

Abstract
Endoplasmic reticulum (ER) stress promotes tumor cell escape from immunosurveillance. However, the underlying mechanisms remain unknown. We hypothesized that ER stress induces hepatocellular carcinoma (HCC) cells to release exosomes, which attenuate antitumor immunity by modulating the expression of programmed death ligand 1 (PD-L1) on macrophages. In this study, we demonstrated that expression of several ER stress markers (glucose-regulated protein 78 [GRP78], activating transcription factor 6 [ATF6], PKR-like endoplasmic reticulum kinase [PERK], inositol-requiring enzyme 1α [IRE1α]) was up-regulated in HCC tissues and negatively correlated with the overall survival and clinicopathological scores in HCC patients. Expression of ER stress-related proteins positively correlated with cluster of differentiation 68-positive (CD68+) macrophage recruitment and PD-L1 expression in HCC tissues. High-throughput sequencing analysis identified microRNA (miRNA/miR)-23a-3p as one of the most abundant miRNAs in exosomes derived from the ER stress inducer tunicamycin treated HCC cells (Exo-TM). miR-23a-3p levels in HCC tissues negatively correlated with overall survival. Treatment with Exo-TM up-regulated the expression of PD-L1 in macrophages in vitro and in vivo. Bioinformatics analysis suggests that miR-23a-3p regulates PD-L1 expression through the phosphatase and tensin homolog (PTEN)- phosphoinositide-4,5-bisphosphate 3-kinase (PI3K)-protein kinase B (AKT) pathway. This notion was confirmed by in vitro transfection and co-culture experiments, which revealed that miR-23a-3p inhibited PTEN expression and subsequently elevated phosphorylated AKT and PD-L1 expression in macrophages. Finally, co-culture of T cells with Exo-TM-stimulated macrophages decreased CD8+ T-cell ratio and interleukin-2 production but increased T-cell apoptosis in vitro.
Conclusion:
ER-stressed HCC cells release exosomes to up-regulate PD-L1expression in macrophages, which subsequently inhibits T-cell function via an exosome miR-23a-PTEN-AKT pathway. Our findings provide a new insight into the mechanism how tumor cells escape from antitumor immunity.
Keywords: hepatocellular carcinoma, endoplasmic reticulum stress, exosome, microRNA, macrophage
Hepatocellular carcinoma (HCC) is a common malignant tumor with very poor overall survival worldwide.(1) Surgery is the most effective treatment strategy for HCC patients, but more than 80% of patients have lost the opportunity by the time they are diagnosed.(2) Due to insensitivity to current treatment strategies, the median overall survival of advanced-stage HCC patients is only 3 to 16 months.(3) Thus, more effective systemic therapies for HCC patients are urgently needed.
HCC is closely associated with immune tolerance and suppression(4); thus, immunotherapy may be a promising strategy against HCC. Recently, immunotherapy has been found to be effective in several types of human cancers, such as melanoma(5) and lung cancer,(6) but only obtained minimal beneficial effect in HCC. The majority of HCC is caused by chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infection, nonalcoholic fatty liver, and alcoholic liver disease.(7) It is generally believed that chronic inflammation contributes to hepatocarcinogenesis and HCC progression.(8,9) Recruitment of several types of immune cells to the tumor area has been correlated with poor outcome in HCC patients, including tumor-associated macrophages (TAMs),(10,11) cluster of differentiation 4-positive (CD4+) regulatory T cells,(12) and myeloid-derived suppressor cells (MDSCs).(13) Furthermore, many cytokines,(14) co-inhibitory signaling and immune inhibitory receptors,(15) and their ligands(15,16) also induce immunosuppression to promote HCC progression. In this setting, modulation of immune cell infiltration in the tumor microenvironment may represent a novel and effective strategy for the treatment of HCC. However, how HCC cells escape from immunosurveillance has not been fully elucidated, which is a critical barrier to developing effective immunotherapeutic intervention for the treatment of HCC.
Endoplasmic reticulum (ER) stress is critical for maintaining cell survival by activating the unfolded protein response (UPR), which is controlled by several ER stress-related proteins, including glucose-regulated protein 78 (GRP78), activating transcription factor 6 (ATF6), inositol essential enzyme 1α (IRE1α), and protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). Activation of ER stress has been reported in various types of tumors, including HCC, and plays an important role in promoting tumor progression.(17,18) We have shown that ER stress contributed to HCC chemotherapy and radiation resistance.(19,20) Other studies have also shown that ER stress promotes tumor cell escape from immunological surveillance. Moreover, activation of ER stress in immune cells has been shown to influence the function of infiltrating immune cells.(21,22) For example, ER stress has been reported to increase a variety of inflammatory factors, including interleukin (IL)-6 and IL-23 in macrophages.(23) ER-stressed tumor cells also impact immune cell functions by releasing ER stress-related molecules and subsequently facilitating tumor survival, progression, and metastasis.(24) Furthermore, cytokines secreted by infiltrating immune cells (predominantly TAMs, MDSCs, and dendritic cells [DC]) can also enhance ER stress and promote tumor cell growth.(25) These studies indicated that ER stress in tumor cells may not only involve intracellular events in controlling tumor cell proliferation but also have extracellular effects in regulating tumor microenvironment and immune responses. However, how ER-stressed tumor cells educate immune cells and dysregulate immune response remains unclear.
Exosomes are a class of 40- to 100-nm membrane vesicles that play an important role in cell-to-cell communication. Recently, microRNAs (miRNAs) were shown to be packaged in the purified exosomes, indicating that these vesicles may deliver genetic information to recipient cells.(26,27) For example, metastatic breast cancer cell-secreted exosomes transfer miR-105 to endothelial cells and promote tumor cell metastasis by destroying tight junctions and the integrity of endothelial monolayers.(28) Furthermore, exosomes derived from HCC cells can transfer specific miRNAs to enhance recipient cell growth.(29) These results suggest that miRNAs are vital molecules in exosome-mediated intercellular communication. However, few studies have examined whether ER stress affects HCC exosomal-miRNA transfer and how these exosomal-miRNAs affect tumor progression. Thus, the purpose of the current study was to investigate whether ER-stressed HCC cells can transmit miRNA-enriched exosomes to macrophages and how these exosomal-miRNAs modulate the immunologic functions of macrophages. To answer these questions, we performed high-throughput sequencing, in vitro and in vivo analyses, and found that exosomes derived from ER-stressed HCC cells (Exo-TM) increased the expression of programmed death ligand 1 (PD-L1) and inflammatory cytokines in macrophages, thereby decreasing CD8+ T-cell ratio and increasing T-cell apoptosis. Mechanistically, Exo-TM up-regulates PD-L1 expression by the transfer of miR-23a-3p, which inhibits phosphatase and tensin homolog (PTEN) and subsequently activates protein kinase B (AKT). Our data suggest that ER stress promotes HCC immune escape by transferring specific miRNAs to infiltrated macrophages in tumor microenvironment. Thus, interference of HCC-macrophage crosstalk may be an effective strategy for the treatment of HCC.
Materials and methods
Patients and tissue samples.
The present protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the ethics committee of the First Affiliated Hospital of Anhui Medical University. All enrolled patients provided informed written consent. Primary HCC tumor tissues were obtained from 169 HCC patients who were hospitalized in the First Affiliated Hospital of Anhui Medical University between 2004 and 2010. The Edmondson-Steiner grading system was employed to define the histologic grade of tumor differentiation. Tumors were categorized based on the World Health Organization (WHO) classification and the International Union Against Cancer tumor-node-metastasis (TNM) classification system. The HCC tumor tissues were formalin-fixed and paraffin-embedded for histopathological diagnosis and construction of paraffin-embedded tissue microarrays. Freshly resected HCC tissues, paired liver tissues, and healthy donor peripheral blood were obtained in the First Affiliated Hospital of Anhui Medical University between August 2017 and October 2018.
Definition of ER stress high and low patients.
