

Abstract
Cold-inducible RNA-binding protein (CIRP) was discovered two decades ago while studying the mechanism of cold stress adaptation in mammals. Since then, the role of intracellular CIRP (iCIRP) as an stress-response protein has been extensively studied. Recently, extracellular CIRP (eCIRP) was discovered to also have an important role, acting as a damage-associated molecular pattern (DAMP), raising critical implications for pathobiology of inflammatory diseases. During hemorrhagic shock and sepsis, inflammation triggers the translocation of CIRP from the nucleus to the cytosol and its released to the extracellular space. eCIRP then induces inflammatory responses in macrophages, neutrophils, lymphocytes and dendritic cells. eCIRP also induces endoplasmic reticulum stress and pyroptosis in endothelial cells by activating the NF-κB and inflammasome pathways, and necroptosis in macrophages via mitochondrial DNA damage. eCIRP works through the TLR4-MD2 receptors. Studies with CIRP−/− mice reveal protection against inflammation, implicating eCIRP to be a novel drug target. Anti-CIRP Ab or CIRP-derived small peptide may have effective therapeutic potentials in sepsis, acute lung injury and organ ischemia/reperfusion injuries. Current review focuses on the pathobiology of eCIRP by emphasizing on signal transduction machineries, leading to discovering novel therapeutic interventions targeting eCIRP in various inflammatory diseases.
Keywords: CIRP, eCIRP, DAMP, neutrophils, macrophage, sepsis, hemorrhage, inflammation, ALI, ischemia/reperfusion
Graphical Abstract
Introduction
Cold-inducible RNA-binding protein (CIRP) was first described in late 90’s as an RNA chaperone controlling the expression of cell cycle proteins in hibernating animals [1]. Since then, intense research on intracellular CIRP (iCIRP) has revealed its role in the regulation of a variety of cellular stress responses, including mRNA stability [2], cell proliferation [3], cell survival [4, 5], circadian clock gene modulation [6], telomerase maintenance [7], stress adaptation [8], and tumor formation and progression [9, 10]. These iCIRP activities, which allow cells to cope with various cellular stress conditions, have been reviewed in detail [8]. In contrast to iCIRP, extracellular CIRP (eCIRP) was recently discovered to act as a damage-associated molecular pattern (DAMP) promoting inflammation and injury [11]. The identification of eCIRP as a new DAMP has originated a new era of investigation in inflammation pathobiology. Serum levels of eCIRP not only were found to be elevated in individuals admitted to the intensive care unit (ICU) with hemorrhagic shock (HS), but also correlated with organ injury [11]. Furthermore, it was demonstrated that eCIRP was able to promote inflammation independently [11]. In cells subjected to hypoxia, CIRP translocates from its usual location in the nucleus to the cytosol [1, 12], and is then released to the extracellular space [11]. eCIRP then acts as a DAMP increasing macrophage production of proinflammatory cytokines [11]. Since its original discovery as a DAMP, eCIRP’s emerging proinflammatory role has been progressively extended to include additional target cells, such as neutrophils, lymphocytes, epithelial cells, and endothelial cells [13–21]. eCIRP promotes inflammation by binding to the TLR4-MD2 receptor complex, leading to the activation and nuclear translocation of the transcription factor nuclear factor-κB (NF-κB) [11, 13]. Accordingly, CIRP−/− mice are protected and exhibit attenuated inflammation and organ injury in preclinical models of inflammatory diseases [11, 13]. These observations suggested that targeting eCIRP could be an effective therapeutic strategy for the treatment of inflammatory diseases. Indeed, we have now shown that treatment with anti-CIRP polyclonal antibodies or with the CIRP-derived small peptide C23 dramatically rescues animals from inflammation [11, 22–26]. In this article, we will review i) eCIRP’s role in pathobiology focusing on CIRP’s induction and release, ii) eCIRP’s mechanism of action, and iii) the development of eCIRP-targeting therapeutic interventions to ameliorate inflammatory diseases.
CIRP
CIRP is an evolutionarily conserved RNA chaperone [8]. It consists of an N-terminal RNA recognition motif (RRM) and a C-terminal arginine-rich (RGG) region structurally similar to stress-induced RNA-binding proteins in various species [1, 8]. CIRP has three protein-coding splice variants which are transcribed during normo- and hypothermic conditions [27]. The precise effects of these variants in the protein structure and function remain to be adequately studied. The shortest transcript is mostly expressed at 37°C, while two other longer transcripts harboring an internal ribosome entry site (IRES) are expressed at 32°C [28]. During hypothermia the core and alternate promoters and an alternative splicing tool become active for generating CIRP’s transcripts [29]. Non-hibernating animals such as hamsters express a long CIRP transcript in their hearts. This transcript contains an additional insert with an internal stop codon at the open reading frame (ORF), leading to a truncated protein and aberrant function. In contrast, hibernating animals predominantly express the short isoform with a complete ORF [9, 29, 30]. Overall, CIRP expression levels are altered in response to stress by versatile mechanisms, suggesting a wide range of adaptation to various external and internal challenges.
CIRP is predominantly found in the nucleus, where it regulates RNA transcription and processing, whereas in the cytoplasm iCIRP regulates mRNA translation and turnover [1, 8]. The cDNA sequence of CIRP has been cloned and characterized in numerous species, with >210 homologues currently identified in fish, amphibians, reptiles, birds and mammals [1, 31–33]. CIRP from human, mouse and rats, in particular, have elevated sequence homology. The human CIRP cDNA encodes an 18-kD protein whose amino acid sequence is 95.3% similar to that of mouse CIRP [31]. Mouse and rat CIRP have a sequence similarity of 100% [32]. In addition to homologues and orthologues, CIRP has at least two paralogues: RNA-binding motif protein 3 (RBM3) – an RNA-binding protein upregulated in response to low temperature [9], and aggrecan promoter-binding protein-1 (APBP-1) – a 19-kDa DNA/RNA binding protein [34]. CIRP is constitutively and ubiquitously expressed at low levels, with studies showing its presence in the brain, lungs, heart, liver, kidneys, and endometrium, as well as in macrophages and neutrophils [11, 13, 35]. Recently, both CIRP expression and release have been shown to be induced in cellular stress and inflammatory conditions [11]. Since CIRP exerts fundamentally different functions depending on its localization, it is critical to clearly establish the unique characteristics and activities of iCIRP and eCIRP.
Intracellular CIRP, an RNA chaperone
iCIRP is synthesized in the cytoplasm and gets back into the nucleus to modulate homeostatic RNA transcription and processing [1, 8]. In cardiomyocytes, for example, iCIRP regulates cardiac repolarization by binding to and inhibits the translation of mRNAs encoding Ito channel proteins [36]. However a fraction of iCIRP can also be found in the cytoplasm. iCIRP’s cytoplasmic fraction is particularly increased by cellular stressors such as hypothermia and ischemia [1, 8, 12]. iCIRP’s nucleocytoplasmic distribution is regulated by methylation of its C-terminal domain [12]. The shift in iCIRP’s subcellular localization, such as that elicited by ischemic and inflammatory states, results in a change in iCIRP’s role. Once in the cytoplasm, iCIRP coordinates translational reprogramming by promoting the synthesis of specific proteins. iCIRP selectively binds to the 3’-untranslated region (UTR) of ribosomal fraction RNA transcripts harboring CIRP’s RNA signature motif [8]. By binding to its target mRNAs, iCIRP inhibits their deadenylation, preserving their poly-A tail length and thus increasing their stability and preventing their degradation [2, 37, 38]. iCIRP also enhances the expression and activity of human antigen R (HuR) protein, which further positively regulates mRNA stability and translation by interacting with AU-rich element located in the 3’-UTR of many mRNAs [39]. Furthermore, iCIRP regulates the translation of its target mRNAs at the translation initiation phase by interacting with eIF4G [40]. All of these mechanisms lead to enhanced translational activity [2, 37, 38]. Indeed, iCIRP has been shown to promote the translation of genes involved in DNA repair [37, 41], cellular redox metabolism [40], cellular adhesion [42], circadian homeostasis [6], reproduction [2], telomerase maintenance [7], and genes associated with the translational machinery [8].
Physiological hypothermia within a scrotum is needed for normal spermatogenesis in mammals. Elevated levels of iCIRP are found in the nucleus of primary spermatocytes which play a pivotal role in spermatogenesis [43]. A recent protein-transcriptome screening study reveals that iCIRP binds to several transcripts encoding circadian oscillator proteins, like CLOCK, implicating iCIRP’s role in maintenance of circadian mRNAs [6]. iCIRP promotes keratinocyte cell survival and proliferation following ultraviolet (UV) exposure via phosphorylated signal transducer and activator of transcription 3 (STAT3)-dependent induction of cyclin D1 and Bag-1/S expression [44]. iCIRP has also been shown to accelerate the cell cycle progression of cultured mouse embryonic fibroblasts by binding to dual specificity tyrosine-phosphorylation regulated kinase 1B (Dyrk1b) [3].
In contrast with its role in keratinocyte proliferation during UV irradiation, iCIRP can inhibit cell proliferation by upregulating the expression of a cell cycle inhibitor protein p27Kip1 during mild hypothermia [4]. In addition to the positive post-transcriptional regulation, upon stress conditions nuclear iCIRP migrates to cytoplasmic stress granules (SGs) where it serves as a translational repressor, thus negatively controlling protein synthesis in general [12]. The genes induced by iCIRP is thought to be a function of the intensity (mild, moderate or severe) and timing (early or late exposure) of the stressors. In a recent study, for example, the neuronal expression of iCIRP was increased in rats subjected to chronic hypoxia, which in turn inhibited HIF-α and, thus, protected neuronal cells from apoptosis [45]. Besides, its role in regulation of mRNA stability and translation in the cytoplasm, nuclear iCIRP has also been shown to play a role in post-transcriptional regulation of its targets by controlling alternative polyadenylation that leads to generating alternative mRNA transcripts [46]. Thus, iCIRP coordinates translational reprogramming involving several post-transcriptional mechanisms to regulate physiological process such as spermatogenesis, circadian rhythm, cell proliferation, differentiation, and survival.
Extracellular CIRP: A novel mediator in the inflammation field
Under physiologic conditions, CIRP is found predominantly in the nucleus [1]. During hypoxia and inflammation, however, CIRP is translocated from the nucleus to the cytoplasm and gradually released to the extracellular space [11]. The recent discovery of eCIRP has led to the investigation of its role as a DAMP in inflammatory diseases.
