

Abstract
The prevalence of autism spectrum disorder (ASD) has been increasing steadily over the last 20 years; however, the molecular basis for the majority of ASD cases remains unknown. Recent advances in next-generation sequencing and detection of DNA modifications have made methylation-dependent regulation of transcription an attractive hypothesis for being a causative factor in ASD etiology. Evidence for abnormal DNA methylation in ASD can be seen on multiple levels, from genetic mutations in epigenetic machinery to loci-specific and genome-wide changes in DNA methylation. Epimutations in DNAmethylation can be acquired throughout life, as global DNA methylation reprogramming is dynamic during embryonic development and the early postnatal period that corresponds to the peak time of synaptogenesis. However, technical advances and causative evidence still need to be established before abnormal DNA methylation and ASD can be confidently associated.
Keywords: DNA methylation, autism spectrum disorder, epimutation
GENETIC AND EPIGENETIC DYSFUNCTION IN AUTISM SPECTRUM DISORDER
Autism spectrum disorder (ASD) is a group of neurodevelopmental disorders characterized by impaired social communication and interaction, as well as increased repetitive behaviors and restricted interests (1). Recent estimates of the prevalence of ASD are one in 59 individuals in the United States. The disorder occurs more frequently in males than females (ratio: 4.5 to 1) and affects all races and all ethnic and socioeconomic groups (2). Although most ASD cases are not inherited, there is a high concordance of ASD among identical twins, indicating a strong genetic component and complex inheritance (3). Genetic discoveries over the last two decades have also provided convincing evidence for the etiologic role of genetic defects in ASD (4, 5). Currently, ~40% of ASD cases can be attributed definitively to genetic mutations that are mostly de novo (6, 7) (Figure 1).
Figure 1.
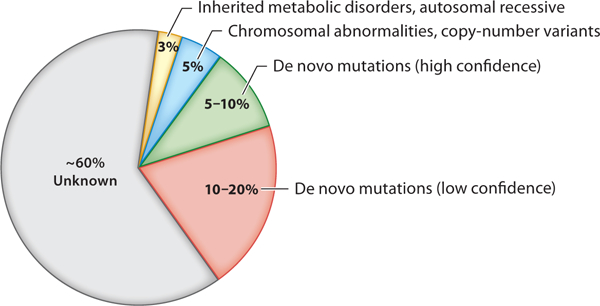
Genetic breakdown of autism spectrum disorder (ASD) (6–9). Although the cause of most ASD cases is unknown, ASD has a strong genetic component. Approximately 40% of cases can be attributed to genetic abnormalities, which can include mutations inherited from a parent (autosomal recessive mutations, yellow), large-scale chromosomal rearrangements or copy-number variants (blue), and de novo mutations in the coding regions of the genome. Approximately 5–10% of de novo mutations are high confidence (green); however, this number may grow in the future as causative evidence of putative mutations is established and understanding of non-protein-coding variants expands (red).
Large-scale genomic studies of >10,000 patients have discovered single-nucleotide variants and copy-number variants in ~100 genes and genetic loci that are highly penetrant in ASD (4– 7, 10–15). The majority of ASD-related genes fall into two distinct functional classes: (a) genes encoding proteins directly implicated in the development and function of synapses and (b) genes encoding enzymes or proteins comprising chromatin regulation and epigenetic machinery (7, 16). The finding of synaptic-related genes in ASD patients is expected because ASD is a neurodevelopmental disorder; however, the significant number of ASD genes involving basic enzymes and proteins involved in chromatin modification and DNA methylation is unexpected. This finding suggests that epigenetic dysfunction is a fundamental contributor to brain development and disease pathogenesis of neurodevelopmental disorders, including ASD.
An alarming increase in the prevalence of ASD over the last two decades has intensified the debate on the etiologic roles of nongenetic factors such as epigenetics (primarily abnormal DNA methylation) and environmental interaction (16, 17). Here, we review the current knowledge of how dysregulation of DNA methylation contributes to ASD and discuss the challenges in testing an epigenetic hypothesis.
EPIGENETIC MACHINERY OF DNA METHYLATION
While the definition of epigenetics is evolving, a widely accepted definition is the study of the modifications of DNA, histones, and chromatin that regulate the expression of genes (18, 19). These modifications include (a) chemical modifications of DNA, (b) modifications of histone tails, and (c) chromatin organization and remodeling within the nucleus (20). The importance of these epigenetic modifications in brain development has been well recognized (21–23). Genetic mutations found in the enzymes and proteins responsible for these modifications have been implicated in epigenetic dysregulation in many human diseases, including neurodevelopmental disorders (20, 24–27).
The cytosine modification 5-methylcytosine (5mC) is the most studied DNA modification and epigenetic mark in the mammalian genome (28). In most somatic cells, 5mC occurs in symmetrical CG dinucleotides (CpG) at 5mCpG sites, and less frequently at 5mCpH (where H can be either A, C, or T) sites. However, the abundance of 5mCpH methylation in neurons is comparable to 5mCpG, suggesting that the epigenome of neurons is uniquely different from other cell types (22). A genomic region enriched with CpGs is referred to as a CpG island. These CpG islands contain at least 50% CG content, are >200 base pairs in length, and are frequently associated with regulatory elements, including the majority of promoters in the genome (29).
The enzymatic “writers” that form 5mC have been studied extensively. Six DNA methyl-transferases, DNMT1, DNMT2, DNMT3A, DNMT3B, DNMT3C, and DNMT3L, have been shown to have either a primary (in the case of DNMT1, DNMT3A, and DNMT3B) or a modifying role in establishing DNA methylation. Both DNMT3A and DNMT3B are de novo methyl-transferases that covalently link a methyl group to the 5’ position of the cytosine ring to create 5mC. DNMT1 is known to function in the maintenance of DNA methylation by remethylating hemimethylated DNA during genome replications (27). All DNMTs use S-adenylsylmethionine (SAM) as a methyl donor from the one-carbon metabolism pathway (30). Passive demethylation of DNA can occur during cell division when DNMT1 fails to remethylate hemimethylated DNA after genome replication (31).