For analysis, the levels of ER stress-related proteins, the percentage of positive cells (0 for negative, 1 for ≤10%, 2 for 11%−50%, 3 for 51%−75%, 4 for >75%), and their staining intensity (0 for negative staining, 1 for light yellow, 2 for claybank, 3 for brown) were analyzed in five random fields of each sample. The expression of ER stress-related proteins was qualitatively scored by the percentages of positive cells multiplied by staining intensity, and the values less than 5 and greater than or equal to 5 indicated low and high levels of GRP78, respectively, whereas values less than 3 and greater than or equal to 3 were defined as low and high levels for ATF6, PERK, and IRE1α, respectively.
Exosome co-culture with macrophages in vitro.
THP-1 monocytes were differentiated into macrophages by incubation with 100 nM of phorbol 12-myristate 13-acetate (PMA) for 48 hours and were thereafter named mTHP-1 cells. RAW264.7 cells and mTHP-1 cells (1×106/well) were plated in 12-well plates and co-cultured with 10 μg/mL of Exo-con or Exo-TM. After co-incubation for 24 hours, cells were collected for flow cytometry, western blot, and quantitative polymerase chain reaction (qPCR) analysis, and the supernatants were used for cytokine measurement.
In vivo assay.
Exosomes were injected intravenously through the tail vein into 6-week-old female nude mice once every other day for 10 times. Peritoneal macrophages were isolated from euthanized mice and allowed to attach at 37°C for 2 hours. The unattached cells were removed and washed with phosphate-buffered saline (PBS) twice; the remaining cells were then incubated overnight. PD-L1 levels were analyzed by flow cytometry and immunohistochemistry (Clone 10F.9G2, BioLegend) and qPCR. Cytokines secreted by peritoneal macrophages were measured by a mouse Cytometric Bead Array (CBA) inflammation kit. Four mice were used per group in the animal experiments.
Statistical Analysis.
SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA) was employed for statistical analysis. Two-tailed Student t test or the Wilcoxon-Mann-Whitney test was applied to evaluate the difference between two groups, and 1-way analysis of variance (ANOVA) multiple comparisons were employed to compare three or more groups. Associations between ER stress-related proteins and PD-L1 expression or miR-23a-3p and overall survival were assessed using Pearson correlation analysis. Representative images and flow cytometric graphs from three independent experiments are shown. Values are presented as the mean ± standard deviation (SD). Differences were considered to be statistically significance when P was less than 0.05.
For more detailed materials and methods information, see Supporting Data.
Results
ER stress is up-regulated and positively correlates with poor survival in HCC patients
To evaluate the role of ER stress in HCC progression, we first tested whether ER stress is up-regulated in HCC patients by measuring the expression of ER stress-related proteins in 169 cases of surgically resected HCC tissues by immunohistochemistry. These ER stress-related proteins include GRP78, ATF6, IRE1α, and PERK. Representative images of low and high expression of GRP78, PERK, ATF6, and IRE1α proteins are shown in Fig. 1A, in which these ER stress-related proteins were predominantly detected in the cytoplasm. Expression of these ER stress-related proteins was much stronger in tumor tissues compared with paired paracarcinoma tissues (65.68% versus 41.42% for GRP78; 63.91% versus 37.87% for ATF6; 60.36% versus 46.75% for PERK; 60.95% versus 43.79% for IRE1α). Based on the percentage of ER stress-related protein positive cells and staining density, we divided patients into two groups with low and high ER stress (see details in Methods). Moreover, we performed western blot analyses to confirm the ER stress-related protein expression in 5 freshly resected HCC tissues, and found that the expression of these ER stress-related proteins was much higher in tumor tissues compared to paracarcinoma tissues (Fig. 1B). We also examined the levels of GRP78, PERK, ATF6, and IRE1α messenger RNAs (mRNAs) in HCC tissues and found that the expression of these genes was significantly up-regulated in tumor tissues compared with paired paracarcinoma tissues (Fig. 1C).The clinicopathological characteristics of HCC patients are shown in Supporting Tables S1-S4, showing that the high ER stress-related patients had larger tumor size and poorly differentiated tumors compared to the low ER stress patients. Finally, the association of GRP78, ATF6, PERK, or IRE1α expression with overall survival of 55 cases of HCC patients was analyzed. As illustrated in Fig. 2, the overall survival of low ER stress patients was significantly longer than the high ER stress patients.
FIG. 1. ER stress is activated in human HCC.
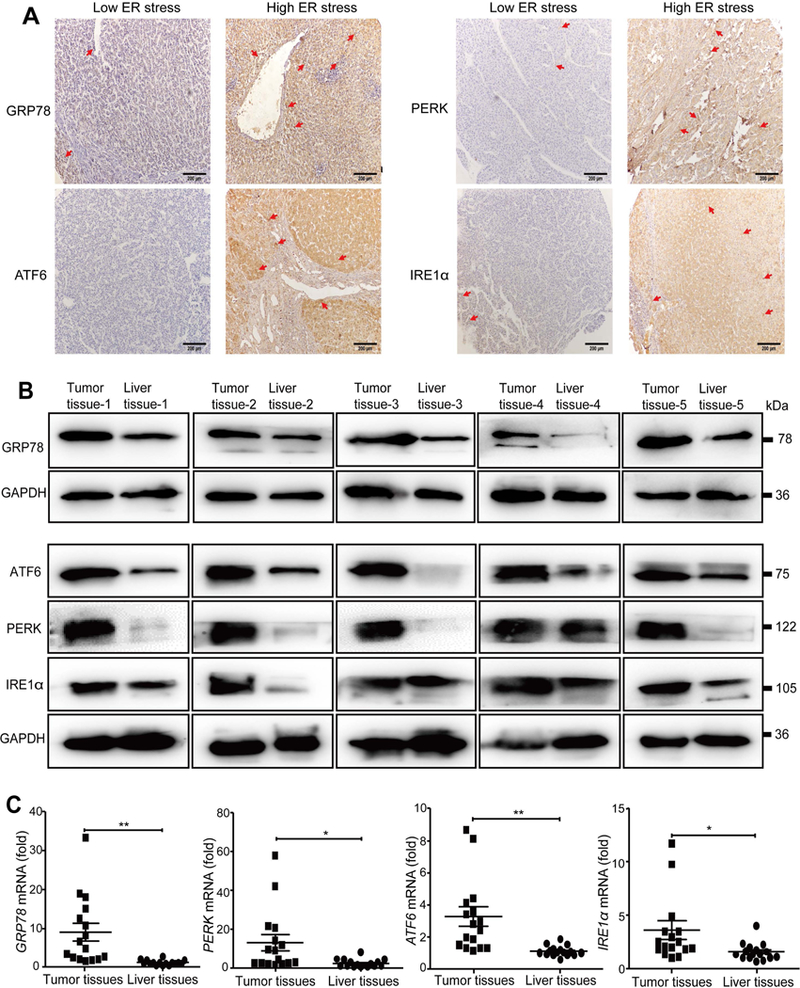
(A) Representative low and high expression of ER stress markers GRP78, PERK, ATF6, and IRE1α in HCC tissue samples (scale bar = 200 μm). (B) Representative western blot analyses of GRP78, PERK, ATF6, and IRE1α protein expression in human HCC tissues. (C) qPCRdetected the mRNA levels of GRP78, PERK, ATF6, and IRE1α in 16 cases of freshly resected human HCC tissues. *P < 0.05, **P < 0.01.
FIG. 2. Activation of ER stress correlates with poor survival in human HCC.
