

Abstract
The sensitivity of Xeroderma pigmentosa (XP) patients to sunlight has spurred the discovery and genetic and biochemical analysis of the eight XP gene products (XPA-XPG plus XPV) responsible for this condition. These studies also have served to elucidate the nucleotide excision repair (NER) process, especially the critical role played by the XPA protein. More recent studies have shown that NER also involves numerous other proteins normally employed in DNA metabolism and cell cycle regulation. Central among these is ataxia telangiectasia and Rad3-related (ATR), a protein kinase involved in intracellular signaling in response to DNA damage, especially replicative and transcription stresses. This review summarizes recent findings on the interplay between ATR as a DNA damage signaling kinase and as a novel ligand for intrinsic cell death proteins to delay damage-induced apoptosis, and on ATR’s regulation of XPA and the NER process for repair of UV-induced DNA adducts. ATR’s regulatory role in the cytosolic-to-nuclear translocation of XPA will be discussed. In addition, recent findings elucidating a non-NER role for XPA in DNA metabolism and genome stabilization at ds-ssDNA junctions, as exemplified in prematurely aging progeroid cells, also will be reviewed.
Introduction
Individuals with mutations in Xeroderma pigmentosa (XP) genes are especially sensitive to the ultraviolet (UV) rays (180–315 nm) in sunlight 1, 2. These individuals accumulate DNA damage in their skin cells after solar irradiation, primarily as a cyclobutane pyrimidine dimer (CPD) and, to a lesser extent, as a (6–4) photoproduct ((6–4) PP). Normally, these intrastrand cross-links of adjacent pyrimidine bases are removed from the DNA by nucleotide excision repair (NER) 1, 3–5. In addition to other repair factors, seven XP gene products are involved in the NER process: Xeroderma pigmentosa complementation groups A through G (XPA – XPG). Mutations in any of these XP gene products reduces the efficiency of this repair process with XPA and XPC mutations being the most frequent 6 and XPA deficiency showing the highest sensitivity to UV 7. If adducts persist they may be bypassed by error-prone translesion synthesis using DNA polymerase eta (Pol η), a product of the XPV (polH) gene 8. The structure and mutational features, plus post-translational modifications of these XP proteins have been reviewed recently by Feltes and Bonatto 9. XPA mutation is the most severe XP deficiency is since this protein is required in both the global genomic NER (GG-NER) and the transcription- coupled NER (TC-NER) sub-pathways of nucleotide excision repair 10–14,15. XPC mutations, though relatively frequent, are less severe since this protein is primarily involved in GG-NER 10,16. Though not an XP protein, the DNA damage checkpoint protein ataxia telangiectasia and Rad3-related (ATR) also is essential for initiation and regulation of the NER process 17, 18. Thus, this review will focus on new information from the last decade on the biochemical roles and cellular mechanisms of XPA and ATR in the nucleotide excision repair process and cell death, and discuss recent findings on possible non-NER functions of XPA in both the nucleus and in the cytoplasm.
ATR signaling mediates the cellular response to DNA damaged induced by ultraviolet radiation
The presence of UV-induced CPD and (6–4) PP adducts in mammalian nuclear DNA generates a cascade of events as part of the DNA damage response (DDR). Generally, these helix-distorting, replication- and transcription-blocking DNA adducts induce activation of the DNA repair process and arrest the cell cycle to allow for repair of the damaged DNA. ATR, a key regulator of these processes, is a member of the phosphatidylinositol 3-kinase (PI3K) family. The PI3K family of protein kinases also includes the other stress-responsive protein kinases ataxia telangiectasia mutated (ATM), DNA-dependent protein kinase (DNA-PK) and mammalian target of rapamycin (mTOR) 19, 20. Although it functions in multiple DDR processes 21 ATR is the primary regulator of the nucleotide excision repair pathway due to its ability to detect the replicative and transcriptional stresses caused by UV-induced damage and other bulky DNA adducts resulting from chemical toxins and some chemotherapeutic agents 22–25.
Induction of CPDs and (6–4) PPs in DNA generates obstacles to DNA replication and transcription. The resulting replicative and transcriptional stresses stall DNA polymerization during replication and pol II progression in RNA synthesis11, 12, respectively, leading to an accumulation of stretches of single-strand DNA (ssDNA), which become coated with the ssDNA-binding replication protein A (RPA) 26. ATR in complex with its nuclear binding partner ATR-interacting protein (ATRIP) binds to this RPA-coated ssDNA via an ATRIP-RPA interaction. ATRIP also serves to activate the checkpoint kinase activity of ATR 4, 27–30. Activated ATR kinase phosphorylates many downstream mediators/effectors which include checkpoint kinase 1 (Chk1), A-kinase-anchoring protein 12 (AKAP12), p38/mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 (MK2), the tumor suppressor protein p53, ATRIP and XPA 27, 31–33. Phosphorylation activates these downstream proteins resulting in arrest of cell cycle progression, activation of DNA repair and, in cases of severe damage, apoptotic cell death 22, 34, 35. ATR is an essential gene for the initiation and regulation of NER and for genome maintenance 17, 36, 37.
Historically, ATR has been described as a necessary protein kinase which functions in the cell nucleus to regulate DNA replication and various responses to DNA damage and cellular stress 38, 39. Possible non-nuclear roles for ATR have received little attention. However, a recent study described an anti-apoptotic, cytoplasmic role for ATR 40, 41. It was demonstrated that in mammals a small fraction of cellular ATR normally exists in the cytoplasm (cytoATR) and that, in response to DNA damaging agents, the amount of this cytoATR increases and changes conformation, resulting in a slower-migrating, higher electrophoretic band (ATR-H) as compared with the faster-migrating, lower electrophoretic band (ATR-L). The most efficient induction of ATR-H formation was by UV irradiation, though it also was induced by camptothecin and hydroxyurea, agents which cause DNA double-strand breaks (DSBs). Interestingly, the increase in cytoATR appears to result from nuclear export and not from new protein biosynthesis 41. This nuclear export of ATR-L and its conversion to cytoplasmic ATR-H by UV irradiation was observed in normal human fibroblasts, transformed skin keratinocytes, multiple human cancer cell lines, and in transformed mouse embryonic fibroblasts 40.
It was found that the ATR-L is a prolyl trans-isomer of cytoplasmic ATR while ATR-H is the cis-isomer 40. The formation of cytoplasmic ATR-L (trans-ATR) from ATR-H (cis-ATR) is mediated by peptidylprolyl cis/trans isomerase NIMA-interacting 1 (Pin1) 40; this enzyme is a critical regulator of many biological processes in both normal and diseased cells 42–48. Since ATR is naturally more stable in its cis-isomeric form, newly-synthesized ATR is in the ATR-H isoform but is quickly converted to the ATR-L isoform by Pin1 isomerization of the phospho- Ser428--Pro429 site of the ATR protein 40. This isomerization converts Pro429 from the cis- (ATR-H) to the trans-isoform (ATR-L). Surprisingly, this conformational change of only one out of 2,644 amino acids is sufficient to reduce the electrophoretic mobility of the ATR protein in 3–8% gradient SDS-polyacrylamide gels, similar to adding ~10 kilodaltons, to generate a clearly distinguishable higher band (ATR-H). The mechanism of this protective response stems from UV-induced changes in the phosphorylation status of the Ser428Pro429 site in ATR and the Ser71 residue in Pin1. UV irradiation induces DAPK1 to phosphorylate Pin1 at Ser71, thus inactivating the isomerase activity 49, 50. The UV irradiation also induces a dephosphorylation of the phospho-Ser428-Pro429 site in ATR, rendering it a non-recognizable Pin1 site 40. Together, these changes in phosphorylation status allow cytoATR to assume the cis isoform, ATR-H. Although the details of the UV-induced changes in DAPK1 kinase and the unknown phosphatase activities remain to be elucidated these observations reveal a very sensitive cellular sensor for ultraviolet damage and ATR isomeric conversion.
