

Abstract
Although epigenetic mechanisms of gene expression regulation have recently been implicated in memory consolidation and persistence, the role of nucleosome-remodeling is largely unexplored. Recent studies show that the functional loss of BAF53b, a postmitotic neuron-specific subunit of the BAF nucleosome-remodeling complex, results in the deficit of consolidation of hippocampus-dependent memory and cocaine-associated memory in the rodent brain. However, it is unclear whether BAF53b expression is regulated during memory formation and how BAF53b regulates fear memory in the amygdala, a key brain site for fear memory encoding and storage. To address these questions, we used viral vector approaches to either decrease or increase BAF53b function specifically in the lateral amygdala of adult mice in auditory fear conditioning paradigm. Knockdown of Baf53b before training disrupted long-term memory formation with no effect on short-term memory, basal synaptic transmission, and spine structures. We observed in our qPCR analysis that BAF53b was induced in the lateral amygdala neurons at the late consolidation phase after fear conditioning. Moreover, transient BAF53b overexpression led to persistently enhanced memory formation, which was accompanied by increase in thin-type spine density. Together, our results provide the evidence that BAF53b is induced after learning, and show that such increase of BAF53b level facilitates memory consolidation likely by regulating learning-related spine structural plasticity.
SIGNIFICANCE STATEMENT Recent works in the rodent brain begin to link nucleosome remodeling-dependent epigenetic mechanism to memory consolidation. Here we show that BAF53b, an epigenetic factor involved in nucleosome remodeling, is induced in the lateral amygdala neurons at the late phase of consolidation after fear conditioning. Using specific gene knockdown or overexpression approaches, we identify the critical role of BAF53b in the lateral amygdala neurons for memory consolidation during long-term memory formation. Our results thus provide an idea about how nucleosome remodeling can be regulated during long-term memory formation and contributes to the permanent storage of associative fear memory in the lateral amygdala, which is relevant to fear and anxiety-related mental disorders.
Keywords: BAF53b, fear conditioning, long-term memory, memory consolidation, nucleosome remodeling, spine structural plasticity
Introduction
How newly acquired memories are converted to stable, long-lasting memories is a central question for our understanding of memory. The memory consolidation hypothesis posits that memory stabilization requires de novo gene expression (Kandel, 2012). In addition to more typical mechanisms of genetic regulation, long-term memory formation may also require epigenetic regulation. Indeed, it has been shown that the regulation of gene expression by both DNA methylation and histone modifications are essential for long-term memory consolidation (Korzus et al., 2004; Levenson et al., 2004; Miller and Sweatt, 2007; Sando et al., 2012; Medrano-Fernández et al., 2016). On the other hand, the possible role of the nucleosome remodeling-dependent epigenetic regulation has only recently begun to be investigated.
Of the ATP-dependent nucleosome remodeling complexes, we focus here on the Brg/Brm-associated factor (BAF) complex, the mammalian homolog of the yeast SWI/SNF complex. The BAF complex comprises various combinations of at least 15 separate subunits (Staahl et al., 2013; Vogel-Ciernia and Wood, 2014), with the embryonic stem cell BAF (esBAF), polybromo-associated BAF (PBAF), neuronal progenitor BAF (npBAF), and neuronal BAF (nBAF) complexes each having unique compositions. As neural progenitor cells differentiate into postmitotic neurons, the expression of the BAF53a subunit falls to be replaced by its postmitotic neuron-specific paralog BAF53b (also known as hArpNα; gene name, Actl6b) (Olave et al., 2002; Yoo et al., 2009).
Cultured primary hippocampal neurons from mice lacking Baf53b (Baf53b−/− neurons) show a severe reduction in activity-dependent dendritic outgrowth (Wu et al., 2007). The fact that activity-dependent dendritic plasticity is important for long-term memory formation (Mauceri et al., 2011) suggested that BAF53b may also be involved in memory formation. Indeed, recent works on BAF53b dominant-negative mutant (BAF53bΔHD) and BAF53b heterozygous null (Baf53b+/−) mice suggest that BAF53b is important for memory consolidation and synaptic plasticity in the hippocampus and nucleus accumbens (Vogel-Ciernia et al., 2013; White et al., 2016). However, many questions remain to be answered. Particularly, it is unknown whether BAF53b expression is regulated during long-term memory formation. In addition, how BAF53b regulates fear memory in the amygdala system is still unclear. To explore these questions, we adopted adeno-associated viral (AAV) shRNA vector for Baf53b knockdown and herpes-simplex viral (HSV) vector for BAF53b overexpression specifically in the lateral amygdala (LA) neurons and investigated their effects on memory formation using auditory fear conditioning paradigm in mice.
Materials and Methods
Mice.
All experiments were performed on adult 129×C57BL/6 hybrid or BAF53b heterozygous knock-out (Baf53b+/−) mice 2–3 months old (Shoji et al., 2016). Mice were group-housed (3–5 mice per cage) with free access to food and water at a constant temperature (22 ± 1°C), 40%–60% humidity, and under a 12/12 h light/dark cycle. All behavioral experiments were performed during the light phase. Baf53b+/− mice (018783) were purchased from The Jackson Laboratory and maintained in a C57BL/6 background. For the behavior experiments, a total of 101 mice (66 males and 35 females) were used for Baf53b knockdown tests and 94 mice (48 males and 46 females) were used for BAF53b overexpression tests. For the Western blot experiments, we used 18 mice (8 males and 10 females) for the shRNA knockdown test, 6 males of Baf53b+/− or wild-type (WT) mice for anti-BAF53b antibody specificity test, 24 mice (12 males and 12 females) for rBAF53b test, and 8 females for BAF53b overexpression test. For the qPCR experiment, 33 male mice were used. For spine analysis experiments, 6 male mice were used for Baf53b knockdown condition, and 15 mice (11 males and 4 females) for BAF53b overexpression condition. Notably, we did not observe any noticeable differences in the results between male and female. All experimental procedures were approved by the Animal Ethics Committee at the Korea Advanced Institute of Science and Technology.
HSV viral vector and packaging.
Genes of interest (i.e., BAF53b and BAF53a) were subcloned into the HSV vector with a dual promoter system to simultaneously express each gene with an EGFP expression marker. The expression of BAF53b or BAF53a was driven by the constitutive promoter for the HSV immediate-early gene IE4/5, and the expression of the EGFP reporter was driven by the cytomegalovirus (CMV) promoter. HSV-EGFP was used to express EGFP alone as a control. HSV was packaged using a replication-defective helper virus 5dl1.2 as previously described (Neve et al., 2005; Kim et al., 2014). Virus stock titers ranged from 1.5 to 5.0 × 107 GFP-forming units per ml.
AAV viral vector and packaging.
For Baf53b knockdown, we designed a shRNA against Baf53b (5′-TACAGCAAGGCATCGTCAA-3′) and a nontarget shRNA (5′-TGGTTTACATGTTGTGTGA-3′) as a control. For shRNA, two 60 bp complementary DNA oligonucleotides, including the sense and antisense strands of the Baf53b sequence and stem loop according to the manufacturer's instruction (OligoEngine), were annealed to produce the double-stranded DNA fragments. These annealed products were subcloned into the pSUPER.neo+gfp vector (OligoEngine), which contains an H1 promoter. Then the H1 promoter and shRNA were subcloned into the AAV vector which expresses DsRed2 under the hSyn promoter. The knockdown-resistant BAF53b cDNA was generated using the QuikChange II XL Site-Directed Mutagenesis Kit (200521, Agilent) by making silent mutations in the Baf53b shRNA target sequence (TACAGCAAGGCATCGTCAA → TTCAACAGGGAATTGTGAA). AAV was packaged into AAV2/1 serotyped virus as previously described (Kwon et al., 2014). Viral titers were determined by quantitative real-time PCR (Rotor-Gene Q, QIAGEN) using SYBR Green (204074, QIAGEN). These ranged between 1 and 3 × 1012 DNA copies per ml.
Transfection.
To transfect AAV-nontarget shRNA or AAV-Baf53b shRNA DNA constructs to the HEK293T cells, we cultured cells on 60 mm culture dishes and used lipofectamine LTX with PLUS reagent (15338–100, Invitrogen) according to the manufacturer's instruction. One day after the transfection, we infected cells with 2 μl of HSV viral vectors and incubated for 1 d.
Surgery.
Mice were anesthetized with pentobarbital (83 mg/kg of body weight) by intraperitoneal injection and placed on a stereotaxic frame. Viral solutions were bilaterally injected into the LA (anteroposterior = −1.6 mm, mediolateral = ±3.45, dorsoventral = −4.55 mm from the bregma) using glass micropipettes and an infusion pump at a rate of 0.1 μl/min for 15 min (total, 1.5 μl per side) and left for an additional 5 min for diffusion.
