

Abstract
Long-term potentiation (LTP) is widely perceived as a memory substrate and in the hippocampal CA3-CA1 pathway, distinct forms of LTP depend on NMDA receptors (nmdaLTP) or L-type voltage-gated calcium channels (vdccLTP). LTP is also known to be effectively regulated by extracellular proteolysis that is mediated by various enzymes. Herein, we investigated whether in mice hippocampal slices these distinct forms of LTP are specifically regulated by different metalloproteinases (MMPs). We found that MMP-3 inhibition or knock-out impaired late-phase LTP in the CA3-CA1 pathway. Interestingly, late-phase LTP was also decreased by MMP-9 blockade. When both MMP-3 and MMP-9 were inhibited, both early- and late-phase LTP was impaired. Using immunoblotting, in situ zymography, and immunofluorescence, we found that LTP induction was associated with an increase in MMP-3 expression and activity in CA1 stratum radiatum. MMP-3 inhibition and knock-out prevented the induction of vdccLTP, with no effect on nmdaLTP. L-type channel-dependent LTP is known to be impaired by hyaluronic acid digestion. We found that slice treatment with hyaluronidase occluded the effect of MMP-3 blockade on LTP, further confirming a critical role for MMP-3 in this form of LTP. In contrast to the CA3-CA1 pathway, LTP in the mossy fiber-CA3 projection did not depend on MMP-3, indicating the pathway specificity of the actions of MMPs. Overall, our study indicates that the activation of perisynaptic MMP-3 supports L-type channel-dependent LTP in the CA1 region, whereas nmdaLTP depends solely on MMP-9.
SIGNIFICANCE STATEMENT Various types of long-term potentiation (LTP) are correlated with distinct phases of memory formation and retrieval, but the underlying molecular signaling pathways remain poorly understood. Extracellular proteases have emerged as key players in neuroplasticity phenomena. The present study found that L-type calcium channel-dependent LTP in the CA3-CA1 hippocampal projection is critically regulated by the activity of matrix metalloprotease 3 (MMP-3), in contrast to NMDAR-dependent LTP regulated by MMP-9. Moreover, the induction of LTP was associated with an increase in MMP-3 expression and activity. Finally, we found that the digestion of hyaluronan, a principal extracellular matrix component, disrupted the MMP-3-dependent component of LTP. These results indicate that distinct MMPs might act as molecular switches for specific types of LTP.
Keywords: extracellular matrix, L-type calcium channels, long-term potentiation, matrix metalloproteinase, proteolysis, synaptic plasticity
Introduction
Long-term potentiation (LTP) at glutamatergic synapses is widely perceived as a memory substrate, but the underlying molecular mechanisms are not fully understood as they involve myriad of participating elements and processes. In the hippocampal CA3-CA1 pathway, specific patterns of stimulation differentially activate NMDARs and L-type voltage-dependent calcium channels (VDCCs), resulting in distinct forms of LTP: nmdaLTP and vdccLTP, respectively (Grover and Teyler, 1990; Blundon and Zakharenko, 2008). Coincidence detecting NMDARs are known for their involvement in memory formation processes, especially episodic-like memory in the hippocampus (for review, see Morris (2013). More recently, also L-type calcium channels have been implicated in the maintenance of long-term spatial memory upon its reactivation (Da Silva et al., 2013). Thus, these channels play fundamental but clearly distinct roles in synaptic plasticity and memory consolidation/reconsolidation processes, and for this reason further elucidation of the molecular signaling pathways that involve these channels is crucially important. Notably, vdccLTP critically depends on the extracellular matrix (ECM), as the digestion of hyaluronic acid, a major ECM component, specifically abolishes vdccLTP in the hippocampus (Kochlamazashvili et al., 2010). This work indicated that specific mechanisms of plasticity can be effectively controlled by the proteolysis of specific ECM components. The enzymatic manipulation of ECM molecules affects distinct types of synaptic plasticity and learning (Senkov et al., 2014), but understanding the roles of specific ECM domains in synaptic functions and signaling events downstream of ECM modifications remains a major challenge in the field. It thus seems interesting to explore the ways in which signaling pathways that are related to nmdaLTP and vdccLTP depend on distinct, endogenous proteolytic activity. It is of note in this context that in several key experiments addressing the effects of hyaluronic acid and proteoglycan digestion on learning and memory (Pizzorusso et al., 2002; Gogolla et al., 2009; Kochlamazashvili et al., 2010) exogenous enzymes were used, which are not present in the mammalian brain. Therefore, studies on endogenous ECM-modifying enzymes are required to shed new light on changes in ECM structure and functions during synaptic plasticity.
In the CA1 region of the hippocampus, LTP consists of early-phase (early LTP) that requires the activity of kinases and late-phase LTP (late LTP) that is known to depend on protein synthesis and proteolysis (Nagy et al., 2006). Several substrates of proteolytic enzymes, such as membrane adhesion proteins and ECM molecules, have emerged as real or putative players in shaping plastic changes at the synaptic level and beyond (Tsien, 2013). Activity of tissue plasminogen activator (tPA), neuropsin, and matrix metalloproteinases (MMPs), has been implicated in synaptic plasticity and learning (Sonderegger and Matsumoto-Miyai, 2014). At least 24 human MMP genes have been broadly divided into classes, including gelatinases (MMP-2, MMP-9) and stromelysins (MMP-3, MMP-10), among others. To date, the role of gelatinases has been the most extensively studied. MMP-9 has been implicated in brain development/neurogenesis (Verslegers et al., 2013), the modification of dendritic spine morphology (Sidhu et al., 2014), synaptic plasticity (Nagy et al., 2006; Wiera et al., 2013), and memory formation (Peixoto et al., 2012; Smith et al., 2014). Although less intensively studied, MMP-3 has also been suggested to play a role in synaptic plasticity and learning (Olson et al., 2008; Conant et al., 2010), but the underlying mechanisms remain more obscure than for MMP-9. Additionally, MMP-3, as opposed to MMP-9, may potentially cleave all brain chondroitin sulfate proteoglycans (Van Hove et al., 2012a), which are known to affect synaptic plasticity and memory (Senkov et al., 2014). We examined the differential roles of MMP-3 and MMP-9 in supporting early and late LTP that was induced by paradigms that depend on NMDARs and VDCCs. We found that the consolidation of vdccLTP in the hippocampal CA3-CA1 projection critically depended on MMP-3, and this process was accompanied by an increase in the expression and activity of this enzyme.
Materials and Methods
Animals.
All of the animal procedures were approved by the Local Ethics Commission, and all efforts were made to minimize the number of animals used for the experiments. The mice were maintained under a standard 12 h/12 h light/dark cycle and socially housed. Brain slices were prepared from male mice. Both male and female wild-type (WT), MMP-3 knock-out (KO), and MMP-9 KO mice (6–8 weeks old, all on a C57BL/6J background) were used. In the analyses that included both male and female animals, sex differences were also tested, but no differences were found, and the data were pooled.
Preparation of hippocampal slices.
Acute mouse hippocampal transverse slices (350 μm) were prepared from C57BL/6 mice (postnatal day 60–100) as described previously (Wiera et al., 2013). In some of the experiments, homozygous MMP-3 or MMP-9 KO mice were used. MMP-3 KO mice are viable, reach adulthood, and present detectable morphological abnormalities in the cortex and cerebellum (Van Hove et al., 2012b; Aerts et al., 2015). The mice were anesthetized with isoflurane. Following brain dissection, slices were cut with a vibratome (VT1200S, Leica) and placed in ice-cold cutting solution that contained 75 mm sucrose, 87 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 0.5 mm CaCl2, 3.5 mm MgCl2, 3.5 mm MgSO4, and 20 mm glucose, pH 7.4. The slices were then incubated in the same solution at 32°C for 20 min. After sectioning, the slices were maintained at room temperature in aCSF that contained 125 mm NaCl, 25 mm NaHCO3, 2.7 mm KCl, 1.25 mm NaH2PO4, 2.5 mm CaCl2, 1.3 mm MgSO4, and 20 mm glucose, pH 7.4. Both the cutting solution and aCSF were saturated with carbogen (95% O2, 5% CO2). In one set of experiments, the slices were incubated at 37°C for 2 h with hyaluronidase from Streptomyces hyalurolyticus (H1136, Sigma) in carbogenated aCSF similarly to Kochlamazashvili et al. (2010).
Field potential recordings in CA3-CA1 and mossy fiber-CA3 pathways.
Field EPSPs (fEPSPs) were recorded with an electrode that was inserted in a glass micropipette (2–3 mΩ, filled with aCSF) in the CA1 in response to stimulation of Schaffer collateral inputs with bipolar tungsten electrodes (FHC). Synaptic transmission in the mossy fiber-CA3 pathway was evoked by stimulating mossy fibers at the border between the suprapyramidal blade of the dentate gyrus and hilus and recorded in the CA3 stratum lucidum. The temperature of the recording chamber was 30°C–31°C. Recordings were amplified and filtered at 3.0 kHz (DAM80, WPI), sampled at 20 kHz using an A/D converter (Digidata 1400, Molecular Devices), and analyzed with Clampfit 10.5. Basal synaptic transmission was initially determined from input–output relationships that were elicited by stimulation with increasing current intensities. Test stimuli (300 μs) were given at a current (20–90 μA) that produced 40% of the maximum amplitude of the fEPSP without population spikes. The paired-pulse ratio (PPR) was investigated by delivering two stimuli with interstimulus intervals of 25, 50, 100, and 250 ms. Basal responses were monitored for at least 20 min before delivering the LTP-inducing stimulation. To generate LTP, we used high-frequency stimulation (HFS) or theta-burst stimulation (TBS). TBS consisted of four theta epochs with eight trains of four 100 Hz pulses that were delivered at 4 Hz. HFS consisted of four trains of 100 pulses that were applied at 100 or 200 Hz, with an intertrain interval of 10 s. The magnitude of LTP was calculated by dividing the average fEPSP slope after HFS by the average fEPSP slope of responses that were evoked during the 15 min before delivering HFS or TBS. In all of the experiments, the fiber volley amplitude was measured relative to preLTP stimulation. Experiments were discarded if the fiber volley amplitude changed >20% during the entire experiment.
Recordings in the mossy fiber-CA3 pathway were performed in the presence of d-APV (25 μm) to eliminate contamination of mossy fiber-CA3 LTP with an NMDAR-dependent component (e.g., from AC/AC synapses). The following a priori criteria were applied to classify recorded fEPSPs as mossy fiber-CA3: (1) the PPR at the 50 ms interval was ≥1.5; (2) the latency of the fEPSP amplitude was <5 ms; and (3) application of the metabotropic glutamate receptor Group II agonist DCG-IV (1 μm) at the end of the experiment reduced the fEPSP amplitude by ≥80%.
Brain tissue processing and immunostaining.