Upon UV irradiation-induced DNA damage ATR initiates the nuclear NER process to repair the genome. To allow time for completion of this repair the cell needs to stall two processes: cell cycle progression, especially through S phase, and the initiation of damage- induced cell death. Cell cycle arrest is needed to allow time for DNA repair and, thus, prevent the introduction of mutations by replication through unrepaired CPD and (6–4) PP damage sites. A classic feature of ATR in response to UV damage is its phosphorylation of Chk1 kinase, which then phosphorylates other proteins to arrest cell cycle progression 51. UV-induced damage also can activate the intrinsic cell death pathway through the release of mitochondrial cytochrome C into the cytosol which activates caspase cleavage and eventual apoptosis 52. But how does ATR stall the onset of apoptotic cell death to allow sufficient time for cell recovery by repair of the CPD and (6–4) PP damage? The answer lies in the interaction of cytosolic ATR-H with the proapoptotic protein tBid (truncated BH3 interacting-domain death agonist) as described by Hilton et al. 40. In response to damage Bid promotes polymerization of proapoptotic proteins Bax (bcl-2-associated X) and Bak (bcl-2 homologous antagonist-killer) at the mitochondrial surface, which induces cytochrome C release leading to apoptotic cell death 52. Hilton et al. surprisingly found that ATR contains a BH3-like domain which allows it to function like a prosurvival Bcl-2 family protein. In the nucleus, ATR remains in the form of ATR-L, regardless of UV, whose BH3 domain appears to be masked in a folded N-terminal region of the trans- isoform protein; however, the N-terminus is unfolded in the cytosolic cis-isoform which exposes this BH3 domain, allowing ATR-H to bind to and sequester tBid protein, thus delaying initiation of the intrinsic cell death pathway 40. Figure 1 illustrates how the cis- and trans-isoforms may affect these changes in the accessibility of the BH3 domain in ATR-L vs. ATR-H isoforms, and how the ATR-L form is necessary for the regulation of XPA import and NER efficiency.
Figure 1. Possible alternative folding conformations of ATR-H vs. ATR-L.
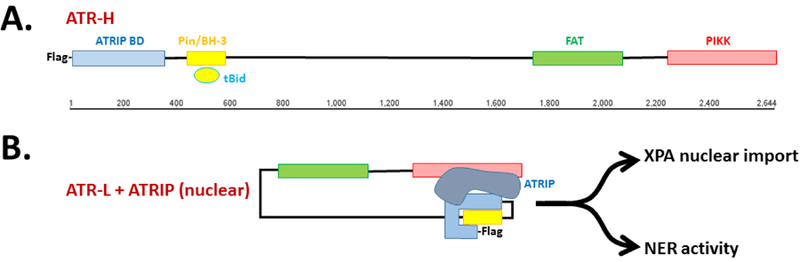
There currently are no 3-demensional structures described for ATR. The diagrammatic representations presented here are based on the predictions of Hilton et al. for the N-terminal regions of ATR-H vs. ATR-L 40. The N-terminal region of ATR-H, which has the cis-Pro429 isomer and an unphosphorylated Ser428, is accessible to both tBid binding and to Flag antibody binding. Thus, ATR-H is presented in an open conformation. In ATR-L, which contains a phosphorylated Ser428 and a trans-Pro429, the BH3 domain is inaccessible to tBid binding as is the Flag tag 40. Thus, ATR-L is drawn with a folded N-terminal region. The N-terminus of ATR contains the ATRIP binding site; binding of ATRIP leads to activation of the ATR kinase via interaction with the C- terminal PIKK region 27–30. Although speculative, the lower two diagrams of ATR-L illustrate this folding of the N-terminal region onto the C-terminal region, perhaps mediated by ATRIP binding.
Nuclear ATR is well known for its association with ATRIP, a necessary interaction which activates the kinase activity of ATR in addition to localizing it to the RPA-coated ssDNA at damage sites 4, 27–30. This kinase activity is essential for ATR’s activation of downstream proteins during the DDR. In contrast, Hilton et al. found that cytoATR is free of ATRIP, which remains sequestered in the nucleus after UV irradiation. Also, the anti-apoptotic function of mitochondrial ATR-H is independent of its checkpoint kinase activity 40, 41. Thus, the regulated cis- vs. trans-isoform switching between ATR-H and ATR-L allows distinct prosurvival functions of ATR in the cytoplasm versus those in the nucleus in response to UV irradiation. Particularly, the cytoplasmic ATR-H prevents premature cell death at mitochondria. This coordination of the cytoplasmic antiapoptotic and the nuclear cell cycle arrest/DNA repair roles provides time for damage repair before any decision on programmed cell death needs to be made. Note that, once formed, ATR-H reaches a maximum within 2 hours but persists in the cytoplasm for over 8 hours, sufficient time for most NER-competent cells to repair all the (6–4) PP adducts and most, if not all, of the CPD adducts 40, 53,41. Thus, this slow re-isomerization of ATR-H to ATR-L may serve as an internal timer of repair efficiency and death.
The novel finding of the cytoplasmic role of ATR as an anti-apoptotic protein at mitochondria highlights that much remains to be discovered about the signaling molecules involved in the DNA damage responses. These observations support previous findings that prolyl isomerization of a single residue in a large protein may have pleotropic effects on a protein’s structure and function 48, 54. Also, these cytoplasmic prosurvival functions are not only novel for ATR since ATM also displays similar stress functions at peroxisomes in response to increased levels of reactive oxygen species 55–59 and at mitochondria in response to DNA damage 60, 61.
ATR-XPA interactions are necessary for the nuclear import of XPA and for efficient nucleotide excision repair
The data sheets accompanying nearly all commercial anti-XPA antibodies recommended for immunofluorescence studies by the suppliers indicate that XPA is a protein located in the nucleus only. This discrepancy stems from the early studies in which formalin (2– 4% para-formaldehyde) was used for cell fixation 62, 63. More recent immunofluorescence studies of XPA’s subcellular distribution confirmed that in para-formaldehyde-fixed cells the endogenous protein was observed to be nuclear 64–66. However, biochemical fractionation of millions of cells into nuclear vs. cytoplasmic fractions revealed that XPA occurs predominantly in the cytoplasm of normal mammalian cells and that it is translocated to the nucleus in response to DNA damage, especially from UV irradiation 67–70. These biochemical findings were confirmed by immunofluorescence observations of methanol-fixed cells 67–70. We have observed that with either fixative the anti-XPA antibodies revealed XPA in the nucleus, but antibody detection of the cytosolic XPA occurred only in cells fixed with cold methanol. Methanol fixation extracts lipids, dehydrates and permeablizes cells causing proteins to denature and precipitate onto the cellular architecture. In contrast, para-formaldehyde fixation cross-links proteins and other macromolecules in place 71. A possible explanation, then, for the reported differences in the subcellular localization of XPA with these two methods is that methanol fixation disrupts the cloaking interaction between XPA and an as yet undescribed cytosolic XPA binding protein (cXBP) which sequesters XPA in the cytoplasm; the methanol fixation with denaturation then exposes XPA’s antigenic site; in contrast, para-formaldehyde fixation locks this XPA-cXBP complex in place, thus masking the XPA epitopes in the cytosol. UV irradiation induces a disruption of this cytosolic XPA-cXBP complex, releasing XPA for nuclear import and detection in nuclei of cells fixed with methanol or para-formaldehyde. This also could be true for other so- called nuclear proteins.
