimine (Sigma). Cultured hippocampal neurons were prepared as described previously (Brewer et al., 1993; Biermann et al., 2010). Briefly, embryonic day 16.5 mouse hippocampi were dissected, digested with 0.25% trypsin (Invitrogen) in 1 × PBS solution for 15 min at 37°C, dissociated by trituration, and plated on glass coverslips coated with 1 mg/ml poly-L-lysine hydrobromide (Sigma) in 0.1 m borate buffer (boric acid/sodium tetraborate). Neurons were seeded at a density of ∼550 cells/mm2 in neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen) and 0.5 mm l-glutamine and cultured in a humidified atmosphere (5% CO2) at 37°C. Cultured hippocampal neurons were transfected at DIV7.
Biochemistry.
Plasmids used for biochemistry experiments encoding FLAG- and Myc-tagged KCTD10, KCTD8, KCTD12, and KCTD16 constructs were described previously (Seddik et al., 2012). pCI-HA-GABAB2-eYFP was a gift from Novartis.
Adult mouse brains were homogenized and lysed in 5 ml NETN buffer (100 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40, 20 mm Tris/HCl, pH 7.4) supplemented with complete EDTA-free protease inhibitor mixture (Roche) using a Dounce homogenizer. Following rotation for 3 h at 4°C, brain lysates were cleared by centrifugation at 15,000 × g for 30 min at 4°C. For affinity-depletion experiments, hippocampi of adult mouse brains were dissected and placed in 100 mg/ml of ice-cold homogenization medium (320 mm sucrose, 4 mm HEPES, pH 7.5, 1 mm EDTA, 1 mm EGTA, supplemented with complete EDTA-free protease inhibitor mixture) and homogenized using a glass-Teflon homogenizer with 20 passes on ice. After homogenization, the material was cleared by centrifugation at 1000 × g (4°C, 15 min). The membrane-enriched fraction was isolated by ultracentrifugation at 48,000 × g (4°C, 45 min) and solubilized for 3 h at 4°C in NETN buffer at 2 mg protein/ml. The solubilized fraction was cleared by ultracentrifugation at 105 × g (4°C, 45 min). Cultured HEK293 cells were lysed 48 h after transfection in NETN buffer or modified RIPA buffer (150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, complete EDTA-free protease inhibitors, pH 7.4) by rotating for 30 min at 4°C. HEK293 cell lysates were then cleared by centrifugation at 10,000 × g for 20 min at 4°C. Brain and cell lysates were directly used for immunoblot analysis (input lanes) or immunoprecipitated with guinea-pig anti-KCTD12 (RRID:AB_2631051), guinea-pig anti-KCTD16 (RRID: AB_2631053), or mouse anti-Myc (9E10, Santa Cruz Biotechnology, RRID: AB_627268) antibodies coupled to a mixture of protein-A and protein-G Sepharose (GE Healthcare). Lysates and immunoprecipitates were resolved using SDS-PAGE and probed with the primary antibodies rabbit anti-KCTD12 (1:2500, RRID:AB_2631049), rabbit anti-KCTD16 (1:2500, RRID:AB_2631050), rabbit anti-Myc (C3956, Sigma, 1:2500, RRID:AB_439680), rabbit anti-FLAG (F7425, Sigma, 1:2500, RRID:AB_439687), rabbit anti-GABAB2 (C44A4, #4819, Cell Signaling Technology, 1:1500, RRID:AB_2108339), and mouse β-tubulin III (T8660, Sigma, 1:2000, RRID:AB_477590) in combination with peroxidase-coupled secondary antibodies goat anti-guinea pig (A7289, Sigma, 1:10,000, RRID:AB_258337), sheep anti-mouse (NA931, GE Healthcare, 1:10,000, RRID:AB_772210), or donkey anti-rabbit (NA934, GE Healthcare, 1:10,000, RRID:AB_772206). Guinea-pig and rabbit KCTD12 and KCTD16 antibodies were raised against synthetic peptides of mouse KCTD12 (amino acid residues 145–167) or KCTD16 (residues 7–23) (Metz et al., 2011). Chemiluminescence was detected using the SuperSignal West kit (Thermo Scientific).
Bimolecular luminescence protein-fragment complementation (BiLC), bimolecular fluorescence complementation (BiFC), and bioluminescence resonance energy transfer (BRET) assays.
The BiLC and BiFC template constructs were published previously (Héroux et al., 2007; Stefan et al., 2007). GATEWAY technology was used to insert the human KCTD cDNA C-terminal to Rluc and Venus fragments contained in a destination vector for eukaryotic expression based on pcDNA3.1(+)/Zeo (Invitrogen): N-terminal (amino acids 1–110, NTRluc) and C-terminal (amino acids 111–311, CTRluc) fragments of a bright Renilla luciferase mutant RlucII (A55T/C124A/M185V-Rluc) and N-terminal (amino acids 1–154, NTVen) and C-terminal (amino acids 146–239, CTVen) fragments of Venus. The construction of pcDNA3.1-Myc-GABAB1 and pcDNA3.1-HA-GABAB2-GFP was reported previously (Villemure et al., 2005). The GFP used for HA-GABAB2-GFP is a blue-shifted GFP known as GFP10 that is a preferred acceptor in BRET2 assays (Mercier et al., 2002). The HA and Myc tags were used to control for protein cell surface expression. The FLAG-Gβ2 and Venus-Gγ2 constructs used for BRET experiments were reported previously (Adelfinger et al., 2014; Turecek et al., 2014; Rajalu et al., 2015).
For BiLC, BiFC, and BRET assays, HEK293 cells were harvested 48 h after transfection, suspended in PBS, and distributed into 96-well microplates at 1–3 × 105 cells/well. To measure reconstituted luciferase activity from BiLC proteins, the luciferase substrate coelenterazine h (Nanolight Technology, final concentration 5 μm) was added to each well and luminescence signals were measured 2 min later using a Mithras LB940 multimode plate reader (Berthold Technologies). The expression levels of fluorescent proteins were measured using a FlexStation 2 fluorimeter (Molecular Devices) with excitation and emission wavelengths set at 410 and 510 nm (GFP10) or 518 and 570 nm (Venus), respectively. BRET2 between KCTD-BiLC and GABAB2-GFP was measured 2 min after addition of the luciferase substrate DeepBlue C (coelenterazine 400A; Nanolight Technology; final concentration of 5 μm). BRET was calculated as the ratio of the light emitted by GFP10 (515 ± 20 nm) over the light emitted by RlucII (400 ± 70 nm) using a Mithras LB940 luminometer. The net BRET signal was defined as the difference between the total BRET and the signal obtained from samples expressing the RlucII-fusion constructs alone (background signal), and plotted against the ratio between the expression levels of the GFP10- and RlucII-fusion constructs as measured by the total fluorescence and luminescence signals, respectively, as previously described (Mercier et al., 2002). BRET between KCTD-BiLC and KCTD-BiFC was measured 2 min after the addition of the luciferase substrate, coelenterazine h (final concentration of 5 μm). BRET was calculated as the ratio of the light emitted by Venus (530 ± 20 nm) over the light emitted by RlucII (480 ± 20 nm) using a Mithras LB940 luminometer. BRET signals between KCTD-BiLC and Venus-Gγ2 were measured on an Infinite F500 microplate reader (Tecan) detecting Venus at 550 ± 25 nm and RlucII at <475 nm.
Electrophysiology.
Whole-cell patch-clamp recordings of GABAB-mediated Kir3 currents from CHO cells and cultured hippocampal neurons were performed 48–72 h and ∼2 weeks (DIV18-DIV21) after transfection, respectively. EGFP-expressing cells were identified via epifluorescence using an FITC filter set and patched under infrared oblique illumination (BX61WI; Olympus). Kir3 currents were recorded at room temperature (22°C–23°C) in aCSF containing the following (in mm): 145 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 25 glucose, pH 7.3. Neurons were superfused with aCSF supplemented with TTX (0.5 μm), DNQX (10 μm), and picrotoxin (100 μm). Patch electrodes were pulled from borosilicate glass capillaries (resistance of 3–5 mΩ) and filled with a solution containing the following (in mm): 107.5 K-gluconate, 32.5 KCl, 10 HEPES, 5 EGTA, 4 Mg-ATP, 10 phosphocreatine, 0.6 Na2-GTP, at pH 7.2 (adjusted with KOH), 292 mOsm. GABAB receptor responses were evoked at −50 mV by fast application of baclofen (100 μm, 1–60 s) (Turecek et al., 2004; Dittert et al., 2006).
IPSCs were recorded in parasagittal hippocampal slices prepared from P25-P30 mice as follows. Animals were decapitated and the brains were excised in ice-cold extracellular solution containing the following (in mm): 130 NaCl, 3.5 KCl, 3.0 MgCl2, 0.5 CaCl2, 10 glucose, 1.25 NaH2PO4, 25 NaHCO2, 0.5 ascorbic acid, 3 myo-inositol, and 3 sodium pyruvate; gassed with 5% CO2/95% O2 to pH 7.3. Slices (300 μm thick) were cut using a VT1200S vibratome (Leica), incubated at 35°C for 45 min and then stored at 31°C–32°C in the extracellular solution in which the concentrations of MgCl2 and CaCl2 were 1 and 2 mm, respectively. For recording, slices were superfused with aCSF containing the following (in mm): 130 NaCl, 3.5 KCl, 1.5 MgCl2, 1.5 CaCl2, 10 glucose, 1.25 NaH2PO4, 24 NaHCO3, pH 7.3, equilibrated with 5% CO2/95% O2. IPSCs were elicited by electric stimulation of stratum radiatum via bipolar electrode and isolated in the presence of DNQX (10 μm), CPP (5 μm), and SR95531 (gabazine; 15 μm, used for recording of sIPSCs). Patch pipette solution contained the following (in mm): 107.5 K-gluconate, 32.5 KCl, 10 HEPES, 0.1 EGTA, 4 Mg-ATP, 10 phosphocreatine, 0.6 Na2-GTP, at pH 7.2, 290 mOsm. Pharmacological GABAB responses or IPSCs were acquired with an Axopatch 200B low-pass filtered at 1 or 5 kHz and digitized at 5 or 20 kHz using a Digidata 1440A interface (Molecular Devices) driven by pClamp 10 software (RRID:SCR_011323). Cell capacitance was calculated using membrane time-constant and cell resistance values obtained from voltage responses of cells upon applying hyperpolarizing −10 pA steps at the resting potential. The holding potential was adjusted for a measured liquid junction potential of 12 mV. Whole-cell currents and voltages were analyzed using Clampfit 10 software (Molecular Devices). All values are given as mean ± SD. Statistical significances were tested using one-way ANOVA or Student's t test (GraphPad Prism, RRID:SCR_002798). TTX was from Latoxan. DNQX, CPP, and SR95531 were from Tocris Bioscience. All other reagents were from Sigma-Aldrich.