Two groups of slices were fixed and used for immunostaining: control slices that were stimulated for 2 h without LTP-inducing tetanus and slices that were tetanically stimulated by 100 Hz and maintained for 2 h upon basal stimulation. After the electrophysiological recordings, the slices were fixed in methanol:ethanol solution (1:3) at 4°C for 20 min and subsequently maintained at −20°C. The slices were then embedded in polyester wax (Science Services) as described by Gawlak et al. (2009), cut into 4-μm-thick sections on a rotary microtome (RM 2255, Leica), and mounted on Superfrost Plus glass slides (Thermo Scientific).
Before in situ zymography (ISZ) or immunolabeling, the sections were dewaxed with ethanol and rehydrated. After blocking for 1 h with 10% normal horse serum (Vector Laboratories) in TBST (TBS with 0.1% Tween 20), the slices were incubated overnight at 4°C with primary antibodies in TBST with 2% normal horse serum (anti-MMP-3, 1:500, catalog #EP1186Y, Abcam; anti-MAP-2, 1:500, catalog #M4403, Sigma; anti-GFAP, 1:500, catalog #G3893, Sigma; anti-synapsin-1, 1:150, catalog #106103, Synaptic Systems). Anti-synapsin-1 antibody recognizes both inhibitory and excitatory synapses. After washing with TBST, the slices were incubated for 2 h at room temperature with secondary antibodies (AlexaFluor-633 goat anti-mouse and AlexaFluor-568 goat anti-rabbit, 1:2000, Invitrogen). Finally, glass slides were mounted with Fluoroshield (Sigma).
Casein ISZ.
The ISZ procedure was performed as in Gawlak et al. (2009) with modifications. Instead of DQ-gelatin that is cleaved by MMP-2/MMP-9, we used fluorogenic substrate BODIPY-casein (E6638, Invitrogen) as a substrate for MMP-3. Sections that were dewaxed in 99.8% ethanol and rehydrated were covered with a BODIPY-casein (with phenylmethylsulfonyl fluoride, PMSF; 0.2 mm, Sigma) at 37°C for 90 min. The slices were then washed with TBS and mounted with Fluoroshield or additionally processed for immunostaining. In a set of control experiments, different protease inhibitors were used during the entire slice processing (hydration, reaction with BODIPY-casein, and washing) to check the specificity of the caseinolytic signal. MMP inhibitors were used at the following concentrations: pan MMP inhibitor phenanthroline (10 mm, Sigma) and NNGH (20 μm).
Image acquisition and analysis.
Confocal microscopy images were captured using an Olympus Fluoview1000S microscope (PlanApo 60× 1.35 NA oil-immersion objective). All confocal parameters (pinhole, offset, brightness) were held constant for all of the datasets from the same experiment. Three to five 60× images of the CA1 stratum radiatum were acquired from sections of one thick (350 μm) slice that was used in the electrophysiological experiment. The images were analyzed using ImageJ software (National Institutes of Health), and the puncta number, size, and intensity were determined. Before each experiment, in a separate set of slices, background ISZ and immunostaining signals were measured and used as a constant threshold that was applied to all images during analysis.
Whole CA1 lysates and immunoblotting.
After the electrophysiological experiments, the CA3-CA1 regions were isolated together from hippocampal slices and then frozen and stored. Control slices were basally stimulated. Slices in the LTP groups were collected 15 min or 1 h after 100 Hz tetanus. The experiments were performed on lysates that were dissociated in RIPA buffer. Membranes were probed with rabbit anti-MMP-3 (1:750; Abcam) and mouse anti-β-actin (1:1000; Abcam) antibodies diluted in TBS with 0.1% Tween 20 and 5% BSA. Secondary anti-mouse and anti-rabbit antibodies (1:2000, Jackson ImmunoResearch Laboratories) conjugated with HRP were used. Immunoreactive chemiluminescence signals were visualized using Luminata Forte Western HRP Substrate (Merck-Millipore) with ChemiDoc MP (Bio-Rad). The levels of immunoreactivity were determined by densitometry (ImageJ). The values were normalized to β-actin and are presented as a percentage change relative to control.
Drugs.
d-APV, DCG-IV, nifedipine, UK356618, and WAY170523 were purchased from Tocris Bioscience. FN-439, NNGH, SB-3CT, and recombinant active MMP-3 protein (SRP7783) were purchased from Sigma-Aldrich. FN-439 was dissolved in water as a stock solution. All of the other MMP inhibitors were dissolved in DMSO. We used the following MMP inhibitors with different specificities against MMP-9 and MMP-3: (1) FN-439, a broad-spectrum MMP inhibitor; Ki for MMP-1 and MMP-8 = 1 μm; Ki for MMP-9 = 30 μm; Ki for MMP-3 = 150 μm (Odake et al., 1994; Franzke et al., 2002); (2) NNGH, a broad-spectrum MMP inhibitor; Ki for MMP-12 and MMP-13 = 4 nm; Ki for MMP-8 = 9 nm; Ki for MMP-1 = 170 nm; Ki for MMP-3 = 130 nm (MacPherson et al., 1997; Calderone et al., 2006); (3) UK356618, MMP-3/MMP-13 inhibitor; Ki for MMP-3 = 5.9 nm; Ki for MMP-13 = 73 nm; Ki for MMP-9 = 840 nm (Fray et al., 2003); (4) WAY-170523, a specific MMP-13 blocker (Ki = 17 nm) that also inhibits MMP-9 (Ki = 945 nm) (Chen et al., 2000); and (5) SB-3CT, a specific inhibitor of gelatinases MMP-9/MMP-2; Ki for MMP-2 = 14 nm; Ki for MMP-9 = 600 nm (Brown et al., 2000).
Statistical analysis.
The analyses were performed using SigmaPlot, and α = 0.05 was chosen for statistical significance. The specific tests that were used are noted in the figure legends. All of the data are presented as mean ± SEM. Significance in single comparisons was calculated using Student's t test (data with a normal distribution) or Mann–Whitney U test (data without a normal distribution). Multiple comparisons were calculated using two-way ANOVA followed by Bonferroni correction. For all of the comparisons, n refers to the number of slices.
Results
Maintenance phase of vdccLTP and nmdaLTP depends on different MMPs
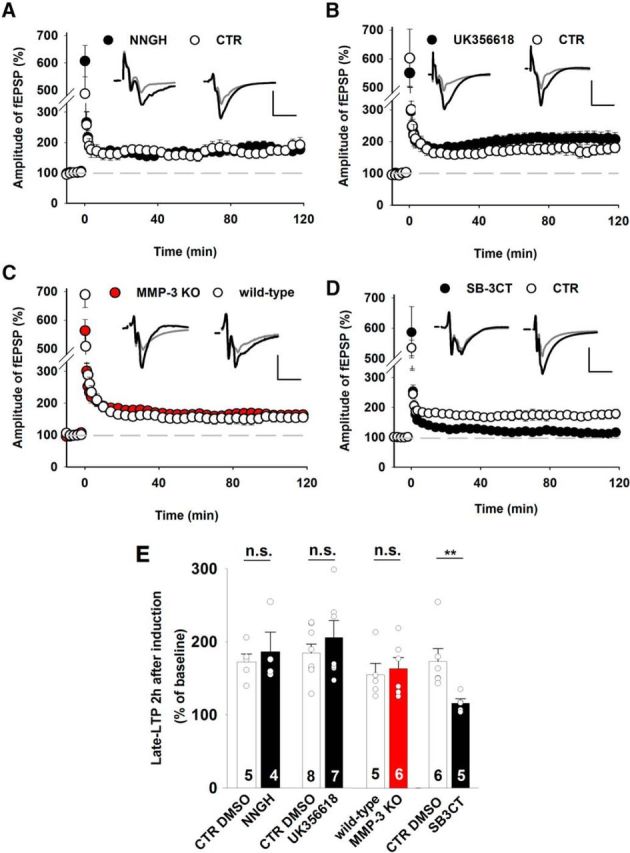
Depending on the route of calcium entry upon stimulation, vdccLTP or nmdaLTP can be evoked. Low-frequency tetanus (25 Hz) is known to result in solely nmdaLTP, whereas higher-frequency stimulation (100–200 Hz) elicits compound LTP that consists of both VDCC- and NMDAR-dependent components (Grover and Teyler, 1990). The induction of LTP with a 200 Hz train allows precise dissection of the two LTP components (Grover and Teyler, 1990). In the presence of nifedipine (a blocker of L-type channels; 100 μm), 200 Hz stimulation induced LTP that entirely depended on NMDAR activity (nmdaLTP; Fig. 1A). In the same stimulation paradigm, treatment with the NMDAR blocker d-APV (50 μm) induced vdccLTP (Fig. 1A). We tested the dependence of nmdaLTP and vdccLTP on extracellular proteolysis using various MMP inhibitors. SB-3CT blocks MMP-9, a protease whose role in synaptic plasticity is particularly well established. We observed a significant reduction of the magnitude of vdccLTP and nmdaLTP after the application of SB-3CT (Fig. 1B,C). We then applied NNGH, a broad-spectrum MMP inhibitor, at a concentration (10 μm) that does not block MMP-9 (Ki for MMP-12 and MMP-13 = 4 nm; Ki for MMP-8 = 9 nm; Ki for MMP-1 = 170 nm; Ki for MMP-3 = 130 nm). Remarkably, in the presence of NNGH, a 200 Hz train (in nifedipine) induced nmdaLTP that was similar to control slices (Fig. 1D), but vdccLTP was completely abolished (Fig. 1E,F). These data indicate that the maintenance phase of vdccLTP depends on specific MMPs that are different from those that are necessary for nmdaLTP. To further support this observation, we applied another protocol to induce vdccLTP (i.e., long TBS in d-APV, with eight trains of four 100 Hz pulses separated by 200 ms and repeated four times) (Morgan and Teyler, 2001). Notably, vdccLTP that was elicited by this protocol was characterized by a slow onset and completely blocked by NNGH (Fig. 1G,H), similar to what was observed in the case of HFS (Fig. 1E). These observations provide evidence that the two types of LTP may depend on the activity of distinct MMPs, as revealed by dramatically different sensitivity to NNGH (Fig. 1E,F). Because of the broad-spectrum profile of NNGH inhibition, subsequent experiments were designed to identify which MMPs, in addition to MMP-9, are involved in the maintenance of compound LTP.
Figure 1.