Auditory fear conditioning.
For Baf53b knockdown experiments, we injected AAV-nontarget shRNA (control), AAV-Baf53b shRNA (Baf53b KD), or AAV-rBAF53b-Baf53b shRNA (rBAF53b) into the LA 3 weeks before training. For training, mice were placed in a conditioning chamber (Coulbourn Instruments) for 2 min, followed by the conditioned stimulus (CS; 2800 Hz, 85 dB pure tone, 30 s duration) coterminating with the unconditioned stimulus (US; 0.5 mA strong foot shock for 2 s). They then were allowed to remain in the chamber for an additional 30 s. For the retention sessions, CS-induced freezing was measured in a novel context (Plexiglas plate and round wall) 1 or 24 h after conditioning to test short-term (control, n = 11; Baf53b KD, n = 12) or long-term (control, n = 9; Baf53b KD, n = 15) memory, respectively. For rBAF53b rescue experiment, long-term memory was tested as above (control, n = 6; rBAF53b, n = 6). During memory test, the CS was presented for 3 min after 2 min of pre-CS. Freezing level during the first minute of tone presentation was used for data analysis.
For BAF53b overexpression experiments, groups of mice were injected with HSV-EGFP, HSV-BAF53b, HSV-BAF53a, or HSV-BAF53(a+b) in the LA 3 d before the training with 0.4 mA shock, and tested for short-term (EGFP, n = 7; BAF53b, n = 7) and long-term (EGFP, n = 9; BAF53b, n = 11) memory as above. For the post-training injection experiment, we injected HSV viral vectors 1 d after the training and tested 3 d later (EGFP, n = 10; BAF53b, n = 10). For remote memory, we tested mice 1 d after the training and then tested them again 28 d later (EGFP, n = 10; BAF53b, n = 8). For subdomain 2 experiment, long-term memory was tested 1 d after the training (EGFP, n = 8; BAF53a, n = 7; BAF53(a+b), n = 7).
Open field test.
Mice were injected with AAV-nontarget shRNA (n = 11) or AAV-Baf53b shRNA (n = 10) into the LA. Three weeks after the injection, mice were placed in the center of an open field box (45 × 45 × 25 cm). Using a video camera, 30 min of movement was recorded per mouse and the total distance each mouse moved and the frequency it crossed the center zone (22.5 × 22.5 cm) during 20 min was quantified using Ethovision XT (Noldus).
Histology.
At the end of behavioral experiments, all the mice were perfused and fixed with 4% PFA and then brains were extracted. The brains were sliced in coronal sections at 40 μm thickness using vibratome (VT1000S, Leica). The virus expression was verified with a fluorescence microscope (ECLIPSE 80i, Nikon). Only mice that showed highly specific expression in the LA region were included in data analysis. In a few cases, we observed small infection along the viral injection track with nice on-target expression in the LA. We also included such animals for the data analysis. Those that showed few infected neurons (<10 in the LA), unilateral expression, or off-target expression in CeA, striatum, and lateral areas to the LA were excluded.
Electrophysiology.
The 250-μm-thick coronal mouse brain slices were prepared with vibratome and incubated in bath solution (124 mm NaCl, 2.5 mm KCl, 1 mm NaH2PO4, 25 mm NaHCO3, 10 mm glucose, 2 mm CaCl2, 2 mm MgSO4 saturated with 95% CO2, 5% O2) at room temperature for 1–2 h. The slices were then transferred to a recording chamber. Recording pipettes were pulled to 3–5 mΩ resistance and filled with internal solution (145 mm K-gluconate, 5 mm NaCl, 0.2 mm EGTA, 10 mm HEPES, 2 mm MgATP, 0.1 mm Na3GTP, 1 mm MgCl2) containing 2 mg/ml biocytin (pH 7.2 with KOH, 280–290 mOsm). Recordings were performed at 32°C-34°C. For miniature EPSC (mEPSC) recordings, picrotoxin (100 μm) and TTX (1 μm) were added to the bath solution. The mCherry-positive neurons in the LA were voltage-clamped at −70 mV using a Multiclamp 700B amplifier and pClamp 10.2 software (Molecular Devices). Data were collected for 3 min and digitized at 10 kHz with a 2 kHz low-pass filter using a Digidata 1440 16 bit A/D converter (Axon Instruments). Series resistances were carefully monitored and recordings that varied significantly (>20%) or reached 20 mΩ were excluded. Data were analyzed using Clampfit 10.4 (Molecular Devices) with a template match threshold of 4.
Spine analysis.
For Baf53b knockdown experiment, we injected AAV-Baf53b shRNA (n = 3) or AAV-nontarget shRNA (n = 3) into the LA and incubated for 6–7 weeks. After electrophysiological recordings as described above, we labeled the infected neurons with biocytin. Recording pipettes were filled with internal solution containing 2 mg/ml biocytin (pH 7.2 with KOH, 280–290 mOsm). The brain slices were fixed with 4% PFA. The fixed slices were then washed three times with PBS, blocked, and permeabilized with 5% goat serum and 0.2% Triton X-100 in PBS for 1 h at room temperature. For staining, the slices were incubated in streptavidin and AlexaFluor-488 conjugate (Invitrogen) at 1:2000 dilution overnight at 4°C. The slices were then washed three times with PBS and mounted with VECTASHIELD Antifade Mounting Media with DAPI (H-1200, Vector Laboratories) on slide glasses and imaged with a LSM780 confocal microscope (Zeiss) using a 100× 1.4 NA objective lens. We focused on secondary or tertiary branches of apical dendrites between 50 and 100 μm from the soma of pyramidal neurons and acquired z-stack images at 0.2 μm intervals. These z-stacks were 3D-reconstructed and analyzed using Imaris FilamentTracer (Bitplane) blindly by an independent experimenter. For BAF53b overexpression experiment, we injected diluted (1:2–1:5) HSV-EGFP (home cage, n = 3; trained, n = 4) or HSV-BAF53b (home cage, n = 4; trained, n = 4) virus into the mice LA to sparsely infect the neurons. Four days later, the mice were subjected to auditory fear conditioning with 0.4 mA foot shock as described above or given foot shock immediately after entering the chamber (0.4 mA foot shock for 2 s) and then left for 3 min for immediate shock condition (EGFP, n = 8; BAF53b, n = 7). One hour after the training or immediate shock, mice were perfused and fixed with 4% PFA. We extracted the brains and prepared coronal sections (20 μm) using a cryostat (CM1850, Leica). Brain sections were immunostained with anti-GFP antibody and imaged with a LSM780 confocal microscope (Zeiss) as described above. The number of spines was manually counted using ImageJ software in a blind manner. Dendritic spines were classified as thin, stubby, or mushroom-type as previously described (Harris et al., 1992; Humeau et al., 2005) with the following modifications: stubby spines were defined as spines without a neck, mushroom spines were defined as spines with heads wider than 0.55 μm, and thin spines were those that could neither be classified as stubby nor mushroom-type.
Antibodies.
The following antibodies were used for immunohistochemical analysis: anti-BAF53b (NeuroMab, 75–311), anti-BAF53a (NeuroMab, 75–330), anti-GAPDH (Abcam, ab9485), anti-GFP (ab290, Abcam), HRP-conjugated anti-mouse IgG (Sigma, A4416), AlexaFlour-488-conjugated goat anti-rabbit antibody (Invitrogen, A-11008), and AlexaFlour-594-conjugated goat anti-mouse (Invitrogen, A-11005) antibodies.
Immunohistochemistry.
For BAF53b staining, heat-induced antigen retrieval was used. Brain sections were incubated in antigen retrieval buffer (100 mm Tris-HCl, pH 10.0) at 95°C for 10 min. After 3 times of washing with PBS, the sections were blocked by blocking solution (100 mm phosphate buffer, 0.1% BSA, 0.2% Triton X-100, 2% goat serum) for 15 min at room temperature. Then primary antibody was applied in the blocking solution (anti-BAF53b, 1:200; anti-GFP, 1:10000) on a shaker at 4°C for 48 h, and then brain sections were washed 4 times with PBS for 10 min. Next, secondary antibody (1:1000) was applied at room temperature for 2 h, and then brain sections were washed 4 times with PBS for 10 min. The slices were mounted with VECTASHIELD Antifade Mounting Media with DAPI on slide glasses.
Western blot.