Extracellular CIRP in ischemic and inflammatory diseases
Increased tissue and serum levels of CIRP have been reported in several ischemic and inflammatory diseases. While serum CIRP was undetectable in the healthy volunteers, its levels were elevated in the serum of the patients admitted to the ICU with hemorrhagic shock [11], which is characterized by a global deficiency in oxygen delivery and low grade inflammation [47]. In the rodent model of hemorrhage, serum CIRP was also elevated, and correlated with increased mRNA and protein expression levels in the heart and liver [11]. Similarly, serum and tissue levels of CIRP were also elevated in a number of organ-targeted ischemia and reperfusion models characterized by sterile inflammation, including rodent models of hepatic ischemia [26], mesenteric ischemia [24], ischemic acute kidney injury (AKI) [48] and stroke [21]. Furthermore, serum and tissue levels of CIRP are also elevated in sepsis [11, 13, 22, 49]. In an ICU-based study, circulating levels of CIRP in septic patients were significantly higher in the non-survivors than in the survivors [49], and correlated with sepsis severity as measured by APACHE II and SOFA scores, serum creatinine level, procalcitonin level, and mortality rate [49]. These observations were corroborated by increased serum and lung levels of CIRP in a murine model of polymicrobial sepsis [11, 13]. Furthermore, increased serum and tissue levels of CIRP have also been determined in chronic inflammatory diseases. Increased expression of CIRP has been shown in the airways and the alveolar epithelium of the lungs from chronic obstructive pulmonary disease (COPD) patients [50]. Similarly, the CIRP expression in alveolar epithelial cells was increased in mice treated with cold air [16]. Increased serum and synovial fluid levels of CIRP were also found in the patients with rheumatoid arthritis (RA) and osteoarthritis (OA), and the increased synovial levels of CIRP correlated with disease activity in two independent studies [51, 52]. In the colonic mucosa of patients with ulcerative colitis, expression of CIRP was correlated with the expression of proinflammatory cytokines, anti-apoptotic proteins and stem cell markers [53, 54]. Using an animal model of colitis, CIRP−/− mice had decreased susceptibility to colonic inflammation through decreased expression of proinflammatory cytokines in the colonic lamina propria cells compared with WT mice [53]. In the murine colitis associated cancer (CAC) model, CIRP deficiency led to decreased expression of proinflammatory, apoptotic and stem cells, leading to attenuated tumorigenic potential [53, 54]. Accordingly, the expression of CIRP was higher in tissue samples from patients with oral squamous cell carcinoma (OSCC) than in matched normal tissues [55]. CIRP’s role in cancer has been recently reviewed [8, 10]. CIRP has also been implicated in binge drinking. Binge drinking is associated with cerebral dysfunction [56]. Mice exposed to binge drinking levels of alcohol have increased brain CIRP mRNA and protein levels [19]. Furthermore, microglial cells exposed to alcohol in vitro increased their expression and release of CIRP [19], suggesting that microglial cells could be the source of the increased levels of CIRP in the brain during binge drinking. Taken collectively, these observations demonstrate that eCIRP levels are elevated and correlate with severity and injury in a wide range of ischemic and inflammatory states (Table 1).
Table 1:
The role of eCIRP in inflammatory diseases.
| Inflammatory diseases |
eCIRP’s source |
eCIRP’s effects on cells/tissues | Inflammatory/ injury markers assessed |
Therapeutic outcomes of targeting eCIRP | References |
|---|---|---|---|---|---|
| sepsis | blood, monocyte, macrophage, liver, heart, lungs | macrophage, lymphocyte, neutrophil, EC, hepatocyte, lung, spleen, kidney | ALT, LDH, TNFα, IL-6, HMGB1, Th1 cell, CD69+ and CD25+ T cell, ICAM-1+ neutrophil, NETs, iNOS, RM neutrophil, NE, JAM-C, histology, apoptosis, E-selectin, ICAM-1, creatinine, BUN, NGAL, KIM-1 | CIRP−/− mice, anti-CIRP Ab and C23 reduce inflammatory cytokines, chemokines, activated immune cells, NETs, and iNOS and improve survival. TLR4−/− mice attenuate eCIRP-induced lymphocyte activation | 11, 13, 14, 15, 22 |
| hemorrhage | blood, macrophage, liver, heart | blood, macrophage, EC, liver, lung | ALT, AST, LDH, TNFα, IL-6, IL-1β, HMGB1, MPO, histology, EC barrier, ICAM-1 | CIRP−/− mice, anti-CIRP Ab and C23 reduce inflammatory markers, MPO,
histology, ICAM-1, EC barrier and improve survival |
11, 23 |
| alcohol-induced brain inflammation | microglial cell, brain | blood, brain | ALT, AST, LDH, TNFα, IL-1β | CIRP−/− mice have reduced levels of ALT, AST, LDH, TNFα, and IL-1β | 19 |
| cerebral ischemia and reperfusion | microglial cell, brain | microglial cell, neuroblastoma cell, brain | TNFα, caspase, Bcl2, NF-κB | CIRP−/− mice have reduced infarct size and volume, less AIF1 expression in microglia | 21 |
| hepatic ischemia and reperfusion | blood | blood, liver | ALT, AST, LDH, histology, IL-6, MIP-2, neutrophils (Gr-1), apoptosis, iNOS, Cox-2 | anti-CIRP Ab reduces ALT, AST, LDH, IL-6, MIP-2, iNOS, Cox-2, neutrophils, apoptosis, and improves survival | 26 |
| ALI | recombinant CIRP injection | EC, lung | TNFα, IL-1β, ICAM-1, L-selectin, NAD(P)H oxidase, ROS, Nlrp3, ASC, casp-1, EC leakage, pyroptosis, BiP, pIRE1α, sXBP1 | CIRP−/− mice have reduced ER stress in lung | 17, 20 |
| sepsis-induced ALI | lung | blood, arterial EC, lung | AST, IL-6, IL-1β, MIP-2, KC, MPO, histology, apoptosis, BiP, pIRE1α, sXBP1, CHOP, c-Casp-12, CD31, NF-κB | CIRP−/− mice have reduced ER stress, NF-κB, IL-6, IL-1β, MIP-2, KC, MPO, histology, and apoptosis | 13, 14, 20 |
| renal I/R | blood, kidney | macrophage, blood, kidney | TNFα, IL-6, IL-1β, KC, NGAL, KIM-1, BUN, creatinine, histology, apoptosis, neutrophils, nitrotyrosine, Cox-2 | CIRP−/− mice, anti-CIRP Ab and C23 attenuate BUN, creatinine, inflammatory and injury markers and improve survival | 25, 48 |
| fracture trauma and hemorrhage (two hits) | blood | macrophage, lung | mtDNA fragmentation, apoptosis, ROS, autophagy, necroptosis | TLR4−/− and MyD88−/− mice protect against CIRP-induced mtDNA fragmentation | 18 |
| intestinal I/R | blood | blood, small intestine, lung | AST, LDH, TNFα, IL-6, histology, apoptosis, casp-3, MIP-2, MPO, Ly6G+ PMN | CIRP−/− mice, C23 attenuate IL-6, LDH, histology, apoptosis, casp-3, and MPO | 24, 65 |
| RA | blood, synovial fluid, monocyte | blood, synovial fluid | ESR, CRP, IL6, TNFα | not studied | 51, 52 |
| COPD | airway epithelial cell | epithelial cells | IL-1β, TNFα, MUC5AC, MAPK | CIRP siRNA attenuates cold-induced expression of IL-1β, TNFα, MUC5AC, and MAPK | 16 |
| UC | colon mucosa | colon | histology, F4/80 cell, apoptosis, TNF, IL-1, IL-17, IL-23, BCL2, BCLXL, Sox2, tumor size | CIRP−/− mice show less colonic inflammation, small tumor size and apoptosis | 53, 54 |
Abbreviations: EC, endothelial cells; ALT, alanine aminotransferase; LDH, lactate dehydrogenase; TNF, tumor necrosis factor; IL, interleukin; HMGB1, high mobility group box 1; ICAM-1, intercellular adhesion molecule-1; NETs, neutrophil extracellular traps; eCIRP, extracellular cold-inducible RNA-binding protein; iNOS, inducible nitric oxide synthase; TLR, Toll-like receptor; RM, reverse migrated; JAM-C, junctional adhesion molecule-C; NE, neutrophil elastase; BUN, blood urea nitrogen; NGAL, neutrophil gelatinase-associated lipocalin; KIM-1, kidney injury molecule-1; MPO, myeloperoxidase; MIP, macrophage inflammatory protein; NF-κB, nuclear factor-κB; Cox, cyclogenase; casp-3, caspase-3; PMN, polymorphonuclear neutrophils; mt, mitochondrial; NLRP3, NACHT, LRR and PYD domains-containing protein 3; ESR, erythrocyte sedimentation rate; CRP, c-reactive protein; MAPK, mitogen activated protein kinase; ALI, acute lung injury; I/R, ischemia/reperfusion; RA, rheumatoid arthritis; COPD, chronic obstructive pulmonary disease; UC, ulcerative colitis.
Mechanism: How does CIRP’s expression, translocation and secretion occur?
As indicated by its name, iCIRP’s transcription and translation are induced by mild hypothermia such as the testicular physiologic temperature and suppressed by elevated temperature [1, 31, 43, 57]. iCIRP is also upregulated by UV radiation [44] and acute hypoxia [11, 21]. In macrophages, hypoxia, endotoxin and alcohol can each upregulate the expression of iCIRP via pathways involving HIF-α, TLR4, NF-κB, Egr-1, AP-1, and STAT1, and cause eCIRP release [11, 19, 58]. The activation of glycogen synthase kinase 3β (GSK3β) and insulin-like growth factor 1 (IGF1) can also increase the expression of iCIRP [59, 60]. To dissect the mechanism of iCIRP expression during mild hypothermia, a recent study revealed the involvement of transient receptor potential vanilloid (TRPV) 3 and transient receptor potential cation channel subfamily M (TRPM) 8 ion channel proteins, but independently of their ion channel activities [61]. The transcription factor specificity protein 1 (sp1) is induced by hypoxia and has been found to bind to the CIRP’s cold responsive element, suggesting that it may mediate hypoxia-induced CIRP transcription [62, 63]. Recently, the overexpression of miRNA-23a has also been shown to induce the expression of CIRP during prolonged hypoxia condition, possibly through its binding to CIRP’s transcriptional enhancer [45]. A recent study also revealed that hypothermia increases intracellular calcium levels via TRPV4 which regulates CIRP’s expression in response to cold stress [64]. CIRP has various transcripts, which possibly differ between species and may have respond differently to inducers, enhancers and repressors [8]. Further studies evaluating each CIRP transcript in homeostasis inflammation will provide valuable insights into the mechanisms of expression, localization and secretion of CIRP with potentially significant implications to CIRP’s effective therapeutic targeting.