Results
KCTD hetero-oligomers in the brain
We used immunoprecipitation (IP) experiments to address whether KCTD hetero-oligomers exist in the brain (Fig. 1A). We limited our analysis to KCTD12 and KCTD16, which in the adult brain are coexpressed in many neuronal populations, including hippocampal pyramidal neurons (Metz et al., 2011). We found that anti-KCTD12 antibodies copurified KCTD16 protein from wild-type (WT) but not from control KCTD12 knock-out (Kctd12−/−) brain lysates (Fig. 1A). Reverse IP experiments with anti-KCTD16 antibodies confirmed the existence of KCTD12/KCTD16 hetero-oligomers in WT but not in control Kctd16−/− brain lysates (Fig. 1A).
Figure 1.

KCTD hetero-oligomers are abundant in the adult mouse brain. A, Anti-KCTD12 and anti-KCTD16 antibodies (Schwenk et al., 2010) copurify KCTD12 and KCTD16 proteins in immunoprecipitation (iP) experiments with brain lysates of WT mice, as shown on immunoblots (IB). Control IPs with brain lysates of Kctd12−/− and Kctd16−/− mice (Metz et al., 2011) show that the antibodies are specific for the KCTD12 and 16 proteins. Asterisks indicate cross-reactions of the anti-KCTD16 antibody in the Input samples. B, Affinity depletion of KCTD12 from hippocampi of adult mouse brains using anti-KCTD12 antibodies leads to a codepletion of KCTD16 engaged in KCTD12/KCTD16 hetero-oligomers (depleted). Control IBs of β-tubulin III show that the neuron-specific marker protein is not depleted by anti-KCTD12 antibodies.
We next depleted KCTD12 from hippocampal lysates to determine the fraction of KCTD16 protein participating in KCTD12/KCTD16 hetero-oligomers. Complete depletion of KCTD12 from lysates with specific anti-KCTD12 antibodies removed 64.9 ± 6.1% (n = 3) of the KCTD16 protein from the supernatant (Fig. 1B). In the hippocampus, approximately two-thirds of the KCTD16 protein is therefore associated with KCTD12. For unknown reasons, anti-KCTD16 antibodies inefficiently depleted KCTD16 from hippocampal lysates, which prevented a meaningful assessment of the fraction of KCTD12 protein involved in KCTD12/KCTD16 hetero-oligomers.
T1 and H1 domains mediate homo- and hetero-oligomerization
The KCTDs are modular proteins consisting of an N-terminal T1 tetramerization domain and one or two C-terminal H1 and H2 homology domains (Fig. 2A). Earlier experiments supported that the T1 domain of KCTD12 forms homo-oligomers (Schwenk et al., 2010; Smaldone et al., 2016). To study whether the KCTDs form hetero-oligomers in transfected HEK293 cells, we performed co-immunoprecipitation experiments with FLAG- and Myc-tagged KCTD proteins. We included KCTD10, which belongs to a different clade of the KCTD family (Seddik et al., 2012), into our analysis. KCTD10 does not associate with GABAB receptors (Schwenk et al., 2010) and lacks H1 and H2 domains (Fig. 2A). We found that KCTD8, KCTD10, KCTD12, and KCTD16 assemble homo-oligomers in HEK293 cells (Fig. 2B); however, only KCTD8, KCTD12, and KCTD16 form hetero-oligomers in all possible dual combinations (Fig. 2C).
Figure 2.
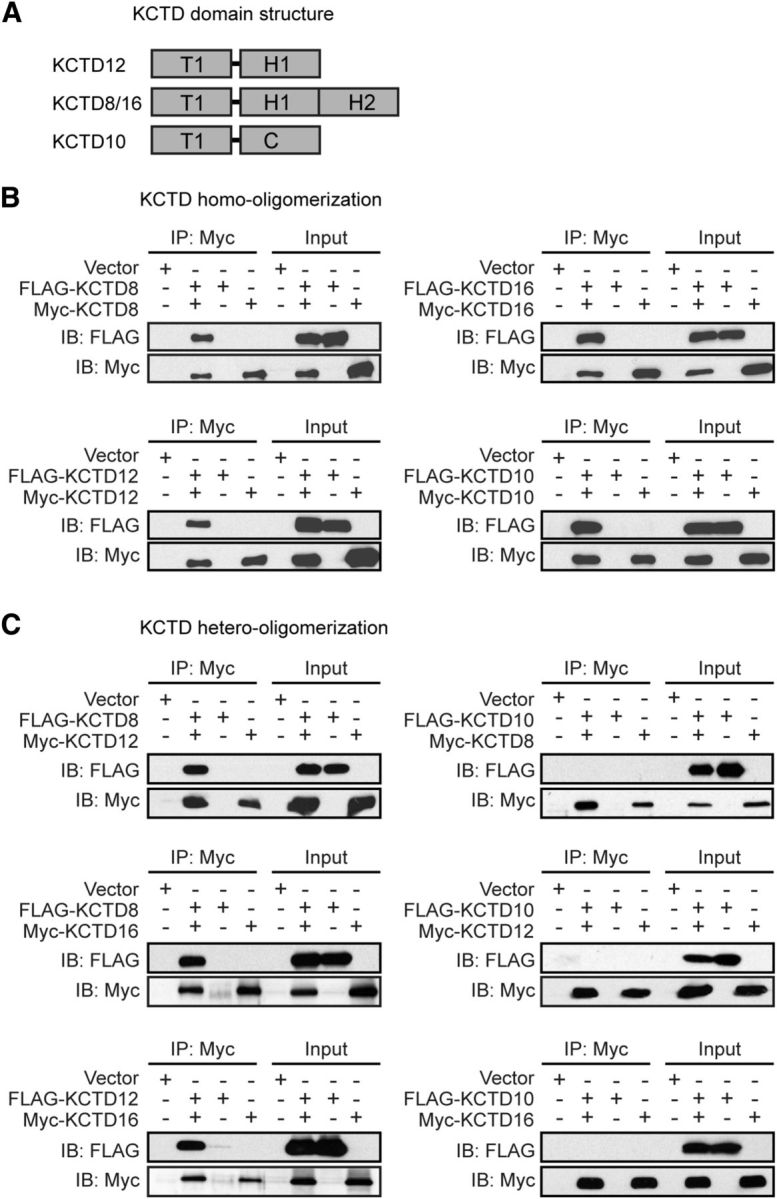
KCTD8, KCTD12, and KCTD16 form homo- and hetero-oligomers in transfected HEK293. A, KCTD domain structure. KCTDs contain T1, H1, and H2 homology domains that are not closely sequence-related with the T1 and C-terminal (C) domains of KCTD10. The H2 domains are selectively present in KCTD8 and KCTD16. B, KCTDs form homo-oligomeric complexes. Input lanes indicate the expression of FLAG- or Myc-tagged KCTD proteins in the cell lysates used for IP with anti-Myc antibodies. Proteins in the IPs were revealed by immunoblotting (IB) with anti-FLAG and anti-Myc antibodies. C, KCTD8, KCTD12, and KCTD16 form hetero-oligomers with each other, but not with KCTD10. Input lanes indicate expression of the FLAG- or Myc-tagged KCTD proteins in the cell lysates used for IPs with anti-Myc antibodies. Proteins in the IPs were revealed by IB with anti-FLAG and anti-Myc antibodies.
IP experiments with the isolated Myc-tagged T1, H1 or H2 domains of KCTD12 and KCTD16 show that both the T1 and H1 domains interact with FLAG-tagged KCTD12 (Fig. 3A) and KCTD16 (Fig. 3B) in transfected HEK293 cells. Neither the H2 domain of KCTD16 nor full-length KCTD10 interacted with KCTD12 (Fig. 3A) or KCTD16 (Fig. 3B). Collectively, these results indicate that both the T1 and H1 domains mediate KCTD homo- and hetero-oligomerization. Consistent with the H1 domains mediating oligomerization, Myc-16ΔT1, a KCTD16 mutant lacking the T1 domain, still forms homo-oligomeric FLAG-KCTD16/Myc-16ΔT1 (Fig. 3B,C) and hetero-oligomeric FLAG-KCTD12/Myc-16ΔT1 complexes (Fig. 3A,B) that interact with GABAB2 through the T1 domain of the full-length KCTD in the complex (Fig. 3C).
Figure 3.

Self-interacting T1 and H1 domains mediate KCTD homo- and hetero-oligomerization in HEK293 cells. IP of FLAG-KCTD12 (A) or FLAG-KCTD16 (B) with the Myc-tagged T1 and H1 domains of KCTD12 or KCTD16. IPs were performed with anti-Myc antibodies from total cell lysates. Proteins in the IPs were revealed by immunoblotting (IB) using anti-FLAG and anti-Myc antibodies. Input lanes indicate expression of the tagged proteins in the cell lysates used for the IPs. The Myc-tagged T1 and H1 domains of KCTD12 and KCTD16, but not the Myc-tagged H2 domain of KCTD16, coprecipitate FLAG-KCTD12 and FLAG-KCTD16. KCTD16 lacking the T1 domain (Myc-16ΔT1) interacts with FLAG-KCTDs via its H1 domain. Myc-KCTD12 and Myc-KCTD16 were used as positive controls, Myc-KCTD10 as a negative control. C, Myc-16ΔT1 copurifies GABAB2 (GB2-YFP) in the presence of KCTD16 or KCTD12, showing that Myc-16ΔT1 can homo- and hetero-oligomerize with KCTD16 and KCTD12 at GABAB2. Myc-16ΔT1 does not directly interact with GABAB2 because it lacks the T1 domain that is a prerequisite necessary for binding to GABAB2. IPs were performed with anti-Myc antibodies from total cell lysates. Proteins in the IPs were revealed by IB using anti-GABAB2, anti-FLAG, and anti-Myc antibodies. Input lanes (bottom) indicate expression of the tagged proteins in the cell lysates used for the IPs.