For the Baf53b knockdown test, naive mice as a control (n = 3) and mice injected with AAV-nontarget shRNA (n = 3) or AAV-Baf53b shRNA (n = 3) into the LA were used. For antibody specificity test, Baf53b+/− (n = 3) or WT (n = 3) mice were used. For rBAF53b experiment, naive mice (n = 3) and mice injected with AAV-nontarget shRNA (n = 3), AAV-Baf53b shRNA (n = 3), or AAV-rBAF53b-Baf53b shRNA (n = 3) into the LA were used. For the BAF53b overexpression test, mice injected with HSV-EGFP (n = 2) or HSV-BAF53b (n = 2) into the LA were used. n values indicate the number of trials. We anesthetized the mice with isoflurane and extracted the brains to prepare 300-μm-thick coronal brain sections using a vibratome. The LA tissue samples were obtained by dissecting LA areas from the brain sections under stereo zoom microscope (S6D, Leica) with guidance by fluorescence signal and mouse brain anatomy. The dissected LA tissues from two mice were pooled and lysed in 20 μl of ice-cold lysis buffer (50 mm HEPES pH 8.0, 400 mm NaCl, 10% glycerol, 1% Triton X-100, 5 mm DTT) containing a protease inhibitor mixture (11836153001, Roche). To measure protein levels in HEK293T cells, HEK293T cells plated on 60 mm culture dishes were harvested and lysed in 100 μl ice-cold lysis buffer. We repeated the experiment 3 times for all experimental groups (n = 3). Total protein concentrations were determined by Bradford assay. Protein samples (30–50 μg per lane) were resolved by SDS-PAGE and transferred to PVDF membranes using the Trans-Blot Turbo Blotting System (Bio-Rad). After blocking with 5% nonfat dried milk in TNTX buffer (50 mm Tris-HCl, pH 7.5, 200 mm NaCl, 0.2% Triton X-100) for 30 min at room temperature, the membranes were incubated with primary antibodies against BAF53b (1:2000) or BAF53a (1:2000) in 3% BSA overnight at 4°C. HRP-conjugated goat anti-mouse IgG was used as secondary antibody (1:2000). Then the antibodies were detected using ECL solution (RPN2232, GE Healthcare). The blotted membranes were stripped using Restore Western Blot Stripping Buffer (21059, Thermo Fisher Scientific) and then stained with anti-GAPDH antibody (1:2000) as loading control. For the comparison and data analysis, protein levels were normalized to GAPDH expression level in every Western blot analysis.
qPCR.
For Baf53b knockdown test, mice injected with AAV-nontarget shRNA (n = 3) or AAV-Baf53b shRNA (n = 2) into the LA were used. To examine BAF53b induction after training, mice were single-housed for a week before the experiments. The mice were subjected to auditory fear conditioning with strong shock and killed 1 (n = 4), 3 (n = 4), 6 (n = 8), or 24 (n = 5) h after the training. Home cage group was used as a control (n = 7). We anesthetized the mice with isoflurane and extracted the brains, then ice-chilled in ACSF. One minute later, the brains were sectioned using vibratome at 300 μm thickness in ice-chilled ACSF. LA tissues were dissected and stored in RNALater (AM7020, Invitrogen) at −20°C until processing. Total RNAs were isolated using the mirVANA miRNA isolation kit (AM1560, Ambion) according to the manufacturer's instructions. cDNAs were reverse-transcribed from 100 to 500 ng of total RNA using the miScript II RT kit (218160, QIAGEN) with the miScript HiFlex buffer and diluted to 5 ng/μl. The following qPCR primers were used for mRNA quantification as published (Bloodgood et al., 2013; Vogel-Ciernia et al., 2013): Baf53b (forward 5′-TCCTGCCTTCTTCTTATGCAA-3′, reverse 5′-CCTGTGGAGCGTCCATTT-3′), Gapdh (forward 5′-ATGGTGAAGGTCGGTGTGA-3′, reverse 5′-AATCTCCACTTTGCCACTGC-3′). qPCR was performed using the miScript SYBR Green PCR kit (218073, QIAGEN) with a Rotor-Gene Q (QIAGEN). Expression of each individual gene was normalized to Gapdh. Relative quantifications between groups were analyzed using the ΔΔCT method (Livak and Schmittgen, 2001). The absolute amount of Gapdh mRNA was determined by producing standard curves with serially diluted (every 10-fold) cDNA (Wang et al., 2000; Schmittgen et al., 2008).
Statistical analysis.
Data are presented as mean ± SEM. Statistical tests included two-tailed Student's t tests, one-way ANOVAs, and two-way ANOVAs followed by Bonferroni or Tukey post hoc tests (Graphpad Prism 6, version 6.05). The threshold for statistical significance was set at p < 0.05.
Results
Specific knockdown of Baf53b by AAV-shRNA
To determine whether BAF53b function in the amygdala is necessary for cued fear memory formation, we used AAV viral vector-mediated delivery of Baf53b shRNA to knockdown endogenous BAF53b expression in the LA neurons. We generated AAV-Baf53b shRNA construct expressing shRNA targeting the mouse Baf53b mRNA under the control of an H1 promoter. We also made a nontarget shRNA construct as a control. To verify their expression, we designed each construct to additionally express DsRed2 under the human synapsin (hSyn) promoter as an expression marker (Fig. 1A). Via quantitative Western blot analysis of mice LA infected with AAV-Baf53b shRNA, AAV-nontarget shRNA, or noninfected LA, we found the Baf53b shRNA, but not the control nontarget shRNA, robustly reduced BAF53b protein level in the LA (Fig. 1B; one-way ANOVA, F(2,6) = 12.23, p < 0.01; Bonferroni post hoc confirmed statistical significance between naive group and Baf53b shRNA group, p < 0.05; nontarget shRNA group and Baf53b shRNA group, p < 0.05).
Figure 1.
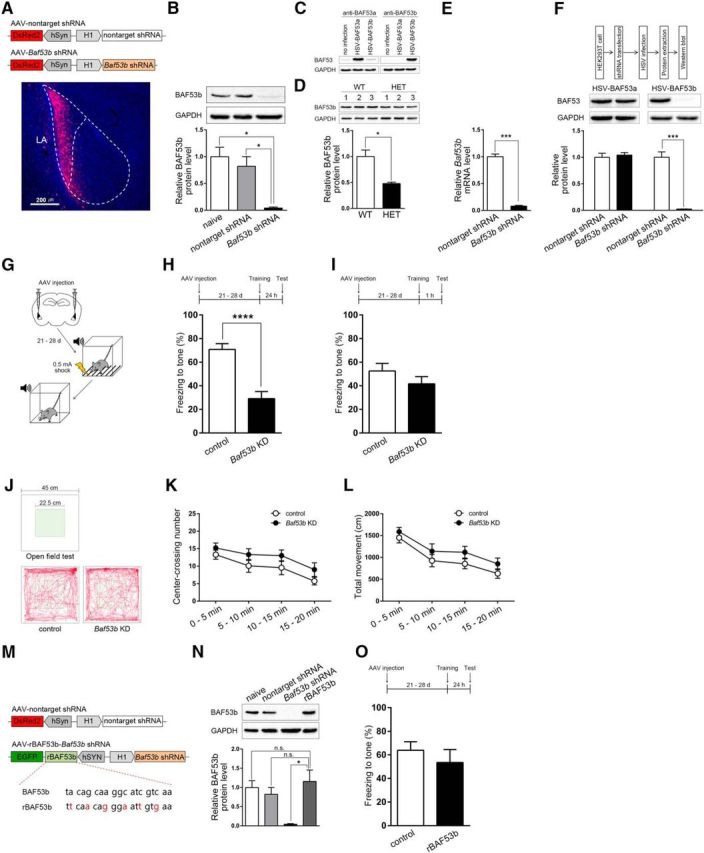
BAF53b function is necessary in the LA for the consolidation of auditory fear memory during long-term memory formation. A, Top, Construct map for AAV-nontarget shRNA and AAV-Baf53b shRNA. Bottom, Representative AAV-infected neurons (red) in the LA. Nuclei (blue) were stained with DAPI. Scale bar, 200 μm. B, Western blot showing BAF53b protein levels in the LA of naive mice (n = 3), nontarget shRNA-expressing mice (n = 3), and Baf53b shRNA-expressing mice (n = 3). Top, Representative Western blot. Bottom, BAF53b expression histograms. The y-axis indicates the BAF53b protein level for each condition relative to naive control. The BAF53b protein level for each condition was normalized to the GAPDH level. n values indicate the number of trials. C, Western blot showing protein levels of BAF53a (left, anti-BAF53a) or BAF53b (right, anti-BAF53b) in HEK293T cells infected with either HSV-BAF53a or HSV-BAF53b. No infection condition was included as a control. GAPDH was used as a loading control. D, Top, Western blot showing BAF53b protein levels in the LA of Baf53b+/− (HET, n = 3) and WT (n = 3) mice. Bottom, BAF53b expression histograms. The y-axis indicates the BAF53b protein level relative to the WT control. The BAF53b protein level for each condition was normalized to the GAPDH protein level. n values indicate the number of trials. E, Baf53b mRNA levels from LAs expressing either the nontarget shRNA (n = 3) or the Baf53b shRNA (n = 2) as measured by qPCR. The y-axis indicates the Baf53b mRNA level relative to the nontarget shRNA control. The Baf53b mRNA level for each condition was normalized to Gapdh mRNA level. F, Top, Experimental