nmdaLTP and vdccLTP show distinct profiles of sensitivity to MMP blockade. A, LTP in the CA3-CA1 pathway was induced by 200 Hz HFS (white). Two components that depend on the activation of NMDARs (nmdaLTP in the presence of 100 μm nifedipine, red) or L-type calcium channels (vdccLTP in the presence of 50 μm APV, gray) were pharmacologically identified. Insets, Representative average fEPSP traces recorded before (gray) and 115–120 min after (black) LTP. Calibration: vertical, 0.5 mV; horizontal, 5 ms. Stimulation artifacts were removed. The mean slope of fEPSPs that were recorded for 15 min before HFS was set at 100%. B, C, The MMP-2/MMP-9 inhibitor SB-3CT (10 μm) reduced the extent of both nmdaLTP (B) (CTR DMSO: 180 ± 18% of baseline 2 h after tetanus; SB-3CT: 132 ± 10%; t test, t(9) = 2.50, p = 0.034) and vdccLTP (C) (CTR DMSO: 169 ± 15%; SB-3CT: 130 ± 10%; t test, t(11) = 2.11, p = 0.047) induced by 200 Hz tetanus. D, The 200 Hz tetanus (delivered in the presence of 100 μm nifedipine) induced nmdaLTP that was similar in control conditions and in the presence of MMP inhibitor NNGH (10 μm, CTR DMSO: 140 ± 6%; NNGH: 141 ± 8%; t test, t(11) = −0.09, p = 0.93). E, Slowly developing L-type-dependent vdccLTP was induced by 200 Hz tetanus in the presence of 50 μm APV, which was impaired by the presence of NNGH (CTR DMSO: 145 ± 10%; NNGH: 107 ± 12%; Mann–Whitney U test, U(11) = 5.0, p = 0.022). F, Summary of the effects of pharmacological MMP inhibition with SB-3CT and NNGH on nmdaLTP and vdccLTP. The induction of vdccLTP, but not nmdaLTP, required MMP-3 activity. G, Extended TBS induced slowly developing vdccLTP when NMDARs were blocked by 50 μm APV (2 h after induction: 177 ± 12%). The inhibition of MMP-3 activity by NNGH (10 μm) abolished TBS-induced vdccLTP (NNGH: 112 ± 8%; t test, t(14) = 4.10, p = 0.001). H, Statistics for vdccLTP that was measured 115–120 min after stimulation. MMP-3 inhibition impaired TBS-induced vdccLTP similarly to vdccLTP that was induced by 200 Hz tetanus. Data are mean ± SEM. *p ≤ 0.05. ***p ≤ 0.001. n.s., not significant.
Impact of MMP-3 and MMP-9 on LTP maintenance in the CA3-CA1 projection
To further characterize the dependence of LTP maintenance on MMP in the CA3-CA1 projection, we recorded compound LTP in response to the most commonly used 100 Hz HFS of Schaffer collaterals. Figure 1 shows that other MMPs beyond MMP-9 (indicated by the use of the specific blocker SB-3CT) must be involved in the maintenance of LTP. To identify these other MMPs, we used various inhibitors and transgenic animals with KO of specific MMPs.
We first characterized LTP that was induced in the CA3-CA1 pathway in hippocampal slices from MMP-9 KO mice. No difference was observed between KO and WT slices with regard to the input–output relationship or short-term plasticity (data not shown), indicating that the loss of MMP-9 did not affect basal synaptic transmission (Nagy et al., 2006). However, the time course of HFS-induced LTP that was recorded in slices from MMP-9 KO mice showed significant fading that began ∼80 min after HFS (Fig. 2A). Likewise, the application of the specific MMP-9 blocker SB-3CT (10 μm) also affected only late LTP (Fig. 2B). These findings further confirm that the late LTP phase depends on MMP-9 (Nagy et al., 2006).
Figure 2.

MMP-3 and MMP-9 are required for the late phase of LTP in the hippocampal CA3-CA1 projection. A–I, Time course of LTP recorded in the CA1 and induced by 100 Hz tetanic stimulation delivered at time = 0 min under control conditions (open circles) and in the presence of different MMP inhibitors (filled circles). Insets, Representative average fEPSP traces recorded before (gray) and 115–120 min after (black) LTP induction. Calibration: vertical, 0.5 mV; horizontal, 5 ms. A, MMP-9 KO slices had a strong deficit in late LTP that was measured 2 h after induction (CTR: 157 ± 11%; MMP-9 KO: 121 ± 7%; Mann–Whitney U test, U(18) = 17, p = 0.014) and normal early LTP (CTR: 168 ± 9% 15–20 min after induction relative to baseline; MMP-9 KO: 159 ± 10%; Mann–Whitney U test, U(18) = 48, p = 0.28) compared with LTP that was recorded in WT controls. B, Only the late phase of LTP was significantly impaired by the specific MMP-2/MMP-9 inhibitor SB-3CT (10 μm, CTR DMSO: 186 ± 15%; SB-3CT: 137 ± 15%; t test, t(12) = 2.29, p = 0.041). Early LTP was unaffected by SB-3CT (CTR DMSO: 188 ± 15%; SB-3CT: 180 ± 11%; t test, t(12) = 0.51, p = 0.62). C, Administration of a high concentration of the broad-spectrum MMP inhibitor FN439 (180 μm) blocked both early LTP that was measured 20 min after induction (CTR: 171 ± 12%; FN439: 129 ± 8%; t test, t(17) = 2.5, p = 0.02) and late LTP that was measured 2 h after HFS (CTR: 171 ± 12%; FN439: 115 ± 8%; Mann–Whitney U test, U(17) = 2.0, p < 0.001). D, A lower concentration of FN439 (25 μm) blocked only late LTP (CTR: 171 ± 12%; FN439: 124 ± 9%; Mann–Whitney U test, U(17) = 9.0, p = 0.006). E, The broad-spectrum MMP inhibitor NNGH at a concentration that blocks MMP-3 (10 μm) impaired late LTP (CTR DMSO: 153 ± 6%; NNGH: 112 ± 11%; t test, t(10) = 3.44, p = 0.006) but not early LTP (CTR DMSO: 162 ± 11%; NNGH: 147 ± 11%; t test, t(10) = 1.0, p = 0.34). F, The specific MMP-3/MMP-13 inhibitor UK356618 at a concentration of 750 nm reduced the magnitude of late LTP (CTR: 160 ± 10%; UK356618: 120 ± 6%; Mann–Whitney U test, U(21) = 29, p = 0.025) but not early LTP (CTR DMSO: 151 ± 8%; UK356618: 164 ± 8%; t test, t(21) = −0.95, p = 0.36). G, A higher concentration of UK356618 (2 μm) yielded similar results as in F. H, The specific MMP-13 inhibitor WAY170523 did not affect LTP (late LTP, CTR DMSO: 182 ± 13%, n = 8; WAY170523: 174 ± 30%, n = 5; t test, t(11) = 0.27, p = 0.79). I, Hippocampal slices that were exposed to a mix of NNGH (10 μm) and SB-3CT (10 μm) showed impairment of both early LTP (CTR DMSO: 181 ± 10%; NNGH+SB-3CT: 129 ± 7%; t test, t(10) = 3.78, p = 0.01) and late LTP beginning during the first minutes after HFS (CTR DMSO: 198 ± 22%; NNGH+SB-3CT: 128 ± 14%; t test, t(10) = 2.73, p = 0.021). J, K, Summary plots that depict the effects of different MMP inhibitors and MMP-9 deficiency on late (J) and early (K) LTP phases. The blockade of MMP-9 or MMP-3 activity similarly impaired late LTP, suggesting that the activity of both proteases is needed for the maintenance of LTP. *p < 0.05. **p < 0.01. ***p ≤ 0.001. n.s., not significant.
NNGH had a high degree of specificity in blocking vdccLTP (Fig. 1D–F). To facilitate the deduction of which MMPs were involved, we used FN439, another broad-spectrum inhibitor with an inhibitory profile that is different from NNGH (Ki for MMP-1 and MMP-8 = 1 μm; Ki for MMP-9 = 30 μm; Ki for MMP-3 = 150 μm). The administration of FN439 at a concentration of 180 μm resulted in strong downregulation of LTP. In contrast to SB3CT, the effect was observed shortly after HFS (Fig. 2C). At 180 μm, FN439 is expected to block both MMP-9 and MMP-3. Therefore, we performed additional recordings at 25 μm to block mainly MMP-9, thus leaving MMP-3 only weakly affected while still saturating the inhibition of MMP-1 and MMP-8. Interestingly, 25 μm FN439 had no effect on early LTP but suppressed the late phase ∼1 h after induction (Fig. 2D).
The abrupt reduction of the extent of LTP in the presence of 180 μm FN439 (Fig. 2C) indicated that another protease, in addition toMMP-9, that is blocked by a high concentration of FN439 is involved in supporting early LTP. MMP-3 appeared to be a good candidate because 180 μm FN439 was expected to block it (MacPherson et al., 1997). Most small-molecule inhibitors have limited selectivity for individual MMPs. We thus used two compounds that block MMP-3 activity with distinct specificities. Bath application of NNGH, a broad-spectrum MMP inhibitor at 10 μm, strongly reduced the amplitude of LTP beginning ∼30 min after HFS (Fig. 2E). Next, we used a more specific MMP-3 blocker, UK356618 (Ki for MMP-3 = 5.9 nm; Ki for MMP-13 = 73 nm; Ki for MMP-9 = 840 nm), which impaired only late LTP in the CA3-CA1 projection at 750 nm (Fig. 2F). Similar results were obtained when we applied 2 μm UK356618, although a slight trend toward earlier interference with the extent of LTP compared with 750 nm was apparent (Fig. 2G). Importantly, the application of a specific blocker of MMP-13, WAY-170523 (Ki for MMP-13 = 17 nm; Ki for MMP-9 = 945 nm) at 750 nm had no effect on LTP (Fig. 2H). Thus, the pharmacological blockade of either MMP-9 or MMP-3 resulted in qualitatively similar effects, namely, impairment of late LTP (Fig. 2J). Remaining unclear, however, was which protease supports the early LTP that was downregulated by 180 μm FN439 (Fig. 2C,K). To test the possibility that early LTP requires the blockade of both MMP-9 and MMP-3, we elicited LTP in the presence of the MMP-3 blocker NNGH (10 μm) and the MMP-9 blocker SB-3CT (10 μm), and indeed we observed that concomitant inhibition of these MMPs impaired both early and late LTP (Fig. 2I–K). Altogether, late LTP was affected by inhibiting either MMP-9 or MMP-3 activity (or both). To impair early LTP, however, the blockade of both MMPs was required (Fig. 2I,K).
MMP-3 KO reduces late LTP in the CA3-CA1 pathway
Considering the pharmacological evidence that indicated the involvement of MMP-3 in vdccLTP, we extended our investigations to MMP-3 KO mice (Van Hove et al., 2012b; Aerts et al., 2015). Because the impact of MMP-3 deficiency on neuronal excitability is unknown, we first measured the fiber volley amplitude, representing the number of Schaffer collaterals that fire an action potential, and observed no significant difference between MMP-3 KO and WT slices (Fig. 3A). The interrelation between the fEPSP amplitude and fiber volley amplitude did not differ between MMP-3 KO and WT slices (Fig. 3B). Thus, the loss of MMP-3 did not affect either excitability or basal synaptic transmission. Next, a paired-pulse facilitation protocol was used to examine short-term plasticity. Again, no difference was observed between MMP-3 KO and WT slices for a broad range of interstimulus intervals (Fig. 3C). Notably, the early phase of LTP that was evoked in slices from MMP-3 KO and WT mice overlapped, but late LTP showed progressive fading in KO group (Fig. 3D).
Figure 3.