Wu et al. reported that ATR regulated XPA nuclear import in response to UV radiation 67,72. More recent studies by Li et al. have revealed further important details of the cytosol-nuclear translocation of XPA. The tumor suppressor protein p53 is a major downstream effector molecule and phosphorylation substrate in the ATR-mediated DDR. In support of earlier observations 53,67, Li et al. demonstrated that the nuclear import of XPA in response to UV irradiation or cisplatin treatment is ATR-dependent in normal fibroblasts and in cancer cells that are p53 proficient; XPA import also is dependent on the transcriptional activity of p53 in these cells 69, 70. In addition, this dependence on ATR checkpoint activity is cell-cycle phase dependent, occurring only during the S phase 69. Most XPA remained sequestered in the cytosol in the G1 phase even after UV treatment; in contrast, in G2-phase cells the nucleus contained the majority of the XPA molecules irrespective of UV irradiation. Consistently, NER recently was found to recruit ATR to the UV-damage sites and to activate ATR in G1-phase but not in S-phase 73–76. Regulation of S-phase cytosolic XPA translocation into the nucleus by ATR is consistent with previous findings that the peak activity of this checkpoint kinase occurs in S phase as part of normal DNA replication and also in response to DNA damage 34, 77. Li et al. observed that the maximum UV-induced phosphorylation of Ser15 of p53 occurred in S phase and that the NER removal of CPD adducts also was most efficient in S phase 69. Recall that ATR binds to XPA via the Lys188 and Ser196 residues in its HTH motif containing 53 and that these residues are important for the efficient repair of CPD adducts.
Interestingly, the p53 status of cells significantly influences the role of ATR in regulating DNA repair after UV or cisplatin damage. Although efficient NER removal of the damage was dependent on ATR kinase activity in p53-proficient (p53+/+) cells the repair process seemed to be ATR-independent in p53-deficient (p53−/−) cells 69, 70, 77. Consistently, nuclear import of cytosolic XPA is dependent on p53 transcriptional activity in p53+/+ cells and occurs much slower in p53−/− cells, but the import stills occurs 70. Thus, damage-induced ATR activation of the p53 tumor suppressor protein appears to be a primary but not the sole mediator of XPA nuclear import in p53+/+ vs. p53−/− cells in S phase. The cell cycle checkpoint kinases ATM, Chk1 and MK2 appear not to have a role in XPA nuclear import in p53+/+ nor p53−/− cells 69, 70.
The phosphorylation of XPA by ATR is the essential for the NER function of XPA 72. Shell et al. found that ATR binds XPA via a specific helix-turn-helix motif in the minimal DNA- binding domain (DBD) and that this XPA motif contains an ATR phosphorylation site (Ser196) 53. In addition, disruption of this phosphorylation site in XPA with a Ser196Ala mutation significantly reduced the repair efficiency of CPDs but not the repair of (6–4) PPs. The nucleotide excision repair of (6–4) PPs is generally much more efficient than the repair of CPDs 78, 79 and the above finding indicates that ATR’s phosphorylation of Ser196 in XPA is mechanistically important in the repair of the more prevalent CPDs which represent persistent UV damage. The phosphorylation of Ser196 in XPA by ATR appears to stabilize XPA against HERC2-mediated ubiquitinylation and degradation 80.
Shell’s structure-function studies also found that the Lys188 residue, which is nearby in the same helical DBD of XPA, was critical since a Lys188Ala mutation disrupted the ATR-XPA interaction, thus significantly reducing DNA repair efficiency 81. Moreover, the normal UV- induced nuclear translocation of cytosolic XPA was lost with the Lys188Ala mutation. However, the Ser196Ala mutation had no effect on XPA’s nuclear translocation. The targeting of XPA to the nucleus occurs via its nuclear localization sequence (NLS) which contains basic residues located at positons 30–34 of the 273 amino acid protein 62, 63, 82, 68. This raises the interesting and important question of how XPA is normally held in the cytoplasm if it contains a NLS sequence and its normal NER function is in the nucleus. One possibility is that XPA is sequestered in the cytosol in normal cells via association with cXBP, from which it is released for nuclear import after a DNA damaging event such as UV irradiation. Perhaps the stability of the XPA-cXBP complex is disrupted by the phosphorylation of XPA at Ser196 and/or by a post-translational modification of the necessary Lys188 (i.e., acetylation). Note that highly over-expressed XPA mutants lacking the NLS site can be detected in the cytoplasm by immunofluorescence microscopy in para-formaldehyde fixed cells 63, indicating that cXBP may occur is physiologically limiting amounts . This as yet uninvestigated cytosolic XPA sequestration and release could be one of the dynamic components of the UV-induced damage response. Also, note that AKAP12 is normally a cytosolic protein associated with protein kinase A (PKA) but becomes phosphorylated by cytosolic ATR after UV irradiation and then is transported into the nucleus in association with ATR 31.
It is obvious that the DNA damage-induced import of cytosolic XPA into the nucleus is a highly regulated process. Mechanistic features of this import process have been resolved in additional studies by Li et al. 68. It was shown that the NLS in the N-terminal region of XPA was required for nuclear localization. In addition, siRNA knockdown revealed that nucleo- cytoplasmic transport proteins importin-α4 and -α7 were required for XPA nuclear import, but not the other importin-α proteins. Co-immunoprecipitation studies demonstrated that importin-α4 and importin-α7 mediate this nuclear import by direct physical interactions with XPA. However, these two carrier proteins appear to serve different functions during the cell cycle. Importin-α4 transport of XPA was activated by UV radiation and required functional ATR kinase activity, consistent with importin-α4 being responsible for the nuclear import of XPA during the S-phase DNA damage response. In contrast, importin-α7 functioned independent of DNA damage and ATR kinase activation, perhaps reflecting the observed nuclear import of XPA in the G2 phase irrespective of UV exposure 69. These features of XPA cytosolic localization and cell cycle- dependent nuclear import in response to UV irradiation are diagrammatically summarized in Figure 2.
Figure 2. Normal and UV-induced redistribution during progression through the cell cycle in p53-competent human cells.