Detection of KCTD homo- and hetero-oligomers in live cells
We next used a bimolecular luminescence protein-fragment complementation (BiLC) assay (Stefan et al., 2007; Armando et al., 2014) to study KCTD oligomerization in live cells. KCTD proteins were tagged at their N termini with the N- or C-terminal fragment of Renilla luciferase (NTRluc and CTRluc). In such a way, oligomerization of KCTD protomers can be quantified by measuring the activity (luminescence) of reconstituted Rluc. The Rluc activity measured in transfected HEK293 cells indicates that KCTD10, KCTD12, and KCTD16 all form homo-oligomers (Fig. 4A). KCTD12 and KCTD16 additionally form hetero-oligomers with each other (Fig. 4A). A 100-fold lower level Rluc activity was observed with KCTD10 in combination with KCTD12 or KCTD16 (Fig. 4A). This may represent weak nonspecific luciferase reconstitution/activity or indicate a very low level of hetero-oligomerization. KCTD homo-oligomer formation also promotes bimolecular fluorescence complementation (BiFC) (Héroux et al., 2007) of Venus (Fig. 4A), a variant of the yellow fluorescent protein that can be used as an energy acceptor in BRET experiments. Of note, we were unable to detect a specific BiFC signal for KCTD16 homo-oligomers, suggesting that KCTD16 protomers have a lower propensity to form homo-oligomers. This is supported by two-component BRET assays using KCTD-RlucII and KCTD-GFP2 constructs, in which KCTD12 homo-oligomers showed ∼9 times lower half BRET saturation values than KCTD16 homo-oligomers (KCTD12: 0.064, and KCTD16: 0.56). BiFC of Rluc and Venus allowed us to investigate whether reconstitution of Rluc and Venus from their N- and C-terminal fragments fused to KCTD monomers yields BRET due to the formation of KCTD oligomers made up from at least four protomers. Expression of increasing amounts of NTVen12 + CTVen12 with fixed amounts of NTRluc12 + CTRluc12 resulted in a hyperbolic BRET donor saturation curve (Fig. 4B), consistent with the assembly of KCTD12 homo-tetramers (or homo-pentamers) in live cells. The complementation experiments support a parallel arrangement of KCTD subunits in the oligomers. Expression of NTRluc12 + CTRluc12 or NTVen10 + CTVen10 with increasing amounts of NTVen10 + CTVen10 or NTRluc12 + CTRluc12, respectively, did not yield clearly saturating BRET donor curves (Fig. 4B). Inefficient complementation of Venus by NTVen16 + CTVen16 (Fig. 4A) prevented us from testing with BiFC/BRET whether KCTD16 homo- and KCTD12/KCTD16 hetero-tetramers are formed.
Figure 4.

KCTD oligomerization in live HEK293 cells. A, BiLC or BiFC through oligomerization of split Rluc- or split Venus-tagged KCTDs, respectively. Cells were transfected with NTRlucKCTD and CTRlucKCTD or NTVenKCTD and CTVenKCTD constructs, and oligomerization between Rluc- or Venus-fragment-tagged KCTDs was monitored by measuring reconstituted luciferase or fluorescence activity, respectively. KCTD10, KCTD12, and KCTD16 all form homo-oligomers. KCTD12 and KCTD16 additionally form hetero-oligomers together. KCTD10 has a weak propensity to also form hetero-oligomers with KCTD12 or KCTD16. Labels in the bar graphs indicate the transfected Rluc- or Venus-fragment-tagged KCTD proteins. Data are mean ± SEM of 3 or 4 independent experiments. B, BRET between reconstituted Rluc and Venus in KCTD oligomers. Cells were transfected with fixed amounts of NTRlucKCTD and CTRlucKCTD constructs and increasing amounts of NTVenKCTD and CTVenKCTD constructs. BRET donor saturation curves were generated by expressing the net BRET signal detected as a function of the ratio between the total fluorescence signal and the luminescence signal (acceptor/donor ratio; expressed in milliBRET units, mBU) and demonstrate that both KCTD12 and KCTD10 can assemble into homo-tetramers or higher-order oligomers from the individually tagged monomers. KCTD12 and KCTD10 have a much weaker propensity to form hetero-tetramers. Data points represent the mean of technical duplicates combined from 4 independent experiments.
KCTD12/KCTD16 hetero-oligomers interact with the receptor and the G-protein
We used BiLC in combination with BRET to study whether KCTD hetero-oligomers interact with GABAB receptors in intact cells. BRET-donor saturation curves were determined with fixed amounts of NTRlucKCTDs + CTRlucKCTDs and increasing amounts of Myc-GABAB1 and the energy acceptor HA-GABAB2-GFP (Fig. 5A). We observed specific BRET between the reconstituted Rluc and HA-GABAB2-GFP with KCTD12 and KCTD16 homo-oligomers as well as with KCTD12/KCTD16 hetero-oligomers. We did not observe specific BRET with KCTD10 homo-oligomers and the receptor, as indicated by low BRET and a nonsaturating BRET to acceptor/donor relationship (Fig. 5A). Similarly, a nonspecific BRET signal with a linear BRET to acceptor/donor relationship (y = 2.4x + 5.7) was obtained in control experiments with KCTD12 homo-oligomers and a GABAB2 acceptor in which tyrosine-902 was mutated to alanine, which prevents KCTD binding to GABAB2 (Schwenk et al., 2010). BRET experiments therefore demonstrate that KCTD12/KCTD16 hetero-oligomers interact with GABAB receptors in live cells.
Figure 5.

KCTD homo- and hetero-oligomers bind to both GABAB receptors and G-proteins in live HEK293 cells. A, BRET between KCTD oligomers and GABAB receptors in living cells. Cells were transfected with fixed amounts of NTRlucKCTD and CTRlucKCTD constructs and increasing amounts of Myc-GABAB1 and HA-GABAB2-GFP constructs. BRET was measured between reconstituted Rluc and GABAB2-GFP. Data points represent the mean of technical duplicates combined from 6 or 7 independent experiments. Donor/acceptor ratios needed to reach half BRET saturation (BRET50) were as follows: KCTD12 homo-oligomer: 0.27 ± 0.03; KCTD16 homo-oligomer: 2.55 ± 0.97; KCTD12/KCTD16 hetero-oligomer: 1.91 ± 0.29; KCTD16/KCTD12 hetero-oligomer: 6.65 ± 0.71 (mean ± SEM, n = 6 or 7). B, BRET between KCTD oligomers and the G-protein in live cells. Cells were transfected with fixed amounts of NTRlucKCTD and CTRlucKCTD constructs and increasing amounts of Gβ2 and Gγ2-YFP constructs. BRET was measured between reconstituted Rluc and Gγ2-YFP. Data points represent the mean of technical quadruplicates from a representative experiment (n = 3).
The KCTDs not only interact with the receptor but also with the G-protein (Turecek et al., 2014). It is unknown whether the KCTDs interact as monomers or oligomers with the G-protein. We therefore used BiLC in combination with BRET to study whether KCTD oligomers can interact with the G-protein. We determined BRET donor saturation curves using fixed amounts of NTRlucKCTDs + CTRlucKCTDs and increasing amounts of the G-protein subunits Gβ2 and Venus-Gγ2 (Fig. 5B). BRET between the reconstituted Rluc and Venus-Gγ2 was observed with KCTD12 and KCTD16 homo-oligomers as well as with KCTD12/KCTD16 hetero-oligomers. Because KCTD12 and KCTD16 exert distinct kinetic effects on G-protein signaling (Schwenk et al., 2010; Turecek et al., 2014), it is possible that KCTD hetero-oligomers endow receptors with novel properties.
KCTD12/KCTD16 hetero-oligomers generate unique receptor-induced K+ current responses in CHO cells
KCTD12, but not KCTD8 or KCTD16, induces strong desensitization of GABAB receptor-activated Kir3 currents (Schwenk et al., 2010; Seddik et al., 2012; Turecek et al., 2014) (Fig. 6A–C). We studied the influence of hetero-oligomeric KCTD12/KCTD16 complexes on K+ current desensitization in transfected CHO cells expressing GABAB receptors together with Kir3 channels. We recorded K+ currents in response to a prolonged application of the GABAB agonist baclofen (100 μm, 1 min). Cells coexpressing KCTD12 and KCTD16 exhibited significantly reduced and slower K+ current desensitization than cells expressing KCTD12 alone (Fig. 6A,C,E). This may be due to the formation of hetero-oligomeric KCTD12/KCTD16 complexes and/or the simultaneous assembly of desensitizing KCTD12 and nondesensitizing KCTD16 homo-oligomers. To avoid association of KCTD16 homo-oligomers with the receptor, we substituted KCTD16 with 16ΔT1, which lacks the T1 domain that is necessary for direct binding to the receptor (Schwenk et al., 2010). However, 16ΔT1 is able to indirectly bind to the receptor through hetero-oligomerization with KCTD12 (Fig. 3C). Cells expressing KCTD12 and 16ΔT1 therefore form receptors associated with KCTD12/KCTD16ΔT1 hetero-oligomers or KCTD12 homo-oligomers but not with 16ΔT1 homo-oligomers. We found that cells coexpressing 16ΔT1 and KCTD12 exhibited reduced and slower K+ current desensitization than cells expressing KCTD12 alone (Fig. 6B–E), which demonstrates that the kinetic properties of KCTD12/KCTD16ΔT1 hetero-oligomers differ from those of KCTD12 homo-oligomers. As a control, expression of 16ΔT1 alone yielded nondesensitizing responses (Fig. 6B,C), similar to expression of KCTD16 alone (Fig. 6A,B). In summary, the electrophysiological data indicate that KCTD12/KCTD16 hetero-oligomers desensitize GABAB-activated K+ currents to a lesser extent and more slowly than KCTD12 homo-oligomers.
Figure 6.

