

Abstract
Development of pharmacological tools for the ionotropic glutamate receptors (iGluRs) is imperative for the study and understanding of the role and function of these receptors in the central nervous system. We report the synthesis of 18 analogues of (2S,3R)-2-carboxy-3-pyrrolidine acetic acid (3a), which explores the effect of introducing a substituent on the ε-carbon (3c−q). A new synthetic method was developed for the efficient synthesis of racemic 3a and applied to give expedited access to 13 racemic analogues of 3a. Pharmacological characterization was carried out at native iGluRs, cloned homomeric kainate receptors (GluK1−3), NMDA receptors (GluN1/GluN2A-D), and excitatory amino acid transporters (EAAT1−3). From the structure−activity relationship studies, several new ligands emerged, exemplified by triazole 3p-d1, GluK3-preferring (GluK1/GluK3 Ki ratio of 15), and the structurally closely related tetrazole 3q-s3−4 that displayed 4.4−100-fold preference as an antagonist for the GluN1/GluN2A receptor (Ki = 0.61 μM) over GluN1/GluN2B-D (Ki = 2.7−62 μM).
Keywords: Glutamate receptors, NMDA receptors, kainate, proline analogues, CNS
Graphical Abstract

INTRODUCTION
Most of the fast excitatory neurotransmission in the central nervous system (CNS) is mediated by the endogenous α-amino acid (S)-glutamate (Glu) through the ionotropic glutamate receptors (iGluRs).1 Based on pharmacological studies of synthetic ligands and natural products, these receptors were early on divided into three groups, named the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), the kainic acid (KA) and the N-methyl-d-aspartate (NMDA) receptors.2 For the AMPA receptors, four subunits have been identified (GluA1−4),3 while five KA receptor subunits have been cloned (GluK1−5).4 For the NMDA receptors, four Glu binding subunits have been identified (GluN2A-D) as well as three glycine (Gly) binding subunits (GluN1 and GluN3A-B).5 The subunits GluA1−4 can all be freely combined in vitro, to form homomeric as well as heteromeric receptors complexes. However, it remains unknown to which extent the conceivable combinations are expressed in vivo.6 The KA receptors may form functional receptors in vitro, which are homomers of one of the three GluK1−3 subunits, but also combinations thereof. The GluK4 and GluK5 subunits only form functional receptors in vitro upon the combination with one of the three subunits GluK1−3.7 Likewise, the NMDA receptors only form Glu-sensitive functional receptors in vitro from two Gly-binding subunits (GluN1) and two Glu-binding subunits (GluN2A-D).8,9 The extracellular domains of the functional receptors are arranged as a dimer of dimers, whereas the transmembrane domain is arranged as a symmetrical tetramer, which forms the ion channel.10
The natural product KA, and other GluK agonists, are known to be potent neuronal excitants and neurotoxins as well as potent convulsants.11 KA receptor knockout animal models have shown that these animals have a higher threshold in the perception of pain, indicating a potential role for antagonists in the treatment of pain.12 Furthermore, GluK antagonists might have a beneficial effect on neurodegenerative diseases such as multiple sclerosis,13 Alzheimer’s disease,14 and Huntington’s disease,15 as well as in several psychiatric disorders including schizophrenia,16 major depression,17 autism,18 bipolar disorder,19 and obsessive-compulsive disorder.20
In the search for subtype selective ligands for iGluRs, a substantial number of Glu analogues have been prepared and characterized pharmacologically, including >200 different 4-substituted Glu analogues by us37–42 and others35,43–47 (see Table 1). Also a number of analogues of the natural product KA have been prepared, including the structurally simplified KA analogue 3a (CPAA) as well as the conformationally restricted analogues 1b27 and 1c23 (Table 1). While several competitive agonists and antagonists are able to differentiate between the three iGluR groups,25,48 the success rate when it comes to the development of subtype-selective iGluR ligands targeting the orthosteric binding site has been low. In fact, only subtype-selective agonists and antagonists for the homomeric GluK1 receptor as well as the selective GluK3 agonist 1c (Table 1) have been disclosed to this date.
Table 1.
Chemical Structures and Affinities of AMPA, NMDA, KA, Selected Published Glu Analogues 1a−e and 2a−d, and New Series of Cε-Substituted Analogues of 3a (CPAA)a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| IC50 |
Ki |
|||||||
| compdb | AMPA | KA | NMDA | GluK1 | GluK2 | GluK3 | GluK1/GluK3 | GluK2/GluK3 |
| AMPA (a) | 0.04 | >100 | >100 | 1.2 | >1000 | 43 | 0.03 | >23 |
| KA (b) | 4 | 0.007 | >100 | 0.08 | 0.01 | 0.03 | 3 | 0.3 |
| NMDA (c) | >100 | >100 | 35 | n.d. | n.d. | n.d. | n.a. | n.a. |
| 1a (d) | >100 | 6 | >100 | 8.18 | 12.3 | 0.38 | 21 | 32 |
| 1b (e) | >100 | 14 | 3 | 0.67 | 2.1 | 0.62 | 1.1 | 3 |
| 1c (f) | 0.54 | 0.23 | >100 | 0.66 | 0.16 | 0.006 | 110 | 27 |
| 1d (g) | 2 | 23 | >100 | 0.002 | >1000 | 2.53 | 0.001 | >400 |
| 1e (h) | 10 | 24 | n.d. | 0.03 | 0.02 | 0.002 | 15 | 10 |
| 2a (i) | 0.43 | 0.36 | 0.33 | 0.14 | 0.33 | 0.49 | 0.3 | 0.7 |
| 2b (j) | 27 | 0.03 | 6 | 0.0007 | 0.02 | 0.006 | 0.1 | 3 |
| 2c (k) | 16 | 0.12 | 28 | 0.002 | 0.07 | 0.03 | 0.07 | 2 |
| 2d (l) | 2 | >100 | >100 | 0.01 | >100 | 2.25 | 0.004 | >45 |
Glutamate is a flexible molecule which can rotate around its C(2)−C(3) and C(3)−C(4) bonds. Nine distinct staggered conformations can be adapted through this rotation.49 The energy differences of these conformations are all in the same order of magnitude.50 From the extensive number of X-ray crystallographic studies of agonists bound in the agonist binding domains of GluA2 and GluK1, it has been shown that a “folded” conformation of the Glu backbone favors iGluR agonist activity over metabotropic Glu receptor (mGluR) agonist activity. Whereas an “extended” conformation is favored by the mGluRs.51,52
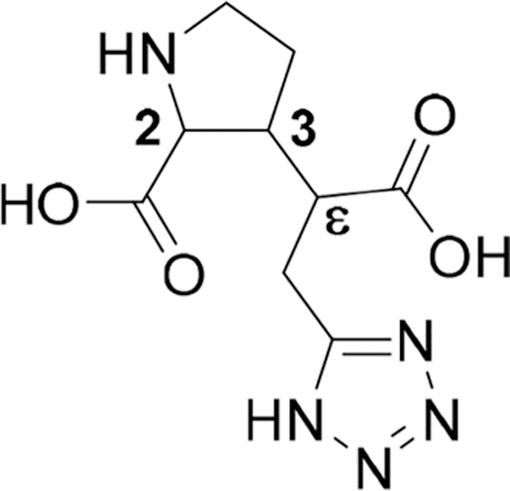
3a (CPAA) is a conformational restricted analogue of Glu, locked in the C(1)−C(3) anti conformation, but it allows for different C(2)−C(5) conformations, by rotation around the C(3)−C(4) bond. This implies that 3a can still adopt both the “folded” as well as the “extended” conformations. Conformational flexibility is reduced in the Glu backbone when substituted on the 4-position, favoring a “folded” conformation for compounds with a syn relative stereochemical relationship.53 In this study, we fused structures of series 2a−d with proline analogue 3a and present the synthesis and pharmacological evaluation of these new conformationally restricted Glu analogues.
RESULTS AND DISCUSSIONS
Chemistry.
Compound 3a and the ethyl and phenyl analogues 3c and 3d, respectively, were prepared following our previous work for the synthesis of methyl analogue 3b.26 The key step is a stereoselective 1,4-addition of the appropriate cuprate to enone 4 (Scheme 1).54 For 3a, the cuprate addition with vinyl bromide gave intermediate 6a in 65% yield as a single trans-diastereomer, determined by 1H NMR and in agreement with literature values for optical rotation.55 Simultaneous hydroboration of the olefin and reduction of the lactam functionality with borane gave alcohol 7a in 52%. Cleavage of the TBS-ether was accomplished with TBAF to yield diol 8a, which was oxidized to diacid 9a, by in situ generated ruthenium(VIII) oxide.56 Finally, removal of the Boc-group by TFA in DCM gave crude 3a, which was purified by preparative HPLC to yield 3a as the HCl salt.
Scheme 1. Synthesis of 3a, 3c-s1, 3c-s2, 3d-s1, and 3d-s2a.

aReagents and conditions: (a) t-butyllithium, CuCN, THF, −78 to −50 °C, followed by 4, TMSCl, −50 °C, 1 h, 61−82%; (b) BH3·THF, THF, rt, 20h, followed by H2O, NaOH, H2O2, 0 °C to rt, 1 h, separation of the two diastereomers by DCVC, 17−52%; (c) TBAF, THF, rt, 56−84%; (d) RuCl3, NaIO4, H2O, MeCN, EtOAc, rt, 7 h, 23−75%; (e) TFA, DCM, rt, 2 h, followed by 1 M HCl, 13−60%.
The ethyl 3c and phenyl 3d analogues (Scheme 1) were prepared, starting from enone 4 and butenyl-2-bromide (5c) or α-bromostyrene (5d), respectively. After the one-pot reduction/hydroboration reaction both intermediate alcohols 7c and 7d were separated into their respective diastereomers 7c-s1, 7c-s2, and 7d-s2. The stereoisomer 7d-s1 could not be purified to more than >90% by dry column vacuum chromatography (DCVC), but it led directly to the free diol 8d-s1 when purified by preparative HPLC.
After cleavage of the silyl ether, oxidation, and Boc-group removal, the optically pure compounds 3c-s1, 3c-s2, 3d-s2, and 3d-s1 were obtained as their corresponding HCl salts. The absolute stereochemistry could be determined for (2S,3R,εS)-9c-s1 and (2S,3R,εS)-9d-s2 by single-crystal X-ray diffraction, as they gave monocrystalline solids from EtOAc/heptane (Figure 1).
Figure 1.

Crystal structures of Boc-protected 3c-s1 (9c-s1, A) and 3d-s2 (9d-s2, B). Displacement ellipsoids of the non-hydrogen atoms are shown at the 50% probability level. Hydrogen atoms are represented by spheres of arbitrary size. Oxygen atoms are in red and nitrogen atoms in blue.
Due to the limited availability of commercial vinyl bromides and the lengthy synthesis, we set out to find a more general and convenient procedure. Several other methods for the preparation of 3a have been reported, however none of these would allow for readily modification of the substituent on the ε-carbon.57–63 We envisioned enone 10 as a suitable key intermediate for conjugate additions of different substituted malonates. Clearly, this strategy comes with the loss of absolute stereochemical control, which we accepted as the cost for a more feasible access to new analogues for a more comprehensive study of the structure−activity relationship (SAR).
Enone 10 was prepared according to literature procedure, starting from proline.64 The feasibility of this route was demonstrated by the synthesis of (±)-3a in 87% overall yield from 2.0 g/7.65 mmol of enone 10. After a short optimization, the most optimal conditions for the conjugated addition were found to be 1 equiv of NaH with 2.5 equiv of the corresponding malonate 11a, 11e−11m, stirred for 3−24h in THF at 50 °C, which yielded the desired Michael products 12a, 12e−12m, in 35−99% isolated yield (Scheme 2). The subsequent global deprotection and decarboxylation was achieved by reflux in 6 M HCl for 4−9 days. The crude of all final products 3a, 3e-3m were purified by preparative HPLC, and the d.r. determined by 1H NMR (Table 2). Exempt to this: For analogue 3i the diastereomeric mixture could be separated into its corresponding two diastereomers 3i-d1 and 3i-d2.
Scheme 2. Synthesis of 3a and 3e−m by Conjugate Addition of Substituted Malonic Ester 11a and 11e−m to Enone 10a.

aReagents and conditions: (a) NaH, THF, 50 °C, 3−24 h, 35−99%; (b) 6 M HCl, reflux, 4−9 days, 6−87%.
Table 2.
Binding Affinities of 3a−q at Native iGluRsa
 | ||||||
|---|---|---|---|---|---|---|
| IC50 |
Ki |
|||||
| compd | R-group | 2,3-transconfiguration | ε-carbon; Abs stereochem or d.r. | AMPA | KA | NMDA |
| 3ab | H | absolute (2S, 3R) | n.a. | 106 | 1.6 | 0.5 |
| 3b-d1c | Me− | absolute (2S, 3R) | 99:1 | >100 | 2.4 | 22 |
| 3b-d2c | Me− | absolute (2S, 3R) | 1:99 | >100 | 1.8 | >100 |
| 3c-s1 | Et− | absolute (2S, 3R) | S | >100 | >100 | 13 [4.88 ± 0.02] |
| 3c-s2 | Et− | absolute (2S, 3R) | R | >100 | >100 | >100 |
| 3d-s1 | Ph− | absolute (2S, 3R) | R | >100 | >100 | >100 |
| 3d-s2 | Ph− | absolute (2S, 3R) | S | >100 | >100 | >100 |
| 3e | nPr− | racemic | 63:37 | >100 | >100 | >100 |
| 3f | iPr− | racemic | 67:33 | >100 | >100 | >100 |
| 3g | Bn− | racemic | 58:42 | >100 | 71 [4.15 ± 0.04] | >100 |
| 3h | Ph(CH2)2− | racemic | n.d. | >100 | >100 | >100 |
| 3i-d1 | F− | racemic | 95:5 | >100 | 48 [4.32 ± 0.03] | 20 [4.71 ± 0.04] |
| 3i-d2 | F− | racemic | 1:99 | >100 | 9.4 [5.04 ± 0.07] | 6.0 [5.23 ± 0.05] |
| 3j-d1 | MeO− | racemic | 1:99 | >100 | >100 | >100 |
| 3k | H2N− | racemic | 50:50 | 31 [4.51 ± 0.03] | >100 | 8.9 [5.06 ± 0.02] |
| 3l | HOOCCH2− | racemic | 63:37 | >100 | 23 [4.63 ± 0.02] | 2.7 [5.58 ± 0.03] |
| 3m | HOOC(CH2)2− | racemic | 67:33 | >100 | >100 | 53 [4.28 ± 0.01] |
| 3n | HO(CH2)2− | racemic | 53:47 | >100c | >100 | 61[4.23 ± 0.06] |
| 3o | HO(CH2)3− | racemic | 56:44 | >100 | >100 | >100 |
| 3p | triazolyl-CH2− | racemic | 60:40 | >100 | 59 [4.23 ± 0.03] | >100 |
| 3p-d1 | triazolyl-CH2− | racemic | 99:1 | >100 | >100 | >100 |
| 3p-d2 | triazolyl-CH2− | racemic | 10:90 | >100 | 58 [4.24 ± 0.01] | 51 [4.30 ± 0.03] |
| 3qd | tetrazolyl-CH2− | racemic | 56:34 | >100 | 93 [4.04 ± 0.06] | 0.21 [6.69 ± 0.07] |
All values in μM. Data are mean values of at least three individual experiments performed in triplicate. For AMPA and KA: IC50 values with mean pIC50 ± SEM in brackets. For NMDA: Ki values with mean pKi ± SEM in brackets.
Data for native receptors taken from ref 67.
Data taken from ref 26.
Ki = > 1000 μM at cloned homomeric GluA2 receptors; −: not tested; n.d.: not determined; n.a.: not applicable.
For 2-hydroxyethyl analogue 3n (Scheme 3), conjugated addition to enone 10 with cyclic malonate 11n gave 12n in 71% yield. Aqueous acidic global hydrolysis yielded the corresponding γ-lactone 13, and a second step with basic hydrolysis was required to obtain the disodium salt of 3n.
Scheme 3. Synthesis of 3n from Enone 10a.

aReagents and conditions: (a) NaH, THF, 50 °C, 4 h, 71%; (b) 6 M HCl, reflux, 4 days. (c) NaOH (42% over two steps).
The homologue of 3n, analogue 3o (Scheme 4), was prepared via the standard protocol for the conjugated addiction of 11o to enone 10, followed by hydroboration of the olefin with borane to give 12o. Hydrolysis and subsequent decarboxylation was done in aq. 6 M HCl to give 3o in 28% after purification by preparative HPLC.
Scheme 4. Synthesis of 3o from Intermediate 14a.

aReagents and conditions: (a) NaH, THF, 50 °C, 4 h, 93%; (b) BH3·THF, THF, 0 °C, 2 h, followed by H2O, NaOH, H2O2, 0 °C, 1 h, 49%; (c) 6 M HCl, reflux, 3 days, 28%.
The introduction of simple five-membered heterocycles was also pursued and analogues 3p (Scheme 5) and3q (Scheme 6, Table 2) were designed. For the synthesis of triazole 3p, conjugated addition of 11p to enone 10 yielded 15 in 89%. The propargyl functionality in 15 was then converted to the corresponding triazole 12p by reaction with azidotrimethylsilane.65 Global deprotection of 12p was readily achieved and gave 3p in 78%.
Scheme 5. Synthesis of 3p from Alkyne 15a.

aReagents and conditions: (a) NaH, THF, 50 °C, 4 h, 89%; (b) TMSN3, CuI, DMF, MeOH, reflux, 18 h, 71%; (c) 6 M HCl, reflux, 3 days, 78%.
Scheme 6. Synthesis of 3q and Separation of Stereoisomersa.

aReagents and conditions: (a) NaH, BrCH2CN, THF, 50 °C, 23 h, 83%; (b) NaN3, Et3NHCl, toluene, 115 °C, 19 h, 55% (c) separation of the 2,3-trans enantiomers by preparative HPLC using a ChiralPak AD column, 26−29%; (d) Pd/C, H2, MeOH, r.t. 24 h, quant.; (e) 6 M HCl, reflux, 4 days, 99%; (f) separation of diastereoisomers by preparative HPLC using Chirobiotic T column, 18−80%.
The synthesis of the tetrazole 3q (Scheme 6, Table 2) commenced with an alkylation of (±)-trans-12a with sodium hydride and bromoacetonitrile to yield nitrile (±)-trans-16 (Scheme 6). Subsequent reaction with sodium azide and triethylammonium chloride converted the nitrile functionality into tetrazole (±)-trans-17.66 At this stage, the enantiomers were separated by chiral preparative HPLC to afford the two optically pure 2,3-trans enantiomers 17-s1 and 17-s2. Hydrogenation over a Pd/C catalyst afforded the free aminetetrazoles 18-s1 and 18-s2, respectively, in quantitative yields. Each enantiomer was subjected to a global deprotection and decarboxylation under aqueous acidic conditions which afforded a mixture of diastereomers at the ε-position. At this stage, the two diastereomers 3q-s1 and 3q-s2 could be separated on chiral preparative HPLC to afford the two optically active enantiomers. Unfortunately, the diastereomeric mixture 3q-s3−4 could not be separated using the HPLC columns available to us.
Pharmacological Characterization.
Binding affinities of 3a,c−q were determined at native iGluRs (rat synaptosomes) (Table 2; Figure 2), and recombinant rat homomeric GluK1−3 receptors expressed in sf9 cells (Table 3) and at recombinant EAAT1−3 expressed in HEK293 cells in a conventional [3H]-d-Asp uptake assay (Table 4). Furthermore, functional studies on recombinant NMDA receptors expressed in Xenopus lævis oocytes were carried out using two-electrode voltage-clamp electrophysiology (Table 5).
Figure 2.