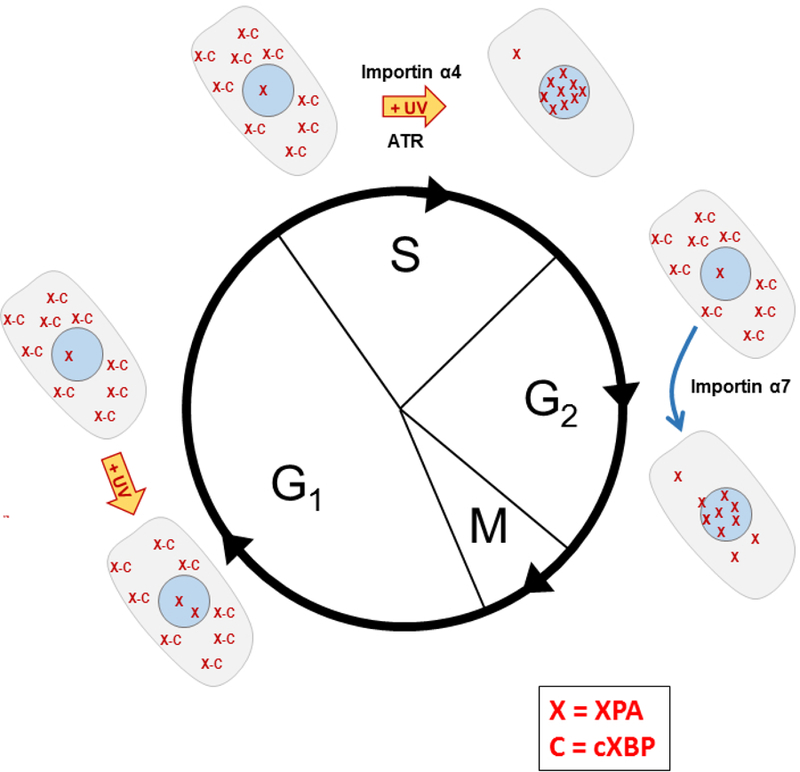
This model is based on the studies of Li et al. 68–70 In non-damaged cells in the G1 phase XPA (X) is mostly located in the cytosol, likely bound to cXBP (C), a hypothetical cytosolic XPA sequestration protein. Exposure of G1 cells to UV does not change this distribution. Likewise, in S phase cells XPA is mostly cytosolic; however, UV exposure induces a release of XPA from cXBP and a translocation of XPA into the nucleus. This XPA nuclear translocation in S phase requires the importin α4 transport protein and is ATR kinase- and p53-dependent in p53-competent cells. XPA is primarily located in the nucleus in G2 phase cells, transported there via importin α7 in a process independent of UV exposure. The XPA redistributes to the cytosol during the M-G1 phase transition and reassociates with cXBP.
Nuclear import of proteins requires a GTPase to coordinate protein-protein interactions 83–85. XAB1 was observed in a yeast two-hybrid system to be an XPA-binding protein with GTPase activity 86. However, Li et al. demonstrated that XAB1 is not the GTPase involved in XPA nuclear import 68. Also, questions remain on how XPA is released from cXBP in the cytosolic sequestration complex to bind to the importin-α4 in S phase cells exposed to UV. These authors demonstrated that there was an increase in the XPA available for importin-α4 binding within 30 minutes after UV exposure; however, the mechanistic details of the cytosolic DDR remain to be resolved. In addition, how the cytosolic XPA sequestered by cXBP during G1 and S phases is released in non-irradiated cells for importin-α7-mediated nuclear import in the G2 phase also remains to be elucidated.
Does XPA have a cytosolic function outside of nucleotide excision repair?
Why is the XPA protein localized in the cytosol of normal (non-DNA damaged) cells during G1 and S phases of the cell cycle, but not in the G2 phase? Does its complex with cXBP provide a cytosolic function in G1 and S phases, and/or is it sequestered there to prevent interference with ongoing nuclear processes?
In addition to high dermatological sensitivity to sunlight XP patients, especially those with an XPA deficiency, often suffer from neurological deficiencies and an early-aging phenotype 2, likely due to non-NER mechanisms as exogenous, genotoxin-induced bulky adducts would not be a concern. XPA interacts with a variety of XP and other proteins during the DNA repair process in the nucleus 68–70, 87, 88,82, 89, but interactions with cytosolic proteins have not been described. Are these non-NER features of XPA deficiency related to XPA’s cytosolic location, especially in the G0/G1 phase status typical of neurons, cardiomyocytes or other differentiated cell types? Other than the descriptions of its UV-induced cytoplasmic-to-nuclear translocation 53, 67–70, possible XPA binding partners and/or functional roles in the cytosol have received little to no attention. One possibility might be that cXBP, the proposed cytoplasmic sequestration factor to which XPA is bound in normal G1 and S phase cells, influences abnormal, dis-regulatory activity in XPA−/− cells leading to deleterious metabolic events. Using a bioinformatics analysis Fang et al. observed that the XPA−/− phenotype includes neurological features similar to mitochondrial diseases, and results in abnormal mitochondrial energy metabolism, even though cytoplasmic XPA in XPA-proficient cells was absent from the mitochondrial matrix 90. They also reported increased poly(ADP-ribose) polymerase 1 (PARP1) activity, resulting in higher parsylation of cellular proteins resulting in NAD+ depletion, thus reducing mitochondrial energy generation. They observed that the reduced level of NAD+ downregulated SIRT1, a NAD+- dependent deacetylase involved in regulating mitochondrial homeostasis and XPA repair activity 91–93. Fang et al. assumed that the PARP1 was activated in XPA−/− cancer cells and neurons by an increased level of basal nuclear DNA damage 90. However, the presence of a basal level and the type of DNA damage occurring in the XPA–/– cells was not demonstrated. In addition, as reviewed by Weaver and Yang 94, PARP1 activation can be induced by stress responses other than DNA damage, including the ERK-1 95, 96 and Notch/HES-1 97 signaling pathways and intracellular calcium overload 98. In addition, XPA and PARP1 appear to have regulatory interactions which would be upset in the XPA−/− cells99. These studies and their interpretation are complicated further by the observed cell-type specificity of PARP1 activation 96–98. Resolution of these ambiguities rest, in part, on an elucidation of the cXBP cytosolic binding partner of XPA which sequesters this NER protein in the cytosol in normal G1 and S phases of the cycling cell and in the G1/G0 states of the non-cycling, highly differentiated cells. There are multiple possibilities since XPA has been described as a highly flexible scaffold protein capable of interacting with numerous proteins simultaneously 82, 89. Future studies also are needed to elucidate XPA’s possible cytosolic binding partner(s) in the G1 and S phase cells, their biochemical properties, and possible normal function after XPA dissociation in G2 and M phases.
Non-NER functions of XPA in the nucleus.
XPA functions as an essential component of the DNA damage repair complexes for both GG-NER and TC-NER. In addition, XPA binds to ds-ssDNA junctions with a significantly higher affinity (Kd = 49.1 ± 5.1 nM) 100 than it’s specific binding to bulky DNA lesions (Kd = 200 nM) 101. This suggests that, in addition to DNA damage recognition/verification, XPA may bind independently to and stabilize such ds-ssDNA junctions during the NER process and/or during other types of DNA metabolism. Hilton et al. recently demonstrated that in binding to ds-ssDNA junctions XPA employs a larger DNA-binding domain 102 than was previously described for repair substrates 103, 104.
