

Abstract
Retinitis pigmentosa (RP) is an inherited retinal degeneration (RD) that leads to blindness for which no treatment is available. RP is frequently caused by mutations in Rhodopsin; in some animal models, RD is exacerbated by light. Valproic acid (VPA) is a proposed treatment for RP and other neurodegenerative disorders, with a phase II trial for RP under way. However, the therapeutic mechanism is unclear, with minimal research supporting its use in RP. We investigated the effects of VPA on Xenopus laevis models of RP expressing human P23H, T17M, T4K, and Q344ter rhodopsins, which are associated with RP in humans. VPA ameliorated RD associated with P23H rhodopsin and promoted clearing of mutant rhodopsin from photoreceptors. The effect was equal to that of dark rearing, with no additive effect observed. Rescue of visual function was confirmed by electroretinography. In contrast, VPA exacerbated RD caused by T17M rhodopsin in light, but had no effect in darkness. Effects in T4K and Q344ter rhodopsin models were also negative. These effects of VPA were paralleled by treatment with three additional histone deacetylase (HDAC) inhibitors, but not other antipsychotics, chemical chaperones, or VPA structural analogues. In WT retinas, VPA treatment increased histone H3 acetylation. In addition, electron microscopy showed increased autophagosomes in rod inner segments with HDAC inhibitor (HDACi) treatment, potentially linking the therapeutic effects in P23H rhodopsin animals and negative effects in other models with autophagy. Our results suggest that the success or failure of VPA treatment is dependent on genotype and that HDACi treatment is contraindicated for some RP cases.
SIGNIFICANCE STATEMENT Retinitis pigmentosa (RP) is an inherited, degenerative retinal disease that leads to blindness for which no therapy is available. We determined that valproic acid (VPA), currently undergoing a phase II trial for RP, has both beneficial and detrimental effects in animal models of RP depending on the underlying disease mechanism and that both effects are due to histone deacetylase (HDAC) inhibition possibly linked to autophagy regulation. Off-label use of VPA and other HDAC inhibitors for the treatment of RP should be limited to the research setting until this effect is understood and can be predicted. Our study suggests that, unless genotype is accounted for, clinical trials for RP treatments may give negative results due to multiple disease mechanisms with differential responses to therapeutic interventions.
Keywords: autophagy, HDAC inhibitor, retinal degeneration, retinitis pigmentosa, rhodopsin, valproic acid
Introduction
Retinitis pigmentosa (RP) is a disorder involving progressive death of retinal rod photoreceptors (Berson, 1993; Hartong et al., 2006; Daiger et al., 2013). Mutations in Rhodopsin, encoding the photosensitive rod pigment, cause 30% of autosomal-dominant RP (ADRP) (Daiger et al., 2013). Some mutant rhodopsins resemble WT in yield and localization, fold correctly, and regenerate with 11-cis-retinal, whereas others have low expression, aggregate, regenerate poorly, and are transported inefficiently to the plasma membrane (Sung et al., 1991, 1993; Kaushal and Khorana, 1994; Kaushal et al., 1994). Rhodopsin misfolding likely causes retinal degeneration (RD) via ER stress or proteasome overload and proteostatic crisis (Lin et al., 2007; Chiang et al., 2012, 2015; Lobanova et al., 2013). In patients, some mutants cause uniform degeneration, whereas others cause sector RP (Cideciyan et al., 1998), suggesting light-exacerbated disease. In animal models, some mutants cause light-exacerbated RD due to destabilization of mutant rhodopsin in the absence of chromophore (Organisciak et al., 2003; Zhu et al., 2004; Paskowitz et al., 2006; Tam and Moritz, 2007; White et al., 2007; Moritz and Tam, 2010; Tam et al., 2010). Using Xenopus laevis models of RP, we demonstrated that rhodopsin mutants cause cell death by at least three pathways involving mutant opsin destabilization, photoactivated rhodopsin destabilization, and rhodopsin mislocalization (Tam et al., 2006, 2014, Tam and Moritz, 2007, 2009).
A controversial study of a cohort of seven patients suggested that valproic acid (VPA) shows promise as an RP treatment (Clemson et al., 2011), but was criticized for lack of supporting research demonstrating a therapeutic mechanism (Sandberg et al., 2011; Tzekov et al., 2011; van Schooneveld et al., 2011). Follow-up reports suggested detrimental effects on acuity (Sisk, 2012; Bhalla et al., 2013). A recent study indicated opposing effects of VPA in mice with PDE6B mutations (Mitton et al., 2014). Nevertheless, a phase II clinical trial is under way.
VPA has anticonvulsant and mood stabilizing activities that are used to treat epilepsy and bipolar disorder. Its mechanisms of action are unclear due to complex pharmacology; it is a histone deacetylase inhibitor (HDACi) (Phiel et al., 2001), a GABA transaminase inhibitor, and a sodium channel blocker (Johannessen, 2000; Löscher, 2002; Owens and Nemeroff, 2003). Its mood stabilizing effects are due to inositol depletion (Williams et al., 2002). It can reduce ER stress via glycogen synthase kinase inhibition (Chen et al., 1999; Bown et al., 2002; Kim et al., 2005) and activate autophagy via PI3K (Sarkar et al., 2005; Williams et al., 2008; Renna et al., 2010). VPA is under investigation as a treatment for neurodegenerative diseases, including Huntington's (Chiu et al., 2011), Parkinson's (Monti et al., 2010) and Alzheimer's (Loy and Tariot, 2002; Tariot and Aisen, 2009; Fleisher et al., 2011) diseases, and is reported to enhance clearance of protein aggregates by autophagy (Renna et al., 2010; Fleming et al., 2011).
Here, we investigated the therapeutic potential of VPA using four transgenic X. laevis models of ADRP-expressing human rhodopsin mutants. Because three of the models show marked light dependence of RD (Tam and Moritz, 2007, 2009), we also assessed the interdependence of the effects of VPA and light exposure. We found that VPA may ameliorate or exacerbate RD depending on the underlying mutation. VPA ameliorated RD and vision deficits caused by P23H rhodopsin, the most common disease-causing RP mutation in North America (Sohocki et al., 2001), but dramatically exacerbated RD caused by T17M rhodopsin in the presence of light. Effects in other models were negative and less pronounced. VPA treatment decreased the burden of misfolded P23H rhodopsin. Similar results were obtained with three other HDACi's, but not with other antipsychotics, VPA structural analogs, or chemical chaperones, indicating that both the positive and negative effects were due to HDACi activity. Our results indicate that applicability of VPA treatment in RP is likely to be dependent on the patient genotype and that VPA treatment will be contraindicated for some RP cases.
Materials and Methods
Generation, rearing, and drug therapy of transgenic X. laevis.
Transgenic X. laevis tadpoles were generated by mating heterozygous or homozygous P23H, T17M, T4K, or Q344ter frogs with WT frogs. Embryos were housed in 4 L tanks in an 18°C incubator on a 12 h light/dark cycle. At 2 d postfertilization (dpf) (corresponding to developmental stage 23), animals of either sex were transferred into 1 L bins of 1× Ringer's solution with (10 μm) or without VPA or other drugs as indicated and grown in either the presence or absence of cyclic light. Fresh drug solution was prepared daily and renewed for 11 d. Animals were killed at 14 dpf (stage 48). For CI-994, this period was shortened to 9 dpf (stage 46). One eye was fixed in 4% formaldehyde buffered with 0.1 m sodium phosphate, pH 7.4; the contralateral eye was solubilized in 100 μl of a 1:1 mixture of PBS and SDS-PAGE loading buffer containing 1 mm EDTA and 100 μg/ml PMSF, as described previously (Tam et al., 2006) for dot blot analyses, and the tail was clipped for PCR genotyping.
Dot blot analyses.
Dot blots of X. laevis eye extracts were performed as described previously (Tam et al., 2006). Blots were probed with primary monoclonal antibody (mAb) B630N (Adamus et al., 1991) at a 1:15 dilution of tissue culture supernatant or mAb 1D4 (MacKenzie et al., 1984) (University of British Columbia, University-Industry Liaison Office) at a 1:5000 dilution of 1 mg/ml solution, followed by IR-dye800-conjugated goat anti-mouse antibody at 1:10,000 of 1 mg/ml solution (Rockland). Each dot blot included standards containing 100% X. laevis rod opsin (from WT retinas) and 100% human rod opsin (from transfected cultured cells).
Immunohistochemistry and confocal microscopy.
Fixed eyes were embedded and cryosectioned as described previously (Tam et al., 2006). Frozen sections were labeled with mAb 2B2 cell culture supernatant (Hicks and Molday, 1986) at a 1:10 dilution or mAb 1D4 at a 1:5000 dilution of 1 mg/ml solution, followed by a 1:750 dilution of Cy3-conjugated secondary antibody (Jackson ImmunoResearch) and counterstained with Alexa Fluor-488-conjugated wheat germ agglutinin (WGA; Invitrogen) and Hoechst 33342 (Sigma-Aldrich) as described previously (Moritz et al., 1999). mAb 2B2 and 1D4 recognize the N and the C terminus of mammalian rhodopsin, respectively. Sections were imaged using a Zeiss 510 laser scanning confocal microscope and 10× (air) or 40× (water-immersion) objectives. Image processing was performed using Adobe Photoshop. For construction of figures, the fluorescent signals derived from antibody labeling were adjusted linearly (i.e., no changes to gamma settings). Signals derived from WGA and Hoechst 33342 staining were adjusted nonlinearly to best demonstrate retinal architecture.
Extraction of genomic DNA and qPCR genotyping.
Clipped tails were immersed in 100 μl of genomic DNA extraction buffer (50 mm Tris, pH 8.5, 0.5% Tween 20, 200 μg/ml proteinase K) and incubated at 55°C for 2 h, 95°C for 10 min. Transgenic DNA was amplified using the primers 5′-TGAACTGCAAGACGAGGC-3′ and 5′-CAAGGTGAGATGACAGGAGATC-3′ and amplification was detected using the FAM-labeled probe 5′-CACTGAAGCGGGAAGGGAC-3′ (Integrated DNA Technologies).
Western blot analyses.
X. laevis whole eye extracts were obtained as described above. Briefly, total protein (5 μl/lane) was electrophoresed through 1D 15% SDS-polyacrylamide gels and transferred to PVDF membranes. Blots were blocked for 30 min at room temperature with 1% milk powder in PBS, followed by overnight incubation with antibody diluted in 0.1% milk powder and 0.05% Tween 20. Antibodies were rabbit anti-acetyl H3 (1:250; Millipore 06-599) and mouse anti-acetylated tubulin (Sigma-Aldrich 6-11B-1). Blots were washed with PBS + 0.05% Tween 20 and similarly incubated for 1 h with secondary antibodies IR-dye700-conjugated goat anti-mouse and IR-dye800-conjugated goat anti-rabbit 1:10,000 of 1 mg/ml solution (Rockland). The anti-acetyl H3 blot was stripped in 67 mm Tris, pH 6.8, 2% SDS, and 100 mm β-mercaptoethanol and reprobed with rabbit anti-H3 (AbCam 1791). A duplicate blot was probed with mouse anti-tubulin (Novus Biologicals NB100-690). Blots were scanned at 700 and 800 nm using a LICOR Odyssey Imaging System.