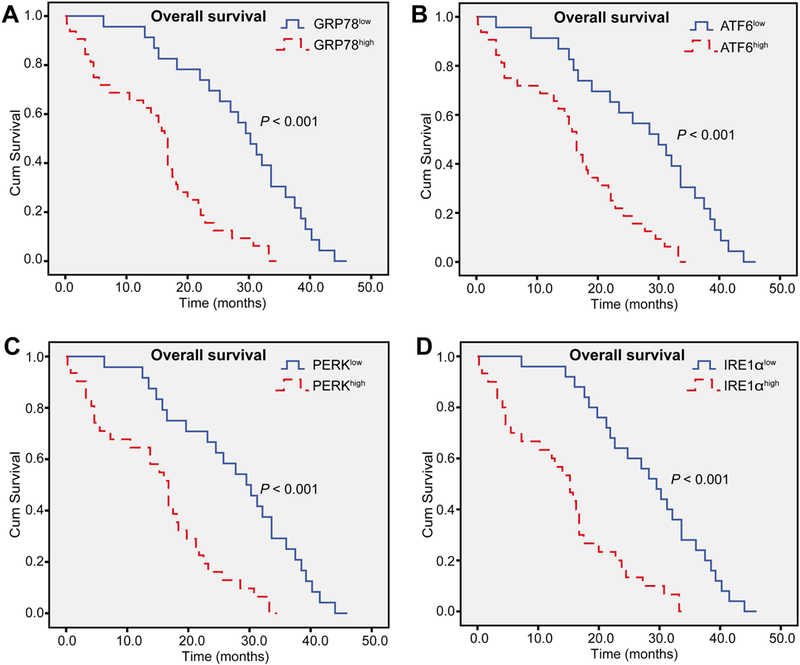
Kaplan-Meier curves demonstrated that the overall survival in patients with overexpression of (A) GRP78, (B) ATF6, (C) PERK, and (D) IRE1α was significantly shorter than those with low expression levels (n = 55). Log-rank test was used to evaluate statistical significance. **P < 0.01 as indicated.
Hepatic ER stress-related protein levels correlate with hepatic macrophage infiltration and PD-L1 expression in HCC patients
ER stress has been shown to play an important role in regulating immune cell functions in tumor microenvironment and antitumor immune response.(24,30) Thus, we tested whether expression of ER stress-related proteins such as GRP78 correlates with macrophage infiltration in HCC patients. As illustrated in Fig. 3A, tumor tissues that expressed a higher level of GRP78 (GRP78high) had massive CD68 staining in the stroma, whereas CD68+ macrophage infiltration was much lower in tumor tissues that expressed a low level of GRP78 (GRP78low). Quantitative analysis revealed that CD68+ areas in GRP78high tumor stroma were significantly higher than those in the GRP78low stroma (Fig. 3A).
FIG. 3. ER stress is associated with macrophage infiltration and PD-L1 expression in HCC patients.
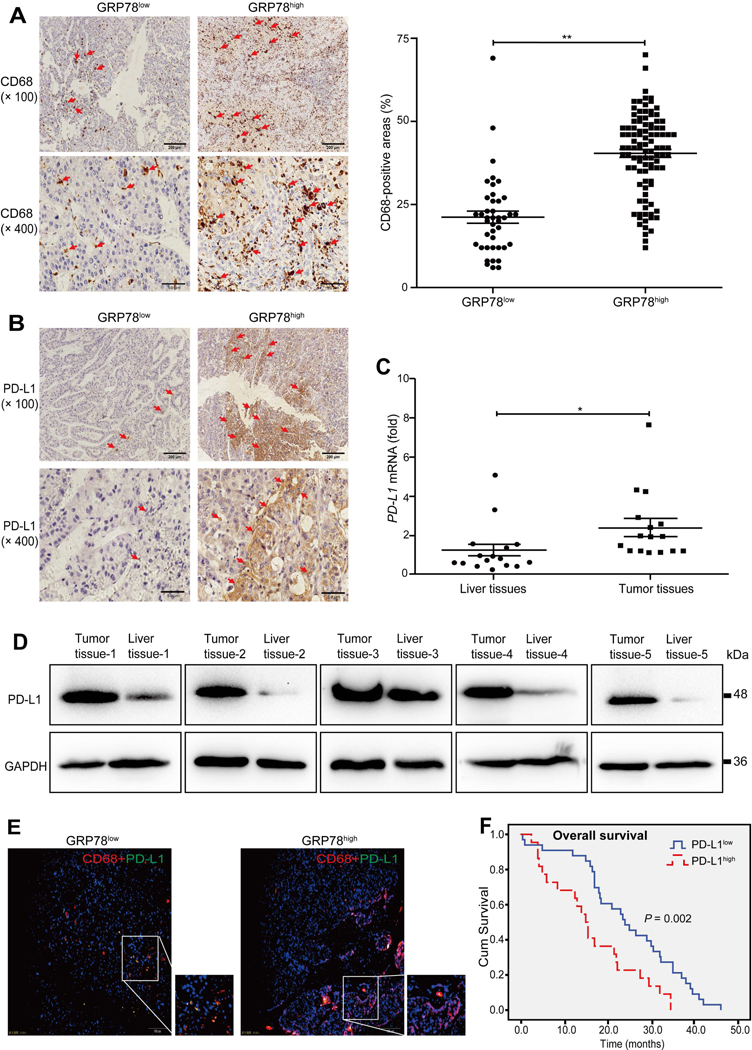
(A) Representative images of the expression pattern of CD68 protein in HCC tissues with GRP78high and GRP78low groups (scale bar = 200 μm for the upper panel; scale bar = 50 μm for the lower panel). Quantitative analysis of CD68-positive areas in the GRP78high and GRP78low group is shown in the right panel. (B) Representative images of the expression pattern of PD-L1 protein in HCC tissues with GRP78high and GRP78low groups (scale bar = 200 μm for the upper panel; scale bar = 50 μm for the lower panel). (C) qPCR analyses of PD-L1 mRNA levels in 16 cases of freshly resected HCC tissues. (D) Western blot analyses of PD-L1 protein levels in human HCC tissues. (E) The expression pattern of PD-L1 on CD68+ cells in GRP78low and GRP78high HCC tissues was measured by immunofluorescence (scale bar = 100 μm). (F) Log-rank test evaluated overall survival and PD-L1 expression in 55 HCC patients. Data are presented as the means ± SD (error bar). *P < 0.05, **P < 0.01 as indicated.
PD-L1 was reported to inhibit T-cell activation and contribute to tumor immune evasion.(31) To examine whether ER stress activation correlates with PD-L1 expression, we measured PD-L1 levels by immunohistochemistry analysis and found in GRP78high tumors, PD-L1 was often distributed in a crumby manner, whereas it sporadically existed in GRP78low tumors (Fig. 3B). Quantitative analysis showed that 51.35% of the GRP78high tumors expressed PD-L1, and only 27.59% of the GRP78low tumors expressed PD-L1, and PD-L1 expression was positively correlated with GRP78 levels (P = 0.003) (Supporting Table S5). Consistently, the expression levels of ATF6 (54.63% versus 22.95%; P < 0.001), PERK (54.90% versus 25.37%; P < 0.001), and IRE1α (55.34% versus 24.24%; P < 0.001) were also positively correlated with PD-L1 expression (Supporting Tables S6-S8). qPCR and western blot analyses showed that PD-L1 mRNA and protein were markedly up-regulated in tumor tissues compared with paired paracarcinoma tissues (Fig. 3C,D).
To test whether infiltrated macrophages expressed PD-L1, we performed immunofluorescence double-staining and found PD-L1 was frequently co-localized with CD68+ macrophages in GRP78high HCC tumor stroma (Fig. 3E). Finally, Kaplan-Meier analysis showed that patients with lower PD-L1 expression had better survival time than those with higher PD-L1 expression (24.9 ± 2.0 months versus 16.4 ± 2.2 months; P = 0.002) (Fig. 3F). Together, these results suggest that ER stress activation is associated with higher macrophage PD-L1 expression and poor prognosis in HCC patients.
Exosomes from ER-stressed HCC cells up-regulate PD-L1 expression on macrophages in vitro
The above data revealed that high ER stress status is associated with up-regulated PD-L1 expression in macrophages on HCC patients, suggesting ER-stressed HCCs may promote PD-L1 expression in macrophages. Because exosomes are an important communicator between different cell types, we hypothesized that ER-stressed HCC cells may release exosomes and subsequently up-regulate PD-L1 expression on macrophages. To test this hypothesis, we first optimized the in vitro cell culture system to mimic the ER stress status by tunicamycin (TM) treatment and found that treatment with TM (2.5 μmol/L) for 24 hours generated significant ER stress in several HCC cell lines, including HepG2, Hep3B, Hepa1–6, and A549 cells (Supporting Fig. S1A-C). Second, exosomes derived from tunicamycin (TM) treated HCC cells (defined as Exo-TM) or without TM (defined as Exo-con). The purity of these exosomes was confirmed by transmission electron microscopy showing a homogeneous population of rounded membrane-bound vesicles with a diameter ranging from 30 to 100 nm (Supporting Fig. S2A,B) and by western blot analysis showing the expression of two exosomal-marker proteins CD63 and tumor susceptibility gene (TSG)-101 (Supporting Fig. S2C). Additionally, ER-stressed HCC cells secreted a higher number of exosomes compared with those without ER stress (Supporting Fig. S2D).