How might this essential biochemical affinity for ds-ssDNA junctions relate to XPA’s cytoplasmic restriction during S phase and XPA’s performance of non-NER functions in cells? Hutchison-Gilford progeria syndrome (HGPS) patients suffer from a variety of laminopathy ailments due to a sporadic deficiency in the proteolytic processing of the precursor form of lamin A into the mature protein. The aberrantly processed protein produced is called progerin, a truncated form of lamin A with a hydrophobic farnesylated C-terminal 105–112. HGPS cells with progerin accumulation exhibit a reduced replicative lifespan plus a deficiency in the repair of endogenous, laminopathy-induced DNA DSBs, which increase with age 113, 114. These DNA metabolism deficiencies also correlate with a proteolytic truncation of replication factor C1 (RFC1) 115 and a sequestration of proliferating cell nuclear antigen (PCNA) in a complex with progerin (Hilton et al., private communication). Both the intact RFC complex and PCNA are essential replication factors and are needed for loading the replicative polymerase onto DNA 116, 117, thus accounting for the reduced replicative lifespan of HGPS cells 113, 118. Interestingly, cellular nucleotide excision repair protein XPA misaccumulates at the DSB sites consisting of ds-ssDNA junctions even though XPA never has had a documented role in DSB repair, causing these breaks to become progressively devoid of DSB repair proteins 114. Those DSBs appear to be generated from stalled and collapsed replication forks in HGPS. Depletion of XPA in these aging HGPS cells significantly relieves the deficiency in DSB repair, possibly by shifting the binding of available free PCNA to these XPA-free junctions (Hilton et al., private communication). These observations suggest that as HGPS cells age progerin accumulates and sequesters PCNA, resulting in collapsed replication forks with DSBs and ds-ssDNA junctions to which XPA binds. Although this XPA binding may limit access to DNA DSBs repair proteins, it appears that the binding could stabilize the forks and prevent the HGPS cells from progerin-induced apoptosis (Hilton et al., private communication). .
These potential non-NER roles allow for interesting speculation concerning XPA’s pleiotropic functions and those of it’s as-yet undescribed binding partners (i.e., cXBP) and will lead to many interesting experimental studies.
Conclusions
XPA is indispensable for both transcription-coupled repair and global genomic repair, and, thus, has a central and critical role in the NER process. Recent studies have revealed that XPA is kept in the cytosol in non-UV irradiated cells where it may be sequestered by a cytosolic XPA-binding protein, here termed cXBP. This subcellular distribution can be easily detected by immunofluorescence microscopy if the cells are fixed in cold methanol but not in cells fixed with p-formaldehyde. In the S phase UV irradiation induces a translocation of XPA into the nucleus for NER of UV-induced adducts. This S phase nuclear import is facilitated by XPA binding to by the transport protein importin-α4 (Fig. 2). In contrast, cells in G1 phase retain XPA in the cytosol while XPA is mostly located in the nucleus in the G2 phase; both the G1 and G2 phase distributions are largely independent of UV irradiation. Importin-α7 facilitates the G2 phase nuclear import of XPA. The S phase nuclear import of XPA is dependent on the kinase activity of ATR and on the tumor suppressor protein p53, which also is activated by the ATR kinase.
The ATR protein has multiple roles in regulating the NER process. In response to UV damage ATR regulates the NER process via its phosphorylation of numerous cell cycle control and DNA repair proteins. One of these is XPA; its phosphorylation by ATR is required for its essential role in NER of persistent CPD adducts. In addition, ATR kinase activity is required for the cytosolic-to-nuclear translocation of XPA by importin-α4 during S phase, the period when ATR kinase activity is at its highest. In addition to these kinase-dependent DDR nuclear functions a recent study reports an important cytosolic, kinase-independent role for ATR in moderating the intrinsic cell death response induced by UV irradiation. Surprisingly, newly formed ATR is a cis-conformer (ATR-H) at the Pro429 residue but the nuclear ATR is isomerized into the trans-isomer (ATR-L) by the proline isomerase Pin1. It is likely that the prolyl isomerization of ATR may change the conformation of ATR between an unfolded structure to expose BH3 domain and a folded structure making BH3 inaccessible; the former is able to bind to and sequester the proapoptotic factor tBid at the mitochondrial surface to prevent initiation of the intrinsic apoptosis, thus allowing time for DNA repair.
XPA binds to ds-ssDNA junctions, such as those found at exposed replication forks and DNA regions undergoing repair. This binding, which is not necessarily unrelated to XPA’s NER activity, is stronger than its binding to bulky DNA adducts. Prematurely-aging progeroid cells accumulate progerin, an abnormal form of lamin A and suffer from an accumulation of DNA DSBs and stalled replication forks. Interestingly, these sites are exposed due to sequestration of PCNA by progerin, allowing XPA to these DSB sites and stalled forks.
These studies have revealed several potential sites for therapeutic intervention to enhance the chemotherapy of cancer cells and/or the survival of progeroid cells.
References
- 1.Cleaver JE, Lam ET, Revet I, and (2009) Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity, Nat Rev Genet 10, 756–768. [DOI] [PubMed] [Google Scholar]
- 2.DiGiovanna JJ, and Kraemer KH (2012) Shining a light on xeroderma pigmentosum, J Invest Dermatol 132, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sancar A (2016) Mechanisms of DNA repair by photolyase and excision nuclease (Nobel lecture), Angewandte Chemie 55, 8502–8527. [DOI] [PubMed] [Google Scholar]
- 4.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, and Linn S (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints, Annu Rev Biochem 73, 39–85. [DOI] [PubMed] [Google Scholar]
- 5.Sugasawa K (2016) Molecular mechanisms of DNA damage recognition for mammalian nucleotide excision repair, DNA Repair 44, 110–117. [DOI] [PubMed] [Google Scholar]
- 6.Hengge UR, and Emmert S (2008) Clinical features of xeroderma pigmentosum, Adv Exp Med Biol 637, 10–18. [DOI] [PubMed] [Google Scholar]
- 7.Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, and Hoeijmakers JH (2006) A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis, Nature 444, 1038–1043. [DOI] [PubMed] [Google Scholar]
- 8.Auclair Y, Rouget R, Belisle J, Costantino S, and Drobetsky EA (2010) Requirement for functional DNA polymerase eta in genome-wide repair of UV-induced DNA damage during S phase, DNA Repair (Amst) 9, 754–764. [DOI] [PubMed] [Google Scholar]
- 9.Feltes BC, and Bonatto D (2015) Overview of xeroderma pigmentosum proteins architecture, mutations and post-translational modifications, Mutat Res Rev Mutat Res 763, 306–320. [DOI] [PubMed] [Google Scholar]
- 10.Gillet LC, and Scharer OD (2006) Molecular mechanisms of mammalian global genome nucleotide excision repair, Chem Rev 106, 253–276. [DOI] [PubMed] [Google Scholar]