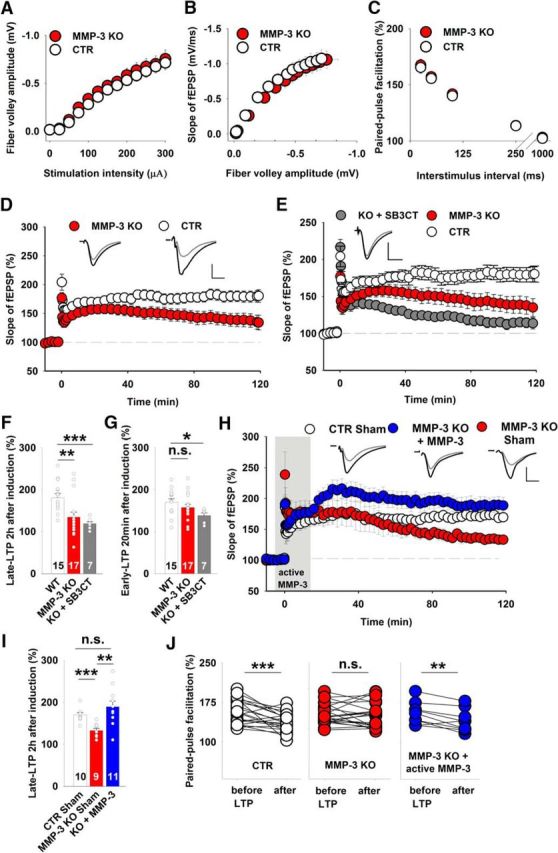
MMP-3 KO mice had impairments of late LTP in the CA3-CA1 pathway but normal basal excitatory synaptic transmission. A, B, Input–output relationships measured for fiber volley amplitudes (A) (WT, white circles, n = 27 slices; MMP-3 KO, red circles, n = 41 slices) and fEPSPs slopes (B), which were not significantly different between the WT and MMP-3 KO groups (p > 0.2 for all data points, t test). C, Short-term plasticity tested as fEPSP paired-pulse facilitation at various interstimulus intervals, showing no difference between WT and MMP-3 KO mice (WT, n = 20 slices; MMP-3 KO, n = 32 slices; for each interstimulus interval; p > 0.3, t test). D, MMP-3 KO mice had strong deficits in late LTP (CTR: 181 ± 10%; MMP-3 KO: 134 ± 12%; t test, t(30) = 2.94, p = 0.006) but not early LTP (CTR: 170 ± 8%; MMP-3 KO: 157 ± 8%; t test, t(30) = 1.16, p = 0.26) 20 min after induction. Insets, Representative fEPSP traces from WT and MMP-3 KO slices before (gray) and 115–120 min after (black) LTP induction. Stimulation artifacts were removed. Calibration: vertical, 0.5 mV; horizontal, 5 ms. E, SB-3CT application to MMP-3 KO slices further impaired late LTP. MMP-3 KO and WT data were similar to those depicted in Figure 2D. Comparisons of LTP that was induced in MMP-3 KO slices in the presence of SB-3CT with LTP that was induced in WT slices revealed impairments of early LTP (MMP-3 KO+SB-3CT: 138 ± 6%; t test vs CTR, t(20) = 2.67, p = 0.014) and late LTP (MMP-3 KO+SB-3CT: 119 ± 5%; Mann–Whitney U test vs CTR, U(20) = 6.0, p = 0.001). F, G, Summary plots that depict the effects of MMP inhibitors and MMP-3 deficiency on late LTP (F) and early LTP (G). Simultaneous inhibition or KO of both MMP-3 and MMP-9 impaired early LTP, whereas MMP-3 KO affected only late LTP. H, Incubation of MMP-3-deficient slices with exogenous active MMP-3 protein during 100 Hz stimulation (from −5 to 15 min, gray area) rescued the impairment of late LTP (CTR sham-treated: 170 ± 5%; MMP-3 KO sham-treated: 132 ± 6%; MMP-3 KO treated with active MMP-3: 189 ± 13%; t test, CTR vs KO sham, t(17) = 5.1, p < 0.001; t test, KO sham vs KO treated, t(18) = 5.1, p = 0.002; Mann–Whitney U test, CTR vs KO treated, U(19) = 33, p = 0.13). I, Summary plot that depicts the effects of infusing the exogenous active MMP-3 protein on late LTP recorded in MMP-3 KO slices. The short exposure to MMP-3 activity restored the magnitude of late LTP in MMP-3-deficient slices. J, Comparison of short-term plasticity before and 2 h after LTP induction. In WT slices, the induction of LTP significantly decreased the paired-pulse facilitation (n = 21 slices, paired t test, p < 0.001). The impairment of late LTP in MMP-3 KO slices was accompanied by a lack of change in the paired-pulse facilitation after LTP (n = 20 slices, paired t test, p = 0.68). Brief infusion of active MMP-3 protein during LTP induction in MMP-3-deficient slices restored the changes in short-term plasticity accompanying LTP that were observed in WT slices (n = 9 slices, paired t test, p = 0.006). *p < 0.05. **p < 0.01. ***p ≤ 0.001. n.s., not significant.
Concurrent MMP-3 KO and MMP-9 blockade impair early LTP
Our pharmacological data showed that early LTP can be impaired by 180 μm FN439 and the concomitant actions of MMP-9 and MMP-3 blockers (NNGH and SB-3CT) but not by the blockade any of these MMPs separately (Fig. 2K). The genetic removal of MMP-3 affected the time course of LTP similarly to the pharmacological suppression of MMP-3 activity (Figs. 2F, 3D). To further explore the specific contributions of MMP-9 and MMP-3, we measured LTP in slices from MMP-3 KO mice in the presence of a specific gelatinase blocker, SB-3CT (10 μm). SB-3CT affected only late LTP in the WT group (Fig. 2B), whereas it impaired both early and late LTP in MMP-3 KO slices (Fig. 3E–G). These results are consistent with our observations after combined application of both NNGH and SB-3CT in WT slices (Fig. 2I) and further confirm that both proteases regulate early LTP.
Administration of an active form of MMP-3 restores the impairment of plasticity in MMP-3 KO slices
Next, we investigated whether restoring MMP-3 activity in MMP-3 KO slices rescues the impairment of LTP. We bath applied exogenous, recombinant active MMP-3 protein (500 ng/ml) 5 min before 100 Hz tetanus and washed it out 15 min after stimulation in MMP-3 KO slices and observed significant rescue of late LTP (Fig. 3H). The extent and time course of the rescue of LTP following MMP-3 washout stabilized at a level that was comparable to control recordings (Fig. 3H,I).
In mammalian synapses, the PPR is commonly used as an index of presynaptic changes (Yang and Calakos, 2013). The induction of LTP in the WT group was accompanied by a significant reduction of the PPR 2 h after HFS (Fig. 3J). Interestingly, in MMP-3 KO slices, the lower magnitude of late LTP was accompanied by a lack of significant changes in the PPR after LTP (Fig. 3J). However, the lack of changes in short-term plasticity after LTP in MMP-3 KO slices was reversible, as an infusion of active MMP-3 during tetanus restored the changes in the PPR 2 h after LTP induction (Fig. 3J). Thus, MMP-3 deficiency markedly, but reversibly, weakened late LTP and affected short-term plasticity that accompanied LTP induction.
MMP-3 KO abolishes vdccLTP, but not nmdaLTP, in the CA3-CA1 pathway
After establishing that MMP-3 deficiency impairs late LTP, we next investigated which components of 200 Hz-induced compound LTP were altered in MMP-3 KO slices. Analogously to NNGH treatment, no difference was observed in nmdaLTP between WT and MMP-3 KO slices (Fig. 4A), but vdccLTP was completely abolished in MMP-3 KO slices (Fig. 4B), similar to previous observations in WT slices (Fig. 1F) that were treated with NNGH at a concentration that blocked MMP-3. Additionally, the application of active MMP-3 protein (500 ng/ml) during 200 Hz stimulation partially restored vdccLTP in MMP-3 KO slices (Fig. 4C). Thus, MMP-3 deficiency reversibly impaired 100 Hz-induced late LTP, causing the complete blockade of vdccLTP but having no effect on nmdaLTP (Fig. 4D). These findings provide evidence that MMP-3 is critically involved in regulating the VDCC-dependent component of compound LTP.
Figure 4.

LTP that is dependent on L-type calcium channels is impaired in MMP-3-deficient slices. A, In WT slices, 200 Hz tetanus that was delivered in the presence of 100 μm nifedipine induced nmdaLTP, which was similar to nmdaLTP in MMP-3 KO slices (CTR: 149 ± 13%; MMP-3 KO: 154 ± 11%; t test, t(11) = −0.26, p = 0.80). Insets, Representative fEPSP traces recorded before (gray) and 115–120 min after (black) LTP induction. Calibration: vertical, 0.5 mV; horizontal, 5 ms. B, Slowly developing L-type-dependent vdccLTP was induced by 200 Hz tetanus in the presence of 50 μm APV. vdccLTP was not present in MMP-3 KO mice (CTR: 164 ± 15%; MMP-3 KO: 102 ± 4%; Mann–Whitney U test, U(10) = 0.0, p = 0.002). C, Incubation of MMP-3-deficient slices with exogenous active MMP-3 protein during 200 Hz stimulation (from −5 to 15 min, gray area) rescued impaired vdccLTP (MMP-3 KO: 102 ± 4%; MMP-3 KO+MMP-3: 135 ± 18%; Mann–Whitney U test, KO vs KO treated, U(12) = 7, p = 0.03). D, Summary of the effects of MMP-3 deficiency on nmdaLTP and vdccLTP. The induction of vdccLTP, but not nmdaLTP, required MMP-3 activity. *p < 0.05. **p < 0.01. n.s., not significant.
Suppression of MMP-3 activity does not affect LTP in the mossy fiber-CA3 pathway
MMP-9 deficiency impaired both early and late LTP in the mossy fiber-CA3 hippocampal pathway (Wiera et al., 2013), which is known for its predominant expression of presynaptic LTP (Castillo et al., 1997; Wiera and Mozrzymas, 2015). Considering that the lack of MMP-3 protease suppressed LTP-induced alterations in the PPR, suggesting presynaptic interference, we investigated the impact of MMP-3 inhibition on LTP in the mossy fiber-CA3 pathway, which precedes the CA3-CA1 projection in trisynaptic hippocampal connections. Interestingly, administration of the MMP-3 inhibitors NNGH or UK356618 had no effect on LTP in this projection up to 2 h after induction (Fig. 5A,B). We also analyzed basal synaptic transmission and plasticity in the mossy fiber-CA3 pathway in MMP-3 KO slices. Neither input–output relationships nor short-term plasticity was affected by MMP-3 deficiency (data not shown), and HFS resulted in LTP that was similar in MMP-3 KO and WT slices (Fig. 5C). Notably, the MMP-2/MMP-9 inhibitor SB-3CT (10 μm) blocked LTP in the mossy fiber-CA3 pathway (Fig. 5D), consistent with our previous study on MMP-9 KO slices (Wiera et al., 2013). Overall, our results indicate that LTP at mossy fiber-CA3 synapses strongly depends on the activity of MMP-9 but not MMP-3 (Fig. 5E), suggesting that the involvement of these MMPs in regulating plasticity-related phenomena in the hippocampus is pathway-specific.
