

Abstract
Angiogenesis is critical to cancer development and metastasis. However, anti-angiogenic agents have only had modest therapeutic success, partly due to an incomplete understanding of tumor endothelial cell (EC) biology. We previously reported that the microRNA (miR)-200 family inhibits metastasis through regulation of tumor angiogenesis, but the underlying molecular mechanisms are poorly characterized. Here, using integrated bioinformatics approaches, we identified the RNA-binding protein (RBP) quaking (QKI) as a leading miR-200b endothelial target with previously unappreciated roles in the tumor microenvironment in lung cancer. In lung cancer samples, both miR-200b suppression and QKI overexpression corresponded with tumor ECs relative to normal ECs, and QKI silencing phenocopied miR-200b-mediated inhibition of sprouting. Additionally, both cancer cell and endothelial QKI expression in patient samples significantly corresponded with poor survival and correlated with angiogenic indices. QKI supported EC function by stabilizing cyclin D1 (CCND1) mRNA to promote EC G1/S cell cycle transition and proliferation. Both nanoparticle-mediated RNA interference of endothelial QKI expression and palbociclib blockade of CCND1 function potently inhibited metastasis in concert with significant effects on tumor vasculature. Altogether, this work demonstrates the clinical relevance and therapeutic potential of a novel, actionable miR/RBP axis in tumor angiogenesis and metastasis.
Keywords: Quaking, microRNA-200b, Cyclin D1, Angiogenesis, Metastasis
INTRODUCTION
Lung cancer is the leading cause of cancer-related deaths in the U.S. and worldwide. Because approximately 90% of cancer-related deaths are due to metastasis (1), developing novel approaches for therapeutic targeting of metastasis is critical for improving patient outcomes. Tumor angiogenesis, which refers to the sprouting of new blood vessels from preexisting host vasculature into the tumor microenvironment (TME), is a critical cancer hallmark for the development of metastasis. It has been demonstrated that tumors cannot grow past 2–3 mm in diameter without promoting angiogenesis to support their metabolic demands and sustain continued growth (2). The newly established vasculature also serves as a conduit for cancer cells to shed from the primary tumor, intravasate into leaky tumor vasculature, and travel through the circulation to distant sites. Once circulating tumor cells extravasate into distant sites, they re-activate pro-angiogenic and proliferation pathways to support continuous growth. This metastatic cascade is an inefficient process largely due to the failure of micro-metastases to continually support their growth in distant sites (3). Thus, angiogenesis has an integral role in regulating both tumorigenesis and cancer spread. Currently, most anti-angiogenic therapeutic strategies focus on inhibition of the vascular endothelial growth factor (VEGF) pathway, which has led to FDA-approval of drugs for several cancer types. However, the therapeutic effects of these agents are often temporary, modest, and susceptible to numerous resistance mechanisms that promote endothelial cell (EC) function (4, 5). Thus, although targeting tumor angiogenesis holds promise as a therapeutic approach, there is a tremendous need to better understand how tumors co-opt pre-programmed EC signals and ultimately to develop more effective therapeutic strategies that target these pathways.
Using nanoparticle-mediated delivery of miR-200 family members to cancer cells or tumor vasculature, we previously discovered that the miR-200 family suppresses cancer cell secretion of interleukin 8 (IL-8) and C-X-C motif chemokine ligand 1 (CXCL1), which significantly alters tumor vasculature and inhibits metastasis development in several cancer types (6). The miR-200 family has also been demonstrated to regulate general EC function, predominately in the context of wound healing and hypoxia, through targeting E26 oncogene homolog 1 (ETS-1), globin transcription factor binding protein 2 (GATA), VEGF receptor 2 (VEGFR2), vasohibin-2 (VASH2), and zinc finger E-box binding homeobox 1 (ZEB1), among others (7–11). However, direct targets regulated by the miR-200 family in ECs specifically in the context of the TME remain largely unknown. To investigate putative miR-200 family targets relevant to tumor EC (TEC) function, we performed integrated bioinformatic analyses using both The Cancer Genome Atlas (TCGA) datasets of several cancer types and ECs transfected with miR-200b. We identified quaking (QKI) as a miR-200 family target gene with previously unappreciated roles in TEC biology.
QKI is a highly conserved member of the signal transduction and activation of RNA (STAR) family of RNA-binding proteins (RBPs) and is expressed as 3 major alternatively spliced isoforms that are 5 (QKI-5), 6 (QKI-6), and 7 (QKI-7) kb in length. The QKI-5 3’UTR is distinct from the QKI-6 and QKI-7 3’UTRs, and the latter two share more homology with each other. All 3 isoforms share similar coding regions, all of which contain 3 conserved functional domains: K homology (KH) RNA-binding domain and QUA1 and QUA2 domains (12). The QUA1 domain is necessary for QKI dimerization, while the KH and QUA2 domains direct specific binding to bipartite sequence RNA motifs (13). QKI-5 contains a nuclear localization signal and remains solely in the nucleus, QKI-6 is cytoplasmic and nuclear, and QKI-7 is primarily cytoplasmic (14, 15). The QKI isoforms have roles in regulating mRNA splicing (16, 17), stability (18–20), translation (21), and nuclear export (22). Intriguingly, mouse embryos homozygous null or mutant for qkI die in utero due to severe vascular defects, including disrupted capillary plexus remodeling (i.e., angiogenesis) as a result of deficient differentiation and recruitment of mural (i.e., pericyte-like) smooth muscle cells (23, 24). qkI loss has also been demonstrated to disrupt the signaling from the visceral endoderm to the mesoderm that is involved in regulating EC maturation and proliferation (25). However, the mechanisms by which QKI exerts its pro-angiogenic effects remain poorly characterized, and whether QKI has a role in tumor angiogenesis is unknown.
Here we identify QKI as an important target of the miR-200 family with a previously unappreciated role in promoting tumor angiogenesis, metastasis, and poor overall survival. We found that QKI stabilizes cyclin D1 (CCND1) mRNA during the G1 to S transition, which promotes EC proliferation and sprouting angiogenesis. Using nanoparticle-mediated delivery of QKI small interfering RNAs (siRs) to target the tumor endothelium, we observed potent effects on tumor vasculature and inhibition of lung cancer metastasis. Intriguingly, repurposing palbociclib to target the CCND1-CDK4/6 axis in TECs recapitulated these therapeutic effects. Our findings highlight a miR-200b/QKI/CCND1 axis of TEC function, which shows promise as a novel target for therapeutic inhibition of metastasis and potential for broad applicability to numerous cancer types.
RESULTS
miR-200b is downregulated in tumor endothelium during lung cancer progression
The miR-200 family consists of 5 members and is split into 2 groups: Group A includes miR-200a and miR-141; Group B includes miR-200b, miR-200c, and miR-429. Group A and B members differ by 1 nucleotide in their seed sequences and have both overlapping and non-overlapping targets (26). We chose to focus on miR-200b, as both our group and others have observed it has the most potent effects on EC function of all miR-200 family members (6–8). To investigate the importance of endothelial miR-200b expression during tumor development, we intranasally delivered adenoviral Cre recombinase to Lox-Stop-Lox KrasG12D; liver kinase B1 (Lkb1)L/L; p53L/L mice to generate an autochthonous model of lung adenocarcinoma (LUAD) (27). We collected tumor-bearing lungs at early and late stages of disease progression (4 and 8 weeks, respectively, post adenoviral Cre delivery), as well as age-matched healthy lungs from non-induced control littermates (Supplementary Fig. 1a). We verified disease progression over time by micro computed tomography (μCT) imaging of the outgrowth of primary tumors in the lung (Supplementary Fig. 1b). Employing Fluorescent Activated Cell Sorting (FACS), we isolated healthy lung normal ECs (LNECs) as well as TECs derived from early and late-stage tumor-bearing mice (Supplementary Fig. 1c). qPCR analysis of the collected ECs revealed that while miR-200b was largely unchanged in early-stage TECs relative to age-matched LNECs, miR-200b expression was significantly down-regulated by more than 50% in TECs isolated from late-stage LUAD tumors compared to age-matched LNECs (Fig. 1a). To determine whether this finding was clinically relevant, we assessed miR-200b expression in surgically resected lung cancer samples from 3 patients and found that miR-200b expression was significantly downregulated in FACS-sorted TECs relative to patient-matched LNECs (Fig. 1b). These data demonstrate that miR-200b is downregulated in TECs during tumor progression and further implicate miR-200b as a modulator of pro-angiogenic pathways.
Figure 1. QKI is a miR-200b target in tumor endothelium.
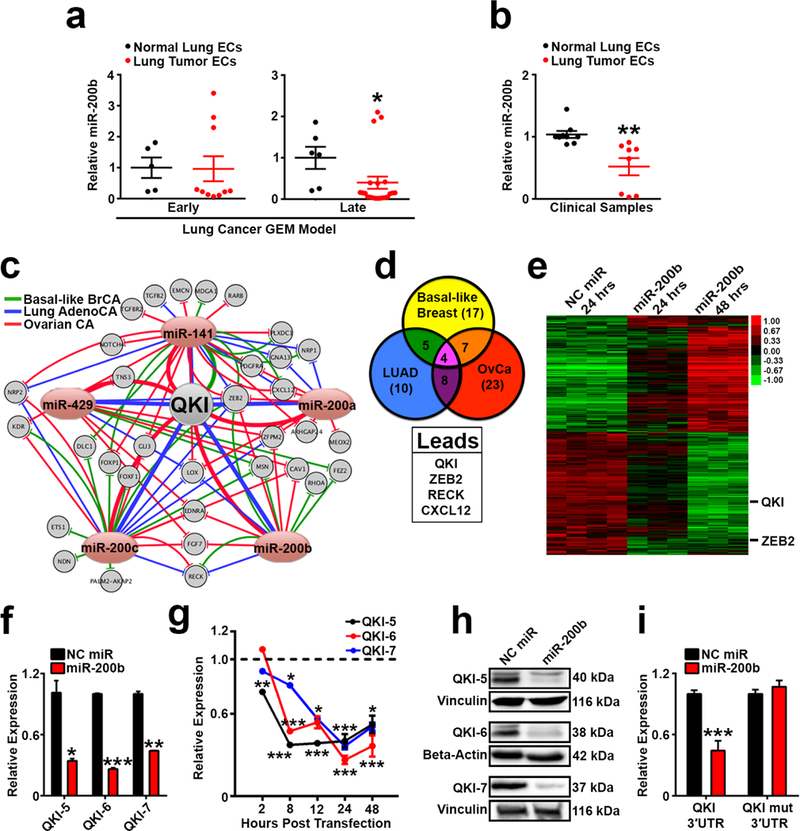
a qPCR analysis for miR-200b in ECs isolated from early and late stage lung tumors from a genetically engineered mouse (GEM) LUAD model and age-matched littermate healthy lungs (early, n=2 control mice, n=4 disease mice; late, n=4 control mice, n=7 disease mice; data points represent qPCR technical duplicates or triplicates per mouse).
b qPCR analysis for miR-200b in tumor and matched healthy lung ECs from lung cancer patients (n=3 patients, data points represent qPCR technical duplicates or triplicates per patient).
c Putative miR-200 family targets identified in TCGA datasets of breast, lung, and ovarian cancer. Connecting lines represent relationships of inverse correlation of expression, and line thickness represents strength of correlation. Pink ovals represent members of the miR-200 family. Gray circles represent putative miR-200 targets that 1) have a strong inverse correlation of expression with at least one miR-200 family member, 2) are predicted targets by TargetScan, and 3) have gene ontology terms associated with angiogenesis or vascular function.
d List of overlapping lead miR-200 candidate targets identified in c in all 3 cancer types.
e Heatmap of the microarray analysis performed on Human Umbilical Vein Endothelial Cells (HUVEC) transfected with Negative Control (NC) miR or miR-200b. Lead candidates that overlap with d are demarcated with black lines. Samples were run in triplicate or quadruplicate.
f – g qPCR analysis of QKI mRNA isoform expression in HUVEC transfected with NC miR or miR-200b at 24 hours or between 2–48 hours.
h Western blot of QKI protein isoform expression in HUVEC following transfection with NC miR or miR-200b.
i QKI 3’UTR or QKI 3’UTR mutated luciferase reporter activity in HEK293 cells transfected with NC miR or miR-200b. Luciferase signal was measured 48 hours post transfection of luciferase-expressing plasmids and miRs.