procedure. HEK293T cells expressing the nontarget shRNA or Baf53b shRNA were infected with either HSV-BAF53a or HSV-BAF53b (n=3 for each condition). Middle, Representative Western blot. Bottom, BAF53a and BAF53b expression histograms. The y-axis indicates the BAF53a or BAF53b protein level relative to nontarget shRNA control. The BAF53a or BAF53b protein level for each condition was normalized to the GAPDH level. n values indicate the number of trials. G, Behavioral procedure for auditory fear conditioning with strong shock (0.5 mA). H, Top, Behavioral procedure. Bottom, Tone-induced freezing during retention test. Long-term memory tested 24 h after training in mice injected with either AAV-nontarget shRNA (control, n = 9) or AAV-Baf53b shRNA (Baf53b KD, n = 15). n values indicate the number of mice. I, Top, Behavioral procedure. Bottom, Tone-induced freezing during retention test. Short-term memory tested 1 h after training (control, n = 11; Baf53b KD, n = 12). n values indicate the number of mice. J–L, Open field test with control (n = 11) or Baf53b KD (n = 10) mice. n values indicate the number of mice. J, Representative activity pattern tracked by EthoVision XT (Noldus). K, L, Center-crossings (K) and total distance moved (L) during 20 min. M, Construct maps for the AAV vectors expressing nontarget shRNA as a control and both knockdown-resistant BAF53b (rBAF53b) and Baf53b shRNA. Red represents mutation sites of rBAF53b. N, BAF53b expression levels from LA of noninfected mice (naive, n = 3), LAs infected with AAV-nontarget shRNA (nontarget shRNA, n = 3), AAV-Baf53b shRNA (Baf53b shRNA, n = 3), or AAV-rBAF53b-Baf53b shRNA (rBAF53b, n = 3) as measured by Western blot. Top, Representative Western blot. Bottom, BAF53b expression histograms. The y-axis indicates the BAF53b protein level relative to the naive control. The BAF53b protein level for each condition was normalized to the GAPDH protein level. n values indicate the number of trials. O, Top, Behavioral procedure. Bottom, Freezing level measured in mice injected with either the AAV-nontarget shRNA (control, n = 6) or the AAV-rBAF53b-Baf53b shRNA (n = 6) during long-term fear memory test. n values indicate the number of mice. Data are mean ± SEM. n.s., Not significant (p > 0.05). *p < 0.05. ***p < 0.001. ****p < 0.0001.
To confirm the specificity of BAF53b antibody, we used two different approaches. We first generated HSV vectors to acutely overexpress BAF53b (HSV-BAF53b) or its homolog BAF53a (HSV-BAF53a). Then we prepared HEK293T cells, which infected with HSV-BAF53a or HSV-BAF53b, and noninfected control. At 24 h after the infection, we lysed the cells to perform Western blot using anti-BAF53b antibody. As expected, we observed robust signal only in the BAF53b-infected cells. When we repeated the Western blot on the same lysates but with anti-BAF53a antibody, we observed strong signal in the BAF53a-infected cells, and weak endogenous signals in the BAF53b-infected cells and noninfected cells, confirming that BAF53a was well overexpressed in the cells infected with HSV-BAF53a. GAPDH was used as a loading control (Fig. 1C). Second, by Western blot with brain lysates of BAF53b heterozygous knock-out (Baf53b+/−) and WT mice, we observed ∼50% reduced signal detected by anti-BAF53b antibody in the Baf53b+/− brain compared with the WT brain (Fig. 1D) (two-tailed Student's t test, t(4) = 4.11, p < 0.05). The expression levels were normalized to GAPDH levels. Together, these results confirmed that the BAF53b antibody we used is highly specific to BAF53b protein.
Using qPCR, we confirmed Baf53b shRNA reduced Baf53b mRNA by >90% compared with the control (Fig. 1E) (two-tailed Student's t test, t(3) = 14.02, p < 0.001). To assess knockdown specificity, we transfected HEK293T cells with AAV-Baf53b shRNA or AAV-nontarget shRNA DNA constructs and infected them with HSV-BAF53b or HSV-BAF53a. As expected, Baf53b shRNA interfered BAF53b expression without affecting BAF53a (BAF53a, two-tailed Student's t test, t(4) = 0.47, p = 0.66; BAF53b, two-tailed Student's t test, t(4) = 9.54, p < 0.001) (Fig. 1F), confirming the specificity of our AAV viral vector-mediated knockdown of Baf53b.
Baf53b knockdown in the LA impairs long-term memory formation of auditory fear conditioning
We first examined the effects of LA-specific Baf53b knockdown on auditory fear memory. Three weeks after bilateral injection of AAV-Baf53b shRNA or AAV-nontarget shRNA into the LA, we subjected mice to auditory fear conditioning that paired a tone (CS) with an aversive foot shock (0.5 mA; US). We then measured conditioned freezing to the CS presentation to assess short- or long-term memories of the fear conditioning (Fig. 1G). When animals were tested 24 h after the conditioning, Baf53b knockdown mice showed significantly less conditioned freezing than control mice (two-tailed Student's t test, t(22) = 4.81, p < 0.0001), indicating the impairment of long-term memory formation (Fig. 1H). Baf53b knockdown mice and control mice showed similar freezing 1 h after conditioning (two-tailed Student's t test, t(21) = 1.22, p = 0.24), suggesting no effect on fear memory acquisition and short-term memory formation (Fig. 1I). As a control, we also tested the possibility that Baf53b knockdown affected basal anxiety level or locomotor activity. In an open field test, Baf53b knockdown and control mice showed similar basal levels of anxiety (center-crossing number, two-way repeated-measures ANOVA, group × time interaction, F(3,57) = 0.13, p = 0.94) and locomotor activity (total movement, two-way repeated-measures ANOVA, group × time interaction, F(3,57) = 0.23, p = 0.87), supporting a specific effect of Baf53b knockdown on long-term memory (Fig. 1J–L).
To further confirm the specificity of Baf53b knockdown effect, we generated an AAV vector that allows coexpression of Baf53b shRNA with a knockdown-resistant BAF53b (hereafter, rBAF53b) containing several silent mutations in the shRNA target sequence (Fig. 1M; see Materials and Methods). We confirmed that the BAF53b protein expression level in mice injected with AAV-rBAF53b-Baf53b shRNA in the LA was comparable to naive or AAV-nontarget shRNA group (one-way ANOVA, F(3,11) = 8.97, p < 0.01; Bonferroni post hoc showed no statistical significant difference between naive group and rBAF53b group, p > 0.99; nontarget shRNA group and rBAF53b group, p > 0.99) (Fig. 1N). For behavioral test, mice injected with AAV-rBAF53b-Baf53b shRNA or AAV-nontarget shRNA as control were trained for auditory fear conditioning and tested 24 h later. During fear memory test, mice injected with AAV-rBAF53b-Baf53b shRNA showed no significant difference in freezing levels compared with control mice (Fig. 1O) (two-tailed Student's t test, t(10) = 0.79, p = 0.45). This behavioral rescue by rBAF53b confirms that the long-term memory impairment caused by expression of Baf53b shRNA is specifically attributed to Baf53b loss of function. Together, these results indicate that BAF53b function is critical in the LA for consolidation of cued fear memory.