Bidirectional modulation of GABAB-activated Kir3 currents by KCTD hetero-oligomers. A, Representative baclofen-activated K+ current traces recorded at −50 mV from CHO cells expressing GABAB receptors and Kir3.1/3.2 channels either without KCTD (without [w/o], gray trace), with KCTD12 alone (black), with KCTD16 alone (blue), or with both KCTDs (red). KCTD12, but not KCTD16, induces pronounced and rapid desensitization of K+ currents. Coexpression of KCTD12 and KCTD16 results in intermediate current desensitization. B, Baclofen-activated K+ currents recorded from CHO cells either expressing KCTD12 (black), a KCTD16 mutant lacking the T1 domain (16ΔT1, blue), or both KCTD isoforms (red). Expression of KCTD12 together with 16ΔT1, which only binds to the receptor in a complex with KCTD12, reduces KCTD12-induced desensitization. C, Bar graph summarizing the relative desensitization of baclofen-activated K+ currents. The relative desensitization was calculated as follows: (1 − (ratio of current amplitude after 60 s vs peak current)) × 100. Values are mean ± SD of 26 (w/o KCTD), 17 (KCTD12), 14 (KCTD16), 11 (16ΔT1), 6 (KCTD12/KCTD16), and 13 (KCTD12/KCTD16ΔT1) cells. D, Normalized traces represent a slower time course of K+ current desensitization in CHO cells coexpressing KCTD12 and 16ΔT1 (red) compared with CHO cells expressing KCTD12 alone (black). Traces are fitted to a double exponential function (gray solid line) with time constants τ1 = 1.0 s (relative contribution to desensitization 71.7%) and τ2 = 9.4 s for KCTD12 and τ1 = 3.9 s (33.8%) and τ2 = 28.4 s for KCTD12/KCTD16ΔT1. E, Bar graph showing mean amplitude-weighted time constants obtained from fits of a double exponential function to K+ current deactivation. F, Superimposed traces of the deactivation phase of K+ currents activated by application of baclofen to CHO cells for 1 min as in A or B displayed with an expanded time scale. KCTD12 and KCTD16 have opposite effects on the time course of the deactivation, with KCTD12 being dominant when coexpressed with KCTD16. G, Bar graph summarizing the time constants obtained from a fit of the K+ current deactivation to a single exponential function. H, Representative traces of K+ currents activated by baclofen (2 s) to CHO cells expressing no KCTD (w/o, gray), KCTD12 alone (black), 16ΔT1 alone (blue), or both KCTDs (red). KCTD12 neither reduces the effect of 16ΔT1 nor accelerates deactivation of brief current responses. I, Bar graph showing mean time constants obtained from fits of current deactivation to a one exponential function. Data are collected from 12 (w/o KCTD), 11 (KCTD12), 6 (16ΔT1), and 6 (KCTD12/KCTD16ΔT1) experiments. *p < 0.05 (Dunnett's multiple-comparison test and Bonferroni pairwise comparison test). **p < 0.01 (Dunnett's multiple-comparison test and Bonferroni pairwise comparison test). ***p < 0.001 (Dunnett's multiple-comparison test and Bonferroni pairwise comparison test).
Analysis of the deactivation kinetics of K+ currents activated by applying baclofen for 1 min revealed a novel regulatory feature of the KCTDs (Fig. 6F,G). KCTD12 and KCTD16 significantly accelerated and slowed current deactivation, respectively, compared with cells lacking KCTDs (Fig. 6F,G). A slowing of current deactivation is also observed with 16ΔT1 (Fig. 6F,G). This indicates that the deactivation does not necessarily require KCTD16 binding to the receptor and supports that the deactivation mechanism operates downstream of the receptor. Coexpression of KCTD12 with 16ΔT1 or KCTD16 yielded fast deactivating currents, showing that KCTD12 overrules the slowing effect of 16ΔT1 or KCTD16 within the hetero-oligomer (Fig. 6F,G). No accelerated current deactivation is observed in the presence of KCTD12 after a baclofen application for 2 s, which is too short to induce pronounced desensitization (Fig. 6H,I). Accelerated current deactivation in the presence of KCTD12 thus correlates with current desensitization. This is corroborated by a similar baclofen concentration dependence of current desensitization and deactivation in the presence of KCTD12 (Fig. 7A). We did not observe a sigmoidal agonist concentration dependence of current deactivation in the absence of KCTD or in the presence of KCTD16 (Fig. 7B), which further supports that accelerated deactivation relates to KCTD12-induced desensitization. The slowing of current deactivation that is observed in the presence of KCTD16 (Fig. 6G) is use-dependent, as indicated by the linear baclofen concentration dependence of the deactivation (Fig. 7B). In summary, the data show that, after prolonged receptor activation, homo-oligomeric KCTD12 complexes yield strongly desensitizing fast deactivating K+ current responses. In contrast, homo-oligomeric KCTD16 complexes yield nondesensitizing slowly deactivating current responses. Hetero-oligomeric KCTD12/KCTD16 complexes produce distinct current K+ responses characterized by intermediate desensitization and fast deactivation. In contrast, during brief receptor activation, KCTD12/KCTD16 hetero-oligomers produce nondesensitizing slowly deactivating current responses.
Figure 7.
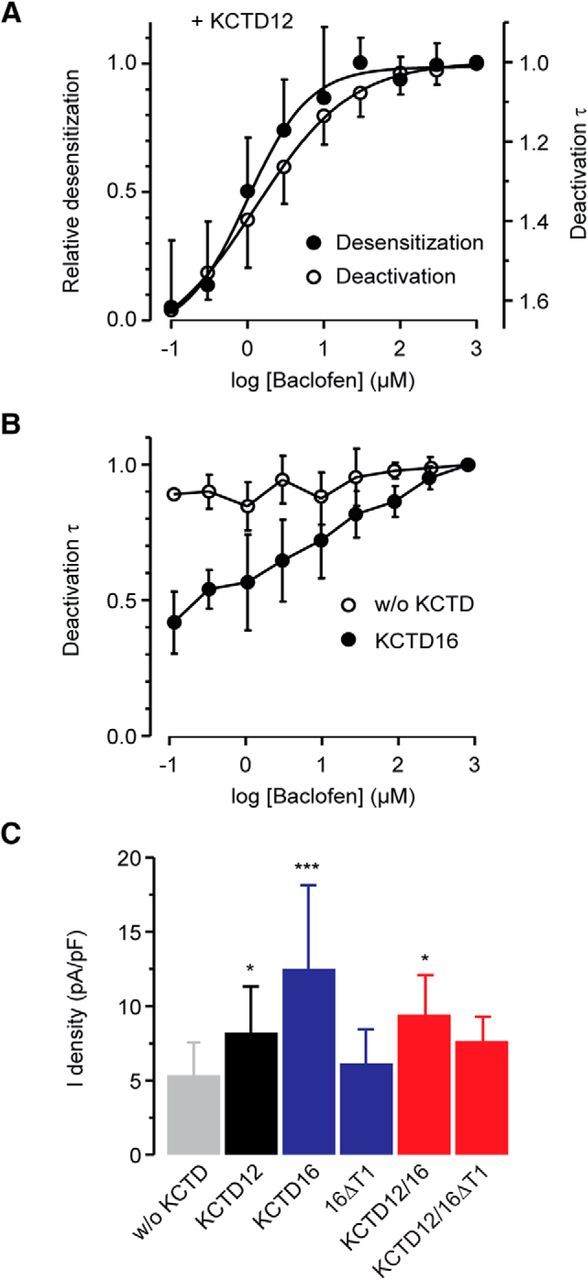
Influence of KCTD12 and KCTD16 on desensitization, deactivation, and amplitude of GABAB-induced Kir3 currents in CHO cells. A, Agonist concentration dependence of the desensitization and deactivation of Kir3 currents in the presence of KCTD12. Each point in the curve represents the relative desensitization (mean ± SD, data from 9 cells) or deactivation time constant (8 cells) of 25-s-long baclofen responses, normalized in each cell to the value obtained at the highest baclofen concentration (1 mm). The log(baclofen)-response curves (solid black lines) were obtained by fitting the experimental data to a sigmoidal function (GraphPad Prism, RRID:SCR_002798). The half-maximal desensitization or deactivation time constant reduction was observed at 1.3 or 1.0 μm of baclofen, respectively. B, Agonist concentration dependence of the deactivation of Kir3 currents in the absence (without [w/o] KCTD) or presence of KCTD16. C, Bar graph summarizing the effects of KCTD12 and KCTD16 on K+ current densities (current normalized to cell capacitance) evoked by 100 μm baclofen. Data are the mean ± SD of 23 (w/o KCTD), 15 (KCTD12), 12 (KCTD16), 11 (16ΔT1), 5 (KCTD12/KCTD16), and 7 (KCTD12/KCTD16ΔT1) cells. KCTD12 and KCTD16 expressed alone or in combination significantly increased the amplitudes of Kir3 currents. *p < 0.05 (Dunnett's multiple-comparison test). ***p < 0.001 (Dunnett's multiple-comparison test). Data are mean ± SD of 10 (w/o KCTD) and 6 (KCTD16) cells.
We observed that individual and combined expression of KCTD12 and KCTD16 in transfected CHO cells significantly increases the peak K+ current amplitudes (presented as current densities) in response to a nearly saturating concentration of baclofen (Fig. 7C). No increase in current density is observed with 16ΔT1, which does not bind to the receptor (Fig. 7C). The increase in current density observed with KCTD12, KCTD16, and KCTD12/KCTD16 may therefore relate to precoupling of the G-protein at the receptor via the KCTD proteins (Turecek et al., 2014; Schwenk et al., 2016). Increased surface expression of GABAB receptors in the presence of KCTD12 (Ivankova et al., 2013) may further contribute to an increase in current amplitude. Receptors assembled with KCTD12/KCTD16ΔT1 show a trend toward increased current amplitudes, which, however, does not reach statistical significance (Fig. 7C). Possibly, precoupling of the G-protein and/or surface expression is less efficient when KCTD12 combines with 16ΔT1 than with full-length KCTD16.