Statistical significance was measured by unpaired t-tests or Mann-Whitney test (specifically for panel a); p-values are indicated as *p<0.05, **p<0.01, ***p<0.001; f and i show representative results from two independent experiments; all qPCR and luciferase assays were run in duplicate or triplicate; error bars represent standard error of the mean.
Quaking is a clinically relevant miR-200b target in tumor endothelium
To investigate clinically relevant targets of miR-200b with roles in tumor angiogenesis, we performed an integrated analysis of TCGA datasets using 3 highly angiogenic and metastatic cancer types: basal-like breast, lung, and high-grade serous ovarian carcinomas. Previously, we identified protein-coding genes that had a strong inverse correlation of expression with any of the 5 members of the miR-200 family (false discovery rate [FDR] <0.01, Benjamini-Hochberg multiple testing correction) and performed in silico enrichment for genes that were predicted to be putative miR-200 target genes as determined by TargetScan (6). Using Ingenuity Pathway Analysis (IPA) of these prior identified genes, we generated a network visualization of all enriched genes associated with angiogenesis based on their Gene Ontology terms (Fig. 1c). We then filtered this list for genes that were predicted to be regulated by miR-200 family members in all 3 cancer types, producing a list of 4 leading candidates (Fig. 1d and Supplementary Table 1). Of these 4 genes, 2 genes were predicted to be regulated by a much larger number of miR-200 family members: QKI and ZEB2 (Supplementary Table 1). Interestingly, ZEB2 is a well-characterized target of the miR-200 family that has been extensively studied for its role in epithelial to mesenchymal transition (EMT) (26, 28). Because our TCGA analyses reflected miR:mRNA interactions across all cellular compartments of the TME, we also transfected ECs with negative control (NC) or miR-200b and performed microarray analyses (Fig. 1e) to identify putative miR-200 targets with direct relevance in endothelium. Both QKI and ZEB2 were potently downregulated upon miR-200b transfection into ECs (Fig. 1e), thus implicating their importance as clinically relevant targets in the context of the endothelial compartment of the TME.
QKI was the top ranked target gene identified in our IPA assessment of the TCGA datasets based on it being a predicted target amongst the majority of miR-200 family members across the 3 cancer types (Supplementary Table 1; predicted miR-200b binding sites in QKI 3’UTR are presented in Supplementary Table 2). Although QKI has been demonstrated to play a vital role in embryologic angiogenesis (23–25) and to regulate EC biology (29–31), the mechanisms by which QKI regulates normal vascular function are poorly understood and remain entirely unknown in the context of tumor angiogenesis. We therefore investigated the role of QKI as a miR-200b target in the context of the TME. We found that transient transfection of miR-200b into ECs dramatically downregulated expression of all QKI mRNA isoforms in a time-dependent manner (Figs. 1f and g) as well as protein expression of all QKI isoforms (Fig. 1h). miR-200b overexpression also inhibited a luciferase reporter driven by expression of a conserved region across the 3’UTR of QKI mRNA, and this suppression was abrogated upon deletion of predicted miR-200b binding sites in the QKI 3’UTR (Fig. 1i). We next performed an Argonaute-2 (AGO2) immunoprecipitation to verify binding of miR-200b to each QKI mRNA isoform within the RNA-Induced Silencing Complex (RISC). We observed an enrichment of all QKI isoforms upon AGO2 pulldown in miR-200b-transfected ECs compared to NC miR-transfected ECs (Supplementary Fig. 2a). In addition, treatment of ECs with an anti-miR inhibiting expression of miR-200b significantly increased expression of all QKI mRNA isoforms relative to treatment with a scrambled anti-miR (Supplementary Fig. 2b). We also observed that transient transfection of ECs with miR mimics of the other group B members of the miR-200 family (miR-200c and miR-429) also significantly downregulated expression of all QKI mRNA isoforms (Supplementary Fig. 2c), further validating QKI is potently regulated by the miR-200 family in endothelium. These results also align with previous findings suggesting QKI-5 is directly regulated by miR-200c during EMT in cancer cells (32). Altogether, these data validate QKI as a direct miR-200b target in ECs with potential clinical relevance in tumor angiogenesis. Intriguingly, we also found that direct QKI suppression using two different pan-QKI siRs resulted in an inhibition of miR-200b expression in ECs (Supplementary Fig. 2d). These results raise the possibility that there may in fact be a regulatory feedback loop between QKI and miR-200b, which needs to be further characterized in future studies.
QKI silencing recapitulates the effects of miR-200b on EC sprouting
We next determined whether QKI knockdown mimics the effects of miR-200b on EC function in vitro. We found that pan-QKI isoform knockdown phenocopies miR-200b activation by reducing the number of elongated sprouting vessels per bead in a 3-dimensional bead assay of sprouting angiogenesis (33) (Fig. 2a). We found that dual treatment with both miR-200b and QKI siR did not further abrogate EC sprouting compared to miR-200b treatment alone (Supplementary Fig. 3). Recently, we optimized a new version of this sprouting assay to also incorporate pericytes, which more accurately models the complex heterotypic cellular interactions involved in angiogenesis in vivo (Supplementary Fig. 4) (34). Both QKI silencing and miR-200b overexpression in the ECs in pericyte-associated sprouts dramatically altered endothelial sprouting (Fig. 2b). Intriguingly, QKI silencing in pericytes alone also resulted in reduction of vessels per bead (Fig. 2c), suggesting QKI may also play a role in modulating the function of mural cells associated with blood vessels. These results demonstrate that QKI expression is important for promoting angiogenesis and that QKI inhibition plays a significant role in the effects of miR-200b-mediated inhibition of EC (and pericyte) function and angiogenesis.
Figure 2. QKI and miR-200b regulate sprouting angiogenesis.
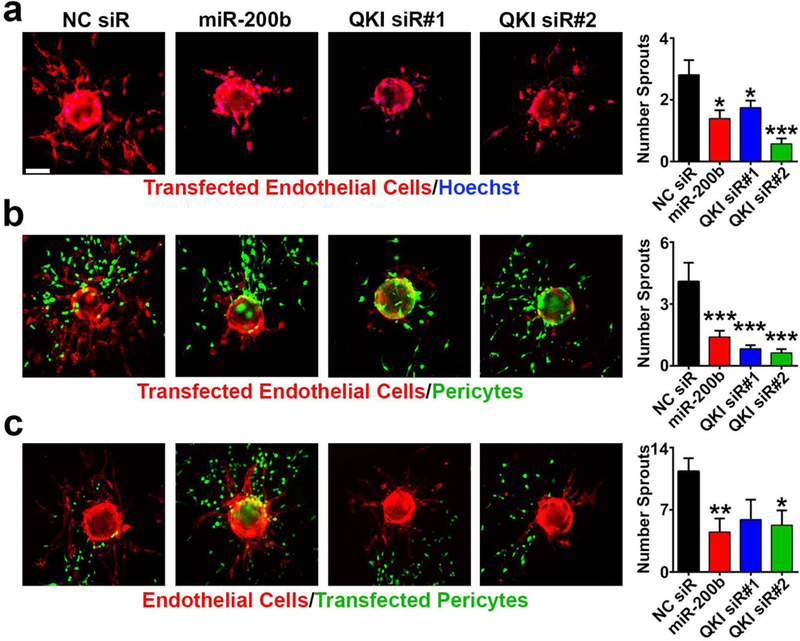
a – c Immunofluorescent imaging and associated quantification of a standard sprouting assay with HUVEC transiently transfected with NC siR, miR-200b, or QKI siRs (n=16 to 27 beads per group); b pericyte-coated sprouting assay with HUVEC transiently transfected with NC siR, miR-200b, or QKI siRs (n=11 to 43 beads per group); and c pericyte-coated sprouting assay with pericytes transiently transfected with NC siR, miR-200b mimic, or QKI siRs (n=6 to 11 beads per group).
Red = CD31; green = Zsgreen labeled 10T½; blue = Hoechst nuclear stain; scale bar = 100 μm; statistical significance was measured by unpaired t-tests; p-values are indicated as *p<0.05, **p<0.01, ***p<0.001; error bars represent standard error of the mean.
QKI expression in tumor endothelium is associated with angiogenesis and poor survival
QKI has previously been demonstrated to play varying and conflicting roles in cancer progression, including promoting metastasis through activating pro-EMT splicing programs (32), inhibiting lung cancer cell invasion and migration through targeting β-catenin (35), inhibiting lung cancer cell proliferation and Notch signaling activation by regulating NUMB alternative splicing (36), and acting as a p53-regulated tumor suppressor in glioblastoma (37). To more thoroughly characterize the clinical relevance of QKI in tumor progression and survival, we assembled a clinically annotated tissue microarray (TMA) consisting of duplicate core biopsies of LUAD (n=150 patients), lung squamous cell carcinoma (LUSC, n=103 patients), and matched normal lung (n=54 patients) cores (Supplementary Table 3). To distinguish the relative contributions of QKI in cancer cells and the tumor endothelium, we employed multiplex immunohistochemistry (IHC) to stain the TMAs for pan-QKI, pan-cytokeratin (pan-CK, cancer cell marker), and cluster of differentiation 31 (CD31, blood vessel marker). Interestingly, although we found that QKI is heterogeneously expressed throughout cancer cells (Figs. 3a – c), in several tumor regions we observed high QKI expression at the leading edge near the vasculature (Fig. 3a-3), further implicating the role of QKI in promoting a more metastatic and invasive phenotype in cancer cells (32). QKI was also abundantly expressed throughout tumor endothelium (Figs. 3a-2, b, and c). The lung TMAs were then analyzed for relationships between QKI expression and indices of tumor angiogenesis and clinical outcome (Supplementary Fig. 5). QKI expression in ECs was significantly and positively correlated with numbers of CD31 positive cells (Spearman r=0.247 and two-tailed p<0.0001, Fig. 3d). In addition, there was a significantly higher percentage of ECs expressing QKI in tumor vasculature compared with normal lung vasculature (Fig. 3e). Intriguingly, there was a significant positive correlation between QKI expression in TECs and cancer cells (Spearman r=0.204 and two-tailed p<0.0001, Supplementary Fig. 6), suggesting QKI expression may be regulated between TME compartments in a coordinated manner. Importantly, an assessment of the clinical features of the TMAs revealed that patients with higher densities of QKI-positive TECs or QKI-positive cancer cells had significantly worse overall survival (Figs. 3f and g). These results highlight the importance of characterizing the function of QKI specifically in the tumor endothelium, as QKI expression in TECs alone is associated with poor survival. Altogether, these data reveal that QKI is a clinically relevant target expressed during tumor angiogenesis and promotes poor survival. Even more intriguingly, these findings highlight the importance of assessing the spatial relationship of QKI expression across various compartments of the TME.
Figure 3. QKI is clinically relevant in tumor angiogenesis and survival.
