

Abstract
Identifying nonaddictive opioid medications is a high priority in medical science, but μ-opioid receptors (MORs) mediate both the analgesic and addictive effects of opioids. We found a significant pharmacodynamic difference between morphine and methadone that is determined entirely by heteromerization of MORs with galanin Gal1 receptors (Gal1Rs), rendering a profound decrease in the potency of methadone. This finding was explained by the weaker proficiency of methadone in activating the dopaminergic system as compared with morphine and predicted a dissociation of the therapeutic and euphoric effects of methadone, which was corroborated by a significantly lower incidence of self-reports of feeling “high” in methadone-medicated patients. These results suggest that μ-opioid–Gal1R heteromers mediate the dopaminergic effects of opioids. The results further suggest a lower addictive liability of some opioids, such as methadone, due to their selective low potency for the μ-opioid–Gal1R heteromer.
Keywords: Neuroscience, Therapeutics
Keywords: Addiction, G-protein coupled receptors

Introduction
The opioid epidemic represents a severe public health crisis (1). Maintenance treatment with the μ-opioid receptor (MOR) agonist methadone is the most highly researched and evidence-based treatment for opioid use disorder (OUD) (2). Yet public perception concerning the substitution of illicit drugs (such as heroin) with medication (such as methadone) has led to stigmatized views of maintenance treatment, stalling the advancement of addiction treatment policy and access to medication-based treatments. MOR agonism also offers the most effective treatment for severe pain, making the search for a nonaddictive opioid drug the holy grail of pain research (1). Opioids also represent an efficacious treatment approach for patients with refractory restless legs syndrome (RLS) (3), but again, their use is tempered by the perceived risk of addiction. The MOR is a GPCR that is essential for opioid-induced analgesia but also responsible for adverse effects, including respiratory depression, reduced gastrointestinal motility, and euphoria that can lead to addiction (4, 5). One recent strategy to design opioids with lower side effects has involved the development of ligands with functional selectivity or biased agonists. This rests on the assumption that a MOR agonist promoting preferential G protein–mediated versus β-arrestin–mediated signaling could dissociate desired from undesired effects (5, 6). However, recent preclinical and clinical findings demonstrating abuse liability in animals and euphoric effects in humans challenge the utility of these biased agonists (7, 8).
Another potential approach to harnessing the therapeutic effects of opioids involves targeting GPCR heteromers (9, 10). Different orthosteric ligands for a specific GPCR can demonstrate different properties (affinity or efficacy) upon GPCR heteromerization (9). This could provide differential effects of ligands for GPCRs localized in different areas of the CNS (9). The neuropeptide galanin acts as a modulator of neurotransmission in the CNS and the PNS (11). It is coexpressed with different neurotransmitters and coreleased by the major ascending noradrenergic, serotoninergic, histaminergic, and cholinergic systems (11). Galanin activates 3 subtypes of GPCRs: Gal1 receptor (Gal1R), Gal2R, and Gal3R. Gal1R and Gal2R are broadly expressed in the CNS, including in cortical, thalamic, and hypothalamic areas, as well as in the spinal cord, whereas Gal3R has more restricted expression in the CNS and is predominantly expressed in peripheral tissues (11). Biochemical and behavioral studies also indicated the functional presence of galanin and galanin receptors in dopaminergic areas, including the ventral tegmental area (VTA) and the nucleus accumbens (NAc), both of which could mediate the antagonistic effect of galanin on the opioid reward response (12). We recently reported the existence of functionally significant heteromers of the MOR and Gal1R in the VTA that could explain these galanin-opioid antagonistic interactions (13).
MOR-Gal1R heteromerization was first demonstrated in mammalian transfected cells using the biophysical techniques bioluminescence resonance energy transfer (BRET) and bimolecular fluorescence complementation (BiFC), whereby the 2 putative interacting receptors are separately fused to 2 biosensors that only interact when in very close proximity. BiFC has allowed examination of the interface of GPCR heteromers by using synthetic peptides with amino acid sequences of the transmembrane domains (TM) possibly involved in the heteromer intermolecular interactions (13, 14). In transfected cells, a synthetic peptide with the amino acid sequence of TM 5 of the MOR (TM5 peptide) selectively destabilized MOR-Gal1R heteromerization and a negative crosstalk between MOR and Gal1R ligands, by which galanin could counteract MAPK activation induced by the endogenous MOR agonist endomorphin-1 (EM1) (13). This heteromer-specific interaction could also be identified in the rat VTA, where TM5 specifically counteracted EM1-induced MAPK activation and dopamine release (13). These results demonstrated that the modulatory effect of galanin was dependent on the integrity of the heteromer and that MOR-Gal1R heteromers represent a main population of MORs in the VTA that modulate dopamine neuronal function (13).
The present study was initially aimed at answering 2 main questions that arose from our study of MOR-Gal1R heteromers: (a) What are the mechanisms involved in the interactions between galanin and opioid ligands within the MOR-Gal1R heteromer? and (b) Do these interactions also involve morphine and synthetic opioids, such as methadone or fentanyl, differentially? We could demonstrate the existence of a significant pharmacodynamic difference between methadone versus morphine and fentanyl that is determined entirely by MOR-Gal1R heteromerization, rendering a profound decrease in the potency of methadone. This would explain the significantly weaker ability of methadone, as compared with morphine and fentanyl, to activate the dopaminergic system and would predict a dissociation of therapeutic versus euphoric effects of methadone.
Results
Allosterism within the MOR-Gal1R heteromer.
We created cell lines with human embryonic kidney cells (FLP-FRT-HEK cells; see Methods) stably transfected with the human MOR alone and with both human MOR and human Gal1R. Two clones, MU cells and MU-GAL cells, were selected according to their functional response to EM1 and the preferential Gal1R agonist M617 (15). We then measured G protein–dependent signaling with dynamic mass redistribution (DMR) (see Methods) assay techniques. The MOR antagonist CTOP (13) and the Gal1R and Gal2R antagonist M40 (13, 15) were used to control agonist specificity. As expected, M40 (0.01–1 μM) dose-dependently counteracted DMR induced by M617 (0.1 μM) in MU-GAL cells (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI126912DS1) (1-way ANOVA: F3,8 = 324, P < 0.001; Dunnett’s multiple comparisons test, versus M617 alone: P < 0.001 for all concentrations of M40). CTOP (1 μM) significantly counteracted signaling induced by EM1 (0.1 μM) in MU and MU-GAL cells and, unexpectedly, M40 (1 μM) also significantly counteracted the effect of EM1 (0.1 μM) in MU-GAL, but not MU, cells (Figure 1A; 1-way ANOVA: F5,24 = 820, P < 0.001; Tukey’s multiple comparisons, versus EM1 in the corresponding cell line: P < 0.001 for all significant differences). The same results were reproduced with morphine: DMR induced by morphine (0.1 μM) was significantly counteracted by CTOP (1 μM) in MU and MU-GAL cells and by M40 (1 μM) in MU-GAL cells (Figure 1B; 1-way ANOVA: F5,30 = 96.4, P < 0.001; Tukey’s multiple comparisons test, versus EM1 in the corresponding cell line: P < 0.001 for all significant differences). In addition, in MU-GAL cells, EM1 (0.1 μM) produced significant MAPK activation (ERK1 and ERK2 [ERK1/2] phosphorylation), and DAMGO (0.1 μM) induced significant MOR internalization, and both were significantly counteracted by CTOP (1 μM) and M40 (1 μM) (Figure 1, C and D; 1-way ANOVA: F4,54 = 18.16, P < 0.001 and F4,25 = 17.2, P < 0.001, respectively; Tukey’s multiple comparisons test, versus control, EM1, or DAMGO: P < 0.001 for all significant differences). M617 (0.1 μM) did not induce MOR internalization in MU-GAL cells (Figure 1D).
Figure 1. Gal1R-dependent allosteric modulation of MOR agonists.

(A and B) Effect of the MOR antagonist CTOP and the Gal1R/Gal2R antagonist M40 on DMR induced by the MOR agonists EM1 (A) and morphine (B) in MU and MU-Gal1R cells. Values are shown as dots and the mean ± SEM (n = 5–6 triplicates/group). ###P < 0.001 versus EM1; 1-way ANOVA with Tukey’s multiple comparisons test. (C) Effect of CTOP and M40 on MAPK activation induced by EM1 in MU-GAL cells. Values are shown as dots and the mean ± SEM n = 6–15 triplicates/group). ***P < 0.001 versus control; ###P < 0.001 versus EM1; 1-way ANOVA with Tukey’s multiple comparisons test. (D) Effect of CTOP and M40 on internalization of MOR induced by the MOR agonist DAMGO and lack of MOR-induced internalization by the Gal1R agonist M617 in MU-GAL cells. Values are shown as dots and the mean ± SEM (n = 6 triplicates/group). ***P < 0.001 versus control; ###P < 0.001 versus control DAMGO; 1-way ANOVA with Tukey’s multiple comparisons test. (E and F), Representative competitive inhibition experiments of [3H]DAMGO versus DAMGO in membrane preparations from MU (E) and MU-GAL (F) cells with or without M617 or M40. Values are expressed as the mean ± SEM of triplicates. See Results and Supplemental Figure 2 for the total number of experiments and statistical comparisons. Concentrations of agonists and antagonists were always 0.1 μM and 1 μM, respectively.