Third, to investigate whether HCC cells transmit ER stress signals to macrophages through exosomes, we performed a co-culture experiment and found that co-culture with Exo-TM derived from HepG2 cells or normal HL-7702 hepatocytes significantly up-regulated the expression of GRP78, ATF6, PERK, or IRE1α proteins compared to co-culture with Exo-con (Supporting Fig. S3A,B). Moreover, co-culture with Exo-con or Exo-TM increased the proliferation of HepG2 cells (Supporting Fig. S3C).
Fourth, we co-cultured macrophages with PKH67-labeled exosomes and found that mTHP-1 cells effectively engulfed exosomes (Fig. 4A). Moreover, flow cytometric analysis showed that co-culture with HepG2-derived Exo-TM significantly up-regulated PD-L1 expression in mTHP-1 cells compared with those in the Exo-con group (Fig. 4B). Such up-regulation of PD-L1 in macrophages by Exo-TM was also confirmed by western blot, immunohistochemistry, and qPCR analyses (Fig. 4C-E). Similarly, Hep3B-derived Exo-TM also significantly up-regulated PD-L1 levels in co-cultured macrophages (Supporting Fig. S4A-D). However, Exo-TM from normal HL-7702 hepatocytes or primary human hepatocytes did not up-regulate PD-L1 expression in mTHP-1 cells compared to the Exo-con group (Supporting Fig. S4E-G). Finally, HepG2-dervied Exo-TM markedly up-regulated PD-L1 expression on mouse RAW264.7 macrophages (Supporting Fig. S5A,B), whereas Hep3B-derived Exo-TM only slightly increased PD-L1 expression in RAW264.7 cells (Supporting Fig. S5C,D).
FIG. 4. Exo-TM increases macrophage PD-L1 expression in vitro.
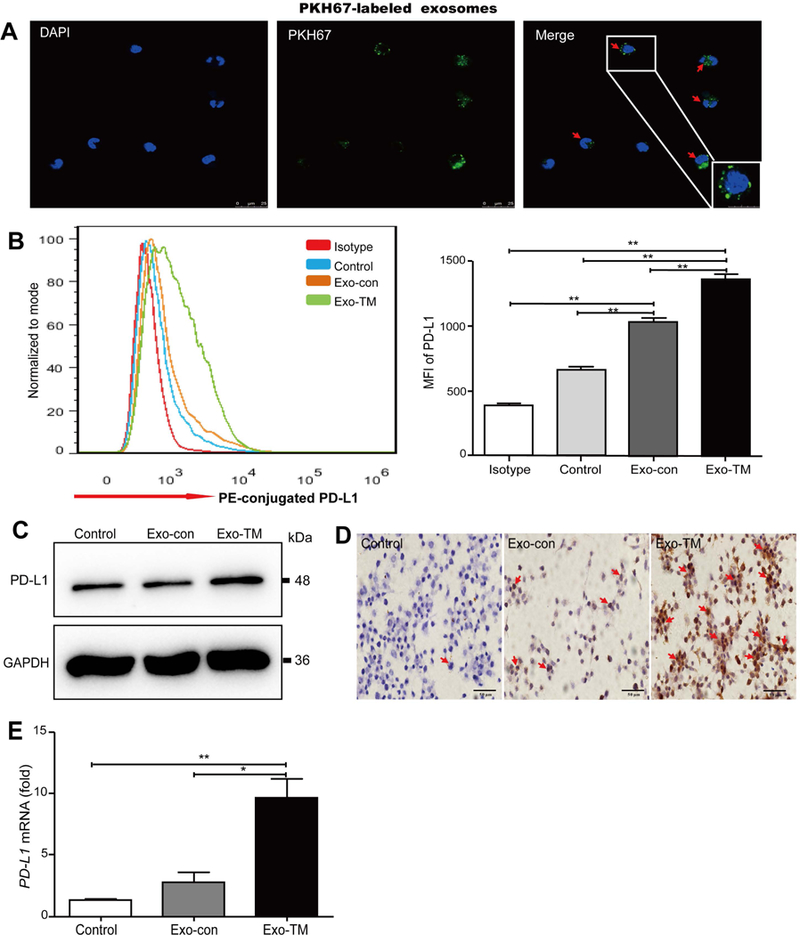
(A) PKH67-labeled exosomes were coincubated with mTHP-1 cells and examined by confocal microscopy (scale bar = 25 μm). Representative images are shown. Red arrows indicate PKH67-labeled exosomes. (B) Macrophages were coincubated with HepG2-derived Exo-con and Exo-TM, the protein levels of PD-L1 were determined by flow cytometry, and the mean fluorescence intensity (MFI) was statistically analyzed. Moreover, the impact of Exo-TM on macrophage PD-L1 protein expression was also determined by (C) western blot and (D) immunohistochemical analysis. PD-L1 mRNA was detected by (E) qPCR. Data are shown as the means ± SD of at least three independent experiments. *P < 0.05, **P < 0.01 with indicated groups.
To rule out whether Exo-TM-induced ER stress in macrophages was a consequence of TM contamination, we performed the following experiments. First, liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) confirmed that no TM existed in Exo-con, whereas Exo-TM contained approximately 20.25 ng/mL of TM (Supporting Fig. S6A). As the exosome used in the current study was diluted 10 times, the final concentration of TM contained in Exo-TM was very low (~2 ng/mL). Second, treatment with up to 50 ng/mL of TM did not up-regulate GRP78 levels in mTHP-1 (Supporting Fig. S6B). Finally, coincubation with TM did not affect PD-L1 expression on mTHP-1 cells (Supporting Fig. S6C). Collectively, Exo-TM-mediated up-regulation of PD-L1 in macrophages is not due to TM contamination.
Exo-TM up-regulates PD-L1 expression on macrophages in vivo
To determine whether Exo-TM also enhances PD-L1 expression in vivo, we injected nude mice with Exo-TM and Exo-con once every 2 days for 10 times, and the peritoneal macrophages were isolated 12 hours after the last injection (Supporting Fig. S7). Confocal microscopy and flow cytometric analyses confirmed that PKH67-labeled exosomes were effectively engulfed by the peritoneal macrophages in vivo (Fig. 5A,B). As illustrated in Fig. 5C-F, flow cytometric, immunohistochemistry, and qPCR analyses demonstrated that peritoneal macrophages isolated from Exo-TM-injected mice had significantly higher levels of PD-L1 compared with the Exo-con and PBS group. In addition, these macrophages also had higher levels of several inflammatory mediators compared to those from the Exo-con or PBS group (Supporting Fig. S8).
FIG. 5. Exo-TM up-regulates the expression of PD-L1 in vivo.