Figure 5.
MMP-3 is not required for the induction of LTP in hippocampal mossy fiber-CA3 synapses. A–D, Time course of LTP recorded in the mossy fiber-CA3 projection in the presence of different MMP inhibitors or in MMP-3 KO slices. Insets, Representative fEPSP traces recorded before (gray) and 115–120 min after (black) LTP induction. Calibration: vertical, 0.5 mV; horizontal, 5 ms. A, The broad-spectrum MMP inhibitor NNGH (10 μm) at a concentration that blocked MMP-3 had no effect on LTP in the mossy fiber-CA3 pathway (CTR DMSO: 172 ± 11% of baseline 2 h after induction; NNGH: 186 ± 27%; t test, t(7) = −4.86, p = 0.81). B, The specific MMP-3/MMP-13 inhibitor UK356618 (2 μm) did not affect the induction of LTP in the mossy fiber-CA3 pathway (CTR DMSO: 193 ± 14%; UK356618: 205 ± 23%; t test, t(13) = −1.31, p = 0.21). C, MMP-3 KO slices showed normal LTP in the mossy fiber-CA3 pathway compared with WT controls (CTR: 155 ± 15%; MMP-3 KO: 163 ± 15%; t test t(9) = −1.04, p = 0.32). D, Exposure to the specific MMP-2/MMP-9 inhibitor SB-3CT (10 μm) impaired LTP in the mossy fiber-CA3 projection beginning during the first minutes after induction (CTR DMSO: 173 ± 17%; SB-3CT: 116 ± 6%; t test, t(9) = 3.34, p = 0.009). E, Summary of the effects of different MMP inhibitors and MMP-3 KO on LTP in the mossy fiber-CA3 projection. The activity of only MMP-9, not MMP-3, was involved in LTP in the mossy fiber-CA3 pathway. **p < 0.01. n.s., not significant.
MMP-3 activity influences late LTP during a short time window after induction
To further evaluate the role of MMP-3 in CA1 synapses, we tested whether MMP-3 activity is required for the consolidation of synaptic plasticity within a specific time window. For this purpose, LTP was induced, and the MMP-3 inhibitor UK356618 (2 μm) was applied at different time points after 100 Hz tetanus. We observed a reduction of the magnitude of LTP when UK356618 was applied immediately (Fig. 6A) or 15 min (Fig. 6B) after tetanus but not after 30 min (Fig. 6C), indicating that MMP-3 activity is required for stable LTP within a relatively narrow time window relative to the stimulation but not necessarily during HFS (Fig. 6D).
Figure 6.

MMP-3 affects LTP within a narrow time window relative to HFS. A–C, LTP recorded in the CA1 and induced by 100 Hz tetanus after administration of the MMP-3 inhibitor UK356618 (2 μm) at different time points. A, Immediately after HFS (CTR DMSO: 205 ± 10% of baseline 2 h after induction; UK356618: 152 ± 7%, t test, t(11) = 3.70, p = 0.004). B, At 15 min after HFS (UK356618: 160 ± 17%; t test vs CTR, t(11) = 2.27, p = 0.044). C, At 30 min after HFS (UK356618: 206 ± 22%, t test vs CTR, t(10) = 0.08, p = 0.94). Insets, Representative average fEPSP traces recorded before (gray) and 115–120 min after (black) LTP. Calibration: vertical, 0.5 mV; horizontal, 5 ms. D, Summary of the effects of UK356618 administration during different time windows on LTP in the CA3-CA1 measured 2 h after induction. The maintenance of LTP required MMP-3 activity within <30 min after HFS. *p < 0.05. **p < 0.01. n.s., not significant.
MMP-3 protein levels and activity are upregulated after LTP induction in the CA1
To gain further insights into the mechanisms that mediate the involvement of MMP-3 in LTP consolidation, we first investigated MMP-3 localization and activity in the hippocampus. MMP-3 protein was reported to be present at low, but detectable, levels in the adult mouse hippocampus (Meighan et al., 2006; Wright et al., 2006). To identify the cellular localization of MMP-3, we used MAP-2 and GFAP as dendritic and astrocytic markers, respectively. We observed MMP-3-positive puncta in both neuronal dendrites (Fig. 7A) and astrocytes (Fig. 7B) in the CA1 stratum radiatum and pyramidale.
Figure 7.

Localization of MMP-3 protein within neurons and astrocytes in the CA1 stratum radiatum. A, Confocal images of the CA1 stratum radiatum in adult mouse slices stained for MMP-3 (magenta) and the pan-neuronal marker MAP-2 (green) revealed that some MMP-3-positive puncta clearly colocalized with MAP-2 (white). Scale bar, 10 μm. Right, High-magnification proximal apical dendrites. Scale bar, 2 μm. All of the images were thresholded. B, Partial colocalization (white) of MMP-3-positive puncta (magenta) in the stratum radiatum with the astrocytic marker GFAP (green). Right, High-magnification astrocyte. Scale bar, 2 μm.
We then investigated whether some of the observed MMP-3-positive puncta in the stratum radiatum contained the active form of this protease. We modified the ISZ method to detect MMP-3 activity in tissue sections. MMP-3 digests casein, and we used BODIPY-casein as a substrate. However, casein is cleaved not only by MMP-3 but also by tPA and other serine proteases. Therefore, to generate data that were more specific for MMP-3, all of the assays were performed in the presence of the serine protease inhibitor PMSF (0.2 mm). ISZ revealed caseinolytic activity in the hippocampal CA1 region (Fig. 8A). To ascribe this activity to MMP-3, we additionally verified the specificity of the caseinolytic signal. The addition of the pan metalloproteinase inhibitor O-phenanthroline (10 mm) or broad-spectrum MMP inhibitor NNGH (20 μm) strongly suppressed the fluorescent ISZ signal (Fig. 8B,C,E). In MMP-3 KO slices, the intensity of the ISZ signal in the CA1 stratum radiatum decreased compared with WT slices (Fig. 8D,F). Furthermore, the ISZ signal colocalized with MMP-3 immunoreactivity (Fig. 8G). These data provide evidence that the vast majority of the caseinolytic signal that was measured in our experiments was attributable to the activity of MMP-3. Finally, a substantial proportion of ISZ puncta in the CA1 stratum radiatum colocalized with the synaptic marker synapsin (Fig. 8H).
Figure 8.

In situ caseinolytic activity in the hippocampal CA1 field. Proteolytic activity was visualized with BODIPY fluorescence (green). In all of the experiments, PMSF was used to block the activity of serine proteases. DAPI (blue) was also used to visualize the strata. A, Representative confocal image of casein ISZ in the hippocampal CA1 stratum pyramidale (SP) and stratum radiatum (SR). B–D, The pan MMP inhibitor phenanthroline (B) (10 mm) and broad-spectrum MMP inhibitor NNGH (C) (20 μm) or genetic KO of MMP-3 (D) decreased ISZ activity relative to sham-treated slices. Scale bar, 40 μm. E, Mean ISZ fluorescence signal in the SR in the presence of phenanthroline with PMSF (n = 3) and NNGH with PMSF (n = 3) relative to the control reaction with PMSF only (n = 6). All of the sections were from the LTP group. F, Mean ISZ fluorescence signal in the presence of PMSF, in the SR in sections from MMP-3 KO mice (n = 8 slices) relative to WT slices (n = 8). A majority of the high-intensity fluorescent puncta that were observed in control slices were absent in the MMP-3 KO group. Differences were tested versus control PMSF group: **p < 0.01 (unpaired t test). G, Caseinolytic activity (green) colocalized with MMP-3-positive puncta (red). The vast majority of the ISZ puncta were positive for MMP-3. Note the predominant yellow in the superimposed image. Scale bar, 40 μm. H, In situ caseinolytic activity (green) and synapsin-positive puncta (magenta) in the stratum radiatum. White represents puncta of ISZ proteolytic activity that colocalized with synapsin-stained presynaptic compartments. Scale bar, 10 μm.
Using immunostaining and ISZ, we investigated potential changes in MMP-3 protein levels and activity in our model. Two hours after LTP induction by 100 Hz tetanus, the intensity and area of MMP-3-positive puncta significantly increased in the CA1 stratum radiatum, but their density remained unaltered (Fig. 9A–D). Moreover, in addition to the LTP-related enhancement of MMP-3 immunoreactivity, casein ISZ revealed the upregulation of total caseinolytic activity in the CA1 stratum radiatum (Fig. 9E,F). To better correlate the observed ISZ signal to MMP-3 activity, we additionally analyzed the intensity of casein ISZ only in MMP-3-positive puncta and found a significant increase in ISZ fluorescence after LTP (Fig. 9G). LTP in the CA1 was accompanied by an increase in the ratio of ISZ fluorescence in MMP-3-positive puncta to ISZ fluorescence in the MMP-3-negative area (Fig. 9H).
Figure 9.

Induction of LTP in the CA3-CA1 increases MMP-3 protein and in situ activity. A–D, The induction of LTP resulted in an increase in the fluorescence intensity and area of MMP-3-positive puncta in the stratum radiatum 2 h after HFS (right) compared with basally stimulated controls (left). The average normalized integrated density of fluorescence in MMP-3-positive puncta (B) (CTR: 100 ± 10%; 2 h after LTP: 138 ± 11%; t test, t(19) = 2.24, p = 0.034) and average area of single MMP-3-positive puncta (C) (CTR: 1.12 ± 0.10 μm2; 2 h after LTP: 1.44 ± 0.083 μm2; t test, t(19) = 2.47, p = 0.023) increased 2 h after LTP induction, but the density of MMP-3-positive puncta was unaltered (D) (CTR: 0.025 ± 0.003 μm−2; 2 h after LTP: 0.026 ± 0.002 μm−2, t test, t(19) = 0.20, p = 0.84). Scale bars, 10 μm. E–H, In situ caseinolytic activity (E, left) and MMP-3 immunostaining (middle) showed substantial colocalization (right), and the extent of colocalization increased 2 h after LTP in the stratum radiatum. The average normalized ISZ fluorescence in the CA1 stratum radiatum (F) (CTR: 100 ± 2%; 2 h after LTP: 115 ± 4%; Mann–Whitney U test, U(19) = 9.0, p = 0.001), average ISZ activity only in MMP-3-positive puncta (G) (CTR: 835 ± 80 AU; 2 h after LTP: 1171 ± 93 AU; t test, t(19) = 2.75, p = 0.016), and ratio of mean ISZ activity in MMP-3-positive puncta to mean ISZ activity in the MMP-3-negative area (H) (CTR: 1.28 ± 0.02; 2 h after LTP: 1.38 ± 0.03; t test, t(19) = 3.18, p = 0.005) were all higher 2 h after LTP. I–K, The induction of LTP increased pro-MMP3 expression and its cleavage to the active form of MMP-3. I, Typical blots of homogenates that were prepared from control slices and slices 15 or 60 min after HFS. J, K, Summary graphs of the semiquantitative analysis of pro-MMP-3 and active MMP-3 protein, respectively, at different time points after HFS (unpaired t test). *p < 0.05. **p < 0.01. ***p ≤ 0.001. n.s., not significant.