The enzymes in the ten-eleven translocation (TET) family of methylcytosine dioxygenases function as “erasers” of 5mC by catalyzing active DNA demethylation (28). TET1, TET2, and TET3 utilize the cofactors iron(II) and 2-oxoglutarate to oxidize 5mC to 5-hydroxymethylcytosine (5hmC). Formation of 5hmC is the first step in the active DNA demethylation pathway. From there, 5hmC can be further oxidized or, at any point in this process, can be excised and repaired using the base excision repair pathway to yield an unmodified cytosine (28, 32). This process is especially important in postmitotic cells such as mature neurons in the brain, as these cells no longer undergo genome replication and cell division, and active demethylation is the primary form of DNA demethylation (33) (Figure 2).
Figure 2.
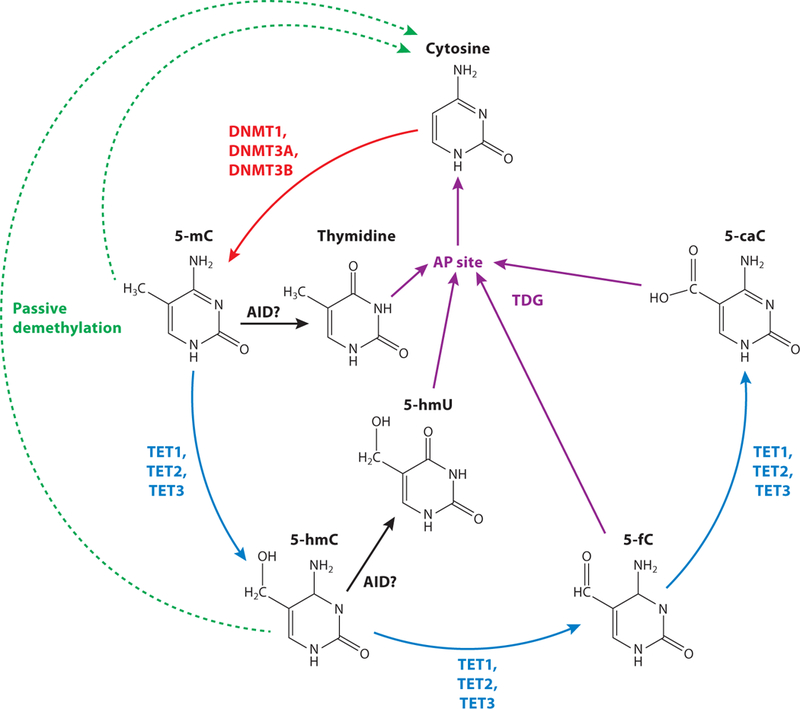
DNA methylation pathway (27, 28, 32). Cytosine can be methylated by DNA methyltransferases (DNMT, red) to create 5-methylcytosine (5mC). The ten-eleven translocation (TET) family of proteins (blue) catalyze the stepwise oxidation of 5mC to create 5-hydroxymethylcytosine (5-hmC), 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC). Methylated DNA can be passively demethylated (green dashed arrows) through cell division. 5-mC and 5-hmC are proposed to be deaminated by activation-induced cytidine deaminase (AID, black) and APOBEC proteins (not shown) to form thymidine and 5-hydroxymethyluracil (5-hmU), respectively. Thymidine, 5-hmU, 5-fC, and 5-CaC can all be excised by thymidine DNA glycosylase (TDG) to create an abasic site (AP site), which is repaired through the base excision repair pathway (purple).
The canonical view of 5mC has been that it represses transcription when it occurs at CpG islands near gene transcription start sites (34). However, this is not a one-size-fits-all rule, as the mechanism by which DNA methylation regulates transcription can be specific to different contexts such as gene content, locus, and developmental timing. DNA methylation is actually more abundant in gene bodies, suggesting a primary role for DNA methylation in fine tuning levels of expression and splicing, rather than acting as an on-off switch at gene promoters. DNA methylation can alter chromatin accessibility and gene expression by modulating binding of DNA methylation “readers,” such as other epigenetic modifiers (histone methyltransferase or demethy-lase enzymes), or by including or excluding the binding of different transcription factors (20, 35). These protein DNA interactions can create repressor or enhancer complexes and alter recruitment and binding of polymerases to the DNA. Increased DNA methylation is correlated with decreased RNA polymerase 2 occupancy. It is also positively correlated with active histone marks such as H3K4me2/3 and negatively correlated with repressive histone marks such as H3K27me3 (36). The functional role of 5hmC in regulating gene transcription is being investigated in ongoing studies and correlates with increased gene expression (22). However, the functions of other oxidative derivatives of 5mC, as well as other DNA modifications, such as N(6)-methyladenine and N(4)-methylcytosine, related to transcription, are less clear or have not been studied extensively in mammalian systems (27, 37).
DYNAMICS AND REPROGRAMMING OF DNA METHYLATION: A HOT SPOT FOR MUTATION ACCUMULATION
Cellular distribution of 5mC, 5hmC, and the proteins that catalyze their formation change greatly during early embryonic development (Figure 3) (38–42). Two major reconfigurations of DNA methylation occur during development, one immediately after fertilization and a second during primordial germ cell specification (43). Imprinted regions across the genome retain 5mC in a parent-of-origin-specific manner, constituting an exception to the global erasure immediately after fertilization (44). This dynamic process of global demethylation and the cell lineage-and gene-specific reestablishment of methyl marks during early embryonic development may be highly susceptible to errors in epigenetic modification. Animal models deficient for DNA methylation writers and erasers have shown that methylation erasure and reestablishment are crucial for cell survival and differentiation (43, 45, 46).
Figure 3.