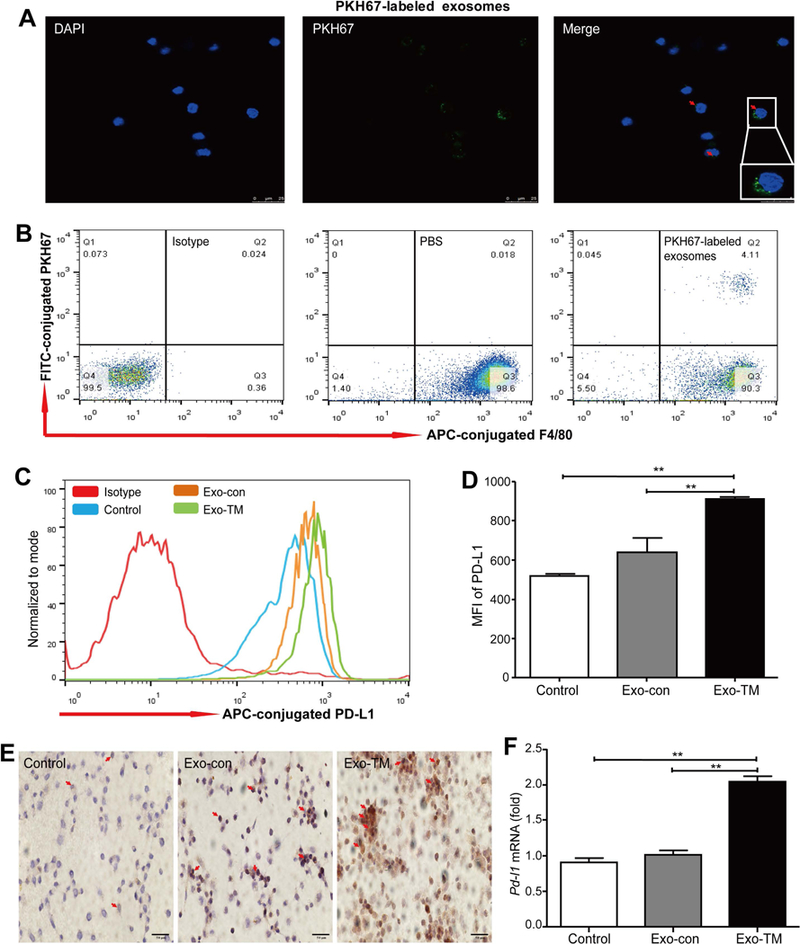
PKH67-labeled exosomes were injected to nude mice intravenously through the tail vein, and the incorporation of PKH67-labeled exosomes by peritoneal macrophages was detected by (A) confocal microscopy (scale bar = 25 μm) and (B) flow cytometric analysis, peritoneal macrophages were isolated from mice treated with PBS, Exo-con, and Exo-TM, and the expression of PD-L1 was evaluated by (C,D) flow cytometric analysis, (E) immunohistochemical assay, and (F) qPCR. Data are shown as the means ± SD (error bar) of at least three independent experiments. *P < 0.05, **P < 0.01 with indicated groups.
Exo-TM-treated macrophages decrease CD8+ T-cell ratio and promote T-cell apoptosis
Macrophage (predominantly M2 macrophage) infiltration in tumor microenvironment has been reported to promote tumor progression by impairing the immune responses of cytotoxic CD8+ T cells.(32) We wondered whether ER-stressed HCC cells also polarized macrophages to M2 phenotype to promote HCC progression by impairing the immune responses of cytotoxic CD8+ T cells. To test this hypothesis, we first examined whether Exo-TM induces macrophage polarization and found co-culture with HepG2-derived Exo-TM markedly increased levels of M2 marker (such CD206) and M2 cytokines (IL-10) in macrophages compared with Exo-con treatment (Supporting Figs. S9-S10). Together, these results suggest that ER stress-related exosomes promote macrophages toward M2 phenotype.
Then, we examined the effects of Exo-TM-treated macrophages on T cells. As illustrated in Fig. 6A-C, Exo-TM-treated macrophages significantly decreased CD8+ T-cell proportion and attenuated IL-2 production in T cells compared with Exo-con-treated macrophages. Moreover, we examined the impact of Exo-TM-treated macrophages on T-cell apoptosis using Annexin V /PI apoptosis detection kit and found that the ratio of apoptotic T cells was significantly higher after incubation with Exo-TM-treated macrophages compared to that with Exo-con-treated macrophages (Fig. 6D,E).
FIG. 6. Exo-TM-treated macrophages inhibit T-cell function and induce T-cell apoptosis.
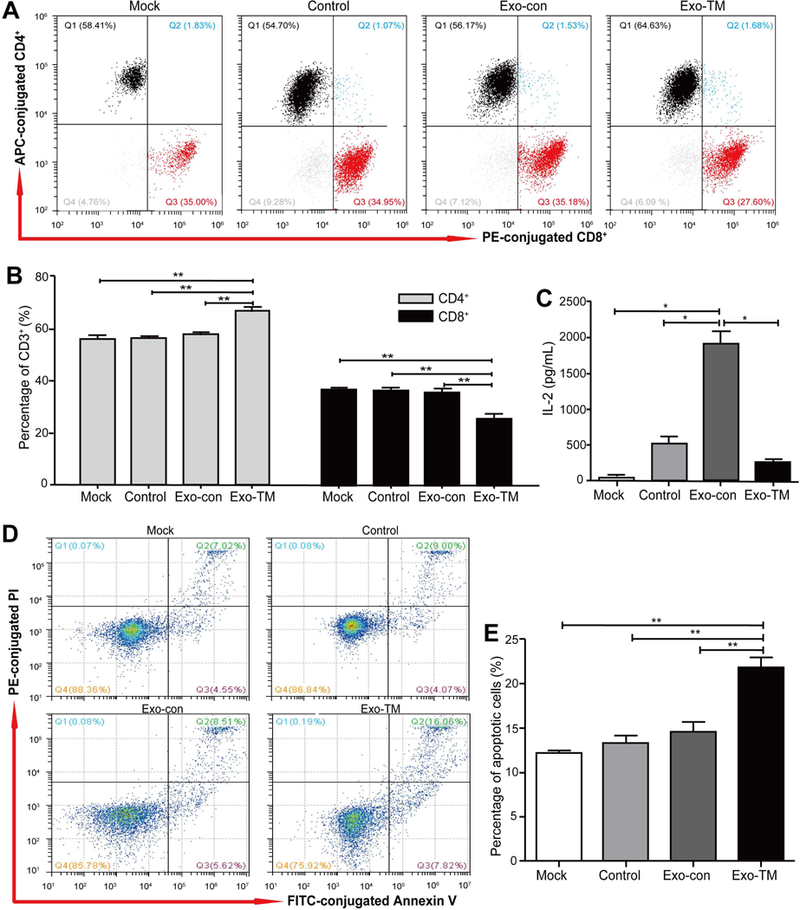
Macrophages were coincubated with Exo-con and Exo-TM for 48 hours, then washed with exosome-free medium, followed by coincubating with CD3+ T cells at a ratio of 1:20 for another 48 hours. (A) CD4+ and CD8+ T cells were measured by flow cytometry and statistically analyzed by (B) SPSS 16.0. (C) The level of IL-2 secreted by T cells was determined by a Cytometric Bead Array (CBA) inflammatory factor kit. (D) Annexin V /PI apoptosis detection kit was used to detect the ratio of apoptotic T cells and statistically analyzed by (E) SPSS 16.0. Data are shown as means ± SD (error bar) of at least three independent experiments. *P < 0.05, **P < 0.01 with indicated groups. Mock= T cells; Con= macrophages + T cells; Exo-con= Exo-con-treated macrophages + T cells; Exo-TM= Exo-TM-treated macrophages + T cells.
ER-stressed HCC-derived exosomes contain high levels of miR-23a-3p, which up-regulates PD-L1 expression in macrophages by regulating the PTEN/AKT pathway
To determine whether ER-stressed HCC cells transmit specific miRNAs to macrophages and impact its immunologic functions, high-throughput sequencing analyses of exosomes were performed. As illustrated in Fig. 7A, 14 miRNAs were differentially expressed between Exo-TM and Exo-con. In comparison with Exo-con, six miRNAs (miR-486–5p, miR-486–3p, miR-1–3p, miR-433–3p, miR-432–5p, and miR-92b-5p) were down-regulated, and eight miRNAs (miR-29a-3p, miR-378a-3p, miR-193b-3p, miR-20a-5p, miR-23a-3p, miR-27a-3p, miR-184, and miR-16–5p) were up-regulated in Exo-TM (Supporting Table S9).
FIG. 7. miR-23a-3p up-regulates PD-L1 expression in macrophages.