We additionally determined the baclofen concentration/K+ current response curves in the presence or absence of KCTD12 and KCTD16. The EC50 values (derived from log(baclofen)/K+ current response) measured in CHO cells expressing KCTD12 (9.7 ± 7.7 μm, n = 8) or KCTD16 (20.8 ± 8.7 μm, n = 7) were significantly smaller than those in cells lacking KCTD proteins (66.4 ± 26.7 μm, n = 8; p < 0.001, Dunnett's multiple-comparison test), consistent with earlier findings (Schwenk et al., 2010). The EC50 values measured in cells expressing KCTD12 or KCTD16 do not significantly differ from each other (Bonferroni pairwise comparison test). Because the KCTD proteins have marginal effects on agonist affinity at the orthosteric binding site (Rajalu et al., 2015), the decrease in EC50 values in the presence of KCTD12 or KCTD16 is best explained by the KCTD-mediated precoupling of the G-protein at the receptor (Turecek et al., 2014; Schwenk et al., 2016). KCTD hetero-oligomers and homo-oligomers therefore likely activate G-proteins to a similar extent.
KCTD12/KCTD16 hetero-oligomers regulate GABAB-activated K+ currents in hippocampal neurons
We next investigated whether KCTD hetero-oligomers influence baclofen-activated K+ currents in cultured neurons. Previous experiments showed that hippocampal neurons express high levels of KCTD12 and KCTD16 (Schwenk et al., 2010; Metz et al., 2011) and that the lack of KCTDs in knock-out neurons influences desensitization of baclofen-evoked K+ currents (Turecek et al., 2014). Accordingly, we found that, during prolonged receptor activation, K+ currents desensitize significantly less in Kctd12−/− and Kctd12/16−/− neurons than in WT neurons, as expected from the lack of KCTD12 (Fig. 8A,B). When directly comparing Kctd12−/− and Kctd12/16−/− neurons, we observe significantly less desensitization in Kctd12/16−/− neurons than in Kctd12−/− neurons (Fig. 8B). Desensitization is activity-dependent and therefore influenced by the kinetic properties of the G-protein activation/deactivation cycle (i.e., the rates of GDP-GTP exchange and GTP hydrolysis at Gα) (Chuang et al., 1998; Leaney et al., 2004). Less desensitization in Kctd12/16−/− than in Kctd12−/− neurons may thus be caused by reduced precoupling of the G-protein at the receptor and a consequent slower K+ current activation. K+ currents displayed significantly increased desensitization in Kctd16−/− neurons versus WT neurons. Considering that two-thirds of the KCTD16 proteins in the hippocampus are associated with KCTD12 (Fig. 1B), the increased desensitization in Kctd16−/− neurons can be explained by the absence of KCTD12/KCTD16 complexes that normally would attenuate KCTD12-induced desensitization. In line with this proposal, expression of exogenous KCTD16 or 16ΔT1 in Kctd16−/− neurons significantly decreased desensitization (Fig. 8C,D), which in the case of 16ΔT1 must relate to oligomerization with endogenous KCTD12. In addition to forming hetero-oligomers with endogenous KCTD12, exogenous KCTD16 may additionally compete with endogenous KCTD12 for binding at the receptor, which further decreases desensitization. K+ current deactivation was significantly slowed in Kctd12−/− neurons compared with WT neurons (Fig. 8E,F), in line with a faster K+ current deactivation observed during prolonged baclofen application in the presence of KCTD12 in transfected CHO cells (Fig. 6F,G). The significantly slower current deactivation in Kctd12−/− compared with Kctd12/16−/− neurons (Fig. 8E,F) is also consistent with the observed slowing of current deactivation in CHO cells expressing KCTD16 (Fig. 6G). We also observed a slowing of current deactivation when overexpressing exogenous KCTD16 in Kctd16−/− neurons (Fig. 8F). In this case, overexpression of exogenous KCTD16 may outcompete endogenous KCTD12 (that accelerates deactivation) at the receptor. In contrast, no slowing of K+ current deactivation is observed when overexpressing exogenous 16ΔT1 in Kctd16−/− neurons, presumably because 16ΔT1 does not compete with endogenous KCTD12 for binding at the receptor. This result is consistent with results obtained in heterologous cells showing that coexpression of KCTD12 with 16ΔT1 produces similar deactivation kinetics as KCTD12 alone during prolonged baclofen application (Fig. 6F,G). Therefore, it appears that KCTD12 dictates the deactivation kinetics in KCTD12/KCTD16ΔT1 hetero-oligomers. In summary, the analysis of hippocampal neurons supports that the KCTD composition of GABAB receptors enables a bidirectional regulation of the desensitization and deactivation kinetics of receptor-activated K+ currents.
Figure 8.

KCTD12/KCTD16 hetero-oligomers modulate baclofen-activated K+ current responses in cultured hippocampal neurons. A, Representative traces of baclofen-evoked K+ currents recorded neurons of WT (black), Kctd12−/− (blue), Kctd16−/− (red), or Kctd12/16−/− (gray) mice. B, Bar graph summarizing K+ current desensitization in neurons of different genotypes. Data are mean ± SD of 18 (WT), 14 (Kctd12−/−), 13 (Kctd16−/−), and 12 (Kctd12/16−/−) neurons. Genetic ablation of KCTD12 or KCTD16 leads to decreased or increased current desensitization, respectively. C, Representative traces of baclofen-evoked K+ currents recorded from Kctd16−/− neurons expressing exogenous 16ΔT1. D, Bar graph summarizing K+ current desensitization in neurons with and without 16ΔT1 or KCTD16. Data are collected of 6 (Kctd16−/−), 7 (Kctd16−/− + 16ΔT1), and 6 (Kctd16−/− + KCTD16) neurons. E, Superimposed traces of the deactivation phase of baclofen-evoked K+ currents shown in A displayed with an expanded time scale. F, Bar graph summarizing the time constants obtained from a fit of the current deactivation to a single exponential function. The current deactivation is similar in Kctd16−/− neurons and WT neurons. However, current deactivation in Kctd12−/− neurons reveals a KCTD16-mediated slowing compared with Kctd12/16−/− neurons. Expression of KCTD16, but not 16ΔT1, in Kctd16−/− neurons significantly prolonged the K+ current deactivation phase, suggesting a competition of exogenous KCTD16 with endogenous KCTD12 at GABAB receptor. Data of 18 (WT), 14 (Kctd12−/−), 12 (Kctd16−/−), 12 (Kctd12/16−/−), 6 (Kctd16−/− + GFP), 7 (Kctd16−/− + 16ΔT1), and 4 (Kctd16−/− + KCTD16) neurons. *p < 0.05 (Dunnett's multiple-comparison test or Bonferroni pairwise comparison test). **p < 0.01 (Dunnett's multiple-comparison test or Bonferroni pairwise comparison test). ***p < 0.001 (Dunnett's multiple-comparison test or Bonferroni pairwise comparison test).
The current densities of baclofen-evoked K+ currents in Kctd12/Kctd16−/− neurons were significantly reduced compared with WT, Kctd12−/−, and Kctd16−/− neurons (WT: 1.7 ± 0.6 pA/pF, n = 18; Kctd12−/−: 1.5 ± 0.7 pA/pF, n = 16, Kctd16−/−: 1.6 ± 0.7 pA/pF, n = 13, Kctd12/Kctd16−/−: 1.0 ± 0.3 pA/pF, n = 13; p < 0.01; Dunnett's multiple-comparison test). This is consistent with the data from heterologous cells showing that KCTD12 and KCTD16 increase the peak amplitude of baclofen-induced K+ currents (Fig. 7C).
Altered GABAB-mediated sIPSCs in Kctd16−/− neurons
The data presented above suggest that the KCTD composition influences the kinetics of sIPSCs during prolonged GABAB receptor activation (Mapelli et al., 2009; Wang et al., 2010). However, whether the KCTD composition influences sIPSCs during phasic short receptor activation (De Koninck and Mody, 1997; Lüscher et al., 1997) is unclear. We therefore analyzed the decay of neuronal GABAB responses following a brief (1 s) pharmacological activation with baclofen (Fig. 9A,B). The deactivation of K+ currents in response to this brief baclofen stimulus is significantly faster in Kctd16−/− neurons compared with WT or Kctd12−/− neurons. This suggests that, in hippocampal neurons, KCTD16, but not KCTD12, influences the duration of GABAB receptor-induced phasic sIPSCs. To test whether KCTD16 indeed regulates the deactivation kinetics of synaptically evoked currents, we recorded sIPSCs elicited by electric stimulation of GABAergic fibers in the stratum radiatum from CA1 pyramidal neurons clamped at −50 mV (Fig. 9C). Fitting of the sIPSC decay phase with a one exponential function revealed that the decay time constant was significantly reduced in Kctd16−/− neurons compared with WT neurons (Fig. 9C–E). The maximal amplitude of sIPSCs (WT: 19.5 ± 8.0 pA, n = 8; Kctd16−/−: 21.1 ± 8.1 pA, n = 8) and the decay phase of GABAA receptor-mediated IPSCs (Fig. 9D,E) are not altered in Kctd16−/− neurons. This makes it unlikely that the changes observed in sIPSC kinetics in Kctd16−/− neurons are due to adaptive changes in GABA uptake or degradation. In Kctd12−/− neurons, the deactivation kinetics of sIPSCs were similar to WT neurons while their maximal amplitudes were significantly reduced (WT: 24.6 ± 9.1 pA, n = 11; Kctd12−/−: 16.8 ± 7.4, n = 11). The reduction in amplitude may relate to a decrease in receptor surface expression in the absence of KCTD12 (Ivankova et al., 2013). In summary, we find that the kinetic properties of sIPSCs in hippocampal neurons are shaped by the KCTD16-mediated slowing of K+ current deactivation, an effect that is retained in hetero-oligomeric complexes with KCTD12.
Figure 9.