The effects of M40 indicated the existence of a robust cross-antagonism by which a Gal1R antagonist counteracts MOR signaling. This type of cross-antagonism usually implies the existence of allosteric interactions between orthosteric ligands within a GPCR heteromer (9). To demonstrate this possibility, we performed competitive inhibition experiments with [3H]DAMGO versus DAMGO in the presence and absence of M617 and M40. We used the 2-state dimer model (see Methods) to analyze the possible presence of cooperativity of DAMGO and the presence of allosteric modulations by M617 and M40. The binding of [3H]DAMGO was not cooperative (monophasic competition curves for both cell lines), and the calculated density of [3H]DAMGO binding sites in MU and MU-GAL cells was (mean ± SEM) 8.7 ± 1.4 (n = 12) and 2.5 ± 0.5 (n = 13) pmol/mg protein, respectively. In MU-GAL cells, both M617 and M40 caused a pronounced decrease of [3H]DAMGO binding (Figure 1, E and F) due to a significant (7- to 9-fold) reduction in the affinity of DAMGO (increase in KDB1 values; see Methods and Supplemental Figure 2; 1-way ANOVA: F2,22 = 49.8, P < 0.001; Tukey’s multiple comparisons, versus DAMGO alone: P < 0.001 in both cases). On the other hand, both ligands were ineffective in MU cells (no significant change in KDB1 values; Supplemental Figure 2; 1-way ANOVA: F2,21 = 1.8, P < 0.195). This represents an example of an effective allosteric modulation of the affinity of an orthosteric ligand of one of the protomers in a GPCR heteromer by orthosteric ligands of the other molecularly different GPCR protomer (9). In addition, the complete cross-antagonism by M40 on the signaling and internalization of different MOR agonists (EM1, morphine, and DAMGO) indicates that M40 also exerts a negative allosteric modulation of the efficacy of MOR agonists.
Selective low potency of methadone at the MOR-Gal1R heteromer.
Our next goal was to explore the possible emergence of different properties of MOR agonists upon MOR-Gal1R heteromerization. BRET experiments can be used to successfully evaluate qualitative differences among different ligands in terms of their ability to induce changes in the interactions between a GPCR and a G protein subtype (equivalent to ligand-induced G protein activation) (16). The BRET donor Renilla luciferase 8 (Rluc) was fused to MOR, and the BRET acceptor yellow fluorescence protein (YFP, the Venus variant) was fused to the α subunit of the Gi1 protein. These constructs were transiently cotransfected to HEK-293T cells with and without the Gal1R, and concentration-response curves of the MOR agonists morphine, EM1, DAMGO, fentanyl, and methadone were evaluated in the presence and absence of M40 (Figure 2, A–E). Gal1R cDNA was transfected in excess of 4 times the amount of MOR cDNA to ensure the highest potential for MORs to form heteromers. In our previous study, the MOR-Rluc/Gal1R cDNA transfection ratio was 1:1.6, and M40 did not significantly counteract EM1-induced ERK1/2 phosphorylation (13). Implementing a 1:4 ratio, both CTOP (1 μM) and M40 (1 μM) significantly counteracted the increase in ERK1/2 phosphorylation induced EM1 (0.1 μM) (Supplemental Figure 3A; 1-way ANOVA: F3,16 = 132, P < 0.001; Tukey’s multiple comparisons, versus control or EM1: P < 0.001 for all significant differences). In agreement with the predominance of MOR-Gal1R heteromers in the VTA, both CTOP (10 μM) and M40 (10 μM) were also able to significantly antagonize ERK1/2 phosphorylation induced by EM1 (1 μM) in rat VTA slices (Supplemental Figure 3B; 1-way ANOVA: F3,16 = 64.2, P < 0.001; Tukey’s multiple comparisons, versus control or EM1: P < 0.001 for all significant differences). In the absence of the Gal1R, the 5 agonists showed very similar potency and efficacy, properties that were not modified by M40 (Figure 2, A–E). However, we obtained significant differential changes in ligand properties with cotransfection of the Gal1R. Morphine, EM1, and fentanyl showed a significant decrease in the maximum efficacy (Emax) (Figure 2F; unpaired, 2-tailed t test: t12 = 4.1, P = 0.001, t16 = 3.8, P = 0.002 and t18 = 5.7, P < 0.001, respectively), whereas methadone showed a significant and very pronounced decrease in its potency (EC50) (Figure 2G; 2-tailed Mann-Whitney U test: U = 2, n1 = n2 = 6, P = 0.002). The right shift of the concentration curve of methadone was of 2 orders of magnitude (Figure 2, E–G). M40 (1 μM) completely counteracted all opioid agonist effects in cells transfected with Gal1Rs (Figure 2, A–E). We did not observe this effect in cells not transfected with Gal1Rs, which substantiated the notion that the pharmacodynamic changes associated with cotransfection of the Gal1R depend on heteromerization with MOR. The flattening of the concentration-response curves of the MOR agonists with M40 confirmed the additional ability of M40 to exert a significant negative allosteric modulation of the intrinsic efficacy of MOR agonists. As an additional control of the dependence of MOR-Gal1R heteromerization, neither the coexpression of Gal2R, which does not heteromerize with MOR (11), nor the presence of M40 (1 μM) modified the concentration-response curve of methadone in cells cotransfected with Gal2R (Supplemental Figure 4).
Figure 2. Gal1R-dependent pharmacodynamic differences of MOR agonists.

(A–E) Representative concentration-response experiments of ligand-induced BRET changes in HEK-293T cells transfected with the MOR fused to Rluc and the α subunit of the Gi1 protein fused to YFP. Values represent the mean ± SEM of triplicates. The effect of increasing concentrations of the MOR agonists morphine (A), EM1 (B), DAMGO (C), fentanyl (D), and methadone (E) were evaluated without cotransfection with Gal1R, in the absence or presence of the Gal1R/Gal2R antagonist M40 (red and green curves, respectively), or with cotransfection with Gal1R, with or without M40 (blue and purple curves, respectively). (F and G) Comparison of the Emax and E50 values obtained with and without cotransfection with the Gal1R. **P < 0.01 versus transfection with MOR alone; unpaired, 2-tailed t test (F); **P < 0.01 versus transfection with MOR alone; 2-tailed Mann-Whitney U test (G). Emax and EC50 values are shown as dots, presented with the mean ± SEM (F) or median with interquartile ranges (G) (n = 5–10 triplicates/group).
Weak dopaminergic activation by methadone.
We then used microdialysis techniques in awake, freely-moving rats to determine whether the results obtained in vitro with morphine and methadone translated to an in vivo preclinical model, with VTA as the target brain structure. As mentioned above, our recent studies indicated that a predominant population of MORs that modulate dopamine cell function in the VTA form heteromers with the Gal1R (13). Furthermore, intracranial self-administration experiments indicated that MORs localized in the VTA and the rostromedial tegmental nucleus (tail of the VTA) are involved with the reinforcing effects of opioids (17–19), effects which depend on the ability of MORs to stimulate the VTA-NAc dopaminergic system (20–23). Perfusion of morphine (1, 3, and 10 μM) within the VTA through a modified microdialysis probe (that allows the controlled local infusion of large peptides) (13, 24) produced significant and sustained concentration-dependent somato-dendritic dopamine release, with the most effective concentration (10 μM) being significantly counteracted by local infusion of 10 μM M40 (Figure 3A; 1-way ANOVA: F3,25 = 6.4, P = 0.002; Dunnett’s multiple comparisons, versus morphine 1 μM: P = 0.046, P < 0.001 and P = 0.199 for 3 μM morphine, 10 μM morphine, and 10 μM morphine plus M40, respectively). Methadone, on the other hand, could only produce a significant increase in the extracellular concentration of dopamine in the VTA at the much higher concentration of 300 μM; at 10 or 100 μM, methadone was ineffective, although at 100 μM, coinfusion with the MOR-Gal1R–destabilizing peptide TM5 (60 μM), but not the control peptide TM1 (60 μM), did produce a significant dopamine increase (Figure 3B; 1-way ANOVA: F4,33 = 6.4, P < 0.001; Dunnett’s multiple comparisons, versus 10 μM methadone: P = 0.872, P = 0.008, P = 0.037 and P = 0.998 for 100 μM methadone, 300 μM methadone, and 100 μM methadone plus TM5 or TM1, respectively). Therefore, in complete agreement with the biochemical data, the minimal concentration of methadone required to elicit a significant VTA dopamine release was 2 orders of magnitude larger than that of morphine (300 μM versus 3 μM, respectively). Furthermore, the selective effect of the MOR-Gal1R heteromer–destabilizing peptide confirmed that the large difference in the potencies of morphine and methadone is determined by the pharmacodynamic changes dependent on MOR-Gal1R heteromerization.
Figure 3. Weaker ability of methadone to stimulate the VTA-NAc dopaminergic system as compared with morphine, fentanyl, and DAMGO.

Microdialysis experiments in rats. Values represent mean dopamine concentrations as a percentage of baseline ± SEM (average of 5 samples before MOR agonist administration). The lined and white rectangles in the x axis indicate the period of MOR agonist perfusion and M40 infusion, respectively; the arrows in D and F indicate the time point of systemic administration. (A and B) Effect of intra-VTA morphine (1–10 μM) or methadone (10–300 μM) on VTA dopamine. *P < 0.05 and ***P < 0.001 versus 1 μM morphine (A); *P < 0.05 and **P < 0.01 versus 10 μM methadone (B); 1-way ANOVA with Dunnett’s multiple comparisons, comparing the average of 8 samples after MOR agonist administration (n = 7–8 animals/group). (C) Effect of intra-NAc morphine (10 μM) or methadone (300 μM) on NAc dopamine. Results were nonsignificant in both cases; paired, 2-tailed t test, comparing the average of 8 samples after MOR agonist administration versus baseline values (n = 8 animals/group). (D) Effect of systemic administration (1 mg/kg, i.p.) of morphine or methadone on VTA and NAc dopamine in the VTA and contralateral NAc. *P < 0.05; paired, 2-tailed t test, comparing the average of 5 samples after MOR administration versus baseline values (n = 6–7 animals/group). (E) Effect of intra-VTA DAMGO (10 μM) or fentanyl (10 μM) on VTA dopamine, respectively (n = 7 and 9 animals/group). **P < 0.01; paired, 2-tailed t test, comparing the average of 8 samples after MOR agonist administration versus baseline values. (F) Effect of systemic administration (0.03 mg/kg, i.p.) of fentanyl on VTA and NAc dopamine in the VTA and contralateral NAc. **P < 0.01; paired, 2-tailed t test, comparing the average of 8 samples after MOR administration versus baseline values (n = 7 animals/group). DA, dopamine.