To determine whether LTP was accompanied by alterations in MMP-3 protein expression, we measured the levels of pro-MMP-3 (55 kDa) and active MMP-3 (46 kDa) by immunoblotting in hippocampal homogenates that were prepared from slices in which LTP was induced by 100 Hz HFS and from control slices that received only basal stimulation. The induction of LTP increased the expression of both pro-MMP-3 and active MMP-3 (Fig. 9I–K). A significant increase in pro-MMP-3 was detected as early as 15 min after HFS, whereas the levels of both pro-MMP-3 and active MMP-3 remained elevated up to 1 h after LTP (Fig. 9I–K). These experiments indicate that the induction of compound LTP (with a VDCC-dependent component) was accompanied by an increase in MMP-3 expression and activity, which was also observed in synaptic puncta.
Digestion of hyaluronan occludes the impact of MMP-3 inhibition on LTP in the CA1
vdccLTP in the CA1 strongly depends on the hyaluronic acid content of the ECM (Kochlamazashvili et al., 2010). We investigated whether the digestion of hyaluronan affects the sensitivity of LTP to MMP-3 inhibition. Incubation of the slices at 37°C for 2 h with hyaluronidase significantly reduced 100 Hz tetanus-induced LTP compared with sham-treated slices (Fig. 10A), with no changes in the input–output relationship or short-term plasticity (Fig. 10B,C). Kochlamazashvili et al. (2010) reported that hyaluronidase treatment suppressed vdccLTP, leaving only the nmdaLTP component. In the present study, we found that nmdaLTP was unaffected by MMP-3 blockers. We expected that after hyaluronidase treatment, the remaining LTP fraction would be resistant to MMP-3 inhibition. Indeed, after hyaluronan digestion, the MMP-3 inhibitor UK356618 failed to suppress LTP (Fig. 10D,E). These results further confirm that MMP-3 activity is strongly involved in regulating the component of LTP that depends on L-type calcium channels.
Figure 10.

Digestion of hyaluronic acid occludes the impact of MMP-3 inhibition on LTP in the CA1. A, Digestion of hyaluronic acid with hyaluronidase decreased the magnitude of LTP that was induced by 100 Hz tetanus (sham: 197 ± 11%; hyaluronidase: 163 ± 10%; t test, t(12) = 2.10, p = 0.041). B, C, Digestion of hyaluronic acid in hippocampal slices using hyaluronidase did not affect the input–output relationship or short-term plasticity in the CA3-CA1 projection. B, Average initial fEPSP amplitudes recorded at CA3-CA1 synapses in slices that were treated for 2 h with hyaluronidase (filled circles, n = 12) and respective controls (open circles, n = 14), plotted against the fiber volley amplitude. The statistical analysis was performed using two-way ANOVA followed by Bonferroni correction, revealing no differences between genotypes (comparison of fiber volley amplitude: F(1,312) = 0.11, p = 0.74; comparison for fEPSP slope: F(1,312) = 0.089, p = 0.77). C, Treatment with hyaluronidase did not affect short-term plasticity, assessed by paired-pulse facilitation, at various interstimulus intervals (n = 6 sham-treated slices, n = 13 hyaluronidase-treated slices; t test between genotypes, p > 0.48 for all interstimulus intervals). D, The hyaluronidase-resistant component of LTP was unaffected by the MMP-3 blocker UK356618 (0.8 μm; hyaluronidase+UK356618: 155 ± 10%; t test vs hyaluronidase, t(9) = 0.58, p = 0.57). E, The statistical analysis of LTP that was induced by 100 Hz HFS showed that UK356618 occluded the effects of hyaluronidase treatment. *p < 0.05. n.s., not significant.
Discussion
In the present study, we tested the hypothesis that the molecular signaling pathways that are involved in the consolidation of vdccLTP and nmdaLTP critically depend on the activity of distinct extracellular proteases. Compared with nmdaLTP, the molecular mechanisms that underlie the L-type-dependent component of LTP maintenance are less clear (Blundon and Zakharenko, 2008). However, the relationship between nmdaLTP and vdccLTP shifts to favor the latter at high tetanus frequencies (≥100 Hz) or when more postsynaptic spikes in a single burst are paired with presynaptic stimulation (Grover and Teyler, 1990; Morgan and Teyler, 2001; Zakharenko et al., 2001). In the present study, these two LTP components (induced by 200 Hz tetanus) were clearly distinct. vdccLTP had a slow onset, whereas nmdaLTP had a time course similar to LTP induced by 100 Hz tetanus but with a lower amplitude. We used several complementary approaches and found that the mechanisms that underlie these two LTP components are related to the activity of distinct MMPs. First, in MMP-3 KO slices, we observed no changes in nmdaLTP and the abolition of vdccLTP. Second, NNGH at a concentration that was expected to inhibit MMP-3 blocked the induction of vdccLTP but had no effect on nmdaLTP. Third, vdccLTP that was induced by extended TBS (Morgan and Teyler, 2001) was also critically sensitive to MMP-3 inhibition. Fourth, vdccLTP has been shown to have a clear presynaptic mechanism (Zakharenko et al., 2001), and we found that MMP-3 deficiency suppressed presynaptic plasticity after LTP induction (Fig. 3J). Finally, the digestion of hyaluronic acid, which blocks vdccLTP but not nmdaLTP in the CA1 (Kochlamazashvili et al., 2010), occluded the LTP component that was sensitive to MMP-3 inhibition.
Considering the limited specificity of the available MMP blockers, we used several pharmacological compounds that enabled us to extract information regarding the involvement of MMP-3. NNGH (broad-spectrum MMP inhibitor) and UK356618 (MMP-3/MMP-13 inhibitor) caused similar effects (i.e., impairment of late LTP; but for NNGH, see Conant et al. (2010)), suggesting that a “common denominator” of their actions was MMP-3 blockade. This view was supported by the use of the specific MMP-13 inhibitor WAY170523, which had no effect on LTP. Additionally, the application of UK356618 30 min after HFS did not affect LTP, arguing against possible nonspecific effects of MMP-3 inhibition on basal synaptic transmission, which was also unaffected by this inhibitor when applied before HFS. The effect of pharmacological MMP-3 blockade was mimicked by MMP-3 KO, further demonstrating that MMP-3 regulates the late phase of LTP in the present model. Moreover, pairing bath application of exogenous active MMP-3 protein with 100 Hz tetanic stimulation in MMP-3-deficient slices restored the magnitude and stability of LTP and changes in short-term plasticity after LTP to the levels that were comparable with those observed in WT slices. Finally, the application of active MMP-3 protein to MMP-3-deficient slices partially, but significantly, (relative to the control) rescued vdccLTP. Unclear is why this form of plasticity was only partially rescued by exogenous MMP-3. One possibility is that the activity of this enzyme and its localization must be properly tuned. This is, to our knowledge, the first demonstration that specific manipulations of MMP-3 shape late LTP. It seems worth reiterating that the multiplicity of MMPs and the limited specificity of their blockers pose major limitations in unequivocally interpreting the data. For example, nonspecific MMP inhibitors that, among other metzincins, block MMP-9, may impair both the early and translation-dependent late phases of LTP (Meighan et al., 2007; Conant et al., 2010), EPSP to spike potentiation (Wójtowicz and Mozrzymas, 2014), the structural plasticity of dendritic spines (Szepesi et al., 2014), ocular dominance plasticity (Spolidoro et al., 2012), and memory formation (Meighan et al., 2006). Attempts to ascribe precise roles to individual MMPs in these numerous phenomena remain a major challenge in the field.
Our findings do not appear to support the direct MMP-3-dependent activation of MMP-9, which was previously suggested by in vitro studies (Ogata et al., 1995). nmdaLTP strongly depends on MMP-9, and significant activation of this MMP by MMP-3 would affect this LTP component, which is contrary to our findings (Fig. 1). Thus, even if the signaling pathways that are induced by MMP-3 and MMP-9 might affect each other, our data do not support any direct interactions between these enzymes.
We found that MMP-3 is involved in shaping the late phase of LTP, which is consistent with observations that long-term plasticity was accompanied by a clear increase in activity in the novel ISZ assay. As described in detail in Results, casein is a substrate for several proteases. To ascribe the zymographic signal to MMP-3 activity, appropriate blockers were used (Fig. 8). We also included a control with MMP-3 KO slices and in a series of experiments performed zymographic analyses that were limited to MMP-3-immunopositive areas. Importantly, increases in caseinolysis were observed in MMP-3-positive regions (Fig. 9G) and colocalized with synaptic puncta (Fig. 8H). Thus, our staining and zymography data showed that LTP induction resulted in the upregulation of MMP-3 activity, which occurs also in the vicinity of the synapses. The results of these morphological analyses were further supported by Western blot, which demonstrated that LTP induction significantly increased the expression of pro-MMP-3 as early as 15 min after HFS. This early appearance of MMP-3 protein expression appears to be compatible with a narrow time window of the involvement of MMP-3 in LTP maintenance. Such a rapid increase in MMP-3 protein expression after LTP is unsurprising. MMP-3 mRNA is dendritically localized (Suzuki et al., 2007) and undergoes activity-dependent translation in a process that depends on fragile X mental retardation 1 protein and eukaryotic initiation factor 4E (Gkogkas et al., 2014). The process of the fast synaptic translation of MMP-3 mRNA may be similar to MMP-9 (Dziembowska et al., 2012). Together, our data indicate that LTP consolidation is correlated with greater expression and activity of MMP-3, and its temporal expression is largely consistent with our functional observations concerning the time window of MMP-3 activity. Notably, this modus operandi of MMP-3 regarding the time window is analogous to the one reported for MMP-9 (Meighan et al., 2007; Wójtowicz and Mozrzymas, 2010). Finally, plasticity-related alterations in MMP-3 expression and activity that were observed in the present study are consistent with previously reported upregulation of this MMP in behavioral tests (Olson et al., 2008).