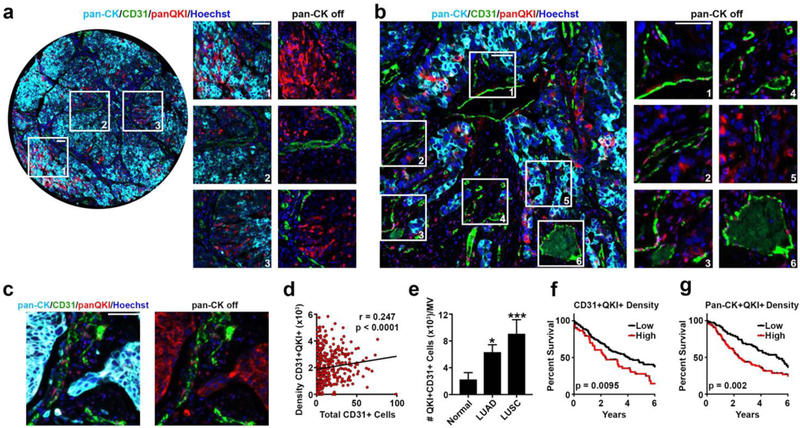
a Immunofluorescence staining of a clinical LUAD tissue section demonstrating QKI expression in (1) cancer cells, (2) TECs, and (3) at the tumor periphery of invading vessels.
b Immunofluorescence staining in clinical LUAD tissue section demonstrating high endothelial QKI expression in highly vascular tumor tissue.
c Immunofluorescent staining in clinical LUSC tissue section demonstrating QKI expression in cancer cells and TECs.
d Two-tailed Spearman’s correlation of the density of pan-QKI low intensity staining ECs per mm2 of tissue versus the total number of CD31+ cells per tissue section.
e Two-tailed Mann Whitney tests comparing the percent of pan-QKI low intensity staining ECs per mm2 of endothelial tissue in normal lung, LUAD, and LUSC tissue sections (p-values are indicated as *p<0.05 and ***p<0.001; error bars represent standard error of the mean).
f Kaplan-Meier curves for overall survival for high and low density of pan-QKI low intensity staining ECs.
g Kaplan-Meier curves for overall survival for high and low density of pan-QKI medium intensity staining in pan-CK positive staining cells.
n=180 patients for f and g; all scale bars 50 μm; cyan = pan-CK, green = CD31, red = pan-QKI isoforms, blue = Hoechst nuclear stain.
QKI regulates EC cell cycle progression
To investigate the molecular mechanisms by which QKI regulates EC function, we performed RNA-sequencing of ECs 24 hours after QKI silencing. We discovered that pan-QKI isoform knockdown leads to both decreases and increases in expression of a large cohort of mRNAs (Fig. 4a). Gene Ontology analysis revealed many of the genes downregulated upon QKI silencing play roles in cell cycle regulation (Figs. 4a and b). Consistent with these findings, we observed that QKI knockdown induces a potent G1 arrest in ECs synchronized by serum starvation (Fig. 4c). Indeed, both miR-200b and QKI siR treatment substantially suppressed EC proliferation (Fig. 4d), and miR-200b-mediated inhibition of proliferation was rescued by overexpression of each QKI isoform (Fig. 4e). Furthermore, inhibition of endothelial sprouting following miR-200b transfection or QKI knockdown was accompanied by a pronounced and significant decrease in Ki-67 proliferative indices amongst sprouting vessels (Fig. 4f). Therefore, QKI-mediated regulation of the EC cell cycle is an important mechanism by which QKI regulates angiogenesis and largely accounts for miR-200b’s effects on EC function.
Figure 4. QKI is an RNA-binding protein that regulates cell cycle progression in ECs.
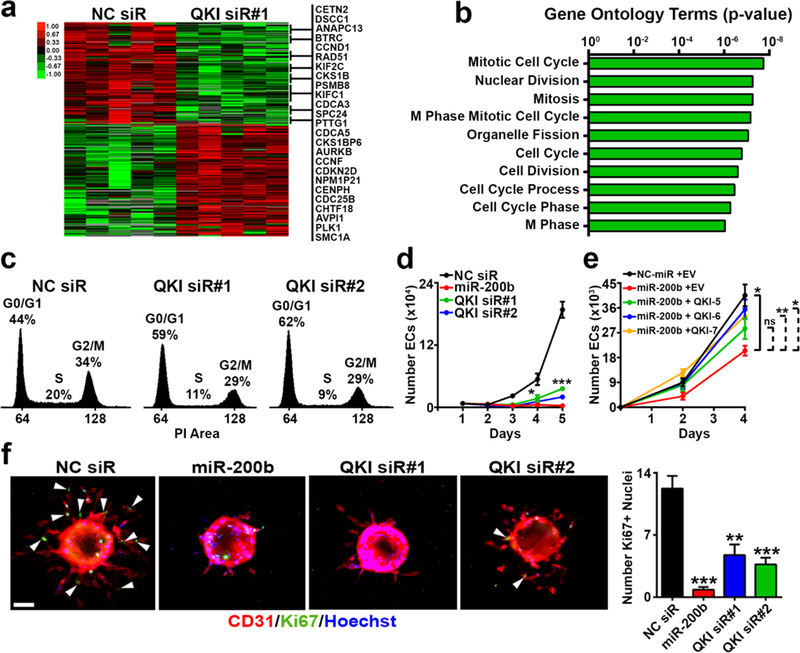
a Heatmap representing the RNA-sequencing analysis performed on HUVEC synchronized by serum starvation with 1% FBS for 24 hours and then transiently transfected with NC siR or QKI siR upon serum starvation release. All genes associated with the Gene Ontology terms presented in b are listed to the right.
b Top 10 Gene Ontology terms with corresponding p-values associated with the cohort of genes downregulated in a (shown in the upper portion of the heat map).
c Cell cycle analysis via propidium iodide staining of HUVEC transiently transfected with NC or QKI siRs.
d In vitro proliferation assay of HUVEC transiently transfected with NC siR, miR-200b, or QKI siRs.
e In vitro proliferation assay of HUVEC serially transfected with NC miR or miR-200b mimic; and empty vector (EV), or QKI-5, QKI-6, or QKI-7 overexpression plasmid.
f Endothelial Ki-67 staining and quantification in standard sprouting assay with HUVEC transiently transfected with NC siR, miR-200b, or QKI siRs; n = 11 to 30 beads per group; red = CD31, green = Ki-67, blue = Hoechst nuclear stain; white arrows demarcate Ki-67+ nuclei; scale bar 75 μm).
c and d show representative results from at least 2 independent experiments; d and e were run in triplicate; statistical significance was measured by unpaired t-tests; p-values are indicated as *p<0.05, **p<0.01, ***p<0.001; error bars represent standard error of the mean.
QKI promotes Cyclin D1 mRNA stability to Regulate EC Growth
To investigate which candidate mRNA targets are most important for modulating the effects of QKI on EC proliferation and cell cycle progression, we identified the cell cycle-related genes most dramatically downregulated in the RNA-sequencing dataset. This revealed CCND1 as the gene most decreased in expression (Supplementary Fig. 7a). Using 2 independent siRs to suppress QKI expression, we assessed the top 9 candidates by qPCR in ECs synchronized by serum starvation (Supplementary Fig. 7b). Although it was not significantly altered in expression in our EC RNA-sequencing dataset, we also added p27 to this qPCR panel for comparison, as it has been previously shown to be a QKI cell cycle target during oligodendrocyte differentiation (18) In the panel of genes investigated, CCND1 was the gene most potently and reproducibly downregulated upon QKI knockdown with both siRs, even more so than the previously published QKI target p27 (Supplementary Fig. 7b). CCND1 mRNA expression remained repressed at both 24 and 48 hours post QKI siR transfection (Fig. 5a). Consistent with these observations, both miR-200b transfection and direct QKI silencing resulted in inhibition of CCND1 protein expression (Fig. 5b). Together, these data support CCND1 mRNA as a novel and important QKI target for its role as a regulator of EC cell cycle progression.
Figure 5. QKI directly regulates CCND1 mRNA in angiogenesis.
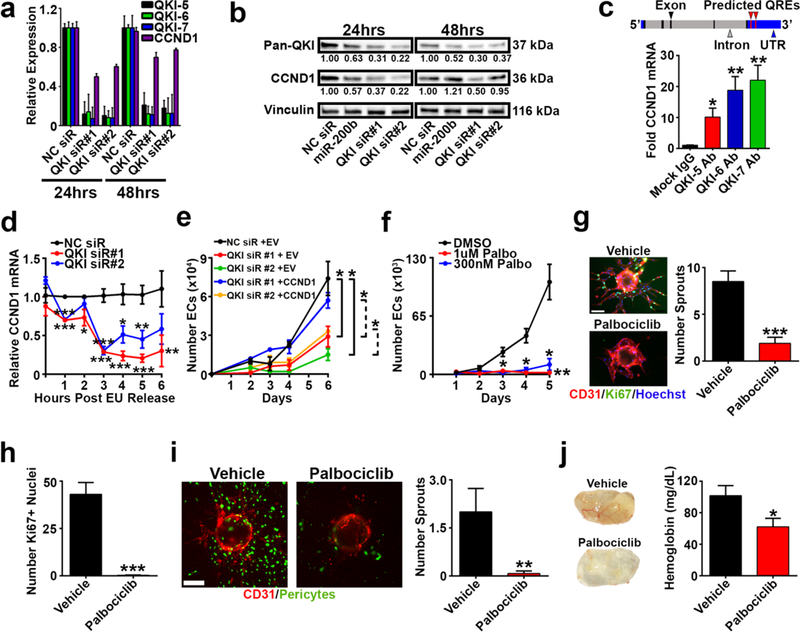
a qPCR analysis for CCND1 mRNA in HUVEC transfected with NC or QKI siRs.
b Western blot for QKI and CCND1 protein expression in HUVEC transfected with NC siR, miR-200b, or QKI siRs. Relative densitometric quantifications (normalized to Vinculin) are presented below the bands.
c Top: CCND1 mRNA schematic; QREs were predicted using RBPmap. Bottom: RNA immunoprecipitation in 2 hours post serum starvation-released HUVEC using mock IgG control or QKI isoform-specific antibody. qPCR was run using CCND1 3’UTR-specific primers.
d qPCR analysis of remaining ethylene uridine ribonucleotide homolog (EU)-labeled CCND1 mRNA in HUVEC transfected with NC or QKI siRs; normalized to the NC siR group at each timepoint.
e Proliferation assay of HUVEC serially transfected with NC siR or QKI siR; and EV or CCND1 overexpression plasmid.
f Proliferation assay of HUVEC treated with DMSO, 1uM, or 300 nM palbociclib.
g Immunofluorescent staining and quantification of sprouting assay treated with DMSO or 1 μM palbociclib (n=22 to 27 beads per group).
h Quantification of Ki-67 positive nuclei in g (n=10 to 14 beads per group).
i Immunofluorescent imaging and quantification of pericyte-coated sprouting assay treated with DMSO or 1 μM palbociclib (n=10 to 13 beads per group).
j Matrigel plugs containing DMSO or 300 nM palbociclib 8 days post subcutaneous implantation and hemoglobin quantification (n=3–4 plugs per group).
Red = CD31; green = Ki-67 in g or ZsGreen-labeled 10T½ in i; blue = Hoechst nuclear stain; scale bars 100 μm. a–f show representative or aggregate results from at least 2 independent experiments; all qPCR assays, proliferation assays, and j (quantification) were performed in triplicate; statistical significance was measured by unpaired t-tests; p-values are indicated as *p<0.05, **p<0.01, ***p<0.001; error bars represent standard error of the mean.
CCND1 mRNA has 4 predicted Quaking Response Elements (QREs) in its 3’UTR based on an in silico prediction using RBPMap (38) (Fig. 5c, top). To directly assess the interaction between QKI and CCND1 mRNA, we performed an RNA immunoprecipitation using isoform-specific antibodies to pull down each QKI protein isoform and probe for CCND1 mRNA association. Relative to IgG control antibody, we observed dramatic enrichment of CCND1 mRNA with each QKI isoform antibody (Fig. 5c, bottom). As a positive control, we also observed enrichment of the previously published QKI mRNA target VE Cadherin (30), while we observed no enrichment of negative control GAPDH mRNA (no predicted QKI binding sites) upon QKI pulldown (Supplementary Fig. 7c). QKI has been shown to bind to the 3’UTR of mRNA targets to regulate their stability (18–20). Therefore, to determine whether QKI modulates CCND1 expression by regulating its mRNA stability, we performed a nascent RNA labeling experiment in synchronized ECs (Supplementary Fig. 7d). QKI silencing in ECs resulted in significantly accelerated CCND1 mRNA degradation (Fig. 5d) relative to control ECs, whereas the rate of degradation of the predicted non-target 18S rRNA was not similarly affected by QKI silencing (Supplementary Fig. 7e). Although QKI has also been demonstrated to regulate the alternative splicing of mRNA targets (16, 17), the absence of predicted QREs in CCND1 intronic regions (Fig. 5c, top), as well as Sashimi plots generated from our RNA-sequencing data set (Supplementary Fig. 8), strongly suggest QKI does not regulate CCND1 alternative splicing. Together, these data demonstrate QKI directly binds to and stabilizes CCND1 mRNA as a mechanism of EC cell cycle progression regulation.