Concentration−inhibition data of 3q-s1−4 and 3q-s2 at NMDA receptor subtypes. (A) Representative two-electrode voltage-clamp recordings showing inhibition of GluN1/2A and GluN1/2D by 3q-s1−4. Receptors were activated by 100 μM glycine and 1 μM glutamate. (B) Concentration−inhibition data for 3q-s1−4 at different NMDA receptor subtypes (GluN1/2A-D). (C) Concentration−inhibition data for 3q-s2 at different NMDA receptor subtypes (GluN1/2A−D). Data points are shown as mean ± SEM from six oocytes. The Ki values were derived from the IC50 values using the Cheng−Prusoff relationship (Table 1).
Table 3.
Binding Affinities of 3a−q at Cloned Homomeric Kainate Receptorsa
 | |||||||
|---|---|---|---|---|---|---|---|
|
Ki |
|||||||
| compd | R-group | 2,3-trans configuration | ε-carbon; Abs stereochem or d.r. | GluK1 | GluK2 | GluK3 | GluK1/K3 |
| 3a | H | absolute (2S, 3R) | n.a. | 2.8 | 2.2 | 0.51 | 6 |
| 3b-d1b | Me− | absolute (2S, 3R) | 99:1 | 0.23 | 3.8 | 0.33 | 1 |
| 3b-d2b | Me− | absolute (2S, 3R) | 1:99 | 0.39 | 0.51 | 0.10 | 4 |
| 3c-s1 | Et− | absolute (2S, 3R) | S | 18 ± 3.8 | >100 | 11 ± 2.4 | 2 |
| 3c-s2 | Et− | absolute (2S, 3R) | R | 41 ± 14 | 70 ± 14 | 6.3 ± 1.3 | 7 |
| 3d-s1 | Ph− | absolute (2S, 3R) | R | >100 | >100 | >100 | n.a. |
| 3d-s2 | Ph− | absolute (2S, 3R) | S | >100 | >100 | >100 | n.a. |
| 3e | nPr− | racemic | 63:37 | >100 | >100 | 87 ± 16 | n.a. |
| 3f | iPr− | racemic | 67:33 | >100 | >100 | >100 | n.a. |
| 3g | Bn− | racemic | 58:42 | 46 ± 2.2 | 67 ± 4.6 | 5.4 ± 0.42 | 9 |
| 3h | Ph(CH2)2− | racemic | n.d. | >100 | >100 | 86 ± 7.3 | n.a. |
| 3i-d1 | F− | racemic | 95:5 | 35 ± 0.26 | 66 ± 20.2 | 5.3 ± 0.13 | 7 |
| 3i-d2 | F− | racemic | 1:99 | 3.3 ± 0.18 | 2.6 ± 0.15 | 0.71 ± 0.08 | 5 |
| 3j-d1 | MeO− | racemic | 1:99 | >100 | >100 | 82 ± 11.2 | n.a. |
| 3k | H2N− | racemic | 50:50 | >100 | >100 | 33 ± 2.4 | >3 |
| 3l | HOOCCH2− | racemic | 63:37 | 2.0 ± 0.16 | 33 ± 1.1 | 6.4 ± 1.2 | 0 |
| 3m | HOOC(CH2)2− | racemic | 67:33 | >100 | >100 | >100 | n.a. |
| 3n | HO(CH2)2− | racemic | 53:47 | >100 | >100 | 52 ± 3.0 | >2 |
| 3o | HO(CH2)3− | racemic | 56:44 | >100 | >100 | >100 | n.a. |
| 3p | triazolyl-CH2− | racemic | 60:40 | 14 ± 2.0 | 55 ± 9.0 | 3.1 ± 0.14 | 5 |
| 3p-d1 | triazolyl-CH2− | racemic | 99:1 | ∼100 | >100 | 6.6 ± 0.09 | ∼15 |
| 3p-d2 | triazolyl-CH2− | racemic | 10:90 | − | 43 ± 5.2 | 2.7 ± 0.32 | n.a. |
| 3qc | tetrazolyl-CH2− | racemic | 56:34 | 2.1 ± 0.1 | 61d | 4.9 ± 0.5 | 0 |
All values in μM. Data are mean values of at least three individual experiments performed in triplicate. Ki values reported are the mean ± SEM
Data taken from ref 26.
Ki = >1000 μM at cloned homomeric GluA2 receptors
Tested once; −: not tested; n.d.: not determined; n.a.: not applicable.
Table 4.
Pharmacological Characterization of 3a,c−q at EAAT1, EAAT2, and EAAT3 Stably Expressed in HEK293 Cellsa
 | ||||||
|---|---|---|---|---|---|---|
| IC50 |
||||||
| compd | R-group | 2,3-trans configuration | ε-carbon; Absstereochem or d.r. | EAAT1 | EAAT2 | EAAT3 |
| 3a | H | absolute (2S, 3R) | n.a. | >300 | >300 | >300 |
| 3c-s1 | Et− | absolute (2S, 3R) | S | ∼100 [∼4.0] | ∼30 [∼4.5] | ∼50 [∼4.3] |
| 3c-s2 | Et− | absolute (2S, 3R) | R | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3d-s1 | Ph− | absolute (2S, 3R) | R | >300 [<3.5] | ∼50 [∼4.3] | >300 [<3.5] |
| 3d-s2 | Ph− | absolute (2S, 3R) | S | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3e | nPr− | racemic | 63:37 | ∼500 [∼3.3] | ∼100 [∼4.0] | ∼1000 [∼3.0] |
| 3f | iPr− | racemic | 67:33 | 45 [4.34 ± 0.13] | 16 [4.80 ± 0.07] | 35 [4.45 ± 0.06] |
| 3g | Bn− | racemic | 58:42 | 34 [4.46 ± 0.05] | 20 [4.70 ± 0.04] | ∼200 [∼4.4] |
| 3h | Ph(CH2)2− | racemic | n.d. | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3i-d1 | F− | racemic | 95:5 | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3i-d2 | F− | racemic | 1:99 | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3j-d1 | MeO− | racemic | 1:99 | >1000 [<3.0] | ∼300 [∼3.5] | >1000 [<3.0] |
| 3k | H2N− | racemic | 50:50 | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3l | HOOCCH2− | racemic | 63:37 | >1000 [<3.0] | >1000 [<3.0] | >1000 [<3.0] |
| 3m | HOOC(CH2)2− | racemic | 67:33 | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3n | HO(CH2)2− | racemic | 53:47 | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3o | HO(CH2)3− | racemic | 56:44 | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3p | triazolyl-CH2− | racemic | 60:40 | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3p-d1 | triazolyl-CH2− | racemic | 99:1 | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
| 3p-d2 | triazolyl-CH2− | racemic | 10:90 | − | − | − |
| 3q | tetrazolyl-CH2− | racemic | 56:34 | >300 [<3.5] | >300 [<3.5] | >300 [<3.5] |
IC50 values are given in μM with mean pIC50 ± SEM in brackets. Data are mean values of at least three individual experiments performed in triplicate. −: not tested; n.d.: not determined.
Table 5.
Binding Affinities of 3q-s1−4 at Native NMDA Receptors and Functional Characterization as Antagonists at Cloned GluN1/GluN2A-D Receptorsa
 | |||||||
|---|---|---|---|---|---|---|---|
|
Ki |
|||||||
| compd | 2,3-trans configuration | Abs stereochem or d.r. at ε-carbon | NMDA (binding affinity) | GluN1/GluN2A | GluN1/GluN2B | GluN1/GluN2C | GluN1/GluN2D |
| 3q-s1 | (−)-Abs (n.d.) | 96:4 | 28 [4.56 ± 0.06] | − | − | − | − |
| 3q-s2 | (−)-Abs (n.d.) | 4:96 | 1.3 [5.89 ± 0.09] | 1.2 ± 0.1 | 2.8 ± 0.1 | 6.5 ± 0.6 | 15 ± 2 |
| 3q-s3,4 | (+)-Abs (n.d.) | 1:2 | 0.25 [6.61 ± 0.07] | 0.61 ± 0.02 | 2.7 ± 0.1 | 12 ± 1 | 62 ± 9 |
All values in μM. Binding data are mean values of at least three individual experiments performed in triplicate. For NMDA: Ki values with mean pKi ± SEM in brackets. For GluN1/GluN2A-D, Ki values are reported as the mean ± SEM from six oocytes and were estimated from IC50 values using the Cheng−Prusoff relationship (see Methods). −: not tested; n.d.: not determined.
Extension of the methyl group of 3b to the corresponding ethyl 3c-s2 and 3c-s1 resulted in an overall decrease of binding affinity for iGluRs, however not all receptor subtypes were equally affected, resulting in a 7-fold selectivity for GluK3 over GluK1. Extending the alkyl chain further to n-propyl, analogue 3e, and i-propyl, analogue 3f, abolishes all affinities for iGluRs. Interestingly, inhibition of the glutamate transporters (EAATs) is restored, with an i-propyl group in the ε-position, making it a selective inhibitor of these transporters. Introduction of a phenyl ring, compound 3d, results in loss of affinity for the iGluRs and inhibitory activity at EAAT1−3. Insertion of a tethering methylene group provides the corresponding racemic benzyl 3g, which then shows affinity for KA receptors, with a selectivity of 9-fold for GluK3 over GluK1. This compound also moderately inhibited EAAT1 and EAAT2, displaying a 10-fold selectivity for these subtypes over EAAT3. Extending the spacer to an ethylene group gives compound 3h, which again shows a decrease in affinity for KA receptors, compared to benzyl analogue 3g.
Introduction of a fluoride atom in to the ε-position of 3a provides analogue 3i, which was isolated as its two separate diastereomers 3i-d1 and 3i-d2. For both, the binding affinities for the iGluRs were improved, compared to 3a, most prominently reflected in the nanomolar affinity exhibited by 3i-d2 at GluK3. Although the GluK1/GluK3 selectivity ratio for 3i-d1 is slightly higher than for 3i-d2 (7 vs 5), both analogues are interesting as chemical tools as they are completely devoid of inhibitory activity at EAAT1−3.
For the methoxy analogue 3j, only one of the two diastereomers could be isolated, and it proved to be without significant binding affinity for iGluRs or functional activity at EAAT1−3. Analogue 3k is distinct from the other analogues in the series, as it comprises two α-amino acid functionalities. Thus, two generally distinct binding modes can be envisaged. While this speculation was not investigated in further detail herein, 3k shows midrange micromolar affinity for both AMPA and NMDA receptors as well as the GluK3 subunit.
The two analogues 3l and 3m both contain additional carboxylic acid functionalities in the ε-position. While 3l displays high-nanomolar to mid-micromolar affinity to all iGluRs tested, 3m is a selective albeit weak NMDA receptor ligand (Ki = 53 μM). Both 3l and 3m display no functional activity at EAAT1−3. The 2′-hydroxyethyl analogue 3n is a low affinity ligand with midrange micromolar affinity for native NMDA receptors as well as GluK3. Extending the side chain by a methylene group, alcohol 3o, leads to an inactive compound in all assays tested here.
The diastereomeric mixture of triazole analogue 3p showed selectivity for native KA receptors as well as for the cloned homomeric GluK1−3 receptors. Most interestingly, upon separation of the two diastereomers, the broad-range KA receptor affinity profile was found to be retained in 3p-d2, while the diastereomer 3p-d1 displays low micromolar binding affinity for the GluK3 subtype (Ki = 6.6 μM) with a GluK1/GluK3 selectivity ratio of 15 and negligible binding affinity for other iGluR subtypes and EAAT1−3. This profile makes it an attractive chemical tool compound for studying the biological role of the GluK3 subtype.
Lastly, the diastereomeric mixture of tetrazole analogue 3q displays high-nanomolar to mid-micromolar affinities for cloned homomeric GluK1−3 receptors. However, this mixture exhibits midnanomolar affinity (Ki = 0.21 μM) for native NMDA receptors, while having no significant binding affinity for the other native iGluRs. Enantiomer 3q-s1 displayed a significant loss in affinity for native NMDA receptors and was not further characterized (Table 5). Enantiomer 3q-s2 retained the affinity for native NMDA receptors, and functional characterization showed this compound to be a low to midrange micromolar antagonist for GluN1/GluN2A-D receptors. A most interesting result was obtained with the diastereomeric mixture 3q-s3−4, which also acted as an antagonist at GluN1/GluN2A-D receptors, showing preference for GluN1/GluN2A receptors (Ki = 0.61 μM) over GluN1/GluN2B-D receptors (Ki = 2.7−62 μM).
CONCLUSION
In conclusion, two synthetic strategies were taken for the synthesis of the herein reported 2,3-trans proline analogues 3a,b−q. Five analogues, including 3a (CPAA), were prepared by a linear five-step enantioselective strategy from common intermediate enone 4. A more efficient racemic route was developed, and starting from common intermediate enone 10 final compounds were obtained in only two or three steps, in overall yields of up to 87%. Furthermore, this procedure allowed for expedited gram scale synthesis of (±)-3a.
Pharmacological characterization at the iGluRs and EAATs show that the added substituents on the ε-carbon of 3a follow a different SAR compared to the 4-substituted Glu analogues and are generally unfavorable, with the following exceptions: (1) Introduction of a triazolyl-CH2− group gave 3p as a mixture of four stereoisomers, with a pharmacological profile of low-to-mid micromolar affinity for GluK1−3. Diastereomers were separated and notably 3p-d1 showed reduced affinity for GluK1,2, i.e., enhanced 15-fold selectivity for GluK3 (Ki = 6.6 μM) with no significant affinity for any of the native Glu receptors and EAAT transporters tested. In comparison with other known GluK3-preferring ligands, the general profile of 3p makes it an attractive tool compound for further exploring and understanding of the structural requirements for GluK3 selectivity and studying the biological role of the GluK3 subtype. (2) Exchanging the triazolyl for a tetrazolyl gave 3q, as a mixture of four stereoisomers, which displayed selective midrange nanomolar binding affinity at native NMDA receptors over native AMPA and KA receptors. (3) The diastereomeric mixture 3q-s3,4 was shown to be an antagonist at the GluN1/GluN2A receptor (Ki = 0.61 μM) with a 4−10-fold preference over GluN1/GluN2B−D, and may serve as a starting point for a fully GluN2A subtype selective antagonist.
METHODS
Chemistry.
All reactions involving dry solvents or sensitive agents were performed under a nitrogen atmosphere and glassware was dried prior to use. Commercially available chemicals were used without further purification. Solvents were dried prior to use with an SG water solvent purification system or dried by standard procedures, and reactions were monitored by analytical thin-layer chromatography (TLC, Merck silica gel 60 F254 aluminum sheets). Dry-column vacuum chromatography (DCVC) was carried out using Merck silica gel 60A (15−40 μm),68 and flash chromatography was carried out using Merck silica gel 60A (35−70 μm). 1H NMR spectra were recorded on a 400 MHz Bruker Avance III or 600 MHz Bruker Avance III HD, and 13C NMR spectra were recorded on a 101 MHz Bruker Avance III or 151 MHz Bruker Avance III HD. Chemical shifts are reported in δ (ppm) relative to the singlet at δ = 7.26 ppm of CDCl3 and to the singlet at 4.79 ppm of D2O for 1H NMR, and to the center line of the triplet at δ = 77.16 ppm of CDCl3 and to the singlet at 67.19 ppm of 1,4-dioxane used as external reference in D2O for 13C NMR. Analytical HPLC was performed using an UltiMate HPLC system consisting of an LPG-3400A pump (1 mL/min), a WPS-3000SL autosampler, and a 3000 Diode Array Detector installed with a Gemini-NX C18 (250 × 4.60 mm, 3 μm) column. Solvent A: H2O + 0.1% TFA; Solvent B: MeCN·H2O 9:1 + 0.1% TFA. For HPLC control, data collection and data handling, Chromeleon software v. 6.80 was used. Preparative HPLC was carried out on an Ultimate HPLC system with an LPG-3200BX pump, a Rheodyne 9721i injector, a 10 mL loop, an MWD-3000SD detector (200, 210, 225, and 254 nm), and a Gemini-NX C18 (250 × 21.2 mm, 5 μm) column for preparative purifications or a Gemini-NX C18 (250 × 10.00 mm, 5 μm) column for semipreparative purifications. Solvent A: H2O + 0.1% TFA; Solvent B: MeCN-H2O 9:1 + 0.1% TFA. For HPLC control, data collection and data handling, Chromeleon software v. 6.80 was used. Chiral preparative HPLC was performed using the same instrumentation as mentioned above. UPLC-MS spectra were recorded using an Acquity UPLC H-Class Waters series solvent delivery system equipped with an autoinjector coupled to an Acquity QDa and TUV detectors installed with an Acquity UPLCBECH C18 (50 × 2.1 mm, 1.7 μm) column. Solvent A: MeCN-H2O 95:5 + 0.1% HCO2H; Solvent B: MeCN + 0.1% HCO2H. For data collection and data handling, MassLynx software was used. Optical rotations were determined in a thermostated cuvette on an Anton Paar MCP300 Modular Circular Polarimeter. Compounds were dried under high vacuum or freeze-dried using a ScanVac Cool Safe Freeze Drier. The purity of compounds submitted for pharmacological characterization was determined by 1H NMR and HPLC to be >95%.
General Procedure I.
NaH (60% in mineral oil) (1.0 equiv) was suspended in THF (4 mL), to which was added dropwise the corresponding malonate (2.5 equiv) and stirred for 15 min at rt. Enone 10, which was prepared according to literature procedure, starting from proline,64 was dissolved in THF (2 mL) and added dropwise to the reaction. The reaction was heated to 50 °C and stirred for 3−24 h. The crude was extracted with EtOAc, and the combined organic fractions were washed with brine and dried over MgSO4. The crude was purified by DCVC or flash chromatography.
General Procedure II.
12e−p (mixture of trans enantiomers) was suspended in excess 6 M HCl(aq.) and stirred under reflux for 4−9 days. The excess of HCl and H2O was evaporated under reduced pressure to give a residue which was dissolved in water or DMSO (2 mL) and without any further purification submitted to preparative HPLC. The isolated fractions were treated with 3 × 1 M HCl(aq.), and the volatiles were evaporated under reduced pressure. The final product was freeze-dried to afford the mixture of trans diastereomers.
(2S,3R)-tert-Butyl 2-(((tert-Butyldimethylsilyl)oxy)methyl)-5-oxo-3-vinylpyrrolidine-1-carboxylate (6a).
Vinyl bromide (1.0 M in THF) (14.05 mL, 14.05 mmol, 2.3 equiv) was dissolved in dry THF (26 mL). The solution was cooled to −78 °C. t-Butyllithium (1.7 M in THF) (17.39 mL, 28.70 mmol, 4.7 equiv) was added dropwise, and the reaction was stirred for 30 min at −78 °C. A suspension of CuCN (656 mg, 7.33 mmol, 1.2 equiv) in dry THF (10 mL) was added dropwise, and the reaction mixture was stirred at −42 °C for 15 min. The reaction mixture was cooled to −78 °C, and a solution of 4 (2.0 g, 6.11 mmol, 1 equiv) in dry THF (24 mL) was added dropwise, followed by chlorotrimethylsilane (1.78 mL, 14.05 mmol, 2.3 equiv). The reaction mixture was stirred at −42 °C for 1 h and was quenched with sat. NH4Cl (20 mL). Brine (40 mL) was added, and the product was extracted with EtOAc (4 × 40 mL). The organic layers were combined, washed with brine (60 mL), and dried over Na2SO4. The crude product was purified by DCVC (0 → 10% EtOAc in heptane), which afforded the product as a colorless oil (1.41 g, 3.97 mmol, 65%). LCMS (ESI) m/z = 356.2 [M + H]+; Rf = 0.45 (3:1 heptane/EtOAc); [α]25589 = −37.3° (c = 0.31, EtOH); 1H NMR (300 MHz, CDCl3) δ 5.91−5.78 (m, 1H), 5.11 (dt, J = 17.10, 1.10 Hz, 1H), 5.08 (dt, J = 10.20, 0.80 Hz, 1H), 3.94 (dd, J = 10.45, 3.85, 1H), 3.90−3.86 (m, 1 H), 3.72 (dd, J = 10.18, 1.93 Hz, 1H), 2.97−2.84 (m, 2H), 2.32−2.19 (m, 1H), 1.52 (s, 9H), 0.87 (s, 9H), 0.05 (s, 3H) 0.03 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 173.6, 149.7, 139.1, 114.8, 82.8, 64.4, 63.3, 38.1, 37.1, 28.0, 25.8, 18.2, 5.5.
tert-Butyl (2S,3R)-3-(But-1-en-2-yl)-2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxopyrrolidine-1-carboxylate (6c).