Electroretinography (ERG).
Tadpoles (13 dpf) were dark adapted overnight and all further procedures were done under red light illumination. Immediately before analyses, tadpoles were anesthetized in 0.01% tricaine in 0.1× Marc's Modified Ringer (MMR) until swimming and twitch responses to stimuli ceased. Tadpoles were mounted on a gold ECG electrode using 2% low-melting-point agarose containing 0.01% tricaine and a silver wire electrode housed in a glass microcapillary filled with 1× MMR was positioned on the cornea using a micromanipulator. The electrodes were connected to the head stage of a Model 1800 AC amplifier (AM Systems) and the output of the amplifier was in turn connected to the input of an Espion electroretinography unit designed for analysis of human subjects (Diagnosys). A Ganzfeld dome was lowered over the tadpole and the responses to a series of blue light flashes of increasing intensity were recorded by the Espion unit. Each intensity was presented five times and the results were averaged. After ERG analysis, tadpoles were immediately fixed for histology and processed for genotyping as described above. In the ERG experiment involving sodium butyrate treatment, animals were derived from a homozygous/WT cross and therefore genotyping was unnecessary. For each ERG experiment, all animals were derived from the same mating and analyzed on the same day using the same electrode.
Electron microscopy.
Procedures for electron microscopy were performed essentially as described previously (Tam et al., 2015). Briefly, tadpoles treated with 10 μm VPA, 300 μm sodium butyrate, or untreated, as described above, were killed at 6 dpf and fixed in 4% paraformaldehyde 1% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4. Eyes were infiltrated with 2.3 m sucrose in 0.1 m phosphate, embedded in optimal cutting temperature medium (TissueTek) and cryosectioned (20 μm). Cryosections were osmicated, dehydrated, infiltrated with Eponate 12 resin, and embedded as described previously (Tam et al., 2015). Sections 70 nm thick were cut, stained with uranyl acetate and lead citrate, and examined in a Hitachi H7600 transmission electron microscope.
Statistics.
Statistical analysis of dot blot data was performed on log-transformed integrated intensity values. Dose–response data were analyzed by one-way ANOVA conducted separately on WT and transgenic data, followed by Dunnet's test. Experiments involving single drug concentrations and measurements of transgenic rhodopsin expression were analyzed by two-way ANOVA. For two-way ANOVA, the reported p-values were as follows: pg, effect of genotype; pt, effect of treatment; and pi, interaction between treatment and genotype. Experiments involving Western blot detection of acetylated proteins were analyzed by Student's t test. ERG data were analyzed by two-way ANOVA (intensity vs condition, where condition is the combination of genotype and treatment), followed by Tukey's test for multiple comparisons or three-way ANOVA (intensity vs genotype vs treatment). Statistical tests were performed using SPSS software.
Results
VPA treatment provides dose-dependent protection from retinal degeneration in transgenic X. laevis expressing P23H rhodopsin
The P23H mutation in rhodopsin is the most common cause of ADRP in North America. Previously, we developed lines of transgenic X. laevis that express human RP-associated rhodopsin mutants such as P23H rhodopsin in rod photoreceptors under control of the X. laevis rod opsin promoter (originally described in Tam and Moritz, 2007). Transgenic tadpoles express human P23H rhodopsin at very low levels (<1% of total rhodopsin) due to a biosynthetic defect involving ER retention. To determine whether VPA is a potentially effective treatment for RP, we exposed P23H-transgenic X. laevis and their WT siblings to 10, 3, 1, and 0 μm VPA in their aqueous environment for 11 d. The animals were reared in normal cyclic light (1700 lux, 12 h on, 12 h off) or dark reared as a positive control for rescue of RD (Tam and Moritz, 2007). Eyes were solubilized for rod opsin dot blot or processed for confocal microscopy. In P23H animals, both VPA treatment and dark rearing resulted in significantly higher rod opsin signals, which were confirmed by histology to represent decreased loss of rod photoreceptors (Fig. 1). The protective effect of VPA treatment was dose dependent, resulting in threefold to sixfold higher levels of total rod opsin compared with untreated P23H animals. Log-transformed dot blot data were analyzed by one-way ANOVA followed by Dunnet's multiple comparisons and showed no effect of VPA on WT animals (ANOVA, p = 0.36) and a significant effect on transgenic animals (ANOVA, p = 1.5 × 10−12; p = 3.0 × 10−8 for 0 vs 10 μm; p = 0.0016 for 0 vs 3 μm). In addition, two-way ANOVA showed a significant interaction between treatment and genotype, indicating that VPA treatment significantly modified the effect of genotype (pi = 7 × 10−19).
Figure 1.
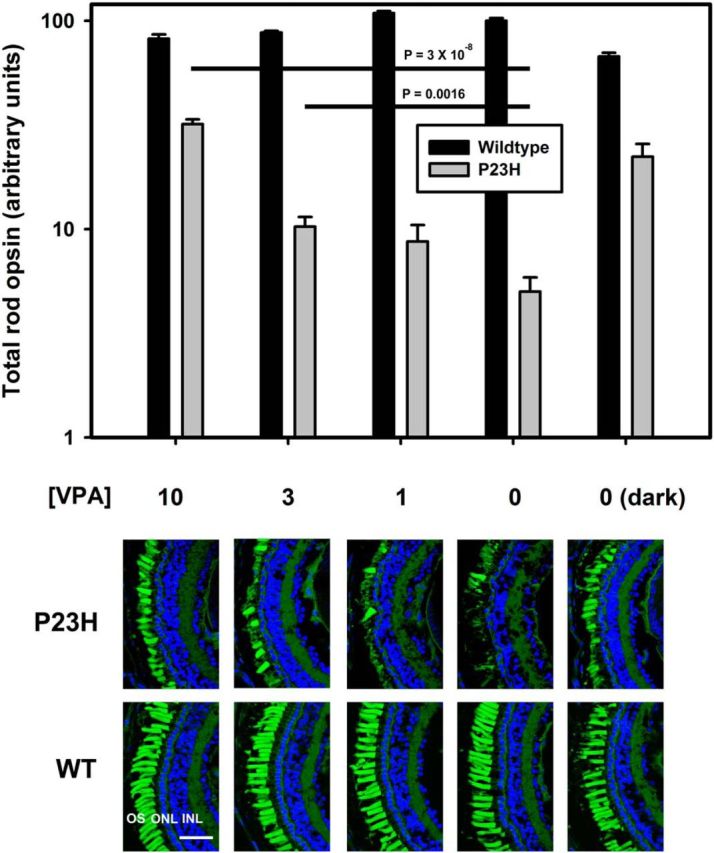
Effect of VPA on RD in transgenic X. laevis expressing P23H rod opsin. Top, Dot blot analysis of total rod opsin from whole eye extracts of P23H X. laevis (gray bars) and their WT siblings (black bars). VPA ameliorates RD significantly in a dose-dependent manner (ANOVA, p = 1.5 × 10−12). At 10 μm, the effect of VPA is equivalent to the effect of dark rearing. WT animals are unaffected by VPA (ANOVA, p = 0.363). p-values shown on chart are Dunnet's multiple comparisons. n = 10–17 animals per condition. Error bars indicate SEM. Bottom, Representative cryosections of contralateral eyes confirm that effects of VPA in dot blot assays are due to reduced RD in P23H-transgenic animals. OSs were stained with WGA (green) and Hoechst dye (blue). VPA treatment resulted in greater density of OSs in P23H X.laevis. WT animals were unaffected. Scale bar, 50 μm.
VPA ameliorates light-induced retinal degeneration in a P23H rhodopsin model of RP
Light-induced RD caused by misfolding of P23H rhodopsin has been well described in the literature (Paskowitz et al., 2006; Tam and Moritz, 2007; Tam et al., 2010). Previously, we determined that dark rearing results in partial rescue of RD in X. laevis expressing human P23H rhodopsin (Tam and Moritz, 2007; Fig. 1) and that the effects of light exposure are equivalent to those of vitamin A deprivation (Tam et al., 2010). To determine whether the rescue mechanism of VPA is analogous to dark rearing, we exposed the animals to four experimental conditions: cyclic light with or without 10 μm VPA and dark rearing with or without 10 μm VPA (Fig. 2). In cyclic light conditions, transgenic animals treated with VPA had significantly less RD as assessed by total rod opsin levels and histology compared with the nontreated group (Fig. 2A–C,). Two-way ANOVA analysis of the results from animals reared in cyclic light shown in Figure 2A indicated significant effects of both genotype and treatment (pg = 6 × 10−9; pt = 2 × 10−5) and a significant interaction between genotype and treatment (pi = 2 × 10−5), indicating that treatment with VPA altered the effects of genotype significantly. In contrast, although the effect of genotype remained significant (pg = 4 × 10−5) in dark reared animals, treatment was marginally significant (pt = 0.047) and there was no significant interaction of treatment and genotype (pi = 0.22). Therefore, the effects of dark rearing and VPA treatment on total rod opsin levels were not additive, indicating that the rescue mechanisms of VPA treatment and dark rearing are likely to be related and redundant (Fig. 2A–C).
Figure 2.