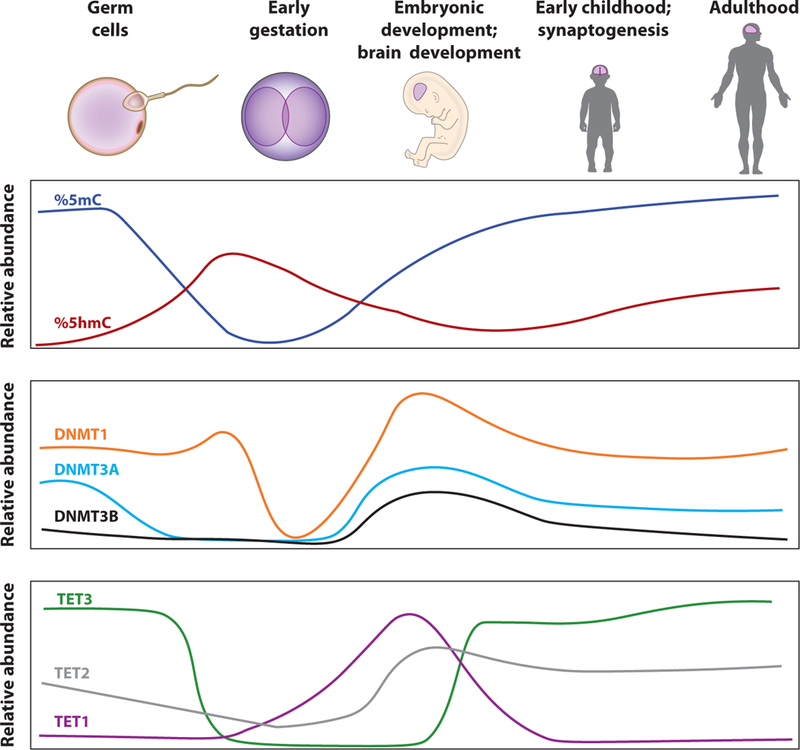
DNA methylation dynamics during brain development (38–42). Levels of 5mC, 5hmC, DNMTs, and TETs fluctuate during development. Although most knowledge of this process has come from mouse models, the overall process is believed to be highly conserved in humans. Relative abundances of 5mC and 5hmC not drawn to scale.
DNA methylation continues to be dynamically regulated during postnatal development and aging. The peak of synaptogenesis in humans (the first five years, varying with brain region) and mice (2–4 weeks) corresponds with an increase in 5mCH methylation in neurons and an increase in DNMT3A transcript abundance (22). In mice, this is also accompanied by an enrichment of 5hmC in actively transcribed gene bodies. This time of rapid 5mC and 5hmC change may be a critical window for brain development and synaptogenesis. It is interesting to speculate whether insults to the DNA methylation process within this critical window of the first five years of human life plays a role in disease onset.
An epimutation, in contrast to a genetic mutation, is a change of gene expression due to abnormal epigenetic modifications, primarily changes in DNA methylation (47, 48). These two reconfigurations of DNA methylation during embryonic and postnatal development support the hypothesis that these points in development may be hot spots for the interface of gene and environmental interaction for any disease mechanism (Figure 4).
Figure 4.
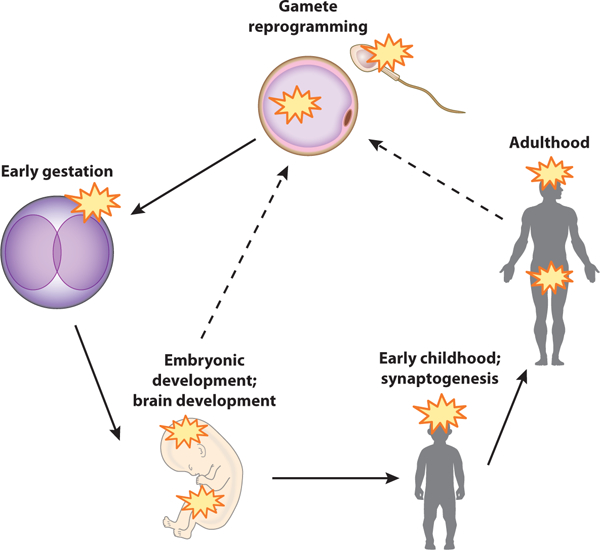
Proposed epimutation hot spot model. DNA methylation epimutations can be acquired at multiple time points in the life cycle. Epimutation hot spots (yellow stars) include pronuclear reprogramming following fertilization, primordial germ cell reprogramming and brain development in the embryo, early childhood and postgestational synaptogenesis, and adult maintenance of synapse function and plasticity. Transgenerational inheritance of epimutations from parental germ cells has also been proposed (dashed lines).
SUPPORT FOR AN EPIGENETIC HYPOTHESIS OF AUTISM SPECTRUM DISORDER
The epigenetic hypothesis of disease refers to causation not by DNA mutations but rather by defects in the epigenome. The epigenetic hypothesis is supported by the understanding of the molecular basis of genomic imprinting disorders such as Angelman and Prader-Willi syndromes (49).In these two disorders, epigenetic defects of DNA methylation in the region of chromosome 15q11-q13 in the cases with uniparental disomy (where both chromosomes have the same parent of origin) can cause clinical features that are indistinguishable from those resulting from genetic deletions. Specially, in the case of uniparental disomy, there is no identifiable genetic defect, but epigenotype assessed by DNA methylation is abnormal. It is also important to underline that a genetic mutation in a gene encoding epigenetic machinery can itself lead to epigenetic instability across the genome. The expanded triplet repeats in the 5’ UTR of FMR1 cause the gene to become hypermethylated and result in the reduced expression of FMR1 in Fragile X syndrome (50),the most common single-gene cause for intellectual disability in males. These historical cases illustrate how epigenetic components can contribute to neurodevelopmental disorders in humans.
The predicted somatic nature of altered DNA methylation may explain the high concordance rate in identical twins and the paradoxically low intergenerational inheritance observed clinically in most ASD cases. As epimutations can arise at any time in any cell lineage in the brain, heterogeneity of methyl marks across individuals is expected. Recent studies of the brain DNA methylome have revealed that DNA methylation patterns in the brain during early prenatal and postnatal development are very dynamic (51–54). Similar to the epigenome reprogramming during early embryonic development, the dynamic epigenomic remodeling in the postnatal brain may present a second hot spot for the accumulation of epimutations and subsequent dysregulation of brain development and function. Furthermore, the epigenetic hypothesis can also integrate environmental factors into molecular pathogenesis, as it is well documented that 5mC can be modified by environmental factors during both prenatal and postnatal periods (55).