- 11.Hanawalt PC, and Spivak G (2008) Transcription-coupled DNA repair: two decades of progress and surprises, Nat Rev Mol Cell Biol 9, 958–970. [DOI] [PubMed] [Google Scholar]
- 12.Lagerwerf S, Vrouwe MG, Overmeer RM, Fousteri MI, and Mullenders LH (2011) DNA damage response and transcription, DNA Repair (Amst) 10, 743–750. [DOI] [PubMed] [Google Scholar]
- 13.Camenisch U, and Nageli H (2008) XPA gene, its product and biological roles, Adv Exp Med Biol 637, 28–38. [DOI] [PubMed] [Google Scholar]
- 14.Ding D, Zhang Y, Yu H, Guo Y, Jiang L, He X, Ma W, and Zheng W (2012) Genetic variation of XPA gene and risk of cancer: a systematic review and pooled analysis, Int J Cancer 131, 488–496. [DOI] [PubMed] [Google Scholar]
- 15.Niedernhofer LJ (2006) A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis, Nature 444, 1038–1043. [DOI] [PubMed] [Google Scholar]
- 16.Sugasawa K (2008) XPC: its product and biological roles, Adv Exp Med Biol 637, 47–56. [DOI] [PubMed] [Google Scholar]
- 17.Cimprich KA, and Cortez D (2008) ATR: an essential regulator of genome integrity, Nat Rev Mol Cell Biol 9, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cortez D (2015) Preventing replication fork collapse to maintain genome integrity, DNA Repair (Amst) 32, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weber AM, and Ryan AJ (2015) ATM and ATR as therapeutic targets in cancer, Pharmacol Ther 149, 124–138. [DOI] [PubMed] [Google Scholar]
- 20.Perry J, and Kleckner N (2003) The ATRs, ATMs, and TORs are giant HEAT repeat proteins, Cell 112, 151–155. [DOI] [PubMed] [Google Scholar]
- 21.Nam EA, and Cortez D (2011) ATR signalling: more than meeting at the fork, Biochem J 436, 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases, Genes Dev 15, 2177–2196. [DOI] [PubMed] [Google Scholar]
- 23.Flynn RL, and Zou L (2011) ATR: a master conductor of cellular responses to DNA replication stress, Trends Biochem Sci 36, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fong Yick W., Cattoglio C, and Tjian R (2013) The Intertwined roles of transcription and repair proteins, Mol Cell 52, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maréchal A, and Zou L (2013) DNA damage sensing by the ATM and ATR kinases, Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeman MK, and Cimprich KA (2014) Causes and consequences of replication stress, Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cortez D, Guntuku S, Qin J, and Elledge SJ (2001) ATR and ATRIP: partners in checkpoint signaling, Science 294, 1713–1716. [DOI] [PubMed] [Google Scholar]
- 28.Choi JH, Sancar A, and Lindsey-Boltz LA (2009) The human ATR-mediated DNA damage checkpoint in a reconstituted system, Methods 48, 3–7. [DOI] [PubMed] [Google Scholar]
- 29.Zou L, and Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA- ssDNA complexes, Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
- 30.Zou L, Liu D, and Elledge SJ (2003) Replication protein A-mediated recruitment and activation of Rad17 complexes, Proc Natl Acad Sci USA 100, 13827–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jarrett SG, Wolf Horrell EM, and D’Orazio JA (2016) AKAP12 mediates PKA- induced phosphorylation of ATR to enhance nucleotide excision repair, Nucleic Acids Res DOI 10.1093/nar/gkw871. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Andrés-León E, Cases I, Arcas A, and Rojas AM (2016) DDRprot: a database of DNA damage response-related proteins, Database, DOI: 10.1093/database/baw1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, and Elledge SJ (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage, Science 316, 1160–1166. [DOI] [PubMed] [Google Scholar]
- 34.Dart DA, Adams KE, Akerman I, and Lakin ND (2004) Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase, J Biol Chem 279, 16433–16440. [DOI] [PubMed] [Google Scholar]
- 35.Roos WP, and Kaina B (2013) DNA damage-induced apoptosis: From specific DNA lesions to the DNA damage response and apoptosis, Cancer Lett 332, 237–248. [DOI] [PubMed] [Google Scholar]
- 36.Brown EJ, and Baltimore D (2000) ATR disruption leads to chromosomal fragmentation and early embryonic lethality, Genes Dev 14, 397–402. [PMC free article] [PubMed] [Google Scholar]
- 37.Brown EJ, and Baltimore D (2003) Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance, Genes Dev 17, 615–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buisson R, Boisvert Jessica L., Benes Cyril H., and Zou L (2015) Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase, Mol Cell 59, 1011–1010-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho YJ, and Liang P (2011) S-phase-coupled apoptosis in tumor suppression, Cell Mol Life Sci 68, 1883–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hilton BA, Li Z, Musich PR, Wang H, Cartwright B, Serrano MA, Zhou XZ, Lu KP, and Zou Y (2015) ATR plays a direct antiapoptotic role at mitochondria which is regulated by prolyl isomerase Pin1, Mol Cell 60, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Z (2013) New insights into the roles of human DNA damage checkpoint protein ATR in the regulation of nucleotide excision repair and DNA damage-induced cell death, In Biomedical Sciences, East Tennessee State University, Johnson City. [Google Scholar]
- 42.Hunter T (1998) Prolyl isomerases and nuclear function, Cell 92, 141–143. [DOI] [PubMed] [Google Scholar]
- 43.Lu KP, Liou YC, and Zhou XZ (2002) Pinning down proline-directed phosphorylation signaling, Trends Cell Biol 12, 164–172. [DOI] [PubMed] [Google Scholar]
- 44.Lu Z, and Hunter T (2014) Prolyl isomerase Pin1 in cancer, Cell Res 24, 1033–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld J-U, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, and Lu KP (1997) Sequence-specific and phosphorylation-dependent proline isomerization: A potential mitotic regulatory mechanism, Science 278, 1957–1960. [DOI] [PubMed] [Google Scholar]
- 46.Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, and Xiao ZX (2002) The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response, Nature 419, 849–853. [DOI] [PubMed] [Google Scholar]
- 47.Zhou XZ, and Lu KP (2016) The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target, Nat Rev Cancer 16, 463–478. [DOI] [PubMed] [Google Scholar]
- 48.Brichkina A, Nguyen NT, Baskar R, Wee S, Gunaratne J, Robinson RC, and Bulavin DV (2016) Proline isomerisation as a novel regulatory mechanism for p38MAPK activation and functions, Cell Death Differ 23, 1592–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim BM, You MH, Chen CH, Lee S, Hong Y, Kimchi A, Zhou XZ, and Lee TH (2014) Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function, Cell Death Dis 5, e1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee TH, Chen CH, Suizu F, Huang P, Schiene-Fischer C, Daum S, Zhang YJ, Goate A, Chen RH, Zhou XZ, and Lu KP (2011) Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function, Mol Cell 42, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Enders GH (2008) Expanded roles for Chk1 in genome maintenance, J Biol Chem 283, 17749–17752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ichim G, Tait SWG, and (2016) A fate worse than death: apoptosis as an oncogenic process, Nat Rev Cancer 16, 539–548. [DOI] [PubMed] [Google Scholar]
- 53.Shell SM, and Zou Y (2009) Protein-protein interactions in Ataxia telangiectesia, In Molecular Mechanisms of Ataxia Telangiectasia (Ahmad S, and Hanaoka F, Eds.), pp 42–51, Landes Bioscience. [Google Scholar]