In addition to being regulated at the transcriptional and translational levels, iCIRP’s function is also regulated by its subcellular translocation. Specifically, stressors such as oxidative stress, endoplasmic reticulum (ER) stress, osmotic shock and heat shock may cause iCIRP methylation and subsequent migration from the nucleus to the SGs in the cytoplasm, where untranslated mRNA becomes sequestered [8, 12]. However, while hypothermia markedly induces CIRP synthesis in various cell types, it does not lead to the accumulation of CIRP in SGs [12]. Likewise, UV irradiation, which induces SGs, promotes CIRP’s nuclear export without further assembly into SGs [12]. These observations suggest that distinct stressors may affect iCIRP translational reprogramming differently via their unique modulation of iCIRP transcript expression and subcellular localization.
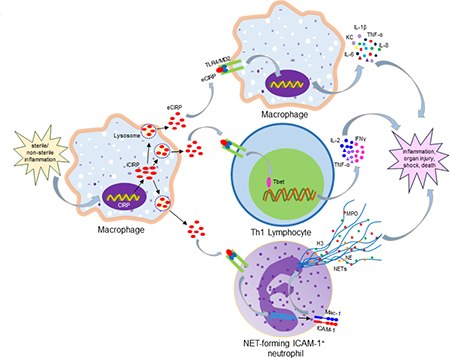
CIRP’s nuclear cytoplasmic translocation and release has been investigated using a green fluorescent protein (GFP)-tagged CIRP expression system in macrophages exposed to hypoxia [11]. Under physiologic normoxia, the GFP-tagged CIRP protein was concentrated in the nucleus, while hypoxia caused its enrichment in the cytoplasm, followed by its release to the extracellular space [11]. CIRP’s release was not attributable to necrosis, as other intracellular proteins were not detected in the culture supernatants after hypoxia [11]. Since CIRP does not contain a signal peptide, its secretion cannot be mediated through the conventional ER-Golgi dependent classical pathway. Biochemical fractionation revealed CIRP’s enrichment at the lysosomal compartment of macrophages undergoing hypoxia, suggesting that CIRP is released by lysosomal secretion [11] (Figure 1). However, it is still unclear exactly how CIRP gets into the lysosomal compartment and is released without disrupting the cell membrane. An in vitro study using macrophages showed that cellular necrosis does not cause CIRP release, thus suggesting that passive release is not an major source of eCIRP during hypoxia or endotoxemia [11]. However, under in vivo condition like other DAMPs, passive release due to cell necrosis might play a role for eCIRP in injury such as I/R and sepsis [13, 25, 65]. Future studies on the involvement of the inflammasome-induced pore forming protein gasdermin D, various carrier/transport proteins and the involvement of ER stress molecules for active release of CIRP during inflammation help understand how eCIRP gets secreted to the extracellular space.
Figure 1: Release of eCIRP.

Sepsis and sterile injury promote global or localized infectious and hypoxic environment which activate TLR4 and HIF-1α. Bacterial LPS binds to TLR4 and recruits MyD88 to transduce downstream signal for activating NF-κB. HIF-1α and NF-κB serve as potent transcription factors to increase the expression of CIRP at mRNA levels. During inflammation or hypoxia cytoplasmic CIRP protein is enriched in the lysosomes. The lysosomes containing CIRP are then excreted outside the cells through exocytosis. eCIRP, extracellular cold-inducible RNA-binding protein; I/R, ischemia/reperfusion; TLR4, Toll-like receptor 4; HIF-1α, hypoxia inducible factor-1α; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; MyD88, myeloid differentiation factor 88.
Post-translational modifications regulate CIRP’s subcellular localization. CIRP’s methylation, for example, is required for its cytoplasmic translocation [12, 66]. Similarly, CIRP’s phosphorylation by GSK3β and CK2 also affects its cellular localization, as demonstrated by the reduced CIRP cytoplasmic translocation in cells exposed to UV and treated with CK2 inhibitors or GSK3β inhibitors [37]. Further studies of CIRP’s post-translational phenotypes after hypoxia, endotoxemia or alcohol exposure are expected to reveal insights into the pathophysiology of inflammatory diseases and data potentially useful for the development of treatments targeting CIRP cytoplasmic translocation and secretion.
Facts and findings: Temperature-dependent and -independent expression of CIRP
In mammalian cells, relative to the body’s steady-state temperature (36.5–37.5°C) CIRP’s transcription levels reach its peak at mild to moderate hypothermia (28–34°C) and drop significantly at extreme hypothermia (15–25°C) [1, 67, 68]. In contrast, hyperthermia (39–42°C) causes substantial decreases in CIRP in cultured cells [43, 69]. These temperature-dependent CIRP expression studies have been conducted mostly in neurons, fibroblasts and male germ cells [43, 67, 68, 70]. CIRP can also be induced by a wide range of stimuli such as endotoxemia, hypoxia, radiation, toxins and drugs in a temperature-independent manner [9, 11, 71–73]. The temperature-independent CIRP’s expression studies such as in hypoxia and endotoxemia were performed mainly on macrophages [11]. During mild hypothermia, the transcription factor Sp1 is recruited to the mild-cold responsive element located in the close proximity of the CIRP gene, leading to increased CIRP expression [62]. Whereas under hypoxia or inflammatory conditions CIRP is upregulated through HIF1α- and NF-κB-dependent pathways [11, 74]. Therefore, temperature-dependent and -independent induction of CIRP’s transcription involve distinct intracellular signaling pathways, transcription factors, and possibly also cell-types.
CIRP’s expression in cold response changes over time. When cells are exposed to hypothermia, CIRP transcription raises within 3 h and reaches maximal expression at 12 h, but drops precipitously within 8 h of rewarming [67, 75]. In normothermia, time-dependent changes in the quantity of CIRP within the cells (iCIRP) and in the extracellular space (eCIRP) have been studied in macrophages following hypoxia and reoxygenation. iCIRP’s expression was dramatically increased at 7 h of reoxygenation, while decreased to its original levels at 24 h of reoxygenation [11]. On the other hand, the highest level of eCIRP was found at 24 h after reoxygenation [11]. In a murine model of hemorrhagic shock, eCIRP serum levels increased after hemorrhage in a time-dependent manner [11]. Although in terms of the kinetics of CIRP’s expression were done in in vitro and in vivo conditions, its stability and degradation statuses were not reported yet. Furthermore, eCIRP’s circulating half-life has not yet been determined, although a single exogenous administration of eCIRP is sufficient to cause persistent inflammation and organ injury [17].
Mechanism: How does extracellular CIRP exaggerate inflammation?
eCIRP’s role as a key inflammatory mediator has been scrutinized in various inflammatory disease conditions. CIRP−/− mice subjected to models of hemorrhagic shock, organ-targeted ischemic/reperfusion injury and sepsis exhibit protective effects, such as the attenuated organ dysfunction and improved survival, suggesting eCIRP plays a critical role as in the development of inflammation and injury in these disease models [11, 13, 24, 26, 65]. Indeed, administration of recombinant CIRP to healthy rats is sufficient to increase serum levels of tumor necrosis factor-α (TNF-α), interleukin (IL)-6 and high mobility group box-1 (HMGB1), and induce liver injury, as assessed by increased levels of the organ injury markers aspartate aminotransferase (AST) and alanine aminotransferase (ALT) [11]. Additionally, healthy mice injected with recombinant CIRP develop lung injury, as indicated by lung histology [17]. eCIRP-induced lung injury is characterized by increased infiltration of neutrophils into the lungs, increased vascular endothelial permeability, and activation of vascular endothelial cells [17, 20]. The effects of eCIRP and its mechanism of action have already been studied in a number of immune cells, including macrophages, lymphocytes, neutrophils and dendritic cells [11, 13–15, 76]. eCIRP’s effects have also been studied in endothelial and epithelial cells [17, 20, 50] (Figure 2).
Figure 2: Proinflammatory effects of eCIRP.

eCIRP binds to its receptor TLR4/MD2 complex expressed in various cell types. In macrophages and lymphocytes, eCIRP activates NF-κB through TLR4/MD2 complex and increases the expression of proinflammatory cytokines and chemokines, and T cell activation markers such as CD69 and CD25. eCIRP skews proinflammatory Th1 type T cells by activating the master transcription factor T-bet. In neutrophils, through the classical TLR4/MD2 and NF-κB pathways eCIRP upregulates the expression of ICAM-1 on their surface. ICAM-1 transduces downstream signaling to activate PAD4-dependent NET formation. Increased NETosis by ICAM-1+ neutrophils in turn causes acute lung injury. In the DCs, a CIRP fusion protein prepared by combining amino acids of CD8 T cell’s TCR specific epitope from OVA and three amino-acid flanking residues linked to the N-terminus of murine CIRP named SIIN-CIRP induces DC maturation, cytokine production and migration in a TLR4-dependent manner. SIIN-CIRP protein also enhances antigen presentation. In the ECs, eCIRP increases the expression of ICAM-1 through TLR4/MD2 and NF-κB pathways. In addition, eCIRP induces NLRP3 inflammasome through TLR4/MD2 and NF-κB-dependent pathways. Increased activation of inflammasome leads to caspase-1 activation and subsequently IL-1β and IL-18 production and pyroptosis. eCIRP, extracellular cold-inducible RNA-binding protein; TLR4, Toll-like receptor 4; NF-κB, nuclear factor-κB; MyD88, myeloid differentiation factor 88; ICAM-1, intercellular cell adhesion molecule-1; DCs, dendritic cells; ECs, endothelial cells; SMC, smooth muscle cells; PAD4, peptidyl arginase deaminase 4; NLRP3, NACHT, LRR and PYD domains-containing protein 3.