Normal basal synaptic transmission and spine structures in Baf53b knockdown neurons in the LA
Because BAF53b has been implicated in dendritic outgrowth and spine structure, it is possible that Baf53b knockdown may alter the functional and structural properties of LA synapses at the basal state. To examine basal synaptic transmission, we measured mEPSC amplitude and frequency by performing patch-clamp recordings from neurons in the LA injected with AAV-Baf53b shRNA or AAV-nontarget shRNA 6–7 weeks after the injection (Fig. 2A,B). We observed no significant changes in mEPSC amplitude (Fig. 2C; two-tailed Student's t test, t(19) = 1.65, p = 0.12) and frequency (Fig. 2D; two-tailed Student's t test, t(19) = 0.25, p = 0.80), indicating that Baf53b knockdown has no significant effect on the basal synaptic transmission in the LA neurons. To visualize the dendritic spines of shRNA-expressing neurons, we injected biocytin into individual DsRed2-positive LA neurons and stained with streptavidin and Alexa Fluor-488 conjugate (Fig. 2E). We then categorized dendritic spines into thin, stubby, or mushroom-type spine using a published criterion with slight modifications (see Materials and Methods) (Harris et al., 1992; Humeau et al., 2005). We found no significant difference between Baf53b knockdown and control neurons (Fig. 2F–I), indicating that Baf53b knockdown in the LA neurons affects neither total spine density nor their subtype-specific distribution (total, two-tailed Student's t test, t(4) = 0.4210, p = 0.70; thin, two-tailed Student's t test, t(4) = 1.48, p = 0.21; stubby, two-tailed Student's t test, t(4) = 0.11, p = 0.92; mushroom, two-tailed Student's t test, t(4) = 0.93, p = 0.40). To confirm whether knockdown efficiency of Baf53b shRNA was still intact 7 weeks after the injection, the time point at which the spine analysis was conducted, we performed Western blot using LAs of noninfected and AAV-Baf53b shRNA infected mice 7 weeks after the injection. As expected, we observed robustly reduced expression of BAF53b level (naive, 1.00 ± 0.02, n = 2; Baf53b shRNA, 0.12 ± 0.03, n = 2; two-tailed Student's t test, t(2) = 23.98, p < 0.01).
Figure 2.

Baf53b knockdown in the LA does not affect basal synaptic transmission or spine structures. A, Experimental procedure. B, Representative mEPSC recording trace. Calibration: 20 pA, 500 ms. C, D, Amplitude (C) and frequency (D) of mEPSCs recorded from DsRed2-positive neurons in the LA expressing either the nontarget shRNA (control, n = 10) or the Baf53b shRNA (Baf53b KD, n = 11). E, A representative biocytin-labeled LA neuron with 3D reconstruction of its dendrites. Thin (blue), stubby (red), and mushroom (green) spines. Scale bar, 50 μm. F–I, Spine density in LA neurons expressing the nontarget shRNA (control, n = 3) or the Baf53b shRNA (Baf53b KD, n = 3). Total (F), thin (G), stubby (H), and mushroom (I) spines are shown separately. n values indicate the number of mice. Data are mean ± SEM.
Baf53b gene expression is induced in the LA after fear conditioning
Given the critical role of BAF53b in memory consolidation, it is possible that Baf53b gene expression is regulated by learning during long-term memory formation. To explore this, we conducted auditory fear conditioning on separate groups of naive mice as before and killed them 1, 3, 6, or 24 h after the training. Then we dissected bilateral LA tissues and determined mRNA levels of Baf53b and Gapdh, a housekeeping gene used as a normalizing control by qPCR. Remarkably, we found that Baf53b mRNA level was gradually increased 3 h and peaked at 6 h after the auditory fear conditioning (Fig. 3A). Notably, an increase of Baf53b expression was observed at 24 h after training, although the level was slightly reduced compared with the level at 6 h after training (one-way ANOVA, F(4,23) = 16.35, p < 0.0001; Bonferroni post hoc confirmed statistical significance between home cage group and 3, 6, and 24 h after fear conditioning group; p < 0.01 for 3 h; p < 0.0001 for 6 h; p < 0.05 for 24 h). Baf53b mRNA level was not significantly changed 1 h after training, and this result is consistent with the previous report that Baf53b expression level in the dorsal hippocampus was not significantly changed 30 min after object location memory training (Vogel-Ciernia et al., 2013). We confirmed that the loading control Gapdh mRNA levels exhibited no significant changes throughout all experimental conditions (Fig. 3B) (one-way ANOVA, F(4,23) = 0.97, p = 0.44). Overall, our results indicate that Baf53b gene expression is induced at the late consolidation phase after fear conditioning.
Figure 3.

BAF53b is induced in the LA after fear conditioning. A, Baf53b mRNA levels measured at intervals after auditory fear conditioning with strong shock (0.5 mA) and normalized to Gapdh (home cage, n = 7; 1 h, n = 4; 3 h, n = 4; 6 h, n = 8; 24 h, n = 5). The y-axis indicates the Baf53b mRNA level relative to the home cage control. The Baf53b mRNA level for each condition was normalized to Gapdh mRNA level. B, The absolute amount of Gapdh mRNA quantified using standard curve measured at different time points after auditory fear conditioning with strong shock (0.5 mA) (home cage, n = 7; 1 h, n = 4; 3 h, n = 4; 6 h, n = 8; 24 h, n = 5). The y-axis indicates the Gapdh mRNA level relative to the home cage control. n values indicate the number of mice. Data are mean ± SEM. *p < 0.05. **p < 0.01. ****p < 0.0001.
Transient BAF53b overexpression leads to persistently enhanced memory formation
Since we found that BAF53b is necessary for long-term memory formation and induced after fear conditioning, we next asked whether increase of BAF53b function can sufficiently facilitate consolidation process. To examine this question, we used HSV viral vector to increase the BAF53b expression specifically in the LA neurons of adult mice (Fig. 4A). The increase of BAF53b expression was confirmed by both immunohistochemistry (Fig. 4B) and Western blot analysis (Fig. 4C) (two-tailed Student's t test, t(2) = 36.18, p < 0.001). Although ∼10%-15% of the cells in the LA were infected with the HSV-BAF53b viral vector, we observed an ∼200% increase in BAF53b protein level. To investigate the role of BAF53b increase in consolidation process, we bilaterally injected either HSV-BAF53b or HSV-EGFP as a control into the LA and trained the mice for auditory fear conditioning using a relatively weak foot shock (0.4 mA) to avoid ceiling effect (Han et al., 2007) (Fig. 4D). We tested mice 24 h after the training for long-term memory. Mice with increased BAF53b expression exhibited greatly increased conditioned freezing to the CS tone than EGFP-expressing control mice (Fig. 4E) (two-tailed Student's t test, t(18) = 2.85, p < 0.05), indicating long-term memory enhancement. In contrast, short-term memory test conducted 1 h after the training showed no significant difference between EGFP- and BAF53b-expressing mice, indicating that the memory-enhancing effect was specific to the long-term memory (Fig. 4F) (two-tailed Student's t test, t(12) = 0.25, p = 0.81). We then asked whether increase of BAF53b expression even after consolidation can enhance memory. We injected the HSV viral vectors in the LA 1 d after the training and tested the mice 3 d later. This time we observed no memory enhancement (two-tailed Student's t test, t(18) < 0.01, p = 0.99), indicating that BAF53b overexpression does not enhance the already consolidated memory (Fig. 4G). Because HSV viral vector-mediated gene expression is transient (∼7 d after injection) (Fink et al., 1996; Neve et al., 2005), we next asked whether transient BAF53b increase is sufficient for persistent memory enhancement even at remote time when BAF53b overexpression disappeared. Separate groups of mice were injected with HSV-BAF53b or HSV-EGFP and trained for auditory fear conditioning. Mice were tested twice 1 d (recent memory) and 29 d (remote memory) after training. Mice injected with HSV-BAF53b, which showed enhanced memory at day 1, still displayed enhanced memory at day 29, indicating the persistence of enhanced memory does not require the existence of BAF53b overexpression per se (Fig. 4H) (two-way repeated-measures ANOVA, group × time interaction, F(1,16) = 0.27, p = 0.61; significant effect of group, F(1,16) = 6.94, p < 0.05; significant effect of time, F(1,16) = 14.69, p < 0.01; Bonferroni post hoc confirmed statistical significance between EGFP group and BAF53b group at day 1, p < 0.01; day 29, p < 0.05). It also suggests that memory enhancement by BAF53b overexpression did not result from its effect on memory retrieval. Together, these results support the idea that the increase of BAF53b expression facilitates persistent memory formation by regulating the memory consolidation process.
Figure 4.