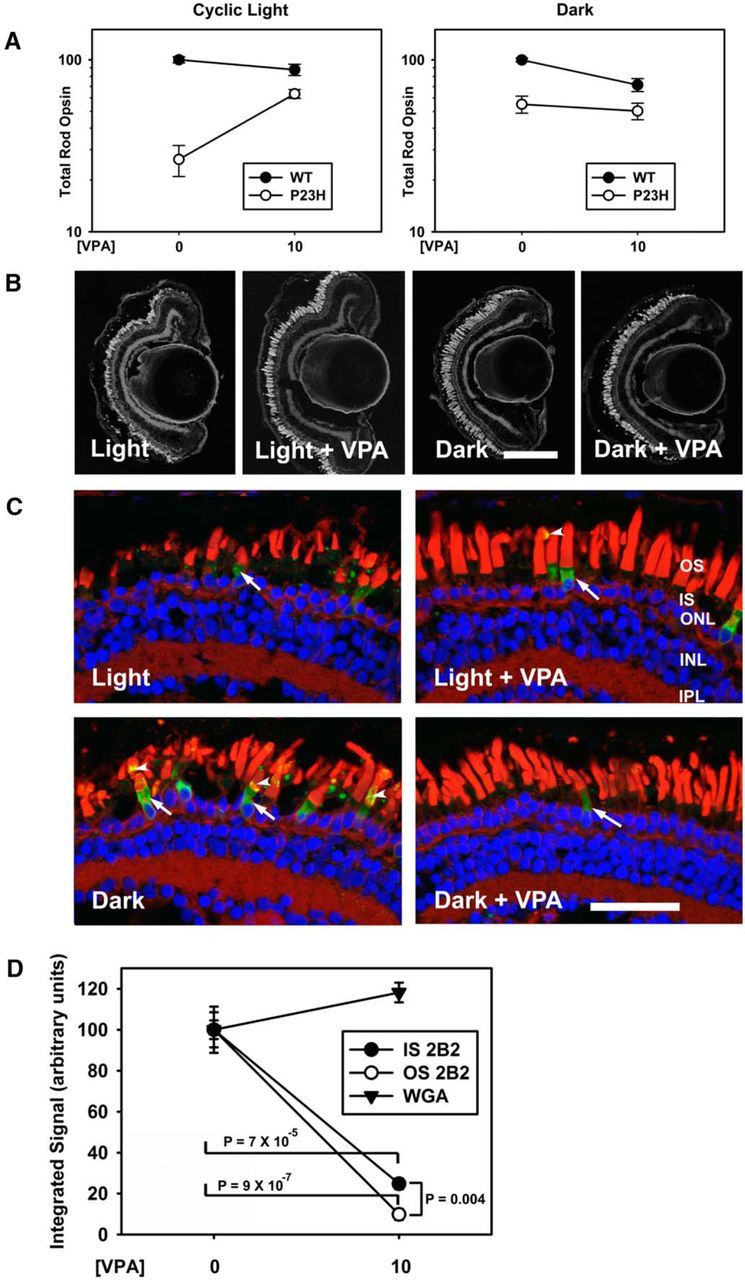
VPA ameliorates light-induced RD in a P23H rod opsin model. A, Dot blot analysis of P23H X. laevis and their WT siblings treated with 10 μm VPA in cyclic light and dark-reared conditions. In cyclic light, the effect of genotype and treatment were significant (two-way ANOVA, pg = 6 × 10−9, pt = 2 × 10−5) and VPA treatment significantly modified the effect of genotype (pi = 2 × 10−5). In the dark, the effect of genotype was significant (pg = 4 × 10−5), the effect of treatment was minimally significant (pt = 0.047), and treatment did not significantly modify the effect of genotype (p = 0.22). n = 5–13 animals per group. Error bars indicate SEM. B, Representative low-magnification confocal micrographs of cryosections from transgenic retinas expressing P23H rod opsin stained with WGA. Scale bar, 200 μm. C, Representative high-magnification confocal micrographs of transgenic retinas expressing P23H rod opsin stained with 2B2 anti-mammalian rhodopsin (green), WGA (red), and Hoechst dye (blue). Dark rearing promotes OS localization of P23H rhodopsin (arrowheads). VPA treatment does not alter P23H rhodopsin localization. ONL, Outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer. Scale bar, 50 μm. D, OS WGA and whole-retina 2B2 (P23H rhodopsin) signals were quantified in confocal microscopy images of dark reared transgenic animals. 2B2 signal was markedly reduced in both IS and OS of treated animals, whereas WGA signal was unchanged (two-way ANOVA pi = 3.5 × 10−9, t tests p = 7 × 10−5, p = 9 × 10−7, p = 0.004).
VPA does not alter P23H rhodopsin distribution, but reduces P23H rhodopsin levels
As reported previously (Tam and Moritz, 2007), immunolabeling of frozen sections using mAb 2B2, which binds mammalian rhodopsin specifically, showed that P23H rhodopsin was transported from the inner segment (IS) to the outer segment (OS) in greater quantities in dark reared animals relative to animals reared in cyclic light (Fig. 2C) as a consequence of the pharmacological chaperone effect of 11-cis-retinal chromophore, which is more abundant in dark-reared retinas (Tam and Moritz, 2007; Tam et al., 2010). Unlike mAb 1D4, mAb 2B2 labels full-length P23H rhodopsin specifically and not proteolytic degradation products such as N-terminally truncated forms (Tam and Moritz, 2007). In contrast, VPA treatment had no appreciable effects on the relative distribution of P23H rhodopsin between outer and ISs (Fig. 2C). Interestingly, comparisons of animals that were dark reared and either treated or untreated with VPA (having indistinguishable levels of RD) demonstrated that VPA treatment reduced the levels of P23H rhodopsin markedly in both inner and OSs of rod photoreceptors (Fig. 2C) to the point where it was difficult to detect the mutant protein in the majority of cells. We quantified the effects of VPA on the mAb 2B2 label in both inner and OSs by confocal microscopy (Fig. 2D) compared with effects on WGA labeling of rod OSs, which served as a measure of rod photoreceptor content of the retina. (Antibody labeling of endogenous rhodopsin was not used because previous experience indicated that the results are extremely nonlinear.) We found a significant effect of VPA treatment on mAb 2B2 labeling in ISs (t test p = 7 × 10−5) and OSs (t test p = 9 × 10−7) in the absence of significant change in WGA labeling (two-way ANOVA Pi = 3.5 × 10−9). Tukey post hoc test after two-way ANOVA did not indicate a significant difference between the effects of VPA on IS versus OS 2B2 labeling (p = 0.817), although the effect was significant by t test (p = 0.004), suggesting a greater decrease in OS labeling relative to IS labeling. Overall, the results do not support a chaperone mechanism in which VPA promotes trafficking of P23H rhodopsin to the OS, but rather indicate that clearing of misfolded P23H rhodopsin in dark-reared animals was enhanced by VPA treatment, suggesting that both dark rearing and VPA treatment act by reducing IS levels of P23H rhodopsin.
VPA exacerbates light-induced retinal degeneration in a T17M rhodopsin model of RP
In transgenic X. laevis, T17M rhodopsin induces RD that is exacerbated by light exposure, but prevented by eliminating chromophore binding, indicating that the underlying mechanism of light-induced cell death requires photoactivation of the mutant rhodopsin (Tam et al., 2014). Therefore, unlike RD caused by P23H rhodopsin, this RD is likely caused by destabilization of the mutant protein in rod OSs (Tam et al., 2014). We also developed lines of transgenic X. laevis expressing human T17M rhodopsin under control of the X. laevis rod opsin promoter, with no evidence of biosynthetic defects such as ER retention (Tam and Moritz, 2009; Tam et al., 2014). In initial experiments examining the effects of VPA on RD caused by T17M rhodopsin in a line expressing at 27% of total rhodopsin, we failed to observe a beneficial effect of VPA (data not shown), but rather noted a reduction of total rod opsin by dot blot analyses in VPA-treated T17M animals compared with the untreated group, the significance of which was potentially limited by a “floor effect.” This led us to reexamine the effect of VPA in a second line of transgenic animals expressing T17M rhodopsin at relatively low levels (5% of total rod opsin) causing minimal baseline RD. Surprisingly, we found that, in cyclic light, VPA dramatically exacerbated RD in these animals (Fig. 3A–C) and the effect occurred at the same doses that protected against RD in P23H animals. This effect was not observed in the absence of light exposure, indicating that VPA exacerbates cell death induced by instability of photoactivated rhodopsin specifically. Two-way ANOVA analysis of the data shown in Figure 3A showed significant effects of genotype and treatment in cyclic light conditions and that treatment modified the effects of genotype significantly (pg = 1.7 × 10−9, pt = 1.8 × 10−9, pi = 1.2 × 10−6), whereas, in dark reared animals, there was no effect of genotype, minimal effect of treatment, and no interaction between the two (pg = 0.56, pt = 0.029, pi = 0.665). Histology of frozen sections showed proper trafficking of T17M rhodopsin to rod OSs in all four experimental groups, indicating that VPA treatment does not introduce an alternate cell death mechanism due to misfolding of T17M rhodopsin in the biosynthetic pathway (Fig. 3C), but rather appears to exacerbate cell death mechanisms that are already present. Furthermore, we examined expression levels of T17M rhodopsin by probing dot blots of dark reared retinal extracts with the antibody B630N, which recognizes both frog and mammalian rod opsins, and 1D4, which recognizes mammalian rod opsin. There was no indication of an alteration in the absolute or relative ratios of transgenic and total rod opsins by two-way ANOVA (pi = 0.353; Fig. 3D).
Figure 3.
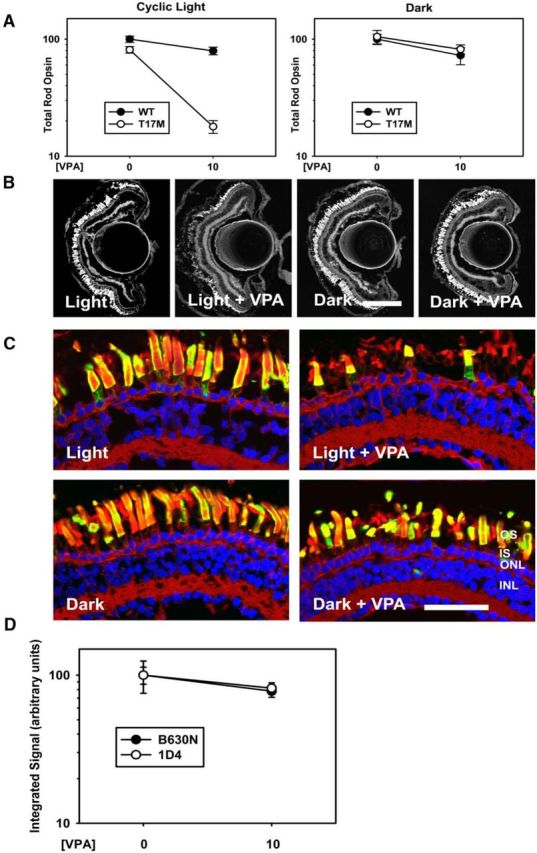
VPA exacerbates light-induced RD in a T17M rod opsin model. A, Dot blot analysis of VPA-treated T17M X. laevis and their WT siblings in cyclic light and dark-reared conditions. In cyclic light, the effects of genotype and treatment were significant (pg = 1.7 × 10−9, pt = 1.8 × 10−9) and VPA treatment modified the effect of genotype significantly (pi = 1.2 × 10−6). In the dark, there was no significant effect of genotype and no interaction between VPA and genotype (pg = 0.56, pt = 0.029, pi = 0.67). n = 6–16 animals per group. Error bars indicate SEM. B, Representative low-magnification confocal micrographs of cryosections from transgenic retinas expressing T17M rod opsin stained with WGA. Scale bar, 200 μm. C, Representative high-magnification confocal micrographs of transgenic retinas expressing P23H rod opsin stained with 2B2 anti-mammalian rhodopsin (green), WGA (red), and Hoechst dye (blue). Neither dark rearing nor VPA treatment altered the distribution of T17M rod opsin significantly. ONL, Outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer. Scale bar, 50 μm. D, Total rod opsin (B630N) and T17M rhodopsin (1D4) signals were quantified in dot blot analyses of dark-reared animals. Two-way ANOVA analysis showed no significant effects of treatment on antibody signals, indicating that VPA treatment did not alter T17M rhodopsin expression levels. n = 11–13 animals for each condition.