Macrophages
Macrophages treated with recombinant CIRP release TNF-α and HMGB-1 [11]. eCIRP-mediated expression of proinflammatory cytokines and chemokines has been determined in murine primary macrophages, RAW264.7 mouse macrophage M1-like cells, THP-1 human monocyte-like cells and BV-2 murine microglial cells [11, 21, 25]. Since the pattern recognition receptors (PRRs) TLR4, TLR2 and RAGE are known to regulate macrophage release of TNF-α, the response to recombinant CIRP was evaluated in macrophages deficient in each of these receptors [11]. While TLR4-deficient macrophages are unable to release TNF-α in response to recombinant CIRP, RAGE- and TLR2-deficient macrophages respond to eCIRP as well as WT macrophages, indicating that the TLR4 pathway is key for macrophage responsiveness to eCIRP [11]. eCIRP’s dependence on TLR4 was further confirmed in vivo [11]. While WT mice injected with recombinant CIRP exhibited a dose-dependent increase in the serum levels of the proinflammatory mediators TNF-α, IL-6 and HMGB1 and the organ injury markers AST and ALT, these effects were not recapitulated in TLR4−/− mice [11]. Surface plasmon resonance (SPR) analysis showed that CIRP binds to the TLR4-MD2 complex, as well as to TLR4 and MD2 individually, with very high affinity, strongly suggesting that eCIRP activates macrophages via its direct binding to the TLR4-MD2 complex [11]. eCIRP’s binding to TLR4 in macrophage causes NF-κB activation and nuclear translocation, ultimately leading to the induction and release of proinflammatory cytokines and chemokines [11] (Figure 2). Recombinant CIRP also showed high affinity binding to RAGE and TLR2, although the precise biological relevance of these two receptors for CIRP-mediated macrophage activation remains to be determined [11].
eCIRP has also been identified to augment trauma-induced sterile inflammation through macrophage necroptosis [18]. Following tissue damage in trauma, eCIRP activates macrophage production of reactive oxygen species (ROS) via the TLR4-myeloid differentiation factor 88 (MyD88) signaling pathway [18]. This ultimately activates endonuclease G, which serves as an executor for mtDNA fragmentation in macrophages, ultimately triggering necroptosis and thus furthering inflammation in trauma [18].
Neutrophils
Neutrophils are the most abundant circulating leukocytes and are critical effector cells of the innate immune system [77]. Neutrophils express a wide range of PRRs, including TLRs, Fc receptors, and complement components [77, 78]. Furthermore, neutrophils have the capacity to kill microorganisms through phagocytosis, degranulation of cytotoxic proteins and peptides, and release of neutrophil extracellular traps (NETs) [79, 80]. Following infection or trauma, neutrophils rapidly migrate to inflamed tissues where they release a large number of microbicidal components such as lactoferrin, lysozyme, proteases, myeloperoxidase (MPO) and ROS, including hydrogen peroxide and superoxide, for elimination of invaded pathogens [81, 82]. However, excessive accumulation of activated neutrophils can lead to deleterious effects by disrupting the endothelial cell barrier, damaging cells and the extracellular matrix and causing unrestrained inflammation [83, 84]. Excessive lung neutrophil accumulation in the interstitial and alveolar spaces, for example, causes lung injury in mouse models of hemorrhagic shock, mesenteric ischemia, and sepsis [20, 84]. CIRP−/− septic mice, however, only develop a mild form of lung injury with mild neutrophil infiltration of the lungs and mostly preserved lung architecture compared with septic WT mice [20], suggesting that eCIRP plays a key role in the development of acute lung injury (ALI) in septic mice. Indeed, injection of healthy mice with recombinant CIRP is sufficient to induce lung injury characterized by increased lung levels of TNF-α and IL-1β and accumulation of neutrophils in the lungs [17]. Similarly, hepatic ischemia results in overproduction of the chemokine macrophage inflammatory protein-2 (MIP-2) in the affected liver tissue, which promotes neutrophil infiltration of the liver ultimately contributing to liver injury [26]. Conversely, treatment of mice subjected to hepatic ischemia and reperfusion with eCIRP neutralizing antibodies significantly reduced levels of proinflammatory chemokines, and decreased associated neutrophil recruitment to the liver [26].
Although neutrophils have traditionally been considered to be a relatively homogeneous population, emerging evidence indicates the existence of phenotypic and functional heterogeneity among neutrophils [85, 86]. eCIRP induces deleterious subsets of neutrophils during sepsis [13, 14]. The effect of eCIRP on the phenotype and function of neutrophils is described below.
NET-forming ICAM-1+ neutrophils
ICAM-1, also known as CD54, is a member of the Ig-like gene family [87] that plays a pivotal role in cell adhesion, migration, and aggregation via the interactions with its ligands, the integrins such as leukocyte functional antigen-1 (LFA-1) and macrophage receptor-1 (Mac-1) [88]. ICAM-1 is predominantly expressed on the endothelial and epithelial cells and to a lesser extent on certain subsets of leukocytes [89, 90]. Its expression is up-regulated by stimulation with endotoxin and proinflammatory cytokines [89–91]. Neutrophil ICAM-1 expression increases after stimulation with LPS and TNF-α, and results in increased neutrophil-neutrophil adherence and aggregation [92]. The ICAM-1 expressing neutrophils have enhanced effector functions through the increase of phagocytosis and ROS generation [93]. A recent study demonstrated increased expression of CIRP and higher frequencies and numbers of ICAM-1+ neutrophils in the lungs of WT mice with sepsis but few ICAM-1+ neutrophils in the lungs of CIRP−/− mice with sepsis, suggesting that eCIRP participates in the induction of ICAM-1+ neutrophils [13]. Indeed, in vitro treatment of primary murine neutrophils with recombinant CIRP significantly induced their surface ICAM-1 expression [13] (Figure 2). The induction of ICAM-1+ neutrophils by eCIRP was significantly attenuated in neutrophils treated with TLR4 neutralizing antibodies or an NF-κB inhibitor, indicating that the induction of ICAM-1+ neutrophils by eCIRP is mediated through the TLR4 and NF-κB pathway [13]. Neutrophils exposed to certain stimulants extrude NETs, which consist of chromatin filaments coated with citrullinated histones and neutrophil enzymes such as MPO and elastase [94]. NETs contribute to pathogen clearance [95] but, in sepsis, excessive NET formation has a deleterious effect by promoting tissue damage and thereby perpetuating inflammation [96]. In the lungs of septic mice, ICAM-1+ neutrophils showed significantly higher levels of iNOS and NETs compared to ICAM-1- neutrophils [13]. Furthermore, eCIRP-induced ICAM-1+ neutrophils had significantly higher frequencies of iNOS and NETs than ICAM-1- neutrophils [13]. Taken together, these observations indicate that ICAM-1+ neutrophils not only are phenotypically distinct, but also functionally relevant. eCIRP upregulation of NET-producing neutrophils is mediated through the induction of peptidylarginine deiminase 4 (PAD4), a rate limiting step in activation of NETs [13, 97]. Thus, eCIRP induces NET-forming ICAM-1+ neutrophils, a novel neutrophil subset participating in the pathogenesis of sepsis-associated lung injury.
Reverse migrated neutrophils
The conventional paradigm consists of neutrophil irreversible and unidirectional migration from the vasculature into the tissue [98]. However, recent studies have reported the ability of neutrophils to return to the bloodstream after migrating to the extravascular space through a process known as reverse transendothelial migration (rTEM) [85, 99–101]. These reverse migrated (RM) neutrophils have prolonged lifespan and are associated with pulmonary inflammation following cremaster muscle ischemia and reperfusion injury in mice, suggesting that RM neutrophils may contribute to dissemination of inflammation [100, 101]. Furthermore, RM neutrophils exhibit a proinflammatory phenotype, with increased levels of superoxide and high surface ICAM-1 expression [101]. In fact, the surface expression of ICAM-1 and CXCR1 have been used to differentiate RM neutrophils (ICAM-1hi CXCR1lo) from circulating (ICAM-1loCXCR1hi) and tissue resident (ICAM-1loCXCR1lo) neutrophils [99]. As discussed in the previous section, eCIRP induces iNOS and NET producing ICAM-1+ neutrophils [13]. A recent study revealed that eCIRP also induces neutrophil rTEM. Septic WT mice had significantly more RM neutrophils in the circulation than septic CIRP−/− mice [14]. Additionally, circulating RM neutrophils increased after intratracheal administration of recombinant CIRP to healthy mice, demonstrating that eCIRP is sufficient to induce neutrophil rTEM [14].
Neutrophil rTEM requires disruption of junctional adhesion molecule (JAM)-C forming endothelial cell (EC) junctions [101]. In the murine cremaster muscle ischemia and reperfusion injury model, lipid chemoattractant leukotriene B4 (LTB4) upregulated in the inflamed tissues induced neutrophil production of the proteolytic enzyme neutrophil elastase (NE), which disrupted JAM-C at EC junction, thus facilitating neutrophil rTEM [100]. This observation is in line with a recent study that showed increased NE and decreased JAM-C in the lungs of mice with sepsis or injected with recombinant CIRP [14]. In vitro neutrophil stimulation with recombinant CIRP significantly induced NE in cell culture supernatants, and the supernatant of eCIRP-stimulated neutrophils dramatically decreased EC surface expression of JAM-C, but not when NE blocking antibodies were added to the conditioned media [14]. These results indicate that eCIRP causes neutrophils to produce NE, which decreases EC surface expression of JAM-C, and provide the mechanism by which eCIRP promotes neutrophil rTEM [14] (Figure 3).
Figure 3: Neutrophil reverse transendothelial migration: role of eCIRP.

During inflammation neutrophils migrate from the blood to the tissues across the EC barrier through transendothelial migration. During inflammation eCIRP is increased in the blood and tissues. The TM neutrophils in the tissues are activated by eCIRP-TLR4 axis to release NE. The EC barrier is maintained by the interaction between JAM-C with its counterpart JAM-B. NE cleaves the JAM-C expressed on the surface of EC and enables TM neutrophils to pass through the disrupted EC barrier towards luminal direction. This process of neutrophil migration from the tissues to the blood during inflammation is called rTEM. The neutrophils that undergo rTEM is called RM neutrophils which further fuel inflammation. eCIRP, extracellular cold-inducible RNA-binding protein; TLR4, Toll-like receptor 4; EC, endothelial cells; TEM, transendothelial migration; TM, transmigrated; NE, neutrophil elastase; JAM-B/C, junctional adhesion molecule-B/C; rTEM, reverse transendothelial migration; RM, reverse migrated.