2-Bromobut-1-ene (1.89 g, 14.05 mmol, 2.3 equiv) was dissolved in dry THF (40 mL). The solution was cooled to −78 °C. tert-Butyllithium (1.45 M in THF) (19.79 mL, 28.70 mmol, 4.7 equiv) was added dropwise, and the reaction was stirred for 30 min at −78 °C. A suspension of CuCN (656 mg, 7.33 mmol, 1.2 equiv) in dry THF (10 mL) was added dropwise, and the reaction mixture was stirred at −42 °C for 15 min. The reaction mixture was cooled to −78 °C, and a solution of 4 (2.0 g, 6.11 mmol, 1 equiv) in dry THF (25 mL) was added dropwise, followed by chlorotrimethylsilane (1.78 mL, 14.05 mmol, 2.3 equiv). The reaction mixture was warmed to −42 °C and stirred for 1 h. The reaction mixture was quenched with sat. NH4Cl (40 mL). Brine (80 mL) was added, and the product was extracted with EtOAc (4 × 80 mL). The organic layers were combined, washed with brine (60 mL), and dried over MgSO4. The crude product was purified by DCVC (0 → 4% EtOAc in heptane), which afforded the product as a colorless oil (1.43 g, 3.73 mmol, 61%). LCMS (ESI) m/z = 384.3 [M + H]+; Rf = 0.52 (3:1 heptane/EtOAc); [α]25589 = −45.4° (c = 0.41, EtOH); 1H NMR (400 MHz, CDCl3) δ 4.82 (t, J = 1.38 Hz, 1H), 4.81 (s, 1H), 3.96 (ddd, J = 4.20, 2.40, 1.50 Hz, 1H), 3.90 (dd, J = 10.30, 4.30 Hz, 1H), 3.73 (dd, J = 10.29, 2.26 Hz, 1H), 2.93 (dd, J = 17.30, 9.54 Hz, 1H), 2.83 (d, J = 9.50 Hz, 1H), 2.34 (dd, J = 17.32, 1.51 Hz, 1H), 2.05 (ttq, J = 15.50, 15.50, 8.80, 8.80, 1.30, 1.30, 1.30 Hz, 2H), 1.54 (s, 9H), 1.07 (t, J = 7.40 Hz, 3H), 0.89 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 174.1, 152.0, 150.1, 108.0, 82.8, 64.0, 63.9, 38.8, 38.0, 28.1, 27.0, 25.8, 18.2, 12.1, −5.5.
tert-Butyl (2S,3R)-2-(((tert-Butyldimethylsilyl)oxy)methyl)-5-oxo-3-(1-phenylvinyl)pyrrolidine-1-carboxylate (6d).
α-Bromostyrene (2.57 g, 14.05 mmol, 2.3 equiv) was dissolved in dry THF (40 mL). The solution was cooled to −78 °C. t-Butyllithium (1.45 M in THF) (19.79 mL, 28.70 mmol, 4.7 equiv) was added dropwise, and the reaction was stirred for 30 min at −78 °C. A suspension of CuCN (656 mg, 7.33 mmol, 1.2 equiv) in dry THF (10 mL) was added dropwise, and the reaction was stirred at −42 °C for 15 min. The reaction mixture was cooled to −78 °C, and a solution of 4 (2.0 g, 6.11 mmol, 1 equiv) in dry THF (25 mL) was added dropwise, followed by chlorotrimethylsilane (1.78 mL, 14.05 mmol, 2.3 equiv). The reaction mixture was warmed to −42 °C and stirred for 1 h. The reaction mixture was quenched with sat. NH4Cl (40 mL). Brine (80 mL) was added, and the product was extracted with EtOAc (4 × 80 mL). The organic layers were combined, washed with brine (60 mL), and dried over MgSO4. The crude product was purified by DCVC (0 → 4% EtOAc in heptane), which yielded the product as a colorless oil (2.16 g, 5.01 mmol, 82%). LCMS (ESI) m/z = 432.3 [M + H]+; Rf = 0.48 (3:1 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.41−7.27 (m, 5H), 5.36 (s, 1H), 5.11 (d, J = 1.0 Hz, 1H), 4.10−4.06 (m, 1H), 3.91 (dd, J = 10.5, 4.5 Hz, 1H), 3.75 (dd, J = 10.5, 2.4 Hz, 1H), 3.42 (dd, J = 9.0, 1.2 Hz, 1H), 3.05 (dd, J = 17.7, 9.2 Hz, 1H), 2.46 (dd, J = 17.6, 1.6 Hz, 1H), 1.51 (s, 9H), 0.89 (s, 8H), 0.05 (s, 3H), 0.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 173.9, 150.2, 150.0, 140.7, 128.7, 128.1, 126.7, 112.3, 83.1, 64.1, 64.1, 38.4, 37.4, 28.2, 26.0, 18.4, −5.4.
(2S,3R)-tert-Butyl 2-(((tert-Butyldimethylsilyl)oxy)methyl)-3-(2-hydroxyethyl)pyrrolidine-1-carboxylate (7a).
6a (600 mg, 1.69 mmol, 1 equiv) was dissolved in dry THF (25 mL). BH3−THF complex (1 M in THF) (10.12 mL, 10.12 mmol, 6 equiv) was added dropwise, and the reaction was stirred for 4 h under reflux. The reaction was allowed to cool down to rt, THF (20 mL) was added, and the reaction was cooled to 0 °C. H2O (1.2 mL) was added carefully, followed by dropwise addition of 2 M NaOH(aq.) (24 mL, 48 mmol, 28.40 equiv) and H2O2 in H2O (30% w/w) (4.8 mL, 47.0 mmol, 28 equiv). The reaction was stirred vigorously for 1 h at rt, it was quenched with sat. NaHCO3 solution (42 mL), and then it was stirred for an additional 30 min. The product was extracted with Et2O/THF 1:1 (3 × 50 mL),and the combined organic layers were washed with brine (120 mL) and dried over MgSO4. The crude product was purified by DCVC (0 → 30% EtOAc in heptane), which afforded the product as a colorless oil (315 mg, 0.88 mmol, 52%). LCMS (ESI) m/z = 260.3 [M + H]+; Rf = 0.58 (2:1 EtOAc/heptane); [α]25589 = −37.7° (c = 0.26, EtOH); 1H NMR (300 MHz, CDCl3) δ 5.01 (d, J = 7.43 Hz, 1H), 3.83−3.50 (m, 5H), 3.25 (ddd, J = 11.00, 8.66, 6.74 Hz, 1H), 2.11−1.90 (m, 2H), 1.90−1.71 (m, 2H), 1.71− 1.53 (m, 1H), 1.49 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 154.3, 79.4, 63.3, 61.2, 46.1, 38.1, 36.8, 30.2, 28.6, 25.9, 18.3, −3.5, −5.3.
tert-Butyl (2S,3R)-2-(((tert-Butyldimethylsilyl)oxy)methyl)-3-((S)-1-hydroxybutan-2-yl)pyrrolidine-1-carboxylate (7c-s1).
6c (1.85 g, 4.82 mmol, 1 equiv) was dissolved in dry THF (75 mL). BH3−THF complex (1 M in THF) (24.1 mL, 24.1 mmol, 5 equiv) was added dropwise, and the reaction was stirred for 20 h at rt. The reaction was cooled to 0 °C, and THF (37 mL) was added. H2O (3.7 mL) was added carefully, followed by dropwise addition of 2 M NaOH (72.3 mL, 144.68 mmol, 30 equiv) and H2O2 in H2O (30% w/w) (13.72 mL, 135.04 mmol, 28 equiv). The reaction was stirred vigorously for 1 h at rt, it was quenched with sat. NaHCO3 solution (150 mL), and then it was stirred for an additional 1 h. The product was extracted with EtOAc (3 × 150 mL), and the combined organic layers were washed with brine (200 mL) and dried over MgSO4. The diastereomers were separated and purified by DCVC (10 → 22% EtOAc in heptane), which afforded the product as a colorless oil (515 mg, 1.33 mmol, 28%). LCMS (ESI) m/z = 388.3 [M + H]+; Rf = 0.61 (2:1 EtOAc/heptane); [α]25589 = −32.3° (c = 1.035, EtOH); 1H NMR (400 MHz, CDCl3) δ 3.71 (br s, 2H), 3.68−3.49 (m, 3H), 3.48−3.39 (m, 1H), 3.26−3.15 (m, 1H), 2.46 (br s, 1H), 2.05−1.94 (m, 2H), 1.74−1.59 (m, 1H), 1.45 (s, 9H), 1.42−1.27 (m, 2H), 0.92 (t, J = 7.30 Hz, 4H), 0.89 (s, 9H), 0.05 (br s, 6H); 13C NMR (100 MHz, CDCl3) (rotamers in brackets) δ 154.2, 79.4 [79.0], 64.4 [63.4], 63.3 [63.1], 60.7, [60.4], 46.6 [45.9], 44.9 [44.8], 41.7 [40.2], 28.5, 27.5 [26.6], 25.9, 21.0 [20.7], 18.3, 11.9 [11.7], −5.5.
tert-Butyl (2S,3R)-2-(((tert-Butyldimethylsilyl)oxy)methyl)-3-((R)-1-hydroxybutan-2-yl)pyrrolidine-1-carboxylate (7c-s2).
From the same reaction as 7c-s1 which afforded the product as a colorless oil (524 mg, 1.35 mmol, 28%). LCMS (ESI) m/z = 388.3 [M + H]+; Rf = 0.65 (2:1 EtOAc/heptane); [α]25589= −35.1° (c = 0.75, EtOH); 1H NMR (400 MHz, CDCl3) δ 3.79 (dd, J = 9.30, 3.50 Hz, 1H), 3.76−3.66 (m, 2H), 3.62 (dd, J = 11.50, 4.00 Hz, 1 H), 3.59−3.50 (m, 1H), 3.50−3.38 (m, 1H), 3.19 (dd, J = 17.60, 7.30 Hz, 1H), 2.30 (br s, 1H), 1.99 (dtd, J = 13.40, 7.40, 7.40, 5.90 Hz, 1H), 1.71−1.59 (m, 1H), 1.46 (s, 9H), 1.44−1.36 (m, 2H), 1.35−1.27 (m, 1H), 0.93 (t, J = 7.40 Hz, 3H), 0.90 (s, 9H), 0.07 (s, 6H); 13C NMR (100 MHz, CDCl3) (rotamers in brackets) δ 154.2, 79.5 [79.1], 64.8 [63.7], 62.5, 61.1 [61.0], 46.2 [45.5], 45.1 [44.9], 42.6 [41.0], 28.5, 27.9 [27.3], 25.9, 21.4 [21.4], 18.3, 11.7, −5.5.
tert-Butyl (2S,3R)-2-(((tert-Butyldimethylsilyl)oxy)methyl)-3-((S)-2-hydroxy-1-phenylethyl)pyrrolidine-1-carboxylate (7d-s2).
6d (4.21 g, 9.75 mmol, 1 equiv) was dissolved in dry THF (160 mL). BH3−THF complex (1 M in THF) (48.8 mL, 48.8 mmol, 5 equiv) was added dropwise, and the reaction was stirred for 20 h at rt. The reaction was cooled to 0 °C, and THF (55 mL) was added. H2O (7.6 mL) was added carefully, followed by dropwise addition of 2 M NaOH(aq.) (146 mL, 292.61 mmol, 30 equiv) and H2O2 in H2O (30% w/w) (27.9 mL, 273.10 mmol, 28 equiv). The reaction was stirred vigorously for 1 h at rt, it was quenched with sat. NaHCO3 solution (320 mL), and it was stirred for an additional hour. The product was extracted with EtOAc (3 × 150 mL), and the combined organic layers were washed with brine (200 mL) and dried over MgSO4. The diastereomers were separated and purified by DCVC (10 → 26% EtOAc in heptane), which afforded the product as a colorless oil (723 mg, 1.66 mmol, 17%). LCMS (ESI) m/z = 436.3 [M + H]+; Rf = 0.68 (1:1 EtOAc/heptane); [α]25589= −35.1° (c = 0.75, EtOH); 1H NMR (400 MHz, CDCl3) δ 7.37−7.27 (m, 2H), 7.26−7.19 (m, 3H), 3.96−3.79 (m, 4H), 3.68−3.40 (m, 1H), 3.31 (dt, J = 8.64, 35.37 Hz, 1H), 3.22−3.09 (m, 1H), 2.68 (dt, J = 5.54, 11.06 Hz, 1H), 2.65−2.54 (m, 1H), 2.18−1.91 (m, 1H), 1.77 (q, J = 7.06, 7.50, 14.04 Hz, 1H), 1.47 (d, J = 12.55 Hz, 9H), 1.39−1.30 (m, 1H), 0.93 (s, 9H), 0.10 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 154.5, 128.9, 128.8, 128.6, 127.1, 62.2, 51.2, 28.7, 26.1, 25.9, 21.2, 18.5, 14.3, 14.2, −5.2.
(2S,3R)-tert-Butyl 3-(2-Hydroxyethyl)-2-(hydroxymethyl)-pyrrolidine-1-carboxylate (8a).
7a (440 mg, 1.23 mmol, 1 equiv) was dissolved in dry THF (12 mL), and TBAF (1 M in THF) (2.09 mL, 2.09 mmol, 1.7 equiv) was added dropwise. The reaction was stirred for 1 h at rt. The reaction was quenched with sat. NaHCO3 (6 mL), and H2O (6 mL) was added. The aqueous phase was extracted with EtOAc (3 × 30 mL). The combined organic fractions were washed with brine (100 mL) and dried over MgSO4. The crude product was purified by DCVC (100% EtOAc), which afforded the product as a colorless oil (253 mg, 1.03 mmol, 84%). LCMS (ESI) m/z = 246.2 [M + H]+; Rf = 0.21 (100% EtOAc); [α]25589 = −38.4° (c = 0.19, EtOH); 1H NMR (300 MHz, CDCl3) δ 5.01 (d, J = 7.43 Hz, 1H), 3.83−3.50 (m, 5H), 3.25 (ddd, J = 11.00, 8.66, 6.74 Hz, 1H), 2.11−1.90 (m, 2H), 1.90−1.71 (m, 2H), 1.71−1.53 (m, 1H), 1.49 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.8, 80.3, 66.7, 65.6, 60.9, 46.5, 38.2, 36.1, 30.2, 28.4
tert-Butyl (2S,3R)-3-((S)-1-Hydroxybutan-2-yl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (8c-s1).
7c-s1 (533 mg, 1.38 mmol, 1 equiv) was dissolved in dry THF (15 mL), and TBAF (1 M in THF) (2.52 mL, 2.34 mmol, 1.7 equiv) was added dropwise. The reaction was stirred for 1 h at rt. The reaction was quenched with sat. NaHCO3 (8 mL), and H2O (8 mL) was added. The aqueous phase was extracted with EtOAc (3 × 40 mL), and the combined organic fractions were washed with brine (75 mL) and dried over MgSO4. The crude product was purified by DCVC (80 → 94% EtOAc in heptane), which yielded the product as a colorless oil (303 mg, 1.11 mmol, 80%). LCMS (ESI) m/z = 274.2 [M + H]+; Rf = 0.30 (2:1 EtOAc/heptane); [α]25589 = −93.0° (c = 0.115, EtOH); 1H NMR (400 MHz, CDCl3) δ 4.82 (br s, 1H) 3.89−3.76 (m, 1H) 3.72−3.51 (m, 5H) 3.19 (ddd, J = 11.00, 8.50, 7.03 Hz, 1 H) 2.12 (br s, 1H) 2.02−1.81 (m, 2H) 1.65 (ddd, J = 16.60, 12.30, 8.30 Hz, 1H) 1.54−1.33 (m, 12H) 0.94 (t, J = 7.50 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 156.8, 80.3, 67.6, 63.0, 62.5, 46.4, 44.1, 42.3, 28.5, 27.4, 22.2, 11.8.
tert-Butyl (2S,3R)-3-((R)-1-Hydroxybutan-2-yl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (8c-s2).
7c-s2 (575.7 mg, 1.49 mmol, 1 equiv) was dissolved in dry THF (15 mL), and TBAF (1 M in THF) (2.52 mL, 2.52 mmol, 1.7 equiv) was added dropwise. The reaction was stirred for 1 h at rt. The reaction was quenched with sat. NaHCO3 (8 mL), and H2O (8 mL) was added. The aqueous phase was extracted with EtOAc (3 × 40 mL). The combined organic fractions were washed with brine (75 mL) and dried over MgSO4. The crude product was purified by DCVC (84 → 92% EtOAc in heptane), which afforded the product as a colorless oil (328 mg, 1.20 mmol, 81%). LCMS (ESI) m/z = 274.2 [M + H]+; Rf = 0.42 (100% EtOAc); [α]25589= −26.2° (c = 0.625, EtOH); 1H NMR (400 MHz, CDCl3) δ 4.82 (br s, 1H), 3.82−3.52 (m, 6H), 3.17 (ddd, J = 11.04, 9.41, 6.65 Hz, 1H), 2.18−1.96 (m, 2H), 1.96−1.68 (m, 2H), 1.66−1.51 (m, 1H), 1.47 (s, 9H), 1.45−1.39 (m, 1H), 1.38−1.24 (m, 1H), 0.95 (t, J = 7.28 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 156.6, 80.2, 67.2, 63.0, 62.8, 46.4, 44.3, 42.2, 28.4, 27.3, 20.5, 11.9.
tert-Butyl (2S,3R)-3-((R)-2-Hydroxy-1-phenylethyl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (8d-s1).
The compound 8d-s1 was obtained as a side-product from the synthesis of 7d-s2. After isolation of 7d-s2 by DCVC, the remaining fractions were combined and submitted to preparative HPLC, which afforded 8d-s1 as a colorless oil (157 mg, 0.49 mmol, 5%). LCMS (ESI) m/z = 322.2 [M + H]+; 1H NMR (400 MHz, CDCl3) δ 7.35−7.24 (m, 3H), 7.21−7.13 (m, 2H), 3.92−3.75 (m, 2H), 3.75−3.65 (m, 1H), 3.42−3.09 (m, 4H), 2.84−2.67 (m, 1H), 2.30−2.15 (m, 1H), 2.15−1.96 (m, 1H), 1.74 (dp, J = 7.4, 13.9 Hz, 1H), 1.40 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 156.6, 140.6, 129.0, 128.5, 127.5, 80.4, 66.3, 64.9, 62.9, 51.4, 46.2, 43.0, 29.5, 28.5.
tert-Butyl (2S, 3R)-3-((S)-2-Hydroxy-1-phenylethyl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (8d-s2).
7d-s2 (700 mg, 1.61 mmol, 1 equiv) was dissolved in dry THF (20 mL), and TBAF (1 M in THF) (2.73 mL, 2.72 mmol, 1.7 equiv) was added dropwise. The reaction was stirred for 1 h at rt. The reaction was quenched with sat. NaHCO3 (10 mL), and H2O (8 mL) was added. The aqueous phase was extracted with EtOAc (3 × 50 mL). The combined organic fractions were washed with brine (100 mL) and dried over MgSO4. The crude product was purified by DCVC (54 → 70% EtOAc in heptane), which afforded the product as a colorless oil (290 mg, 0.90 mmol, 56%). LCMS (ESI) m/z = 322.2 [M + H]+; Rf = 0.26 (1:1 EtOAc/heptane); 1H NMR (400 MHz, CDCl3) δ 7.35−7.29 (m, 2H), 7.27−7.19 (m, 3H), 3.96−3.83 (m, 3H), 3.79−3.67 (m, 2H), 3.37 (br. s, 1H), 3.14 (dt, J = 7.2, 11. Hz, 1H), 2.74 (br. s, 1H), 2.32 (br. s, 1H), 1.70 (dq, J = 6.8, 13.0 Hz, 1H), 1.45 (s, 9H), 1.36 (dq, J = 7.2, 14.2 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 156.5, 141.0, 128.8, 128.7, 127.1, 80.4, 66.7, 65.6, 63.4, 51.0, 45.8, 42.8, 28.4, 14.3.