VPA exacerbates light-induced retinal degeneration in T4K and Q344ter rhodopsin models of RP
Previously, we developed lines of transgenic X. laevis expressing human T4K rhodopsin in rod photoreceptors under control of the X. laevis rod opsin promoter (Tam et al., 2014). Based on our previous studies, light-exacerbated RD caused by T4K rhodopsin and T17M rhodopsin share a common pathological mechanism involving photoactivated mutant rhodopsin (Tam et al., 2014). To determine whether VPA consistently exacerbates RD caused by this mechanism, we treated animals expressing human T4K rhodopsin (at 4% of total rhodopsin) reared in either cyclic light or darkness with 10 μm VPA using an identical experimental paradigm. We found that VPA treatment reduced total rod opsin levels in T4K animals (Fig. 4A–D). However, the lack of a significant interaction between treatment and genotype (p = 0.452) indicates that the effect is not equivalent to that seen in T17M animals and may mirror a general downward trend in rod opsin levels in treated WT animals also seen in several other experiments. Nevertheless, the effect on total rod opsin in light-exposed transgenic animals was unfavorable (pt = 0.00012 for effect of treatment in two-way ANOVA, p = 0.006 for effect of treatment on T4K animals by t test). Histology indicated that RD is higher in these animals (Fig. 4B,C). Overall, we conclude that here was a detrimental effect of VPA in this model of RP, although it may be a generalized effect that is not specific to the T4K genotype. Furthermore, despite a common requirement for photoactivated rhodopsin, these results suggest that there may be significant differences in the pathologies underlying RD caused by T4K and T17M rhodopsin.
Figure 4.
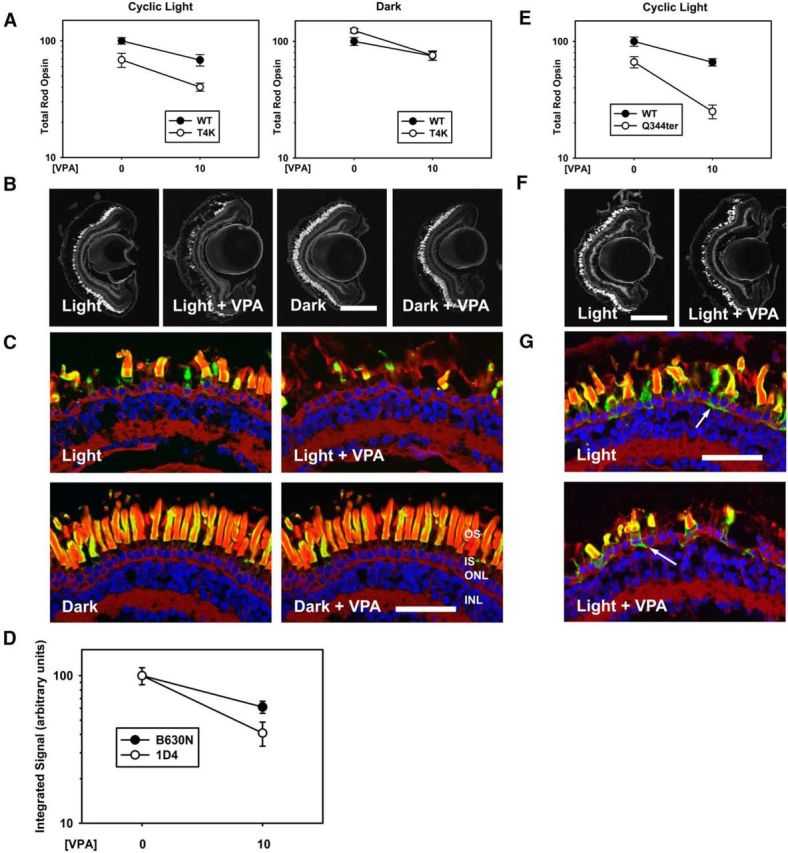
VPA has negative effects in a T4K rod opsin model and exacerbates RD in a Q344ter rod opsin model. A–D, Effects of VPA in a T4K rod opsin model. A, In cyclic light, effects of genotype and treatment were significant (pg = 5.8 × 10–5, pt = 1.2 × 10–4) and negative, but treatment did not modify the effect of genotype (pi = 0.45). In the dark, there was no effect of genotype (p = 0.111) and no interaction between treatment and genotype (p = 0.133). n = 6–11 animals per group. B, Representative low-magnification confocal micrographs of transgenic retinas expressing T4K rod opsin stained with WGA. C, Representative high-magnification confocal micrographs of transgenic retinas expressing T4K rod opsin stained with 2B2 anti-mammalian rhodopsin (green), WGA (red), and Hoechst dye (blue). VPA treatment did not alter T4K rod opsin distribution. D, Total rod opsin (B630N) and T4K rhodopsin (1D4) signals were quantified in dark reared animals. Two-way ANOVA analysis showed that VPA treatment reduced the antibody signals (p = 1.0 × 10−4), but the effect was not significantly different between total and transgenic rhodopsins (p = 0.099). n = 6–13 animals for each condition. E–G, Effects of VPA in a Q344ter rod opsin model. E, In cyclic light, the effects of genotype and treatment were significant (pg = 2 × 10−7, pt = 1 × 10−6) and treatment modified the effect of genotype significantly (pi = 0.016). n = 8–11 animals for each condition. F, Representative low-magnification confocal micrographs of transgenic retinas expressing Q344ter rod opsin stained with WGA. Scale bar, 200 μm. G, Representative high-magnification confocal micrographs of transgenic retinas expressing Q344ter rod opsin stained with 2B2 anti-mammalian rhodopsin (green), WGA (red), and Hoechst dye (blue). VPA treatment did not alter the distribution of Q344ter rod opsin. Scale bar, 50 μm. Error bars indicate SEM.
Q344ter rhodopsin lacks a C-terminal OS localization signal and mislocalizes throughout the rod photoreceptor plasma membrane. Based on our previous results using the equivalent mutation (Q350ter) in X. laevis rhodopsin, this mutation likely causes RD via a mechanism distinct from P23H rhodopsin and T17M/T4K rhodopsins and the phenotype is not exacerbated by light exposure (Tam et al., 2006). We subsequently developed a line of transgenic animals expressing human Q344ter rhodopsin in rods under control of the X. laevis rod opsin promoter at 10% of total rod opsin levels. To determine whether VPA can influence this form of RD, we treated Q344ter animals reared in cyclic light with 10 μm VPA. We found that VPA similarly exacerbated RD in Q344ter animals (Fig. 4E–G; two-way ANOVA, pg = 2 × 10−7, pt = 1 × 10−6, pi = 0.016). This suggests that, despite differences in the initiating events of retinal cell death (rhodopsin mislocalization vs rhodopsin activation), RD caused by T17M and Q344ter rhodopsins may share a common mechanism.
Similar to results obtained with T17M rhodopsin, VPA did not alter the abundance or distribution of T4K rhodopsin (Fig. 4B,C). Dot blot data suggested a trend toward decreased transgenic rhodopsin, but this was not significantly different from the effect observed on endogenous rhodopsin (pi = 0.099). Similarly, the distribution of Q344ter rhodopsin was unaltered by VPA treatment (Fig. 4G). Due to the absence of the C-terminal 1D4 epitope and saturating 2B2 signals in OSs, Q344ter expression levels could not be quantified as in Figures 2D, 3D, and 4D.
HDAC inhibitors (HDACi's) ameliorate light-induced retinal degeneration in a P23H rhodopsin model of RP
The wide spectrum of pharmacological activities ascribed to VPA makes it difficult to determine which activity is responsible when an effect is observed. To elucidate the mechanism of action of VPA in our models of RP, we tested nine compounds separately using the experimental paradigm shown in Figure 1. The compounds all share pharmacological properties with VPA and comprise four pharmacological groups: mood stabilizers, HDACi's, molecular chaperones, and structural analogs of VPA (Fig. 5A, Table 1). We found that the HDACi's vorinostat, CI-994, and sodium butyrate ameliorated light-induced RD in P23H animals (Fig. 5; vorinostat data not shown). CI-994 was tested using a simplified dose–response protocol involving transgenic animals only and a total of 7 d of drug exposure (Fig. 5D,E,I–K). Other compounds shown in Figure 5A and Table 1 did not have statistically significant effects. The data indicate that HDAC inhibition is the common property underlying the efficacy of these compounds as treatments for RD in P23H rhodopsin-transgenic X. laevis. To our knowledge, this is the first report of beneficial effects of sodium butyrate (Fig. 5B,C,F–H), vorinostat, and CI-994 in an RP model.
Figure 5.