In contrast to the results obtained in the VTA, perfusion of effective intra-VTA concentrations of morphine (10 μM) or methadone (300 μM) in the NAc did not produce local dopamine release (Figure 3C; paired, 2-tailed t test: t7 = 0.9, P = 0.384, and t7 = 1.4, P = 0.215, respectively). Finally, systemic administration of morphine (1 mg/kg, i.p.) produced significant dopamine release in both the VTA and the NAc (Figure 3D; paired, 2-tailed t test: t5 = 2.8, P = 0.040, and t7 = 2.6, P = 0.036, respectively), while the same dose of methadone (1 mg/kg, i.p.) did not produce significant changes in either brain area (Figure 3D; paired, 2-tailed t test: t7 = 0.5, P = 0.624, and t6 = 1.6, P = 0.169, respectively). Collectively, the results of these microdialysis experiments with morphine and methadone indicate, first, that MOR agonists stimulate the mesolimbic dopaminergic system by activating MORs in the VTA, which induces simultaneous somato-dendritic and terminal release of dopamine, findings that are supported by previous seminal studies (20–23). But more specifically, these findings show that opioids stimulate the VTA-NAc dopaminergic system by activating VTA MOR-Gal1R heteromers.
Methadone is also pharmacodynamically different from morphine in that it has a greater ability to promote MOR internalization and the consequent recycling of nondesensitized MOR (25, 26). This difference may explain the reduced tolerance of methadone relative to morphine (morphine characteristically induces little MOR internalization; refs. 25, 26). But this difference would predict weaker dopaminergic activation by morphine, which is the opposite of what we observed in the present study. Nonetheless, to discard a possible role of differential internalizing properties in VTA-NAc dopaminergic activation, we evaluated the effect of the intra-VTA infusion of DAMGO and fentanyl on somato-dendritic dopamine release. It should be noted that, in terms of chemical structure, morphine, methadone, and fentanyl belong to different chemical classes of opioids (27). Like methadone and in contrast to morphine, DAMGO and fentanyl promote pronounced internalization (28). As with morphine and in contrast to methadone, the potencies of these drugs for MORs are not modified upon heteromerization with the Gal1R (Figure 2G). Yet both DAMGO and fentanyl produced a significant somato-dendritic dopamine release when perfused into the VTA at a much lower concentration (10 μM) than the minimal effective concentration of methadone (300 μM) (Figure 3E; paired, 2-tailed t test: t6 = 3.7, P = 0.009, and t9 = 4.1, P = 0.003, respectively). In agreement with the well-known lipophilicity-dependent pharmacokinetic profile of fentanyl (29), the low dose of 0.03 mg/kg (i.p.) produced a response similar to that of 1 mg/kg morphine, with significant dopamine release detected in both the VTA and the NAc (Figure 3F; paired, 2-tailed t test: t6 = 4.0, P = 0.007, and t6 = 3.8, P = 0.009, respectively).
Methadone has substantially higher brain penetrability than does morphine (30). It follows that at sufficiently high doses, methadone concentration levels in the VTA should surmount its selective low potency for the MOR-GalR1 heteromer. Indeed, methadone doses higher than 1 mg/kg (i.p.) have been reported to produce significant dopamine release in the NAc (31, 32). Nonetheless, lower methadone doses should still be sufficient to activate MORs localized in other areas of the CNS, which might mediate its therapeutic effects (see Discussion). In fact, methadone is more potent than morphine at producing analgesic effects in rodents upon acute systemic administration (33, 34). We should then be able to demonstrate that methadone, at a dose of 1 mg/kg (i.p.), although it does not activate the VTA-NAc dopaminergic system, is able to influence MORs localized in areas of the CNS other than the VTA.
To substantiate this hypothesis, we conducted a series of in vivo PET imaging experiments in the same rat strain. The uptake of [18F]2-fluoro-2-deoxy-D-glucose ([18F]FDG) was measured to monitor whole-brain metabolic activity changes induced by methadone and morphine (1 mg/kg for each, i.p.) during the interval with maximal morphine-induced dopamine release (between 30 and 80 minutes after drug administration) (Figure 4A). We observed a predominant increase in baseline metabolic activity in the frontal cortex, dorsal striatum, thalamus, inferior colliculus, and deep cerebellar nuclei when analyzing the average standardized uptake value ratio (SUVR) calculated using the whole brain as a reference region (Figure 4B). Both morphine and (more visibly) methadone induced an apparent decrease in the cortical and striatal metabolic activity (Figure 4B). In agreement with the microdialysis experiments, voxel-based statistical parametric mapping revealed that morphine, but not methadone, significantly modified the metabolic activity in the ventral striatum in 2 separate analyses. First, administration of morphine, but not methadone, significantly decreased metabolic activity in a basal forebrain region that comprises the NAc and its projecting areas, which include the ventral pallidum, the lateral preoptic area, the lateral hypothalamus, and the lateral septum (35, 36), as compared with baseline values (Figure 4C; statistical parametric maps of significant decreases of [18F]FDG uptake, according to paired t tests, P < 0.05, filtered by clusters of more than 100 contiguous voxels). Second, volume of interest (VOI) analysis of selected regions revealed a remarkable differential pattern of [18F]FDG uptake: morphine significantly decreased metabolic activity in the basal forebrain but did not significantly modify metabolic activity in the frontal cortex or dorsal striatum (Figure 4D; Wilcoxon 2-tailed, matched-pairs test [n = 7 pairs], P = 0.016, P = 0.078, and P = 0.469, respectively). On the other hand, methadone did not significantly modify metabolic activity in the basal forebrain, but it significantly decreased [18F]FDG uptake in both the frontal cortex and dorsal striatum (Figure 4E; Wilcoxon 2-tailed, matched-pairs test [n = 7 pairs], P = 0.218, P = 0.016, and P = 0.031, respectively). Although this method could not provide sufficient resolution to visualize changes in the VTA, the results support our conclusions from the biochemical and microdialysis experiments regarding the pharmacodynamic-dependent weak influence of methadone on the VTA-NAc dopaminergic system. In addition, as expected because of its higher brain penetrability, methadone was able to produce a more significant effect than did morphine in brain areas other than the NAc.
Figure 4. Differential ability of morphine and methadone to influence metabolic activity in the basal forebrain.

Metabolic mapping using [18F]FDG PET scanning in rats. (A) Timeline of the experiment. (B) [18F]FDG uptake after administration of saline (baseline, n = 14), morphine (1 mg/kg, n = 7), or methadone (1 mg/kg, n = 7). Coronal and sagittal images (1.5 mm anterior to bregma and 1.4 mm lateral from the midline, respectively) show the average SUVR calculated using the whole brain as a reference region. (C) Voxel-based parametric mapping analyses revealed significantly decreased metabolic activity from baseline values in a basal forebrain region that included the NAc and its projecting areas after morphine, but not methadone, treatment. Statistical parametric maps of significant decreases of [18F]FDG uptake (P < 0.05, paired t test). (D and E) VOI analyses of the frontal cortex (FCx), dorsal striatum (DS), and basal forebrain (BF) region, showing a significant differential pattern of [18F]FDG uptake after administration of morphine (D) or methadone (E). Values are shown as dots and as the median with interquartile ranges. *P < 0.05 versus the corresponding baseline value; 2-tailed Wilcoxon matched-pairs test (n = 7 animals/group).
Lower incidence of euphoric effects with methadone.
The MOR-Gal1R heteromer–dependent weak dopaminergic activation by methadone would predict that methadone should not promote self-administration in animals. Counter to this prediction, several early studies showed self-administration of methadone by rodents and monkeys (37–39). However, an important, little-acknowledged factor in studies of methadone self-administration is previous exposure to other opioids (37), and methadone, as an efficacious MOR ligand, could promote self-administration by attenuating aversive effects associated with morphine or heroin withdrawal. Remarkably, a more recent study in rats showed that methadone is weakly reinforcing, even with a previous history of heroin self-administration (32). Thus, methadone resulted in only partial maintenance of self-administration and, in sharp contrast to equivalent doses of heroin, failed to induce reinstatement of the drug-seeking behavior in these rodents (32). It is conceivable that in the previous experiments, the doses, route, and schedule of administration of methadone favored its capacity to activate the VTA-NAc dopaminergic system. In fact, methadone was administered at doses very similar to those of morphine (37–39). Yet our results from in vivo microdialysis and PET experiments would predict that lower doses of methadone would dissociate the effects of the dopaminergic system from other CNS effects.
An additional extension of our findings is that methadone should be associated with a significantly lower incidence of euphoria in humans, since the subjective feelings of a “high” associated with drugs of abuse correlate with the activation of the canonical VTA-NAc dopamine-addiction circuit (40, 41). At a minimum, we would expect a dissociation of the euphoric effects of methadone from its therapeutic effects. The study of methadone-associated euphoria has received little attention in the literature. Outside the context of addiction (and thus in opioid-naive patients), there is little evidence to suggest that methadone produces euphoria (42, 43). And in the context of OUD, the few relevant references we found that explore methadone-associated euphoria either do not take into account previous opioid exposure (44, 45), or do not disambiguate off-label isolated methadone use from off-label methadone taken in combination with other drugs (46, 47). Nevertheless, a careful analysis of some of these studies (44, 45) finds support for subjective feelings of euphoria at relatively high doses, depending on the subject’s level of tolerance, as well as for the dissociation of this euphoria from other effects of methadone (see Discussion).
Anecdotally, patients being treated with daily methadone for OUD rarely report a methadone-associated “high.” To explore this more deeply, we took advantage of data being collected as part of 2 separate ongoing clinical studies (see Supplemental Table 1 for the characteristics of the study participants in these clinical trials). These unique populations of patients were being treated with daily regimens of methadone and include: (a) data from a large registry of patients with RLS, and (b) patients diagnosed with OUD enrolled in an urban medication-assisted treatment program. The rationale for addressing the question of methadone-associated euphoria in 2 separate clinical settings was that methadone represents a very effective therapeutic treatment for the 2 conditions (2, 3), each of which possesses a different etiology, symptomatology, and accepted treatment course.