To assess QKI isoform specific contributions to EC proliferation regulation, we used QKI isoform siRs. Silencing of each QKI isoform resulted in inhibition of CCND1 expression in ECs (Supplementary Fig. 9a), however the effects of QKI-6 silencing on CCND1 expression were not as potent. Interestingly, both QKI-5 and QKI-7 silencing, but not QKI-6, recapitulated the effects of pan-QKI isoform silencing on inhibiting EC proliferation (Supplementary Fig. 9b) and sprouting (Supplementary Fig. 9c). These results suggest multiple QKI isoforms are important for CCND1 regulation.
Palbociclib recapitulates the effects of QKI silencing on EC function
We next hypothesized that CCND1-mediated promotion of cell cycle progression is important for the miR-200b/QKI regulatory axis of EC function and angiogenesis. Importantly, we found that CCND1 overexpression rescued the effects of QKI siR-mediated inhibition of EC proliferation (Fig. 5e), suggesting QKI regulation of CCND1 largely accounts for QKI’s effects on EC growth. To determine the implications for angiogenesis regulation upon direct, pharmacologic inhibition of CCND1 function, we used palbociclib to inhibit the CDK4/6-cyclin D complex (39). As with miR-200b over-expression or QKI silencing (Fig. 4d), we observed a potent and significant reduction in EC proliferation (Fig. 5f). Palbociclib treatment also dramatically decreased the number of sprouting vessels per bead (Fig. 5g), as well as the Ki-67 proliferative index of the sprouting ECs (Fig. 5h). In addition, palbociclib elicited potent effects on EC sprouting in the context of pericyte coverage (Fig. 5i) and disrupted blood flow in an angiogenesis Matrigel plug assay in vivo (Fig. 5j).
Inhibition of QKI or the CCND1-CDK4/6 complex potently affects tumor angiogenesis and metastasis
To assess the therapeutic relevance of targeting the miR-200b/QKI/CCND1 regulatory axis on tumor angiogenesis and metastasis in vivo, we used the metastatic 344SQ LUAD model (6, 40). First, following orthotopic injection and tumor formation within the left lung, we performed immunofluorescence staining and verified QKI over-expression in TECs compared with LNECs (Supplementary Fig. 10), consistent with our findings in clinical LUAD samples (Fig. 3e). To target the miR-200b/QKI/CCND1 regulatory axis in the tumor endothelium, we utilized chitosan nanoparticles (CNP), which we have previously demonstrated can deliver oligonucleotide payloads to TECs (6, 41, 42). We found that in established orthotopic 344SQ tumors, CNPs carrying Cy3-labeled miR payloads predominately localized to TECs and peri-vascular regions (Fig. 6a). Next, following orthotopic injection of luciferase-labeled 344SQ cells, mice were randomized into the following groups (n=12 mice/group): 1) NC siR-CNP, 2) miR-200b-CNP, 3) QKI siR#1-CNP, or 4) QKI siR #2-CNP. Four days after 344SQ injection, mice received twice weekly treatments and were euthanized after 4 treatments. Disease burden was assessed, and tumors were collected for staining. There was no evident toxicity from the CNP treatments. Relative to NC siR-CNP treatment, miR-200b-CNP led to modest decreases in metastases (16% reduction in mass and 38% reduction in ex vivo bioluminescence [BLI] signal), while direct QKI silencing led to a more robust inhibition of metastasis (QKI siR#1-CNP 40% reduction in mass, p=0.0247, Student’s t-test and 65% reduction in BLI signal; QKI siR#2-CNP 42% reduction in mass and 64% reduction in BLI signal, p=0.0405, Student’s t-test, Figs. 6b and c). Intriguingly, inhibition of metastasis was associated with statistically significant increases in micro-vessel density (MVD) in tumors scored for CD31 staining, particularly in the QKI siR-CNP-treated groups (52–69% increase in MVD, both p≤0.001, Fig. 6d). Next, we assessed whether palbociclib could recapitulate the effects of QKI siR-CNP in vivo. One week following orthotopic injection of luciferase-expressing 344SQ cells, mice were randomized into the following treatment groups (n=10 mice/group): 1) vehicle or 2) palbociclib. After 1 week of treatment, we assessed disease burden using ex vivo BLI. Similar to treatment with QKI siR-CNPs, we observed that direct pharmacologic inhibition of the CCND1-CDK4/6 pathway resulted in a 73% reduction in metastatic disease burden (p=0.0086, Student’s t-test, Fig. 7a). To directly compare the therapeutic effects of QKI siR-CNP delivery and palbociclib treatment, we orthotopically injected 344SQ cells and 5 days later randomized mice into 4 treatment groups: 1) vehicle + NC siR-CNP, 2) palbociclib, 3) QKI siR#1-CNP, or 4) QKI siR#2-CNP. After 10 days of vehicle or palbociclib treatment or 3 doses of CNP, mice were euthanized and assessed for disease burden. Relative to the control group, palbociclib treatment and QKI siR-CNP delivery inhibited the development of distant metastases (Fig. 7b). Intriguingly, the inhibitory effects of QKI siR-CNP and palbociclib on metastasis did not correlate with an inhibition of primary orthotopic lung tumor growth (Supplementary Fig. 11a).
Figure 6. Endothelial QKI and miR-200b targeting alters tumor vasculature and inhibits metastasis.
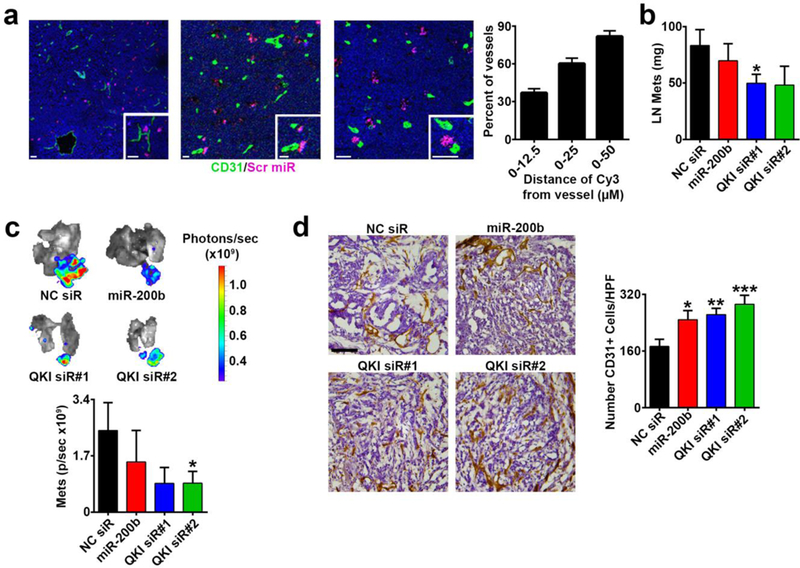
a Immunofluorescent imaging of orthotopic 344SQ LUAD tumor sections from mice intravenously injected with CNPs carrying a scrambled, Cy3-labeled miR and associated quantification of proximity of CNP payload signal to tumor vessels (n=21 images; green = CD31; purple = scrambled miR-CNP; all scale bars 20 μm).
b – c Mass of 344SQ gross lymph node (LN) metastases and ex vivo bioluminescent imaging and quantification of luciferase-labeled LN metastases from mice treated with NC siR-CNP, miR-200b-CNP, QKI siR#1-CNP, or QKI siR#2-CNP (n=12 mice/group).
d Immunohistochemistry 3,3’-Diaminobenzidine (DAB) stain for CD31 in 344SQ lymph node metastases from mice in the treatment groups described in b – c (n=4–5 mice per group and 72–94 images per group). Scale bar 100 μm.
In a, mice were necropsied 12 days post orthotopic 344SQ injection, with CNP deliveries 48 and 24 hours before sacrifice. In b – d, mice were necropsied 15 days post 344SQ orthotopic injection and mice received 4 CNP deliveries. Statistical significance was measured by unpaired t-tests; p-values are indicated as *p<0.05, **p<0.01, ***p<0.001; error bars represent standard error of the mean.
Figure 7. Palbociclib treatment recapitulates the effects of endothelial QKI siR treatment on the tumor microenvironment and metastasis.
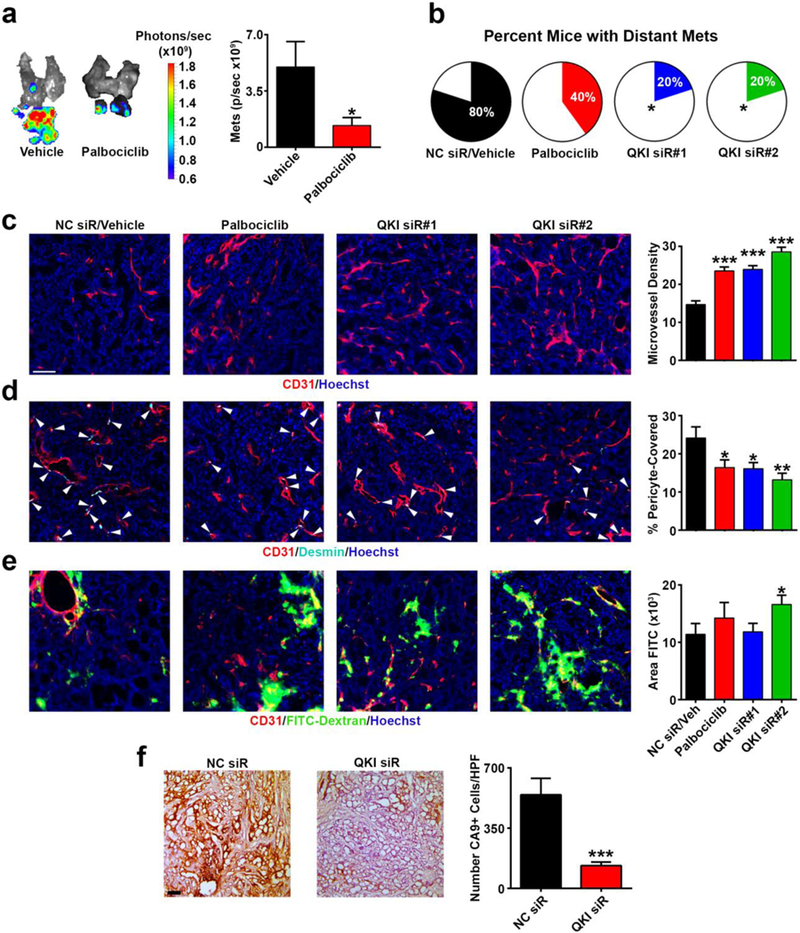
a Ex vivo bioluminescent imaging and quantification of luciferase-labeled LN metastases from mice treated with vehicle or palbociclib.
b Number of contralateral 344SQ metastases from mice treated with the following groups: vehicle/NC siR-CNP, palbociclib, QKI siR#1-CNP, or QKI siR#2-CNP.
c – e Immunofluorescent stained 344SQ subcutaneous tumors from mice treated with the same groups as described in b (n=44 to 62 images per group; red = CD31; cyan = desmin; green = FITC-dextran; blue = Hoechst nuclear stain; scale bar 75 μm).
f Immunohistochemistry 3,3’-Diaminobenzidine (DAB) stain for CA9 in 344SQ subcutaneous tumors in mice treated with either NC siR-CNP or QKI siR#2-CNP (n=45 to 46 images per group). Scale bar 100 μm.