The involvement of MMP-3 in regulating vdccLTP suggests the existence of unknown specific MMP-3 substrates whose cleavage modulates dendritic L-type calcium channels. Calcium influx through these channels is known to induce signaling that activates the translation of plasticity-related immediate early genes (Wheeler et al., 2012) that, in turn, drive the consolidation phase of LTP (Magee and Johnston, 1997). This possibility may explain the impairment of the LTP maintenance phase in MMP-3-deficient slices. MMP-3 may cleave NMDARs in vitro (Pauly et al., 2008), but we found that MMP-3 did not affect nmdaLTP, thus arguing against this possibility in our model. The identity of MMP-3 substrates that affect L-type channels remains unknown, although some likely candidates can be proposed. In vitro studies indicated that MMP-3 can cleave almost all constituents of perineuronal nets (PNNs), brain-specific ECM structures that are composed of proteins, hyaluronan, and proteoglycans (Van Hove et al., 2012a). Notably, hyaluronic acid and tenascin C (i.e., two well-known PNN constituents) act as permissive factors for the induction of vdccLTP in the hippocampus (Evers et al., 2002; Kochlamazashvili et al., 2010). Considering that both hyaluronidase and MMP-3 cleave PNN components, at first glance, it might look surprising that whereas MMP-3 activity supports vdccLTP, hyaluronidase disrupts it. However, hyaluronidase digests polysaccharide hyaluronic acid, the backbone of PNNs, whereas MMP-3 cleaves proteins at sites that are characterized by specific sequences, leading to their gain or loss of function. Extracellular proteolytic cleavage that is mediated by MMP-3 may reveal protein–protein interaction sites or release signaling peptides (e.g., RGD peptide for integrins). Notably, MMP-3 does not cause global protein disintegration; instead, it acts as a fine proteolytic scalpel that modifies protein functions, thereby shaping their signaling properties. In contrast, hyaluronidase treatment causes the global disruption of PNN backbone and blocks the possibility of vdccLTP induction. Interestingly, specific sequences of heparan sulfates that are present on syndecans, glypicans, perlecan, and agrin bind to and modulate neuronal L-type channels (Garau et al., 2015). Thus, the interaction between heparin sulfates and L-type channels may act as a permissive factor for vdccLTP. Additionally, almost all protein-bearing heparan sulfates are well-known MMP-3 substrates (Stegemann et al., 2013). Thus, vdccLTP induction may require the MMP-3-mediated cleavage of heparan sulfate-bearing proteoglycans. The MMP-3-dependent cleavage of agrin was previously shown to be essential for motor endplate remodeling and ischemia-induced plasticity (Solé et al., 2004; Chao et al., 2013).
Further investigations are needed to verify the involvement of heparan sulfate proteoglycans and tenascin C in the MMP-3-sensitive LTP component. Semiquantitative proteomic studies have revealed the lack of bulk changes in PPN composition in the MMP-3 KO brain (Van Hove et al., 2015). New high-resolution tools may be needed to visualize the fine remodeling and processing of the ECM by MMP-3 in the vicinity of synapses after LTP or learning (Tsien, 2013).
Presynaptic and postsynaptic forms of LTP are dissociable phenomena that are activated by different patterns of neuronal activity and at least partially mediated by distinct signaling pathways. Especially vdccLTP is known to be associated with presynaptic forms of CA1 LTP (Bayazitov et al., 2007). Presynaptic changes that occur in response to 200 Hz stimulation develop slowly, similar to our observations regarding vdccLTP in the present study (Fig. 1), and resemble the late phase of LTP (Bayazitov et al., 2007). Thus, the observed decrease in 100 Hz stimulation-induced late LTP in MMP-3-deficient slices may have resulted from the selective inhibition of vdccLTP. Additionally, synaptic stimulation that successfully activates L-type channels was found to recruit a presynaptic component of LTP expression that involves the retrograde signaling of nitric oxide (Johnstone and Raymond, 2011; Padamsey and Emptage, 2014). These findings suggest that presynaptic enhancement of vdccLTP might result from direct pro-MMP-3 activation by nitric oxide (Gu et al., 2002).
The role of MMP-3 in neuroplasticity phenomena have only started to emerge. In addition to above-mentioned reports that MMP-3 expression is affected by behavioral training (Olson et al., 2008), a more recent study found that MMP-3 KO mice exhibited substantial impairments of cross-modal plasticity in the visual cortex after monocular enucleation (Aerts et al., 2015). Considering our findings, it is interesting to note that ocular dominance plasticity is highly dependent on L-type calcium channels (Frank, 2014) and the integrity of hyaluronic acid-containing perineuronal nets (Pizzorusso et al., 2002; Happel et al., 2014). Therefore, synaptic mechanisms that are activated in response to MMP-3 activity might affect the local opening of the plasticity window.
A novel finding of the present study was that MMP-3 was abundantly present in astrocytes in the hippocampus. The physiological role of astrocytic MMP-3 remains unknown, but a tempting hypothesis is that it may couple neuronal activity and glia-driven ECM remodeling (Dzyubenko et al., 2016). However, the identity of signals that induce the secretion of astrocytic MMP-3 upon increases in neuronal activity awaits investigations.
Our findings indicate that proteolysis that is mediated by MMP-3 may regulate plasticity in synaptic networks. In particular, our study underscores the role of MMP-3 in LTP in the hippocampus and provides strong evidence that the mechanism by which MMP-3 shapes plasticity involves L-type calcium channels. An important, but unresolved, issue is the way in which the activities of different extracellular proteases in excitatory and inhibitory synapses converge on local networks and single neurons to shape signaling and affect functional and structural plasticity.
Footnotes
This work was supported by Polish National Science Centre Grant 2013/08/T/NZ3/00999 scholarship to G.W. and Grant 2014/15/B/NZ4/01689 to J.W.M. This work was supported in part by the KU Leuven Research Council, KU Leuven, Belgium, BOF-OT/14/064, to L.M., the Research Foundation Flanders, Belgium, FWO G.0054.12, G0B2315N, to L.M., and the Flemish government agency for Innovation by Science and Technology, Belgium, postdoctoral fellowship to I.V.H. G.W. was additionally supported by Stipend for Young Researchers START from Foundation for Polish Science. We thank Leszek Kaczmarek (Nencki Institute, Warsaw) for providing the MMP-9 KO mice.
The authors declare no competing financial interests.
References
- Aerts J, Nys J, Moons L, Hu TT, Arckens L (2015) Altered neuronal architecture and plasticity in the visual cortex of adult MMP-3-deficient mice. Brain Struct Funct 220:2675–2689. 10.1007/s00429-014-0819-4 [DOI] [PubMed] [Google Scholar]
- Bayazitov IT, Richardson RJ, Fricke RG, Zakharenko SS (2007) Slow presynaptic and fast postsynaptic components of compound long-term potentiation. J Neurosci 27:11510–11521. 10.1523/JNEUROSCI.3077-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundon JA, Zakharenko SS (2008) Dissecting the components of long-term potentiation. Neuroscientist 14:598–608. 10.1177/1073858408320643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S, Bernardo MM, Li Z, Kotra LP, Tanaka Y, Fridman R (2000) Potent and selective mechanism-based inhibition of gelatinases. J Am Chem Soc 122:6799–6800. 10.1021/ja001461n [DOI] [Google Scholar]
- Calderone V, Fragai M, Luchinat C, Nativi C, Richichi B, Roelens S (2006) A high-affinity carbohydrate-containing inhibitor of matrix metalloproteinases. Chem Med Chem 1:598–601. 10.1002/cmdc.200600020 [DOI] [PubMed] [Google Scholar]
- Castillo PE, Janz R, Südhof TC, Tzounopoulos T, Malenka RC, Nicoll RA (1997) Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature 388:590–593. 10.1038/41574 [DOI] [PubMed] [Google Scholar]
- Chao T, Frump D, Lin M, Caiozzo VJ, Mozaffar T, Steward O, Gupta R (2013) Matrix metalloproteinase 3 deletion preserves denervated motor endplates after traumatic nerve injury. Ann Neurol 73:210–223. 10.1002/ana.23781 [DOI] [PubMed] [Google Scholar]
- Chen JM, Nelson FC, Levin JI, Mobilio D, Moy FJ, Nilakantan R, Zask A, Powers R, May RV, Recei M, July V (2000) Structure-based design of a novel, potent, and selective inhibitor for MMP-13 utilizing NMR spectroscopy and computer-aided molecular design. J Am Chem Soc 122:9648–9654. 10.1021/ja001547g [DOI] [Google Scholar]
- Conant K, Wang Y, Szklarczyk A, Dudak A, Mattson MP, Lim ST (2010) Matrix metalloproteinase-dependent shedding of intercellular adhesion molecule-5 occurs with long-term potentiation. Neuroscience 166:508–521. 10.1016/j.neuroscience.2009.12.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva WC, Cardoso G, Bonini JS, Benetti F, Izquierdo I (2013) Memory reconsolidation and its maintenance depend on L-voltage-dependent calcium channels and CaMKII functions regulating protein turnover in the hippocampus. Proc Natl Acad Sci U S A 110:6566–6570. 10.1073/pnas.1302356110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziembowska M, Milek J, Janusz A, Rejmak E, Romanowska E, Gorkiewicz T, Tiron A, Bramham CR, Kaczmarek L (2012) Activity-dependent local translation of matrix metalloproteinase-9. J Neurosci 32:14538–14547. 10.1523/JNEUROSCI.6028-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzyubenko E, Gottschling C, Faissner A (2016) Neuron-glia interactions in neural plasticity: contributions of neural extracellular matrix and perineuronal nets. Neural Plast 2016:5214961. 10.1155/2016/5214961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers MR, Salmen B, Bukalo O, Rollenhagen A, Bösl MR, Morellini F, Bartsch U, Dityatev A, Schachner M (2002) Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. J Neurosci 22:7177–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA. (2014) How voltage-gated calcium channels gate forms of homeostatic synaptic plasticity. Front Cell Neurosci 8:40. 10.3389/fncel.2014.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzke CW, Tasanen K, Schäcke H, Zhou Z, Tryggvason K, Mauch C, Zigrino P, Sunnarborg S, Lee DC, Fahrenholz F, Bruckner-Tuderman L (2002) Transmembrane collagen XVII, an epithelial adhesion protein, is shed from the cell surface by ADAMs. EMBO J 21:5026–5035. 10.1093/emboj/cdf532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fray MJ, Dickinson RP, Huggins JP, Occleston NL (2003) A potent, selective inhibitor of matrix metalloproteinase-3 for the topical treatment of chronic dermal ulcers. J Med Chem 46:3514–3525. 10.1021/jm0308038 [DOI] [PubMed] [Google Scholar]