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(carboxymethyl)pyrrolidine-2-carboxylic Acid (9a).
8a (275 mg, 1.12 mmol, 1.0 equiv) was dissolved in EtOAc (9.3 mL), and MeCN (9.3 mL) was added. NaIO4 (1960 mg, 9.19 mmol, 8.2 equiv) was suspended in H2O (8.5 mL), followed by addition of RuCl3·H2O (4.65 mg, 0.02 mmol, 0.02 equiv). The aqueous phase was transferred to the organic phase, and H2O (4 mL) was added. The reaction was vigorously stirred for 4 h at rt. The reaction mixture was filtered through Celite, and the residue was washed with EtOAc (30 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 30 mL). The organic fractions were combined, washed with brine (75 mL), and dried over MgSO4. The crude product was purified by DCVC (10 → 50% EtOAc in heptane % AcOH). All fractions were combined and evaporated. The residue was dissolved in a minimum volume of warm EtOAc (approximately 1.5 mL). Heptane (approximately 7 mL) was added until the solution stayed turbid, and the mixture was placed at 5 °C overnight, which afforded a crystalline white solid (228 mg, 0.83 mmol, 75%); LCMS (ESI) m/z = 274.1 [M + H]+; 1H NMR (300 MHz, CDCl3) δ 10.21 (br. s, 2H), 4.01 (dd, J = 50.90, 6.30 Hz, 1H), 3.71−3.39 (m, 2 H), 2.92−2.64 (m, 2H), 2.51 (dt, J = 15.75, 7.67 Hz, 1H), 2.24 (dq, J = 11.86, 6.04 Hz, 1H), 1.78−1.59 (m, 1H), 1.47 (d, J = 15.68 Hz, 9H); 13C NMR (100 MHz, CDCl3) δ 176.2, 153.8, 80.8, 64.1, 45.9, 40.7, 38.9, 37.3, 30.3, 28.2.
(2S,3R)-1-(tert-Butoxycarbonyl)-3-((S)-1-carboxypropyl)-pyrrolidine-2-carboxylic Acid (9c-s1).
8c-s1 (325 mg, 1.19 mmol, 1.0 equiv) was dissolved in EtOAc (10.0 mL), and MeCN (10.0 mL) was added. NaIO4 (2085 mg, 9.75 mmol, 8.2 equiv) was suspended in H2O (9.0 mL), followed by the addition of RuCl3·H2O (4.93 mg, 0.02 mmol, 0.02 equiv). The aqueous phase was transferred to the organic phase, and H2O (4.3 mL) was added. The reaction was vigorously stirred for 7 h at rt. The reaction mixture was filtered through Celite, and the residue was washed with EtOAc (35 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 25 mL). The organic fractions were combined, washed with brine (75 mL), and dried over MgSO4. The crude product was purified by DCVC (34 → 50% EtOAc in heptane + 1% AcOH), which afforded the product as a white solid (110 mg). The impure fractions were combined and together crystallized in minimum volume of warm EtOAc (approximately 1 mL). Heptane (approximately 6 mL) was added until the solution stayed turbid, and the mixture was placed at 5 °C and was left overnight, which afforded additional product as a crystalline white solid (27 mg). This was combined with the already obtained product (138 mg, 0.46 mmol 38%). LCMS (ESI) m/z = 302.1 [M + H]+; mp: 133.7−135.3 °C (decomposition); [α]25589 = +5.1° (c = 0.33, EtOH); 1H NMR (400 MHz, CDCl3) (rotamers in brackets) δ 11.21 (br s, 2H), 4.22 [4.08] (d, J = 5.27 Hz, 1H), 3.66−3.37 (m, 2H), 2.71−2.54 (m, 1H), 2.53−2.37 (m, 1H), 2.17−2.06 (m, 1H), 1.85−1.68 (m, 3H), 1.47 [1.42] (s, 9H), 0.98 (t, J = 7.28 Hz, 3H); 13C NMR (100 MHz, CDCl3) (rotamers in brackets) δ 179.5 [179.4], 178.6 [175.8], 155.7 [153.8], 81.4 [80.9], 62.0, 49.3 [49.2], 45.9 [45.7], 45.3 [44.0], 28.4, 28.2, 23.8 [23.4], 11.8 [11.7].
(2S,3R)-1-(tert-Butoxycarbonyl)-3-((R)-1-carboxypropyl)-pyrrolidine-2-carboxylic Acid (9c-s2).
8c-s2 (325 mg, 1.19 mmol, 1.0 equiv) was dissolved in EtOAc (10.0 mL), and MeCN (10.0 mL) was added. NaIO4 (2085 mg, 9.75 mmol, 8.2 equiv) was suspended in H2O (9.0 mL), followed by the addition of RuCl3·H2O (4.93 mg, 0.02 mmol, 0.02 equiv). The aqueous phase was transferred to the organic phase, and H2O (4.3 mL) was added. The reaction was vigorously stirred for 7 h at rt. The reaction mixture was filtered through Celite, and the residue was washed with EtOAc (35 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 25 mL). The organic fractions were combined, washed with brine (75 mL), and dried over MgSO4. The crude product was purified by DCVC (34 → 50% EtOAc in heptane + 1% AcOH), which afforded the product as a white solid (110 mg, 31%). The impure fractions were combined and together crystallized in minimum volume of warm EtOAc (approximately 1 mL). Heptane (approximately 6 mL) was added until the solution stayed turbid, and the mixture was placed at 5 °C and was left overnight, which afforded the product as a white solid (27 mg). This was combined with the already obtained product (138 mg, 0.46 mmol, 38%). LCMS (ESI) m/z = 302.1 [M + H]+; mp: 128.0−129.4 °C (decomposition); [α]25589 = −3.0° (c = 0.32, EtOH); 1H NMR (400 MHz, CDCl3) (rotamers in brackets) δ 9.21 (br s, 2H), 4.14 [3.93] (d, J = 7.53 Hz, 1H), 3.70−3.62 [3.59−3.51] (m, 1H), 3.47−3.33 (m, 1H), 2.74−2.59 (m, 1H), 2.42−2.29 (m, 1H), 2.18−2.06 (m, 1H), 1.79−1.55 (m, 3H), 1.45 [1.41] (s, 9H) 0.97 (t, J = 7.28 Hz, 3H); 13C NMR (100 MHz, CDCl3) (rotamers in brackets) δ 179.7 [179.4], 178.0 [176.1], 155.0 [153.7], 81.1 [81.0], 63.7 [63.2], 50.2, 46.0 [45.8], 45.5 [44.5], 28.4, 28.2, 23.6 [23.4], 11.5 [11.4].
(2S,3R)-1-(tert-Butoxycarbonyl)-3-((R)-Carboxy(phenyl)methyl)-pyrrolidine-2-carboxylic Acid (9d-s1).
8d-s1 (130 mg, 0.40 mmol, 1.0 equiv) was dissolved in EtOAc (3.4 mL), and MeCN (3.4 mL) was added. NaIO4 (709 mg, 3.32 mmol, 8.2 equiv) was suspended in H2O (3.0 mL), followed by the addition of RuCl3·H2O (1.68 mg, 0.02 mmol, 0.01 equiv). The aqueous phase was transferred to the organic phase, and H2O (1.5 mL) was added. The reaction was vigorously stirred for 4 h at rt. The reaction mixture was filtered through Celite, and the residue was washed with EtOAc (10 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 10 mL). The organic fractions were combined, washed with brine (25 mL), and dried over MgSO4. The crude product was purified by DCVC (40 → 55% EtOAc in heptane + 1% AcOH), which afforded the product as a white solid. The product was crystallized in minimum volume of warm iPOH (approximately 4 mL). H2O (approximately 1 mL) was added until the solution stayed turbid, and the mixture was placed at rt and was left overnight, which afforded the product as a crystalline white solid (32 mg, 0.09 mmol, 23%). LCMS (ESI) m/z = 350.2 [M + H]+; 1H NMR (400 MHz, acetone) δ 7.43−7.38 (m, 2H), 7.38−7.32 (m, 2H), 7.32−7.27 (m, 1H), 3.86 (dd, J = 2.3, 36.8 Hz, 1H), 3.60 (dd, J = 6.6, 11.2 Hz, 1H), 3.57−3.47 (m, 2H), 3.10 (ddd, J = 3.0, 6.6, 10.9 Hz, 1H), 2.29−2.13 (m, 1H), 1.93−1.79 (m, 1H), 1.37 (d, J = 43.6 Hz, 9H).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-((S)-carboxy(phenyl)methyl)-pyrrolidine-2-carboxylic Acid (9d-s2).
8d-s2 (290 mg, 0.90 mmol, 1.0 equiv) was dissolved in EtOAc (7.6 mL), and MeCN (7.6 mL) was added. NaIO4 (1583 mg, 7.40 mmol, 8.2 equiv) was suspended in H2O (6.8 mL), followed by the addition of RuCl3·H2O (3.74 mg, 0.02 mmol, 0.02 equiv). The aqueous phase was transferred to the organic phase, and H2O (3.3 mL) was added. The reaction was vigorously stirred for 4 h at rt. The reaction mixture was filtered through Celite, and the residue was washed with EtOAc (20 mL). The layers were separated, and the aqueous phase was extracted with EtOAc (3 × 15 mL). The organic fractions were combined, washed with brine (50 mL), and dried over MgSO4. The crude product was purified by DCVC (40 → 55% EtOAc in heptane + 1% AcOH), which afforded the product as a white solid (129 mg, 0.37 mmol, 41%). LCMS (ESI) m/z = 350.2 [M + H]+; 1H NMR (400 MHz, acetone) δ 11.13 (br. s, 2H)7.47−7.41 (m, 2H), 7.41−7.35 (m, 2H), 7.35−7.29 (m, 1H), 4.11 (dd, J = 3.6, 28.2 Hz, 1H), 3.68 (dd, J = 4.0, 10.6 Hz, 1H), 3.49−3.32 (m, 2H), 3.14−3.03 (m, 1H), 1.85 (ddq, J = 7.5, 12.7, 25.3 Hz, 1H), 1.45−1.36 (m, 10H).
(2S,3R)-3-(Carboxymethyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3a).
9a (205 mg, 0.75 mmol, 1 equiv) was dissolved in DCM (25 mL), and TFA (25 mL) was added dropwise. The reaction was stirred for 2 h. The solvents were removed under reduced pressure. The crude was dissolved in 1 M HCl(aq.) (3 × 30 mL), and the solvent was evaporated. The crude was purified by preparative HPLC. The combined fractions were evaporated and redissolved in 1 M HCl(aq.) (3 × 30 mL), and the solvent was evaporated. The final compound was freeze-dried, which afforded the product as a hygroscopic solid (21 mg, 0.10 mmol, 13%). LCMS (ESI) m/z = 174.1 [M + H]+; [α]25589 = +18.3° (c = 0.61, H2O); 1H NMR (300 MHz, D2O) δ 3.81 (d, J = 7.70 Hz, 1H), 3.50−3.34 (m, 2H), 2.85 (dd, J = 15.70, 4.70 Hz, 1H), 2.79−2.64 (m, 1H), 2.54 (dd, J = 15.70, 8.80 Hz, 1H), 2.38−2.25 (m, 1H), 1.81 (dq, J = 13.34, 8.30 Hz, 1H); 13C NMR (100 MHz, D2O) δ 177.1, 174.0, 65.4, 45.5, 39.9, 38.1, 30.5.
(2S,3R)-3-((S)-1-Carboxypropyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3c-s1).
9c-s1 (105 mg, 0.34 mmol, 1 equiv) was dissolved in DCM (6.8 mL), and the solution was cooled to 0 °C. TFA (6.8 mL) was added dropwise, and the reaction was allowed to warm to rt and stirred for 2 h. The solvents were removed under reduced pressure. The crude was purified by preparative HPLC. The purified compound was dissolved in 1 M HCl in H2O (3 × 30 mL), and solvent was evaporated. The final compound was freeze-dried, which afforded the product as a white solid (38 mg, 0.16 mmol, 47%). LCMS (ESI) m/z = 202.1 [M + H]+; mp: 184.1−188.7 °C (decomposition); [α]25589 = +18.3° (c = 0.57, H2O); 1H NMR (300 MHz, D2O) δ 4.31 (d, J = 7.28 Hz, 1H), 3.50 (ddd, J = 11.92, 7.40, 4.77 Hz, 1H), 3.40 (ddd, J = 11.50, 8.80, 7.00 Hz, 1H), 2.82 (quin, J = 7.65 Hz, 1H), 2.69 (dd, J = 14.30, 7.50 Hz, 1H), 2.34−2.24 (m, 1H), 1.93 (dq, J = 13.55, 8.28 Hz, 1H), 1.78−1.68 (m, 2H), 0.95 (t, J = 7.40 Hz, 3H); 13C NMR (100 MHz, D2O) δ 178.5, 171.9, 62.0, 49.3, 46.2, 44.0, 28.0, 24.2, 11.6.
(2S,3R)-3-((R)-1-Carboxypropyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3c-s2).
9c-s2 (89 mg, 0.29 mmol, 1 equiv) was dissolved in DCM (5.8 mL), and the solution was cooled to 0 °C. TFA (5.8 mL) was added dropwise, and the reaction was allowed to warm to rt and stirred for 2 h. The solvents were removed under reduced pressure. The crude was purified by preparative HPLC. The combined fractions were evaporated and redissolved in 1 M HCl (3 × 30 mL), and the solvent was evaporated. The final compound was freeze-dried, which afforded the product as a white solid (27 mg, 0.11 mmol, 39%). LCMS (ESI) m/z = 202.1 [M + H]+; mp: 68.7−71.2 °C (decomposition); [α]25589 = +12.9° (c = 0.23, H2O); 1H NMR (300 MHz, D2O) δ 4.24 (d, J = 7.28 Hz, 1H), 3.51 (ddd, J = 11.73, 7.84, 4.02 Hz, 1H), 3.39 (ddd, J = 11.54, 9.91, 7.15 Hz, 1H), 2.82 (quin, J = 8.09 Hz, 1H), 2.57 (td, J = 9.16, 4.27 Hz, 1H), 2.42−2.28 (m, 1H), 1.90 (dtd, J = 13.50, 9.00, 9.00, 8.20 Hz, 1H), 1.80−1.56 (m, 2H), 0.94 (t, J = 7.40 Hz, 3H); 13C NMR (100 MHz, D2O) δ 179.0, 171.8, 62.6, 49.9, 46.1, 44.3, 28.6, 23.5, 11.3.
(2S,3R)-3-((R)-Carboxy(phenyl)methyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3d-s1).
9d-s1 (25 mg, 0.07 mmol, 1 equiv) was dissolved in DCM (1.4 mL), and the solution was cooled to 0 °C. TFA (1.4 mL) was added dropwise, and the reaction was allowed to warm to rt and stirred for 2 h. The solvents were removed under reduced pressure. The crude was purified by preparative HPLC. The combined fractions were evaporated and redissolved in 1 M HCl (3 × 30 mL), and the solvent was evaporated. The final compound was freeze-dried, which afforded the product as a white solid (12 mg, 0.04 mmol, 60%). LCMS (ESI) m/z = 250.1 [M + H]+; 1H NMR (600 MHz, D2O) δ 7.49−7.42 (m, 5H), 3.96 (d, J = 5.4 Hz, 1H), 3.88 (d, J = 10.1 Hz, 1H), 3.53 (td, J = 2.8, 7.3 Hz, 2H), 3.34 (ddt, J = 5.8, 7.92, 10.5 Hz, 1H), 2.36 (dq, J = 7.6, 14.9 Hz, 1H), 2.08 (dq, J = 7.1, 13.6 Hz, 1H); 13C NMR (151 MHz, D2O) δ 175.6, 170.8, 135.7, 129.2, 128.8, 128.5, 61.4, 53.4, 45.3, 44.9, 28.3.
(2S,3R)-3-((S)-Carboxy(phenyl)methyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3d-s2).
9d-s2 (25 mg, 0.07 mmol, 1 equiv) was dissolved in DCM (1.4 mL), and the solution was cooled to 0 °C. TFA (1.4 mL) was added dropwise, and the reaction was allowed to warm to rt and stirred for 2 h. The solvents were removed under reduced pressure. The crude was purified by preparative HPLC. The combined fractions were evaporated and redissolved in 1 M HCl (3 × 30 mL), and the solvent was evaporated. The final compound was freeze-dried, which afforded the product as a white solid (10 mg, 0.035 mmol, 50%). LCMS (ESI) m/z = 250.1 [M + H]+; 1H NMR (600 MHz, D2O) δ 7.48−7.44 (m, 2H), 7.44−7.40 (m, 3H), 4.18 (d, J = 7.4 Hz, 1H), 3.93 (d, J = 9.3 Hz, 1H), 3.41−3.36 (m, 2H), 3.32 (p, J = 7.8 Hz, 1H), 2.03 (dq, J = 6.8, 13.5 Hz, 1H), 1.69 (dq, J = 8.4, 13.7 Hz, 1H). 13C NMR (151 MHz, D2O) δ 175.9, 171.2, 135.8, 129.2, 128.7, 128.4, 62.8, 52.9, 44.7, 44.1, 27.4.
trans-1-Benzyl 2-Methyl 3-(1,3-Dimethoxy-1,3-dioxopropan-2-yl)pyrrolidine-1,2-dicarboxylate (12a).
The title compound was prepared according to general procedure I, starting from 10 (2.0 g, 7.65 mmol) and dimethyl malonate (2.53 g, 2.19 mL, 19.14 mmol), which afforded the title compound as a colorless oil (2.98 g, 7.57 mmol, 99%). LCMS (ESI) m/z = 394.1 [M + H]+; Rf = 0.41 (1:1 heptane/EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.38−7.28 (m, 5H), 5.18 (dd, J = 12.4, 15.5 Hz, 1H), 5.05 (dd, J = 12.4, 56.3 Hz, 1H), 4.24 (dd, J = 4.8, 25.0 Hz, 1H), 3.77−3.73 (m, 7.5H), 3.69−3.63 (m, 1H), 3.60−3.58 (m, 1H), 3.57 (s, 1.5H), 3.51 (dd, J = 1.4, 8.9 Hz, 1H), 3.05−2.97 (m, 1H), 2.20 (tdd, J = 6.4, 10.2, 12.9 Hz, 1H), 1.78 (dp, J = 7.0, 21.1 Hz, 1H); 13C NMR (151 MHz, CDCl3) (rotamers) δ 172.1, 172.0, 168.0, 168.0, 167.9, 167.9, 154.7, 154.1, 136.5, 136.4, 128.5, 128.4, 128.06, 128.04, 128.00, 127.96, 127.90, 67.24, 67.18, 62.5, 62.1, 53.8, 53.7, 52.85, 52.81, 52.79, 52.5, 52.3, 45.6, 45.1, 43.3, 42.3, 28.2, 27.6.
trans-1-Benzyl 2-Methyl 3-(1-Ethoxy-2-(ethoxycarbonyl)-1-oxopentan-2-yl)pyrrolidine-1,2-dicarboxylate (12e).