Transient BAF53b overexpression leads to persistently enhanced memory. A, Top, Construct maps of HSV-EGFP and HSV-BAF53b. Bottom, Representative HSV-infected neurons (green) in the LA. Nuclei (blue) were stained with DAPI. Scale bar, 200 μm. B, Top, Experimental procedure. Bottom, Representative HSV-infected neurons (green) immunostained with an anti-BAF53b antibody (red). Nuclei (white) were stained with DAPI. Scale bar, 10 μm. C, Top, Experimental procedure. Middle, Representative Western blot. Bottom, BAF53b expression in LAs injected with HSV-EGFP (EGFP, n = 2) or HSV-BAF53b (BAF53b, n = 2). The y-axis indicates the BAF53b protein level relative to the EGFP control. The BAF53b protein level for each condition was normalized to the GAPDH protein level. D, Behavioral procedure for auditory fear conditioning with weak shock (0.4 mA). E–H, Top, Behavioral procedure. Bottom, Tone-induced freezing during retention test. E, Long-term memory of mice injected with either the HSV-EGFP (EGFP, n = 9) or the HSV-BAF53b (BAF53b, n = 11) tested 24 h after training. F, Short-term memory tested 1 h after training (EGFP, n = 7; BAF53b, n = 7). G, Long-term memory of mice injected with either the HSV-EGFP (EGFP, n = 10) or the HSV-BAF53b (BAF53b, n = 10) 24 h after training was tested 3 d after injection. H, Long-term memory (Recent) and remote memory (Remote) of mice injected with either the HSV-EGFP (EGFP, n = 10) or the HSV-BAF53b (BAF53b, n = 8) tested 24 h (Recent) and 29 d (Remote) after training. I, Top, 3D structures of BAF53a and BAF53b proteins deduced with SWISS-Model and visualized with the PyMOL Molecular Graphics System (Oma et al., 2003). Bottom, Chimeric BAF53(a+b) schematic, in which the BAF53a subdomain 2 is swapped for that of BAF53b. J, Top, Experimental procedure. Bottom, Tone-induced freezing in mice expressing EGFP (n = 8), BAF53a (n = 7), or BAF53(a+b) (n = 7). n values indicate the number of mice. Data are mean ± SEM. *p < 0.05. **p < 0.01. ***p < 0.001.
BAF53b function mediated by subdomain 2 is critical for the memory enhancement
Because BAF53a, a BAF53b homolog, has a high sequence similarity with BAF53b (Oma et al., 2003), we asked whether the memory-enhancing effect is specific to BAF53b and how such specificity occurs (Fig. 4I). We first determined the effect of BAF53a expression in the LA on memory enhancement. We injected mice with either HSV-BAF53a or HSV-EGFP bilaterally into the LA and trained them for auditory fear conditioning with a weak training protocol (0.4 mA shock as an US). Unlike BAF53b, BAF53a expression showed no memory enhancement (Fig. 4J), indicating that memory-enhancing effect is specific to BAF53b. The actin fold subdomain 2 (hereafter subdomain 2) is the most divergent sequence between BAF53b and its homolog BAF53a (Fig. 4I) (Oma et al., 2003), which is known to be essential for BAF53b's promotion of dendritic spine outgrowth in cultured neurons (Wu et al., 2007). Based on these findings, we speculated that subdomain 2 may be important for BAF53b-mediated memory enhancement. To test this idea, we tested a chimeric form of BAF53a, BAF53(a+b), which swaps the BAF53a subdomain 2 for that of BAF53b (Fig. 4I). Interestingly, mice expressing the chimeric BAF53(a+b) displayed significantly enhanced long-term memory compared with EGFP control mice similar to the BAF53b-expressing condition (Fig. 4J) (one-way ANOVA, F(2,19) = 6.59, p < 0.01; Bonferroni post hoc confirmed statistical significance between EGFP group and BAF53(a+b) group, p < 0.01; BAF53a group and BAF53(a+b) group, p < 0.05; no significance between EGFP group and BAF53a group, p = 0.88). These results thus suggest that BAF53b function mediated by subdomain 2 is critical for the enhancement of long-term memory formation.
BAF53b overexpression promotes thin spine outgrowth following training
Given the role of BAF53b in dendritic outgrowth and spine plasticity (Wu et al., 2007; Vogel-Ciernia et al., 2013), we reasoned that learning-related spine plasticity may also be affected by BAF53b increase. To investigate this possibility, we measured spine density in HSV-injected LA neurons at basal state and 1 h after fear conditioning conducted with a weak training protocol (0.4 mA shock as an US) (Fig. 5A). Immediate shock group was included as a control to rule out any stress-induced effects. Dendritic spine morphology was visualized by EGFP fluorescence signal amplified with immunostaining with an anti-GFP antibody (Fig. 5B). We observed a significant increase in total spine density after fear conditioning in LA neurons infected with HSV-BAF53b compared with the EGFP control neurons, whereas the total spine density at basal state was unchanged (Fig. 5C) (two-way ANOVA, virus × behavior interaction, F(1,11) = 4.12, p = 0.07; significant effect of virus, F(1,11) = 22.81, p < 0.001; Tukey post hoc confirmed statistical significance between trained groups of EGFP and BAF53b, p < 0.05). Spine subtype analysis revealed that the increase of total spine density was due to the increase of thin-type spine density (Fig. 5D–F) (thin, two-way ANOVA, virus × behavior interaction, F(1,11) = 3.34, p = 0.09; significant effect of virus, F(1,11) = 24.73, p < 0.001, Tukey post hoc confirmed statistical significance between trained groups of EGFP and BAF53b, p < 0.05; stubby, two-way ANOVA, virus × behavior interaction, F(1,11) = 0.02, p = 0.88; mushroom, two-way ANOVA, virus × behavior interaction, F(1,11) = 0.59, p = 0.46). Notably, we also observed that BAF53b overexpression slightly increased mushroom-type spine density in the immediate shock condition without affecting total, thin-type, and stubby-type spine density, likely reflecting a shock-induced stress effect (Fig. 5G–J) (two-tailed Student's t test, total, t(13) = 1.28, p = 0.22; thin, t(13) = 0.44, p = 0.67; stubby, t(13) = 0.38, p = 0.71; mushroom, t(13) = 2.19, p < 0.05). Together, our spine analysis data indicate that increase of BAF53b function promotes new spine outgrowth after training, providing a possible mechanism for persistently enhanced memory formation by BAF53b overexpression.
Figure 5.

Transient BAF53b overexpression promotes learning-induced outgrowth of thin spines in the LA neurons. A, Experimental procedure. B, Representative dendritic segments and spines. Scale bar, 5 μm. C–F, Spine density of neurons infected with HSV-EGFP (EGFP) or HSV-BAF53b (BAF53b) from mice taken from their home cage as a control (home cage) or 1 h after auditory fear conditioning (trained). Total (C), thin (D), stubby (E), and mushroom (F) spines are presented separately (EGFP, home cage, n = 3; EGFP, trained, n = 4; BAF53b, home cage, n = 4; BAF53b, trained, n = 4). G–J, Spine density of neurons infected with HSV-EGFP (EGFP, n = 8) or HSV-BAF53b (BAF53b, n = 7) 1 h after 0.4 mA immediate shock. Total (G), thin (H), stubby (I), and mushroom (J) spines are presented separately. n values indicate the number of mice. Data are mean ± SEM. *p < 0.05. **p < 0.01.
Discussion
BAF53b in the LA neurons is required to form long-term cued fear memory
In this study, we show BAF53b, a postmitotic neuron-specific subunit of BAF nucleosome remodeling complex, is induced in the LA after fear conditioning and plays a critical role for long-term memory formation by facilitating memory consolidation. Previously, Vogel-Ciernia et al. (2013) generated mice expressing a dominant-negative form of BAF53b (BAF53bΔHDlow) and heterozygous Baf53b-null mutant mice (Baf53b+/−) that led them to conclude BAF53b is required for hippocampus-dependent memory, but not amygdala-dependent memory. This is inconsistent with our findings, but our unique approach to manipulating BAF53b levels may explain this discrepancy. With our AAV viral-vector mediated RNAi method, we saw ∼90% reduction in BAF53b. While Baf53b+/− mice produce 50% of the normal level of Baf53b, the extent of BAF53b loss of function in BAF53bΔHDlow mice is unclear. Thus, as suggested by Vogel-Ciernia et al. (2013), it is possible amygdala-dependent memory is simply less sensitive to the loss of BAF53b than hippocampus-dependent memory (Vogel-Ciernia et al., 2013). In addition, genetic knock-outs often show less severe phenotypes than expected because of functional compensation (Marie et al., 2005; Pulvers et al., 2010; Sharf et al., 2010; Freudenberg et al., 2012; Kim et al., 2015). Perhaps, our acute knockdown of Baf53b using RNAi allowed us to identify the function of BAF53b in memory formation in the adult brain without any potentially confounding effects on brain development. When we tested Baf53b+/− mice with our own behavioral protocols, we also found no significant defect in long-term memory formation of auditory fear conditioning (WT, 49.81 ± 8.68%; Baf53b+/−, 45.18 ± 12.28%; two-tailed Student's t test, t(8) = 0.32, p = 0.76), consistent with the published study. Thus, it seems that shRNA-mediated knockdown of Baf53b causes more severe loss-of-function phenotype than heterozygous knock-out or dominant-negative inhibition of BAF53b function, suggesting that our unique result is unlikely attributable to differences in behavioral experimental condition. Regardless of its merits, a major issue with the RNAi approach is whether the knockdown is specific to the target. We first tried to test knockdown specificity in primary neuron culture before using HEK293T cells. However, it was technically infeasible in our hand. First, because it took >2 weeks for the AAV virus to induce gene expression in transduced neurons, we always ended up observing only few infected neurons with AAV-shRNA virus (generally 1 or 2 cells/FOV) before the cultured neurons died (∼DIV 21). Second, we noticed that it is very difficult to cotransfect HSV-BAF53a and AAV-shRNA vector DNA in the same neurons due to the generally low DNA transfection efficiency in primary cultured neurons (<10% in our condition). Because of these technical limitations, we could not test the shRNA specificity in neurons. Nevertheless, given our analysis with HEK293T cells that showed unaffected BAF53a expression by AAV-Baf53b shRNA, and the well-known fact that BAF53a is undetectable in postmitotic neurons in the mouse adult brain (Olave et al., 2002), we believe that the knockdown by AAV-Baf53b shRNA in our experimental condition was specific to BAF53b.