- Garau G, Magotti P, Heine M, Korotchenko S, Lievens PM, Berezin V, Dityatev A (2015) Heparin/heparan sulfates bind to and modulate neuronal L-type (Cav1.2) voltage-dependent Ca(2+) channels. Exp Neurol 274:156–165. 10.1016/j.expneurol.2015.08.006 [DOI] [PubMed] [Google Scholar]
- Gawlak M, Górkiewicz T, Gorlewicz A, Konopacki FA, Kaczmarek L, Wilczynski GM (2009) High resolution in situ zymography reveals matrix metalloproteinase activity at glutamatergic synapses. Neuroscience 158:167–176. 10.1016/j.neuroscience.2008.05.045 [DOI] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Cao R, Jafarnejad SM, Prager-Khoutorsky M, Giannakas N, Kaminari A, Fragkouli A, Nader K, Price TJ, Konicek BW, Graff JR, Tzinia AK, Lacaille JC, Sonenberg N (2014) Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep 9:1742–1755. 10.1016/j.celrep.2014.10.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogolla N, Caroni P, Lüthi A, Herry C (2009) Perineuronal nets protect fear memories from erasure. Science 325:1258–1261. 10.1126/science.1174146 [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ (1990) Two components of long-term potentiation induced by different patterns of afferent activation. Nature 347:477–479. 10.1038/347477a0 [DOI] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA (2002) S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297:1186–1190. 10.1126/science.1073634 [DOI] [PubMed] [Google Scholar]
- Happel MF, Niekisch H, Castiblanco Rivera LL, Ohl FW, Deliano M, Frischknecht R (2014) Enhanced cognitive flexibility in reversal learning induced by removal of the extracellular matrix in auditory cortex. Proc Natl Acad Sci U S A 111:2800–2805. 10.1073/pnas.1310272111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone VP, Raymond CR (2011) A protein synthesis and nitric oxide-dependent presynaptic enhancement in persistent forms of long-term potentiation. Learn Mem 18:625–633. 10.1101/lm.2245911 [DOI] [PubMed] [Google Scholar]
- Kochlamazashvili G, Henneberger C, Bukalo O, Dvoretskova E, Senkov O, Lievens PM, Westenbroek R, Engel AK, Catterall WA, Rusakov DA, Schachner M, Dityatev A (2010) The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca(2+) channels. Neuron 67:116–128. 10.1016/j.neuron.2010.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson LJ, Bayburt EK, Capparelli MP, Carroll BJ, Goldstein R, Justice MR, Zhu L, Hu S, Melton RA, Fryer L, Goldberg RL, Doughty JR, Spirito S, Blancuzzi V, Wilson D, O'Byrne EM, Ganu V, Parker DT (1997) Discovery of CGS 27023A, a non-peptidic, potent, and orally active stromelysin inhibitor that blocks cartilage degradation in rabbits. J Med Chem 40:2525–2532. 10.1021/jm960871c [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D (1997) A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275:209–213. 10.1126/science.275.5297.209 [DOI] [PubMed] [Google Scholar]
- Meighan PC, Meighan SE, Davis CJ, Wright JW, Harding JW (2007) Effects of matrix metalloproteinase inhibition on short- and long-term plasticity of schaffer collateral/CA1 synapses. J Neurochem 102:2085–2096. 10.1111/j.1471-4159.2007.04682.x [DOI] [PubMed] [Google Scholar]
- Meighan SE, Meighan PC, Choudhury P, Davis CJ, Olson ML, Zornes PA, Wright JW, Harding JW (2006) Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J Neurochem 96:1227–1241. 10.1111/j.1471-4159.2005.03565.x [DOI] [PubMed] [Google Scholar]
- Morgan SL, Teyler TJ (2001) Electrical stimuli patterned after the theta-rhythm induce multiple forms of LTP. J Neurophysiol 86:1289–1296. [DOI] [PubMed] [Google Scholar]
- Morris RG. (2013) NMDA receptors and memory encoding. Neuropharmacology 74:32–40. 10.1016/j.neuropharm.2013.04.014 [DOI] [PubMed] [Google Scholar]
- Nagy V, Bozdagi O, Matynia A, Balcerzyk M, Okulski P, Dzwonek J, Costa RM, Silva AJ, Kaczmarek L, Huntley GW (2006) Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J Neurosci 26:1923–1934. 10.1523/JNEUROSCI.4359-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odake S, Morita Y, Morikawa T, Yoshida N, Hori H, Nagai Y (1994) Inhibition of matrix metalloproteinases by peptidyl hydroxamic acids. Biochem Biophys Res Commun 199:1442–1446. 10.1006/bbrc.1994.1392 [DOI] [PubMed] [Google Scholar]
- Ogata Y, Itoh Y, Nagase H (1995) Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinases-1 complex by 4-aminophenylmercuric acetate and proteinases. J Biol Chem 270:18506–18511. 10.1074/jbc.270.31.18506 [DOI] [PubMed] [Google Scholar]
- Olson ML, Meighan PC, Brown TE, Asay AL, Benoist CC, Harding JW, Wright JW (2008) Hippocampal MMP-3 elevation is associated with passive avoidance conditioning. Regul Pept 146:19–25. 10.1016/j.regpep.2007.07.004 [DOI] [PubMed] [Google Scholar]
- Padamsey Z, Emptage N (2014) Two sides to long-term potentiation: a view towards reconciliation. Philos Trans R Soc Lond B Biol Sci 369:20130154. 10.1098/rstb.2013.0154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly T, Ratliff M, Pietrowski E, Neugebauer R, Schlicksupp A, Kirsch J, Kuhse J (2008) Activity-dependent shedding of the NMDA receptor glycine binding site by matrix metalloproteinase 3: a PUTATIVE mechanism of postsynaptic plasticity. PLoS One 3:e2681. 10.1371/journal.pone.0002681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto RT, Kunz PA, Kwon H, Mabb AM, Sabatini BL, Philpot BD, Ehlers MD (2012) Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron 76:396–409. 10.1016/j.neuron.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L (2002) Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298:1248–1251. 10.1126/science.1072699 [DOI] [PubMed] [Google Scholar]
- Senkov O, Andjus P, Radenovic L, Soriano E, Dityatev A (2014) Neural ECM molecules in synaptic plasticity, learning, and memory. Prog Brain Res 214:53–80. 10.1016/B978-0-444-63486-3.00003-7 [DOI] [PubMed] [Google Scholar]
- Sidhu H, Dansie LE, Hickmott PW, Ethell DW, Ethell IM (2014) Genetic removal of matrix metalloproteinase 9 rescues the symptoms of fragile X syndrome in a mouse model. J Neurosci 34:9867–9879. 10.1523/JNEUROSCI.1162-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AC, Kupchik YM, Scofield MD, Gipson CD, Wiggins A, Thomas CA, Kalivas PW (2014) Synaptic plasticity mediating cocaine relapse requires matrix metalloproteinases. Nat Neurosci 17:1655–1657. 10.1038/nn.3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé S, Petegnief V, Gorina R, Chamorro A, Planas AM (2004) Activation of matrix metalloproteinase-3 and agrin cleavage in cerebral ischemia/reperfusion. J Neuropathol Exp Neurol 63:338–349. 10.1093/jnen/63.4.338 [DOI] [PubMed] [Google Scholar]
- Sonderegger P, Matsumoto-Miyai K (2014) Activity-controlled proteolytic cleavage at the synapse. Trends Neurosci 37:413–423. 10.1016/j.tins.2014.05.007 [DOI] [PubMed] [Google Scholar]
- Spolidoro M, Putignano E, Munafò C, Maffei L, Pizzorusso T (2012) Inhibition of matrix metalloproteinases prevents the potentiation of nondeprived-eye responses after monocular deprivation in juvenile rats. Cereb Cortex 22:725–734. 10.1093/cercor/bhr158 [DOI] [PubMed] [Google Scholar]
- Stegemann C, Didangelos A, Barallobre-Barreiro J, Langley SR, Mandal K, Jahangiri M, Mayr M (2013) Proteomic identification of matrix metalloproteinase substrates in the human vasculature. Circ Cardiovasc Genet 6:106–117. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Tian QB, Kuromitsu J, Kawai T, Endo S (2007) Characterization of mRNA species that are associated with postsynaptic density fraction by gene chip microarray analysis. Neurosci Res 57:61–85. 10.1016/j.neures.2006.09.009 [DOI] [PubMed] [Google Scholar]
- Szepesi Z, Hosy E, Ruszczycki B, Bijata M, Pyskaty M, Bikbaev A, Heine M, Choquet D, Kaczmarek L, Wlodarczyk J (2014) Synaptically released matrix metalloproteinase activity in control of structural plasticity and the cell surface distribution of GluA1-AMPA receptors. PLoS One 9:e98274. 10.1371/journal.pone.0098274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. (2013) Very long-term memories may be stored in the pattern of holes in the perineuronal net. Proc Natl Acad Sci U S A 110:12456–12461. 10.1073/pnas.1310158110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hove I, Lemmens K, Van de Velde S, Verslegers M, Moons L (2012a) Matrix metalloproteinase-3 in the central nervous system: a look on the bright side. J Neurochem 123:203–216. 10.1111/j.1471-4159.2012.07900.x [DOI] [PubMed] [Google Scholar]
- Van Hove I, Verslegers M, Buyens T, Delorme N, Lemmens K, Stroobants S, Gantois I, D'Hooge R, Moons L (2012b) An aberrant cerebellar development in mice lacking matrix metalloproteinase-3. Mol Neurobiol 45:17–29. 10.1007/s12035-011-8215-z [DOI] [PubMed] [Google Scholar]
- Van Hove I, Verslegers M, Hu TT, Carden M, Arckens L, Moons L (2015) A proteomic approach to understand MMP-3-driven developmental processes in the postnatal cerebellum: Chaperonin CCT6A and MAP kinase as contributing factors. Dev Neurobiol 75:1033–1048. [DOI] [PubMed] [Google Scholar]
- Verslegers M, Lemmens K, Van Hove I, Moons L (2013) Matrix metalloproteinase-2 and -9 as promising benefactors in development, plasticity and repair of the nervous system. Prog Neurobiol 105:60–78. 10.1016/j.pneurobio.2013.03.004 [DOI] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW (2012) Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 149:1112–1124. 10.1016/j.cell.2012.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiera G, Mozrzymas JW (2015) Extracellular proteolysis in structural and functional plasticity of mossy fiber synapses in hippocampus. Front Cell Neurosci 9:427. 10.3389/fncel.2015.00427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiera G, Wozniak G, Bajor M, Kaczmarek L, Mozrzymas JW (2013) Maintenance of long-term potentiation in hippocampal mossy fiber-CA3 pathway requires fine-tuned MMP-9 proteolytic activity. Hippocampus 23:529–543. 10.1002/hipo.22112 [DOI] [PubMed] [Google Scholar]
- Wójtowicz T, Mozrzymas JW (2010) Late phase of long-term potentiation in the mossy fiber-CA3 hippocampal pathway is critically dependent on metalloproteinases activity. Hippocampus 20:917–921. 10.1002/hipo.20787 [DOI] [PubMed] [Google Scholar]
- Wójtowicz T, Mozrzymas J (2014) Matrix metalloprotease activity shapes the magnitude of EPSPs and spike plasticity within the hippocampal CA3 network. Hippocampus 24:135–153. 10.1002/hipo.22205 [DOI] [PubMed] [Google Scholar]
- Wright JW, Meighan SE, Murphy ES, Holtfreter KL, Davis CJ, Olson ML, Benoist CC, Muhunthan K, Harding JW (2006) Habituation of the head-shake response induces changes in brain matrix metalloproteinases-3 (MMP-3) and -9. Behav Brain Res 174:78–85. 10.1016/j.bbr.2006.07.006 [DOI] [PubMed] [Google Scholar]
- Yang Y, Calakos N (2013) Presynaptic long-term plasticity. Front Synaptic Neurosci 5:8. 10.3389/fnsyn.2013.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA (2001) Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci 4:711–717. 10.1038/89498 [DOI] [PubMed] [Google Scholar]