Lymphocytes and dendritic cells
During sepsis, T cells are activated, as evidenced by increased expression of CD69 and CD25 in CD4+ splenic T cells and of CD69 in CD8+ splenic T cells [15]. Excessive T cell activation, however, leads to T cell exhaustion via activation-induced cell death (AICD) [102]. eCIRP has been found to promote T lymphocyte activation in sepsis (Figure 2). Mice injected recombinant CIRP showed significantly increased the expression of activation markers on CD4+ and CD8+ splenic T cells [15]. This effect, however, was not seen in TLR4-deficient mice [15]. Additionally, treatment with recombinant CIRP promoted CD4+ T cell polarization towards a Th1 hyperinflammatory profile [15]. Thus, eCIRP plays an important role in T-cell dysregulation during sepsis in a TLR4-dependent manner. eCIRP has also been shown to induce dendritic cell activation and migration, as well as to enhance presentation of eCIRP-bound antigens to T-cells, revealing a novel role for eCIRP in the activation of professional antigen presenting cells [76].
Lung endothelial and epithelial cells
eCIRP has been shown to bind with high affinity to TLR4 to increase the release of pro-inflammatory cytokines in vitro and in vivo [11]. It is not yet known whether eCIRP can be internalized by itself or after binding to TLR4. Another unexplored possibility is that eCIRP or iCIRP could interact with intracellular PRRs such as nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and the RIG-like helicases (RLHs), which would give a potential new direction on its effect on innate immune system.
The inflammasome is a multimolecular platform which assembles as a complex of NLR proteins, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC), and caspase-1 (or caspase-5) to release proinflammatory cytokines IL-1β and IL-18 [103]. NAD(P)H oxidase-derived ROS has been shown to contribute to the formation of the NLRP3 inflammasome in ECs [104]. A recent study focused on the role of eCIRP on endothelial cells [17] (Figure 2). Healthy mice injected with recombinant CIRP developed lung injury, as evidenced by vascular leakage, edema, leukocyte infiltration and cytokine production in the lungs [17]. The lung damage induced by eCIRP was accompanied by marked EC activation characterized by upregulation of the cell-surface adhesion molecules E-selectin and ICAM-1 [17]. Using in vitro primary mouse lung vascular ECs (MLVECs), recombinant CIRP was further shown to directly induce MLVEC ICAM-1 surface expression and NAD(P)H oxidase activation [17]. Increased levels of NAD(P)H oxidase in eCIRP-treated MLVECs caused increased ROS production to aggravate lung injury [17]. Importantly, eCIRP promoted the assembly and activation of the Nlrp3 inflammasome in MLVECs, leading to caspase-1 activation, IL-1β release and induction of EC pyroptosis, thus worsening the EC barrier dysfunction [17]. Thus, eCIRP can directly activate ECs, inducing inflammasome activation and EC pyroptosis, resulting in injury to the lung.
Another mechanism of eCIRP-induced lung injury during sepsis is the induction of ER stress [20]. In sepsis, the lung ER stress markers (BiP, pIRE1α, sXBP1, CHOP, cleaved caspase-12) were increased in septic WT mice, but not in septic CIRP−/− mice [20]. The effector pathways downstream from ER stress such as apoptosis, NF-κB, IL-6, IL-1β, neutrophil chemoattractants (MIP-2, KC), neutrophil infiltration, lipid peroxidation, and iNOS were also significantly increased in WT mice, but only mildly elevated in CIRP−/− mice [20]. These ER stress markers increased in the lungs of healthy WT mice injected with recombinant CIRP, but not in the lungs of TLR4−/− mice, suggesting that eCIRP induces lung ER stress and downstream responses to cause sepsis-associated ALI [20].
Mucus overproduction by bronchial epithelial cells is an important feature in patients with chronic obstructive pulmonary disease (COPD) and cold air stimulation has been shown to be associated with the severity of these diseases. A recent study revealed that cold air stimulation induced mucin expression in WT mice but not in CIRP−/− mice [16]. In vitro, cold air stress significantly elevated the transcriptional and protein expression levels of mucin in human bronchial epithelial cells [16]. CIRP, TLR4 and phosphorylated NF-κB p65 increased significantly in response to cold stress, while CIRP siRNA, TLR4 neutralizing antibodies and an NF-κB inhibitor attenuated cold stress induced mucin expression in epithelial cells [16]. As such, eCIRP contributes to the activation of bronchial epithelial cells.
Translational approaches targeting extracellular CIRP
eCIRP released in various inflammatory diseases fuels inflammation by binding to the TLR4-MD2 receptor complex to induce the expression of proinflammatory cytokines, activate leukocytes and ECs, and promote systemic inflammation and lung injury [11, 13, 17, 26]. In contrast, CIRP−/− mice are protected against inflammatory diseases [11, 20, 48], demonstrating that eCIRP plays a causal role in the infection and sterile inflammation. Therefore, several therapeutic approaches have been devised and employed to ameliorate the deleterious effect of eCIRP in inflammatory diseases.
Neutralizing antibody against eCIRP
The first study of therapeutic approach based on targeting eCIRP was performed using polyclonal CIRP neutralizing antibodies raised in rabbit [11]. The antibodies effectively inhibited recombinant CIRP-induced TNF-α production in macrophages [11]. Administration of eCIRP neutralizing antibodies during fluid resuscitation to rats with hemorrhagic shock significantly reduced inflammation, measured by serum and hepatic levels of TNF-α and IL-6, neutrophil infiltration of the liver, and organ injury measured by serum AST and ALT [11]. Additionally, the survival rate of hemorrhaged rats administered CIRP neutralizing antibodies was significantly higher than that of hemorrhaged rats administered control IgG [11]. eCIRP neutralizing antibodies were also administered to mice with polymicrobial sepsis. Similarly to the hemorrhaged rats, septic mice treated with CIRP neutralizing antibodies had attenuated inflammation, less organ injury, and improved survival [11].
The beneficial effects of treatment with CIRP neutralizing antibodies have also been documented in a model of hepatic ischemia. Mice with hepatic ischemia treated with CIRP neutralizing antibodies exhibited markedly reduced inflammation, neutrophil infiltration of the liver, hepatocellular damage, and liver histological damage [26]. CIRP neutralizing antibodies also dramatically decreased the amount of apoptosis and nitrosative stress [26]. Ultimately, treatment with CIRP neutralizing antibodies lead to significant improvement in the survival after hepatic ischemia [26].
CIRP-derived small peptide
Recombinant CIRP binds to the TLR4-MD2 complex as well as to TLR4 and MD2 independently [11]. In order to identify small molecular inhibitors for eCIRP, overlapping 15-mer peptides covering the entire length of the human CIRP protein sequence were screened for their binding affinity for MD2 using SPR technology. Compound #23 (C23) peptide, covering human CIRP amino acid residues 111–125 was identified to have the highest affinity for MD2 [11]. Computational modeling predicted that C23 fits into MD2’s pocket. Initial studies showed that C23 was able to reduce ICAM-1 expression and IL-1β release in ECs stimulated with recombinant CIRP [11]. Moreover, administration of C23 to rats with hemorrhagic shock significantly reduced EC activation in the lungs [11]. These observations revealed the potential for developing C23 as a novel adjunctive therapeutic for hemorrhage resuscitation.
Further studies showed that C23 dose-dependently inhibited the production of TNF-α in rat peritoneal macrophages and in THP-1 cells exposed to recombinant CIRP, by protecting the degradation of IκBα, and inhibiting NF-κB nuclear translocation [22, 25]. When administered to septic mice, C23 was protective and attenuated inflammation (IL-6, TNF-α, and IL-1β) and liver injury (LDH, ALT) and kidney injury (creatinine, BUN, NGAL, KIM-1) [22]. C23-treated mice also had improved lung histology, less TUNEL-positive cells, and decreased pulmonary EC activation (E-selectin, ICAM). Importantly, C23 improved the survival rate after sepsis [22]. Similar studies have shown that treatment with C23 also protects mice from hemorrhagic shock [11] and ischemic AKI [25] and mesenteric ischemia [24], underscoring the eCIRP’s critical role in inflammation and suggesting that C23 be developed as an effective treatment for sepsis and other inflammatory diseases.
CIRP-based vaccination strategy
Immunotherapy is an active area of cancer research [105], and many approaches are being developed to target tumor-induced immune suppression. A recent study suggests that eCIRP indirectly exerts anti-tumor effect by serving as an adjuvant enhancing T-cell reaction to tumor antigens [76]. To prove this concept, the investigators created a fusion protein combining amino acids of CD8 T cell’s TCR specific epitope from OVA and three amino-acid flanking residues linked to the N-terminus of murine CIRP and named it SIIN-CIRP [76]. SIIN-CIRP protein induces DC maturation, cytokine production and migration in a TLR4-dependent manner [76] (Figure 2). SIIN-CIRP protein also enhances antigen presentation. When DC were pre-incubated with SIIN-CIRP and co-cultured with OT-I CD8 T-cells, strong interactions were observed in SIIN-CIRP-treated DC with CD8 T cells, which further led to T cell proliferation and differentiation [76]. Thus, eCIRP improves tumor antigen presentation by DC and activate effector T cells to promote tumor rejection. Additionally, CIRP-enhanced tumor immunogenicity was further improved by combination with conventional adjuvants as well as with antibodies neutralizing PD-1/PD-L1, CTLA-4 and IL-10 [76]. Therefore, CIRP-based vaccination strategies are able to circumvent tumor-induced immune suppression and may further improve the anti-tumor effects of checkpoint inhibitors.