In addition to the cued fear memory, the effect on spine morphology also somewhat differs between two studies. Baf53b knockdown had no effect on spine morphology in LA neurons in our study, whereas hippocampal neurons of BAF53bΔHDhigh mice showed significant alterations, such as decrease of thin spine density. This difference may also be explained by the possibilities discussed above. Still, although we do not yet know how to explain the discrepancy, our results indicate that BAF53b is required in the LA neurons for the formation of long-term memories of cued fear conditioning.
BAF53b is induced after learning at the late phase of memory consolidation
Learning induces expression of many genes, which are considered as important molecules for memory consolidation (Hall et al., 2000; Josselyn et al., 2001). Our results provide the first evidence that an epigenetic factor involved in nucleosome remodeling can be induced after learning at the late phase of memory consolidation. Previous RNA-sequencing data in the hippocampus fail to see BAF53b induction after learning (Vogel-Ciernia et al., 2013). It may be because they monitored gene expression at relatively early phase (30 min after training) after training or because different learning paradigm (object location memory task) was used. Alternatively, BAF53b induction may differ in different brain regions, as was the case for other memory-related genes shown in a previous study (Ressler et al., 2002). It is unknown how BAF53b is induced in the LA after fear conditioning. Given that BAF53b induction occurs at the late consolidation phase, perhaps the transcription factors induced or activated at the early phase may regulate BAF53b induction at the late phase. Interestingly, previous study shows that PKA activity and P-CREB (phosphorylated form of CREB) level increase 3 and 6 h after inhibitory avoidance learning in rats (Bernabeu et al., 1997). Thus, it is also possible that BAF53b induction is regulated by the delayed activation of cell signaling or CREB activation. We found that Baf53b mRNA level was also increased 24 h after training. Although we do not know in this study what could be the role of such delayed BAF53b induction, it may suggest a possible role of BAF53b in memory persistence, which needs to be investigated in the future study. Regardless of these possibilities, the gene expression induction of BAF53b at the late consolidation phase suggests that BAF53b and likely nucleosome remodeling mechanism may have more important roles for memory regulation than previously considered. It is unknown how the increased expression of single subunit of nucleosome remodeling complex composed of multiple subunits might affect nucleosome remodeling-dependent gene expression regulation. Given that BAF53b is a stoichiometric component of combinatorial assembly of BAF complex expressed exclusively in postmitotic neuron (Olave et al., 2002; Wu et al., 2007), we speculate that BAF53b induction after learning may elevate the portion of BAF complex containing BAF53b to enhance the BAF53b-dependent gene expression regulation during memory formation.
Transient increase of BAF53b in the LA enhances long-term memory formation
Transient BAF53b overexpression led to persistently enhanced memory formation under subthreshold learning condition. A number of studies have reported long-term memory enhancement by manipulating factors involved in gene regulation. For instance, the increased expression of transcription factor CREB enhances long-term memory formation (Yin et al., 1995; Josselyn et al., 2001; Han et al., 2007; Suzuki et al., 2011). It has also been shown that epigenetic mechanisms can induce memory enhancement. DNMT3a2 restores the cognitive declines of aged mice (Oliveira et al., 2012). Treatment with a nonspecific HDAC inhibitor (Levenson et al., 2004; Lattal et al., 2007) and specific deletion of either HDAC2 (Guan et al., 2009) or HDAC3 (McQuown et al., 2011) also reportedly enhance long-term memory. Here, to the best of our knowledge, we show the first evidence of persistent memory enhancement via transient modulation of an epigenetic factor involved in chromatin remodeling. We found that BAF53b overexpression also promotes the formation of new dendritic spines after learning, particularly of the thin subtype. The resulting increase in thin-type spine density may explain how transient BAF53b overexpression produced persistently enhanced memory. Thin-type spines represent synapses with low synaptic strength and a low AMPAR/NMDAR ratio (Harris and Stevens, 1989; Das et al., 2008). Thin spines are highly plastic and can be converted by LTP and learning to mushroom-type spines or “memory spines” that have a higher AMPAR/NMDAR ratio (Holtmaat et al., 2005; Yasumatsu et al., 2008; Kasai et al., 2010a, b). Increases in small-head spine formation correlate well with enhanced memory (Enriquez-Barreto et al., 2014). Moreover, CREB overexpression (Sargin et al., 2013) and HDAC2 deletion (Guan et al., 2009), which lead to long-term memory enhancement, also increase spine density.
In conclusion, BAF53b induced in the LA after fear conditioning facilitates memory consolidation during long-term fear memory formation likely by promoting learning-related spine structural changes. Our study thus provides an idea about how nucleosome remodeling can likely be regulated during long-term memory formation and contributes to the permanent storage of associative fear memory in the LA, which is relevant to fear and anxiety-related mental disorders, such as post-traumatic stress disorder.
Footnotes
This work was supported by National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (MSIP) of the Korean government Grants 2014R1A2A1A10053821 and 2011-0005755; Brain Research Program through NRF Grant 2015M3C7A1027351; the Intelligent Synthetic Biology Center of the Global Frontier Project funded by the MSIP Grant 2011-0031955; and the Korea Advanced Institute of Science and Technology, Future Systems Healthcare Project. B.-K.K. was supported by Korea Government (MSIP) National Honor Scientist Program Grant NRF 2012R1A3A1050385. We thank all our laboratory members for critical discussions and helpful comments; and Dr. Andrew Yoo for BAF53b constructs.
The authors declare no competing financial interests.