- 54.Lu KP, and Zhou XZ (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease, Nat Rev Mol Cell Biol 8, 904–916. [DOI] [PubMed] [Google Scholar]
- 55.Alexander A, Cai S-L, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, and Walker CL (2010) ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS, Proc Natl Acad Sci U S A 107, 4153–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexander A, Kim J, and Walker CL (2010) ATM engages the TSC2/mTORC1 signaling node to regulate autophagy, Autophagy 6, 672–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alexander A, and Walker CL (2010) Differential localization of ATM is correlated with activation of distinct downstream signaling pathways, Cell Cycle 9, 3685–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tripathi DN, Zhang J, Jing J, Dere R, and Walker CL (2016) A new role for ATM in selective autophagy of peroxisomes (pexophagy), Autophagy 12, 711–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait- Mulder J, Lee JH, Paull TT, Pandita RK, Charaka K, Pandita TK, Kastan MB, and Walker CL (2015) ATM functions at the peroxisome to induce pexophagy in response to ROS, Nat Cell Biol 17, 1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barroso-Gonzalez J, Auclair S, Luan S, Thomas L, Atkins KM, Aslan JE, Thomas LL, Zhao J, Zhao Y, and Thomas G (2016) PACS-2 mediates the ATM and NF- [kappa]B-dependent induction of anti-apoptotic Bcl-xL in response to DNA damage, Cell Death Differ 23, 1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Valentin-Vega YA, MacLean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, Cleveland JL, Green DR, and Kastan MB (2012) Mitochondrial dysfunction in ataxia-telangiectasia, Blood 119, 1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miura N, Miyamoto I, Asahina H, Satokata I, Tanaka K, and Okada Y (1991) Identification and characterization of xpac protein, the gene product of the human XPAC (xeroderma pigmentosum group A complementing) gene, J Biol Chem 266, 19786–19789. [PubMed] [Google Scholar]
- 63.Miyamoto I, Miura N, Niwa H, Miyazaki J, and Tanaka K (1992) Mutational analysis of the structure and function of the xeroderma pigmentosum group A complementing protein. Identification of essential domains for nuclear localization and DNA excision repair, J Biol Chem 267, 12182–12187. [PubMed] [Google Scholar]
- 64.Bomgarden RD, Lupardus PJ, Soni DV, Yee MC, Ford JM, and Cimprich KA (2006) Opposing effects of the UV lesion repair protein XPA and UV bypass polymerase eta on ATR checkpoint signaling, EMBO J 25, 2605–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rademakers S, Volker M, Hoogstraten D, Nigg AL, Mone MJ, Van Zeeland AA, Hoeijmakers JH, Houtsmuller AB, and Vermeulen W (2003) Xeroderma pigmentosum group A protein loads as a separate factor onto DNA lesions, Mol Cell Biol 23, 5755–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Solimando L, Luijsterburg MS, Vecchio L, Vermeulen W, van Driel R, and Fakan S (2009) Spatial organization of nucleotide excision repair proteins after UV-induced DNA damage in the human cell nucleus, J Cell Sci 122, 83–91. [DOI] [PubMed] [Google Scholar]
- 67.Wu X, Shell SM, Liu Y, and Zou Y (2007) ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation, Oncogene 26, 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Z, Musich PR, Cartwright BM, Wang H, and Zou Y (2013) UV-induced nuclear import of XPA is mediated by importin-alpha4 in an ATR-dependent manner, PloS One 8, e68297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Z, Musich PR, Serrano MA, Dong Z, and Zou Y (2011) XPA-mediated regulation of global nucleotide excision repair by ATR Is p53-dependent and occurs primarily in S- phase, PloS One 6, e28326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Z, Musich PR, and Zou Y (2011) Differential DNA damage responses in p53 proficient and deficient cells: cisplatin-induced nuclear import of XPA is independent on ATR checkpoint in p53-deficient lung cancer cells, Int J Biochem Mol Biol 2, 138–145. [PMC free article] [PubMed] [Google Scholar]
- 71.Schnell U, Dijk F, Sjollema KA, and Giepmans BNG (2012) Immunolabeling artifacts and the need for live-cell imaging, Nat Meth 9, 152–158. [DOI] [PubMed] [Google Scholar]
- 72.Wu X, Shell SM, Yang Z, and Zou Y (2006) Phosphorylation of nucleotide excision repair factor Xeroderma pigmentosum group A by ataxia telangiectasia mutated and Rad3-related-dependent checkpoint pathway promotes cell survival in response to UV irradiation, Cancer Res 66, 2997–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ray A, Blevins C, Wani G, and Wani AA (2016) ATR- and ATM-Mediated DNA damage response is dependent on excision repair assembly during G1 but bot in S phase of cell cycle, PLoS One 11, e0159344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ray A, Milum K, Battu A, Wani G, and Wani AA (2013) NER initiation factors, DDB2 and XPC, regulate UV radiation response by recruiting ATR and ATM kinases to DNA damage sites, DNA Repair 12, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lindsey-Boltz LA, Kemp MG, Reardon JT, DeRocco V, Iyer RR, Modrich P, and Sancar A (2014) Coupling of human DNA excision repair and the DNA damage checkpoint in a defined in vitro system, J Biol Chem 289, 5074–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kemp MG, Gaddameedhi S, Choi JH, Hu J, and Sancar A (2014) DNA repair synthesis and ligation affect the processing of excised oligonucleotides generated by human nucleotide excision repair, J Biol Chem 289, 26574–26583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Auclair Y, Rouget R, Affar el B, and Drobetsky EA (2008) ATR kinase is required for global genomic nucleotide excision repair exclusively during S phase in human cells, Proc Natl Acad Sci U S A 105, 17896–17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rouget R, Auclair Y, Loignon M, Affar EB, and Drobetsky EA (2008) A sensitive flow cytometry-based nucleotide excision repair assay unexpectedly reveals that mitogen-activated protein kinase signaling does not regulate the removal of UV-induced DNA damage in human cells, J Biol Chem 283, 5533–5541. [DOI] [PubMed] [Google Scholar]
- 79.Lima-Bessa K. M. d., Armelini MG, Chiganças V, Jacysyn JF, Amarante-Mendes GP, Sarasin A, and Menck CFM (2008) CPDs and 6–4PPs play different roles in UV- induced cell death in normal and NER-deficient human cells, DNA Repair (Amst) 7, 303–312. [DOI] [PubMed] [Google Scholar]
- 80.Lee TH, Park JM, Leem SH, and Kang TH (2012) Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair, Oncogene 33, 19–25. [DOI] [PubMed] [Google Scholar]
- 81.Shell SM, Li Z, Shkriabai N, Kvaratskhelia M, Brosey C, Serrano MA, Chazin WJ, Musich PR, and Zou Y (2009) Checkpoint kinase ATR promotes nucleotide excision repair of UV-induced DNA damage via physical interaction with Xeroderma Pigmentosum Group A, J Biol Chem 284, 24213–24222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sugitani N, Sivley RM, Perry KE, Capra JA, and Chazin WJ (2016) XPA: A key scaffold for human nucleotide excision repair, DNA Repair (Amst) 44, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stewart M (2007) Molecular mechanism of the nuclear protein import cycle, Nat Rev Mol Cell Biol 8, 195–208. [DOI] [PubMed] [Google Scholar]