For the first patient population (RLS; Supplemental Table 1), we compared self-reports of past-2-week drug treatment–associated euphoria between patients prescribed methadone (n = 122) and patients (n = 101) prescribed other MOR agonists (morphine, codeine, oxycodone, hydrocodone, or hydromorphone). A significantly lower proportion of patients with RLS receiving methadone (0.8%) endorsed subjective feelings of a methadone-associated “high” compared with patients receiving other MOR agonist treatment (8%) (Figure 5A; 2-sided χ21 = 6.9, P = 0.008). This finding was mirrored in a separate population of OUD patients prescribed methadone (n = 30), who, despite significant increases in the methadone dose (Supplemental Table 1), reported no perceived methadone-associated euphoria at either 2 weeks or 3 months after entry into a medication-assisted treatment program (Figure 5B and Supplemental Table 1). Importantly, withdrawal scores (using the subjective opioid withdrawal scale [SOWS]) for these same patients were significantly decreased (Figure 5B; Wilcoxon 2-tailed, matched-pairs test [n = 30 pairs], P = 0.009), indicating a dissociation between the therapeutic and euphoric effects of methadone.
Figure 5. Very low reporting of feeling “high” by methadone-treated subjects.

(A) Total number and proportion of patients with RLS reporting feeling “high” upon treatment with methadone or with other MOR agonists (other opioids). **P < 0.01, significantly different proportion of subjects versus patients treated with methadone; 2-sided χ2 test. (B) Assessment of symptoms of craving and withdrawal (according to the SOWS) and perceived severity of methadone-associated euphoria (“high”). Both measurements (SOWS and “high”) were obtained after 14 and 84 days of methadone treatment. Values are shown as dots and as the median with interquartile ranges. **P < 0.01 versus 14 days of treatment; 2-tailed Wilcoxon matched-pairs test (n = 30). (C) Total number and proportion of patients with OUD reporting first-time nonmedical use of methadone or other MOR agonists (other opioids) with the express intent of achieving a “high” (and not for alleviating withdrawal symptoms or for other purposes; see Methods). **P < 0.01, significantly different proportion of subjects seeking a “high” with other opioids versus with methadone; 2-sided χ2 test.
The ongoing clinical trial of patients with OUD afforded another, separate measurement of methadone-associated euphoria in the form of self-reporting of motivation for seeking first-time off-label use of a comprehensive list of drugs, including methadone (Supplemental Table 1). We found that a significantly lower proportion of patients with OUD endorsed having sought methadone nonmedically for the express intent of achieving a “high” (3.8%; n = 26), relative to initial nonmedical use of other MOR agonists (35.4%; n = 48, Figure 5C; χ21 = 9.1, 2-sided, P = 0.002). Indeed, in our study, the most common reason why these patients sought off-label methadone was for “control of withdrawal symptoms.” This finding is in accord with a now-classic literature demonstrating that, although opioid-addicted individuals use methadone illicitly, they do so not for the purpose of achieving a “high” (as they do with other prescription opioids), but for self-medication (48, 49). Collectively, these data support our hypothesis that methadone may be less euphorigenic than other opioids because of its weak ability to activate the VTA-NAc dopaminergic system.
Discussion
Morphine and methadone have long been known to have very similar pharmacological properties, as both are selective MOR agonists with similar affinities and high efficacy for the human MOR (50–53). Indeed, methadone has been reported to be a more efficacious MOR ligand than morphine (52, 53). Also taking into account the higher brain penetrability of methadone (30), it could be predicted that methadone is a more efficacious analgesic than morphine. In fact, in experimental animals, methadone has been reported to be a more potent analgesic and to produce significantly less antinociceptive tolerance than does morphine (25, 34). The reduced tolerance of methadone has been attributed to its superior ability to induce MOR internalization and the consequent recycling of nondesensitized MORs (25, 26). Yet, methadone is almost never given as a first-line analgesic in human clinical settings. Here, we propose a new basis for promoting methadone as a prescription opioid of choice, especially for patients who may be more vulnerable to OUD, as methadone possesses a specific pharmacodynamic property that endows it with a higher therapeutic index compared with other prescription opioids, with a higher ratio of therapeutic versus euphoric effects.
This pharmacodynamic property of methadone is determined by heteromerization of the MOR with the Gal1R in the VTA, which renders methadone significantly less potent than other MOR agonists at activating the VTA-NAc dopaminergic system. Thus, we found that a much higher concentration of methadone was needed to directly activate the MOR in the VTA relative to morphine, fentanyl, and DAMGO. In agreement with the local effective dose differences between methadone and morphine, when systemically administered at a relatively high dose, only morphine was effective at activating the VTA-NAc dopaminergic system, which was associated with significant metabolic changes in the NAc and main output areas, as demonstrated in PET imaging experiments. On the other hand, methadone, at the same dose, produced a significantly different qualitative pattern than did morphine and was only effective at producing significant metabolic changes in the cortex and dorsal striatum. This dissociation of the potential dopaminergic effects of methadone from other central effects also implies that MORs do not form heteromers with Gal1Rs outside of the VTA. In fact, in other areas of the CNS, including the spinal cord, MORs form heteromers with δ-opioid receptors (DORs), which seem to be significantly involved in the antinociceptive effects of opioids (54, 55), including methadone (56).
Given its high brain penetrability, depending on the dose, route of administration, and the subject’s level of tolerance, methadone could still surmount its selective decrease in potency for the MOR-Gal1R heteromer and activate the VTA-NAc dopaminergic system. In fact, previous studies reported mild dose- and tolerance-dependent euphoric symptoms upon oral methadone treatment in subjects maintained on methadone (45). This could also explain the results of the present study, in which a small proportion of methadone-treated patients reported a “high.” In addition, methadone was also reported to produce euphoria with an intensity similar to that obtained with heroin or morphine upon i.v. administration (44). In that study, methadone was found to have a slightly lower potency and efficacy in eliciting euphoria and positive symptoms compared with morphine. This implies that, because of its higher brain penetrability, higher concentrations of methadone in the brain may be necessary to induce euphoria as compared with morphine. In the same study, the miotic effects of morphine and heroin paralleled the observer-rated liking, while methadone produced very long-lasting miosis, which was still present when the subjective effects were markedly attenuated or absent. The authors concluded that “these findings may indicate that there are differences in mechanisms of actions between the various effects of methadone” (44). The present results provide an explanation for those apparently different mechanisms, by the existence of different pharmacodynamic properties of methadone determined by the MOR-Gal1R heteromers, localized in the VTA, versus MORs localized in other areas of the CNS, including those involved in pupillary constriction (57).
On a more general level, we believe the present study provides compelling evidence for the use of GPCR heteromers as targets for drug development (9, 10). Heteromerization represents a biological mechanism that allows allosteric modulations between endogenous ligands (such as the ability of galanin to modulate the potency and efficacy of endogenous opioids to activate the VTA-NAc dopaminergic system) (11). But heteromerization may also reveal new properties of exogenous compounds, such as the cross-antagonism of M40, which could be used as a new therapeutic strategy to counteract the dopaminergic effects of opioids. Selective Gal1R agonists, which also counteract MOR signaling in the MOR-Gal1R heteromer (13), could have a more important application in analgesia. This is because Gal1Rs are also colocalized in the spinal dorsal horn with MORs and have been shown to be a significant player in the well-demonstrated antinociceptive role of galanin (58). In addition, there is clear preclinical evidence for a synergistic spinal antinociceptive effect with opioids (59), which indicates that colocalization of spinal MORs and Gal1Rs does not imply heteromerization (which would predict an antagonistic interaction between MOR and Gal1R ligands). Therefore, a new clinical strategy for analgesia could be the coadministration of Gal1R agonists with opioids, which would allow for a decrease of the effective analgesic doses of opioids (synergistic spinal effect), while counteracting their euphorigenic effects (antagonistic VTA effect). Several unsuccessful attempts have been made to develop safer analgesics, such as one involving the nonselective galanin receptor agonist galnon (12, 60), but the use of selective, potent, nonpeptidic small molecules targeting the Gal1R that are suitable for clinical applications may represent a promising therapeutic strategy for the future.
Also on a more general level, but of more relevance, given the results obtained in the present preclinical and clinical studies, heteromerization determines potential pharmacodynamic differences between exogenous compounds, such as between morphine and methadone, which endows methadone with a less addictive opioid profile. Taken together, the current findings provide a powerful rationale for the development of potentially safer methadone-like compounds that preferentially target MORs not forming heteromers with Gal1Rs. Additionally, and most importantly from an OUD perspective, these findings argue against the misconception that methadone maintenance treatment is simply the substitution of a licit opioid for an illicit one.
Methods
Stable cell lines.
The cDNAs for the human MOR and Gal1R were obtained from the Missouri S&T cDNA Resource Center and were modified N-terminally with in-frame fusion of a signal peptide for enhanced cell-surface expression (61), followed by a Flag epitope tag and subcloned into the pcDNA™5⁄FRT plasmid. All constructs were confirmed by sequencing analysis. These plasmids were cotransfected into Flp-In 293 cells with the Flp recombinase expression vector pOG44 (1 μg/9 μg) to obtain FLP-FRT-HEK stable cell lines expressing the MOR or the Gal1R. Transfection was performed using the Lipofectamine method following the manufacturer’s instructions (Invitrogen, Thermo Fisher Scientific). Clones resistant to hygromycin B (50 μg/ml) were isolated, and a cell line expressing MORs (MU cells) and another expressing Gal1Rs (GAL cells) were selected according to a significant functional response to the MOR agonist EM1 and the Gal1R agonist M617, respectively, as measured by a DMR assay (see below). A stable cell line expressing both MORs and Gal1Rs (MU-GAL cells) was then generated using GAL cells. cDNA of the MOR subcloned into the pcDNA3.1 vector was fused at its N-terminus with a signal peptide followed by a Myc epitope tag and transfected into GAL cells. Clones resistant to Geneticin (400 μg/ml; Gibco, Thermo Fisher Scientific) were isolated, and a cell line expressing MORs and Gal1Rs (MU-GAL cells) was selected according to a significant functional response to both EM1 and M617 (see Results).