EVIDENCE OF EPIGENETIC DYSREGULATION OF DNA METHYLATION IN AUTISM SPECTRUM DISORDER
It has been challenging to find definitive evidence to support the epigenetic hypothesis for ASD. However, evidence of dysregulation of DNA methylation in ASD exists on multiple levels: genetic mutations in genes encoding epigenetic machinery, abnormally methylated loci, and genome-wide correlations of hyper-and hypomethylation. We summarize the major findings and discuss the caveats of interpreting these results below.
Genetic Mutations in DNA Methylation Writers, Readers, and Erasers
The growing body of whole-exome and -genome sequencing data from ASD patients has revealed mutations in genes encoding DNA methylation machinery (Table 1, Supplemental Table 1) (56). De novo mutations in DNA methylation writers, readers, and erasers (DNMT3A, TET2, MECP2, MBD5) have been identified in ASD cases through genomic studies. Mutations in these genes can cause loss or gain of function of epigenetic modifying enzymes. These genetic discoveries from patients provide substantial molecular evidence to support epigenetic dysfunction and the epigenetic hypothesis in ASD.
Table 1.
Genes encoding DNA methylation machinery which correlate with autism spectrum disorder
| Genea | Description | SFARI scoreb |
Comorbidities | References | |
|---|---|---|---|---|---|
| DNA methylation writers and erasers |
DNMT3A | De novo methyltransferase | 3 | Tatton-Brown-Rahman syndrome, acute myeloid leukemia |
6, 7, 14 |
| TET2 | DNA methylcytosine dioxygenase |
3 | Myelodysplastic syndrome (somatic) |
6 | |
| GADD45B | DNA demethylation through base excision repair pathway |
5 | Unknown | 7 | |
| DNA methylation readers known to be influenced by or to influence DNA methylation |
ATRX | Binds DNA, interacts with DAXX in histone chaperone complex, chromatin remodeler |
4 |
α-Thalassemia, intellectual disability |
57 |
| CTCF | Master regulator recruited by zinc finger proteins |
3 | Intellectual disability | 15 | |
| MECP2 | Methylation-dependent transcriptional repressor |
2 | Rett syndrome, X-linked intellectual disability, encephalopathy |
58 | |
| MBD1 | Binds methylated DNA, represses transcription |
4 | Unknown | 59 | |
| MBD3 | Can bind methylated DNA with additional proteins |
4 | Unknown | 59 | |
| MBD4 | Complex with MLH1, binds methyl-CpG DNA, has thymidine glycosylase activity |
4 | Unknown | 15 | |
| MBD5 | Methyl-CpG-binding domain family member, interacts with the polycomb repressive complex |
3 | Intellectual disability, microcephaly, intellectual disabilities, severe speech impairment, and seizure |
59, 60 |
Abbreviations: DAXX, Death domain-associated protein; SFARI, Simons Foundation Autism Reference Initiative.
Genes are separated by function of (a) DNA methylation writers and erasers and (b) proteins known to read DNA methylation or to be impacted by DNA methylation.
SFARI score based on clinical data: 1–2, strong evidence; 3–4, suggestive/minimal evidence; 5, hypothesized but untested.
Targeted Gene-Specific DNA Methylation Changes
Studies have identified abnormal methylation, primarily hypermethylation of promoter CpG islands, in patients with ASD when compared to normal controls. Initial studies found promoter hypermethylation of MECP2 and UBE3A, two genes in which loss-of-function mutations are known to cause syndromic ASD, in post mortem ASD brains in humans (61, 62). Subsequently, many other genes implicated in ASD have been found to have hypermethylated transcription start sites in ASD patients compared to control brains, resulting in decreased expression of the gene product. These genes include OXTR, SNRPN, MAGEL2, RELN, and GAD1 (63–67). In contrast, hypomethylation of targeted candidate genes such as RORA, ERMN, USP24, METTL21C, PDE10A, STX16, and DBT has also been seen in peripheral blood DNA of ASD subjects (68). Conversely, accumulation of 5hmC in promoters has been correlated with increase of transcript, as in Engrailed-2, GAD1, and MECP2 (58, 69). While these findings provide some support for the epigenetic hypothesis, the origins and causal roles of these changes remain unknown.
Aside from the traditional hypothesis that promoter methylation results in increased or decreased amounts of gene product, DNA methylation can also alter the sequence of gene products by influencing isoform generation and splicing of genes. Our group has shown that hypermeth-ylation in select intragenic CpG islands regulates the use of alternative transcription start sites to create isoform-specific expression of SHANK3 in ASD brains (70). This finding suggests that DNA methylation within the gene body helps to prevent aberrant transcription initiation, thereby regulating usage of alternative promoters of gene products in the brain (71,72). Intragenic or gene body DNA methylation has also been implicated in regulating alternative splicing by slowing RNA polymerase II elongation rate through the recruitment of methylation-dependent splicing factors (73). Several studies have reported the dysregulation of splicing pre-mRNAs in ASD brains (74, 75). Isoform-specific data and DNA methylation data should be integrated in future studies, as >95% of neuronal genes (including ASD-causing and candidate genes) undergo alterative splicing (76, 77).
Genome-Wide DNA Methylation Changes
While a genetic or epigenetic change in a single gene is conceptually simple, it may not occur frequently. Genome-wide DNA methylation profiling in ASD patients is considered an ideal experimental approach to identify candidate loci with abnormal methylation. However, due to the limited availability of brain tissues, many studies have used blood samples to investigate 5mC in ASD (63, 78, 79). Interestingly, the genome-wide methylation pattern in sperm DNA has been associated with early signs of ASD risk in one ASD cohort (80). Additional studies have attempted to use placental tissue, rather than blood samples, to assess DNA methylation levels at birth (81).