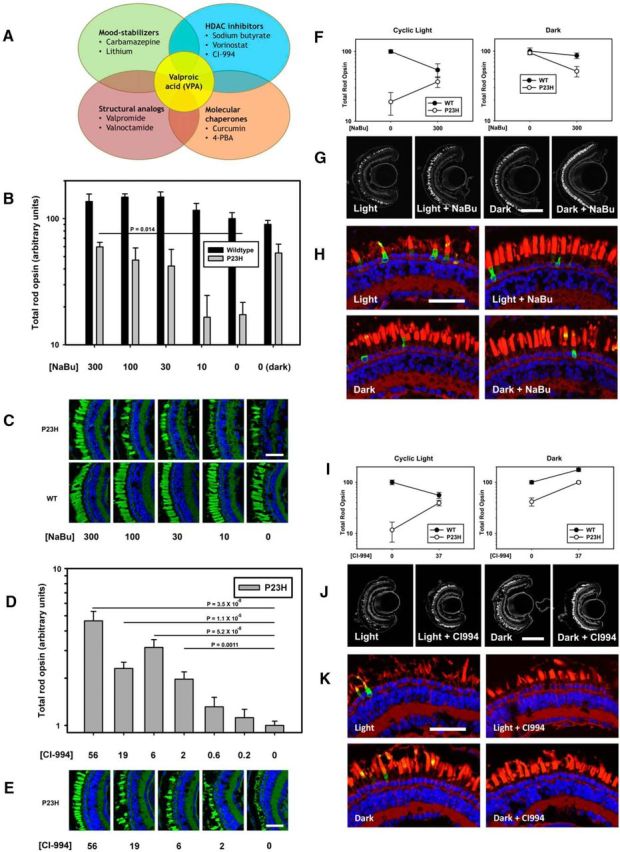
The HDACi's sodium butyrate (NaBu) and CI-994 reproduce the effects of VPA. A, Venn diagram showing overlapping pharmacological activities of compounds examined. B, Effect of NaBu on total rod opsin levels. NaBu ameliorates RD in P23H X. laevis in a dose-dependent manner (ANOVA, p = 0.0022). Treatment with 300 μm sodium butyrate is equivalent to dark rearing. WT animals are unaffected (ANOVA, p = 0.10). p-values shown are Dunnet's multiple comparisons. n = 7–10 animals per group. C, Representative cryosections of contralateral eyes confirm that effects of NaBu in dot blot assays are due to reduced RD in P23H-transgenic animals. OSs were stained with WGA (green) and Hoechst dye (blue). NaBu treatment resulted in greater density of OSs in P23H X. laevis. WT animals were unaffected. D, Dot blot analysis of total rod opsin from whole-eye extracts of P23H X. laevis treated with varying concentrations of CI-994. CI-994 increased total rhodopsin significantly and in a dose-dependent manner (ANOVA, p = 4.5 × 10−13). p-values shown on chart are Dunnet's multiple comparisons. n = 7–10 animals per group. E, Representative cryosections of contralateral eyes confirm that effects of CI-994 in dot blot assays are due to reduced RD in P23H-transgenic animals. OSs were stained with WGA (green) and Hoechst dye (blue). CI-994 treatment resulted in greater density of OSs in P23H X. laevis. Scale bar, 50 um. F–K, Same as described in Figure 2, A–C. F–H, Effects of NaBu on light-exacerbated RD in P23H animals were identical to the effects of VPA (two-way ANOVA, cyclic light: pg = 3.7 × 10−15, pt = 1.6 × 10−9, pi = 4.3 × 10−6. Two-way ANOVA, dark: pg = 0.03, pt = 0.005, pi = 0.11), n = 5–14 animals per group. I–K, Effects of CI-994 on light-exacerbated RD were essentially identical to the effects of VPA (two-way ANOVA, cyclic light: pg = 1.9 × 10−7, pt = 0.038, pi = 5.0 × 10−6; two-way ANOVA, dark: pg = 2.2 × 10−10, pt = 1.6 × 10−10, pi = 0.036.). N = 8–12 animals per group. Error bars indicate SEM.
Table 1.
Additional compounds tested on P23H rhodopsin-transgenic X. laevis and results obtained
| Compound | Category | Concentrations tested | n | p-valuea | Most effective concentration |
|---|---|---|---|---|---|
| VPA | MS, GSK3I, CC, HDACi | 1, 3, 10 μmb | 13–15 | 1.5 × 10−12 | 10 μm |
| Valpromide | MS, SA | 0.1, 0.3, 0.5, 1 mm | 6–11 | NS | — |
| Valnoctamide | MS, SA | 0.5 mm | 10–15 | NS | — |
| Lithium chloride | MS, GSK3I | 0.3, 1 mmb | 13–15 | NS | — |
| Carbamazepine | MS | 100 μmb | 7–12 | NS | — |
| 4-PBA | CC | 3, 10, 30 μmb | 11–16 | NS | — |
| Curcumin | CC | 333 μmb | 7–9 | NS | — |
| Sodium butyrate | HDACi | 10, 30, 100, 300 μmb | 8–9 | 0.0022 | 300 μm |
| Vorinostat | HDACi | 3, 10, 30, 100 μmb | 6–7 | 0.007 | 30 μm |
| CI-994 | HDACi | 0.2, 0.6, 2, 6, 19, 37, 56 μmc | 7–10 | 4.5 × 10−13 | 56 μmd |
ap-value associated with one-way ANOVA in an experiment comparable to that shown in Figure 1. For compounds tested at a single concentration, a t test (treated vs untreated) was used. p > 0.05, NS.
bHigher concentrations were toxic.
cHighest readily achievable concentration.
dAlthough 56 μm resulted in the highest total rod opsin levels, it did show some toxicity, so 37 μm was used in Figure 5I–K.
SA, Structural analog of VPA; MS, mood stabilizer; CC, chemical chaperone; GSK3I, glycogen synthase kinase 3 inhibitor.
HDACi's exacerbate light-induced retinal degeneration in a T17M rhodopsin model of RP
To determine whether the negative effect of VPA on RD in the T17M model of RP was also due to HDAC inhibition, we similarly treated T17M rhodopsin tadpoles with the HDACi's sodium butyrate (Fig. 6A–C) and CI-994 (Fig. 6D–F). Sodium butyrate exacerbated RD caused by T17M rhodopsin in the presence of light in a manner indistinguishable from the previously observed effects of VPA (Figs. 3B,C, 6B,C). Again, results from animals reared in cyclic light were highly significant by two-way ANOVA (pg = 3.7 × 10−15, pt = 1.6 × 10−9, pi = 4.3 × 10−6) and, in the absence of light, there was no significant degeneration and no interaction of treatment and genotype (pi = 0.774). Similarly, CI-994 also exacerbated RD caused by T17M rhodopsin in the presence of light (pi = 2.6 × 10−5), but not in darkness (pi = 0.538; Fig. 6D–F). Therefore, HDAC activity was responsible for both the beneficial (Fig. 5B–K) and detrimental (Fig. 6) effects of these compounds.
Figure 6.

Effects of VPA on T17M-transgenic retinas are reproduced by the HDACi's sodium butyrate (NaBu) and CI-994. A–C, Effects of NaBu on light exacerbated RD in T17M animals were essentially identical to the effects of VPA. Panels are as described in Figure 3, A–C (two-way ANOVA, cyclic light: pg = 3.7 × 10−15, pt = 1.6 × 10−9, pi = 4.2 × 10−6; two-way ANOVA, dark: pg = 0.002, pt = 0.29, pi = 0.77). n = 8–14 animals per group. D–F, Effects of CI-994 on light-exacerbated RD in T17M animals were essentially identical to the effects of VPA. Panels are as described in Figure 3, A–C (two-way ANOVA, cyclic light: pg = 6.1 × 10−11, pt = 5.2 × 10−8, pi = 2.6 × 10−5; two-way ANOVA, dark: pg = 0.156, pt = 0.013, pi = 0.538). n = 8–13 animals per group.
VPA treatment increases retinal histone H3 acetylation, but does not increase retinal tubulin acetylation
If HDACi activity mediates the beneficial effects of VPA activity in the retina, then evidence of altered histone acetylation should be present. We therefore examined the acetylation of histone H3 in WT retinas treated with VPA using Western blots of retinal extracts probed with anti-acetyl H3 and anti-H3 antibodies. We found that, in retinas treated with 10 μm VPA, acetylation of histone H3 (normalized to H3 levels) was increased by 2.5-fold relative to untreated animals (t test, p = 0.001; Fig. 7A). We further confirmed that acetylation of histone H3 was upregulated in rod photoreceptors by probing retinal cryosections with anti-acetylated H3 antibodies and found that histone H3 acetylation was uniformly upregulated in all retinal cell layers, including photoreceptors (Fig. 7B). Because histone deacetylases can also alter the acetylation of non-nuclear proteins, we also examined tubulin acetylation by a similar assay, which is influenced by the activity of HDAC6 and may be linked to alterations in protein degradation pathways (Hubbert et al., 2002; Zhang et al., 2003; Pandey et al., 2007). However, we found no evidence for increased tubulin acetylation (normalized to tubulin levels) relative to untreated animals (Fig. 7A), consistent with reports that VPA has relatively low HDAC6 inhibitory activity (Gurvich et al., 2004).
Figure 7.
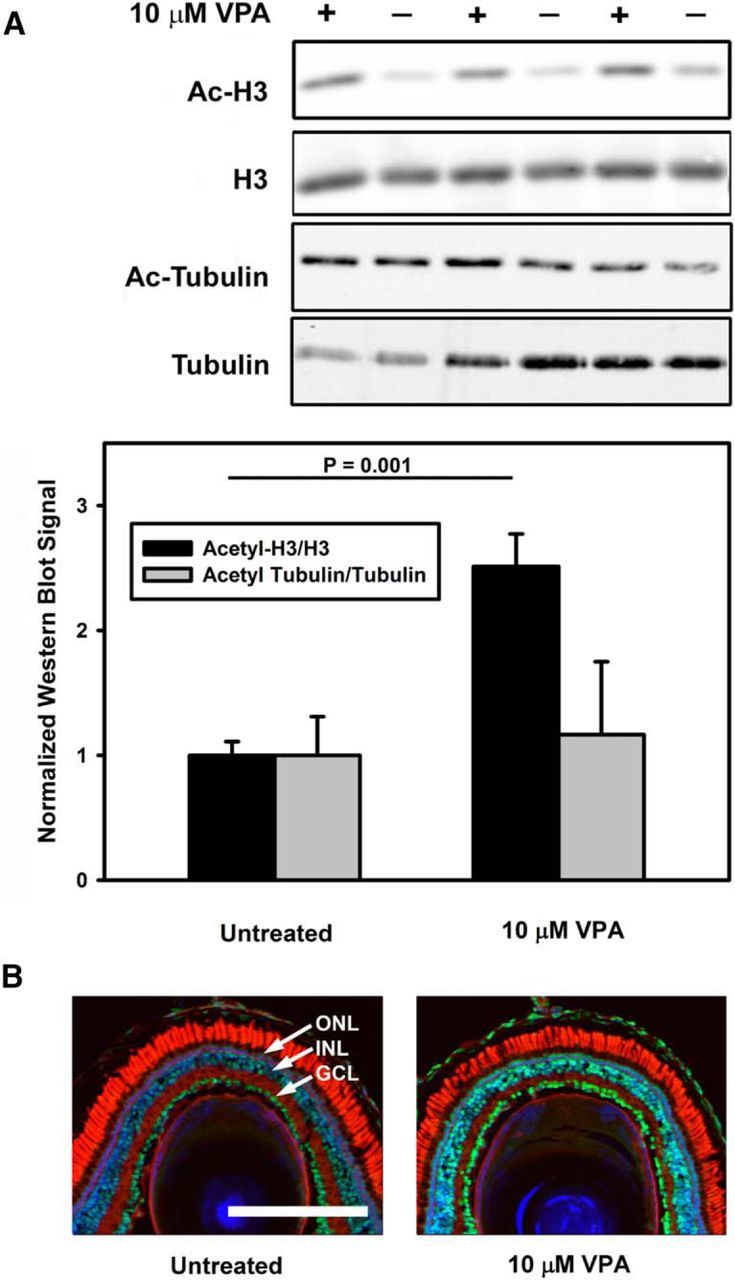
VPA treatment increases histone H3 acetylation in WT X. laevis eyes. A, Western blots of eye extracts probed with anti-acetyl H3, anti-H3, anti-acetyl tubulin, and anti-tubulin (top) were quantified (bottom) and showed an increase in relative levels of H3 acetylation on treatment with 10 μm VPA (p = 0.001, t test), but no significant change in relative levels of tubulin acetylation. n = 6–7 animals per condition. Error bars indicate SEM. B, Immunolabeling with anti-acetyl H3 (green) shows that the effect observed in A is due to increased anti-acetyl H3 labeling in all retinal layers, including photoreceptors. Blue indicates Hoechst 33342; red, WGA; ONL, outer nuclear layer; INL, inner nuclear layer; IPL, inner plexiform layer.