DMR.
A global cell-signaling profile or DMR was measured using the EnSpire Multimode Plate Reader (PerkinElmer) (62). This label-free approach uses refractive waveguide grating optical biosensors, integrated into 96-well microplates. Changes in local OD are measured in a detection zone up to 150 nm above the surface of the sensor. Cellular mass movements induced upon receptor activation are detected by illuminating the underside of the biosensor with polychromatic light and measured as changes in the wavelength of the reflected monochromatic light. These changes are a function of the refraction index. The magnitude of this wavelength shift (in picometers) is directly proportional to the amount of DMR. All DMR assays were performed using the EnSpire Plate Reader 2300 (PerkinElmer). MU cells or MU-GAL cells were directly seeded in EnSpire-LFC 96-well plates and cultured overnight to form a confluent monolayer in the cell culture medium. The cell seeding density was 30,000 cells per well/100 μl. After washing 4 times in HBSS (Invitrogen, Thermo Fisher Scientific), the cells were maintained with 60 μl HBSS and further incubated inside the plate reader for 2 hours. Agonists (EM1, morphine, or M617) were added after a 10-minute baseline reading, and DMR was measured for 1 hour. The antagonists (CTOP or M40) were added 10 minutes before the baseline reading. All data reported were calculated on the basis of the amplitudes of DMR signals 10 minutes after agonist-induced stimulation and the background. Statistical analysis was performed with GraphPad Prism 7 (GraphPad Software).
ERK1/2 phosphorylation in cells in culture.
MU-GAL cells or HEK-293T cells (with or without transient transfection with MORs and Gal1Rs) were maintained in culture with DMEM supplemented with 10% FBS (Atlanta Biologicals), 2 mM l-glutamine (Gibco, Thermo Fisher Scientific), and 1% penicillin-streptomycin (Gibco, Thermo Fisher Scientific) and kept in an incubator at 37°C and 5% CO2 with selection antibiotics (hygromycin B and Geneticin). Cells were seeded onto 12-well plates (0.25 × 106/well) in full growth medium. The day before the assay, the medium was changed to DMEM without FBS for approximately 16 hours before the addition of ligands. Next, cells were incubated or not with the antagonists (CTOP or M40) in HBSS (1 μM final concentration) or the same volume of HBSS for 15 minutes and then with EM1 (0.1 μM) for 7 minutes. Cells were rinsed with ice-cold PBS and lysed by the addition of 100 μl ice-cold lysis buffer (provided in the Cell Signaling Technology kit). Phosphorylated MAPK levels were determined using an enzyme-linked sandwich ELISA kit (Cell Signaling Technology) following the manufacturer’s recommended protocol. Statistical analysis was performed with GraphPad Prism 7.
FACS.
MOR agonist–induced internalization in MU-GAL cells, which express a Myc-tagged MOR, was performed using FACS with the FACSCanto II system (BD Biosciences). Briefly, MU-GAL cells were harvested and incubated at room temperature with the MOR agonist DAMGO. The antagonists CTOP or M40 were added 10 minutes before the agonist. After drug treatment at room temperature, cells were chilled to 4°C and washed using FACS buffer (DPBS with 1% BSA and 0.1% sodium azide). The anti-Myc monoclonal antibody (1:500; Cell Signaling Technology) was added, following by the anti-mouse antibody conjugated with Alexa Fluor 647 (Invitrogen, Thermo Fisher Scientific). After an additional wash, cell-surface mean fluorescence of 10,000 live cells was analyzed on a FACSCanto II system (BD Biosciences). Statistical analysis was performed with GraphPad Prism 7.
Radioligand-binding experiments.
Upon reaching 80%–90% confluence, MU and MU-GAL cells were harvested using premixed Earle’s Balanced Salt Solution (EBSS) with 5 mM EDTA (Life Technologies, Thermo Fisher Scientific) and centrifuged at 1489 g for 10 minutes at 21°C. The supernatant was removed, and the pellet was resuspended in 10 ml hypotonic lysis buffer (5 mM MgCl2, 5 mM Tris, pH 7.4, at 4°C) and centrifuged at 24,226 g for 30 minutes at 4°C. The pellet was then resuspended in fresh binding buffer. A Bradford protein assay (Bio-Rad) was used to determine the protein concentration, and membrane aliquots were frozen in fresh binding buffer at –80°C for future use. The binding buffer consisted of 50 mM Tris and 5 mM MgCl2 at pH 7.4. On the test day, the test compound was diluted into half-log serial dilutions using 30% DMSO vehicle. Membranes were diluted in fresh binding buffer at a stock concentration of 300 to 500 μg/ml. Radioligand competition experiments were conducted in 96-well plates containing 300 μl fresh binding buffer, 50 μl diluted test compound, 100-μl membranes (final amount of 30 μg/well for MOR cells and 30–50 μg/well for MOR-GAL cells, respectively), and 50 μl radioligand diluted in binding buffer ([3H]DAMGO, 3 nM final concentration). Aliquots of [3H]DAMGO solution were also quantified accurately to determine the added radioactivity. Nonspecific binding was determined using 10 μM CTOP, and total binding was determined with 30% DMSO vehicle in the presence or absence of 1 μM M40 or M617. All dilutions were tested in triplicate and the reactions incubated for 60 minutes at room temperature. The reaction was terminated by filtration through a PerkinElmer Uni-Filter-96 GF/B, presoaked for 60 minutes in 0.5% polyethylenimine, using a Brandel 96-Well Plate Harvester Manifold (Brandel Instruments). The filters were washed 3 times with 3 ml (3 × 1 ml/well) ice-cold binding buffer and water, and 65 μl PerkinElmer MicroScint 20 Scintillation Cocktail was added to each well, and the filters were counted using a PerkinElmer MicroBeta Microplate Counter.
Radioligand-binding data analysis.
Radioligand competition curves were analyzed by nonlinear regression using GraFit curve-fitting software (Erithacus), by fitting the binding data to the mechanistic 2-state dimer receptor model, as described in detail elsewhere (63). To calculate the macroscopic equilibrium dissociation constants from the competition experiments, the following general equation was applied:
(Equation 1)
In the above equation, A and B represent the assayed radioligand and competitor concentration, respectively. KD1 and KD2 are, respectively, the equilibrium dissociation constants of the first and second binding of A or B to the receptor homodimer, and KDAB is the dissociation constant of B binding to a receptor dimer semioccupied by A and shows the allosteric modulation between A and B. Because A and B are the same noncooperative compound, we considered the experiment an autocompetition, and the general equation was simplified, as shown below, given that KDA2 = 4KDA1, KDB2 = 4KDB1, KDA1 = KDB1, KDA2 = KDB2 and KDAB = KDB2, to provide KDB1 values as the measure of affinity of DAMGO, in the presence or absence of Gal1R ligands.
(Equation 2)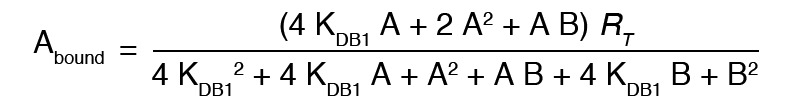
Moreover, for a more accurate fit, when adjusting the MU-GAL cell data in the presence of Gal1R ligands, we fixed the number of receptors obtained in the absence of these ligands. This is because, despite the fact that the curves in the presence of Gal1R compounds started from below, the number of receptors had to be the same each day. Statistical analysis was performed with GraphPad Prism 7.
Transient transfections.
The cDNAs for the human MOR and Gal1R were modified N-terminally with in-frame fusion of a signal peptide for enhanced cell-surface expression (61), followed by a Flag or Myc epitope tag. The cDNA encoding full-length Renilla luciferase 8 (Rluc) (64) was fused in-frame to the C-terminus of the MOR using the pcDNA3.1 vector. For BRET assays, plasmid cDNAs with MOR-Rluc, GALR1, and G protein subunits (Gαi-Venus, Gβ1 and Gγ2) were cotransfected into human embryonic kidney 293T (HEK-293T) cells using polyethylenimine (PEI) (MilliporeSigma) at a 1:2 weight ratio in 10-cm plates. The ratio of MOR-Rluc to Gal1R cDNA plasmid was 1:4, and the total amount of plasmid cDNA was 15.75 μg (see Results). All constructs were confirmed by sequencing analysis. Cells were maintained in culture with DMEM supplemented with 10% FBS (Atlanta Biologicals), 2 mM l-glutamine (Gibco, Thermo Fisher Scientific), and 1 % penicillin-streptomycin (Gibco, Thermo Fisher Scientific) and kept in an incubator at 37°C and 5% CO2.
BRET.
A BRET assay was performed to detect ligand-induced changes in the interaction between MOR (fused to Rluc) and the Gαi subunit (fused to Venus, a variant of YFP) in the presence and absence of the Gal1R. Experiments were performed approximately 48 hours after transfection. The transient transfected cells were collected, washed, and resuspended in Dulbecco’s PBS (DPBS) with 0.1% glucose and 200 μM sodium bisulfite. Approximately 200,000 cells/well were distributed in 96-well plates, and 5 μM coelenterazine H (NanoLight Technology) was added. Two minutes after the addition of coelenterazine, increasing concentrations of different MOR agonists were added to different wells in the presence and absence of M40 (added 10 minutes before the agonist). The plate was read after agonist addition using a Mithras LB940 microplate reader (Berthold Technologies). BRET signal from cells was calculated as the ratio of the light emitted by Rluc8 at 485 nm to that emitted by Venus at 530 nm. A BRET change was defined as the BRET ratio for the corresponding drug minus the BRET ratio in the absence of the drug. Emax and EC50 values are expressed as the basal subtracted BRET change in the concentration-response graphs. Nonlinear fitting to obtain Emax and EC50 values and statistical analysis were performed with GraphPad Prism 7.