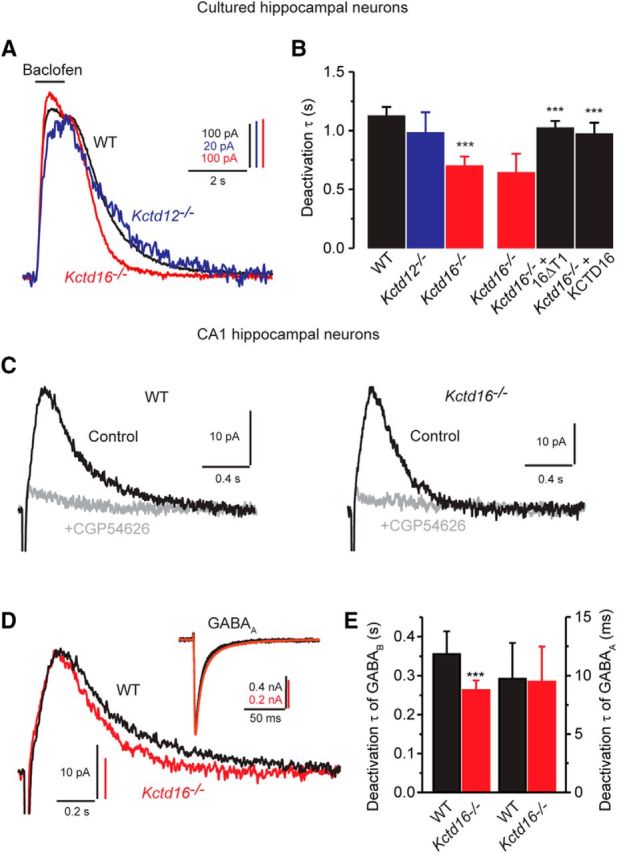
Rapid deactivation kinetics of sIPSCs in Kctd16−/− neurons. A, Superimposed traces showing K+ currents evoked by application of baclofen for 1 s to cultured hippocampal neurons of WT, Kctd12−/−, or Kctd16−/− mice. B, Deactivation time constants obtained from fitting the deactivation phase to a single exponential function. The deactivation time constant was similar in WT and Kctd12−/− neurons but significantly reduced in Kctd16−/− neurons. The slow deactivation of the currents was restored in Kctd16−/− neurons transfected with 16ΔT1 or KCTD16. Data of 6 (WT), 7 (Kctd12−/−), 8 (Kctd16−/−), 5 (Kctd16−/− + 16ΔT1), and 5 (Kctd16−/− + KCTD16) neurons. ***p < 0.001 (Dunnett's multiple-comparison test). C, Examples of sIPSCs recorded from CA1 hippocampal neurons of WT or Kctd16−/− mice in the absence (Control) or presence of the specific GABAB receptor antagonist CGP54626 (holding potential −60 mV). Traces are averages of 10 sIPSCs. D, Superimposed traces of control sIPSCs shown in C displayed with an expanded time scale. Note the faster deactivation kinetics obtained from Kctd16−/− neurons. Inset, GABAA receptor-mediated IPSCs recorded from CA1 hippocampal neurons as in C (in the absence of gabazine), clamped at −60 mV. E, Bar graph summarizing the time constants obtained from a fit of the sIPSC deactivation phase to a single exponential function (left) and the amplitude-weighted mean time constants obtained from a fit of GABAA IPSC deactivation phase to a double exponential function (right). Data are of 8 (WT) or 8 (Kctd16−/−) experiments. ***p < 0.001 (unpaired Student's t test).
Discussion
It is well established that KCTD homo-oligomers bind to GABAB receptors and regulate G-protein signaling of the receptor (Schwenk et al., 2010; Turecek et al., 2014; Rajalu et al., 2015). Here we demonstrate that KCTDs also form hetero-oligomers in all possible dual combinations. We further show that GABAB receptors assembled with KCTD12/KCTD16 hetero-oligomers combine regulatory properties of the individual KCTDs to generate receptors with novel signaling characteristics. In response to prolonged receptor stimulation for 1 min, KCTD12/KCTD16 hetero-oligomers produce moderately desensitizing fast deactivating currents whereas KCTD12 and KCTD16 homo-oligomers produce strongly desensitizing fast deactivating currents and nondesensitizing slowly deactivating currents, respectively. In response to brief receptor stimulation (2 s), hetero-oligomers produce nondesensitizing slowly deactivating currents. Accordingly, we show that, in hippocampal neurons, KCTD12/KCTD16 hetero-oligomers significantly slow the deactivation kinetics of phasic GABAB receptor-induced sIPSCs.
KCTD hetero-oligomers in the brain
Hetero-oligomerization of KCTD8, KCTD12, and KCTD16 is mediated by the self-interacting T1 and H1 homology domains that also mediate homo-oligomerization (Schwenk et al., 2010; Correale et al., 2013). Hetero-oligomerization was observed within other KCTD clades (Sowa et al., 2009; De Smaele et al., 2011). Based on our BiLC experiments we conclude that KCTD oligomers arrange in parallel, similar as with KCTD5 (Dementieva et al., 2009). BiLC experiments are consistent with the proposed tetrameric or pentameric assembly of native KCTD oligomers (Schwenk et al., 2010; Correale et al., 2013; Smaldone et al., 2016). Because many neurons in the brain simultaneously express several KCTDs and some possibly all KCTDs (Metz et al., 2011), hetero-oligomers are expected to be abundant. In the hippocampus, we estimate that approximately two-thirds of the KCTD16 proteins are involved in KCTD12/KCTD16 hetero-oligomers. The KCTDs assemble with GABAB receptors at the cytoplasmic side of the endoplasmic reticulum membrane (Ivankova et al., 2013). It is currently unknown whether KCTD hetero-oligomers assemble randomly or combine by a regulated process.
Effects on G-protein signaling
KCTD12 promotes current desensitization by uncoupling Gβγ subunits from effector K+ channels via its H1 domain (Seddik et al., 2012; Turecek et al., 2014). In contrast, KCTD16 prevents desensitization via its H2 domain (Seddik et al., 2012). We now show that KCTD hetero-oligomers not only bind to the receptor but also to the G-protein. Simultaneous incorporation of distinct KCTD proteins into GABAB receptors therefore allows combining regulatory effects on G-protein signaling into individual receptors. We found that, in KCTD12/KCTD16 hetero-oligomers, both KCTDs retain their regulatory effect on K+ current desensitization. Reconstitution experiments in CHO cells reveal that KCTD12/KCTD16 hetero-oligomers yield K+ currents with intermediate desensitization kinetics. We also observed that KCTD12 and KCTD16 have opposite effects on the deactivation of GABAB receptor-activated K+ currents; KCTD12 accelerates, whereas KCTD16 decelerates, deactivation. Interestingly, faster deactivation kinetics appear to depend on the extent of KCTD12-induced desensitization. In the presence of KCTD12, prolonged receptor activation results in desensitized K+ currents exhibiting fast deactivation while short activation produces nondesensitized K+ currents with slow deactivation. The underlying molecular mechanism is unknown but might relate to scavenging of Gβγ from the Kir3 channel by KCTD12. At steady-state desensitization, scavenging of Gβγ will result in lower Gβγ occupancy at the channel (Sadja et al., 2002; Whorton and MacKinnon, 2013; Yakubovich et al., 2015), which will accelerate dissociation of Gβγ and deactivation of the response. Similarly, the exact mechanism by which KCTD16 slows Kir3 current deactivation is unknown. Because the mutant 16ΔT1 slows the deactivation in the absence of binding to the receptor, the deactivation mechanism likely operates at the G-protein and/or at the effector channel. KCTD16 may, for example, slow current deactivation by facilitating binding of G-protein βγ subunits at Kir3 channels. This would explain why KCTD16 slows deactivation in the absence (short receptor activation) but not in the presence of βγ scavenging by KCTD12 (prolonged activation). Alternatively, it is also possible that KCTD16 slows current deactivation by interfering with the activity of endogenous regulator of G-protein signaling (RGS) proteins at the G-protein α subunit (Xie et al., 2010; Zhou et al., 2012; Ostrovskaya et al., 2014).
Physiological relevance of KCTD hetero-oligomers
Our electrophysiological experiments in hippocampal neurons support that KCTD12/KCTD16 hetero-oligomers slow the deactivation kinetics of GABAB-induced K+ currents during brief activation of the receptor. The deactivation kinetics of K+ currents is known to be a limiting step for the termination of GABAB-mediated sIPSCs (Xie et al., 2010). Accordingly, we found that, in Kctd16−/− hippocampal neurons, the decay of GABAB-activated sIPSCs is significantly accelerated. Conversely, a recent study reported that the decay of GABAB receptor-activated sIPSCs in Kctd12−/− cholecystokinin-containing interneurons, which normally express high amounts of KCTD12, is significantly decelerated (Booker et al., 2016). These studies highlight that the KCTDs can directly influence the kinetics of sIPSCs in select neuronal populations.
Of note, our experiments show that the kinetic changes measured in electrophysiological experiments with knock-out neurons are less pronounced than those measured in cells in which receptor assemblies are forced through overexpression of KCTD subunits. Earlier proteomic experiments revealed that ∼60% of all GABAB receptors in the adult mouse brain are devoid of KCTD proteins (Turecek et al., 2014). In KCTD knock-out neurons, the responses of affected GABAB receptors are partly masked by the responses of unaffected GABAB receptors. Electrophysiological whole-cell recordings from knock-out neurons thus underestimate the kinetic changes at individual receptors. Nevertheless, we observe significant kinetic changes in knock-out neurons. These changes are likely to be of physiological relevance, given that Kctd12−/− and Kctd16−/− mice exhibit behavioral phenotypes (Cathomas et al., 2015, 2017). Of note, behavioral phenotypes in Kctd12−/− mice can be directly related to GABAB receptor signaling because KCTD12 in the adult brain is exclusively associated with GABAB receptors (Turecek et al., 2014). Modulatory effects on the kinetics of GABAB-activated K+ currents were also observed for RGS proteins. For example, RGS7 was shown to act in concert with Gβ5 to accelerate GABAB receptor induced K+ current deactivation (Xie et al., 2010; Zhou et al., 2012; Ostrovskaya et al., 2014), whereas RGS4 enhances K+ current desensitization (Mutneja et al., 2005). Likewise, phosphorylation of GABAB2 by cAMP-dependent protein kinase (PKA) also influences the kinetics of the receptor response (Couve et al., 2002; Adelfinger et al., 2014).
Based on our findings, we propose that KCTD hetero-oligomers serve as regulatory building blocks that enable a fine-tuning of GABAB receptor-induced K+ currents. In this context, it will be important to determine the KCTD composition of GABAB receptors in axonal, somatic, and dendritic compartments of identified neurons. Both the distribution of KCTD proteins (Metz et al., 2011) and the temporal dynamics of receptor activation may vary between these compartments and, as we show here, will influence the receptor response. Mounting evidence also suggests that the level of inhibition mediated by GABAB-Kir3 signaling is tuned to changes in neuronal excitability and modified by drugs of abuse (Huang et al., 2005; Arora et al., 2011; Padgett et al., 2012; Hearing et al., 2013). It will therefore be important to address whether the composition of GABAB/KCTD receptor complexes is regulated, as has been suggested for other GPCRs and their associated proteins (Maurice et al., 2012). For example, changes in KCTD12 expression during development (Resendes et al., 2004) or in disease (Glatt et al., 2005; Miller et al., 2008; Sibille et al., 2009; Benes, 2010; Lee et al., 2011; Cathomas et al., 2015) might alter the kinetics of receptor responses through influencing the composition of KCTD hetero-oligomers.