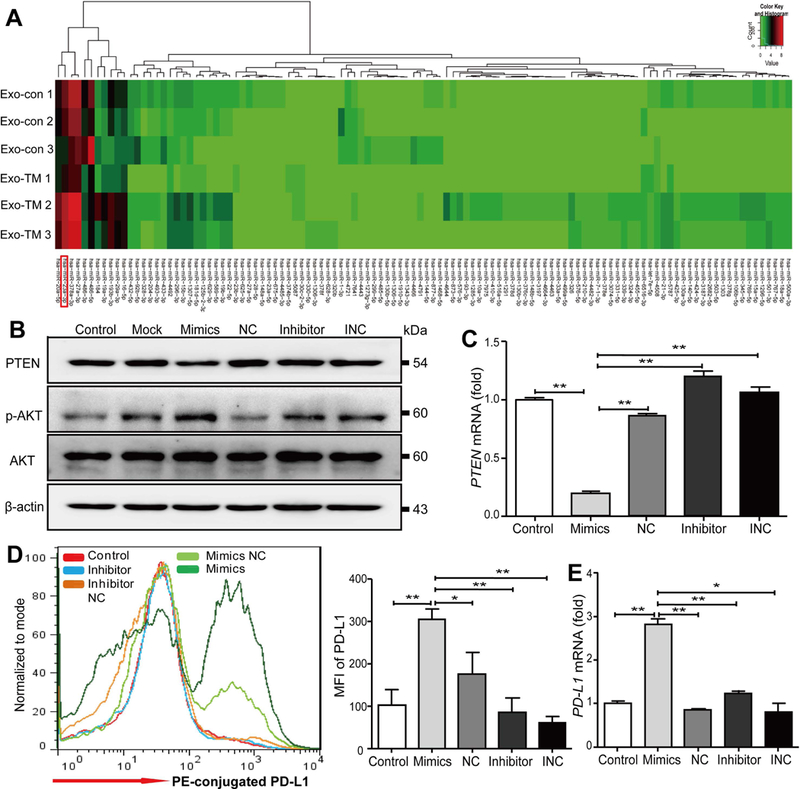
(A) The heat map of miRNAs expressed in Exo-con and Exo-TM. (B-D) Macrophages were transfected with miR-23a-3p mimics, inhibitors, and their corresponding controls (NC, INC) for 48 hours. (B) Western blot and (C) qPCR assay were used to analyze the protein and mRNA levels of PTEN, respectively. The expression of PD-L1 on macrophages following miR-23a-3p transfection was analyzed by (D) flow cytometry and (E) qPCR. Data are shown as the means ± SD of at least three independent experiments. *P < 0.05, **P < 0.01 with indicated groups. NC=Negative control; INC= Inhibitor negative control.
Subsequently, bioinformatics analysis revealed that miR-29a-3p, miR-23a-3p, miR-486–3p, and miR-486–5p were predicted to regulate PD-L1 expression through the PTEN-phosphatidylinositol 3-kinase (PI3K)/AKT pathway (Supporting Fig. S11A). Moreover, qPCR analysis (plus cel-miR-39–3p as external control) demonstrated that miR-23a-3p had the highest fold induction (2.44-fold) in Exo-TM, followed by miR-29a-3p with a 1.95-fold induction; in contrast, miR-486–5p expression was down-regulated by 1.96-fold, and miR-486–3p had no changes (Supporting Fig. S11B). Furthermore, we explored the relationship between miR-23a-3p levels and survival times in 55 cases of HCC patients and found that miR-23a-3p levels were negatively correlated with overall survival (r = −0.306; P = 0.023) (Supporting Table S10).
According to TargetScan, microRNA Database (miRDB), and miRanda, we found PTEN is a putative target of miR-23a-3p, and this notion was confirmed by a dual luciferase reporter gene assay (Supporting Fig. S11C,D). Western blot analysis shows that transfection of miR-23a-3p mimics down-regulated PTEN expression in macrophages (Supporting Fig. S11E). PTEN is known to regulate PD-L1 expression(33,34); therefore we hypothesized that miR-23a-3p may up-regulate PD-L1 expression through the down-regulation of PTEN expression in macrophages. To confirm whether miR-23a-3p regulates PD-L1 expression by modulating the PTEN pathway in macrophages, mTHP-1 cells were transfected with miR-23a-3p mimics, inhibitors, and their relative controls for 48 hours, and we found miR-23a-3p mimics significantly down-regulated PTEN at both protein and mRNA levels (Fig. 7B,C) and up-regulated phosphorylated-AKT (p-AKT) protein expression (Fig. 7B). Moreover, miR-23a-3p mimics significantly up-regulated PD-L1 expression on macrophages, whereas the miR-23a-3p inhibitor and control had no influence on PD-L1 protein and mRNA expression (Fig. 7D,E). However, transfection of macrophages with either miR-23a-3p mimics or inhibitors did not influence the expression of IL-6, IL-10, and tumor necrosis factor-α (TNF-α) (data not shown).
To further confirm whether miR-23a-3p regulates PD-L1 expression through the activation of the PTEN/AKT pathway, we first co-cultured macrophages with Exo-TM or Exo-con and found that, compared to Exo-con, Exo-TM significantly down-regulated PTEN levels at both protein and mRNA levels (Fig. 8A,B) and up-regulated phosphorylated-AKT (p-AKT) protein expression (Fig. 8A). Moreover, qPCR analysis demonstrated that Exo-TM significantly increased miR-23a-3p expression in coincubated macrophages compared to Exo-con (Fig. 8C). Transfection of macrophages with miR-23a-3p inhibitors abolished Exo-TM-mediated PTEN down-regulation and p-AKT up-regulation (Fig. 8D). Next, we knocked down PTEN using RNA interference (RNAi) and found PTEN significantly reduced at both protein and mRNA levels, whereas p-AKT protein expression was up-regulated (Supporting Fig. S12A,B). Furthermore, knockdown of PTEN significantly up-regulated PD-L1 protein and mRNA expression on macrophages (Fig. 8E,F). However, knockdown of PTEN had no impact on expression of inflammatory cytokines, such as IL-6, IL-10, and TNF-α (data not shown).
FIG. 8. Exo-TM up-regulates expression of PD-L1 in macrophages through the inhibition of PTEN and subsequent activation of AKT pathway.
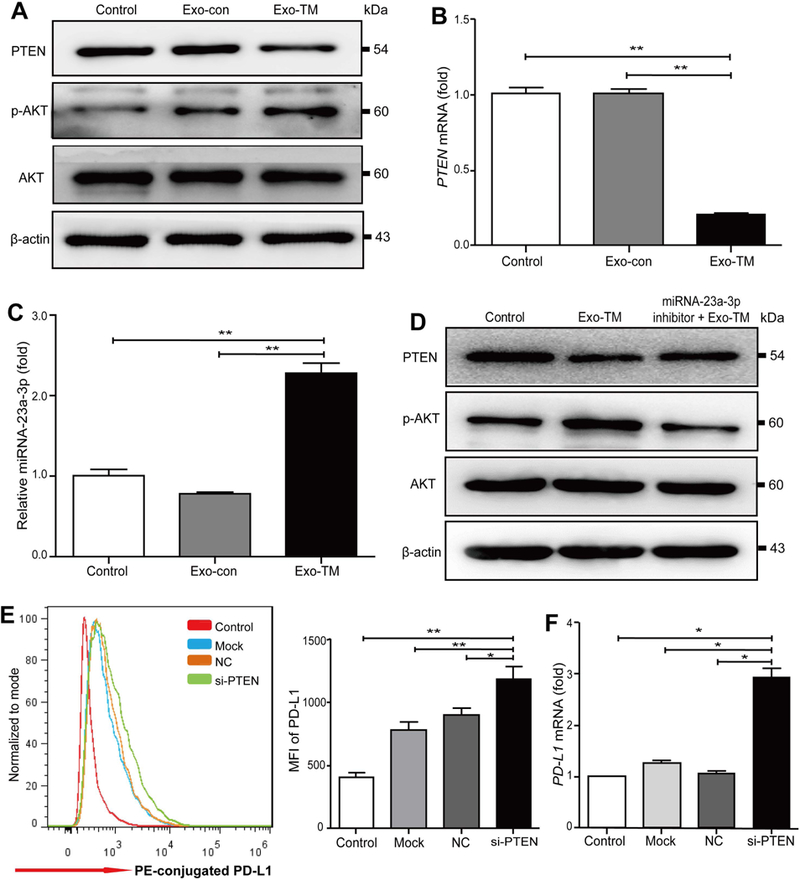
mTHP-1 cells were incubated with Exo-TM or Exo-Con, and the expression of PTEN and AKT proteins (A), PTEN mRNA (B), miR-23a-3p (C) levels in mTHP-1 were determined. (D) Transfection of mTHP-1 cells with miR-23a-3p inhibitor significantly reversed Exo-TM-mediated down-regulation of PTEN and up-regulation of p-AKT proteins. (E,F) PTEN in mTHP cells was knocked down by transfection of PTEN siRNA (si-PTEN). PD-L1 protein and mRNA expression on mTHP-1 cells were measured by flow cytometry and qPCR, respectively. Data are presented as the means ± SD of at least three independent experiments. *P < 0.05, **P < 0.01 as indicated. Abbreviation: siRNA, small interfering RNA.