VPA treatment increases autophagy in rod photoreceptors
VPA has been reported to be an activator of autophagy (Sarkar et al., 2005) and this activity has been suggested to mediate its beneficial effects in models of neurodegenerative disorders such as Huntington's disease (Renna et al., 2010). To investigate whether VPA increased autophagy in rod photoreceptor cells, we examined rod photoreceptors in WT retinas treated with VPA by electron microscopy and found that their ISs contained increased numbers of autophagic structures, identifiable as vesicular structures with resolvable membranous or cytoplasmic contents, sometimes surrounded by a double membrane (Fig. 8A–D). Sodium butyrate has also been reported to increase autophagy (Lee and Lee, 2012) and we observed similar effects in sodium-butyrate-treated retinas (Fig. 8E,E′). In addition, we found signs of increased autophagy in untreated retinas expressing human P23H rhodopsin (Fig. 8F,F′), similar to results obtained previously with bovine P23H rhodopsin (Bogéa et al., 2015), suggesting that RD caused by human P23H rhodopsin is associated with increased autophagy, potentially as a protective response to large quantities of misfolded rod opsin.
Figure 8.
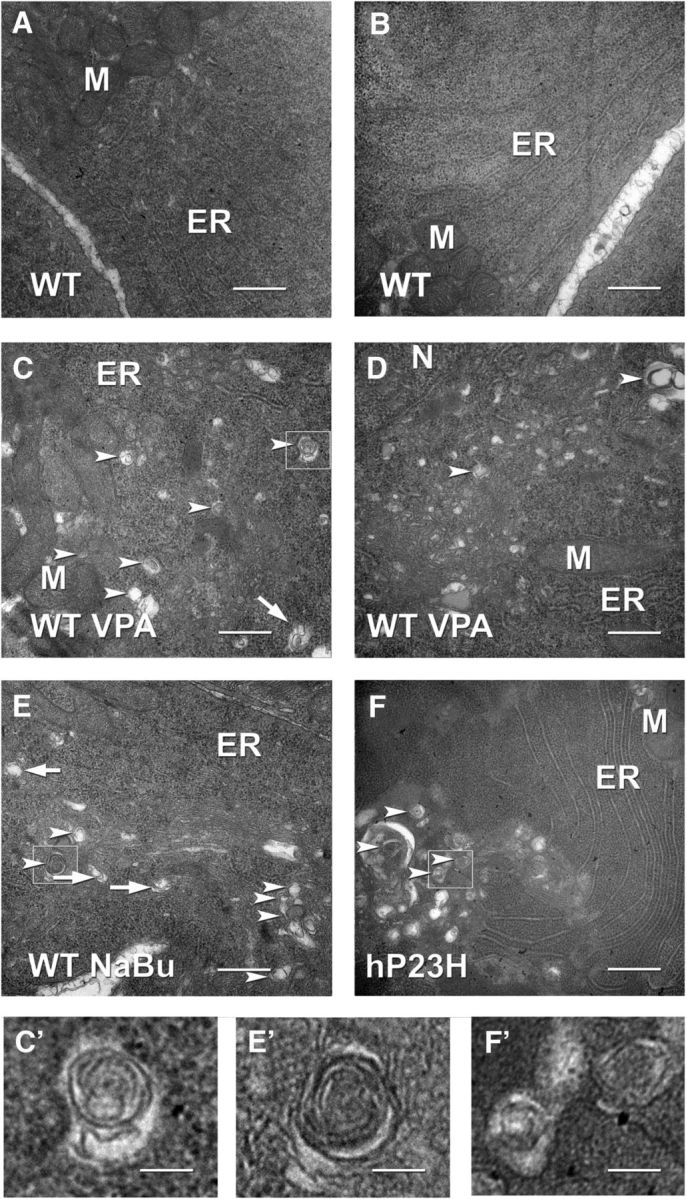
Transmission electron microscopy of X. laevis rod photoreceptors. A, B, Untreated WT. C, D, VPA-treated WT. Structures indicated by arrowheads are consistent with autophagosomes or autolysosomes. Structures indicated by arrows are consistent with newly forming autophagosomes (phagophores). Small vesicular structures morphologically consistent with autophagosomes and autolysosomes increased with VPA treatment consistent with an increase in autophagy. E, Vesicular structures also increased in rods treated with sodium butyrate. F, Vesicular structures were increased in photoreceptors expressing P23H rhodopsin, consistent with previous studies suggesting induction of autophagy during retinal degeneration. Larger vesicular structures were also apparent in these samples. C′, E′, F′, Boxed structures from C, E, and F shown at higher magnification. M, Mitochondria; ER, endoplasmic reticulum; N, nucleus. Scale bar, 500 nm.
HDACi treatment rescues visual function measured by electroretinography (ERG)
To determine whether VPA treatment promoted functional rescue of visual responses in addition to anatomical rescue of RD, we examined the effects of VPA treatment on the ERG of WT and P23H tadpoles. We found that untreated P23H tadpoles reared in cyclic light had a significantly reduced ERG response relative to WT siblings, particularly at intermediate flash intensities, and that this phenotype was ameliorated by treatment with 10 μm VPA (Fig. 9A,B). Two-way ANOVA analysis of the data (log intensity vs condition) showed a significant effect of condition and subsequent multiple-comparisons tests (Tukey's tests) indicated that the untreated P23H animals were significantly different from all other conditions (p < 0.02) and that no other comparisons were significant. In addition, three-way ANOVA (intensity vs treatment vs genotype) showed a significant interaction between treatment and genotype (p = 0.022). We performed a similar analysis using transgenic P23H tadpoles treated with sodium butyrate and found a similar increase in ERG responses in treated animals (Fig. 9C,D). Two-way ANOVA analysis (log intensity vs condition) showed a significant effect of sodium butyrate treatment (p = 0.012).
Figure 9.

Electroretinography (ERG) of X. laevis tadpoles treated with HDACi's. A, B-wave amplitudes obtained from VPA-treated and untreated WT and P23H X. laevis tadpoles in response to flashes of increasing intensities. Two-way ANOVA shows a significant effect of intensity and group (p = 7 × 10−17, p = 4.1 × 10−4). n = 5–7 animals per condition. Multiple comparisons (Tukey's test) showed that P23H untreated responses were significantly different from all other groups (p < 0.019) and no other differences between groups were significant. B, Representative ERG traces from each condition in A. C, B-wave amplitudes obtained from sodium butyrate (NaBu)-treated and untreated P23H X. laevis tadpoles in response to increasing flash intensities. Two-way ANOVA showed a significant effect of intensity and group (p = 1.1 × 10−6, p = 0.012). Responses from treated P23H X. laevis improved significantly. D, Representative ERG traces from each condition in C. n = 4–8 animals per condition. Error bars indicate SEM.
Discussion
Here, we provide evidence that VPA can promote the clearance of misfolded P23H rhodopsin from rod photoreceptors, preventing RD and promoting visual responses as assessed by histology and electroretinography and supporting the use of VPA as a treatment for RP involving misfolding of rhodopsin in the biosynthetic pathway. Our results indicate that the beneficial effects of VPA are mediated by its HDACi activity because the HDACi's vorinostat, CI-994, and sodium butyrate had identical beneficial effects, whereas structural analogs or compounds sharing other non-HDACi properties with VPA were ineffective treatments. We did not find any evidence for a mechanism associated with pharmacological chaperone activity, such as increased ER exit and delivery of mutant rhodopsin to the OS, as originally suggested (Clemson et al., 2011). In fact, we observed the opposite effect, because treatment with VPA reduced the abundance of mutant rhodopsin in rod photoreceptors. Interestingly, VPA treatment did not provide complete rescue of RD either alone or in combination with dark rearing. There was no added protective effect of dark rearing, which also protects against RD dramatically in these animals by promoting ER exit of the mutant rhodopsin (Tam and Moritz, 2007; Tam et al., 2010; Fig. 2A–C). The lack of a synergistic effect of VPA treatment and dark rearing further suggests that VPA acts by decreasing the burden of misfolded rhodopsin in the biosynthetic pathway. Because dark rearing, VPA treatment, or the combination did not completely protect against RD, it is possible that a second, uncharacterized mechanism is responsible for the remainder of the RD observed in these animals.
We also found that, in models of ADRP caused by mutations that do not promote misfolding of rhodopsin, the effects of VPA were detrimental and this was particularly significant in the case of T17M rhodopsin, a mutation that also occurs frequently in RP patients (Sullivan et al., 2013), in which light-exacerbated RD is likely associated with photoactivation of the mutant rhodopsin rather than misfolding in the biosynthetic pathway (Tam et al., 2014). Notably, the detrimental effects of VPA were not associated with altered localization or expression levels of the mutant protein, indicating that VPA does not interfere with normal biosynthesis of rhodopsin or alter transgene expression levels. The mechanism underlying this detrimental effect is not clear, but it was also associated with the HDACi activity of VPA because it was similarly reproduced by treatment with sodium butyrate and CI-994.
Our previous investigations suggest that T4K and T17M rhodopsins cause light-exacerbated cell death by similar mechanisms involving photoactivation of rhodopsin (Tam and Moritz, 2009; Tam et al., 2014). However, RD caused by T17M rhodopsin was much more dramatically exacerbated by HDACi treatment. It is possible that the cell death mechanisms associated with these mutant rhodopsins diverge after rhodopsin activation, although further research is required.
We confirmed that systemic treatment with VPA can alter histone acetylation in the retina, including photoreceptors, by Western blot and microscopy analysis. However, levels of acetylated tubulin were unchanged, suggesting that the effects of VPA were not mediated by inhibition of HDAC6, an HDAC of particular interest due to its involvement in regulation of proteolytic degradation pathways, including regulation of aggresome formation (Kawaguchi et al., 2003) and hsp90 chaperone activity (Kovacs et al., 2005). The HDACi's we tested are reported to inhibit class I HDACs (1, 2, 3, and 8) with limited activity toward class II HDACs (4, 5, 6, 7, 9, and 10), although complete data on each inhibitor are not present in the literature (Table 2). Our results are most consistent with the beneficial effect of VPA and other HDACi's mediated by a class I HDAC, with HDAC1 and HDAC2 being prime candidates. The pharmacological effect responsible for the detrimental effects of VPA in T17M retinas is similar and possibly identical.
Table 2.