Footnotes
This work was supported by National Center for Competences in Research “Synapsy, Synaptic Bases of Mental Health Disease” Grant to B.B., Swiss National Science Foundation Grant 31003A-152970 to B.B., Canadian Institute for Health Research Grant MOP-10501 to M.B., and Grant Agency of the Czech Republic Grant GACR P303/14/28334S to M.K. M.B. holds a Canada Research Chair in Signal Transduction and Molecular Pharmacology. We thank Bolette Christiansen, Alessandra Porcu, and Klara Ivankova for contributions to biochemical or electrophysiological experiments; and Bernd Fakler, Uwe Schulte, and Jochen Schwenk for helpful discussions.
The authors declare no competing financial interests.
References
- Adelfinger L, Turecek R, Ivankova K, Jensen AA, Moss SJ, Gassmann M, Bettler B (2014) GABAB receptor phosphorylation regulates KCTD12-induced K+ current desensitization. Biochem Pharmacol 91:369–379. 10.1016/j.bcp.2014.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armando S, Quoyer J, Lukashova V, Maiga A, Percherancier Y, Heveker N, Pin JP, Prezeau L, Bouvier M (2014) The chemokine CXC4 and CC2 receptors form homo- and heterooligomers that can engage their signaling G-protein effectors and βarrestin. FASEB J 10:4509–4523. 10.1096/fj.13-242446 [DOI] [PubMed] [Google Scholar]
- Arora D, Hearing M, Haluk DM, Mirkovic K, Fajardo-Serrano A, Wessendorf MW, Watanabe M, Luján R, Wickman K (2011) Acute cocaine exposure weakens GABAB receptor-dependent G-protein-gated inwardly rectifying K+ signaling in dopamine neurons of the ventral tegmental area. J Neurosci 31:12251–12257. 10.1523/JNEUROSCI.0494-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoi T, Rigbolt KT, Du D, Köhr G, Blagoev B, Kornau HC (2010) GABAB receptor constituents revealed by tandem affinity purification from transgenic mice. J Biol Chem 285:20625–20633. 10.1074/jbc.M109.049700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM. (2010) Amygdalocortical circuitry in schizophrenia: from circuits to molecules. Neuropsychopharmacology 35:239–257. 10.1038/npp.2009.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biermann B, Ivankova-Susankova K, Bradaia A, Abdel Aziz S, Besseyrias V, Kapfhammer JP, Missler M, Gassmann M, Bettler B (2010) The Sushi domains of GABAB receptors function as axonal targeting signals. J Neurosci 30:1385–1394. 10.1523/JNEUROSCI.3172-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker SA, Gross A, Althof D, Shigemoto R, Bettler B, Frotscher M, Hearing M, Wickman K, Watanabe M, Kulik Á, Vida I (2013) Differential GABAB-receptor-mediated effects in perisomatic- and dendrite-targeting parvalbumin interneurons. J Neurosci 33:7961–7974. 10.1523/JNEUROSCI.1186-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker SA, Althof D, Gross A, Loreth D, Muller J, Unger A, Fakler B, Varro A, Watanabe M, Gassmann M, Bettler B, Shigemoto R, Vida I, Kulik A (2016) KCTD12 auxiliary proteins modulate kinetics of GABAB receptor-mediated inhibition in cholecystokinin-containing interneurons. Cereb Cortex. Advance online publication. Retrieved Apr. 12, 2016. doi: 10.1093/cercor/bhw090. 10.1093/cercor/bhw090 [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35:567–576. 10.1002/jnr.490350513 [DOI] [PubMed] [Google Scholar]
- Cathomas F, Stegen M, Sigrist H, Schmid L, Seifritz E, Gassmann M, Bettler B, Pryce CR (2015) Altered emotionality and neuronal excitability in mice lacking KCTD12, an auxiliary subunit of GABAB receptors associated with mood disorders. Transl Psychiatry 5:e510. 10.1038/tp.2015.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathomas F, Sigrist H, Schmid L, Seifritz E, Gassmann M, Bettler B, Pryce CR (2017) Behavioural endophenotypes in mice lacking the auxiliary GABAB receptor subunit KCTD16. Behav Brain Res 317:393–400. 10.1016/j.bbr.2016.10.006 [DOI] [PubMed] [Google Scholar]
- Chalifoux JR, Carter AG (2011) GABAB receptor modulation of synaptic function. Curr Opin Neurobiol 21:339–344. 10.1016/j.conb.2011.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Yu M, Jan YN, Jan LY (1998) Evidence that the nucleotide exchange and hydrolysis cycle of G proteins causes acute desensitization of G-protein gated inward rectifier K+ channels. Proc Natl Acad Sci U S A 95:11727–11732. 10.1073/pnas.95.20.11727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correale S, Esposito C, Pirone L, Vitagliano L, Di Gaetano S, Pedone E (2013) A biophysical characterization of the folded domains of KCTD12: insights into interaction with the GABAB2 receptor. J Mol Recognit 26:488–495. 10.1002/jmr.2291 [DOI] [PubMed] [Google Scholar]
- Couve A, Moss SJ, Pangalos MN (2000) GABAB receptors: a new paradigm in G protein signaling. Mol Cell Neurosci 16:296–312. 10.1006/mcne.2000.0908 [DOI] [PubMed] [Google Scholar]
- Couve A, Thomas P, Calver AR, Hirst WD, Pangalos MN, Walsh FS, Smart TG, Moss SJ (2002) Cyclic AMP-dependent protein kinase phosphorylation facilitates GABAB receptor-effector coupling. Nat Neurosci 5:415–424. 10.1038/nn833 [DOI] [PubMed] [Google Scholar]
- De Koninck Y, Mody I (1997) Endogenous GABA activates small-conductance K+ channels underlying slow IPSCs in rat hippocampal neurons. J Neurophysiol 77:2202–2208. [DOI] [PubMed] [Google Scholar]
- Dementieva IS, Tereshko V, McCrossan ZA, Solomaha E, Araki D, Xu C, Grigorieff N, Goldstein SA (2009) Pentameric assembly of potassium channel tetramerization domain-containing protein 5. J Mol Biol 387:175–191. 10.1016/j.jmb.2009.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smaele E, Di Marcotullio L, Moretti M, Pelloni M, Occhione MA, Infante P, Cucchi D, Greco A, Pietrosanti L, Todorovic J, Coni S, Canettieri G, Ferretti E, Bei R, Maroder M, Screpanti I, Gulino A (2011) Identification and characterization of KCASH2 and KCASH3, 2 novel Cullin3 adaptors suppressing histone deacetylase and Hedgehog activity in medulloblastoma. Neoplasia 13:374–385. 10.1593/neo.101630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittert I, Benedikt J, Vyklický L, Zimmermann K, Reeh PW, Vlachová V (2006) Improved superfusion technique for rapid cooling or heating of cultured cells under patch-clamp conditions. J Neurosci Methods 151:178–185. 10.1016/j.jneumeth.2005.07.005 [DOI] [PubMed] [Google Scholar]
- Gassmann M, Bettler B (2012) Regulation of neuronal GABAB receptor functions by subunit composition. Nat Rev Neurosci 13:380–394. 10.1038/nrn3249 [DOI] [PubMed] [Google Scholar]
- Glatt SJ, Everall IP, Kremen WS, Corbeil J, Sásik R, Khanlou N, Han M, Liew CC, Tsuang MT (2005) Comparative gene expression analysis of blood and brain provides concurrent validation of SELENBP1 up-regulation in schizophrenia. Proc Natl Acad Sci U S A 102:15533–15538. 10.1073/pnas.0507666102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayasaki H, Sohma Y, Kanbara K, Otsuki Y (2012) Heterogenous GABAB receptor-mediated pathways are involved in the local GABAergic system of the rat trigeminal ganglion: possible involvement of KCTD proteins. Neuroscience 218:344–358. 10.1016/j.neuroscience.2012.05.038 [DOI] [PubMed] [Google Scholar]
- Hearing M, Kotecki L, Marron Fernandez de Velasco E, Fajardo-Serrano A, Chung HJ, Luján R, Wickman K (2013) Repeated cocaine weakens GABAB-Girk signaling in layer 5/6 pyramidal neurons in the prelimbic cortex. Neuron 80:159–170. 10.1016/j.neuron.2013.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Héroux M, Hogue M, Lemieux S, Bouvier M (2007) Functional calcitonin gene-related peptide receptors are formed by the asymmetric assembly of a calcitonin receptor-like receptor homo-oligomer and a monomer of receptor activity-modifying protein-1. J Biol Chem 282:31610–31620. 10.1074/jbc.M701790200 [DOI] [PubMed] [Google Scholar]
- Huang CS, Shi SH, Ule J, Ruggiu M, Barker LA, Darnell RB, Jan YN, Jan LY (2005) Common molecular pathways mediate long-term potentiation of synaptic excitation and slow synaptic inhibition. Cell 123:105–118. 10.1016/j.cell.2005.07.033 [DOI] [PubMed] [Google Scholar]
- Ivankova K, Turecek R, Fritzius T, Seddik R, Prezeau L, Comps-Agrar L, Pin JP, Fakler B, Besseyrias V, Gassmann M, Bettler B (2013) Up-regulation of GABAB receptor signaling by constitutive assembly with the K+ channel tetramerization domain-containing protein 12 (KCTD12). J Biol Chem 288:24848–24856. 10.1074/jbc.M113.476770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaney JL, Benians A, Brown S, Nobles M, Kelly D, Tinker A (2004) Rapid desensitization of G protein-gated inwardly rectifying K+ currents is determined by G protein cycle. Am J Physiol Cell Physiol 287:C182–C191. 10.1152/ajpcell.00540.2003 [DOI] [PubMed] [Google Scholar]
- Lee MT, Chen CH, Lee CS, Chen CC, Chong MY, Ouyang WC, Chiu NY, Chuo LJ, Chen CY, Tan HK, Lane HY, Chang TJ, Lin CH, Jou SH, Hou YM, Feng J, Lai TJ, Tung CL, Chen TJ, Chang CJ, et al. (2011) Genome-wide association study of bipolar I disorder in the Han Chinese population. Mol Psychiatry 16:548–556. 10.1038/mp.2010.43 [DOI] [PubMed] [Google Scholar]