Future perspectives and conclusions
iCIRP and eCIRP are now known to have very different functions. iCIRP acts as an RNA chaperone to coordinate stressor-related translational reprogramming [1, 8], while eCIRP functions as a DAMP to promote and amplify the inflammatory response [11, 17]. The marked improvement of inflammation and tissue injury in animal models treated with eCIRP targeting strategies underscores eCIRP’s critical role in the pathogenesis of inflammation, and provides a rational framework for further developing these therapeutic agents as treatments for patients with inflammatory diseases [11, 22, 24]. Although studies have considerably expanded our understanding of eCIRP’s role in acute inflammatory diseases, its role in chronic inflammatory diseases, Alzheimer’s disease, fibrosis, asthma and autoimmunity remains to be explored. Likewise, studies have focused on eCIRP’s role in macrophage, neutrophil and T cell biology, but it remains to be investigated in other cell types such as B and NK cells. eCIRP induction of NETs [13, 97] suggests that it may also influence extracellular traps in other cells, e.g, macrophages (METosis) and B cells (BETosis). Other knowledge gaps in eCIRP pathobiology include the biological significance of its binding to RAGE and TLR2, as well as the possibility of activation of additional receptors, such as MARCO, FcRs and P2X7 [106–108]. eCIRP has been shown to activate NF-κB and the Nrlp3 inflammasome [17, 22], raising questions about eCIRP’s connection to autophagy, which is linked to innate immune responses [109]. In addition, since miRNAs play an important role in regulating gene expression and function [110], it is worthwhile to investigate iCIRP’s regulation by miRNA in both health and diseased states. Various miRNAs are transported by RNA-binding proteins towards multivesicular bodies (MVBs) for exosome loading and to the plasma membrane for secretion [111]. As such, iCIRP’s role in miRNA processing, exosomal loading or the secretion of miRNA by MVBs may also be worth investigating. A better understanding of eCIRP’s secretion through exosomes, microvesicles or exocytosis is likely to generate additional targets to treat inflammation. A better understanding of each post-translational form of iCIRP is also needed. Identification of various forms of extracellular CIRP and their putative receptors will help reveal the pathophysiology of diseases and provide the rationale for the design of novel therapeutics. The recent studies focused on therapeutic approaches by targeting eCIRP using its neutralizing Abs or by developing small peptide that can mask its receptor [11, 22, 23]. Since there is still a lack of adequate information about how this molecule is released during inflammation, therapeutic targeting by interfering with its release would be promising. However, since lysosomal mediated CIRP release was discovered [11], therefore the use of molecules/drugs that inhibit lysosome release and ER stress inhibitors could be of importance for better clinical use in various diseases.
In conclusion, the discovery of eCIRP’s role as a new DAMP has led to a new era in inflammation research, and numerous diseases and cells have been found to be profoundly affected by this molecule. eCIRP targeting strategies underline how critical eCIRP’s proinflammatory activities are. Many knowledge gaps still exist and will need to be addressed. Many more disease conditions are likely to benefit from research targeting eCIRP. Thus, further discovering eCIRP’s black box will help change CIRP-oriented research from bench to bedside.
Grants/Acknowledgments
This study was supported by the National Institutes of Health (NIH) grants R35GM118337 and R01HL076179 (to P.W.), R01GM129633 and a Shock Society Faculty Research Award grant (to M.A.).
List of abbreviations
- iCIRP
intracellular cold-inducible RNA-binding protein
- eCIRP
extracellular cold-inducible RNA-binding protein
- DAMP
damage-associated molecular pattern
- TLR
Toll-like receptor
- MD2
myeloid differentiation factor 2
- NF-κB
nuclear factor-κB
- C23
compound#23
- STAT3
signal transducer and activator of transcription 3
- COPD
chronic obstructive pulmonary disease
- EC
endothelial cells
- ALT
alanine aminotransferase
- LDH
lactate dehydrogenase
- TNF
tumor necrosis factor
- IL
interleukin
- HMGB1
high mobility group box 1
- TRPV
transient receptor potential vanilloid
- ICAM-1
intercellular adhesion molecule-1
- NETs
neutrophil extracellular traps
- iNOS
inducible nitric oxide synthase
- rTEM
reverse transendothelial migration
- RM
reverse migrated
- JAM-C
junctional adhesion molecule-C
- NE
neutrophil elastase
- BUN
blood urea nitrogen
- NGAL
neutrophil gelatinase-associated lipocalin
- KIM-1
kidney injury molecule-1
- MPO
myeloperoxidase
- MIP
macrophage inflammatory protein
- Cox
cyclogenase
- casp-3
caspase-3
- PMN
polymorphonuclear neutrophils
- mt
mitochondrial
- NLRP3
NACHT, LRR and PYD domains-containing protein 3
- ESR
erythrocyte sedimentation rate
- CRP
c-reactive protein
- MAPK
mitogen activated protein kinase
- ALI
acute lung injury
- I/R
ischemia/reperfusion
- RA
rheumatoid arthritis
- UC
ulcerative colitis
Footnotes
Disclosures
MA has no financial conflicts of interests. One of the authors (PW) is an inventor of patent applications covering the fundamental concept of targeting CIRP for the treatment of inflammatory diseases, licensed by TheraSource LLC. PW is a co-founder of and MB is a director at TheraSource LLC.
References
- 1.Nishiyama H, Itoh K, Kaneko Y, Kishishita M, Yoshida O, Fujita J (1997) A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J Cell Biol 137, 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xia Z, Zheng X, Zheng H, Liu X, Yang Z, Wang X (2012) Cold-inducible RNA-binding protein (CIRP) regulates target mRNA stabilization in the mouse testis. FEBS Lett 586, 3299–308. [DOI] [PubMed] [Google Scholar]
- 3.Masuda T, Itoh K, Higashitsuji H, Nakazawa N, Sakurai T, Liu Y, Tokuchi H, Fujita T, Zhao Y, Nishiyama H, Tanaka T, Fukumoto M, Ikawa M, Okabe M, Fujita J (2012) Cold-inducible RNA-binding protein (Cirp) interacts with Dyrk1b/Mirk and promotes proliferation of immature male germ cells in mice. Proc Natl Acad Sci U S A 109, 10885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roilo M, Kullmann MK, Hengst L (2018) Cold-inducible RNA-binding protein (CIRP) induces translation of the cell-cycle inhibitor p27Kip1. Nucleic Acids Res 46, 3198–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakurai T, Itoh K, Higashitsuji H, Nonoguchi K, Liu Y, Watanabe H, Nakano T, Fukumoto M, Chiba T, Fujita J (2006) Cirp protects against tumor necrosis factor-alpha-induced apoptosis via activation of extracellular signal-regulated kinase. Biochim Biophys Acta 1763, 290–5. [DOI] [PubMed] [Google Scholar]
- 6.Morf J, Rey G, Schneider K, Stratmann M, Fujita J, Naef F, Schibler U (2012) Cold-inducible RNA-binding protein modulates circadian gene expression posttranscriptionally. Science 338, 379–83. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Wu Y, Mao P, Li F, Han X, Jiang S, Chen Y, Huang J, Liu D, Zhao Y, Ma W, Songyang Z (2016) Cold-inducible RNA-binding protein CIRP/hnRNP A18 regulates telomerase activity in a temperature-dependent manner. Nucleic Acids Res 44, 761–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong P and Huang H (2017) Recent progress in the research of cold-inducible RNA-binding protein. Future Sci OA 3, FSO246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu X, Bührer C, Wellmann S (2016) Cold-inducible proteins CIRP and RBM3, a unique couple with activities far beyond the cold. Cell Mol Life Sci 73, 3839–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lujan DA, Ochoa JL, Hartley RS (2018) Cold-inducible RNA binding protein in cancer and inflammation. Wiley Interdiscip Rev RNA 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, Kuncewitch M, Ji Y, Yang H, Wang H, Fujita J, Nicastro J, Coppa GF, Tracey KJ, Wang P (2013) Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med 19, 1489–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Leeuw F, Zhang T, Wauquier C, Huez G, Kruys V, Gueydan C (2007) The cold-inducible RNA-binding protein migrates from the nucleus to cytoplasmic stress granules by a methylation-dependent mechanism and acts as a translational repressor. Exp Cell Res 313, 4130–44. [DOI] [PubMed] [Google Scholar]
- 13.Ode Y, Aziz M, Wang P (2018) CIRP increases ICAM-1+ phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J Leukoc Biol 103, 693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin H, Aziz M, Ode Y, Wang P (2018) Cirp induces neutrophil reverse transendothelial migration in sepsis. Shock. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolognese AC, Sharma A, Yang WL, Nicastro J, Coppa GF, Wang P (2016) Cold-inducible RNA-binding protein activates splenic T cells during sepsis in a TLR4-dependent manner. Cell Mol Immunol 15, 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen L, Ran D, Xie W, Xu Q, Zhou X (2016) Cold-inducible RNA-binding protein mediates cold air inducible airway mucin production through TLR4/NF-κB signaling pathway. Int Immunopharmacol 39, 48–56. [DOI] [PubMed] [Google Scholar]