In a and b, mice were necropsied 15 days post 344SQ orthotopic injection. In a, mice started daily drug treatments 8 days post 344SQ orthotopic injection (n=10 mice/group). In b, mice started daily drug treatments 6 days post orthotopic injections or received 3 CNP deliveries (n=5 mice/group). In c – f, mice were necropsied 16 days post subcutaneous 344SQ injection, after 2 CNP deliveries or 6 days of drug treatment (n=4–5 mice/group). Statistical significance was measured by unpaired t-tests except by Chi-square test in b; p-values are indicated as *p<0.05, **p<0.01, ***p<0.001; error bars represent standard error of the mean.
To more thoroughly assess the effects of QKI silencing and CCND1-CDK4/6 blockade on TECs in the TME, we subcutaneously injected 344SQ cells, and once tumors reached ~100 mm3, mice were randomized into 4 treatment groups (n=15 mice/group): 1) vehicle + NC siR-CNP, 2) palbociclib, 3) QKI siR#1-CNP, or 4) QKI siR#2-CNP. Only palbociclib treatment significantly slowed tumor growth, while QKI siR#1-CNP and QKI siR#2-CNP did not have any consistent effects (Supplementary Fig. 11b). These results reflected the direct effects of palbociclib and QKI siR treatments on 344SQ cell proliferation in vitro (Supplementary Figs. 11c and d). The lack of an effect of direct QKI silencing on 344SQ growth strongly suggests that the phenotypic effects of QKI siR-CNP-mediated metastasis inhibition in vivo are specifically due to effects on the TME and not on cancer cell growth. After 1 week of treatment (6 days of oral gavage, 2 deliveries of CNP), mice were euthanized and tumors were collected for staining (n=5 mice/group). Immunofluorescence analysis of the stained tumors revealed that both the palbociclib and QKI siR-CNP treatment groups displayed pronounced effects on the TME. Intriguingly, relative to the control group, there were significant increases (60–94%, all p<0.001) in MVD in the tumors for all treatment groups (Fig. 7c), similar to our observations in the orthotopic QKI siR-CNP experiment (Fig. 6d). These increases in MVD were associated with consistent, statistically significant decreases (ranging from 32 to 45%) in the percentage of pericyte-covered vessels relative to the control group (Fig. 7d) but inconsistent effects on vessel leakiness as determined by FITC-dextran extravasation (Fig. 7e).
We hypothesized that the increases in MVD and associated decreases in metastasis induced by QKI siR-CNP and palbociclib treatments may in part be due to decreased tumor hypoxia. To test this, we compared carbonic anhydrous IX (CA9, a marker of hypoxia) staining in subcutaneous tumors treated twice with NC siR-CNP or QKI siR#2-CNP (treatments started when tumors were ~100 mm3, n=5 mice/group); the latter treatment of which consistently produced the greatest MVD increases compared to all other treatment groups (Figs. 6d and 7c). Consistent with decreases in hypoxia leading to less metastatic tumors, we observed a significant reduction in hypoxia upon QKI siR#2-CNP delivery (Fig. 7f). In summary, these results demonstrate the importance and therapeutic potential of the miR-200b/QKI/CCND1 axis in regulating tumor angiogenesis and inhibiting metastasis.
DISCUSSION
We sought to investigate targets in the tumor endothelium that may be responsible for the effects of miR-200b on the TME. We identified QKI as a potent target gene of miR-200b in ECs and found that QKI silencing recapitulates the effects of miR-200b overexpression on endothelial sprouting. We report a novel mechanism whereby QKI promotes CCND1 mRNA stability to facilitate EC proliferation and highlight an exciting new approach for therapeutic inhibition of metastasis through targeting of tumor vasculature.
Apart from QKI, miR-200b likely also regulates a complex network of angiogenesis-related genes and pathways. For example, known miR-200b targets associated with EMT, such as ZEB2, have also been implicated in promoting the endothelial to mesenchymal transition, which has been speculated to contribute to angiogenesis (43). Therefore, miR-200b may also inhibit angiogenesis through suppression of ZEB2 and the endothelial to mesenchymal transition. Future studies expanding our understanding of the miR-200b mediated network of endothelial function will lend further insight into such pathways.
In this study we focused on the role of QKI in promoting angiogenesis downstream of miR-200b. QKI has been demonstrated to play numerous roles in RNA metabolism and has been most thoroughly characterized for its roles in promoting oligodendrocyte differentiation during myelination (16, 18, 20). We characterized a novel role of QKI in regulating the TME by driving EC cell cycle progression in angiogenesis through the promotion of CCND1 mRNA stability. Notably, all 3 major QKI isoforms immunoprecipitated with CCND1 mRNA, and silencing of each isoform led to loss of CCND1 expression. In addition, overexpression of each isoform rescued the effects of miR-200b-mediated inhibition of EC proliferation. However, isoform-specific silencing demonstrated that only QKI-5 and QKI-7 silencing recapitulated inhibitory effects on EC proliferation and sprouting, while QKI-6 silencing did not. Interestingly, however, QKI-5 silencing also resulted in inhibition of QKI-6 and QKI-7 expression, suggesting QKI-5 may play a role in regulating expression of the other isoforms, which has also been previously hypothesized (25). Therefore, it may be reasonable to speculate from these results that the predominately cytoplasmic isoform QKI-7 is most important for EC proliferation and angiogenesis. This aligns well with the proposed mechanism of CCND1 regulation through promoting its mRNA stability, which occurs in the cytoplasm. The effects of QKI-5 loss or overexpression on EC function may be explained as primarily through its role as a regulator of QKI-7 expression. Finally, although QKI-6 loss did not affect angiogenesis, QKI-6 overexpression was able to rescue EC proliferation in the presence of miR-200b. One possibility is that in the absence of QKI-7, QKI-6 is able to compensate for QKI-7 function as another QKI cytoplasmic isoform.
QKI-mediated promotion of angiogenesis is likely much more complex and extensive than only through regulation of CCND1. Remaining unanswered questions to address include what other targets are modulated by QKI to promote EC cell cycle progression and what other mechanisms of QKI are important for regulating ECs and other cell types in the TME? Intriguingly, we observed that QKI expression itself is dependent on EC cell cycle progression (data not shown) and that the physical interaction between QKI protein and CCND1 mRNA is cell cycle-specific. QKI is regulated by Src family protein tyrosine kinases (Src-PTKs) and undergoes tyrosine phosphorylation during CNS myelination to modulate its ability to interact with myelin basic protein (MBP) mRNA (44). Thus, we speculate that the phosphorylation status of QKI makes this RBP susceptible to external stimuli that regulate its ability to interact with mRNA targets in specific cellular and molecular contexts. In addition, the different QKI isoforms have unusually long 3’UTRs (approximately 8,000 to 14,000 nucleotides), which carry many predicted miR seed sequences (over 250 according to TargetScan), suggesting QKI may be largely susceptible to complex networks of miR regulation. As an RBP, QKI may play a pivotal role in varying biological contexts, and these roles may be largely contingent upon extrinsic stimuli acting through phosphorylating kinases and miRs. A better understanding of the complexity of QKI regulation and its roles in various cell types is needed.
QKI has recently been shown to be targeted by microRNAs specifically in the context of promoting alternative splicing programs during EMT in cancer cells (32). Intriguingly, EMT-induced QKI has also been implicated in generating circular RNAs, a recently emerging class of noncoding RNAs whose biological functions are largely unknown (45). In this study, we demonstrate the importance of a novel biological context for miR-200/QKI regulation in ECs. QKI was highly expressed in both cancer cells and TECs in clinical samples of lung cancer, and QKI expression in both TME compartments was independently associated with poor overall survival. These results highlight the clinical relevance of studying QKI-mediated tumor progression and stress the importance of understanding the unique mechanisms of QKI function in different cellular compartments within the TME. Our work demonstrates that the role of QKI in promoting cancer progression extends beyond its effects in cancer cells and is also critical in regulating patient survival and metastasis through its functions in the tumor endothelium. To our knowledge, this is the first report of distinguishing QKI’s roles within different spatial regions of the TME, which may be key to clarifying the apparent conflicting reports of QKI’s roles in cancer progression (32, 35–37).
In addition, we also demonstrate the importance of cellular context of miR-200b regulation in cancer. In previous work we demonstrate that miR-200b delivery directly to cancer cells via DOPC liposomal nanoparticles in 344SQ tumors exert paracrine effects on decreasing tumor angiogenesis via inhibition of cancer cell secretion of IL-8 (6). DOPC liposomes deliver payloads far beyond the tumor vasculature and into cancer cells due to their neutral charge (46), whereas the positively-charged chitosan nanoparticles used in this study have been demonstrated to have robust delivery to endothelium (41, 42). In this study, we reported the effects of delivery of miR-200b directly to the ECs within 344SQ lung cancer cell line tumors, as opposed to the cancer cells themselves, further highlighting the importance of cellular context in characterizing the miR-200b/QKI/CCND1 in tumor biology. In addition, in our previous study, we also tested the effects of chitosan-mediated delivery of miR-200 directly to ECs within HeyA8 and A2774 ovarian cancer cell line tumors and observed the opposite effects from what we observe in this study (6), also emphasizing the importance of cancer model and cancer type when investigating mechanisms of tumor microenvironment regulation.
RNA interference is a promising therapeutic strategy due to its potential for silencing targets independent of their molecular function or localization (41). QKI siR delivery to tumor endothelium using CNPs dramatically inhibited distant metastasis. Intriguingly, these therapeutic effects were accompanied by increases in tumor MVD and decreased pericyte coverage. Vessel density within tumors is dynamic and constantly changing in response to the metabolic demands of the tumor (47, 48). MVD has previously been demonstrated to increase in tumors treated with angiogenesis-targeting therapies due to decreases in tumor size (therefore increasing the ratio of pre-existing tumor vessels to tumor area); normalization of tumor vasculature and resultant inhibition of hypoxia; or conversely, conversion to nonfunctional vasculature and a resulting uncoupling of tumor growth from tumor MVD (47, 49–52). Our results suggest that the reduction in metastases upon CNP-QKI siR delivery to tumor endothelium was due in part to alleviation of tumor hypoxia by increased MVD. Indeed, excessive vessel pruning by anti-VEGF therapies has been shown to promote hypoxia and augment metastasis (4, 53–55), while a more balanced alteration of the TME has been suggested to alleviate the activation of aggressive resistance mechanisms (including those stimulated by excessive hypoxia) (56, 57). Future studies assessing the effects of endothelial QKI silencing on tumor vessel development over time, as well as effects on vessel function, perfusion, and morphology are needed to further characterize the mechanisms by which QKI regulates tumor endothelium in vivo.
CNP delivery of QKI siRs also resulted in decreases in pericyte coverage. We observed perivascular QKI expression in the 344SQ orthotopic lung tumors, and QKI expression in pericytes may also have important roles in tumor vasculature. Thus, we cannot exclude the possibility that “off-target” (or non-TEC) Chitosan delivery of QKI siRs to pericytes resulted in the phenotype of decreased pericyte coverage. Indeed, we observed that QKI silencing in pericytes alone in the bead sprouting assay was sufficient to significantly alter sprouting. However, the in vivo decreases in pericyte coverage were not associated with consistent increases in vessel extravasation, suggesting that QKI siR-mediated inhibition of pericyte function may not compromise vessel integrity. All together, these results suggest QKI may directly regulate pericyte function independent of its roles in EC function during angiogenesis and vessel formation. The distinct roles of QKI in these 2 vessel-associated cell types remains to be further investigated.
The pleiotropic role(s) of QKI in different cell types may also contribute to cancer progression through other mechanisms. For example, apart from targeting QKI in vascular-associated ECs and pericytes, QKI siR-CNP delivery may have also elicited “off-target” QKI silencing in cancer cells and therefore may have contributed to the inhibition of metastasis through the proposed role of QKI in EMT (32). As supported by our TMA analyses, QKI expression is highly coordinated in at least cancer cells and TECs, and thus targeting QKI may have potent therapeutic effects due to the ability to inhibit multiple components of tumor progression simultaneously. Although QKI has previously been reported to be a tumor suppressor in different cancer types, we observed a lack of an effect on cancer cell proliferation upon QKI silencing in our metastatic model of LUAD, suggesting that the role of QKI in both cancer cells and the TME in metastasis may be more critical for modulating its effects on cancer progression. A better understanding of the complex mechanisms with which QKI regulates EC, pericyte, and cancer cell function and its ability to modulate interactions among these cell types is needed before further therapeutic approaches can be developed.