- Liu Z, Xiang Y, Sun G (2013) The KCTD family of proteins: structure, function, disease relevance. Cell Biosci 3:45. 10.1186/2045-3701-3-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Slesinger PA (2010) Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11:301–315. 10.1038/nrn2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA (1997) G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19:687–695. 10.1016/S0896-6273(00)80381-5 [DOI] [PubMed] [Google Scholar]
- Mapelli L, Rossi P, Nieus T, D'Angelo E (2009) Tonic activation of GABAB receptors reduces release probability at inhibitory connections in the cerebellar glomerulus. J Neurophysiol 101:3089–3099. 10.1152/jn.91190.2008 [DOI] [PubMed] [Google Scholar]
- Maurice P, Benleulmi-Chaachoua A, Jockers R (2012) Differential assembly of GPCR signaling complexes determines signaling specificity. Subcell Biochem 63:225–240. 10.1007/978-94-007-4765-4_12 [DOI] [PubMed] [Google Scholar]
- Mercier JF, Salahpour A, Angers S, Breit A, Bouvier M (2002) Quantitative assessment of β1- and β2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J Biol Chem 277:44925–44931. 10.1074/jbc.M205767200 [DOI] [PubMed] [Google Scholar]
- Metz M, Gassmann M, Fakler B, Schaeren-Wiemers N, Bettler B (2011) Distribution of the auxiliary GABAB receptor subunits KCTD8, 12, 12b, and 16 in the mouse brain. J Comp Neurol 519:1435–1454. 10.1002/cne.22610 [DOI] [PubMed] [Google Scholar]
- Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, Ma R, Cole SW (2008) A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-κB signaling. Biol Psychiatry 64:266–272. 10.1016/j.biopsych.2008.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möhler H, Fritschy JM (1999) GABAB receptors make it to the top: as dimers. Trends Pharmacol Sci 20:87–89. 10.1016/S0165-6147(99)01323-1 [DOI] [PubMed] [Google Scholar]
- Monnier C, Tu H, Bourrier E, Vol C, Lamarque L, Trinquet E, Pin JP, Rondard P (2011) Trans-activation between 7TM domains: implication in heterodimeric GABAB receptor activation. EMBO J 30:32–42. 10.1038/emboj.2010.270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutneja M, Berton F, Suen KF, Lüscher C, Slesinger PA (2005) Endogenous RGS proteins enhance acute desensitization of GABAB receptor-activated GIRK currents in HEK-293T cells. Pflugers Arch 450:61–73. 10.1007/s00424-004-1367-1 [DOI] [PubMed] [Google Scholar]
- Ostrovskaya O, Xie K, Masuho I, Fajardo-Serrano A, Lujan R, Wickman K, Martemyanov KA (2014) RGS7/Gβ5/R7BP complex regulates synaptic plasticity and memory by modulating hippocampal GABABR-GIRK signaling. Elife 3:e02053. 10.7554/eLife.02053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett CL, Lalive AL, Tan KR, Terunuma M, Munoz MB, Pangalos MN, Martínez-Hernández J, Watanabe M, Moss SJ, Luján R, Lüscher C, Slesinger PA (2012) Methamphetamine-evoked depression of GABAB receptor signaling in GABA neurons of the VTA. Neuron 73:978–989. 10.1016/j.neuron.2011.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Kniazeff J, Binet V, Liu J, Maurel D, Galvez T, Duthey B, Havlickova M, Blahos J, Prézeau L, Rondard P (2004) Activation mechanism of the heterodimeric GABAB receptor. Biochem Pharmacol 68:1565–1572. 10.1016/j.bcp.2004.06.035 [DOI] [PubMed] [Google Scholar]
- Rajalu M, Fritzius T, Adelfinger L, Jacquier V, Besseyrias V, Gassmann M, Bettler B (2015) Pharmacological characterization of GABAB receptor subtypes assembled with auxiliary KCTD subunits. Neuropharmacology 88:145–154. 10.1016/j.neuropharm.2014.08.020 [DOI] [PubMed] [Google Scholar]
- Resendes BL, Kuo SF, Robertson NG, Giersch AB, Honrubia D, Ohara O, Adams JC, Morton CC (2004) Isolation from cochlea of a novel human intronless gene with predominant fetal expression. J Assoc Res Otolaryngol 5:185–202. 10.1007/s10162-003-4042-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadja R, Alagem N, Reuveny E (2002) Graded contribution of the Gβγ binding domains to GIRK channel activation. Proc Natl Acad Sci U S A 99:10783–10788. 10.1073/pnas.162346199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Metz M, Zolles G, Turecek R, Fritzius T, Bildl W, Tarusawa E, Kulik A, Unger A, Ivankova K, Seddik R, Tiao JY, Rajalu M, Trojanova J, Rohde V, Gassmann M, Schulte U, Fakler B, Bettler B (2010) Native GABAB receptors are heteromultimers with a family of auxiliary subunits. Nature 465:231–235. 10.1038/nature08964 [DOI] [PubMed] [Google Scholar]
- Schwenk J, Pérez-Garci E, Schneider A, Kollewe A, Gauthier-Kemper A, Fritzius T, Raveh A, Dinamarca MC, Hanuschkin A, Bildl W, Klingauf J, Gassmann M, Schulte U, Bettler B, Fakler B (2016) Modular composition and dynamics of native GABAB receptors identified by high-resolution proteomics. Nat Neurosci 19:233–242. 10.1038/nn.4198 [DOI] [PubMed] [Google Scholar]
- Seddik R, Jungblut SP, Silander OK, Rajalu M, Fritzius T, Besseyrias V, Jacquier V, Fakler B, Gassmann M, Bettler B (2012) Opposite effects of KCTD subunit domains on GABAB receptor-mediated desensitization. J Biol Chem 287:39869–39877. 10.1074/jbc.M112.412767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibille E, Wang Y, Joeyen-Waldorf J, Gaiteri C, Surget A, Oh S, Belzung C, Tseng GC, Lewis DA (2009) A molecular signature of depression in the amygdala. Am J Psychiatry 166:1011–1024. 10.1176/appi.ajp.2009.08121760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoblov M, Marakhonov A, Marakasova E, Guskova A, Chandhoke V, Birerdinc A, Baranova A (2013) Protein partners of KCTD proteins provide insights about their functional roles in cell differentiation and vertebrate development. Bioessays 35:586–596. 10.1002/bies.201300002 [DOI] [PubMed] [Google Scholar]
- Smaldone G, Pirone L, Pedone E, Marlovits T, Vitagliano L, Ciccarelli L (2016) The BTB domains of the potassium channel tetramerization domain proteins prevalently assume pentameric states. FEBS Lett 590:1663–1671. 10.1002/1873-3468.12203 [DOI] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403. 10.1016/j.cell.2009.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan E, Aquin S, Berger N, Landry CR, Nyfeler B, Bouvier M, Michnick SW (2007) Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc Natl Acad Sci U S A 104:16916–16921. 10.1073/pnas.0704257104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek R, Vlcek K, Petrovic M, Horak M, Vlachova V, Vyklicky L Jr (2004) Intracellular spermine decreases open probability of N-methyl-D-aspartate receptor channels. Neuroscience 125:879–887. 10.1016/j.neuroscience.2004.03.003 [DOI] [PubMed] [Google Scholar]
- Turecek R, Schwenk J, Fritzius T, Ivankova K, Zolles G, Adelfinger L, Jacquier V, Besseyrias V, Gassmann M, Schulte U, Fakler B, Bettler B (2014) Auxiliary GABAB receptor subunits uncouple G protein βγ subunits from effector channels to induce desensitization. Neuron 82:1032–1044. 10.1016/j.neuron.2014.04.015 [DOI] [PubMed] [Google Scholar]
- Urwyler S, Mosbacher J, Lingenhoehl K, Heid J, Hofstetter K, Froestl W, Bettler B, Kaupmann K (2001) Positive allosteric modulation of native and recombinant γ-aminobutyric acidB receptors by 2,6-di-tert-butyl-4-(3-hydroxy-2,2-dimethyl-propyl)-phenol (CGP7930) and its aldehyde analog CGP13501. Mol Pharmacol 60:963–971. 10.1124/mol.60.5.963 [DOI] [PubMed] [Google Scholar]
- Villemure JF, Adam L, Bevan NJ, Gearing K, Chénier S, Bouvier M (2005) Subcellular distribution of GABAB receptor homo- and hetero-dimers. Biochem J 388:47–55. 10.1042/BJ20041435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Neubauer FB, Lüscher HR, Thurley K (2010) GABAB receptor-dependent modulation of network activity in the rat prefrontal cortex in vitro. Eur J Neurosci 31:1582–1594. 10.1111/j.1460-9568.2010.07191.x [DOI] [PubMed] [Google Scholar]
- Whorton MR, MacKinnon R (2013) X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature 498:190–197. 10.1038/nature12241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie K, Allen KL, Kourrich S, Colón-Saez J, Thomas MJ, Wickman K, Martemyanov KA (2010) Gβ5 recruits R7 RGS proteins to GIRK channels to regulate the timing of neuronal inhibitory signaling. Nat Neurosci 13:661–663. 10.1038/nn.2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakubovich D, Berlin S, Kahanovitch U, Rubinstein M, Farhy-Tselnicker I, Styr B, Keren-Raifman T, Dessauer CW, Dascal N (2015) A quantitative model of the GIRK1/2 channel reveals that its basal and evoked activities are controlled by unequal stoichiometry of Gα and Gβγ. PLoS Comput Biol 11:e1004598. 10.1371/journal.pcbi.1004598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Chisari M, Raehal KM, Kaltenbronn KM, Bohn LM, Mennerick SJ, Blumer KJ (2012) GIRK channel modulation by assembly with allosterically regulated RGS proteins. Proc Natl Acad Sci U S A 109:19977–19982. 10.1073/pnas.1214337109 [DOI] [PMC free article] [PubMed] [Google Scholar]