ERK1/2 phosphorylation in VTA slices.
Two-month-old male Sprague-Dawley rats (from the animal facility of the Faculty of Biology, University of Barcelona) were used. The animals were housed 2 per cage and kept on a 12-hour light/12-hour dark cycle with food and water available ad libitum, and experiments were performed during the light cycle. Animals were euthanized by decapitation under 4% isoflurane anesthesia, and brains were rapidly removed, placed in ice-cold oxygenated (O2/CO2, 95%/5%) Krebs-HCO3– buffer (124 mM NaCl, 4 mM KCl, 1.25 mM KH2PO4, 1.5 mM MgCl2, 1.5 mM CaCl2, 10 mM glucose, and 26 mM NaHCO3, pH 7.4), and sliced at 4°C using a brain matrix (Zivic Instruments). VTA slices (500-μm thick) were dissected at 4°C in Krebs-HCO3– buffer; each slice was transferred onto a 12-well plate with Corning Netwell inserts containing 2 ml ice-cold Krebs-HCO3– buffer. The temperature was raised to 23°C, and after 30 minutes, the medium was replaced by 2 ml fresh buffer (23°C). Slices were incubated under constant oxygenation (O2/CO2, 95%/5%) at 30°C for 4 hours in an Eppendorf ThermoMixer (5 PRIME), and the medium was replaced by fresh buffer and incubated for 30 minutes before the addition of any agent. After incubation, the solution was discarded, and slices were frozen on dry ice and stored at –80°C until ERK1/2 phosphorylation was determined. VTA slices were incubated for 20 minutes with medium, CTOP, or M40 (10 μM) and treated for 12 minutes with medium or EM1 (1 μM). Then, the slices were lysed by the addition of 300 μl ice-cold lysis buffer (50 mM Tris-HCl, pH 7.4, 50 mM NaF, 150 mM NaCl, 45 mM β-glycerophosphate, 1% Triton X-100, 20 μM phenyl-arsine oxide, 0.4 mM NaVO4, and protease inhibitor mixture). Cellular debris was removed by centrifugation at 13,000 g for 5 minutes at 4°C, and the protein was quantified by the bicinchoninic acid method using BSA dilutions as the standard. Phosphorylated proteins were then determined by Western blotting using a mouse anti–phosphorylated ERK1/2 antibody (1:2500; MilliporeSigma) and rabbit anti–total ERK1/2 antibody (1:40,000; MilliporeSigma). Bands were visualized by the addition of a mixture of IRDye 800 (anti-mouse) antibody (1:10,000; LI-COR) and IRDye 680 (anti-rabbit) antibody (1:10,000; LI-COR) and scanned with a LI-COR Odyssey infrared scanner. Band densities were quantified using the scanner software exported to Microsoft Excel. The level of phosphorylated proteins was normalized for differences in loading using the total (phosphorylated plus nonphosphorylated) protein band intensities. Statistical analysis was performed with GraphPad Prism 7.
In vivo microdialysis.
Three-month-old male Sprague Dawley rats (Charles River Laboratories) were used. Animals were housed 2 per cage and kept on a 12-hour light/12-hour dark cycle with food and water available ad libitum. Experiments were performed during the light cycle. Rats were deeply anesthetized with 3 ml/kg Equithesin (4.44 g chloral hydrate, 0.972 g Na pentobarbital, 2.124 g MgSO4, 44.4 ml propylene glycol, 12 ml ethanol, and distilled H2O up to 100 ml of the final solution; NIDA Pharmacy) and implanted unilaterally into the VTA (coordinates from bregma with a 10° angle in the coronal plane: anterior, –5.6 mm; lateral, 2.4 mm; vertical, –9 mm) or in the shell of the NAc (anterior, 1.6 mm; lateral, 0.5 mm; vertical, –5.1 mm) with a regular microdialysis probe or with a specially designed microdialysis probe that allows the direct infusion of large peptides within the sampling area (24). Some animals were implanted simultaneously with 2 regular probes, 1 in the VTA and the other in the contralateral NAc (same coordinates as above). After surgery, the rats were allowed to recover in hemispherical CMA-120 cages (CMA Microdialysis) equipped with 2-channel overhead fluid swivels (Instech) connected to a sample collector (CMA 470, CMA). Twenty-four hours after implantation of the probes, experiments were performed on freely moving rats in the same hemispherical home cages in which they recovered overnight from surgery. An artificial cerebrospinal solution containing 144 mM NaCl, 4.8 mM KCl, 1.7 mM CaCl2, and 1.2 mM MgCl2 was pumped through the probe at a constant rate of 1 μl/min. After a washout period of 90 minutes, dialysate samples were collected at 20-minute intervals. For peptide infusion, M40 was dissolved in artificial cerebrospinal fluid (ACSF) to a final concentration of 10 μM, whereas TM peptides were dissolved in 0.1% DMSO in ACSF to a final concentration of 60 μM. All peptides were injected with a 1-μl syringe (Hamilton) driven by an infusion pump and coupled with silica tubing (73-μm inner diameter; Polymicro Technologies) to the microdialysis probe infusion cannula (dead volume, 40 nl), which was primed with ACSF and plugged during implantation. All peptides were delivered at a rate of 16.6 nl/min. Some animals received an i.p. dose of either morphine or methadone (1 mg/kg). At the end of the experiment, rats were given an overdose of Equithesin, the brains were extracted and fixed in formaldehyde, and probe placement was verified using cresyl violet staining. Dopamine content was measured by HPLC coupled with a coulometric detector (5200a Coulochem III; ESA). Statistical analysis was performed with GraphPad Prism 7.
[18F]FDG PET imaging.
The rats (same strain, sex, and age as those used in the microdialysis experiments) were fasted overnight. The next day, the rats received an i.p. injection of saline (1 ml/kg) and were placed in their home cages. Thirty minutes later, the animals were i.p. injected with [18F]FDG (Cardinal Health) and placed back into their home cages. After 30 minutes, the rats were anesthetized with 1.5% isoflurane, placed on a custom-made bed of a NanoScan small animal PET/CT scanner (Mediso Medical Imaging Systems), and scanned for 20 minutes according to a static acquisition protocol. A CT scan was acquired at the end of the PET scan, and the rats were returned to their home cage. Two days later, the animals were fasted overnight and the next day received an i.p. injection of morphine (1 mg/kg) or methadone (1 mg/kg), after which the FDG-PET procedure was conducted as described above. In all cases, the PET data were reconstructed using the NanoScan’s built-in algorithm (Teratomo-3D), correcting for attenuation and radioactive decay with a voxel size of 0.4 mm. Images were coregistered to an MRI template using PMOD software (PMOD Technologies) and then analyzed using MATLAB R2016 (MathWorks) and SPM12 (University College London). Voxel-based repeated-measures Student’s t tests were performed, and the resulting parametric images were filtered for statistically significant (P < 0.05) clusters larger than 100 contiguous voxels. Additionally, VOI values corresponding to the frontal cortex, dorsal striatum, and basal forebrain were drawn using PMOD. The VOI values (kBq/cc) were extracted, SUVR were calculated using the whole brain as a reference region, and statistical analysis was performed with GraphPad Prism 7.
Clinical assessment of methadone-associated “high” and symptoms of craving and withdrawal.
For the RLS patient population, patients treated with methadone or other MOR agonists were first confirmed not to have a prior history or diagnosis of substance use disorder and were largely opioid naive when treatment commenced. Additionally, the treatment course for RLS involves low-to-moderate doses of methadone, with the median dose being 10 mg/d (p.o.), or other opioids. On the other hand, patients with OUD treated with methadone often present with a long history of exposure to opioids (and thus probably have a tolerance to narcotics), and the usual treatment course involves daily treatment with a much higher methadone dose of approximately 90 mg/d (also p.o.). A commonality between these 2 very different clinical studies is that for both, albeit with different methods, participants give subjective responses regarding whether they feel a “high” from the methadone — a response that can be assessed quantitatively. The National RLS Opioid Registry is a multisite registry of patients who are prescribed opioid drugs for the treatment of symptoms of RLS. Data for 226 patients were included in this report; 122 of these patients were prescribed methadone, and the other 104 were prescribed other MOR agonists, which included morphine, codeine, oxycodone, hydrocodone, and hydromorphone. As part of the battery of assessments, participants were asked, “In an average week, do you ever feel high from the opioid you are taking; for example, feelings of floating, warmth, intense relaxation, or giddiness?”
The second population of individuals represented in this study receive daily methadone as part of a medication-assisted treatment regimen for OUD. As participants in a 3-month clinical trial designed to test the efficacy of a behavioral intervention, these patients respond to a series of questionnaires that allowed us to assess the potential for methadone to produce feelings of a “high.” In the first subset of patients, we measured responses on the SOWS, a 16-item self-report instrument designed to assess common subjective symptoms of craving and withdrawal (65). Additionally, we assessed the participants’ responses to a question included in The Methadone Side Effects Checklist (66) regarding the perceived severity of methadone-associated symptoms. Symptom severity in this checklist is rated on a 5-point scale, with 1 equaling not at all severe to 5 equaling very severe. One of these questions asks the patient to identify how severe the symptom of feeling “high” or “loaded” on methadone has been in the past week.
An additional questionnaire given in the context of the OUD clinical trial is a tool that allows for comprehensive assessment of substance use history and treatment, environmental and psychosocial risk factors, and recent use of 34 commonly used licit and illicit drugs. Self-reporting of off-label use and initial motivation to use drugs are assessed individually for all 34 drugs, 5 of which are MOR agonists: heroin, prescription opioids/narcotics (e.g., Vicodin, OxyContin, Percocet, Oxy, Percs, etc.), methadone, prescription fentanyl (e.g., Actiq, Fentora, Duragesic, etc.), and street fentanyl (distinguished from prescription fentanyl, given the differences in mode of access: prescription fentanyl from a medicine cabinet versus fentanyl from a street dealer).
Statistics.