The mechanism we identified by which QKI regulates EC function through CCND1 mRNA stabilization also led us to consider the general importance of EC cell cycle in angiogenesis and tumor progression. We observed that direct targeting of the functional CCND1-CDK4/6 complex with palbociclib recapitulated the effects of QKI TEC silencing on angiogenesis and metastasis inhibition. These exciting results suggest that CDK4/6 inhibitors developed for targeting cancer cells (58), several of which already have FDA-approval for use in women with hormone-positive breast cancer, can be repurposed for targeting the TME, thus broadening their therapeutic potential and scope. Targeting the cell cycle machinery of ECs provides a more direct pharmacological approach by which to perturb the miR-200b/QKI/CCND1 axis of tumor angiogenesis regulation.
Altogether, we have discovered a clinically relevant, novel miR/RBP regulatory mechanism of tumor endothelial function that shows promise as a therapeutic target for regulating tumor angiogenesis and inhibiting distant metastasis.
MATERIALS AND METHODS
Cell lines, maintenance, transfection reagents, and in vitro drug use.
All cell lines were maintained in 5% CO2/95% air at 37°C. 344SQ lung adenocarcinoma cells were kindly provided by Dr. John Kurie (M.D. Anderson Cancer Center; Houston, TX). 344SQ luciferase-expressing cells were generated by stably transducing 344SQ cells with a lentivirus expressing a GFP/firefly luciferase construct kindly provided by Dr. Shawn D Hingtgen (University of North Carolina at Chapel Hill, Chapel Hill, NC). 10T½ pericyte-like cells and Normal Human Lung Fibroblast (NHLF) cells were obtained from the ATCC. 10T½ ZsGreen cells were generated by stably transducing 10T½ cells with a ZsGreen construct. Human Umbilical Vein Endothelial Cells (HUVEC) were obtained from Lonza. 344SQ cells were maintained in RPMI 1640, NHLF cells in DMEM, and 10T½ cells in Basal Medium Eagle, all supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin Streptomycin. HUVEC were maintained in complete EGM2 supplemented with the growth factor bullet kit (Lonza) and 10% FBS. All cell lines were tested to confirm the absence of Mycoplasma, and all in vitro experiments were conducted with 60–80% confluent cultures. All cells were transiently reverse-transfected with RNAiMax reagent (Invitrogen) using mirVana® mature miR mimic (Ambion, miR-200b, miR-200c, miR-249, or scrambled), anti-miRs (Ambion, negative control anti-miR or anti miR-200b), QKI siRNAs (Sigma or Ambion Silencer Select, QKI siR #1 sense: 5’CAGGCUGCUCCAAGGAUCAdTdT3’, antisense: 5’UGAUCCUUGGAGCAGCCUGdTdT3’; Sigma, QKI siR #2 sense: 5’CUACAGAGAUGCCAACAUUdTdT3’, antisense: 5’AAUGUUGGCAUCUCUGUAGdTdT3’; Ambion Silencer Select, QKI-5 siR sense: 5’CUAUUAACCCACAGCAUUAdTdT3’, antisense: 5’UAAUGCUGUGGGUUAAUAGdTdT3’; Ambion Silencer Select QKI-6 siR sense: 5’GUGUAUUAGGUAUGGCUUUdTdT3’, antisense: 5’AAAGCCAUACCUAAUACACdTdT3’; Ambion Silencer Select QKI-7 siR sense: 5’GAGUGGAUUGAAAUGCCAdTdT3’, antisense: 5’UGGCAUUUCAAUCCACUC3dTdT’) or a negative control siR (Sigma or Ambion Silencer Select, sense: 5’UUCUCCGAACGUGUCACGUdTdT3’, antisense: 5’ACGUGACACGUUCGGAGAAdTdT3’) at a final concentration of 20 nM. QKI siRs #1 and #2 targeted sequences that were conserved across all QKI isoforms in both mice and humans; QKI-5, QKI-6, and QKI-7 siRs targeted sequences unique to each mRNA isoform. For rescue experiments, cells were reverse transfected with miR or siR using RNAimax as described above, then the next day cells were forward transfected at a ratio of 2.5 uL of Lipofectamine 2000 (Invitrogen) per 1 μg of plasmid DNA. DNA plasmids used for the CCND1 orf experiments were pCMV-Cyclin D1 (Addgene #19927) and pmCherry-C1 (as empty vector control), DNA plasmids (GeneCopoeia) used for the QKI orf experiments were EX-NEG-Lv224 (as empty vector control), EX-T4215-Lv224 (QKI-5), EX-H2552-Lv224 (QKI-6), and EX-H2553-Lv225 (QKI-7). Media was changed 4–6 hours following transfections to minimize toxicity. For all in vitro experiments, palbociclib HCl (MedKoo Biosciences, Chapel Hill, NC, # 202173) was reconstituted at a concentration of 300 nM or 1 μM in DMSO. For all serum starve synchronization experiments, HUVEC were serum starved for 24 hours in 1% FBS (59).
Chitosan nanoparticle (CNP) preparation.
miRs or siRs for in vivo EC delivery was incorporated into chitosan (molecular weight 50–190 KDa). CNP was prepared based on ionic gelation of anionic tripolyphosphate and oligos. Briefly, predetermined tripolyphosphate (0.25% w/v) and miRs or siRs (1 μg μl−1) were added in chitosan solution, and the miR or siR-CNP were spontaneously formed under constant stirring at room temperature. After incubation at 4 °C for 40 min, miR or siR-CNP was collected by centrifugation (Thermo Biofuge, Germany) at 13,000 r.p.m. for 40 min at 4 °C. The pellet was washed by sterile water 3 times to isolate miR or siR-CNP, which was stored at 4 °C until used.
Animals, in vivo models and tissue processing.
Female athymic nude mice were purchased from Jackson Labs and from the UNC Chapel Hill Animal Studies Core. These animals were cared for according to guidelines set forth by the American Association for Accreditation of Laboratory Animal Care and the U.S. Public Health Service policy on Human Care and Use of Laboratory Animals. All mouse studies were approved and supervised by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee. All animals used were between 6–10 weeks of age at the time of injection. For all animal experiments, cells were trypsinized, washed, and resuspended in Hanks balanced salt solution (HBSS; Gibco) prior to injection. 344SQ cancer cells were injected either subcutaneously over the posterior flank (1×105 cells in 100 μL HBSS) or by an intra-pulmonary technique (5×103 cells in 50 μL for therapeutic experiments or 1×105 in 100 μL for the biodistribution experiment, both in 1:1 mixture of HBSS and BD Matrigel (BD Biosciences). For the intra-pulmonary injections, mice were anesthetized with ketamine (80 mg/kg) + xylazine (8 mg/kg) + acepromazine (1 mg/kg) and placed in the right lateral recumbency. Following sterile skin preparation, an incision parallel to the rib cage between ribs 10–11 was made to visualize the lung through the intact thoracic pleura. A 1 mL tuberculin syringe with a 30-g needle was used to inject the cell suspension directly into the lung parenchyma at the left lateral dorsal axillary line. After injection, the skin incision was closed using surgery clips, and the mice were turned on the left lateral recumbency and observed until fully recovered. Mice were randomized a few days after orthotopic or subcutaneous injection of cancer cells, before initiating treatment. Mice were all combined into one or two cages and then randomly redistributed into treatment groups. Caliper measurements of subcutaneous tumor growth were taken twice weekly and tumor volume was calculated as L X W2 where L is the greatest cross-sectional length across the tumor and W is the length perpendicular to L. Luciferase-labeled tumor progression was monitored once or twice weekly using an IVIS Lumina optical imaging system and D-Luciferin K+ substrate (Perkin Elmer, # 122796) as per the manufacturer’s instructions. Briefly, substrate was reconstituted at a concentration of 15 mg/mL in PBS and 200 μL of substrate were injected IP. Mice were imaged after a 10-minute incubation. Palbociclib HCl (MedKoo Biosciences, Chapel Hill, NC, # 202173) was reconstituted in 50 mM sodium lactate buffer (pH 4.0, Sigma, # 71718) at a concentration of 37.5 mg/mL and was administered at a dose of 150 mg/kg/mouse, daily by oral gavage. Chitosan nanoparticles packaged with miR or siR were administered at a dose of 150 ug of oligo/kg/mouse and intravenously, twice a week.
A biodistribution study was performed to assess chitosan nanoparticle delivery of oligonucleotides to tumor endothelium in the orthotopic 344SQ lung model. 12 days after cell injection, mice were intravenously administered two doses (24 hours apart) at 200 μg/kg/mouse of Cy3-labeled oligonucleotides packaged in chitosan nanoparticles. 24 hours after the second dose, mice were sacrificed and their primary tumors were snap frozen in OCT. Tumors were assessed for co-localization of Cy3 signal with endothelium.
In all therapeutic experiments 5–15 mice per group were used. Once mice in any group became moribund, they were all sacrificed, necropsied, and tumors were harvested. Tumor weights and number and location of lymphatic and distant metastases were recorded. For TME assessments, mice were injected intravenously with 100 μL of FITC-dextran at 10 mg/mL (2,000,000 molecular weight, Life Technologies, # D7137) 1 hour before sacrifice. All tissues used for immunostaining analysis were fixed in 10% neutral buffered formalin, cryoprotected in 30% sucrose, and then slowly frozen at −20°C in optimal cutting temperature (OCT, Fisher Healthcare) media.
Quantitative real-time PCR.
For mRNA quantification, total RNA was extracted from cultured cells using the Quick RNA MiniPrep Zymo Research Kit (Genesee Scientific, # 11–328) or from ex vivo sorted cells using the Quick RNA MicroPrep Zymo Research Kit (Genesee Scientific, # 11–328M). For coding mRNA gene expression analyses, cDNA was synthesized using an iScript cDNA Synthesis Kit (Bio-Rad, # 1708891) as per the manufacturer’s instructions. Analysis of mRNA levels was performed on a StepOnePlus Real-Time PCR System (Applied Biosystems). Specific primers for [QKI-5 (forward)-TACCCTATTGAACCCAGTGGTGT, (reverse)-TCGAACTTTAGTAGCCACCGC; QKI-6 (forward)-ACCCTATTGAACCCAGTGGTGT, (reverse)-TAGCCTTTCGTTGGGAAAGC; QKI-7 (forward)-CCCAGTGGTGTGTTAGAGTGGA, (reverse)-TGAAATATCAGGCATGACTGGC; CCND1 (forward)-GGCGGATTGGAAATGAACTT, (reverse)-TCCTCTCCAAAATGCCAGAG; CCND1 3’UTR (forward)-GGAGGAGGGTTGTGCTACAG, (reverse)-CGCCTCCTTTGTGTTAATGC; ANAPC13 (forward)-GCCACCATTGTCTTGTTCAG, (reverse)-TTTGATTGATGATGCTTGGC; CETN2 (forward)-AGGGCTCATTCTTTTTCGCT, (reverse)-TGTACACGTCGGTTGCCTAA; CKS1B (forward)-GCCAAGATTCCTCCATTCAG, (reverse)-CGACGACGAGGAGTTTGAGT; CDKN2D (forward)-GTCCTGGACATTGGGGCT, (reverse)-AACCGCTTCGGCAAGAC; RAD51 (forward)-TATCCAGGACATCACTGCCA, (reverse)-GGTGAAGGAAAGGCCATGTA; AURKB (forward)-AGATGGGGTGACAGGCTCTT, (reverse)-AGGAGAACTCCTACCCCTGG; CHTF18 (forward)-ACGCAGGAAGTTGTCAAACA, (reverse)-CTCACAGCGATTCTACCGTG; AVPl1 (forward)-GGTACCCATTGTGGATGCTC, (reverse)-TGAAGGCTGTGGAAGAGGTT; p27 (forward)-AACGTGCGAGTGTCTAACGG, (reverse)-CCCTCTAGGGGTTTGTGATTCT; GAPDH (forward)-TTCCAGGAGCGAGATCCCT, (reverse)-CACCCATGACGAACATGGG; VE Cadherin (forward)-GCACCAGTTTGGCCAATATA, (reverse)-GGGTTTTTGCATAATAAGCAGG] were used for SYBR Green-based real-time PCR, and 18S rRNA (or GAPDH for Supplementary Fig. 2a) was used as a housekeeping gene. PCR was done with reverse-transcribed RNA, 1 μL each of 20 μM forward and reverse primers, and 2X PowerUp SYBR Green Master Mix (Life Technologies, # 100029284) in a total volume of 25 μL. TaqMan Assays (Applied Biosystems) were used for miR-200b expression (Assay ID # 002251), and U6 snRNA (Asay ID #001973) was used as a housekeeping gene. PCR was done with reverse-transcribed RNA, 20x TaqMan probe, and TaqMan Universal Master Mix II as per the manufacturer’s instructions. For both SYBR and TaqMan-based PCR, each cycle consisted of 15 seconds of denaturation at 95°C and 1 min of annealing and extension at 60°C (40 cycles). Reactions were run in duplicate or triplicate.