Studies have reported several differentially methylated regions (DMRs) distinguishing ASD patients from controls (82–85). The DMRs have been found in regions associated with promoters, gene bodies, and the 3’ untranslated region of a list of ASD-related genes. However, concrete evidence for the direction of global fluctuations in 5mC and the number and location of DMRs in ASDs remains elusive, as some studies report higher genome-wide amounts of 5mC and others report lower (82–86). Pathway analysis and gene enrichment analysis indicate that differentially methylated sites are enriched for genes which undergo changes in methylation during embryonic to late fetal development or are tied to synaptic function (83). It still remains to be determined whether synaptic function and DNA methylation are two distinct etiologies that converge in ASD or whether one directly influences the other.
Many recent studies are also integrating human genetic variation. Several have identified methylation quantitative trait loci (meQTLs) across the genome to be associated with ASD (78, 87). It is valuable to examine how common genetic variants in the population may affect the DNA methylation status of potentially pathogenic variants, especially variants in noncoding regions of the genome.
Genome-wide profiling of 5hmC has also reported been in ASD cerebellums by using β-glucosyltransferase to enrich for fragments of DNA containing 5hmC (88, 89). Differently hy-droxymethylated regions (DhMRs) between ASD patients and unaffected controls were identified only in a young group (age ≤ 18), suggesting that 5hmC dysregulation may be specific to a developmental time point. Furthermore, these DhMRs were associated with genes that are important in psychiatric disease and development, such as those involved in cell-cell communication and neurological disorders (89). In addition, 5hmC is enriched in intragenic noncoding regions, for which a functional role is poorly understood. Going forward, it is important to elucidate potential regulatory functions for 5hmC in these noncoding regions.
Interpreting the Results of DNA Methylation Analysis
Numerous technological and biological factors prevent any definitive conclusion from individual candidate genes or genome-wide DNA methylation profiling studies in ASD (Table 2). These factors will continue posing significant challenges for experimental design and data analysis in future studies.
Table 2.
Issues complicating interpretation of DNA methylation data in autism spectrum disorder (ASD) studies
| Issues | Solutions |
|---|---|
| Working with post mortem ASD brain samples: ◾ Clinical heterogeneity of patients ◾Brain tissue availability; age of proband, sample quality, post mortem time, limited brain regions ◾ Small sample size (typically n = 10–50) ◾Choice of brain subregions: The exact neuroanatomic basis for the pathophysiology underlying ASD remains unclear. This creates a challenge to select the brain subregion (or neuron subtype) for study |
◾Use brain tissues from young (< 10 years) ASD subjects to minimize the impact of other environmental factors that may result in secondary change of DNA methylation ◾Include multiple brain regions, rather than just neocortex, in studies |
| Working with DNA from blood samples: DNA methylation profiles vary across different tissue types |
Validate findings from blood studies in brain tissues |
| Cell type specificity of DNA methylation: ◾Cell lineage-specific DNA methylation profiles ◾Many different neuronal cell subtypes |
◾Use single-cell DNA methylation assays ◾Purify or sort cells into subpopulations before analysis |
| Coverage for non-CpG methylation: 5mCH can account for up to 25% of methylated cytosines in the mammalian frontal cortex |
Include 5mCH in genome-wide methylation studies |
| Establish the relationship between changes in DNA methylation and molecular pathogenesis: ◾Correlation of changes in DNA methylation to changes in transcription, ectopic expression from alternative promoters, or cryptic splicing of pre-mRNAs ◾Modifier effects of other cell intrinsic epigenetic programs ◾Characterization of DNA methylation reader binding and functional consequences ◾Extending mechanisms from model systems to humans |
Utilize tools: ◾Genome-wide methylation inhibitors, such as 5-aza-2‘-deoxycytidine ◾dCas9 fusion protein-mediated epigenome editing at specific loci ◾Advancements in brain organoid cultures that allow use of human “brain” samples in vitro Exercise caution when comparing animal in vivo data and human data Pay attention to isoform generation in neuronal tissues |
| Method of DNA methylation profiling: ◾Bisulfite based method does not distinguish between 5mC and 5hmC ◾Lack of data on other 5mC oxidative derivatives ◾Depth/coverage of genome-wide next-generation sequencing approaches |
Develop new methods: ◾DNA immunoprecipitation and ELISA can detect methylation at a locus-specific level. TAB-seq, β-glucosyltransferase, and oxidative bisulfite sequencing allow base resolution-specific detection of 5mC and 5hmC ◾Technical advancements and dropping costs can aid in integrating these methods into a clinical setting |
WHAT CAUSES ABNORMAL DNA METHYLATION?
Although the key readers and writers and developmental time points of DNA methylation have been discovered, much about DNA methylation dynamics may remain unknown. The delicate interplay between enzymatic kinetics and metabolic reactions plays a role. Furthermore, many DNA methylation readers, such as chromatin modifiers, are themselves known to modulate DNA methylation readers and writers in a feedback loop. The confounding effects of these dynamics are yet to be determined.
Several environmental factors have been shown to influence DNAmethylation in mammals. In mice, supplementation of maternal diet with methyl-donor precursors is capable of increasing the methylation in the agouti locus and altering the coat color in offspring (90), which provides direct experimental evidence that environmental factors (such as maternal diet in utero) can have lasting effects on DNA methylation of offspring. In rats, maternal care of pups (licking and grooming) has been shown to prevent accumulation of 5mC at the glucocorticoid receptor gene promoter in the hippocampus of the offspring, and these modifications can be reversed by changes in care and persist into adulthood (91). In humans, in utero exposure to environmental burdens including organic pollutants, tobacco, alcohol, obesity, asthma, and maternal stress and care have been shown to impact DNA methylation of offspring (16, 92–96). It is interesting to speculate whether aberrant methyl marks can be passed from parents to offspring in humans and how environmental insults during early development hot spots may cause accumulations of epimutations.
HOW DO EPIMUTATIONS CONTRIBUTE TO THE MOLECULAR PATHOGENESIS OF AUTISM SPECTRUM DISORDER?