- 17.Yang WL, Sharma A, Wang Z, Li Z, Fan J, Wang P (2016) Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci Rep 6, 26571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, Fan EK, Liu J, Scott MJ, Li Y, Li S, Xie W, Billiar TR, Wilson MA, Jiang Y, Wang P, Fan J (2017) Cold-inducible RNA-binding protein through TLR4 signaling induces mitochondrial DNA fragmentation and regulates macrophage cell death after trauma. Cell Death Dis 8, e2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajayer SR, Jacob A, Yang WL, Zhou M, Chaung W, Wang P (2013) Cold-inducible RNA-binding protein is an important mediator of alcohol-induced brain inflammation. PLoS One 8, e79430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan MM, Yang WL, Brenner M, Bolognese AC, Wang P (2017) Cold-inducible RNA-binding protein (CIRP) causes sepsis-associated acute lung injury via induction of endoplasmic reticulum stress. Sci Rep 7, 41363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou M, Yang WL, Ji Y, Qiang X, Wang P (2014) Cold-inducible RNA-binding protein mediates neuroinflammation in cerebral ischemia. Biochim Biophys Acta 1840, 2253–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang F, Brenner M, Yang WL, Wang P (2018) A cold-inducible RNA-binding protein (CIRP)-derived peptide attenuates inflammation and organ injury in septic mice. Sci Rep 8, 3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang F, Yang WL, Brenner M, Wang P (2017) Attenuation of hemorrhage-associated lung injury by adjuvant treatment with C23, an oligopeptide derived from cold-inducible RNA-binding protein (CIRP). J Trauma Acute Care Surg 83, 690–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGinn JT, Aziz M, Zhang F, Yang WL, Nicastro JM, Coppa GF, Wang P (2018) Cold-inducible RNA-binding protein-derived peptide C23 attenuates inflammation and tissue injury in a murine model of intestinal ischemia-reperfusion. Surgery 164, 1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McGinn J, Zhang F, Aziz M, Yang WL, Nicastro J, Coppa GF, Wang P (2017) The protective effect of a short peptide derived from cold-inducible RNA-binding protein in renal ischemia-reperfusion injury. Shock 49, 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godwin A, Yang WL, Sharma A, Khader A, Wang Z, Zhang F, Nicastro J, Coppa GF, Wang P (2015) Blocking cold-inducible RNA-binding protein protects liver from ischemia-reperfusion injury. Shock 43, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Fageeh MB and Smales CM (2009) Cold-inducible RNA binding protein (CIRP) expression is modulated by alternative mRNAs. RNA 15, 1164–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu L, Sun HL, Gao Y, Hui KL, Xu MM, Zhong H, Duan ML (2017) Therapeutic hypothermia enhances cold-inducible RNA-binding protein expression and inhibits mitochondrial apoptosis in a rat model of cardiac arrest. Mol Neurobiol 54, 2697–2705. [DOI] [PubMed] [Google Scholar]
- 29.Al-Fageeh MB and Smales CM (2013) Alternative promoters regulate cold inducible RNA-binding (CIRP) gene expression and enhance transgene expression in mammalian cells. Mol Biotechnol 54, 238–49. [DOI] [PubMed] [Google Scholar]
- 30.Sano Y, Shiina T, Naitou K, Nakamori H, Shimizu Y (2015) Hibernation-specific alternative splicing of the mRNA encoding cold-inducible RNA-binding protein in the hearts of hamsters. Biochem Biophys Res Commun 462, 322–5. [DOI] [PubMed] [Google Scholar]
- 31.Nishiyama H, Higashitsuji H, Yokoi H, Itoh K, Danno S, Matsuda T, Fujita J (1997) Cloning and characterization of human CIRP (cold-inducible RNA-binding protein) cDNA and chromosomal assignment of the gene. Gene 204, 115–20. [DOI] [PubMed] [Google Scholar]
- 32.Xue JH, Nonoguchi K, Fukumoto M, Sato T, Nishiyama H, Higashitsuji H, Itoh K, Fujita J (1999) Effects of ischemia and H2O2 on the cold stress protein CIRP expression in rat neuronal cells. Free Radic Biol Med 27, 1238–44. [DOI] [PubMed] [Google Scholar]
- 33.Saito T, Sugimoto K, Adachi Y, Wu Q, Mori KJ (2000) Cloning and characterization of amphibian cold inducible RNA-binding protein. Comp Biochem Physiol B Biochem Mol Biol 125, 237–45. [DOI] [PubMed] [Google Scholar]
- 34.Pirok EW, Domowicz MS, Henry J, Wang Y, Santore M, Mueller MM, Schwartz NB (2005) APBP-1, a DNA/RNA-binding protein, interacts with the chick aggrecan regulatory region. J Biol Chem 280, 35606–16. [DOI] [PubMed] [Google Scholar]
- 35.Larrayoz IM, Rey-Funes M, Contartese DS, Rolón F, Sarotto A, Dorfman VB, Loidl CF, Martínez A (2016) Cold shock proteins are expressed in the retina following exposure to low temperatures. PLoS One 11, e0161458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Xie D, Huang J, Lv F, Shi D, Liu Y, Lin L, Geng L, Wu Y, Liang D, Chen YH (2015) Cold-inducible RNA-binding protein regulates cardiac repolarization by targeting transient outward potassium channels. Circ Res 116, 1655–9. [DOI] [PubMed] [Google Scholar]
- 37.Yang R, Zhan M, Nalabothula NR, Yang Q, Indig FE, Carrier F (2010) Functional significance for a heterogenous ribonucleoprotein A18 signature RNA motif in the 3’-untranslated region of ataxia telangiectasia mutated and Rad3-related (ATR) transcript. J Biol Chem 285, 8887–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aoki K, Matsumoto K, Tsujimoto M (2003) Xenopus cold-inducible RNA-binding protein 2 interacts with ElrA, the Xenopus homolog of HuR, and inhibits deadenylation of specific mRNAs. J Biol Chem 278, 48491–7. [DOI] [PubMed] [Google Scholar]
- 39.Guo X, Wu Y, Hartley RS (2010) Cold-inducible RNA-binding protein contributes to human antigen R and cyclin E1 deregulation in breast cancer. Mol Carcinog 49, 130–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang R, Weber DJ, Carrier F (2006) Post-transcriptional regulation of thioredoxin by the stress inducible heterogenous ribonucleoprotein A18. Nucleic Acids Res 34, 1224–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haley B, Paunesku T, Protić M, Woloschak GE (2009) Response of heterogeneous ribonuclear proteins (hnRNP) to ionising radiation and their involvement in DNA damage repair. Int J Radiat Biol 85, 643–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng Y, Yang PH, Tanner JA, Huang JD, Li M, Lee HF, Xu RH, Kung HF, Lin MC (2006) Cold-inducible RNA binding protein is required for the expression of adhesion molecules and embryonic cell movement in Xenopus laevis. Biochem Biophys Res Commun 344, 416–24. [DOI] [PubMed] [Google Scholar]
- 43.Nishiyama H, Danno S, Kaneko Y, Itoh K, Yokoi H, Fukumoto M, Okuno H, Millán JL, Matsuda T, Yoshida O, Fujita J (1998) Decreased expression of cold-inducible RNA-binding protein (CIRP) in male germ cells at elevated temperature. Am J Pathol 152, 289–96. [PMC free article] [PubMed] [Google Scholar]
- 44.Liao Y, Feng J, Zhang Y, Tang L, Wu S (2017) The mechanism of CIRP in inhibition of keratinocytes growth arrest and apoptosis following low dose UVB radiation. Mol Carcinog 56, 1554–1569. [DOI] [PubMed] [Google Scholar]
- 45.Chen X, Liu X, Li B, Zhang Q, Wang J, Zhang W, Luo W, Chen J (2017) Cold inducible RNA binding protein is involved in chronic hypoxia induced neuron apoptosis by down-regulating HIF-1α expression and regulated by microRNA-23a. Int J Biol Sci 13, 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Hu W, Murakawa Y, Yin J, Wang G, Landthaler M, Yan J (2013) Cold-induced RNA-binding proteins regulate circadian gene expression by controlling alternative polyadenylation. Sci Rep 3, 2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cannon JW (2018) Hemorrhagic shock. N Engl J Med 378, 370–379. [DOI] [PubMed] [Google Scholar]
- 48.Cen C, Yang WL, Yen HT, Nicastro JM, Coppa GF, Wang P (2016) Deficiency of cold-inducible ribonucleic acid-binding protein reduces renal injury after ischemia-reperfusion. Surgery 160, 473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y, Dong H, Zhong Y, Huang J, Lv J, Li J (2015) The cold-inducible RNA-binding protein (CIRP) level in peripheral blood predicts sepsis outcome. PLoS One 10, e0137721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ran D, Chen L, Xie W, Xu Q, Han Z, Huang H, Zhou X (2016) Cold-inducible RNA binding protein regulates mucin expression induced by cold temperatures in human airway epithelial cells. Arch Biochem Biophys 603, 81–90. [DOI] [PubMed] [Google Scholar]
- 51.Yoo IS, Lee SY, Park CK, Lee JC, Kim Y, Yoo SJ, Shim SC, Choi YS, Lee Y, Kang SW (2018) Serum and synovial fluid concentrations of cold-inducible RNA-binding protein in patients with rheumatoid arthritis. Int J Rheum Dis 21, 148–154. [DOI] [PubMed] [Google Scholar]
- 52.Yu L, Li QH, Deng F, Yu ZW, Luo XZ, Sun JL (2017) Synovial fluid concentrations of cold-inducible RNA-binding protein are associated with severity in knee osteoarthritis. Clin Chim Acta 464, 44–49. [DOI] [PubMed] [Google Scholar]
- 53.Sakurai T, Kashida H, Watanabe T, Hagiwara S, Mizushima T, Iijima H, Nishida N, Higashitsuji H, Fujita J, Kudo M (2014) Stress response protein cirp links inflammation and tumorigenesis in colitis-associated cancer. Cancer Res 74, 6119–28. [DOI] [PubMed] [Google Scholar]
- 54.Sakurai T, Kashida H, Komeda Y, Nagai T, Hagiwara S, Watanabe T, Kitano M, Nishida N, Fujita J, Kudo M (2017) Stress response protein RBM3 promotes the development of colitis-associated cancer. Inflamm Bowel Dis 23, 57–65. [DOI] [PubMed] [Google Scholar]
- 55.Ren WH, Zhang LM, Liu HQ, Gao L, Chen C, Qiang C, Wang XL, Liu CY, Li SM, Huang C, Qi H, Zhi KQ (2014) Protein overexpression of CIRP and TLR4 in oral squamous cell carcinoma: an immunohistochemical and clinical correlation analysis. Med Oncol 31, 120. [DOI] [PubMed] [Google Scholar]
- 56.Petit G, Maurage P, Kornreich C, Verbanck P, Campanella S (2014) Binge drinking in adolescents: a review of neurophysiological and neuroimaging research. Alcohol Alcohol 49, 198–206. [DOI] [PubMed] [Google Scholar]
- 57.Wang X, Che H, Zhang W, Wang J, Ke T, Cao R, Meng S, Li D, Weiming O, Chen J, Luo W (2015) Effects of Mild Chronic Intermittent Cold Exposure on Rat Organs. Int J Biol Sci 11, 1171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mandrekar P and Szabo G (2009) Signalling pathways in alcohol-induced liver inflammation. J Hepatol 50, 1258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu GP, Zhang Y, Yao XQ, Zhang CE, Fang J, Wang Q, Wang JZ (2008) Activation of glycogen synthase kinase-3 inhibits protein phosphatase-2A and the underlying mechanisms. Neurobiol Aging 29, 1348–58. [DOI] [PubMed] [Google Scholar]
- 60.Pan Y, Cui Y, He H, Baloch AR, Fan J, Xu G, He J, Yang K, Li G, Yu S (2015) Developmental competence of mature yak vitrified-warmed oocytes is enhanced by IGF-I via modulation of CIRP during in vitro maturation. Cryobiology 71, 493–8. [DOI] [PubMed] [Google Scholar]
- 61.Fujita T, Liu Y, Higashitsuji H, Itoh K, Shibasaki K, Fujita J, Nishiyama H (2018) Involvement of TRPV3 and TRPM8 ion channel proteins in induction of mammalian cold-inducible proteins. Biochem Biophys Res Commun 495, 935–940. [DOI] [PubMed] [Google Scholar]