Previously documented activities (IC50, μm) of HDACi's investigated in the present study
| Class I |
Class II |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC4 | HDAC5 | HDAC6 | HDAC7 | HDAC9 | HDAC10 | |
| VPA (Gurvich et al., 2004; Khan et al., 2008) | 1584, 700 | 3068, 800 | 3071, 1000 | 7442, ND | ND, 1500 | ND, 1000 | >10000, >20000 | >10000, 1300 | >10000, ND | ND, >20000 |
| NaBu (Gurvich et al., 2004) | 300 | 400 | ND | ND | ND | ND | ND | 300 | ND | ND |
| SAHA (Beckers et al., 2007; Khan et al., 2008) | 68, 21 | 164, ND | 48, 37 | 1524, 1200 | 101, ND | ND | 90, 25 | 104, ND | 107, ND | ND |
| CI-994 (Beckers et al., 2007; Moradei et al., 2007) | 900, 410 | 900, ND | 1200, 750 | 20000, >100000 | — | — | ND, >100000 | — | — | — |
Previous studies have suggested an influence of HDACi's on RD caused by mutations in the PDE6B gene (Sancho-Pelluz et al., 2010; Sancho-Pelluz and Paquet-Durand, 2012; Mitton et al., 2014). Sancho-Pelluz et al. found that treatment with trichostatin A, an inhibitor of class I and II HDACs, was neuroprotective in the rd1 mouse (Sancho-Pelluz et al., 2010; Sancho-Pelluz and Paquet-Durand, 2012), a naturally occurring PDE6B knock-out (Bowes et al., 1990; Pittler and Baehr, 1991). The rd1 mouse is a well characterized model with aggressive RD that is associated with overabundance of cGMP, the second messenger of the phototransduction cascade. Mitton et al., 2014 found a similar protective effect of VPA in the rd1 mouse, but exacerbation of RD in the rd10 mouse, which carries a point mutation in PDE6B that is not a null allele and has a slower rate of RD (Chang et al., 2007). Mitton et al., 2014 also found altered expression of several neurotrophic factors on VPA treatment, but the expression of rhodopsin and PDE6B were unaffected.
We have not examined the effects of VPA on secondary degeneration of cones, which does not occur to a significant extent in our models in the two week timeframe of these experiments, but is responsible for the most debilitating symptoms of RP in patients (Papermaster, 1995). VPA did not promote cone degeneration dramatically because abundant cone OSs were apparent in treated retinas of all genotypes investigated.
Previous studies have identified beneficial effects of VPA in other models of neurodegenerative disease, possibly mediated by an effect on autophagy (Chiu et al., 2013; Frake et al., 2015), which can promote the clearance of protein aggregates and recycle damaged organelles and membranes (Boya et al., 2013). VPA has also been reported to induce autophagy in yeast (Robert et al., 2011). P23H rhodopsin is a substrate for autophagy in cultured cells (Kaushal, 2006). We observed an increase in autophagy in rod photoreceptors of VPA-treated retinas and decreased P23H rhodopsin, supporting, but not proving, a link between the beneficial effects of VPA and autophagy. Other HDACi's, including vorinostat, can also increase autophagy (Shao et al., 2004; Koeneke et al., 2015; Zhang et al., 2015) and this increase in autophagy can modulate cell death pathways (Gammoh et al., 2012). Interestingly, recent findings suggest that the mood stabilizers carbamazepine and lithium act by modulating autophagy through the PI3K pathway (Sarkar et al., 2005; Hidvegi et al., 2010; Schiebler et al., 2014), a mechanism shared by VPA (Williams et al., 2002). However, neither lithium nor carbamazepine had beneficial effects in our assays. Further investigations (e.g., testing the effects of VPA in combination with autophagy knock-out) are required to determine conclusively whether modulation of autophagy pathways contributes to the mechanism of action of HDACi's in our system.
HDACi's such as sodium butyrate and vorinostat can also induce caspase-dependent and caspase-independent cell death (Shao et al., 2004) and vorinostat can also increase the sensitivity of cells to cell death stimuli such as DNA-damaging agents, an effect that is also mediated by autophagy (Robert et al., 2011; Gammoh et al., 2012), prompting the investigation and use of HDACi's as anticancer agents (West and Johnstone, 2014). It is possible that the enhanced cell death that we observed in T17M retinas is related to these HDACi properties. Further investigations of the beneficial and detrimental effects of HDACi's in our system will include the use of more specific HDACi's, knock-out of individual HDAC and autophagy genes, and investigations of gene regulation by HDAC inhibition in tadpole retina.
Critically, our results suggest that clinical trials of VPA and other treatments for RP should take into account genetic variation, including specific gene mutations (i.e., “precision medicine”) and small trials may be more informative if they are restricted to specific genotypes. Moreover, both the beneficial and detrimental effects of VPA were only detected under specific environmental conditions for two of the four mutations examined, namely cyclic light of intensity comparable to daylight. This is an additional level of complication in the investigation of mechanisms of and treatments for RP in that both genotype and environment could strongly influence the outcome of preclinical and clinical investigations. This is consistent with previous reports of disparate phenotypes between family members that suggest a potentially large environmental influence in some cases (Heckenlively et al., 1991; Berson, 1993). Our results suggest that genotyping of RP patients should be standard practice in clinical trials. Our results do not support indiscriminate treatment of RP patients with VPA or other HDACi's and are consistent with case reports that VPA treatment may be detrimental in some individuals (Sisk, 2012; Bhalla et al., 2013). Experiments conducted by others indicate that similar issues may occur with genes other than rhodopsin (Mitton et al., 2014) and other disorders such as Alzheimer's disease (Fleisher et al., 2011).
The fact that VPA treatment is beneficial in an animal model of RP involving defective biosynthesis of rhodopsin is a notable success and supports the possibility of HDACi treatment in RP and other neurodegenerative disorders. If we can dissect the beneficial mechanism of VPA action from its detrimental effects using more specific inhibitors or confidently associate cell death mechanisms with specific patient genotypes, it may be possible to develop an RP treatment that could be applied without fear of harming patients. Furthermore, such treatments could potentially be beneficial for other neurodegenerative disorders. In addition, the finding that VPA exacerbates RD caused by T17M and Q344ter rhodopsins provides a novel “handle” that will aid in the characterization of these alternate RD mechanisms.
Footnotes
This study was funded by operating grants from the Foundation Fighting Blindness–Canada and the Canadian Institutes of Health Research (Grant MOP-64400). R.Y.V.-S. is a Natural Sciences and Engineering Research Council of Canada Postgraduate Scholarship-Doctoral Program fellow and a University of British Columbia Four Year Doctoral Fellowship scholar.
The authors declare no competing financial interests.
References
- Adamus G, Zam ZS, Arendt A, Palczewski K, McDowell JH, Hargrave PA. Anti-rhodopsin monoclonal antibodies of defined specificity: characterization and application. Vis Res. 1991;31:17–31. doi: 10.1016/0042-6989(91)90069-H. [DOI] [PubMed] [Google Scholar]
- Beckers T, Burkhardt C, Wieland H, Gimmnich P, Ciossek T, Maier T, Sanders K. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int J Cancer. 2007;121:1138–1148. doi: 10.1002/ijc.22751. [DOI] [PubMed] [Google Scholar]
- Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993;34:1659–1676. [PubMed] [Google Scholar]
- Bhalla S, Joshi D, Bhullar S, Kasuga D, Park Y, Kay CN. Long-term follow-up for efficacy and safety of treatment of retinitis pigmentosa with valproic acid. Br J Ophthalmol. 2013;97:895–899. doi: 10.1136/bjophthalmol-2013-303084. [DOI] [PubMed] [Google Scholar]
- Bogéa TH, Wen RH, Moritz OL. Light induces ultrastructural changes in rod outer and inner segments, including autophagy, in a transgenic Xenopus laevis P23H rhodopsin model of retinitis pigmentosa autophagy in P23H light-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2015;56:7947–7955. doi: 10.1167/iovs.15-16799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Bown CD, Wang JF, Chen B, Young LT. Regulation of ER stress proteins by valproate: therapeutic implications. Bipolar Disord. 2002;4:145–151. doi: 10.1034/j.1399-5618.2002.t01-1-40201.x. [DOI] [PubMed] [Google Scholar]
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–720. doi: 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Pardue MT, German AM, Hurd RE, Davisson MT, Nusinowitz S, Rengarajan K, Boyd AP, Sidney SS, Phillips MJ, Stewart RE, Chaudhury R, Nickerson JM, Heckenlively JR, Boatright JH. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res. 2007;47:624–633. doi: 10.1016/j.visres.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999;72:1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- Chiang WC, Hiramatsu N, Messah C, Kroeger H, Lin JH. Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Invest Ophthalmol Vis Sci. 2012;53:7159–7166. doi: 10.1167/iovs.12-10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang WC, Kroeger H, Sakami S, Messah C, Yasumura D, Matthes MT, Coppinger JA, Palczewski K, LaVail MM, Lin JH. Robust endoplasmic reticulum-associated degradation of rhodopsin precedes retinal degeneration. Mol Neurobiol. 2015;52:679–695. doi: 10.1007/s12035-014-8881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CT, Liu G, Leeds P, Chuang DM. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington's disease. Neuropsychopharmacology. 2011;36:2406–2421. doi: 10.1038/npp.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65:105–142. doi: 10.1124/pr.111.005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Hood DC, Huang Y, Banin E, Li ZY, Stone EM, Milam AH, Jacobson SG. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A. 1998;95:7103–7108. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Tzekov R, Krebs M, Checchi JM, Bigelow C, Kaushal S. Therapeutic potential of valproic acid for retinitis pigmentosa. Br J Ophthalmol. 2011;95:89–93. doi: 10.1136/bjo.2009.175356. [DOI] [PubMed] [Google Scholar]
- Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84:132–141. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Truran D, Mai JT, Langbaum JBS, Aisen PS, Cummings JL, Jack CR, Jr, Weiner MW, Thomas RG, Schneider LS, Tariot PN. Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology. 2011;77:1263–1271. doi: 10.1212/WNL.0b013e318230a16c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- Frake RA, Ricketts T, Menzies FM, Rubinsztein DC. Autophagy and neurodegeneration. J Clin Invest. 2015;125:65–74. doi: 10.1172/JCI73944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl Acad Sci U S A. 2012;109:6561–6565. doi: 10.1073/pnas.1204429109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004;64:1079–1086. doi: 10.1158/0008-5472.CAN-03-0799. [DOI] [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- Heckenlively JR, Rodriguez JA, Daiger SP. Autosomal dominant sectoral retinitis pigmentosa. Two families with transversion mutation in codon 23 of rhodopsin. Arch Ophthalmol. 1991;109:84–91. doi: 10.1001/archopht.1991.01080010086038. [DOI] [PubMed] [Google Scholar]