Target Gene Binding Sites, Luciferase Reporter Assays and 3’ UTR Site Mutagenesis.
The putative binding sites for miR-200b were predicted bioinformatically using the TargetScan algorithm (publically available at http://www.targetscan.org) for predicting miR targets and binding sites for QKI. GoClone pLightSwitch luciferase reporters for the QKI 3’ UTR reporter was obtained from SwitchGear Genomics (Menlo Park, CA). HEK293 cells (obtained from the ATCC) were transfected with FuGENE HD TFX reagent in a 96-well plate with scrambled control or miR-200b mimics along with the 3’ UTR reporter gene and Cypridina TK control construct (pTK-Cluc). After 48 hours of transfection, luciferase activity was obtained with the LightSwitch Dual Luciferase assay kits using a microplate luminometer per manufacturer guidelines. Two independent experiments were performed with 3 replicates each. An empty luciferase reporter vector was used as a negative control. The ratios obtained were further normalized according to the scrambled control. Mutants of the QKI 3’ UTR were generated using the QuikChange Lightning Multi Site-direct Mutagenesis kit (Agilent Technologies, La Jolla, CA) using the following primers to delete 7 nucleotides in a miR-200b binding site: (forward) cttttgaaatctctgaatgccttggttctatcattctttattgaatttatttcttatta; (reverse) taataagaaataaattcaataaagaatgatagaaccaaggcattcagagatttcaaaag. Proper site mutagenesis was confirmed with sequencing prior to luciferase assays.
Western blotting.
After reverse transfection, cells were lysed by scraping in RIPA buffer (ThermoFisher, # PI89901) containing 1x cOmplete Mini protease inhibitor cocktail (Roche, # 11836153001). Equal amounts of lysates (10–40 μg of total protein) were run on 4–20% SDS-PAGE gradient gels, after which protein was transferred to nitrocellulose membranes (BioRad, Hercules, CA). Membranes were blocked in 5% milk/Tris-buffered saline-Tween 20 (TBS-T) for one hour at room temperature prior to probing with primary antibodies overnight at 4°C. Primary antibodies included pan-QKI (Clone N147/6, # 75–168, Neuromab), QKI-5 (rabbit anti-QKI, Millipore, # AB9904), QKI-6 (rabbit anti-QKI, Millipore, # AB9906), QKI-7 (mouse anti-QKI, Neuromab, # 75–200), Cyclin D1 (# 2922s, Cell Signaling Technology, Danvers, MA), and anti-vinculin (clone hVIN-1, # V9131, Sigma), all at a 1:1000 dilution in blocking buffer. After probing with primary antibodies, membranes were washed three times in TBS-T and then probed with the appropriate horseradish peroxidase-conjugated secondary antibodies (anti-mouse [# 115–035-003] or anti-rabbit [# 111–035-003] from Jackson ImmunoResearch) diluted 1:5,000 to 1:10,000. Then, the membranes were washed four times in TBS-T and developed using Clarity Western ECL substrate (BioRad, Hercules, CA, # 1705060). Membranes were visualized using a BioRad ChemiDoc MP system (BioRad, Hercules, CA). Densitometry quantifications were determined using Fiji software.
Proliferation Assays.
Transfected or treated HUVEC were seeded at a density of 5,000 or 7,500 cells per well in 12-well plates in triplicate and counted on a hemocytometer using a Trypan Blue counterstain.
Cell Cycle Analysis.
Cells were fixed in cold 70% EtOH and stored at −20°C until ready for staining. Cells were washed with PBS and incubated with staining solution (Propidium iodide [Roche, # 11348639001 stock concentration 500 μg/mL, working concentration 50 μg/mL] and RNAse A [Qiagen, stock concentration 100 mg/mL, working concentration 100 μg/mL] diluted in PBS) for 30 minutes at room temperature in the dark. Stained cells were analyzed on a Beckman Coulter CyAn ADP.
RNA Immunoprecipitation.
RNA immunoprecipitation assay was performed using the Magna Rip Kit (Millipore Sigma, # 17–700) per the manufacturer’s instructions using the following antibodies for QKI isoform pulldown: QKI-5 (rabbit anti-QKI, Millipore, # AB9904), QKI-6 (rabbit anti-QKI, Millipore, # AB9906), and QKI-7 (rabbit anti-QKI, Millipore, #AB9908).
Argonaute-2 Immunoprecipitation.
One or two confluent 10 cm plates of cells were washed with cold PBS and UV crosslinked at 400J. Cells were washed with cold PBS again, scraped into 15 mL conical tubes, and centrifuged at 810 g for 5 minutes. Cell pellet was resuspended in 1 mL cold Ago IP buffer (50 μM HEPES, 100 μM NaCl, 0.5% NP40), vortexed, and incubated on ice for 20 minutes with intermittent vortexing. Samples were centrifuged at maximum speed for 30 seconds. Supernatants were collected and incubated with 10 μL of Ago2 Mab (Diagenode, #c15200167) on ice for 1 hour. Next, 25 μL of Protein A dynabeads (Invitrogen, #10001d) and 25 μL of Protein G dynabeads (Invitrogen, #10003d) were mixed together and washed 2 times with cold Ago IP buffer. Samples were then added to the beads and rotated at 4°C for 30 minutes. Beads were then washed 2 times with cold Ago IP buffer and then 2 times with 0.5 M NaCl Ago IP buffer. Beads were then resuspended in 100 μL Ago IP buffer, 200 μL Trizol, and 50 μL of chloroform. Samples were vigorously vortexed and centrifuged at 12000g 4°C for 15 minutes. The top aqueous layer was removed and mixed with 5 μL glycogen and100 μL isopropanol and centrifuged at 12000g 4°C for 10 minutes. The pellet was washed 1X in 75% EtOH and then resuspended in 10 μL. All the resulting RNA was loaded into an iScript CDNA reaction and subjected to qPCR analysis.
mRNA Stability Assay.
CCND1 mRNA stability was assessed using the Click-it Nascent RNA capture kit (Life Technologies, # C10365) according to the manufacturer’s instructions.
In vivo Angiogenesis Plug Assay.
A 450–500 μL solution of Matrigel containing 500 ng/mL rVEGF 165 contianing DMSO or 300 nM palbociclib was subcutaneously implanted on to the flanks of nude mice. 8 days after implantation, the plugs were extracted and snap frozen. Plugs were then dissociated using Dispase (Worthington Biochemical, # LS02104) and hemoglobin content was quantified using the QuantiChrom Hemoglobin Assay Kit (BioAssay Systems, # DIHB-250) according to the manufacturer’s instructions.
Sprouting Assay.
Both the standard and pericyte-coated sprouting assays were performed, quantified, and immunostained as previously described (34). The number of sprouts per bead was determined by counting the number of endothelial protrusions greater than or equal to 100 μm in length growing off each bead. In general, 10–20 beads per experimental group were quantified. Beads in proximity to one another or near the edge of the imaging well were excluded from analysis due to possible confounding effects on sprouting. Primary antibodies used for immunofluorescence staining include CD31 (mouse anti-human, Dako, clone # JC70A, 1:1000) and Ki-67 (rabbit anti-mouse, 1:200, Abcam ab15580). Secondary antibodies used include goat anti-rabbit Alexa 488 (Molecular Probes, #A10034, 1:600) and goat anti-mouse Alexa 594 (Molecular Probes, #A11005, 1:600).
Immunostaining.
Staining was performed in OCT-embedded frozen tissue sections (10–20 μm thick) either with or without prior formalin fixation and 30% sucrose cryoprotection. Sections were permeabilized with cold acetone for 10 minutes. For immunohistochemistry, 3% H2O2 was used to block endogenous peroxidase activity for 10 minutes. Avidin Biotin blocking was performed using an Avidin/Biotin blocking kit (Vector Labs, # sp-2001) per the manufacturer’s instructions. Protein blocking of non-specific epitopes was performed using 4% fish gelatin in PBS for 20 minutes. Slides were incubated with primary antibody for overnight at 4ºC. After washing with PBS, the appropriate amount of biotinylated secondary antibody was added, and the signal was amplified using an ABC kit (Vector Labs, # PK-6200) per the manufacturer’s instructions. The slides were then visualized with 3,3’-diaminobenzidine chromogen (Vector Labs, # SK-4105), counterstained with Gill’s hematoxylin #3, and mounted with Permount medium. For immunofluorescence, after acetone permeabilization, protein blocking using 4% fish gelatin was performed, and slides were incubated with primary antibody overnight at 4ºC. After washing with PBS, secondary antibody staining was performed in blocking buffer for 1 hour at RT. Hoechst 33342 (BD Pharmingen, 1:10,000, # 561908) was used for nuclear staining. Slides were mounted with Prolong Gold (Invitrogen). A Leica DMi8 inverted microscope was used for both fluorescent and light microscopy.
Primary antibodies used include the following: Desmin (rabbit anti-mouse, 1:200, Abcam, # 15200), CD31 (rat anti-mouse, 1:400, BD Pharmingen # 553370 for non-fixed tissues or rat anti-mouse, 1:50, BD Pharmingen # 550274 for fixed tissues), and CA9 (Novus Biologicals, 1:1000, # NB100–417SS). Immunofluorescent secondary antibodies used include the following: goat anti-mouse Alexa 594 (Molecular Probes, # A11005,1:600); goat anti-rat Alexa 488 (Molecular Probes, # A11006, 1:500); and goat anti-rat Alexa 594 (Molecular Probes, # A11007, 1:600). The horseradish peroxidase-conjugated secondary antibody used was Biotinylated anti-Rat IgG (Vector Labs, # BA-9401, 1:100)
To quantify microvessel density (MVD), we examined fields at 20x magnification for each tumor in a blinded fashion and counted the microvessels within those fields. A vessel was defined as an open lumen with at least one adjacent CD31-positive cell. Multiple positive cells beside a single lumen were counted as one vessel. To quantify pericyte-covered vessels, we determined the percent of vessels in each field that had at least 50% pericyte-coverage. To quantify FITC-dextran extravasation, we measured the total area of FITC signal per field of view. Enumeration of CD31-and CAIX-positive cells per high powered field were determined using a pipelines in CellProfiler 3.0 software (60). In general, 4–5 mice were analyzed per experimental group for all immunofluorescent and immunohistochemistry analyses, and 10–20 images were analyzed per mouse.
Statistical analyses.