The title compound was prepared according to general procedure I, starting from 10 (200 mg, 0.77 mmol) and diethyl 2-propylmalonate (387 mg, 0.39 mL, 1.91 mmol), which afforded the title compound as a colorless oil (146 mg, 0.31 mmol, 41%). LCMS (ESI) m/z = 464.2 [M + H]+; Rf = 0.31 (2:1 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.39− 7.27 (m, 6H), 5.24−4.96 (m, 2H), 4.55 (dd, J = 3.6, 18.8 Hz, 1H), 4.26−3.94 (m, 4H), 3.77 (s, 1.5H), 3.58 (s, 1.5H), 3.51 (ddd, J = 6.4, 8.8, 10.5 Hz, 2H), 3.06−2.93 (m, 1H), 2.30−2.18 (m, 1H), 2.06− 1.87 (m, 3H), 1.28−1.12 (m, 8H), 0.91 (t, J = 7.2 Hz, 3H) ; 13C NMR (101 MHz, CDCl3) (rotamers) δ 173.4, 173.3, 170.7, 170.62, 170.57, 170.5, 154.6, 154.2, 136.8, 136.7, 128.7, 128.5, 128.2, 128.1, 128.0, 127.9, 67.2, 61.7, 61.6, 61.4, 60.9, 59.9, 59.8, 52.5, 52.3, 46.9, 46.0, 45.7, 45.4, 36.4, 26.5, 25.8, 17.74, 17.72, 14.5, 14.12, 14.07, 14.05, 13.99.
trans-1-Benzyl 2-Methyl 3-(1-Ethoxy-2-(ethoxycarbonyl)-3-methyl-1-oxobutan-2-yl)pyrrolidine-1,2-dicarboxylate (12f).
The title compound was prepared according to general procedure I, starting from 10 (200 mg, 0.77 mmol) and diethyl 2-isopropylmalonate (387 mg, 0.39 mL, 1.91 mmol), which afforded the title compound as a colorless oil (125 mg, 0.27 mmol, 35%). LCMS (ESI) m/z = 464.2 [M + H]+; Rf = 0.32 (2:1 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.37−7.27 (m, 5H), 5.18 (dd, J = 5.9, 12.5 Hz, 1H), 5.02 (dd, J = 12.4, 14.2 Hz, 1H), 4.51 (dd, J = 3.4, 19.3 Hz, 1H), 4.26−3.95 (m, 4H), 3.76 (s, 1.5H), 3.58 (s, 1.5H), 3.55−3.38 (m, 2H), 3.25−3.12 (m, 1H), 2.54 (dp, J = 6.6, 10.0 Hz, 1H), 2.31−2.17 (m, 1H), 2.07−1.90 (m, 1H), 1.31−1.10 (m, 6H), 1.03−0.91 (m, 6H). 13C NMR (101 MHz, CDCl3) (rotamers) δ 172.9, 169.8, 169.60, 169.57, 169.5, 154.6, 154.2, 136.8, 136.7, 128.55, 128.49, 128.23, 128.19, 128.12, 128.05, 67.2, 67.1, 64.3, 64.2, 61.5, 61.5, 61.4, 61.3, 60.8, 46.8, 46.0, 45.7, 45.3, 31.9, 31.8, 26.8, 26.1, 18.9, 18.7, 18.5, 18.45 14.3, 14.2, 14.1, 14.0.
trans-1-Benzyl 2-Methyl 3-(2-Benzyl-1,3-diethoxy-1,3-dioxopropan-2-yl)pyrrolidine-1,2-dicarboxylate (12g).
The title compound was prepared according to general procedure I (5 h reaction time), starting from 10 (180 mg, 0.69 mmol) and diethyl benzylmalonate (431 mg, 0.41 mL, 1.72 mmol), which afforded the title compound as a colorless oil (210 mg, 0.41 mmol, 60%). LCMS (ESI) m/z = 512.2 [M + H]+; Rf = 0.10 (8:2 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.37−7.28 (m, 5H), 7.25−7.18 (m, 3H), 7.12−7.07 (m, 2H), 5.20 (d, J = 12.3 Hz, 1H), 5.01 (d, J = 12.9 Hz, 1H), 4.67 (br d, J = 21.1 Hz, 1H), 4.20−3.79 (m, 4H), 3.73 (s, 1.5H), 3.57 (s, 1.5H), 3.56−3.41 (m, 2H), 3.32 (s, 2H), 2.94−2.83 (m, 1H), 2.34−2.11 (m, 2H), 1.17 (t, J = 7.2 Hz, 3H), 1.08 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) (rotamers) δ 173.2, 173.1, 170.3, 170.2, 170.13, 170.10, 154.5, 154.2, 136.7, 136.6, 135.4, 130.1, 128.6, 128.54, 128.49, 128.3, 128.2, 128.1, 127.4, 67.2, 61.94, 61.89, 61.8, 61.7, 61.4, 61.3, 61.2, 52.5, 52.3, 46.7, 45.9, 45.5, 45.2, 40.0, 26.2, 25.5, 14.0, 13.9, 13.83, 13.79.
trans-1-Benzyl 2-Methyl 3-(1-Ethoxy-2-(ethoxycarbonyl)-1-oxo-4-phenylbutan-2-yl)pyrrolidine-1,2-dicarboxylate (12h).
The title compound was prepared according to general procedure I, starting from 10 (200 mg, 0.77 mmol) and diethyl 2-phenethylmalonate (506 mg, 1.91 mmol), which afforded the title compound as a colorless oil (242 mg, 0.46 mmol, 60%). LCMS (ESI) m/z = 526.3 [M + H]+; Rf = 0.39 (2:1 heptane/EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.36− 7.26 (m, 7H), 7.22−7.18 (m, 1H), 7.18−7.14 (m, 2H), 5.23−5.15 (m, 1H), 5.02 (dd, J = 12.4, 29.2 Hz, 1H), 4.60 (dd, J = 3.5, 33.1 Hz, 1H), 4.25−4.16 (m, 2H), 4.16−4.00 (m, 2H), 3.77 (s, 1.5H), 3.59 (s, 1.5H), 3.58−3.45 (m, 2H), 3.09 (ddq, J = 3.8, 8.3, 12.3 Hz, 1H), 2.63−2.54 (m, 1H), 2.50−2.43 (m, 1H), 2.32−2.20 (m, 3H), 2.13−1.99 (m, 1H), 1.28−1.18 (m, 6H); 13C NMR (151 MHz, CDCl3) (rotamers) δ 173.3, 173.2, 172.7, 170.5, 170.4, 170.32, 170.30, 170.27, 170.25, 170.22, 170.18, 154.59, 154.57, 154.23, 154.18, 141.18, 141.15, 141.11, 136.8, 136.71, 136.66, 128.67, 128.65, 128.62, 128.57, 128.52, 128.51, 128.46, 128.45, 128.20, 128.16, 128.13, 128.10, 128.08, 128.06, 126.37, 126.35, 67.22, 67.18, 67.16, 61.90, 61.88, 61.86, 61.83, 61.80, 61.78, 61.6, 61.5, 61.44, 61.41, 61.0, 60.9, 59.9, 59.81, 59.80, 52.6, 52.4, 47.22, 47.14, 46.05, 46.01, 45.98, 45.4, 36.5, 36.3, 30.89, 30.87, 26.7, 26.6, 26.0, 25.9, 14.3, 14.16, 14.14, 14.12, 14.10, 14.08.
trans-1-Benzyl 2-Methyl 3-(2-Fluoro-1,3-dimethoxy-1,3-dioxopropan-2-yl)pyrrolidine-1,2-dicarboxylate (12i).
The title compound was prepared according to general procedure I, starting from 10 (200 mg, 0.77 mmol) and dimethyl 2-fluoromalonate (287 mg, 1.91 mmol), which afforded the title compound as a colorless oil (198 mg, 0.48 mmol, 63%). LCMS (ESI) m/z = 412.1 [M + H]+; Rf = 0.32 (2:1 heptane/EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.37−7.27 (m, 5H), 5.18 (dd, J = 12.3, 34.5 Hz, 1H), 5.03 (dd, J = 12.4, 67.1 Hz, 1H), 4.37 (dd, J = 5.5, 40.4 Hz, 1H), 3.84−3.80 (m, 6H), 3.81−3.72 (m, 1H), 3.74 (s, 1.5H), 3.57−3.52 (m, 1H), 3.51 (s, 1.5H), 3.39− 3.26 (m, 1H), 2.22−2.08 (m, 1H), 2.03−1.90 (m, 1H); 13C NMR (151 MHz, CDCl3) (rotamers) δ 172.1, 165.5, 165.4, 165.33, 165.31, 165.27, 165.23, 165.16, 165.1, 154.5, 153.9, 136.5, 136.4, 128.6, 128.5, 128.2, 128.14, 128.07, 128.0, 95.4, 95.1, 94.1, 93.7, 67.4, 67.3, 59.52, 59.50, 58.85, 58.83, 53.84, 53.81, 53.7, 52.7, 52.4, 48.3, 48.1, 47.1, 46.9, 46.2, 45.8, 32.0, 26.03, 26.01, 25.3, 25.2, 22.8, 14.3, 14.2.
trans-1-Benzyl 2-Methyl 3-(1,2,3-Trimethoxy-1,3-dioxopropan-2-yl)pyrrolidine-1,2-dicarboxylate (12j).
The title compound was prepared according to general procedure I (24 h reaction time), starting from 10 (135 mg, 0.52 mmol) and dimethyl methoxymalonate (209 mg, 0.18 mL, 1.29 mmol), which afforded the title compound as a colorless oil (149 mg, 0.35 mmol, 68%). LCMS (ESI) m/z = 424.2 [M + H]+; Rf = 0.40 (1:1 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.38−7.27 (m, 5H), 5.18 (d, J = 12.5 Hz, 1H), 5.05 (d, J = 12.5 Hz, 1H), 4.61 (br d, J = 23.3, 1H), 3.77−3.71 (4s, 7.5H), 3.61−3.45 (m, 2H), 3.57 (s, 1.5H), 3.48 (br s, 3H), 3.17−3.09 (m, 1H), 2.17−2.05 (m, 2H); 13C NMR (101 MHz, CDCl3) (rotamers) δ 172.93, 172.91, 168.3, 168.14, 168.09, 154.6, 154.2, 136.7, 128.50, 128.47, 128.1, 128.0, 85.3, 85.2, 67.24, 67.21, 60.3, 59.7, 55.4, 53.0, 52.9, 52.2, 52.80, 52.6, 52.4, 48.9, 47.8, 46.2, 45.7, 29.8, 26.0, 25.2.
trans-1-Benzyl 2-Methyl 3-(2-((tert-Butoxycarbonyl)amino)-1,3-diethoxy-1,3-dioxopropan-2-yl)pyrrolidine-1,2-dicarboxylate (12k).
The title compound was prepared according to general procedure I, starting from 10 (200 mg, 0.77 mmol) and diethyl 2-((tert-butoxycarbonyl)amino)malonate (527 mg, 1.91 mmol), which afforded the title compound as a colorless oil (347 mg, 0.65 mmol, 84%). LCMS (ESI) m/z = 537.2 [M + H]+; Rf = 0.28 (2:1 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.35−7.27 (m, 5H), 5.99 (d, J = 6.17 Hz, 1H), 5.18 (dd, J = 12.3, 23.3 Hz, 1H), 5.00 (dd, J = 12.5, 47.7 Hz, 1H), 4.58 (dd, J = 2.77, 22.91 Hz, 1H), 4.33−4.14 (m, 3H), 4.14−3.99 (m, 1H), 3.73 (s, 1.5H), 3.56 (s, 1.5H), 3.59−3.49 (m, 2H), 3.44−3.34 (m, 1H), 2.32−2.18 (m, 1H), 2.12−2.03 (m, 1H), 1.40 (s, 9H), 1.22 (dt, J = 7.10, 15.46 Hz, 6H) ; 13C NMR (101 MHz, CDCl3) (rotamers) δ 172.8, 172.7, 167.5, 167.4, 167.3, 167.1, 154.6, 154.3, 154.2, 136.7, 136.7, 128.54, 128.47, 128.08, 128.03, 128.00, 80.9, 80.8, 67.2, 67.13, 67.07, 63.1, 62.9, 62.8, 60.9, 60.5, 52.5, 52.3, 48.4, 47.3, 46.3, 45.6, 31.0, 28.23, 28.20, 26.1, 25.2, 14.04, 14.00, 13.90, 13.86.
trans-Triethyl 1-(1-((Benzyloxy)carbonyl)-2-(methoxycarbonyl)-pyrrolidin-3-yl)ethane-1,1,2-tricarboxylate (12l).
The title compound was prepared according to general procedure I, starting from 10 (200 mg, 0.77 mmol) and triethylethane-1,1,2-tricarboxylate (471 mg, 0.44 mL, 1.91 mmol), which afforded the title compound as a colorless oil (285 mg, 0.56 mmol, 73%). LCMS (ESI) m/z = 508.2 [M + H]+; Rf = 0.23 (2:1 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.37−7.27 (m, 5H), 5.18 (dd, J = 12.41, 15.72 Hz, 1H), 5.03 (dd, J = 12.44, 30.17 Hz, 1H), 4.60 (dd, J = 4.21, 10.41 Hz, 1H), 4.26−4.14 (m, 3H), 4.14−4.03 (m, 3H), 3.74 (s, 1.5H), 3.67−3.59 (m, 1H), 3.55 (s, 1.5H), 3.54−3.46 (m, 1H), 3.19−3.10 (m, 1H), 3.02 (d, J = 7.95 Hz, 2H), 2.29−2.17 (m, 1H), 2.08−1.88 (m, 1H), 1.29−1.13 (m, 9H) ; 13C NMR (101 MHz, CDCl3) δ 173,0, 170.10, 170.08, 169.4, 169.3, 154.6, 154.1, 136.7, 136.6, 128.6, 128.5, 128.14, 128.09, 67.2, 62.19, 62.16, 61.4, 61.0, 60.8, 57.5, 57.49, 52.5, 52.3, 47.4, 46.3, 46.2, 45.6, 38.5, 38.3, 27.1, 26.4, 14.2, 14.00, 13.98, 13.93, 13.91.
trans-1-Benzyl 2-Methyl 3-(4-Cyano-1-ethoxy-2-(ethoxycarbonyl)-1-oxobutan-2-yl)pyrrolidine-1,2-dicarboxylate (12m).
The title compound was prepared according to general procedure I, starting from 10 (200 mg, 0.77 mmol) and diethyl 2-(2-cyanoethyl)malonate (408 mg, 0.38 mL, 1.91 mmol), which afforded the title compound as a colorless oil (354 mg, 0.74 mmol, 97%). LCMS (ESI) m/z = 475.2 [M + H]+; Rf = 0.39 (2:1 heptane/EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.37−7.28 (m, 5H), 5.19 (dd, J = 12.3, 16.8 Hz, 1H), 5.03 (dd, J = 12.4, 33.4 Hz, 1H), 4.49 (dd, J = 3.8, 36.7 Hz, 1H), 4.26−4.05 (m, 4H), 3.77 (s, 1.5H), 3.58 (s, 1.5H), 3.64−3.48 (m, 2H), 2.97−2.89 (m, 1H), 2.54−2.46 (m, 1H), 2.46−2.39 (m, 1H), 2.33−2.26 (m, 2H), 2.25−2.15 (m, 1H), 2.04−1.92 (m, 1H), 1.27−1.19 (m, 6H) ; 13C NMR (151 MHz, CDCl3) (rotamers) δ 172.81, 172.78, 169.2, 169.1, 169.04, 169.03, 154.5, 154.0, 136.55, 136.47, 128.60, 128.57, 128.24, 128.21, 128.1, 118.9, 67.37, 67.36, 62.50, 62.47, 62.4, 61.2, 60.7, 59.1, 59.0, 52.7, 52.5, 48.0, 46.8, 46.0, 45.4, 30.2, 30.1, 26.8, 26.2, 14.01, 13.99, 13.9, 13.4.
trans-3-(Carboxymethyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3a).
The title compound was prepared according to general procedure II, starting from 12a (1.50 g, 4.13 mmol) and stirred for 5 days, which afforded the title compound as a white solid (753 mg, 3.59 mmol, 87%). LCMS (ESI) m/z = 174.1 [M + H]+; 1H NMR (600 MHz, D2O) δ 4.15 (d, J = 8.46 Hz, 1H), 3.51 (ddd, J = 4.48, 8.29, 11.84 Hz, 1H), 3.43 (ddd, J = 7.34, 9.03, 11.80 Hz, 1H), 2.91−2.81 (m, 2H), 2.72−2.65 (m, 1H), 2.39 (dtd, J = 4.43, 7.27, 13.26 Hz, 1H), 1.87 (dq, J = 8.74, 13.48 Hz, 1H); 13C NMR (151 MHz, D2O) δ 175.4, 171.0, 62.9, 45.2, 38.5, 36.2, 29.7.
trans-3-(1-Carboxybutyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3e).
The title compound was prepared according to general procedure II, starting from 12e (140 mg, 0.30 mmol) and stirred for 6 days. The diastereomers could not be separated on preparative HPLC, which afforded a mixture of the trans-diastereomers 3e as a white solid (21 mg, 0.08 mmol, 28%). d.r. 63:37; LCMS (ESI) m/z = 216.1[M + H]+; 1H NMR (600 MHz, D2O) (minor trans-diastereomer in brackets) δ 4.21 (d, J = 6.8 Hz, 1H), [4.16 (d, J = 7.34 Hz, 1H)], 3.45−3.37 (m, 1H), [3.45−3.37 (m, 1H)], 3.35−3.26 (m, 1H), [3.35−3.26 (m, 1H)], 2.76−2.65 (m, 2H), [2.76−2.65 (m, 1H)], [2.71−2.65 (m, 1H)], [2.26 (dtd, J = 3.92, 7.35, 14.31 Hz, 1H)], 2.23−2.16 (m, 1H), [1.90−1.77 (m, 1H)], 1.90−1.77 (m, 1H), 1.62 (tdd, J = 5.1, 9.7, 14.0 Hz, 1H), [1.58−1.51 (m, 2H)], 1.58−1.51 (m, 1H), [1.34−1.17 (m, 2H)], 1.34−1.17 (m, 2H), [0.82 (t, J = 7.33 Hz, 3H)], 0.82 (t, J = 7.4 Hz, 3H); 13C NMR (151 MHz, D2O) (minor trans-diastereomer in brackets) δ [178.5], 178.0, 171.4, [171.3], [61.9], 61.6, [47.6], 46.8, 45.6, [45.5], [43.9], 43.6, 32.3, [31.6], [28.0], 27.3, 19.9, [19.8], [13.1], 13.0.
trans-3-(1-Carboxy-2-methylpropyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3f).
The title compound was prepared according to general procedure II, starting from 12f (120 mg, 0.25 mmol) and stirred for 8 days. The diastereomers could not be separated on preparative HPLC, which afforded a mixture of the trans-diastereomers 3f as a white solid (27 mg, 0.11 mmol, 43%). d.r. 67:33; LCMS (ESI) m/z = 216.1 [M + H]+; 1H NMR (600 MHz, D2O) (minor trans-diastereomer in brackets) δ [4.25 (d, J = 7.2 Hz, 1H)], 4.17 (d, J = 8.2 Hz, 1H), [3.56−3.44 (m, 1H)], 3.56−3.44 (m, 1H), [3.44−3.36 (m, 1H)], 3.44−3.36 (m, 1H), [2.97−2.86 (m, 1H)], 2.97−2.86 (m, 1H), 2.61 (t, J = 7.3 Hz, 1H), [2.49 (dd, J = 56.0, 8.8 Hz, 1H)], [2.37 (dtd, J = 4.1, 7.3, 13.6 Hz, 1H)], 2.34−2.25 (m, 1H), [2.09−2.00 (m, 1H)], 2.09−2.00 (m, 2H), [1.94−1.84 (m, 1H)], 0.99 (dd, J = 6.7, 24.9 Hz, 6H), [0.98 (dd, J = 6.9, 51.2 Hz, 6H)]; 13C NMR (151 MHz, D2O) (minor trans-diastereomer in brackets) δ [177.2], 177.1, [171.3], 171.1, [62.2], 61.6, [53.6], 53.1, 45.5, [45.2], [42.2], 41.5, 30.2, [28.4], [28.1], 26.7, [20.5], 19.8, 19.1, [17.3].
trans-3-(1-Carboxy-2-phenylethyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3g).