- 62.Sumitomo Y, Higashitsuji H, Liu Y, Fujita T, Sakurai T, Candeias MM, Itoh K, Chiba T, Fujita J (2012) Identification of a novel enhancer that binds Sp1 and contributes to induction of cold-inducible RNA-binding protein (cirp) expression in mammalian cells. BMC Biotechnol 12, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin HO, An S, Lee HC, Woo SH, Seo SK, Choe TB, Yoo DH, Lee SB, Um HD, Lee SJ, Park MJ, Kim JI, Hong SI, Rhee CH, Park IC (2007) Hypoxic condition- and high cell density-induced expression of Redd1 is regulated by activation of hypoxia-inducible factor-1alpha and Sp1 through the phosphatidylinositol 3-kinase/Akt signaling pathway. Cell Signal 19, 1393–403. [DOI] [PubMed] [Google Scholar]
- 64.Fujita T, Higashitsuji H, Liu Y, Itoh K, Sakurai T, Kojima T, Kandori S, Nishiyama H, Fukumoto M, Shibasaki K, Fujita J (2017) TRPV4-dependent induction of a novel mammalian cold-inducible protein SRSF5 as well as CIRP and RBM3. Sci Rep 7, 2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cen C, McGinn J, Aziz M, Yang WL, Cagliani J, Nicastro JM, Coppa GF, Wang P (2017) Deficiency in cold-inducible RNA-binding protein attenuates acute respiratory distress syndrome induced by intestinal ischemia-reperfusion. Surgery 162, 917–927. [DOI] [PubMed] [Google Scholar]
- 66.Aoki K, Ishii Y, Matsumoto K, Tsujimoto M (2002) Methylation of Xenopus CIRP2 regulates its arginine- and glycine-rich region-mediated nucleocytoplasmic distribution. Nucleic Acids Res 30, 5182–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tong G, Endersfelder S, Rosenthal LM, Wollersheim S, Sauer IM, Bührer C, Berger F, Schmitt KR (2013) Effects of moderate and deep hypothermia on RNA-binding proteins RBM3 and CIRP expressions in murine hippocampal brain slices. Brain Res 1504, 74–84. [DOI] [PubMed] [Google Scholar]
- 68.Rzechorzek NM, Connick P, Patani R, Selvaraj BT, Chandran S (2015) Hypothermic preconditioning of human cortical neurons requires proteostatic priming. EBioMedicine 2, 528–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Danno S, Nishiyama H, Higashitsuji H, Yokoi H, Xue JH, Itoh K, Matsuda T, Fujita J (1997) Increased transcript level of RBM3, a member of the glycine-rich RNA-binding protein family, in human cells in response to cold stress. Biochem Biophys Res Commun 236, 804–7. [DOI] [PubMed] [Google Scholar]
- 70.Jackson TC, Manole MD, Kotermanski SE, Jackson EK, Clark RS, Kochanek PM (2015) Cold stress protein RBM3 responds to temperature change in an ultra-sensitive manner in young neurons. Neuroscience 305, 268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheikh MS, Carrier F, Papathanasiou MA, Hollander MC, Zhan Q, Yu K, Fornace AJ (1997) Identification of several human homologs of hamster DNA damage-inducible transcripts. Cloning and characterization of a novel UV-inducible cDNA that codes for a putative RNA-binding protein. J Biol Chem 272, 26720–6. [DOI] [PubMed] [Google Scholar]
- 72.Ryan JC, Morey JS, Ramsdell JS, Van Dolah FM (2005) Acute phase gene expression in mice exposed to the marine neurotoxin domoic acid. Neuroscience 136, 1121–32. [DOI] [PubMed] [Google Scholar]
- 73.Prieto-Alamo MJ, Abril N, Osuna-Jiménez I, Pueyo C (2009) Solea senegalensis genes responding to lipopolysaccharide and copper sulphate challenges: large-scale identification by suppression subtractive hybridization and absolute quantification of transcriptional profiles by real-time RT-PCR. Aquat Toxicol 91, 312–9. [DOI] [PubMed] [Google Scholar]
- 74.Wellmann S, Bührer C, Moderegger E, Zelmer A, Kirschner R, Koehne P, Fujita J, Seeger K (2004) Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J Cell Sci 117, 1785–94. [DOI] [PubMed] [Google Scholar]
- 75.Neutelings T, Lambert CA, Nusgens BV, Colige AC (2013) Effects of mild cold shock (25°C) followed by warming up at 37°C on the cellular stress response. PLoS One 8, e69687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Villanueva L, Silva L, Llopiz D, Ruiz M, Iglesias T, Lozano T, Casares N, Hervas-Stubbs S, Rodríguez MJ, Carrascosa JL, Lasarte JJ, Sarobe P (2018) The Toll like receptor 4 ligand cold-inducible RNA-binding protein as vaccination platform against cancer. Oncoimmunology 7, e1409321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A (2012) Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30, 459–89. [DOI] [PubMed] [Google Scholar]
- 78.Thomas CJ and Schroder K (2013) Pattern recognition receptor function in neutrophils. Trends Immunol 34, 317–28. [DOI] [PubMed] [Google Scholar]
- 79.Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, Benarafa C, Roos D, Skokowa J, Hartl D (2015) Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog 11, e1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M (2017) Neutrophil extracellular traps and its implications in inflammation: An Overview. Front Immunol 8, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rosales C (2018) Neutrophil: A cell with many roles in inflammation or several cell types? Front Physiol 9, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB (2014) Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20, 1126–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mócsai A (2013) Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med 210, 1283–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kolaczkowska E and Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13, 159–75. [DOI] [PubMed] [Google Scholar]
- 85.de Oliveira S, Rosowski EE, Huttenlocher A (2016) Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol 16, 378–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, Burk RD, Kunisaki Y, Jang JE, Scheiermann C, Merad M, Frenette PS (2015) Neutrophil ageing is regulated by the microbiome. Nature 525, 528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Staunton DE, Marlin SD, Stratowa C, Dustin ML, Springer TA (1988) Primary structure of ICAM-1 demonstrates interaction between members of the immunoglobulin and integrin supergene families. Cell 52, 925–33. [DOI] [PubMed] [Google Scholar]
- 88.Springer TA (1990) Adhesion receptors of the immune system. Nature 346, 425–34. [DOI] [PubMed] [Google Scholar]
- 89.Hubbard AK and Rothlein R (2000) Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic Biol Med 28, 1379–86. [DOI] [PubMed] [Google Scholar]
- 90.Roy J, Audette M, Tremblay MJ (2001) Intercellular adhesion molecule-1 (ICAM-1) gene expression in human T cells is regulated by phosphotyrosyl phosphatase activity. Involvement of NF-kappaB, Ets, and palindromic interferon-gamma-responsive element-binding sites. J Biol Chem 276, 14553–61. [DOI] [PubMed] [Google Scholar]
- 91.Madjdpour C, Oertli B, Ziegler U, Bonvini JM, Pasch T, Beck-Schimmer B (2000) Lipopolysaccharide induces functional ICAM-1 expression in rat alveolar epithelial cells in vitro. Am J Physiol Lung Cell Mol Physiol 278, L572–9. [DOI] [PubMed] [Google Scholar]
- 92.Wang JH, Sexton DM, Redmond HP, Watson RW, Croke DT, Bouchier-Hayes D (1997) Intercellular adhesion molecule-1 (ICAM-1) is expressed on human neutrophils and is essential for neutrophil adherence and aggregation. Shock 8, 357–61. [DOI] [PubMed] [Google Scholar]
- 93.Woodfin A, Beyrau M, Voisin MB, Ma B, Whiteford JR, Hordijk PL, Hogg N, Nourshargh S (2016) ICAM-1-expressing neutrophils exhibit enhanced effector functions in murine models of endotoxemia. Blood 127, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zawrotniak M and Rapala-Kozik M (2013) Neutrophil extracellular traps (NETs) - formation and implications. Acta Biochim Pol 60, 277–84. [PubMed] [Google Scholar]
- 95.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–5. [DOI] [PubMed] [Google Scholar]
- 96.Camicia G, Pozner R, de Larrañaga G (2014) Neutrophil extracellular traps in sepsis. Shock 42, 286–94. [DOI] [PubMed] [Google Scholar]
- 97.Ode Y A. M. a. W. P. (2018) CIRP induces NETosis in lungs during sepsis., Volume 49 (6) 19–20. [Google Scholar]
- 98.Nourshargh S and Alon R (2014) Leukocyte migration into inflamed tissues. Immunity 41, 694–707. [DOI] [PubMed] [Google Scholar]
- 99.Buckley CD, Ross EA, McGettrick HM, Osborne CE, Haworth O, Schmutz C, Stone PC, Salmon M, Matharu NM, Vohra RK, Nash GB, Rainger GE (2006) Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J Leukoc Biol 79, 303–11. [DOI] [PubMed] [Google Scholar]
- 100.Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, Chavakis T, Brohi K, Imhof BA, Nourshargh S (2015) Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity 42, 1075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S (2011) The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol 12, 761–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Green DR, Droin N, Pinkoski M (2003) Activation-induced cell death in T cells. Immunol Rev 193, 70–81. [DOI] [PubMed] [Google Scholar]
- 103.Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, Scott MJ, Billiar TR, Wilson MA, Fan J (2011) Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol 187, 4809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chesney JA, Mitchell RA, Yaddanapudi K (2017) Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J Leukoc Biol 102, 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roh JS and Sohn DH (2018) Damage-associated molecular patterns in inflammatory diseases. Immune Netw 18, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lotze MT and Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5, 331–42. [DOI] [PubMed] [Google Scholar]
- 108.Bianchi ME (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81, 1–5. [DOI] [PubMed] [Google Scholar]
- 109.Deretic V (2016) Autophagy in leukocytes and other cells: mechanisms, subsystem organization, selectivity, and links to innate immunity. J Leukoc Biol 100, 969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Quinn EM, Wang J, Redmond HP (2012) The emerging role of microRNA in regulation of endotoxin tolerance. J Leukoc Biol 91, 721–7. [DOI] [PubMed] [Google Scholar]
- 111.Janas T, Janas MM, Sapoń K (2015) Mechanisms of RNA loading into exosomes. FEBS Lett 589, 1391–8. [DOI] [PubMed] [Google Scholar]