An unpaired t test or a nonparametric Mann-Whitney U test was used when comparing results of 2 independently treated groups of subjects (cells, rats, or patients). A paired t test or a nonparametric Wilcoxon matched-pairs test was used when comparing results for the same subjects before and after a specific treatment. Mann-Whitney U and Wilcoxon tests were applied when data did not show a Gaussian distribution (Shapiro-Wilk normality test P > 0.05). A 1-way ANOVA with Tukey’s or Dunnett’s multiple comparisons test was used when comparing more than 2 independently treated groups of subjects. The Dunnett’s test was preferred when comparing statistical differences for each of a number of differently treated groups with a single control group. A χ2 test was used to analyze categorical clinical data from patients with RLS or OUD. All statistical analyses between 2 groups of noncategorical data were 2-tailed, and, for all the analyses, the level of statistical significance was set at a P value of less than 0.05.
Study approval.
All animals used in the study were maintained in accordance with the NIH’s Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011), and the animal research conducted to perform this study was reviewed and approved by the NIDA Intramural Research Program Animal Care and Use Committee (protocol nos. 18-MTMD-13 and 18-NRB-43). All procedures using rodents were also approved by the Catalan Ethical Committee for Animal Use (CEAA/DMAH 4049 and 5664). Clinical data were obtained from The National RLS Opioid Registry (a multisite registry of patients who are prescribed opioid drugs for the treatment of symptoms of RLS) and from patients diagnosed with OUD who were enrolled in an urban medication-assisted treatment program and were participants in a 3-month clinical trial designed to test the efficacy of a behavioral intervention (ClinicalTrials.gov identifier: NCT02941809).
Author contributions
SF and AMB designed the project and wrote the manuscript, with input from all authors. EDW, ADG, AEJ, KCR, VC, AHN, JWW, MM, EW, NDV, AMB, and SF designed or supervised the experiments and analyzed the clinical data. NSC, CQ, JB, AB, XG, WR, SL, E. Moreno, and VCA performed experiments. NSC, CQ, JB, AB, TOC, JP, ASB, E. Massey, XG, WR, E. Moreno, MW, and VCA analyzed data.
Supplementary Material
Acknowledgments
This study was supported by intramural funds from the NIDA and by grants from the Restless Legs Foundation; the Foundation for Science of the Therapeutic Experience; the University of Maryland MPowering the State Opioid Use Disorder initiative; the “Ministerio de Economía y Competitividad” with FEDER funds (SAF2017-87629-R); and the “Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas” (CB06/05/0064).
Version 1. 03/26/2019
In-Press Preview
Version 2. 05/28/2019
Electronic publication
Version 3. 07/01/2019
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(7):2730–2744.https://doi.org/10.1172/JCI126912.
See the related Commentary at Opioid–galanin receptor heteromers differentiate the dopaminergic effects of morphine and methadone.
Contributor Information
Ning-Sheng Cai, Email: ncai@intra.nida.nih.gov.
César Quiroz, Email: cesar.quiroz-molina@nih.gov.
Jordi Bonaventura, Email: jordi.bonaventura@nih.gov.
Alessandro Bonifazi, Email: alessandro.bonifazi2@nih.gov.
Thomas O. Cole, Email: ThCole@som.umaryland.edu.
Julia Purks, Email: JPURKS@mgh.harvard.edu.
Amy S. Billing, Email: billing@umd.edu.
Ebonie Massey, Email: ebonie711@live.com.
Michael Wagner, Email: Mike@cesar.umd.edu.
Eric D. Wish, Email: ewish@umd.edu.
Xavier Guitart, Email: guitartx@mail.nih.gov.
William Rea, Email: wrea@intra.nida.nih.gov.
Sherry Lam, Email: sherry.lam@nih.gov.
Estefanía Moreno, Email: fifa877@hotmail.com.
Verònica Casadó-Anguera, Email: vcasadoanguera@gmail.com.
Aaron D. Greenblatt, Email: Agreenbl@som.umaryland.edu.
Arthur E. Jacobson, Email: arthurj@nida.nih.gov.
Kenner C. Rice, Email: kennerr@nida.nih.gov.
Vicent Casadó, Email: vcasado@ub.edu.
Amy H. Newman, Email: anewman@intra.nida.nih.gov.
John W. Winkelman, Email: JWWINKELMAN@PARTNERS.ORG.
Michael Michaelides, Email: mike.michaelides@nih.gov.
Eric Weintraub, Email: Eweintra@som.umaryland.edu.
Nora D. Volkow, Email: nvolkow@nida.nih.gov.
Annabelle M. Belcher, Email: Abelcher@som.umaryland.edu.
Sergi Ferré, Email: SFerre@intra.nida.nih.gov.
References
- 1.Volkow ND, Collins FS. The role of science in addressing the opioid crisis. N Engl J Med. 2017;377(4):391–394. doi: 10.1056/NEJMsr1706626. [DOI] [PubMed] [Google Scholar]
- 2.Volkow ND, et al. Medication-assisted therapies--tackling the opioid-overdose epidemic. N Engl J Med. 2014;370(22):2063–2066. doi: 10.1056/NEJMp1402780. [DOI] [PubMed] [Google Scholar]
- 3.Mackie SE, Winkelman JW. Therapeutic utility of opioids for restless legs syndrome. Drugs. 2017;77(12):1337–1344. doi: 10.1007/s40265-017-0773-6. [DOI] [PubMed] [Google Scholar]
- 4.Manglik A, et al. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485(7398):321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manglik A, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537(7619):185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmid CL, et al. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell. 2017;171(5):1165–1175. doi: 10.1016/j.cell.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Viscusi ER, et al. A randomized, phase 2 study investigating TRV130, a biased ligand of the μ-opioid receptor, for the intravenous treatment of acute pain. Pain. 2016;157(1):264–272. doi: 10.1097/j.pain.0000000000000363. [DOI] [PubMed] [Google Scholar]
- 8.Altarifi AA, David B, Muchhala KH, Blough BE, Akbarali H, Negus SS. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J Psychopharmacol (Oxford) 2017;31(6):730–739. doi: 10.1177/0269881116689257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferré S, et al. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev. 2014;66(2):413–434. doi: 10.1124/pr.113.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomes I, et al. G Protein-Coupled Receptor Heteromers. Annu Rev Pharmacol Toxicol. 2016;56:403–425. doi: 10.1146/annurev-pharmtox-011613-135952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang R, et al. Physiology, signaling, and pharmacology of galanin peptides and receptors: three decades of emerging diversity. Pharmacol Rev. 2015;67(1):118–175. doi: 10.1124/pr.112.006536. [DOI] [PubMed] [Google Scholar]
- 12.Hawes JJ, et al. Galanin protects against behavioral and neurochemical correlates of opiate reward. Neuropsychopharmacology. 2008;33(8):1864–1873. doi: 10.1038/sj.npp.1301579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moreno E, et al. Functional μ-opioid-galanin receptor heteromers in the ventral tegmental area. J Neurosci. 2017;37(5):1176–1186. doi: 10.1523/JNEUROSCI.2442-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navarro G, et al. Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. Nat Commun. 2018;9(1):1242. doi: 10.1038/s41467-018-03522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langel Ü. Galanin receptor ligands. Springerplus. 2015;4(Suppl 1):L18. doi: 10.1186/2193-1801-4-S1-L18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yano H, et al. Gs- versus Golf-dependent functional selectivity mediated by the dopamine D(1) receptor. Nat Commun. 2018;9(1):486. doi: 10.1038/s41467-017-02606-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bozarth MA, Wise RA. Intracranial self-administration of morphine into the ventral tegmental area in rats. Life Sci. 1981;28(5):551–555. doi: 10.1016/0024-3205(81)90148-X. [DOI] [PubMed] [Google Scholar]
- 18.Zangen A, Ikemoto S, Zadina JE, Wise RA. Rewarding and psychomotor stimulant effects of endomorphin-1: anteroposterior differences within the ventral tegmental area and lack of effect in nucleus accumbens. J Neurosci. 2002;22(16):7225–7233. doi: 10.1523/JNEUROSCI.22-16-07225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jhou TC, Xu SP, Lee MR, Gallen CL, Ikemoto S. Mapping of reinforcing and analgesic effects of the mu opioid agonist endomorphin-1 in the ventral midbrain of the rat. Psychopharmacology. 2012;224(2):303–312. doi: 10.1007/s00213-012-2753-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leone P, Pocock D, Wise RA. Morphine-dopamine interaction: ventral tegmental morphine increases nucleus accumbens dopamine release. Pharmacol Biochem Behav. 1991;39(2):469–472. doi: 10.1016/0091-3057(91)90210-S. [DOI] [PubMed] [Google Scholar]
- 21.Devine DP, Leone P, Pocock D, Wise RA. Differential involvement of ventral tegmental mu, delta and kappa opioid receptors in modulation of basal mesolimbic dopamine release: in vivo microdialysis studies. J Pharmacol Exp Ther. 1993;266(3):1236–1246. [PubMed] [Google Scholar]