The title compound was prepared according to general procedure II, starting from 12g (184 mg, 0.36 mmol) and stirred for 7 days. The diastereomers could not be separated on preparative HPLC, which afforded a mixture of the trans-diastereomers 3g as a white solid (24 mg, 0.080 mmol, 22%). d.r. 58:42; LCMS (ESI) m/z = 264.1 [M + H]+ 1H NMR (400 MHz, D2O) δ 7.43−7.38 (m, 2H), 7.36−7.31 (m, 3H), 4.34 (d, J = 7.5 Hz, 1H), 3.60−3.37 (m, 2H), 3.22−2.93 (m, 3H), 2.83 (p, J = 7.6 Hz, 1H), 2.49−2.29 (2m, 1H), 2.10−1.96 (m, 1H); 13C NMR (151 MHz, D2O) δ 178.4, 178.2, 173.9, 173.6, 139.44, 139.41, 129.72, 129.69, 129.65, 129.61, 127.7, 127.6, 64.0, 63.9, 51.1, 50.4, 46.0, 45.9, 45.0, 44.8, 37.3, 36.4, 28.8, 28.1.
trans-3-(1-Carboxy-3-phenylpropyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3h).
The title compound was prepared according to general procedure II, starting from 12h (225 mg, 0.43 mmol) and stirred for 5 days. The diastereomers could not be separated on preparative HPLC, which afforded a mixture of the trans-diastereomers 3h as a white solid (48 mg, 0.15 mmol, 36%). LCMS (ESI) m/z = 278.1 [M + H]+; 1H NMR (600 MHz, D2O) δ 7.39 (t, J = 7.5 Hz, 2H), 7.33−7.28 (m, 3H), 4.21 (d, J = 7.5 Hz, 1H), 3.50−3.44 (m, 1H), 3.42−3.33 (m, 1H), 2.86−2.72 (m, 3H), 2.72−2.60 (m, 1H), 2.34−2.24 (m, 1H), 2.13−1.83 (m, 3H); 13C NMR (151 MHz, D2O) δ 178.0, 171.2, 141.4, 128.7, 128.6, 126.3, 61.7, 47.1, 45.4, 43.7, 32.6, 31.1, 28.0.
trans-3-(Carboxyfluoromethyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3i-d1).
The title compound was prepared according to general procedure II, starting from 12i (150 mg, 0.36 mmol) and stirred for 4 days. The diastereomers were separated on preparative HPLC, which afforded the trans-diastereomer 3i-d1 as a white solid (4 mg, 0.02 mmol, 5%). d.r. 95:5; LCMS (ESI) m/z = 192.1 [M + H]+; 1H NMR (600 MHz, D2O) δ 5.29 (dd, J = 2.8, 47.5 Hz, 1H), 4.41 (d, J = 6.6 Hz, 1H), 3.50 (ddd, J = 5.4, 7.7, 12.6 Hz, 1H), 3.41 (dt, J = 7.9, 11.5 Hz, 1H), 3.26 (dqd, J = 2.7, 8.9, 31.7 Hz, 1H), 2.43 (dtd, J = 5.5, 7.5, 13.8 Hz, 1H), 2.14 (dq, J = 7.9, 13.7 Hz, 1H); 13C NMR (151 MHz, D2O) δ 171.8, 171.6, 170.5, 88.8, 87.6, 58.3, 58.2, 45.9, 43.8, 43.7, 27.12, 27.10.
And trans-diastereomer 3i-d2 as a white solid (35 mg, 0.15 mmol, 43%). d.r. 1:99; LCMS (ESI) m/z = 192.1 [M + H]+; 1H NMR (600 MHz, D2O) δ 5.41 (dd, J = 2.6, 49.0 Hz, 1H), 4.41 (d, J = 7.9 Hz, 1H), 3.53 (ddd, J = 5.2, 8.2, 11.7 Hz, 1H), 3.46 (dt, J = 8.0, 11.7 Hz, 1H), 3.21 (dqd, J = 2.7, 8.2, 31.9 Hz, 1H), 2.21 (dtd, J = 5.1, 7.8, 13.2 Hz, 1H), 2.10 (dq, J = 8.3, 13.6 Hz, 1H); 13C NMR (151 MHz, D2O) δ 172.5, 172.4, 171.1, 88.5, 87.3, 60.22, 60.19, 45.6, 44.1, 44.0, 23.82, 23.80.
trans-3-(Carboxy(methoxy)methyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3j-d1).
The title compound was prepared according to general procedure II, starting from 12j (189 mg, 0.45 mmol) and stirred for 5 days. One diastereomer was purified on preparative HPLC, which afforded the trans-diastereomer 3j-d1 as a white solid (6 mg, 0.025 mmol, 6%). d.r. 1:99; LCMS (ESI) m/z = 204.1 [M + H]+; 1H NMR (600 MHz, D2O) δ 4.41 (d, J = 5.7 Hz, 1H), 4.12 (d, J = 3.5 Hz, 1H), 3.55−3.50 (m, 1H), 3.48 (s, 3H), 3.46−3.35 (m, 1H), 3.14−3.09 (m, 1H), 2.40−2.33 (m, 1H), 2.13−2.06 (m, 1H); 13C NMR (151 MHz, D2O) δ 175.6, 172.1, 80.7, 60.3, 59.6, 46.8, 45.4, 28.3.
trans-3-(Amino(carboxy)methyl)pyrrolidine-2-carboxylic Acid Dihydrochloride (3k).
The title compound was prepared according to general procedure II, starting from 12k (300 mg, 0.59 mmol) and stirred for 5 days. The diastereomers could not be separated on preparative HPLC, which afforded a mixture of the trans-diastereomers 3k as a white solid (57 mg, 0.22 mmol, 37%). d.r. 50:50; LCMS (ESI) m/z = 189.1 [M + H]+; 1H NMR (600 MHz, D2O) (two diastereomers) δ 4.53 (d, J = 9.2 Hz, 1H), 4.32 (d, J = 5.7 Hz, 1H), 4.30−4.24 (m, 1H), 3.48 (tdd, J = 3.6, 7.9, 12.0 Hz, 1H), 3.38−3.29 (m, 1H), 2.99 (qd, J = 4.8, 8.9 Hz, 1H), 2.95−2.85 (m, 1H), 2.42−2.30 (m, 1H), 2.24−2.15 (m, 1H), 1.98−1.88 (m, 1H); 13C NMR (151 MHz, D2O) (two diastereomers) δ 170.79, 170.75, 169.9, 169.8, 61.8, 60.5, 54.1, 53.6, 45.3, 44.9, 42.1, 41.6, 27.8, 27.0.
trans-2-(2-Carboxypyrrolidin-3-yl)succinic Acid Hydrochloride (3l).
The title compound was prepared according to general procedure II, starting from 12l (275 mg, 0.54 mmol) and stirred for 9 days. The diastereomers could not be separated on preparative HPLC, which afforded a mixture of the trans-diastereomers 3l as a white solid (48 mg, 0.18 mmol, 33%). d.r. 63:37; LCMS (ESI) m/z = 232.1 [M + H]+; 1H NMR (400 MHz, D2O) (minor trans-diastereomer in brackets) δ 4.36 (d, J = 8.5 Hz, 1H), [4.33 (d, J = 7.4 Hz, 1H)], 3.49−3.39 (m, 1H), [3.49−3.39 (m, 1H)], 3.35−3.25 (m, 1H), [3.35−3.25 (m, 1H)], 3.22 (ddd, J = 4.6, 5.6, 10.1 Hz, 1H), [3.02 (ddd, J = 4.8, 7.4, 9.7 Hz, 1H)], 2.86−2.62 (m, 3H), [2.86−2.62 (m, 3H)], 2.31−2.19 (m, 1H), [2.31−2.19 (m, 1H)], 1.90−1.76 (m, 1H), [1.90−1.76 (m, 1H)]; 13C NMR (151 MHz, D2O) (minor trans-diastereomer in brackets) δ [176.7], 176.0, 175.53, [175.51], 171.0, [170.9], 61.2, [61.0], 45.6, [45.5], [43.2], 43.0, [42.8], 42.2, 34.6, [33.7], [28.1], 27.1.
trans-2-(2-Carboxypyrrolidin-3-yl)pentanedioic Acid Hydrochloride (3m).
The title compound was prepared according to general procedure II, starting from 12m (300 mg, 0.63 mmol) and stirred for 9 days. The diastereomers could not be separated on preparative HPLC, which afforded a mixture of the trans-diastereomers 3m as a white solid (52 mg, 0.18 mmol, 29%). d.r. 67:33; LCMS (ESI) m/z = 246.1 [M + H]+; 1H NMR (600 MHz, D2O) (minor trans-diastereomer in brackets) δ 4.33 (d, J = 6.9 Hz, 1H), [4.28 (d, J = 7.4 Hz, 1H)], 3.56−3.48 (m, 1H), [3.56−3.48 (m, 1H)], 3.45−3.36 (m, 1H), [3.45−3.36 (m, 1H)], 2.90−2.80 (m, 2H), [2.90−2.80 (m, 1H)], [2.72−2.69 (m, 1H)], 2.58−2.43 (m, 2H), [2.58−2.43 (m, 2H)], [2.40−2.35 (m, 1H)], 2.32 (dtd, J = 4.5, 7.3, 13.8 Hz, 1H), 2.08−1.89 (m, 3H), [2.08−1.89 (m, 3H)]; 13C NMR (151 MHz, D2O) (minor trans-diastereomer in brackets) δ [177.4], 177.33, [177.27], 176.9, 171.6, [171.4], [61.9], 61.7, [46.6], 46.1, 45.5, [45.4], [43.7], 43.6, 31.34, [31.29], [27.8], 27.3, 25.2, [24.4].
trans-1-Benzyl 2-Methyl 3-(3-(Ethoxycarbonyl)-2-oxotetrahydrofuran-3-yl)pyrrolidine-1,2-dicarboxylate (12n).
The title compound was prepared according to general procedure I, starting from 10 (200 mg, 0.77 mmol) and ethyl 2-oxotetrahydrofuran-3-carboxylate (303 mg, 1.91 mmol), which afforded the title compound as a colorless oil (228 mg, 0.54 mmol, 71%). LCMS (ESI) m/z = 420.2 [M + H]+; Rf = 0.25 (1:1 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.38− 7.27 (m, 5H), 5.27−4.93 (m, 2H), 4.44−4.11 (m, 5H), 3.75 (d, J = 5.95 Hz, 1.5H), 3.72−3.64 (m, 1H), 3.64−3.55 (m, 1H), 3.53 (d, J = 4.05 Hz, 1.5H), 3.32 (td, J = 5.79, 7.84 Hz, 0.5H), 3.15 (dtd, J = 5.42, 7.66, 13.28 Hz, 0.5H), 2.74 (dqd, J = 2.21, 6.87, 14.18 Hz, 1H), 2.37−2.17 (m, 2H), 1.89−1.71 (m, 1H), 1.33−1.21 (m, 3H); 13C NMR (101 MHz, CDCl3) (rotamers) δ 173.0, 172.93, 172.87, 172.6, 172.3, 172.2, 168.3, 168.2, 168.11, 168.07, 154.7, 154.6, 154.0, 136.5, 136.35, 136.31, 128.60, 128.56, 128.54, 128.21, 128.15, 128.08, 128.05, 67.5, 67.4, 66.5, 66.4, 66.1, 63.03, 62.97, 62.9, 61.6, 61.22, 61.19, 60.7, 56.89, 56.87, 56.77, 56.69, 52.66, 52.5, 52.4, 52.3, 46.9, 46.6, 46.3, 46.1, 46.0, 45.9, 45.7, 45.5, 31.0, 29.8, 29.8, 28.71, 28.66, 27.7, 27.1, 26.9, 26.3, 14.1, 14.0.
Disodium trans-3-(1-Carboxylato-3-hydroxypropyl)pyrrolidine-2-carboxylate (3n).
The title compound was prepared according to general procedure II, starting from 12n (200 mg, 0.48 mmol) and stirred for 4 days. The diastereomers could not be separated on preparative HPLC, but afforded a mixture of the trans-diastereomers of the corresponding lactone·HCl salt 13, which was submitted to 3 equiv of 0.5 M NaOH(aq.) and afforded a mixture of the trans-diastereomers 3n as a white solid (52 mg, 0.20 mmol, 42%). d.r. 53:47; 1H NMR (600 MHz, D2O) (minor trans-diastereomer in brackets) δ [4.05 (d, J = 5.2 Hz, 1H)], 3.98 (d, J = 4.6 Hz, 1H), [3.74−3.64 (m, 2H)], 3.65−3.55 (m, 2H), [3.49−3.38 (m, 2H)], 3.49−3.38 (m, 2H), [2.73 (p, J = 6.3 Hz, 1H)], [2.62−2.55 (m, 1H)], 2.53 (ddd, J = 3.7, 6.6, 10.7 Hz, 1H), 2.41 (td, J = 3.8, 10.4 Hz, 1H), [2.19−2.09 (m, 1H)], 2.19−2.09 (m, 1H), [2.01−1.91 (m, 1H)], 2.01−1.91 (m, 2H), [1.91−1.82 (m, 1H)], 1.91−1.82 (m, 1H), [1.82−1.74 (m, 1H)]; 13C NMR (151 MHz, D2O) (minor trans-diastereomer in brackets) δ [181.9], 181.7, [174.5], 174.4, [64.1], 63.4, [60.2], 60.1, 48.8, [46.9], 45.4, [45.10], 45.08, [45.0], 33.9, [31.8], [28.2], 27.0.
trans-1-Benzyl 2-Methyl 3-(1-Ethoxy-2-(ethoxycarbonyl)-1-oxopent-4-en-2-yl)pyrrolidine-1,2-dicarboxylate (14).
The title compound was prepared according to general procedure I (3 h reaction time), starting from 10 (270 mg, 1.03 mmol) and diethyl allylmalonate (517 mg, 0.54 mL, 2.58 mmol), which afforded the title compound as a colorless oil (444 mg, 0.96 mmol, 93%). LCMS (ESI) m/z = 462.2 [M + H]+; Rf = 0.18 (7:3 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.38−7.28 (m, 5H), 5.71−5.57 (m, 1H), 5.19 (d, J = 12.3 Hz, 1H), 5.15−5.07 (m, 2H), 5.01 (d, J = 12.5 Hz, 1H), 4.58 (br d, J = 16.2 Hz, 1H), 4.21−3.93 (m, 4H), 3.77 (s, 1.5H), 3.59 (s, 1.5H), 3.57−3.45 (m, 2H), 3.02−2.92 (m, 1H), 2.74 (br d, J = 6.1 Hz, 2H), 2.28−2.14 (m, 1H), 2.09−1.94 (m, 1H), 1.21 (t, J = 7.2 Hz, 3H), 1.17 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) (rotamers) δ 173.3, 173.2, 170.1, 170.0, 169.9, 154.5, 154.2, 136.7, 136.6, 131.9, 128.6, 128.5, 128.2, 128.14, 128.11, 128.08, 119.8, 67.2, 61.8, 61.5, 61.0, 59.8, 52.5, 52.3, 46.7, 46.0, 45.6, 45.4, 38.6, 26.3, 25.6, 14.13, 14.11, 14.01, 13.98.
trans-1-Benzyl 2-Methyl 3-(1-Ethoxy-2-(ethoxycarbonyl)-5-hydroxy-1-oxopentan-2-yl)pyrrolidine-1,2-dicarboxylate (12o).
To a solution of trans-enantiomers 14 (410 mg, 0.89 mmol, 1 equiv) in anhydrous THF (9 mL) at 0 °C, 1 M borane tetrahydrofuran complex solution (1.3 mL, 1.33 mmol, 1.5 equiv) was added and the resulting solution was stirred at this temperature for 2 h. Then, water (80 μL, 4.44 mmol, 5 equiv), 4 M aq NaOH (933 μL, 3.73 mmol, 4.2 equiv), and 30% aq peroxide solution (483 μL, 4.26 mmol, 4.8 equiv) were added to the previous mixture, which was stirred at 0 °C for an additional hour. After that, the reaction mixture was diluted with EtOAc (60 mL) and water (60 mL). The aqueous layer was extracted with EtOAc (3 × 60 mL), and the collected organic layers were washed with brine (60 mL), dried over MgSO4, and filtered. The solvent of the filtrate was removed under reduced pressure to give a residue that was purified by flash chromatography (silica gel, heptane/ EtOAc 1:1, isocratic) to afford the desired mixture of trans-enantiomers as a colorless oil (211 mg, 0.44 mmol, 50%). LCMS (ESI) m/z = 480.2 [M + H]+; Rf = 0.10 (1:1 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.37−7.27 (m, 5H), 5.19 (d, J = 12.7 Hz, 1H), 5.02 (d, J = 12.4 Hz, 1H), 4.56 (br d, J = 19.0 Hz, 1H), 4.23−3.96 (m, 4H), 3.76 (s, 1.5H), 3.62 (t, J = 6.2 Hz, 2H), 3.58 (s, 1.5H), 3.55−3.44 (m, 2H), 3.05−2.95 (m, 1H), 2.29−2.15 (m, 1H), 2.05 (t, J = 8.2 Hz, 2H), 2.03−1.95 (m, 1H), 1.52−1.42 (m, 2H), 1.26−1.16 (m, 6H); 13C NMR (101 MHz, CDCl3) (rotamers) δ 173.31, 173.27, 170.5, 170.44, 170.42, 170.3, 154.5, 154.2, 136.7, 136.6, 129.1, 128.51, 128.47, 128.3, 128.15, 128.13, 128.10, 127,99, 125.4, 67.2, 62.5, 61.8, 61.8, 61.4, 60.9, 59.51, 59.50, 52.6, 52.3, 47.1, 46.0, 45.4, 30.7, 30.6, 27.6, 26.5, 25.8, 14.1, 14.03, 14.00.
trans-3-(1-Carboxy-4-hydroxybutyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3o).
The title compound was prepared according to general procedure II, starting from 12o (119 mg, 0.25 mmol) and stirred for 3 days. The diastereomers could not be separated on preparative HPLC, which afforded a mixture of the trans-diastereomers 3o as a white solid (19 mg, 0.071 mmol, 29%). d.r. 56:44; LCMS (ESI) m/z = 250.1 [M + H]+; 1H NMR (600 MHz, D2O) (two diastereomers) δ 4.33−4.20 (2m, 1H), 3.71−3.61 (m, 2H), 3.54−3.48 (m, 1H), 3.44−3.38 (m, 1H), 2.88−2.68 (2m, 2H), 2.40−2.28 (m, 1H), 1.98−1.90 (m, 1H), 1.90−1.76 (m, 4H); 13C NMR (151 MHz, D2O) (two diastereomers) δ 178.6, 178.1, 172.4, 172.1, 62.7, 62.5, 47.6, 47.0, 46.1, 46.0, 45.5, 44.4, 44.3, 30.2, 30.1, 28.5, 28.2, 28.0, 27.4.
trans-1-Benzyl 2-Methyl 3-(1-Methoxy-2-(methoxycarbonyl)-1-oxopent-4-yn-2-yl)pyrrolidine-1,2-dicarboxylate (15).