If the relationship between DNA methylation and gene expression were linear and monoepigenetic, it would be clear how methylation can lead to ASD pathogenesis. However, it is more challenging to understand how genome-wide changes in methylation are related to ASD. Several hypotheses could explain how stochastic mutations can lead to ASD. It is possible that an epimutation acquired during a critical window of reprogramming may lie dormant in the genome and not cause a phenotype until later in development, when the genetic loci containing the epimutation are utilized. Furthermore, abnormal DNA methylation may not be a driving factor for pathogenesis but may play a secondary role, contributing to the expressivity of ASD phenotypes by regulating transcription levels and splicing of key genomic elements or ASD-related genes. It is also interesting to speculate whether the correlation between DNA methylation and synaptic function extends further and influences other aspects of the epigenome. It was recently reported that, in the developing human prefrontal cortex, three-dimensional chromatin loops are located around synapse-related genes, suggesting that synaptic gene expression is regulated by chromatin looping (21, 97, 98). Perhaps the CCCTC-binding factor (CTCF) modulated formation of these loops is regulated by DNA methylation. More broadly, many things are still unknown about how DNA methylation regulates brain development and how it influences the pathogenicity of neurodevelopmental disorders such as ASD.
CONCLUDING REMARKS
Historical examples of genomic imprinting disorders and ASD syndromes such as fragile X and Angelman syndrome have provided initial support for epimutations in human disease etiology and a hypothesis of altered DNA methylation in neurodevelopmental disorders like ASD and intellectual disability. Recent genomic discoveries of highly penetrant mutations in genes encoding epigenetic machinery, including DNA methylation writers, readers, and erasers, offer convincing evidence of the importance of proper epigenetic regulation during neurodevelopment. Altered DNA methylation in ASD patients has been reported in individual genes and genome wide. Technical and biological difficulties/caveats within these studies will need to be addressed in the future. Nonetheless, the mounting evidence for an epigenetic hypothesis of disease should be incorporated into future studies investigating the molecular pathogenesis of ASD.
The observed heterogeneity of abnormal DNA methylation in ASD patients may support a more plausible hypothesis in which epigenetic dysregulation occurs in a stochastic manner, rather than in a predictable and distinct pattern. A similar phenomenon for stochastic allelic specific methylation has also been revealed by a genome-wide bisulfite sequencing study in normal population samples (99). These stochastic events could result from random errors during hot spots of epigenetic reprogramming (100) or from environmental insults during this process.
Understanding the significance of DNA methylation in ASD pathophysiology can shed light on other neuropsychiatric disorders in which DNA methylation may also play a causal role. A growing number of studies report abnormal methylation at individual genes and genome wide in schizophrenia, bipolar disorder, depression, Alzheimer’s disease, and dementia (25). Defining epimutations in ASD may also help with current diagnostic standards by proving to be a biomarker for mental disorders. Finally, DNA methylation is an attractive therapeutic avenue for ASD because a hot spot for epimutation during development provides a clear time window during which interventional therapeutic approaches could be utilized.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Samuel W. Hulbert and Anne E. West for careful reading of this review. Y.H.J. is supported by grants from the National Institutes of Health (MH098114, MH104316, HD087795, and HD088626).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.American Psychiatric Association. 2013. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Washington, DC: Am. Psychiatr. Assoc. [Google Scholar]
- 2.Baio J, Wiggins L, Christensen DL, et al. 2018. Prevalence of autism spectrum disorder among children aged 8 years—Autism and Developmental Disabilities Monitoring Network, 11 sites, United States, 2014. MMWR Surveill. Summ. 67:1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folstein SE, Rosen-Sheidley B. 2001. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat. Rev. Genet. 2:943–55 [DOI] [PubMed] [Google Scholar]
- 4.Jiang YH, Wang Y, Xiu X, et al. 2014. Genetic diagnosis of autism spectrum disorders: the opportunity and challenge in the genomics era. Crit. Rev. Clin. Lab. Sci. 51:249–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willsey AJ, State MW. 2015. Autism spectrum disorders: from genes to neurobiology. Curr. Opin. Neurobiol. 30C:92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iossifov I, O’Roak BJ, Sanders SJ, et al. 2014. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515:216–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Rubeis S, He X, Goldberg AP, et al. 2014. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515:209–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaugler T, Klei L, Sanders SJ, et al. 2014. Most genetic risk for autism resides with common variation. Nat. Genet. 46:881–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu TW, Chahrour MH, Coulter ME, et al. 2013. Using whole-exome sequencing to identify inherited causes of autism. Neuron 77:259–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buxbaum JD, Daly MJ, Devlin B, et al. 2012. The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron 76:1052–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neale BM, Kou Y, Liu L, et al. 2012. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485:242–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Roak BJ, Deriziotis P, Lee C, et al. 2011. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43:585–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Roak BJ, Vives L, Girirajan S, et al. 2012. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485:246–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanders SJ,Murtha MT, Gupta AR, et al. 2012. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485:237–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.IossifovI, Ronemus M, Levy D, et al. 2012. Denovo gene disruptions in children on the autistic spectrum. Neuron 74:285–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran NQV, Miyake K. 2017. Neurodevelopmental disorders and environmental toxicants: epigenetics as an underlying mechanism. Int.J. Genom. 2017:7526592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keil KP, Lein PJ. 2016. DNA methylation: a mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ. Epigenet. 2:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bird A 2007. Perceptions of epigenetics. Nature 447:396–98 [DOI] [PubMed] [Google Scholar]
- 19.Henikoff S, Greally JM. 2016. Epigenetics, cellular memory and gene regulation. Curr. Biol. 26:R644–48 [DOI] [PubMed] [Google Scholar]