- 22.Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci USA. 1992;89(6):2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chefer VI, Denoroy L, Zapata A, Shippenberg TS. Mu opioid receptor modulation of somatodendritic dopamine overflow: GABAergic and glutamatergic mechanisms. Eur J Neurosci. 2009;30(2):272–278. doi: 10.1111/j.1460-9568.2009.06827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Navarro G, et al. Orexin-corticotropin-releasing factor receptor heteromers in the ventral tegmental area as targets for cocaine. J Neurosci. 2016;35(17):6639–6653. doi: 10.1523/JNEUROSCI.4364-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Enquist J, Ferwerda M, Milan-Lobo L, Whistler JL. Chronic methadone treatment shows a better cost/benefit ratio than chronic morphine in mice. J Pharmacol Exp Ther. 2012;340(2):386–392. doi: 10.1124/jpet.111.187583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32(5):829–839. doi: 10.1016/S0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 27.Drewes AM, et al. Differences between opioids: pharmacological, experimental, clinical and economical perspectives. Br J Clin Pharmacol. 2013;75(1):60–78. doi: 10.1111/j.1365-2125.2012.04317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Celver J, Xu M, Jin W, Lowe J, Chavkin C. Distinct domains of the mu-opioid receptor control uncoupling and internalization. Mol Pharmacol. 2004;65(3):528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- 29.Kalvass JC, Olson ER, Cassidy MP, Selley DE, Pollack GM. Pharmacokinetics and pharmacodynamics of seven opioids in P-glycoprotein-competent mice: assessment of unbound brain EC50,u and correlation of in vitro, preclinical, and clinical data. J Pharmacol Exp Ther. 2007;323(1):346–355. doi: 10.1124/jpet.107.119560. [DOI] [PubMed] [Google Scholar]
- 30.Oldendorf WH, Hyman S, Braun L, Oldendorf SZ. Blood-brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. Science. 1972;178(4064):984–986. doi: 10.1126/science.178.4064.984. [DOI] [PubMed] [Google Scholar]
- 31.Di Chiara G, Imperato A. Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J Pharmacol Exp Ther. 1988;244(3):1067–1080. [PubMed] [Google Scholar]
- 32.Peng XQ, et al. Is slow-onset long-acting monoamine transport blockade to cocaine as methadone is to heroin? Implication for anti-addiction medications. Neuropsychopharmacology. 2010;35(13):2564–2578. doi: 10.1038/npp.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemberg K, Kontinen VK, Viljakka K, Kylänlahti I, Yli-Kauhaluoma J, Kalso E. Morphine, oxycodone, methadone and its enantiomers in different models of nociception in the rat. Anesth Analg. 2006;102(6):1768–1774. doi: 10.1213/01.ane.0000205751.88422.41. [DOI] [PubMed] [Google Scholar]
- 34.Altarifi AA, Rice KC, Negus SS. Effects of μ-opioid receptor agonists in assays of acute pain-stimulated and pain-depressed behavior in male rats: role of μ-agonist efficacy and noxious stimulus intensity. J Pharmacol Exp Ther. 2015;352(2):208–217. doi: 10.1124/jpet.114.219873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zahm DS, Jensen SL, Williams ES, Martin JR., 3rd Direct comparison of projections from the central amygdaloid region and nucleus accumbens shell. Eur J Neurosci. 1999;11(4):1119–1126. doi: 10.1046/j.1460-9568.1999.00524.x. [DOI] [PubMed] [Google Scholar]
- 36.Zahm DS, Parsley KP, Schwartz ZM, Cheng AY. On lateral septum-like characteristics of outputs from the accumbal hedonic “hotspot” of Peciña and Berridge with commentary on the transitional nature of basal forebrain “boundaries”. J Comp Neurol. 2013;521(1):50–68. doi: 10.1002/cne.23157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Woods JH, Schuster CR. Opiates as reinforcing stimuli. In: Thompson T, Pickens R, eds. Stimulus Properties of Drugs. Boston, MA; Springer; 1971:163–175. [Google Scholar]
- 38.Werner TE, Smith SG, Davis WM. A dose-response comparison between methadone and morphine self-administration. Psychopharmacologia. 1976;47(2):209–211. doi: 10.1007/BF00735825. [DOI] [PubMed] [Google Scholar]
- 39.Harrigan SE, Downs DA. Self-administration of heroin, acetylmethadol, morphine, and methadone in rhesus monkeys. Life Sci. 1978;22(7):619–623. doi: 10.1016/0024-3205(78)90342-9. [DOI] [PubMed] [Google Scholar]
- 40.Drevets WC, et al. Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol Psychiatry. 2001;49(2):81–96. doi: 10.1016/S0006-3223(00)01038-6. [DOI] [PubMed] [Google Scholar]
- 41.Volkow ND, et al. Stimulant-induced dopamine increases are markedly blunted in active cocaine abusers. Mol Psychiatry. 2014;19(9):1037–1043. doi: 10.1038/mp.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Judd LL, et al. The effect of methadone on the behavioral and neuroendocrine responses of manic patients. Psychiatry Res. 1982;7(2):163–170. doi: 10.1016/0165-1781(92)90089-L. [DOI] [PubMed] [Google Scholar]
- 43.Juver JP, Figueiredo NV, Barrucand L, Tostes Mde A. Methadone to treat non-oncologic neuropathic pain. Case reports. Rev Bras Anestesiol. 2005;55(4):450–459. doi: 10.1590/s0034-70942005000400010. [DOI] [PubMed] [Google Scholar]
- 44.Jasinski DR, Preston KL. Comparison of intravenously administered methadone, morphine and heroin. Drug Alcohol Depend. 1986;17(4):301–310. doi: 10.1016/0376-8716(86)90079-7. [DOI] [PubMed] [Google Scholar]
- 45.Walsh SL, June HL, Schuh KJ, Preston KL, Bigelow GE, Stitzer ML. Effects of buprenorphine and methadone in methadone-maintained subjects. Psychopharmacology. 1995;119(3):268–276. doi: 10.1007/BF02246290. [DOI] [PubMed] [Google Scholar]
- 46.Cohen MJ, Hanbury R, Stimmel B. Abuse of amitriptyline. JAMA. 1978;240(13):1372–1373. [PubMed] [Google Scholar]
- 47.Davis WR, Johnson BD. Prescription opioid use, misuse, and diversion among street drug users in New York City. Drug Alcohol Depend. 2008;92(1-3):267–276. doi: 10.1016/j.drugalcdep.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greene MH, Brown BS, DuPont RL. Controlling the abuse of illicit methadone in Washington, DC. Arch Gen Psychiatry. 1975;32(2):221–226. doi: 10.1001/archpsyc.1975.01760200085008. [DOI] [PubMed] [Google Scholar]
- 49.Gwin Mitchell S, et al. Uses of diverted methadone and buprenorphine by opioid-addicted individuals in Baltimore, Maryland. Am J Addict. 2009;18(5):346–355. doi: 10.3109/10550490903077820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raynor K, et al. Characterization of the cloned human mu opioid receptor. J Pharmacol Exp Ther. 1995;272(1):423–428. [PubMed] [Google Scholar]
- 51.Yu Y, Zhang L, Yin X, Sun H, Uhl GR, Wang JB. Mu opioid receptor phosphorylation, desensitization, and ligand efficacy. J Biol Chem. 1997;272(46):28869–28874. doi: 10.1074/jbc.272.46.28869. [DOI] [PubMed] [Google Scholar]
- 52.Selley DE, Liu Q, Childers SR. Signal transduction correlates of mu opioid agonist intrinsic efficacy: receptor-stimulated [35S]GTP gamma S binding in mMOR-CHO cells and rat thalamus. J Pharmacol Exp Ther. 1998;285(2):496–505. [PubMed] [Google Scholar]
- 53.Borgland SL, Connor M, Osborne PB, Furness JB, Christie MJ. Opioid agonists have different efficacy profiles for G protein activation, rapid desensitization, and endocytosis of mu-opioid receptors. J Biol Chem. 2003;278(21):18776–18784. doi: 10.1074/jbc.M300525200. [DOI] [PubMed] [Google Scholar]
- 54.Fujita W, Gomes I, Devi LA. Heteromers of μ-δ opioid receptors: new pharmacology and novel therapeutic possibilities. Br J Pharmacol. 2015;172(2):375–387. doi: 10.1111/bph.12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yekkirala AS, Kalyuzhny AE, Portoghese PS. Standard opioid agonists activate heteromeric opioid receptors: evidence for morphine and [d-Ala(2)-MePhe(4)-Glyol(5)]enkephalin as selective μ-δ agonists. ACS Chem Neurosci. 2010;1(2):146–154. doi: 10.1021/cn9000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yekkirala AS, et al. Clinically employed opioid analgesics produce antinociception via μ-δ opioid receptor heteromers in Rhesus monkeys. ACS Chem Neurosci. 2012;3(9):720–727. doi: 10.1021/cn300049m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Larson MD. Mechanism of opioid-induced pupillary effects. Clin Neurophysiol. 2008;119(6):1358–1364. doi: 10.1016/j.clinph.2008.01.106. [DOI] [PubMed] [Google Scholar]
- 58.Liu HX, et al. Receptor subtype-specific pronociceptive and analgesic actions of galanin in the spinal cord: selective actions via GalR1 and GalR2 receptors. Proc Natl Acad Sci U S A. 2001;98(17):9960–9964. doi: 10.1073/pnas.161293598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hua XY, et al. Galanin acts at GalR1 receptors in spinal antinociception: synergy with morphine and AP-5. J Pharmacol Exp Ther. 2004;308(2):574–582. doi: 10.1124/jpet.103.058289. [DOI] [PubMed] [Google Scholar]
- 60.Florén A, et al. Multiple interaction sites of galnon trigger its biological effects. Neuropeptides. 2005;39(6):547–558. doi: 10.1016/j.npep.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 61.Guan XM, Tong Sun K, Kobilka BK. Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. J Biol Chem. 1992;267(31):21995–21998. [PubMed] [Google Scholar]
- 62.Schröder R, et al. Applying label-free dynamic mass redistribution technology to frame signaling of G protein-coupled receptors noninvasively in living cells. Nat Protoc. 2011;6(11):1748–1760. doi: 10.1038/nprot.2011.386. [DOI] [PubMed] [Google Scholar]
- 63.Casadó V, et al. Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: the receptor-dimer cooperativity index. Pharmacol Ther. 2007;116(3):343–354. doi: 10.1016/j.pharmthera.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 64.Urizar E, et al. CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat Chem Biol. 2011;7(9):624–630. doi: 10.1038/nchembio.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Handelsman L, Cochrane KJ, Aronson MJ, Ness R, Rubinstein KJ, Kanof PD. Two new rating scales for opiate withdrawal. Am J Drug Alcohol Abuse. 1987;13(3):293–308. doi: 10.3109/00952998709001515. [DOI] [PubMed] [Google Scholar]
- 66.Longwell B, Kestler RJ, Cox TJ. Side effects in methadone patients: a survey of self-reported complaints. Int J Addict. 1979;14(4):485–494. doi: 10.3109/10826087909054596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.