Discussion
Multiple studies have shown that ER stress can dysregulate antitumor immunity through various mechanisms,(17,21) including stimulating protumor inflammatory factors(35) and inducing MDSCs.(23) In the current study, we identified a novel mechanism by which ER stress promotes HCC cells to release miR-23a-3p-enriched exosomes which up-regulate PD-L1 expression on macrophages and subsequently suppress T-cell functions. We found that most of human HCC samples highly express ER stress-related proteins and genes, and their expression levels negatively correlated with patients’ overall survival. Furthermore, GRP78high tumors had more CD68+ macrophages and PD-L1+ cells in the stroma, and PD-L1 expression also correlated with poor overall survival in HCC patients. Finally, ER-stressed HCC cells up-regulated macrophage PD-L1 expression and consequently impaired T-cell function through the transmission of exosomal-miR-23a-3p, which inhibits PTEN and consequently activates the AKT pathway, resulting in elevation of PD-L1 expression. Together, our results suggest that HCC cells can transmit ER stress signals to infiltrated macrophages through miR-23a-3p-enriched exosomes and promote tumor progression, which is summarized in Supporting Fig. S13.
ER stress promotes HCC cells to release exosomes.
ER stress has been reported to promote tumor cell immune escaping by constructing an immunosuppressive microenvironment, but the precise mechanism remains unclear. Exosomes have been reported to play an important role in intercellular communication by delivering their contents (such as miRNAs) to the “recipient” cells.(28,36,37) However, whether ER-stressed HCC cells influence the function of immune cells by transmitting exosomes and whether this immunomodulation is mediated by specific exosomal miRNAs is not clear. Recently, Kanemoto et al. reported that ER stress promoted exosome release in IRE1α and PERK-dependent manners.(38) Moreover, ER stress increased the levels of cytochrome P450 2E1 (CYP2E1) and other P450 isoforms in exosomes of alcohol-exposed rodents and patients with alcoholism.(39) Our results demonstrated that ER stress significantly increased HCC cell exosome secretion, and exosome RNA sequencing found that 14 miRNAs were differentially packaged in Exo-TM, and bioinformatics analysis revealed these miRNAs are closely related to several cancer-associated pathways, such as the PTEN-PI3K-AKT pathway. Moreover, these exosomes were effectively transferred into macrophages and transmitted ER stress signals to macrophages, suggesting that ER stress influences HCC cell exosome release and transmits miRNAs-enriched exosomes to macrophages that resided in the tumor microenvironment.
ER stress-related exosomes modulate PD-L1 expression through the PTEN-AKT pathway.
Clinical and preclinical evidence showed that loss of PTEN was associated with enhanced PD-L1 expression in human glioma,(40) breast cancer,(41) and colorectal cancer.(42) A recent study revealed that PTEN mutation resulted in an increase of PD-L1 protein expression through the activation of the PI3K-AKT- mammalian rapamycin target protein (mTOR)- ribosomal S6 kinase 1 (S6K1) pathway.(40) In the present study, we found that miR-184 is the most up-regulated miRNA in Exo-TM, however, bioinformatics analysis revealed miR-184 was not associated with PD-L1 expression while other miRNAs including miR-23a-3p, miR-29a-3p, miR-486–3p and miR-486–5p did. Moreover, qPCR assays demonstrated that miR-23a-3p was the most significantly up-regulated miRNA packaged in Exo-TM, and we provided several lines of evidence suggesting that miR-23a-3p up-regulates PD-L1 expression through the inhibition of PTEN. First, miR-23a-3p was elevated in ER stress-related exosomes, and Exo-TM significantly reduced PTEN expression but augmented PD-L1 expression in macrophages. Second, bioinformatics analysis and dual luciferase reporter assay revealed that miR-23a-3p specifically targeted PTEN.(33,34) Third, overexpression of miR-23a-3p in macrophages inhibited PTEN expression but significantly up-regulated PD-L1 expression. Finally, knockdown of PTEN elevated PD-L1 expression in macrophages. Taken together, our results suggest that ER-stressed HCC cells down-regulate PTEN in macrophages by transmitting exosomal-miR-23a-3p to macrophages and subsequently increasing PD-L1 expression to promote immune escaping.
Macrophage expressed PD-L1 influences T-cell functions.
Macrophages are the prominent components in tumor stroma, and high macrophage density was associated with unfavorable prognosis in many types of tumors.(35) Several studies have reported that macrophages directly communicate with tumor cells, thereby promoting tumor progression.(43) We found that GRP78high HCC tissues recruited more CD68+ macrophages in the stroma, and in vitro studies demonstrated that Exo-TM polarized macrophages to M2 phenotype. Moreover, there is evidence suggesting that cancer cells promote macrophage production of inflammatory mediators, such as IL-6 and TNF-α, to facilitate tumor metastasis.(44,45) Here, we found Exo-TM not only increased macrophage IL-6 and TNF-α expression but also up-regulated protumor factor IL-10 expression, suggesting that ER-stressed HCC cells may “educate” macrophages toward TAMs to facilitate disease progression.
PD-L1, also called B7-H1 or CD274, inhibits T-cell proliferation and promotes T-cell dysfunction by binding to programmed death 1 (PD-1).(31) PD-L1 is expressed by both hematopoietic cells and nonhematopoietic cells, and up-regulation of PD-L1 has been observed in various tumors and contributes to immune evasion. High levels of PD-L1 have been observed in macrophages and been shown to play an important role in suppressing antitumor immunity.(46–48) Macrophage infiltration in the tumor microenvironment has been reported to promote tumor progression by impairing the immune responses of cytotoxic CD8+ T cells.(32) Several studies have also demonstrated that Kupffer cells(15) or activated monocytes/macrophages(16) in the HCC stroma express higher levels of PD-L1 than that in the surrounding nontumor liver tissues. Furthermore, blocking PD-L1 using a specific antibody improved T-cell immune response, suggesting that increased PD-L1 expression on macrophages helps HCC cells escape from cytotoxic T cells. However, it is unclear how PD-L1 expression is up-regulated in macrophages. In this study, we found that macrophages infiltrated in ER-stressed HCC tissues expressed high levels of PDL-1, and PD-L1 levels negatively correlated with overall survival. Finally, we provided evidence that ER stress promotes HCC cells to release miR-23a-3p-enriched exosomes, and these exosome-treated macrophages significantly up-regulate PD-L1 expression that reduced the proportion of CD8+ T cells and IL-2 production and increased T-cell apoptosis in a co-culture system. Collectively, our data suggest that exosomes released by ER-stressed HCCs induce immunosuppression through the up-regulation of PD-L1 on macrophages.
In summary, we found ER stress is activated in HCC tissues, and activation of ER stress contributes to CD68+PD-L1+ macrophage recruitment and poor prognosis. Moreover, ER-stressed HCC cells transmit exosomal-miR-23a-3p to macrophages and activate the PI3K-AKT pathway by inhibiting PTEN, thereafter increasing PD-L1 expression and inhibiting T-cell function. Our findings suggest that the transfer of exosomal-miR-23a-3p from ER-stressed HCC cells to macrophages plays an important role in tumor cell escape from immunologic cytotoxicity, and blockade of the HCC-macrophage communication may be a novel strategy to treat HCC progression.
Supplementary Material
Acknowledgments
This study was supported by the National Nature and Science Foundation of China (81872047 and 81572430 to G.S.; 81770588, 81522009, and 81372577 to H.W.; and 81602115 to F.W.) and the Anhui Provincial Natural Science Foundation (1608085QH195 to J.L.). B.G., Y.H., and D.F. were supported by the intramural program of the National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health (AA000368, AA000369 to B.G.).