- Hicks D, Molday RS. Differential labeling frog rod and cone cells using monoclonal against bovine rhodopsin of bovine and antibodies. Exp Eye Res. 1986;42:55–71. doi: 10.1016/0014-4835(86)90017-5. [DOI] [PubMed] [Google Scholar]
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, Michalopoulos G, Perlmutter DH. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Johannessen CU. Mechanisms of action of valproate: a commentatory. Neurochem Int. 2000;37:103–110. doi: 10.1016/S0197-0186(00)00013-9. [DOI] [PubMed] [Google Scholar]
- Kaushal S. Effect of rapamycin on the fate of P23H opsin associated with retinitis pigmentosa (an American Ophthalmological Society thesis) Trans Am Ophthalmol Soc. 2006;104:517–529. [PMC free article] [PubMed] [Google Scholar]
- Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33:6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- Kaushal S, Ridge KD, Khorana HG. Structure and function in rhodopsin: the role of asparagine-linked glycosylation. Proc Natl Acad Sci U S A. 1994;91:4024–4028. doi: 10.1073/pnas.91.9.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/S0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Kim AJ, Shi Y, Austin RC, Werstuck GH. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J Cell Sci. 2005;118:89–99. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- Koeneke E, Witt O, Oehme I. HDAC family members intertwined in the regulation of autophagy: a druggable vulnerability in aggressive tumor entities. Cells. 2015;4:135–168. doi: 10.3390/cells4020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Lee JS, Lee GM. Effect of sodium butyrate on autophagy and apoptosis in Chinese hamster ovary cells. Biotechnol Prog. 2012;28:349–357. doi: 10.1002/btpr.1512. [DOI] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanova ES, Finkelstein S, Skiba NP, Arshavsky VY. Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc Natl Acad Sci U S A. 2013;110:9986–9991. doi: 10.1073/pnas.1305521110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs. 2002;16:669–694. doi: 10.2165/00023210-200216100-00003. [DOI] [PubMed] [Google Scholar]
- Loy R, Tariot PN. Neuroprotective properties of valproate: potential benefit for AD and tauopathies. J Mol Neurosci. 2002;19:303–307. doi: 10.1385/jmn:19:3:301. [DOI] [PubMed] [Google Scholar]
- MacKenzie D, Arendt A, Hargrave P, McDowell JH, Molday RS. Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry. 1984;23:6544–6549. doi: 10.1021/bi00321a041. [DOI] [PubMed] [Google Scholar]
- Mitton KP, Guzman AE, Deshpande M, Byrd D, DeLooff C, Mkoyan K, Zlojutro P, Wallace A, Metcalf B, Laux K, Sotzen J, Tran T. Different effects of valproic acid on photoreceptor loss in Rd1 and Rd10 retinal degeneration mice. Mol Vis. 2014;20:1527–1544. [PMC free article] [PubMed] [Google Scholar]
- Monti B, Gatta V, Piretti F, Raffaelli SS, Virgili M, Contestabile A. Valproic acid is neuroprotective in the rotenone rat model of Parkinson's disease: involvement of alpha-synuclein. Neurotox Res. 2010;17:130–141. doi: 10.1007/s12640-009-9090-5. [DOI] [PubMed] [Google Scholar]
- Moradei OM, Mallais TC, Frechette S, Paquin I, Tessier PE, Leit SM, Fournel M, Bonfils C, Trachy-Bourget MC, Liu J, Yan TP, Lu AH, Rahil J, Wang J, Lefebvre S, Li Z, Vaisburg AF, Besterman JM. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J Med Chem. 2007;50:5543–5546. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Tam BM. Recent insights into the mechanisms underlying light-dependent retinal degeneration from X. laevis models of retinitis pigmentosa. Adv Exp Med Biol. 2010;664:509–515. doi: 10.1007/978-1-4419-1399-9_58. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Tam BM, Knox BE, Papermaster DS. Fluorescent photoreceptors of transgenic Xenopus laevis imaged in vivo by two microscopy techniques. Invest Ophthalmol Vis Sci. 1999;40:3276–3280. [PubMed] [Google Scholar]
- Organisciak DT, Darrow RM, Barsalou L, Kutty RK, Wiggert B. Susceptibility to retinal light damage in transgenic rats with rhodopsin mutations. Invest Ophthalmol Vis Sci. 2003;44:486–492. doi: 10.1167/iovs.02-0708. [DOI] [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB. Pharmacology of valproate. Psychopharmacol Bull. 2003;37:17–24. [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Papermaster DS. Necessary but insufficient. Nat Med. 1995;1:874–875. doi: 10.1038/nm0995-874. [DOI] [PubMed] [Google Scholar]
- Paskowitz DM, LaVail MM, Duncan JL. Light and inherited retinal degeneration. Br J Ophthalmol. 2006;90:1060–1066. doi: 10.1136/bjo.2006.097436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276:36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Pittler SJ, Baehr W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc Natl Acad Sci U S A. 1991;88:8322–8326. doi: 10.1073/pnas.88.19.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renna M, Jimenez-Sanchez M, Sarkar S, Rubinsztein DC. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J Biol Chem. 2010;285:11061–11067. doi: 10.1074/jbc.R109.072181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, Botrugno OA, Parazzoli D, Oldani A, Minucci S, Foiani M. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471:74–79. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Pelluz J, Paquet-Durand F. HDAC inhibition prevents Rd1 mouse photoreceptor degeneration. Adv Exp Med Biol. 2012;723:107–113. doi: 10.1007/978-1-4614-0631-0_15. [DOI] [PubMed] [Google Scholar]
- Sancho-Pelluz J, Alavi MV, Sahaboglu A, Kustermann S, Farinelli P, Azadi S, van Veen T, Romero FJ, Paquet-Durand F, Ekström P. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 2010;1:e24. doi: 10.1038/cddis.2010.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg MA, Rosner B, Weigel-DiFranco C, Berson EL. Lack of scientific rationale for use of valproic acid for retinitis pigmentosa. Br J Ophthalmol. 2011;95:744. doi: 10.1136/bjo.2010.198176. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–1111. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebler M, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med. 2014;7:127–139. doi: 10.15252/emmm.201404137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2004;101:18030–18035. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisk RA. Valproic acid treatment may be harmful in non-dominant forms of retinitis pigmentosa. Br J Ophthalmol. 2012;96:1154–1155. doi: 10.1136/bjophthalmol-2012-301950. [DOI] [PubMed] [Google Scholar]
- Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H, Heckenlively JR, Birch DG, Mintz-Hittner H, Ruiz RS, Lewis RA, Saperstein DA, Sullivan LS. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17:42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LS, Bowne SJ, Reeves MJ, Blain D, Goetz K, NDifor V, Vitez S, Wang X, Tumminia SJ, Daiger SP. Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Invest Opthalmol Vis Sci. 2013;54:6255–6261. doi: 10.1167/iovs.13-12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Davenport CM, Nathans J. Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa: clustering of functional classes along the polypeptide chain. J Biol Chem. 1993;268:26645–26649. [PubMed] [Google Scholar]
- Tam BM, Moritz OL. Dark rearing rescues P23H rhodopsin-induced retinal degeneration in a transgenic Xenopus laevis model of retinitis pigmentosa: a chromophore-dependent mechanism characterized by production of N-terminally truncated mutant rhodopsin. J Neurosci. 2007;27:9043–9053. doi: 10.1523/JNEUROSCI.2245-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Moritz OL. The role of rhodopsin glycosylation in protein folding, trafficking, and light-sensitive retinal degeneration. J Neurosci. 2009;29:15145–15154. doi: 10.1523/JNEUROSCI.4259-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Xie G, Oprian DD, Moritz OL. Mislocalized rhodopsin does not require activation to cause retinal degeneration and neurite outgrowth in Xenopus laevis. J Neurosci. 2006;26:203–209. doi: 10.1523/JNEUROSCI.3849-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Qazalbash A, Lee HC, Moritz OL. The dependence of retinal degeneration caused by the rhodopsin P23H mutation on light exposure and vitamin a deprivation. Invest Ophthalmol Vis Sci. 2010;51:1327–1334. doi: 10.1167/iovs.09-4123. [DOI] [PubMed] [Google Scholar]
- Tam BM, Noorwez SM, Kaushal S, Kono M, Moritz OL. Photoactivation-induced instability of rhodopsin mutants T4K and T17M in rod outer segments underlies retinal degeneration in X. laevis transgenic models of retinitis pigmentosa. J Neurosci. 2014;34:13336–13348. doi: 10.1523/JNEUROSCI.1655-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Yang LL, Bogėa TH, Ross B, Martens G, Moritz OL. Preparation of Xenopus laevis retinal cryosections for electron microscopy. Exp Eye Res. 2015;136:86–90. doi: 10.1016/j.exer.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Tariot PN, Aisen PS. Can lithium or valproate untie tangles in Alzheimer's disease? J Clin Psychiatry. 2009;70:919–921. doi: 10.4088/JCP.09com05331. [DOI] [PubMed] [Google Scholar]
- Tzekov R, Bigelow C, Clemson C, Checchi J, Krebs M, Kaushal S. Authors' response. Br J Ophthalmol. 2011;95:1177–1179. doi: 10.1136/bjo.2010.199893. [DOI] [PubMed] [Google Scholar]
- van Schooneveld MJ, van den Born LI, van Genderen M, Bollemeijer JG. The conclusions of Clemson et al concerning valproic acid are premature. Br J Ophthalmol. 2011;95:153. doi: 10.1136/bjo.2010.194373. author reply 153–154. [DOI] [PubMed] [Google Scholar]
- West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DA, Fritz JJ, Hauswirth WW, Kaushal S, Lewin AS. Increased sensitivity to light-induced damage in a mouse model of autosomal dominant retinal disease. Invest Ophthalmol Vis Sci. 2007;48:1942–1951. doi: 10.1167/iovs.06-1131. [DOI] [PubMed] [Google Scholar]
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O'Kane CJ, Floto RA, Rubinsztein DC. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Cheng L, Mudge AW, Harwood AJ. A common mechanism of action for three mood-stabilizing drugs. Nature. 2002;417:292–295. doi: 10.1038/417292a. [DOI] [PubMed] [Google Scholar]
- Zhang J, Ng S, Wang J, Zhou J, Tan SH, Yang N, Lin Q, Xia D, Shen HM. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy. 2015;11:629–642. doi: 10.1080/15548627.2015.1023981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Jang GF, Jastrzebska B, Filipek S, Pearce-Kelling SE, Aguirre GD, Stenkamp RE, Acland GM, Palczewski K. A naturally occurring mutation of the opsin gene (T4R) in dogs affects glycosylation and stability of the G-protein-coupled receptor. J Biol Chem. 2004;279:53828–53839. doi: 10.1074/jbc.M408472200. [DOI] [PMC free article] [PubMed] [Google Scholar]