Between 5 and 10 mice were assigned per treatment group; this sample size gave approximately 80% power to detect a 50% reduction in tumor weight with 95% confidence. Results for each group were compared using a Student’s t test (for comparisons of two groups) and analysis of variance (for multiple group comparisons). For values that were not normally distributed (as determined by the Kolmogorov-Smirnov test), the Mann–Whitney rank sum test was used. A P-value less than 0.05 was deemed statistically significant. All other statistical tests for in vitro and in vivo experiments were performed using GraphPad Prism 7 (GraphPad Software, Inc., San Diego, CA). All line and bar graphs represent mean values, and all error bars represent standard error of the mean.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to especially thank members of the Victoria Bautch laboratory for their advice on performing the endothelial sprouting assay, the UNC Animal Histopathology Core, the UNC Biomedical Research Imaging Center, Janet Dow from the UNC Flow Cytometry Core Facility, and the Tissue Pathology Lab Core. The authors would also like to thank Adolfo Alfonso for providing the ZsGreen fluorescent labeled 10T½ (pericyte) cells used in the pericyte-coated sprouting assay. C.V.P. was supported in part by the National Institutes of Health R01-CA215075, R01-CA042978 and U54-CA198999, a Mentored Research Scholar Grants in Applied and Clinical Research (MRSG-14–222-01-RMC) from the American Cancer Society, the Jimmy V Foundation Scholar award, the UCRF Innovator Award, the Stuart Scott V Foundation/Lung Cancer Initiative Award for Clinical Research, the University Cancer Research Fund, the Lung Cancer Research Foundation, the Free to Breathe Metastasis Research Award and the Susan G. Komen Career Catalyst Award. S.H.A. was supported in part by a grant from the National Institute of General Medical Sciences under award 5T32 GM007092. E.B.H. was supported in part by a grant from the National Cancer Institute of the National Institutes of Health under award number T32CA196589. AKS was supported in part by The American Cancer Society Research Professor Award and grants from the National Cancer Institute (P50 CA217685 and R35 CA209904). The UNC Flow Cytometry Core Facility and Lineberger Comprehensive Cancer Center Animal Histopathology and Animal Studies Cores are all supported in part by a National Cancer Institute Center Core Support Grant (CA016086) to the UNC Lineberger Comprehensive Cancer Center. The UNC Flow Cytometry Core Facility is also supported in part by the North Carolina Biotech Center Institutional Support Grant 2012-IDG-1006.
Footnotes
Declaration of Interests: The authors declare no competing interests.
Supplementary information is available at Oncogene’s website.
Please see Supplemental Experimental Procedures for additional methods.
REFERENCES
- 1.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–95. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6. [DOI] [PubMed] [Google Scholar]
- 3.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–72. [DOI] [PubMed] [Google Scholar]
- 4.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bottsford-Miller JN, Coleman RL, Sood AK. Resistance and escape from antiangiogenesis therapy: clinical implications and future strategies. J Clin Oncol. 2012;30(32):4026–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pecot CV, Rupaimoole R, Yang D, Akbani R, Ivan C, Lu C, et al. Tumour angiogenesis regulation by the miR-200 family. Nat Commun. 2013;4:2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan YC, Khanna S, Roy S, Sen CK. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J Biol Chem. 2011;286(3):2047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan YC, Roy S, Khanna S, Sen CK. Downregulation of endothelial microRNA-200b supports cutaneous wound angiogenesis by desilencing GATA binding protein 2 and vascular endothelial growth factor receptor 2. Arterioscler Thromb Vasc Biol. 2012;32(6):1372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding Y, Hu Z, Luan J, Lv X, Yuan D, Xie P, et al. Protective effect of miR-200b/c by inhibiting vasohibin-2 in human retinal microvascular endothelial cells. Life Sci. 2017;191:245–52. [DOI] [PubMed] [Google Scholar]
- 10.Gill JG, Langer EM, Lindsley RC, Cai M, Murphy TL, Murphy KM. Snail promotes the cell-autonomous generation of Flk1(+) endothelial cells through the repression of the microRNA-200 family. Stem Cells Dev. 2012;21(2):167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Magenta A, Cencioni C, Fasanaro P, Zaccagnini G, Greco S, Sarra-Ferraris G, et al. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011;18(10):1628–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kondo T, Furuta T, Mitsunaga K, Ebersole TA, Shichiri M, Wu J, et al. Genomic organization and expression analysis of the mouse qkI locus. Mamm Genome. 1999;10(7):662–9. [DOI] [PubMed] [Google Scholar]
- 13.Teplova M, Hafner M, Teplov D, Essig K, Tuschl T, Patel DJ. Structure-function studies of STAR family Quaking proteins bound to their in vivo RNA target sites. Genes Dev. 2013;27(8):928–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy RJ, Loushin CL, Friedrich VL Jr., Chen Q, Ebersole TA, Lazzarini RA, et al. Neural cell type-specific expression of QKI proteins is altered in quakingviable mutant mice. J Neurosci. 1996;16(24):7941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J, Zhou L, Tonissen K, Tee R, Artzt K. The quaking I-5 protein (QKI-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J Biol Chem. 1999;274(41):29202–10. [DOI] [PubMed] [Google Scholar]
- 16.Wu JI, Reed RB, Grabowski PJ, Artzt K. Function of quaking in myelination: regulation of alternative splicing. Proc Natl Acad Sci U S A. 2002;99(7):4233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Bruin RG, Shiue L, Prins J, de Boer HC, Singh A, Fagg WS, et al. Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat Commun. 2016;7:10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larocque D, Galarneau A, Liu HN, Scott M, Almazan G, Richard S. Protection of p27(Kip1) mRNA by quaking RNA binding proteins promotes oligodendrocyte differentiation. Nat Neurosci. 2005;8(1):27–33. [DOI] [PubMed] [Google Scholar]
- 19.Zearfoss NR, Clingman CC, Farley BM, McCoig LM, Ryder SP. Quaking regulates Hnrnpa1 expression through its 3’ UTR in oligodendrocyte precursor cells. PLoS Genet. 2011;7(1):e1001269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Zhang Y, Li D, Feng Y. Destabilization and mislocalization of myelin basic protein mRNAs in quaking dysmyelination lacking the QKI RNA-binding proteins. J Neurosci. 2000;20(13):4944–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saccomanno L, Loushin C, Jan E, Punkay E, Artzt K, Goodwin EB. The STAR protein QKI-6 is a translational repressor. Proc Natl Acad Sci U S A. 1999;96(22):12605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larocque D, Pilotte J, Chen T, Cloutier F, Massie B, Pedraza L, et al. Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron. 2002;36(5):815–29. [DOI] [PubMed] [Google Scholar]
- 23.Noveroske JK, Lai L, Gaussin V, Northrop JL, Nakamura H, Hirschi KK, et al. Quaking is essential for blood vessel development. Genesis. 2002;32(3):218–30. [DOI] [PubMed] [Google Scholar]
- 24.Li Z, Takakura N, Oike Y, Imanaka T, Araki K, Suda T, et al. Defective smooth muscle development in qkI-deficient mice. Dev Growth Differ. 2003;45(5–6):449–62. [DOI] [PubMed] [Google Scholar]
- 25.Bohnsack BL, Lai L, Northrop JL, Justice MJ, Hirschi KK. Visceral endoderm function is regulated by quaking and required for vascular development. Genesis. 2006;44(2):93–104. [DOI] [PubMed] [Google Scholar]
- 26.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22(7):894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448(7155):807–10. [DOI] [PubMed] [Google Scholar]
- 28.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. [DOI] [PubMed] [Google Scholar]
- 29.Cochrane A, Kelaini S, Tsifaki M, Bojdo J, Vila-Gonzalez M, Drehmer D, et al. Quaking Is a Key Regulator of Endothelial Cell Differentiation, Neovascularization, and Angiogenesis. Stem Cells. 2017;35(4):952–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Bruin RG, van der Veer EP, Prins J, Lee DH, Dane MJ, Zhang H, et al. The RNA-binding protein quaking maintains endothelial barrier function and affects VE-cadherin and beta-catenin protein expression. Sci Rep. 2016;6:21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Mil A, Grundmann S, Goumans MJ, Lei Z, Oerlemans MI, Jaksani S, et al. MicroRNA-214 inhibits angiogenesis by targeting Quaking and reducing angiogenic growth factor release. Cardiovasc Res. 2012;93(4):655–65. [DOI] [PubMed] [Google Scholar]
- 32.Pillman KA, Phillips CA, Roslan S, Toubia J, Dredge BK, Bert AG, et al. miR-200/375 control epithelial plasticity-associated alternative splicing by repressing the RNA-binding protein Quaking. EMBO J. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakatsu MN, Davis J, Hughes CC. Optimized fibrin gel bead assay for the study of angiogenesis. J Vis Exp. 2007(3):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Azam SH, Smith M, Somasundaram V, Pecot CV. Incorporating Pericytes into an Endothelial Cell Bead Sprouting Assay. J Vis Exp. 2018(132). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou X, Li X, Sun C, Shi C, Hua D, Yu L, et al. Quaking-5 suppresses aggressiveness of lung cancer cells through inhibiting beta-catenin signaling pathway. Oncotarget. 2017;8(47):82174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zong FY, Fu X, Wei WJ, Luo YG, Heiner M, Cao LJ, et al. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014;10(4):e1004289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen AJ, Paik JH, Zhang H, Shukla SA, Mortensen R, Hu J, et al. STAR RNA-binding protein Quaking suppresses cancer via stabilization of specific miRNA. Genes Dev. 2012;26(13):1459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akerman M, David-Eden H, Pinter RY, Mandel-Gutfreund Y. A computational approach for genome-wide mapping of splicing factor binding sites. Genome Biol. 2009;10(3):R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–38. [PubMed] [Google Scholar]
- 40.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23(18):2140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harrison EB, Azam SH, Pecot CV. Targeting Accessories to the Crime: Nanoparticle Nucleic Acid Delivery to the Tumor Microenvironment. Front Pharmacol. 2018;9:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu C, Han HD, Mangala LS, Ali-Fehmi R, Newton CS, Ozbun L, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010;18(2):185–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welch-Reardon KM, Wu N, Hughes CC. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol. 2015;35(2):303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Lu Z, Ku L, Chen Y, Wang H, Feng Y. Tyrosine phosphorylation of QKI mediates developmental signals to regulate mRNA metabolism. EMBO J. 2003;22(8):1801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, et al. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160(6):1125–34. [DOI] [PubMed] [Google Scholar]
- 46.Landen CN Jr., Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65(15):6910–8. [DOI] [PubMed] [Google Scholar]
- 47.Hlatky L, Hahnfeldt P, Folkman J. Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn’t tell us. J Natl Cancer Inst. 2002;94(12):883–93. [DOI] [PubMed] [Google Scholar]
- 48.Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004;6(6):553–63. [DOI] [PubMed] [Google Scholar]
- 49.Goel S, Gupta N, Walcott BP, Snuderl M, Kesler CT, Kirkpatrick ND, et al. Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J Natl Cancer Inst. 2013;105(16):1188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wong PP, Demircioglu F, Ghazaly E, Alrawashdeh W, Stratford MR, Scudamore CL, et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell. 2015;27(1):123–37. [DOI] [PubMed] [Google Scholar]
- 51.Wong PP, Bodrug N, Hodivala-Dilke KM. Exploring Novel Methods for Modulating Tumor Blood Vessels in Cancer Treatment. Curr Biol. 2016;26(21):R1161–R6. [DOI] [PubMed] [Google Scholar]
- 52.Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444(7122):1032–7. [DOI] [PubMed] [Google Scholar]
- 53.Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. 2014;17(3):471–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15(3):220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10(6):417–27. [DOI] [PubMed] [Google Scholar]
- 57.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Santo L, Siu KT, Raje N. Targeting Cyclin-Dependent Kinases and Cell Cycle Progression in Human Cancers. Semin Oncol. 2015;42(6):788–800. [DOI] [PubMed] [Google Scholar]
- 59.Griffin MJ. Synchronization of some human cell strains by serum and calcium starvation. In Vitro. 1976;12(5):393–8. [DOI] [PubMed] [Google Scholar]
- 60.Lamprecht MR, Sabatini DM, Carpenter AE. CellProfiler: free, versatile software for automated biological image analysis. Biotechniques. 2007;42(1):71–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.