- 20.Allis CD, Jenuwein T. 2016. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17:487–500 [DOI] [PubMed] [Google Scholar]
- 21.de la Torre-Ubieta L, Stein JL, Won H, et al. 2018. The dynamic landscape of open chromatin during human cortical neurogenesis. Cell 172:289–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lister R, Mukamel EA, Nery JR, et al. 2013. Global epigenomic reconfiguration during mammalian brain development. Science 341:1237905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bale TL. 2015. Epigenetic and transgenerational reprogramming of brain development. Nat. Rev. Neurosci. 16:332–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madrid A, Papale LA, Alisch RS. 2016. New hope: the emerging role of 5-hydroxymethylcytosine in mental health and disease. Epigenomics 8:981–91 [DOI] [PubMed] [Google Scholar]
- 25.Jiang YH, Bressler J, Beaudet AL. 2004. Epigenetics and human disease. Annu. Rev. Genom. Hum. Genet. 5:479–510 [DOI] [PubMed] [Google Scholar]
- 26.Brookes E, Shi Y. 2014. Diverse epigenetic mechanisms of human disease. Annu. Rev. Genet. 48:237–68 [DOI] [PubMed] [Google Scholar]
- 27.Lyko F 2018. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 19:81–92 [DOI] [PubMed] [Google Scholar]
- 28.Wu X, Zhang Y. 2017. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 18:517–34 [DOI] [PubMed] [Google Scholar]
- 29.Bird A 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16:6–21 [DOI] [PubMed] [Google Scholar]
- 30.Mentch SJ, Locasale JW. 2016. One-carbon metabolism and epigenetics: understanding the specificity. Ann. N. Y. Acad. Sci. 1363:91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen T, Li E. 2004. Structure and function of eukaryotic DNA methyltransferases. Curr. Top. Dev. Biol. 60:55–89 [DOI] [PubMed] [Google Scholar]
- 32.Cortellino S, Xu J, Sannai M,et al. 2011. Thymine DNA glycosylase is essential for active DNAdemethy-lation by linked deamination-base excision repair. Cell 146:67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu SC, Zhang Y. 2010. Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. CellBiol. 11:607–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev. 25:1010–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yin Y, Morgunova E, Jolma A, et al. 2017. Impact of cytosine methylation on DNA binding specificities ofhuman transcription factors. Science 356:eaaj2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lorincz MC, Dickerson DR, Schmitt M, et al. 2004. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 11:1068–75 [DOI] [PubMed] [Google Scholar]
- 37.Luo GZ, Blanco MA, Greer EL, et al. 2015. DNA N(6)-methyladenine: a new epigenetic mark in eukaryotes? Nat. Rev. Mol. Cell Biol. 16:705–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng J, Chang H, Li E, et al. 2005. Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res. 79:734–46 [DOI] [PubMed] [Google Scholar]
- 39.Feng J, Zhou Y, Campbell SL, et al. 2010. Dnmtl and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13:423–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ko YG, Nishino K, Hattori N, et al. 2005. Stage-by-stage change in DNA methylation status ofDnmtl locus during mouse early development. J. Biol. Chem. 280:9627–34 [DOI] [PubMed] [Google Scholar]
- 41.Wossidlo M, Nakamura T, Lepikhov K,et al. 2011. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2:241. [DOI] [PubMed] [Google Scholar]
- 42.Szwagierczak A, Bultmann S, Schmidt CS, et al. 2010. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 38:e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan HD, Santos F, Green K, et al. 2005. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 14(Spec. No. 1):R47–58 [DOI] [PubMed] [Google Scholar]
- 44.Reik W, Walter J. 2001. Genomic imprinting: parental influence on the genome. Nat. Rev. Genet. 2:21–32 [DOI] [PubMed] [Google Scholar]
- 45.Gu TP, Guo F, Yang H, et al. 2011. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477:606–10 [DOI] [PubMed] [Google Scholar]
- 46.von Meyenn F, Iurlaro M, Habibi E, et al. 2016. Impairment of DNA methylation maintenance is the main cause of global demethylation in naive embryonic stem cells. Mol. Cell 62:848–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oey H, Whitelaw E. 2014. On the meaning of the word ‘epimutation’. Trends Genet. 30:519–20 [DOI] [PubMed] [Google Scholar]
- 48.Horsthemke B 2006. Epimutations in human disease. Curr. Top. Microbiol. Immunol. 310:45–59 [DOI] [PubMed] [Google Scholar]
- 49.Jiang Y, Tsai TF, Bressler J, et al. 1998. Imprinting in Angelman and Prader-Willi syndromes. Curr. Opin. Genet. Dev. 8:334–42 [DOI] [PubMed] [Google Scholar]
- 50.Penagarikano O, Mulle JG, Warren ST. 2007. The pathophysiology of fragile X syndrome. Annu. Rev. Genom. Hum. Genet. 8:109–29 [DOI] [PubMed] [Google Scholar]
- 51.Lister R, Pelizzola M, Dowen RH, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462:315–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spiers H, Hannon E, Schalkwyk LC, et al. 2017. 5-hydroxymethylcytosine is highly dynamic across human fetal brain development. BMC Genom. 18:738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pidsley R, Viana J, Hannon E, et al. 2014. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol. 15:483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spiers H, Hannon E, Schalkwyk LC, et al. 2015. Methylomic trajectories across human fetal brain development. Genome Res. 25:338–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jirtle RL, Skinner MK. 2007. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 8:253–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abrahams BS,Arking DE, Campbell DB, et al. 2013. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deciphering Developmental Disorders Study. 2015. Large-scale discovery of novel genetic causes of developmental disorders. Nature 519:223–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhubi A, Chen Y, Dong E, et al. 2014. Increased binding ofMeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry 4:e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cukier HN, Rabionet R, Konidari I, et al. 2010. Novel variants identified in methyl-CpG-binding domain genes in autistic individuals. Neurogenetics 11:291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Talkowski ME, Mullegama SV, Rosenfeld JA, et al. 2011. Assessment of2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus ofintellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 89:551–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang YH, Sahoo T, Michaelis RC, et al. 2004. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am. J. Med. Genet. A 131:1–10 [DOI] [PubMed] [Google Scholar]