- 84.Wickner W, and Schekman R (2005) Protein translocation across biological membranes, Science 310, 1452–1456. [DOI] [PubMed] [Google Scholar]
- 85.Knudsen NO, Andersen SD, Lutzen A, Nielsen FC, and Rasmussen LJ (2009) Nuclear translocation contributes to regulation of DNA excision repair activities, DNA Repair (Amst) 8, 682–689. [DOI] [PubMed] [Google Scholar]
- 86.Nitta M, Saijo M, Kodo N, Matsuda T, Nakatsu Y, Tamai H, and Tanaka K (2000) A novel cytoplasmic GTPase XAB1 interacts with DNA repair protein XPA, Nucl Acids Res 28, 4212–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shell SM, and Zou Y (2008) Other proteins interacting with XP proteins, Adv Exp Med Biol 637, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jiang G, Zou Y, and Wu X (2012) Replication mediated disassociation of replication protein A-XPA complex upon DNA damage: Implications for RPA handing off, Cell Biol Int 36, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fadda E (2016) Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly, Comput Struct Biotech J 14, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, Mitchell JR, Croteau DL, and Bohr VA (2014) Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction, Cell 157, 882–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fan W, and Luo J (2010) SIRT1 regulates UV-induced DNA repair through deacetylating XPA, Mol Cell 39, 247–258. [DOI] [PubMed] [Google Scholar]
- 92.Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, and Auwerx J (2013) The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling, Cell 154, 430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mouchiroud L, Houtkooper RH, and Auwerx J (2013) NAD(+) metabolism: a therapeutic target for age-related metabolic disease, Crit Rev Biochem Mol Biol 48, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weaver AN, and Yang ES (2013) Beyond DNA repair: Additional functions of PARP-1 in cancer, Front Oncol 3, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cohen-Armon M (2007) PARP-1 activation in the ERK signaling pathway, Trends Pharmacol Sci 28, 556–560. [DOI] [PubMed] [Google Scholar]
- 96.Cohen-Armon M, Visochek L, Rozensal D, Kalal A, Geistrikh I, Klein R, Bendetz- Nezer S, Yao Z, and Seger R (2007) DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: A link to histone acetylation, Mol Cell 25, 297–308. [DOI] [PubMed] [Google Scholar]
- 97.Kannan S, Fang W, Song G, Mullighan CG, Hammitt R, McMurray J, and Zweidler- McKay PA (2016) Notch/HES1-mediated PARP1 activation: a cell type–specific mechanism for tumor suppression, Blood 117, 2891–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Geistrikh I, Visochek L, Klein R, Miller L, Mittelman L, Shainberg A, and Cohen- Armon M (2011) Ca+2-induced PARP-1 activation and ANF expression are coupled events in cardiomyocytes, Biochem J 438, 337–347. [DOI] [PubMed] [Google Scholar]
- 99.Fischer JM, Popp O, Gebhard D, Veith S, Fischbach A, Beneke S, Leitenstorfer A, Bergemann J, Scheffner M, Ferrando-May E, Mangerich A, and Burkle A (2014) Poly(ADP-ribose)-mediated interplay of XPA and PARP1 leads to reciprocal regulation of protein function, FEBS J 281, 3625–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang Z, Roginskaya M, Colis LC, Basu AK, Shell SM, Liu Y, Musich PR, Harris CM, Harris TM, and Zou Y (2006) Specific and efficient binding of Xeroderma pigmentosum complementation group A to double-strand/single-strand DNA junctions with 3’- and/or 5’-ssDNA branches, Biochemistry 45, 15921–15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu Y, Liu Y, Yang Z, Utzat C, Wang G, Basu AK, and Zou Y (2005) Cooperative interaction of human XPA stabilizes and enhances specific binding of XPA to DNA damage, Biochemistry 44, 7361–7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hilton B, Shkriabai N, Musich PR, Kvaratskhelia M, Shell S, and Zou Y (2014) A New Structural Insight into XPA-DNA Interaction, Biosci Rep 34, 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Buchko GW, Daughdrill GW, de Lorimier R, Rao BK, Isern NG, Lingbeck JM, Taylor JS, Wold MS, Gochin M, Spicer LD, Lowry DF, and Kennedy MA (1999) Interactions of human nucleotide excision repair protein XPA with DNA and RPA70 Delta C327: chemical shift mapping and 15N NMR relaxation studies, Biochemistry 38, 15116–15128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ikegami T, Kuraoka I, Saijo M, Kodo N, Kyogoku Y, Morikawa K, Tanaka K, and Shirakawa M (1998) Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA, Nat Struct Mol Biol 5, 701–706. [DOI] [PubMed] [Google Scholar]
- 105.Dechat T, Adam SA, Taimen P, Shimi T, and Goldman RD (2010) Nuclear Lamins, Cold Spring Harb Perspect Biol 2, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, and Collins FS (2004) Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome, Proc Natl Acad Sci U S A 101, 8963–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kudlow BA, Kennedy BK, and Monnat RJ Jr. (2007) Werner and Hutchinson- Gilford progeria syndromes: mechanistic basis of human progeroid diseases, Nat Rev Mol Cell Biol 8, 394–404. [DOI] [PubMed] [Google Scholar]
- 108.Misteli T, and Scaffidi P (2005) Genome instability in progeria: when repair gets old, Nat Med 11, 718–719. [DOI] [PubMed] [Google Scholar]
- 109.Musich PR, and Zou Y (2009) Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A, Impact Aging 1, 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pereira S, Bourgeois P, Navarro C, Esteves-Vieira V, Cau P, De Sandre-Giovannoli A, and Lévy N (2008) HGPS and related premature aging disorders: From genomic identification to the first therapeutic approaches, Mech Ageing Dev 129, 449–459. [DOI] [PubMed] [Google Scholar]
- 111.Smith ED, Kudlow BA, Frock RL, and Kennedy BK (2005) A-type nuclear lamins, progerias and other degenerative disorders, Mech Ageing Dev 126, 447–460. [DOI] [PubMed] [Google Scholar]
- 112.Wiesel N, Mattout A, Melcer S, Melamed-Book N, Herrmann H, Medalia O, Aebi U, and Gruenbaum Y (2008) Laminopathic mutations interfere with the assembly, localization, and dynamics of nuclear lamins, Proc Natl Acad Sci U S A 105, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu Y, Rusinol A, Sinensky M, Wang Y, and Zou Y (2006) DNA damage responses in progeroid syndromes arise from defective maturation of prelamin A, J Cell Sci 119, 4644–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, and Zou Y (2008) Involvement of Xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A, FASEB J 22, 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tang H, Hilton B, Musich PR, Fang DZ, and Zou Y (2011) Replication factor C1, the large subunit of replication factor C, is proteolytically truncated in Hutchinson-Gilford Progeria Syndrome, Aging Cell 11, 363–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thompson JA, Marzahn MR, O’Donnell M, and Bloom LB (2012) Replication factor C is a more effective PCNA opener than the checkpoint clamp loader, RAD24- RFC, J Biol Chem 287, 2203–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang S-C (2014) PCNA: a silent housekeeper or a potential therapeutic target?, Trends Pharmacol Sci 35, 178–186. [DOI] [PubMed] [Google Scholar]
- 118.Musich PR, and Zou Y (2011) DNA-damage accumulation and replicative arrest in Hutchinson-Gilford progeria syndrome, Biochem Soc Trans 39, 1764–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]