Abbreviations:
- Akt
protein kinase B
- APC
antigen-presenting cell
- ATF6
activating transcription factor 6
- CD
cluster of differentiation
- Cum
cumulative
- DAPI, 4´
6-diamidino-2-phenylindole
- ER
endoplasmic reticulum
- Exo-con
exosomes derived from untreated HCC cells
- Exo-TM
exosomes derived from tunicamycin treated HCC cells
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GRP78
glucose-regulated protein 78
- HCC
hepatocellular carcinoma
- IRE1α
inositol-requiring enzyme 1α
- IL
interleukin
- Max
maximum
- MDSC
myeloid-derived suppressor cell
- MIP-1
macrophage inflammatory protein 1
- miRNA/miR
microRNA
- mRNA
messenger RNA
- p-AKT
phosphorylated protein kinase B
- PBS
phosphate-buffered saline
- PD-1
programmed death 1
- PD-L1
programmed death ligand 1
- PERK
protein kinase R (PKR)-like endoplasmic reticulum kinase
- PI3K
phosphoinositide 3-kinase
- PTEN
phosphatase and tensin homolog
- Q
quadrant
- qPCR
quantitative polymerase chain reaction
- TAM
tumor-associated macrophage
- TNF-α
tumor necrosis factor-α
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1).Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2).Mazzoccoli G, Tarquini R, Valoriani A, Oben J, Vinciguerra M, Marra F. Management strategies for hepatocellular carcinoma: old certainties and new realities. Clin Exp Med 2016;16:243–256. [DOI] [PubMed] [Google Scholar]
- 3).Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003;362:1907–1917. [DOI] [PubMed] [Google Scholar]
- 4).Li G, Liu D, Cooper TK, Kimchi ET, Qi X, Avella DM, et al. Successful chemoimmunotherapy against hepatocellular cancer in a novel murine model. J Hepatol 2017;66:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Lipson EJ, Drake CG. Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res 2011;17:6958–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Smit EF, van den Heuvel MM. PD-L1 in non-small-cell lung cancer: the third target for immunotherapy. Lancet 2016;387:1795–1796. [DOI] [PubMed] [Google Scholar]
- 7).Shlomai A, de Jong YP, Rice CM. Virus associated malignancies: the role of viral hepatitis in hepatocellular carcinoma. Semin Cancer Biol 2014;26:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Zhang HH, Mei MH, Fei R, Liu F, Wang JH, Liao WJ, et al. Regulatory T cells in chronic hepatitis B patients affect the immunopathogenesis of hepatocellular carcinoma by suppressing the anti-tumour immune responses. J Viral Hepat 2010;17 (Suppl. 1):34–43. [DOI] [PubMed] [Google Scholar]
- 9).Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007;46:48–57. [DOI] [PubMed] [Google Scholar]
- 10).Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014;147:1393–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol 2015;62:607–616. [DOI] [PubMed] [Google Scholar]
- 12).Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007;132:2328–2339. [DOI] [PubMed] [Google Scholar]
- 13).Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, et al. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother 2013;62:1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Budhu A, Forgues M, Ye QH, Jia HL, He P, Zanetti KA, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006;10:99–111. [DOI] [PubMed] [Google Scholar]
- 15).Wu K, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res 2009;69:8067–8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med 2009;206:1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell 2014;25:563–573. [DOI] [PubMed] [Google Scholar]
- 18).Rachidi S, Sun S, Wu BX, Jones E, Drake RR, Ogretmen B, ]et al. Endoplasmic reticulum heat shock protein gp96 maintains liver homeostasis and promotes hepatocellular carcinogenesis. J Hepatol 2015;62:879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Fan L, Sun G, Ma T, Zhong F, Lei Y, Li X, et al. Melatonin reverses tunicamycin-induced endoplasmic reticulum stress in human hepatocellular carcinoma cells and improves cytotoxic response to doxorubicin by increasing CHOP and decreasing survivin. J Pineal Res 2013;55:184–194. [DOI] [PubMed] [Google Scholar]
- 20).Zha L, Fan L, Sun G, Wang H, Ma T, Zhong F, et al. Melatonin sensitizes human hepatoma cells to endoplasmic reticulum stress-induced apoptosis. J Pineal Res 2012;52:322–331. [DOI] [PubMed] [Google Scholar]
- 21).Lee BR, Chang SY, Hong EH, Kwon BE, Kim HM, Kim YJ, et al. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget 2014;5:12331–12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017;168:692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci U S A 2011;108:6561–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Zanetti M Cell-extrinsic effects of the tumor unfolded protein response on myeloid cells and T cells. Ann N Y Acad Sci 2013;1284:6–11. [DOI] [PubMed] [Google Scholar]
- 25).Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008;454:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Hannafon BN, Ding WQ. Intercellular communication by exosome-derived microRNAs in cancer. Int J Mol Sci 2013;14:14240–14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007;9:654–659. [DOI] [PubMed] [Google Scholar]
- 28).Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014;25:501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Kogure T, Lin WL, Yan IK, Braconi C, Patel T. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology 2011;54:1237–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol 2016;16:469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002;8:793–800. [DOI] [PubMed] [Google Scholar]
- 32).Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010;141:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Lin ST, Huang Y, Zhang L, Heng MY, Ptacek LJ, Fu YH. MicroRNA-23a promotes myelination in the central nervous system. Proc Natl Acad Sci U S A 2013;110:17468–17473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Tan X, Wang S, Zhu L, Wu C, Yin B, Zhao J, et al. cAMP response element-binding protein promotes gliomagenesis by modulating the expression of oncogenic microRNA-23a. Proc Natl Acad Sci U S A 2012;109:15805–15810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Gurzov EN, Ortis F, Cunha DA, Gosset G, Li M, Cardozo AK, et al. Signaling by IL-1beta+IFN-gamma and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic beta-cell apoptosis. Cell Death Differ 2009;16:1539–1550. [DOI] [PubMed] [Google Scholar]
- 36).Penfornis P, Vallabhaneni KC, Whitt J, Pochampally R. Extracellular vesicles as carriers of microRNA, proteins and lipids in tumor microenvironment. Int J Cancer 2016;138:14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs bind to toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 2012;109:E2110–E2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Kanemoto S, Nitani R, Murakami T, Kaneko M, Asada R, Matsuhisa K, et al. Multivesicular body formation enhancement and exosome release during endoplasmic reticulum stress. Biochem Biophys Res Commun 2016;480:166–172. [DOI] [PubMed] [Google Scholar]
- 39).Cho YE, Mezey E, Hardwick JP, Salem N Jr., Clemens DL, Song BJ. Increased ethanol-inducible cytochrome P450–2E1 and cytochrome P450 isoforms in exosomes of alcohol-exposed rodents and patients with alcoholism through oxidative and endoplasmic reticulum stress. Hepatol Commun 2017;1:675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007;13:84–88. [DOI] [PubMed] [Google Scholar]
- 41).Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res 2014;2:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, et al. MiR-20b, −21, and −130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol 2014;75:348–353. [DOI] [PubMed] [Google Scholar]
- 43).Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res 2005;65:5278–5283. [DOI] [PubMed] [Google Scholar]
- 44).Chang LY, Lin YC, Chiang JM, Mahalingam J, Su SH, Huang CT, et al. Blockade of TNF-alpha signaling benefits cancer therapy by suppressing effector regulatory T cell expansion. Oncoimmunology 2015;4:e1040215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Lanton T, Shriki A, Nechemia-Arbely Y, Abramovitch R, Levkovitch O, Adar R, et al. Interleukin 6-dependent genomic instability heralds accelerated carcinogenesis following liver regeneration on a background of chronic hepatitis. Hepatology 2017;65:1600–1611. [DOI] [PubMed] [Google Scholar]
- 46).Staples KJ, Nicholas B, McKendry RT, Spalluto CM, Wallington JC, Bragg CW, et al. Viral infection of human lung macrophages increases PDL1 expression via IFNbeta. PLoS One 2015;10:e0121527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47).Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med 2016;8:328rv324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.