The title compound was prepared according to general procedure I, starting from 10 (180 mg, 0.69 mmol) and dimethyl propargylmalonate (293 mg, 0.28 mL 1.72 mmol), which afforded the title compound as a colorless oil (264 mg, 0.61 mmol, 89%). LCMS (ESI) m/z = 432.2 [M + H]+; Rf = 0.17 (7:3 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.39−7.27 (m, 5H), 5.18 (d, J = 12.5 Hz, 1H), 5.05 (d, J = 12.0 Hz, 1H), 4.58−4.49 (br d, J = 11.4 Hz, 1H), 3.78 (s, 1.5H), 3.74 (s, 1.5H), 3.72 (s, 1.5H), 3.66 (s, 1.5H), 3.63 (s, 1.5H), 3.61 (s, 1.5H), 3.60−3.49 (m, 2H), 3.32−3.23 (m, 1H), 3.01−2.86 (m, 2H), 2.37−2.23 (m, 1H), 2.10−1.94 (m, 2H); 13C NMR (151 MHz, CDCl3) (rotamers) δ 173.0, 172.9, 169.7, 169.63, 169.61, 169.5, 154.6, 154.2, 136.65, 136.63, 128.60, 128.5, 128.21, 128.18, 78.10, 78.07, 72.4, 67.34, 67.30, 61.3, 60.8, 58.9, 53.2, 53.1, 53.0, 52.7, 52.5, 46.5, 46.0, 45.5, 45.4, 26.6, 25.9, 24.34, 24.32.
trans-1-Benzyl 2-Methyl 3-(2-((1H-1,2,3-Triazol-4-yl)methyl)-1,3-dimethoxy-1,3-dioxopropan-2-yl)pyrrolidine-1,2-dicarboxylate (12p).
In a 9 mL vessel, a solution of 15 (391 mg, 0.91 mmol), copper(I) iodide (17 mg, 0.091 mmol), and azidotrimethylsilane (95%, 380 μL, 2.72 mmol) in DMF·MeOH 1:1 (5 mL) was prepared under argon atmosphere. The vessel was sealed with a septum, heated to reflux, and stirred overnight. Next day, the reaction mixture was allowed to reach rt and then diluted with water (50 mL) and EtOAc (50 mL). The aqueous layer was extracted with EtOAc (3 × 50 mL), and the collected organic layers were washed with water (3 × 50 mL), dried over MgSO4, and filtered. After that, the solvent of the filtrate was removed under reduced pressure to give a brown oil that was purified by flash chromatography (silica gel, heptane/EtOAc 1:1 to 2:8, gradient) to afford the desired mixture of trans-enantiomers as a white solid (307 mg, 0.65 mmol, 71%). LCMS (ESI) m/z = 475.2 [M + H]+; 1H NMR (400 MHz, CDCl3) (rotamers) δ 7.48 (s, 1H), 7.39−7.26 (m, 5H), 5.19 (d, J = 12.5 Hz, 1H), 5.04 (br t, J = 11.9 Hz, 1H), 4.61 (br d, J = 25.0, 1H), 3.74−3.72 (2s, 1.5H), 3.69 (s, 1.5H), 3.66 (s, 1.5H), 3.63 (s, 1.5H), 3.60 (s, 1.5H), 3.56 (s, 1.5H), 3.55−3.38 (m, 4H), 3.01−2.91 (m, 1H), 2.32−2.18 (m, 1H), 2.17−2.01 (m, 1H); 13C NMR (101 MHz, CDCl3) (rotamers) δ 173.2, 173.1, 170.4, 170.3, 170.1, 154.8, 154.4, 140.8, 136.6, 132.1, 128.72, 128.70, 128.34, 128.29, 128.26, 67.5, 61.6, 61.1, 60.3, 60.2, 53.2, 53.14, 53.10, 53.0, 52.8, 52.6, 47.0, 46.0, 45.9, 45.4, 30.03, 30.00, 26.7, 25.9.
trans-3-(1-Carboxy-2-(1H-1,2,3-triazol-4-yl)ethyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3p).
The title compound was prepared according to general procedure II, starting from 12p (289 mg, 0.61 mmol) and stirred for 3 days. The two epsilon diastereomers were separated on preparative HPLC. For the trans-diastereomer 3p-d1: as a white solid (18 mg, 0.06 mmol, 10%). d.r. 99:1; LCMS (ESI) m/z = 255.1 [M + H]+; 1H NMR (600 MHz, D2O) δ 8.02 (br s, 1H), 4.49 (br d, J = 8.1 Hz, 1H), 3.56−3.50 (m, 1H), 3.43−3.36 (m, 1H), 3.34−3.28 (m, 2H), 3.22−3.15 (m, 1H), 2.87−2.80 (m, 1H), 2.37−2.31 (m, 1H), 2.00−1.92 (m, 1H); 13C NMR (151 MHz, D2O) δ 176.1, 171.6, 141.9, 127.3, 62.0, 46.8, 46.3, 43.7, 27.6, 25.5. For the trans-diastereomer 3p-d2: as a white solid (8 mg, 0.03 mmol, 5%). d.r. 10:90; LCMS (ESI) m/z = 255.1 [M + H]+; 1H NMR (400 MHz, D2O) δ 7.92 (br s, 1H), 4.39 (br d, J = 7.4 Hz, 1H), 3.64−3.53 (m, 1H), 3.49−3.42 (m, 1H), 3.26−3.19 (m, 2H), 3.15−3.07 (m, 1H), 2.98−2.88 (m, 1H), 2.51−2.41 (m, 1H), 2.11−2.00 (m, 1H); 13C NMR (151 MHz, D2O) δ 177.1, 171.9, 142.2, 127.4, 62.3, 48.3, 46.1, 44.0, 28.8, 25.2. In addition, a mixture of the trans-diastereomers 3p-d1/3p-d2 was isolated as a white solid (113 mg, 0.39 mmol, 64%). LCMS (ESI) m/z = 255.1 [M + H]+.
trans-1-Benzyl 2-Methyl 3-(2-(Cyanomethyl)-1,3-dimethoxy-1,3-dioxopropan-2-yl)pyrrolidine-1,2-dicarboxylate (16).
To a solution of 12a (5.23 g, 13.3 mmol, 1 equiv) in THF (150 mL) was added NaH (532 mg, 13.3 mmol, 1 equiv) at 0 °C, and the reaction mixture was stirred for 15 min. Then bromoacetonitrile (7.92 g, 66.6 mmol, 5 equiv) was added, and the mixture was stirred at 50 °C for 23 h. The reaction was quenched with NH4Cl (100 mL). The mixture was concentrated under reduced pressure, and following purified by DCVC (0 → 50% EtOAc in heptane), to afford the title compound as a colorless oil (4.80 g, 11.11 mmol, 84%). LCMS (ESI) m/z = 433.2 [M + H]+; Rf = 0.27 (2:1 heptane/EtOAc); 1H NMR (600 MHz, CDCl3) δ 7.37−7.29 (m, 5H), 5.19 (dd, J = 12.4, 19.0 Hz, 1H), 5.05 (dd, J = 12.3, 41.0 Hz, 1H), 4.46 (dd, J = 4.4, 25.1 Hz, 1H), 3.80−3.73 (m, 6H), 3.72 (s, 1.5H), 3.71−3.61 (m, 1H), 3.60 (s, 1.5H), 3.53 (dt, J = 7.8, 10.8 Hz, 1H), 3.20−3.14 (m, 1H), 3.12−2.97 (m, 2H), 2.35−2.23 (m, 1H), 2.04 (ddt, J = 7.4, 14.5, 47.9 Hz, 1H) ; 13C NMR (151 MHz, CDCl3) (rotamers) δ 172.3, 172.2, 168.3, 168.2, 168.13, 168.08, 154.6, 154.0, 136.5, 136.4, 128.64, 128.61, 128.34, 128.29, 128.25, 128.21, 115.9, 115.8, 67.5, 67.4, 60.6, 60.5, 57.54, 57.50, 53.8, 53.72, 53.68, 52.9, 52.7, 47.3, 46.2, 46.0, 45.5, 27.0, 26.3, 22.92, 22.90, 21.2.
trans-1-Benzyl 2-Methyl 3-(2-((2H-Tetrazol-5-yl)methyl)-1,3-dimethoxy-1,3-dioxopropan-2-yl)pyrrolidine-1,2-dicarboxylate (17-s1 and 17-s2).
To a vial with a solution of 16 (1.31 g, 3.03 mmol, 1 equiv) in dry toluene (17.5 mL) were added sodium azide (1.77 g, 27.3 mmol, 9 equiv) and triethylammonium chloride (3.76 g, 27.3 mmol, 9 equiv) under N2 atmosphere. The vial was capped and stirred for 19 h at 115 °C using a sand bath. The reaction mixture was quenched using sat. Na2CO3 (0.30 mL) with H2O (15 mL) added in addition. The separated aqueous phase was acidified using 1 M HCl to about pH 2 and extracted with EtOAc (3 × 20 mL). The combined organic phases were concentrated in vacuo and purified using preperative HPLC to give the racemic product (0.796 mg, 55%). The enantiomers were obtained using a ChiralPak AD (250 × 20 mm, 10 μm) column (isocratic elution: 70:30:0.1 n-heptane/EtOH/TFA, 20 mL/min) after four injections of the racemic mixture. The fractions of first eluting enantiomer were pooled, concentrated in vacuo, and freeze-dried to afford pure enantiomer 17-s1 as a clear oil (419 mg, 0.88 mmol, 29%). LCMS (ESI) m/z = 476.2 [M + H]+; 1H NMR (600 MHz, CDCl3) δ 7.40−7.27 (m, 5H), 5.21 (dd, J = 19.2, 12.3 Hz, 1H), 5.07 (dd, J = 53.9, 12.3 Hz, 1H), 4.59 (dd, J = 33.4, 3.7 Hz, 1H), 3.74−3.65 (m, 8H), 3.64−3.61 (m, 2H), 3.60−3.54 (m, 2H), 3.54−3.46 (m, 1H), 3.10−2.99 (m, 1H), 2.38−2.22 (m, 1H), 2.18−1.98 (m, 1H); 13C NMR (151 MHz, CDCl3) (rotamers) δ 173.2, 173.1, 173.0, 172.93, 172.92, 169.6, 169.54, 169.51, 169.4, 155.1, 154.4, 136.13, 136.09, 136.06, 128.7, 128.6, 128.44, 128.42, 128.2, 128.1, 67.9, 67.7, 61.2, 60.91, 60.87, 59.7, 59.6, 59.63, 59.61, 53.6, 53.55, 53.53, 53.47, 53.12, 53.11, 52.9, 47.53, 47.50, 46.48, 46.46, 46.0, 45.5, 28.2, 27.2, 26.2. The fractions of the second eluting enantiomer were pooled, concentrated in vacuo, and freeze-dried to afford the pure enantiomer 17-s2 as a clear oil (370 mg, 0.78 mmol, 26%). LCMS (ESI) m/z = 476.2 [M + H]+; 1H NMR (600 MHz, CDCl3) δ 7.38−7.27 (m, 5H), 5.20 (dd, J = 17.4, 12.4 Hz, 1H), 5.06 (dd, J = 52.2, 12.4 Hz, 1H), 4.60 (dd, J = 29.6, 3.8 Hz, 1H), 3.72−3.64 (m, 7H), 3.64−3.58 (m, 3H), 3.56 (s, 2H), 3.53−3.46 (m, 1H), 3.10−3.00 (m, 1H), 2.29 (m, 1H), 2.18−1.96 (m, 1H); 13C NMR (151 MHz, CDCl3) (rotamers) δ 173.1, 172.9, 169.6, 169.5, 169.4, 154.9, 154.3, 136.23, 136.22, 128.65, 128.61, 128.33, 128.29, 128.14, 128.10, 67.7, 67.6, 61.3, 60.9, 59.53, 59.49, 53.52, 53.50, 53.4, 53.0, 52.8, 47.4, 46.4, 45.9, 45.5, 28.4, 28.3, 27.1, 26.1, 25.3.
trans-Dimethyl 2-((2H-Tetrazol-5-yl)methyl)-2-(2-(methoxycarbonyl)pyrrolidin-3-yl)malonate (18-s1).
To a round-bottomed flask with a three-way valve connected were added 17-s1 (419 mg, 0.88 mmol, 1 equiv) and 10 mol % Pd/C (9.3 mg, 0.1 equiv). The flask was evacuated and refilled with nitrogen three times, and MeOH (5 mL) was added. The flask was evacuated and refilled once with nitrogen and three times with hydrogen. The reaction was stirred for 1 day at rt. The reaction flask was evacuated and refilled with nitrogen three times. The reaction mixture was filtered over a pad of Celite with MeOH (25 mL) under a flow of nitrogen. The catalyst was washed with water and disposed, while the MeOH solution was concentrated in vacuo to give the crude of the desired enantiomer 18-s1 as a clear oil (313 mg, 0.92 mmol). The crude product was used in next reaction without further purification. LCMS (ESI) m/z = 342.1 [M + H]+.
trans-Dimethyl 2-((2H-Tetrazol-5-)methyl)-2-(2-(methoxycarbonyl)pyrrolidin-3-yl)malonate (18-s2).
To a round-bottomed flask with a three-way valve connected were added 17-s2 (370 mg, 0.78 mmol, 1 equiv) and 10 mol % Pd/C (8.2 mg, 0.1 equiv). The flask was evacuated and refilled with nitrogen three times, and MeOH (4 mL) was added. The flask was evacuated and refilled once with nitrogen and three times with hydrogen. The reaction was stirred for 1 day at rt. The reaction flask was evacuated and refilled with nitrogen three times. The reaction mixture was filtered over a pad of Celite with MeOH (25 mL) under a flow of nitrogen. The catalyst was washed with water and disposed, while the MeOH solution was concentrated in vacuo to give the crude of the desired enantiomer 18-s2 as a clear oil (267 mg, 0.78 mmol). Product used in next reaction without further purification. LCMS (ESI) m/z = 342.1 [M + H]+.
trans-3-(1-Carboxy-2-(2H-tetrazol-5-yl)ethyl)pyrrolidine-2-carboxylic Acid (3q-s1 and 3q-s2).
A stirring solution of crude 18-s1 (301 mg, 0.88 mmol, 1 equiv) in excess 6 M HCl was stirred under reflux for 4 days, under an argon atmosphere. The acidic solution was concentrated in vacuo, and the crude mixture of diastereomers was resolved using a chiral Chirobiotic T column (500 × 10 mm) (Astec) (isocratic 20% B; solvent A: NH4OAc (15 mM) + AcOH to pH 4; solvent B: EtOH, 5 mL/min) to afford 3q-s1 (contains 2.5 equiv of AcONH4) (the first eluting diastereomer) as semisolid (hygroscopic), after removal of the solvent in vacuo (181 mg, 0.40 mmol, 46% over two steps). d.r. 96:4; LCMS (ESI) m/z 256.1 [M + H]+; [α]25589 = −20.6° (c = 0.369, 0.1 M HCl); 1H NMR (600 MHz, D2O) δ 4.17 (d, J = 5.7 Hz, 1H), 3.45 (dt, J = 12.9, 6.6 Hz, 1H), 3.42−3.36 (m, 1H), 3.28 (d, J = 8.2 Hz, 2H), 2.91 (q, J = 7.9 Hz, 1H), 2.60 (p, J = 7.3 Hz, 1H), 2.20 (dt, J = 13.9, 7.0 Hz, 1H), 1.91 (dt, J = 14.5, 7.2 Hz, 1H); 13C NMR (151 MHz, D2O) δ 179.1, 173.8, 157.3, 63.8, 50.2, 45.2, 44.7, 27.7, 25.9. The second eluting diastereomer, 3q-s2 (contains 0.14 of equiv AcONH4), was obtained as a white solid after removal of the solvent in vacuo (41 mg, 0.15 mmol, 18% over two steps). d.r. 4:96; LCMS (ESI) m/z 256.1 [M + H]+; [α]25589 = −28.6° (c = 0.273, 0.1 M HCl); 1H NMR (600 MHz, D2O) δ 4.08 (d, J = 4.9 Hz, 1H), 3.43 (t, J = 7.5 Hz, 2H), 3.25 (dd, J = 15.0, 9.6 Hz, 1H), 3.06 (dd, J = 14.9, 5.0 Hz, 1H), 2.93 (ddd, J = 9.6, 6.9, 5.0 Hz, 1H), 2.87−2.79 (m, 1H), 2.14 (dt, J = 14.8, 7.5 Hz, 1H), 1.96 (dt, J = 13.6, 7.0 Hz, 1H); 13C NMR (151 MHz, D2O) δ 180.4, 173.8, 160.6, 63.4, 49.3, 45.0, 44.9, 26.9, 25.2.
trans-3-(1-Carboxy-2-(1H-tetrazol-5-yl)ethyl)pyrrolidine-2-carboxylic Acid Hydrochloride (3q-s3–4).
A stirring solution of crude 18-s2 (265 mg, 0.78 mmol, 1 equiv) in excess 6 M HCl(aq.) was stirred under reflux for 4 days, under argon. The acidic solution was concentrated in vacuo to afford a mixture of the trans-diastereomers 3q-s3/3q-s4 as a white solid (226 mg, 0.77 mmol, 99%). d.r. 2:1. LCMS (ESI) m/z 256.1 [M + H]+; 1H NMR (600 MHz, D2O) (two diastereomers) δ 4.48 (dd, J = 38.3, 7.9 Hz, 1H), 3.64−3.24 (m, 5H), 3.01−2.81 (m, 1H), 2.49−2.29 (m, 1H), 2.10−1.90 (m, 1H); 13C NMR (151 MHz, D2O) (two diastereomers) δ 175.5, 174.9, 171.1, 171.0, 154.6, 154.5, 61.4, 61.2, 45.54, 45.51, 45.4, 44.5, 43.2, 43.1, 28.2, 26.9, 24.0, 23.3.
Pharmacological Characterization.
Ligand binding affinities for native AMPA, KA, and NMDA receptors in rat cortical synaptosomes were determined as previously described using [3H]-AMPA, [3H]-KA, and [3H]-CGP 39653, respectively.40 CPM values were fitted by nonlinear regression using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA). Ligand affinities at recombinant rat homomeric GluK1−3 were determined as previously described21,52 using [3H]-KA and the newly developed radioligand [3H]-NF608 for GluK1.69 Competition curves (n ≥ 3) were conducted in triplicate at 12−16 ligand concentrations. Data were analyzed using GraphPad Prism v6 (GraphPad Software, San Diego, CA) to determine ligand affinity (Ki one site equation) and Hill slope (four-parameter logistic equation) values. The inhibitory concentrations of the compounds were determined at human EAAT1, EAAT2, and EAAT3 stably expressed in HEK293 cells in a conventional [3H]-d-Asp uptake assay essentially as described previously.70 The cell lines were cultured at 37 °C in a humidified 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium Glutamax-I supplemented with 5% dialyzed fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 1 mg/mL G-418. The day before the assay, cells were split into poly-d-lysinecoated white 96-well plates. At 16−24 h later, the culture medium was aspirated and cells were washed twice with 100 μL of assay buffer (Hank’s buffered saline solution supplemented with 20 mM HEPES, 1 mM CaCl2 and 1 mM MgCl2, pH 7.4). Then 50 μL of assay buffer supplemented with 100 nM [3H]-d-Asp (PerkinElmer, Boston, MA) and various concentrations of test compounds were added to the wells, and the plate was incubated at 37 °C for 3 min. Nonspecific [3H]-d-Asp uptake/binding in the cells was determined in the presence of 3 mM (S)-Glu. The assay mixtures were quickly removed from the wells, and the wells were washed with 2 × 100 μL ice-cold assay buffer, after which 150 μL of Microscint-20 scintillation fluid (PerkinElmer, Boston, MA) was added to each well. Then the plate was shaken for at least 1 h and counted in a TopCounter (PerkinElmer, Boston, MA). The experiments were performed in duplicate at least three times for each compound. Functional characterization at cloned GluN1/GluN2A-D receptors was done using rat cDNAs for GluN1–1a (GenBank U08261; hereafter GluN1), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), and GluN2D (L31611) were provided by S. Heinemann (Salk Institute), S. Nakanishi (Osaka Bioscience Institute), and P. Seeburg (University of Heidelberg). The cDNA encoding rat GluN2B was modified by removing a T7 RNA polymerase termination site located in the C-terminal domain, without changing the amino acid sequence.71 DNA constructs were first linearized by restriction enzymes and then used as templates for in vitro cRNA transcription. The cRNA encoding GluN1 was coinjected with different GluN2 subunit cRNA at a volume of 50 nL (0.5−5 ng) into Xenopus oocytes (purchased from Rob Weymouth at Xenopus 1, Dexter, MI). The injected oocytes were incubated at 17 °C for at least 2 days before recording in modified Barth’s solution containing (in mM) 88 NaCl, 1.0 KCl, 15 HEPES, 2.4 NaHCO3, 0.41 CaCl2, 0.82 MgSO4, and 0.3 Ca(NO3)2 (pH 7.5; osmolarity 0.22 Osm/kg) supplemented with 0.1 U/L penicillin and 0.1 mg/L streptomycin. Two-electrode voltage-clamp recordings were performed essentially as previously described.71 Briefly, a gravity-driven perfusion system was used with extracellular recording solution containing (in mM) 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2, and 0.01 EDTA (pH 7.4; osmolarity 0.20 Osm/kg) at a flow rate of 5 mL/min. Oocytes expressing GluN1/2A or GluN1/2B were injected with 50 nL or 30 nL of 50 mM BAPTA at least 10 min before recording to prevent activity-dependent increases in response amplitude.72,73 For antagonist concentration−inhibition experiments, 100 μM glycine and 1 μM glutamate were coapplied to elicit current response followed by increasing concentrations of compound to inhibit current the response. Concentration−response data were analyzed using GraphPad Prism (GraphPad Software). Antagonist concentration−inhibition data were fitted to , where Imin is the minimum current in response to the agonist plus a saturating concentration of antagonist. For some combinations of antagonist and NMDA receptor subtype, Imin was constrained to be 0. For graphical presentation, the data from individual oocytes were normalized to the current response to glutamate plus glycine in the same recording and averaged. Antagonist binding affinities were estimated using published glutamate EC50 values for GluN1/2A−D,74 IC50 values for individual oocytes, the agonist concentration [A], and the Cheng−Prusoff relationship: Ki = IC50/(1 + [A]/EC50).