- 62.Nagarajan RP, Patzel KA, Martin M, et al. 2008. MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res. 1:169–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wong CC, Meaburn EL, Ronald A, et al. 2014. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol. Psychiatry 19:495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gregory SG, Connelly JJ, Towers AJ,et al. 2009. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 7:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Labouesse MA, Dong E, Grayson DR, et al. 2015. Maternal immune activation induces GAD1 and GAD2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 10:1143–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hogart A, Leung KN, Wang NJ, et al. 2009. Chromosome 15q11–13 duplication syndrome brain reveals epigenetic alterations in gene expression not predicted from copy number. J. Med. Genet. 46:86–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lintas C, Sacco R, Persico AM. 2016. Differential methylation at the RELN gene promoter in temporal cortex from autistic and typically developing post-puberal subjects. J. Neurodev. Disord. 8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Homs A, Codina-Sola M, Rodriguez-Santiago B, et al. 2016. Genetic and epigenetic methylation defects and implication of the ERMN gene in autism spectrum disorders. Transl. Psychiatry 6:e855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.James SJ, Shpyleva S, Melnyk S, et al. 2014. Elevated 5-hydroxymethylcytosine in the Engrailed-2 (EN-2) promoter is associated with increased gene expression and decreased MeCP2 binding in autism cerebellum. Transl. Psychiatry 4:e460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu L, Wang X, Li XL, et al. 2014. Epigenetic dysregulation ofSHANK3 in brain tissues from individuals with autism spectrum disorders. Hum. Mol. Genet. 23:1563–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maunakea AK, Chepelev I, Cui K, et al. 2013. Intragenic DNAmethylation modulates alternative splicing byrecruiting MeCP2 to promote exon recognition. CellRes. 23:1256–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neri F, Rapelli S, Krepelova A, et al. 2017. Intragenic DNA methylation prevents spurious transcription initiation. Nature 543:72–77 [DOI] [PubMed] [Google Scholar]
- 73.Lev Maor G, Yearim A, Ast G. 2015. The alternative role of DNA methylation in splicing regulation. Trends Genet. 31:274–80 [DOI] [PubMed] [Google Scholar]
- 74.Quesnel-Vallieres M, Dargaei Z, Irimia M, et al. 2016. Misregulation of an activity-dependent splicing network as a common mechanism underlying autism spectrum disorders. Mol. Cell 64:1023–34 [DOI] [PubMed] [Google Scholar]
- 75.Irimia M, Weatheritt RJ, Ellis JD, et al. 2014. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159:1511–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Q, Lee JA, Black DL. 2007. Neuronal regulation of alternative pre-mRNA splicing. Nat. Rev. Neurosci. 8:819–31 [DOI] [PubMed] [Google Scholar]
- 77.Vuong CK, Black DL, Zheng S. 2016. The neurogenetics of alternative splicing. Nat. Rev. Neurosci. 17:265–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Andrews SV, Ellis SE, Bakulski KM, et al. 2017. Cross-tissue integration ofgenetic and epigenetic data offers insight into autism spectrum disorder. Nat. Commun. 8:1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsang SY, Ahmad T, Mat FW, et al. 2016. Variation of global DNA methylation levels with age and in autistic children. Hum. Genom. 10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feinberg JI, Bakulski KM, Jaffe AE, et al. 2015. Paternal sperm DNA methylation associated with early signs of autism risk in an autism-enriched cohort. Int. J. Epidemiol. 44:1199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schroeder DI, Schmidt RJ, Crary-Dooley FK, et al. 2016. Placental methylome analysis from a prospective autism study. Mol. Autism 7:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nardone S, Sams DS, Reuveni E, et al. 2014. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 4:e433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nardone S, Sams DS, Zito A, et al. 2017. Dysregulation of cortical neuron DNA methylation profile in autism spectrum disorder. Cereb. Cortex 27:5739–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ladd-Acosta C, Hansen KD, Briem E, et al. 2014. Common DNA methylation alterations in multiple brain regions in autism. Mol. Psychiatry 19:862–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dunaway KW, Islam MS, Coulson RL, et al. 2016. Cumulative impact of polychlorinated biphenyl and large chromosomal duplications on DNA methylation, chromatin, and expression of autism candidate genes. Cell Rep. 17:3035–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ellis SE, Gupta S, Moes A, et al. 2017. Exaggerated CpH methylation in the autism-affected brain. Mol. Autism 8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hannon E, Schendel D, Ladd-Acosta C, et al. 2018. Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Med. 10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheng Y, Li Z, Manupipatpong S, et al. 2018. 5-Hydroxymethylcytosine alterations in the human postmortem brains of autism spectrum disorder. Hum. Mol. Genet. 17:2955–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang T, Pan Q, Lin L, et al. 2012. Genome-wide DNA hydroxymethylation changes are associated with neurodevelopmental genes in the developing human cerebellum. Hum. Mol. Genet. 21:5500–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Waterland RA, Jirtle RL. 2003. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 23:5293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Weaver IC, Cervoni N, Champagne FA, et al. 2004. Epigenetic programming by maternal behavior. Nat. Neurosci. 7:847–54 [DOI] [PubMed] [Google Scholar]
- 92.Dolinoy DC, Huang D, Jirtle RL. 2007. Maternal nutrient supplementation counteracts bisphenol A-induced DNAhypomethylation in early development. PNAS 104:13056–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ruiz-Hernandez A, Kuo CC, Rentero-Garrido P,et al. 2015. Environmental chemicals and DNA meth-ylation in adults: a systematic review of the epidemiologic evidence. Clin. Epigenet. 7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharp GC, Salas LA, Monnereau C, et al. 2017. Maternal BMI at the start of pregnancy and offspring epigenome-wide DNA methylation: findings from the pregnancy and childhood epigenetics (PACE) consortium. Hum. Mol. Genet. 26:4067–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maccani JZ, Maccani MA. 2015. Altered placental DNA methylation patterns associated with maternal smoking: current perspectives. Adv. Genom. Genet. 2015:205–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McGowan PO, Sasaki A, D’Alessio AC, et al. 2009. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12:342–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Won H, de la Torre-Ubieta L, Stein JL, et al. 2016. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538:523–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Parikshak NN, Luo R, Zhang A, et al. 2013. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155:1008–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Onuchic V, Lurie E, Carrero I, et al. 2018. Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci. Science 23:aar3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmitz RJ, Schultz MD, Lewsey MG, et al. 2011. Transgenerational epigenetic instability is a source of novel methylation variants. Science 334:369–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.