References
- Bernabeu R, Bevilaqua L, Ardenghi P, Bromberg E, Schmitz P, Bianchin M, Izquierdo I, Medina JH (1997) Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc Natl Acad Sci U S A 94:7041–7046. 10.1073/pnas.94.13.7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloodgood BL, Sharma N, Browne HA, Trepman AZ, Greenberg ME (2013) The activity-dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503:121–125. 10.1038/nature12743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Lilly SM, Zerda R, Gunning WT 3rd, Alvarez FJ, Tietz EI (2008) Increased AMPA receptor GluR1 subunit incorporation in rat hippocampal CA1 synapses during benzodiazepine withdrawal. J Comp Neurol 511:832–846. 10.1002/cne.21866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriquez-Barreto L, Cuesto G, Dominguez-Iturza N, Gavilán E, Ruano D, Sandi C, Fernández-Ruiz A, Martín-Vázquez G, Herreras O, Morales M (2014) Learning improvement after PI3K activation correlates with de novo formation of functional small spines. Front Mol Neurosci 6:54. 10.3389/fnmol.2013.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink DJ, DeLuca NA, Goins WF, Glorioso JC (1996) Gene transfer to neurons using herpes simplex virus-based vectors. Annu Rev Neurosci 19:265–287. 10.1146/annurev.ne.19.030196.001405 [DOI] [PubMed] [Google Scholar]
- Freudenberg JM, Ghosh S, Lackford BL, Yellaboina S, Zheng X, Li R, Cuddapah S, Wade PA, Hu G, Jothi R (2012) Acute depletion of Tet1-dependent 5-hydroxymethylcytosine levels impairs LIF/Stat3 signaling and results in loss of embryonic stem cell identity. Nucleic Acids Res 40:3364–3377. 10.1093/nar/gkr1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459:55–60. 10.1038/nature07925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ (2000) Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci 3:533–535. 10.1038/75698 [DOI] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, Neve RL, Guzowski JF, Silva AJ, Josselyn SA (2007) Neuronal competition and selection during memory formation. Science 316:457–460. 10.1126/science.1139438 [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK (1989) Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci 9:2982–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B (1992) Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci 12:2685–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K (2005) Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45:279–291. 10.1016/j.neuron.2005.01.003 [DOI] [PubMed] [Google Scholar]
- Humeau Y, Herry C, Kemp N, Shaban H, Fourcaudot E, Bissière S, Luthi A (2005) Dendritic spine heterogeneity determines afferent-specific Hebbian plasticity in the amygdala. Neuron 45:119–131. 10.1016/j.neuron.2004.12.019 [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA Jr, Neve RL, Nestler EJ, Davis M (2001) Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci 21:2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. (2012) The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5:14. 10.1186/1756-6606-5-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, Noguchi J (2010a) Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci 33:121–129. 10.1016/j.tins.2010.01.001 [DOI] [PubMed] [Google Scholar]
- Kasai H, Hayama T, Ishikawa M, Watanabe S, Yagishita S, Noguchi J (2010b) Learning rules and persistence of dendritic spines. Eur J Neurosci 32:241–249. 10.1111/j.1460-9568.2010.07344.x [DOI] [PubMed] [Google Scholar]
- Kim J, Kwon JT, Kim HS, Josselyn SA, Han JH (2014) Memory recall and modifications by activating neurons with elevated CREB. Nat Neurosci 17:65–72. 10.1038/nn.3592 [DOI] [PubMed] [Google Scholar]
- Kim S, Titcombe RF, Zhang H, Khatri L, Girma HK, Hofmann F, Arancio O, Ziff EB (2015) Network compensation of cyclic GMP-dependent protein kinase II knockout in the hippocampus by Ca2+-permeable AMPA receptors. Proc Natl Acad Sci U S A 112:3122–3127. 10.1073/pnas.1417498112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M (2004) CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42:961–972. 10.1016/j.neuron.2004.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JT, Nakajima R, Kim HS, Jeong Y, Augustine GJ, Han JH (2014) Optogenetic activation of presynaptic inputs in lateral amygdala forms associative fear memory. Learn Mem 21:627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Barrett RM, Wood MA (2007) Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci 121:1125–1131. 10.1037/0735-7044.121.5.1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD (2004) Regulation of histone acetylation during memory formation in the hippocampus. J Biochem 279:40545–40559. 10.1074/jbc.M402229200 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC (2005) Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron 45:741–752. 10.1016/j.neuron.2005.01.039 [DOI] [PubMed] [Google Scholar]
- Mauceri D, Freitag HE, Oliveira AM, Bengtson CP, Bading H (2011) Nuclear calcium-VEGFD signaling controls maintenance of dendrite arborization necessary for memory formation. Neuron 71:117–130. 10.1016/j.neuron.2011.04.022 [DOI] [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, Wood MA (2011) HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci 31:764–774. 10.1523/JNEUROSCI.5052-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrano-Fernández A, Barco A (2016) Nuclear organization and 3D chromatin architecture in cognition and neuropsychiatric disorders. Mol Brain 9:83. 10.1186/s13041-016-0263-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD (2007) Covalent modification of DNA regulates memory formation. Neuron 53:857–869. 10.1016/j.neuron.2007.02.022 [DOI] [PubMed] [Google Scholar]
- Neve RL, Neve KA, Nestler EJ, Carlezon WA Jr (2005) Use of herpes virus amplicon vectors to study brain disorders. Biotechniques 39:381–391. 10.2144/05393PS01 [DOI] [PubMed] [Google Scholar]
- Olave I, Wang W, Xue Y, Kuo A, Crabtree GR (2002) Identification of a polymorphic, neuron-specific chromatin remodeling complex. Gene Dev 16:2509–2517. 10.1101/gad.992102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AM, Hemstedt TJ, Bading H (2012) Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat Neurosci 15:1111–1113. 10.1038/nn.3151 [DOI] [PubMed] [Google Scholar]
- Oma Y, Nishimori K, Harata M (2003) The brain-specific actin-related protein ArpN alpha interacts with the transcriptional co-repressor CtBP. Biochem Biophys Res Commun 301:521–528. 10.1016/S0006-291X(02)03073-5 [DOI] [PubMed] [Google Scholar]
- Pulvers JN, Bryk J, Fish JL, Wilsch-Bräuninger M, Arai Y, Schreier D, Naumann R, Helppi J, Habermann B, Vogt J, Nitsch R, Tóth A, Enard W, Pääbo S, Huttner WB (2010) Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc Natl Acad Sci U S A 107:16595–16600. 10.1073/pnas.1010494107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Paschall G, Zhou XL, Davis M (2002) Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci 22:7892–7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sando R 3rd, Gounko N, Pieraut S, Liao L, Yates J 3rd, Maximov A (2012) HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151:821–834. 10.1016/j.cell.2012.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargin D, Mercaldo V, Yiu AP, Higgs G, Han JH, Frankland PW, Josselyn SA (2013) CREB regulates spine density of lateral amygdala neurons: implications for memory allocation. Front Behav Neurosci 7:209. 10.3389/fnbeh.2013.00209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Lee EJ, Jiang J, Sarkar A, Yang L, Elton TS, Chen C (2008) Real-time PCR quantification of precursor and mature microRNA. Methods 44:31–38. 10.1016/j.ymeth.2007.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharf R, Sarhan M, Brayton CE, Guarnieri DJ, Taylor JR, DiLeone RJ (2010) Orexin signaling via the orexin 1 receptor mediates operant responding for food reinforcement. Biol Psychiatry 67:753–760. 10.1016/j.biopsych.2009.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji H, Takao K, Hattori S, Miyakawa T (2016) Age-related changes in behavior in C57BL/6J mice from young adulthood to middle age. Mol Brain 9:11. 10.1186/s13041-016-0191-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staahl BT, Tang J, Wu W, Sun A, Gitler AD, Yoo AS, Crabtree GR (2013) Kinetic analysis of npBAF to nBAF switching reveals exchange of SS18 with CREST and integration with neural developmental pathways. J Neurosci 33:10348–10361. 10.1523/JNEUROSCI.1258-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Fukushima H, Mukawa T, Toyoda H, Wu LJ, Zhao MG, Xu H, Shang Y, Endoh K, Iwamoto T, Mamiya N, Okano E, Hasegawa S, Mercaldo V, Zhang Y, Maeda R, Ohta M, Josselyn SA, Zhuo M, Kida S (2011) Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J Neurosci 31:8786–8802. 10.1523/JNEUROSCI.3257-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Wood MA (2014) Neuron-specific chromatin remodeling: a missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology 80:18–27. 10.1016/j.neuropharm.2013.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Matheos DP, Barrett RM, Kramár EA, Azzawi S, Chen Y, Magnan CN, Zeller M, Sylvain A, Haettig J, Jia Y, Tran A, Dang R, Post RJ, Chabrier M, Babayan AH, Wu JI, Crabtree GR, Baldi P, Baram TZ, Lynch G, Wood MA (2013) The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat Neurosci 16:552–561. 10.1038/nn.3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li X, Currie RW, Willette RN, Barone FC, Feuerstein GZ (2000) Application of real-time polymerase chain reaction to quantitate induced expression of interleukin-1beta mRNA in ischemic brain tolerance. J Neurosci Res 59:238–246. [DOI] [PubMed] [Google Scholar]
- White AO, Kramar EA, Lopez AJ, Kwapis JL, Doan J, Saldana D, Davatolhagh MF, Alaghband Y, Blurton-Jone M, Matheos DP, Wood MA (2016) BDNF rescues BAF53b-dependent synaptic plasticity and cocaine-associated memory in the nucleus accumbens. Nat Comm 7:11725. 10.1038/ncomms11725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, Crabtree GR (2007) Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56:94–108. 10.1016/j.neuron.2007.08.021 [DOI] [PubMed] [Google Scholar]
- Yasumatsu N, Matsuzaki M, Miyazaki T, Noguchi J, Kasai H (2008) Principles of long-term dynamics of dendritic spines. J Neurosci 28:13592–13608. 10.1523/JNEUROSCI.0603-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JC, Del Vecchio M, Zhou H, Tully T (1995) CREB as a memory modulator: induced expression of a dCREB2 activator isoform enhances long-term memory in Drosophila. Cell 81:107–115. 10.1016/0092-8674(95)90375-5 [DOI] [PubMed] [Google Scholar]
- Yoo AS, Staahl BT, Chen L, Crabtree GR (2009) MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature 460:642–646. 10.1038/nature08139 [DOI] [PMC free article] [PubMed] [Google Scholar]