Supplementary Material
ACKNOWLEDGMENTS
The technical assistance of Niels Vissing Holst and Anders Kadziola, Department of Chemistry, University of Copenhagen, with collecting X-ray data and data reduction is gratefully acknowledged.
Funding
This work was financially supported by the Lundbeck Foundation (grant given to L.B.) and NIH (GM103546 and NS097536 grants given to K.B.H.).
ABBREVIATIONS
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CPAA
(2S,3R)-2-carboxy-3-pyrrolidine acetic acid
- DCVC
dry column vacuum chromatography
- DHK
dihydrokainic acid
- EAATs
excitatory amino acid transporters
- Glu
(S)-glutamic acid
- GluK1−3
Kainate receptor subunits 1−3
- iGluRs
ionotropic Glu receptors
- KA
kainic acid
- NMDA
N-methyl-d-aspartate
- SAR
structure−activity relationship
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.9b00205.
Experimental details for crystal structure determination of compound 9c-s1 and compound 9d-s2 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Reiner A, and Levitz J (2018) Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 98, 1080–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Traynelis SF, et al. (2010) Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gan Q, Salussolia CL, and Wollmuth LP (2015) Assembly of AMPA receptors: mechanisms and regulation. J. Physiol 593, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhu S, and Gouaux E (2017) Structure and symmetry inform gating principles of ionotropic glutamate receptors. Neuropharmacology 112, 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hansen KB, et al. (2018) Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol 150, 1081–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Greger IH, Watson JF, and Cull-Candy SG (2017) Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron 94, 713–730. [DOI] [PubMed] [Google Scholar]
- (7).Chen PE, and Wyllie D. J. a. (2006) Pharmacological insights obtained from structure-function studies of ionotropic glutamate receptors. Br. J. Pharmacol 147, 839–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lee C-H, et al. (2014) NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 511, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Karakas E, and Furukawa H (2014) Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 344, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sobolevsky AI, Rosconi MP, and Gouaux E (2009) X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462, 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Farooqui AA, Ong WY, Lu X-R, Halliwell B, and Horrocks LA (2001) Neurochemical consequences of kainate-induced toxicity in brain: involvement of arachidonic acid release and prevention of toxicity by phospholipase A2 inhibitors. Brain Res. Rev 38, 61–78. [DOI] [PubMed] [Google Scholar]
- (12).Ko S, Zhao M-G, Toyoda H, Qiu C-S, and Zhuo M (2005) Altered Behavioral Responses to Noxious Stimuli and Fear in Glutamate Receptor 5 (GluR5)- or GluR6-Deficient Mice. J. Neurosci 25, 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pitt D, Werner P, and Raine CS (2000) Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med 6, 67–70. [DOI] [PubMed] [Google Scholar]
- (14).Matute C (2011) Therapeutic potential of kainate receptors. CNS Neurosci. Ther 17, 661–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lerma J, and Marques JM (2013) Kainate receptors in health and disease. Neuron 80, 292–311. [DOI] [PubMed] [Google Scholar]
- (16).Meador-Woodruff JH, Davis KL, and Haroutunian V (2001) Abnormal kainate receptor expression in prefrontal cortex in schizophrenia. Neuropsychopharmacology 24, 545–52. [DOI] [PubMed] [Google Scholar]
- (17).Contractor A, Mulle C, and Swanson GT (2011) Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci 34, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jamain S, et al. (2002) Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry 7, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Woo TU, et al. (2007) Differential alterations of kainate receptor subunits in inhibitory interneurons in the anterior cingulate cortex in schizophrenia and bipolar disorder. Schizophr. Res 96, 46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Xu J, et al. (2017) Complete Disruption of the Kainate Receptor Gene Family Results in Corticostriatal Dysfunction in Mice. Cell Rep 18, 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Frydenvang K, et al. (2010) Biostructural and Pharmacological Studies of Bicyclic Analogues of the 3-Isoxazolol Glutamate Receptor Agonist Ibotenic Acid. J. Med. Chem 53, 8354–8361. [DOI] [PubMed] [Google Scholar]
- (22).Madsen U, et al. (1996) N-Methyl-d-aspartic Acid Receptor Agonists: Resolution, Absolute Stereochemistry, and Pharmacology of the Enantiomers of 2-Amino-2-(3-hydroxy-5-methyl-4-isoxazolyl)-acetic Acid. J. Med. Chem 39, 183–190. [DOI] [PubMed] [Google Scholar]
- (23).Conti P, et al. (1999) Synthesis and Enantiopharmacology of New AMPA-Kainate Receptor Agonists. J. Med. Chem 42, 4099–4107. [DOI] [PubMed] [Google Scholar]
- (24).Sagot E, et al. (2008) Chemo-Enzymatic Synthesis of a Series of 2,4-Syn -Functionalized (S)-Glutamate Analogues: New Insight into the Structure–Activity Relation of Ionotropic Glutamate Receptor Subtypes 5, 6, and 7. J. Med. Chem 51, 4093–4103. [DOI] [PubMed] [Google Scholar]
- (25).Bunch L, and Krogsgaard-Larsen P (2009) Subtype selective kainic acid receptor agonists: Discovery and approaches to rational design. Med. Res. Rev 29, 3–28. [DOI] [PubMed] [Google Scholar]
- (26).Rasmussen JL, Storgaard M, Pickering DS, and Bunch L (2011) Rational Design, Synthesis and Pharmacological Evaluation of the (2R)- and (2S)-Stereoisomers of 3-(2-Carboxypyrrolidinyl)-2-methyl Acetic Acid as Ligands for the Ionotropic Glutamate Receptors. ChemMedChem 6, 498–504. [DOI] [PubMed] [Google Scholar]
- (27).Bunch L, Nielsen B, Jensen AA, and Bräuner-Osborne H (2006) Rational Design and Enantioselective Synthesis of (1R,4S,5R,6S)-3-Azabicyclo[3.3.0]octane-4,6-dicarboxylic Acid A Novel Inhibitor at Human Glutamate Transporter Subtypes 1, 2, and 3. J. Med. Chem 49, 172–178. [DOI] [PubMed] [Google Scholar]
- (28).Møllerud S, et al. (2017) Structure and Affinity of Two Bicyclic Glutamate Analogues at AMPA and Kainate Receptors. ACS Chem. Neurosci 8, 2056–2064. [DOI] [PubMed] [Google Scholar]
- (29).Stensbøl TB, et al. (1999) Resolution, absolute stereo-chemistry and molecular pharmacology of the enantiomers of ATPA. Eur. J. Pharmacol 380, 153–162. [DOI] [PubMed] [Google Scholar]
- (30).Clausen RP, et al. (2009) The Glutamate Receptor GluR5 Agonist (S)-2-Amino-3-(3-hydroxy-7,8-dihydro-6H-cyclohepta[d]-isoxazol-4-yl)propionic Acid and the 8-Methyl Analogue: Synthesis, Molecular Pharmacology, and Biostructural Characterization. J. Med. Chem 52, 4911–4922. [DOI] [PubMed] [Google Scholar]
- (31).Lash LL, et al. (2007) Novel Analogs and Stereoisomers of the Marine Toxin Neodysiherbaine with Specificity for Kainate Receptors. J. Pharmacol. Exp. Ther 324, 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Shoji M, et al. (2006) Total Synthesis and Biological Evaluation of Neodysiherbaine A and Analogues. J. Org. Chem 71, 5208–5220. [DOI] [PubMed] [Google Scholar]
- (33).Butini S, et al. (2008) 1H-Cyclopentapyrimidine-2,4(1H,3H)-dione-Related Ionotropic Glutamate Receptors Ligands. Structure–Activity Relationships and Identification of Potent and Selective iGluR5Modulators. J. Med. Chem 51, 6614–6618. [DOI] [PubMed] [Google Scholar]
- (34).Zhou LM, et al. (1997) (2S,4R)-4-methylglutamic acid (SYM 2081): a selective, high-affinity ligand for kainate receptors. J. Pharmacol. Exp. Ther 280, 422–7. [PubMed] [Google Scholar]
- (35).Gu Z-Q, et al. (1995) Synthesis, Resolution, and Biological Evaluation of the Four Stereoisomers of 4-Methylglutamic Acid: Selective Probes of Kainate Receptors. J. Med. Chem 38, 2518–2520. [DOI] [PubMed] [Google Scholar]
- (36).Alaux S, et al. (2005) Chemoenzymatic Synthesis of a Series of 4-Substituted Glutamate Analogues and Pharmacological Characterization at Human Glutamate Transporters Subtypes 1–3. J. Med. Chem 48, 7980–7992. [DOI] [PubMed] [Google Scholar]
- (37).Bunch L, et al. (2009) Pharmacological characterization of (4R)-alkyl glutamate analogues at the ionotropic glutamate receptors — Focus on subtypes iGlu5–7. Eur. J. Pharmacol 609, 1–4. [DOI] [PubMed] [Google Scholar]
- (38).Bunch L, et al. (2001) Synthesis and receptor binding affinity of new selective GluR5 ligands. Bioorg. Med. Chem 9, 875–879. [DOI] [PubMed] [Google Scholar]
- (39).Bunch L, et al. (2009) 4,4-Dimethyl- and diastereomeric 4-hydroxy-4-methyl- (2S)-glutamate analogues display distinct pharmacological profiles at ionotropic glutamate receptors and excitatory amino acid transporters. ChemMedChem 4, 1925–9. [DOI] [PubMed] [Google Scholar]
- (40).Assaf Z, et al. (2013) Chemoenzymatic Synthesis of New 2,4-syn-Functionalized (S)-Glutamate Analogues and Structure–Activity Relationship Studies at Ionotropic Glutamate Receptors and Excitatory Amino Acid Transporters. J. Med. Chem 56, 1614–1628. [DOI] [PubMed] [Google Scholar]
- (41).Venskutonyte R, et al. (2014) Molecular Recognition of Two 2,4-syn-Functionalized (S)-Glutamate Analogues by the Kainate Receptor GluK3 Ligand Binding Domain. ChemMedChem 9, 2254–2259. [DOI] [PubMed] [Google Scholar]
- (42).Sagot E, et al. (2008) Chemo-enzymatic synthesis of a series of 2,4-syn-functionalized (S)-glutamate analogues: new insight into the structure-activity relation of ionotropic glutamate receptor subtypes 5, 6, and 7. J. Med. Chem 51, 4093–103. [DOI] [PubMed] [Google Scholar]
- (43).Escribano AM, et al. (1998) (2S,4S)-2-Amino-4-(2,2-diphenylethyl)pentanedioic acid selective group 2 metabotropic glutamate receptor antagonist. Bioorg. Med. Chem. Lett 8, 765–770. [DOI] [PubMed] [Google Scholar]
- (44).Erreger K, et al. (2007) Subunit-Specific Agonist Activity at NR2A-, NR2B-, NR2C-, and NR2D-Containing N-Methyl-D-aspartate Glutamate Receptors. Mol. Pharmacol 72, 907–920. [DOI] [PubMed] [Google Scholar]
- (45).Small B, et al. (1998) LY339434, a GluR5 kainate receptor agonist. Neuropharmacology 37, 1261–1267. [DOI] [PubMed] [Google Scholar]
- (46).Baker SRR, et al. (2000) 4-Alkylidenyl glutamic acids, potent and selective GluR5 agonists. Bioorg. Med. Chem. Lett 10, 1807–1810. [DOI] [PubMed] [Google Scholar]
- (47).Pedregal C, et al. (2000) 4-Alkyl- and 4-Cinnamylglutamic Acid Analogues Are Potent GluR5 Kainate Receptor Agonists. J. Med. Chem 43, 1958–1968. [DOI] [PubMed] [Google Scholar]
- (48).Larsen AM, and Bunch L (2011) Medicinal Chemistry of Competitive Kainate Receptor Antagonists. ACS Chem. Neurosci 2, 60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Byskov Vogensen S, Greenwood JR, Bunch L, and Clausen RP (2011) Glutamate receptor agonists: stereochemical aspects. Curr. Top. Med. Chem 11, 887–906. [DOI] [PubMed] [Google Scholar]
- (50).Todeschi N, et al. (1997) Conformational analysis of glutamic acid analogues as probes of glutamate receptors using molecular modelling and NMR methods. Comparison with specific agonists. Bioorg. Med. Chem 5, 335–352. [DOI] [PubMed] [Google Scholar]
- (51).Stawski P, Janovjak H, and Trauner D (2010) Pharmacology of ionotropic glutamate receptors: A structural perspective. Bioorg. Med. Chem 18, 7759–7772. [DOI] [PubMed] [Google Scholar]
- (52).Møllerud S, Frydenvang K, Pickering DS, and Kastrup JS (2017) Lessons from crystal structures of kainate receptors. Neuropharmacology 112, 16–28. [DOI] [PubMed] [Google Scholar]
- (53).Todeschi N, Gharbi-Benarous J, Acher F, Azerad R, and Girault J-P (1996) Conformational analysis by NMR spectroscopy and molecular simulation in water of methylated glutamic acids, agonists at glutamate receptors. J. Chem. Soc., Perkin Trans. 2 2 (7), 1337–1351. [Google Scholar]
- (54).Acevedo CM, Kogut EF, and Lipton MA (2001) Synthesis and analysis of the sterically constrained l-glutamine analogues (3S,4R)-3,4-dimethyl-l-glutamine and (3S,4R)-3,4-dimethyl-l-pyroglutamic acid. Tetrahedron 57, 6353–6359. [Google Scholar]
- (55).Coldham I, Fernández J-C, Price KN, and Snowden DJ (2000) Intramolecular Carbolithiation Reactions for the Preparation of Azabicyclo[2.2.1]heptanes. J. Org. Chem 65, 3788–3795. [DOI] [PubMed] [Google Scholar]
- (56).Tojo G, and Fernández M (2007) Ruthenium Tetroxide and Other Ruthenium Compounds. In Oxidation of Primary Alcohols to Carboxylic Acids, pp 61–78, Springer; 10.1007/0-387-35432-8_5. [DOI] [Google Scholar]
- (57).Yoo S-E, Lee S-H, and Kim N-J (1988) An efficient synthesis of the basic pyrrolidine ring for the kainoids. Tetrahedron Lett 29, 2195–2196. [Google Scholar]
- (58).Moss WO, Bradbury RH, Hales NJ, and Gallagher T (1990) Generation of α-amino acid homoenolate equivalents. Synthesis of 3-substituted prolines. Tetrahedron Lett 31, 5653–5656. [Google Scholar]
- (59).Lunglois N, and Andriamialisoa RZ (1991) Diastereoselective synthesis of (2S,3R) 2-carboxy-3-pyrrolidine acetic acid, a simple kainic acid analogue. Tetrahedron Lett 32, 3057–3058. [Google Scholar]
- (60).Carpes MJ, Miranda PCML, and Correia CRD (1997) Stereoselective synthesis of conformationally restricted analogues of aspartic and glutamic acids from endocyclic enecarbamates. Tetrahedron Lett 38, 1869–1872. [Google Scholar]
- (61).Karoyan P, and Chassaing G (2002) Short asymmetric synthesis of (2S,3S)- and (2S,3R)-3-prolinoglutamic acids: 2-carboxy-3-pyrrolidine-acetic acids (CPAA). Tetrahedron Lett 43, 253–255. [Google Scholar]
- (62).Oba M, Saegusa T, Nishiyama N, and Nishiyama K (2009) Synthesis of non-proteinogenic amino acids using Michael addition to unsaturated orthopyroglutamate derivative. Tetrahedron 65, 128–133. [Google Scholar]
- (63).Felluga F, et al. (2012) First chemoenzymatic synthesis of (+)-2-carboxypyrrolidine-3-acetic acid, the nucleus of kainoid amino acids. Chirality 24, 112–118. [DOI] [PubMed] [Google Scholar]
- (64).Huy P, Neudörfl J-M, and Schmalz H-G (2011) A Practical Synthesis of Trans –3-Substituted Proline Derivatives through 1,4-Addition. Org. Lett 13, 216–219. [DOI] [PubMed] [Google Scholar]
- (65).Tay NES, and Nicewicz DA (2017) Cation Radical Accelerated Nucleophilic Aromatic Substitution via Organic Photoredox Catalysis. J. Am. Chem. Soc 139, 16100–16104. [DOI] [PubMed] [Google Scholar]
- (66).Pallandre J-R, et al. (2015) Novel aminotetrazole derivatives as selective STAT3 non-peptide inhibitors. Eur. J. Med. Chem 103, 163–174. [DOI] [PubMed] [Google Scholar]
- (67).Sonnenberg JD, et al. (1996) The role of the C-4 side chain of kainate and dihydrokainate in EAA receptor and transporter selectivity. Bioorg. Med. Chem. Lett 6, 1607–1612. [Google Scholar]
- (68).Pedersen D, and Rosenbohm C (2001) Dry Column Vacuum Chromatography. Synthesis 2001, 2431–2434. [Google Scholar]
- (69).Alcaide A, et al. (2016) Synthesis and pharmacological characterization of the selective GluK1 radioligand (S)-2-amino-3-(6-[3H]-2,4-dioxo-3,4-dihydrothieno[3,2-d]pyrimidin-1(2H)-yl)-propanoic acid ([3H]-NF608). MedChemComm 7, 2136–2144. [Google Scholar]
- (70).Jensen AA, and Bräuner-Osborne H (2004) Pharmacological characterization of human excitatory amino acid transporters EAAT1, EAAT2 and EAAT3 in a fluorescence-based membrane potential assay. Biochem. Pharmacol 67, 2115–2127. [DOI] [PubMed] [Google Scholar]
- (71).Hansen KB, Ogden KK, Yuan H, and Traynelis SF (2014) Distinct Functional and Pharmacological Properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA Receptors. Neuron 81, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Yi F, et al. (2016) Structural Basis for Negative Allosteric Modulation of GluN2A-Containing NMDA Receptors. Neuron 91, 1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Williams K (1993) Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharmacol 44, 851–859. [PubMed] [Google Scholar]
- (74).Lind GEE, et al. (2017) Structural basis of subunit selectivity for competitive NMDA receptor antagonists with preference for GluN2A over GluN2B subunits. Proc. Natl. Acad. Sci. U. S. A 114, E6942–E6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.